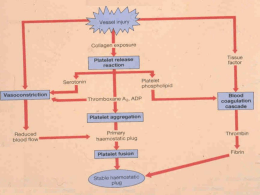
Monografia apresentada ao Programa de Residência Médica em Pediatria Hospital Regional da Asa Sul (HRAS)/ SES DF Alterações Congênitas das Plaquetas: Revisão de Literatura Residente: Dominique Bastos Sasaki Orientador: Jefferson Augusto Piemonte Pinheiro Brasília-DF Brasília, 28 de outubro de 2010 www.paulomargotto.com.br Introdução As alterações congênitas das plaquetas representam um grupo raro e heterogêneo que abrangem alterações tanto qualitativas quanto quantitativas. Possuem uma grande dificuldade diagnóstica e diferentes estratégias de tratamento. Objetivos Realizar revisão da literatura sobre as alterações congênitas das plaquetas visando atualizar as informações aos profissionais de saúde, a fim de melhorar o diagnóstico e manejo da trombocitopenia e trombocitopatia congênitas Materiais e Métodos Revisão da literatura nacional e internacional Bancos de dados: MEDLINE, HIGHWIRE, PUBMED, LILACS-BIREME E COCHRANE Artigos publicados no últimos 10 anos Língua inglesa e portuguesa Formação das Plaquetas Fragmentos citoplasmáticos anucleados presentes no sangue Formados na medula óssea a partir dos megacariócitos 70% estão na circulação e 30% no baço Vida média de 10 dias Formação das Plaquetas Possuem estrutura discóide complexa, com divisão de sua estrutura interna: FONTE: Castro HC, Ferreira BLA, Nagashima T, Schueler A, Rueff C. Plaquetas: ainda um alvo terapêutico. J Bras Patol Med Lab 2006; 5: 321-332 Fisiologia FONTE: Castro HC, Ferreira BLA, Nagashima T, Schueler A, Rueff C. Plaquetas: ainda um alvo terapêutico. J Bras Patol Med Lab 2006; 5: 321-332 ALTERAÇÕES QUALITATIVAS Síndrome de Hermansky-Pudlak Rara doença autossômica recessiva Incidência de 1:500.000 a 1:1.000.000 na população mundial Curiosamente mais prevalente na região norte de Porto Rico (1:1.800) Tríade ◦ Albinismo oculocutâneo ◦ Tendência a sangramento ◦ Complicações sistêmicas associadas ao acúmulo de lipofucsina Síndrome de Hermansky-Pudlak Falência renal, cardiomiopatia, fibrose pulmonar (complicação mais séria com 50% de morbidade), colite granulomatosa, neutropenia Contagem plaquetária dentro da normalidade Diagnóstico definitivo: ausência de grânulos densos na microscopia eletrônica Síndrome de Bernard-Soulier Doença autossômica recessiva ◦ Disfunção plaquetária ◦ Macrotrombocitopenia (“plaquetas gigantes”) ◦ Sangramento FONTE: Mhawech P, Saleem A. Inherited giant platelet disorders classification and literature review. American Society of Clinical Pathologists 2000; 113:176-190 Síndrome de Bernard-Soulier Ausência de glicoproteína Ib/IX/V no receptor de membrana plaquetário responsável pela ligação com o FVW (quantitativamente e qualitativamente normal) Incidência homozigótica1:1.000.000 e incidência heterozigótica 1:500 Heterozigotos são assintomáticos em sua maioria Diagnóstico: a citometria de fluxo para a quantificação de glicoproteínas plaquetárias é confirmatório Tratamento: transfusão plaquetária. Transplante de células tronco? Síndrome da Plaqueta Cinzenta Raro distúrbio da hemostasia primária Trombocitopenia Diminuição ou ausência de grânulos α Aumento do tamanho plaquetário Tendência a sangramento Inabilidade dos megacariócitos em envolver as enzimas secretórias sintetizadas endogenamente para os grânulos α maduros Síndrome da Plaqueta Cinzenta Defeito específico na linhagem megacariocítica e seletivo para os grânulos α Tempo de sangramento prolongado, de 10 a até mais de 30 minutos O esfregaço do sangue periférico mostra plaquetas grandes agranulares na coloração de Wright-Giemsa. Síndrome da Plaqueta Cinzenta Não sãoo FVW, fibrinogênio, Os grânulos α contém liberadosderivado na fator de crescimento de plaquetas injúria tecidual e β tromboglobulina Tendência ao sangramento Diagnóstico: a microscopia eletrônica mostra escassez de grânulos α e de vacúolos onde esses grânulos costumam estar localizados. A avaliação da medula óssea pode revelar mielofibrose. Síndrome de Chediak-Higashi Doença autossômica recessiva Comumente diagnosticada em torno de 5 anos de idade Achados: ◦ ◦ ◦ ◦ Albinismo oculocutâneo parcial Sangramento excessivo Infecções bacterianas recorrentes Fase acelerada ou sd. Hemofagocítica Síndrome de Chediak-Higashi Imunodeficiência causada por mutações em um gene regulador do tráfico lisossomal (LYST) ou gene CHS1 Diminuição do número de grânulos densos com número de plaquetas normal Achados neurológicos por infiltrados linfohistiocíticos do sistema nervoso Infecções recorrentes: predominantemente por S. aureus e Streptococcus β-hemolítico Síndrome de Chediak-Higashi A maioria dos pacientes que não se submetem a transplante de medula óssea morrem de sd. Linfoproliferativa Fase acelerada: acometimento difuso dos órgãos encontrada em 85% dos casos Diagnóstico: confirmado pela demonstração de grânulos peroxidase-positivos em diferentes linhas celulares Diagnóstico precoce: amniocentese com amostra de vilo coriônico, e por meio de biópsias de couro cabeludo Síndrome de Paris-Trousseau Síndrome autossômica dominante Achados: ◦ ◦ ◦ ◦ Trombocitopenia Grânulos α gigantes Dismegacariopoiese Sangramento geralmente leve Comprometimento na liberação do conteúdo do grânulo α nas células com grânulos gigantes Síndrome de Paris-Trousseau Cardiopatias, trigonocefalia, dismorfismos faciais, baixa estatura, retardo mental, infecções respiratórias, atraso no desenvolvimento e mal funcionamento de múltiplos órgãos Síndrome de Quebec Doença autossômica dominante Aumento da expressão megacariocítica Armazenamento de ativador de plasminogênio uroquinase-like (u-PA). O u-PA gera plasmina, gerando degradação do fibrinogênio plaquetário e de outras proteínas dos grânulos α importantes para a homeostase Síndrome de Quebec Contagem plaquetária normal ou reduzida Adequado armazenamento de proteínas plaquetárias Alta concentração de u-PA Hemorragia Proteólise precoce Síndrome de Quebec Diagnóstico: avaliação direta das proteínas plaquetárias pelo ELISA Diferentemente das outras doenças relacionadas aos grânulos α, os pacientes não tem o sangramento resolvido através da transfusão plaquetária, mas respondem bem a inibidores fibrinolíticos Trombastenia Glanzmann Defeito na Doença autossômica recessiva agregação plaquetária Deficiência de glicoproteína IIb/IIIa Número de plaquetas normal, assim como sua estrutura e meia-vida Trombastenia de Glanzmann Classificada de acordo com a quantidade e qualidade de glicoproteína presente (tipo I mais grave, com menos de 5% de glicoproteína normal presente) Diagnóstico: análise proteica por citometria de fluxo e Western Blot ou análise genética direta Tratamento: transfusão de plaquetas ou fator recombinante VIIa ALTERAÇÕES QUANTITATIVAS Desordens relacionadas ao MYH9 Grupo de macrotrombocitopenias autossômicas dominantes, com uma base genética comum Desordens relacionadas ao MYH9 Todas envolvem mutações no gene MYH9 O sangramento não é frequente e raramente leva ao risco de morte, sendo muitas vezes desproporcional a trombocitopenia Diagnóstico: quadro clínico, esfregaço periférico, identificação da mutação no gene MYH9 Síndrome de Wiskott-Aldrich Doença recessiva de imunodeficiência primária ligada ao X Causada por mutações no gene WASP (citoesqueleto de actina) Incidência estimada de 10:1.000.000 de nascidos vivos, sendo que na população européia é de 1:250.000 Síndrome de Wiskott-Aldrich - Risco de infecções bacterianas, virais e fúngicas Comprometimento das imunidades celular - Predisposição ao surgimento de eczema humoral - Doença proliferativa e Complicações auto-imunes ou inflamatórias: anemia hemolítica, neutropenia, artrite, vasculite da pele, vasculite cerebral, doença inflamatória intestinal, doença renal Sobrevida média de 14,5 anos Síndrome de Wiskott-Aldrich Se desenvolvimento de auto-imunidade, considerar o transplante de medula óssea Diagnóstico: clínica, contagem plaquetária inferior a 70x109/L, redução do volume da plaqueta (metade do normal), citometria de fluxo e anticorpos anti-WASP Tratamento: transfusão de plaquetas, transplante de células-tronco Trombocitopenias amegacariocíticas Trombocitopenias congênitas graves que se manifestam nos primeiros dias de vida Aspirado de medula óssea e biópsia mostram ausência ou redução significativa da produção megacariocítica Síndrome de trombocitopenia com ausência de rádio (TARS) Trombocitopenia amegacariocítica congênita (CAMT) TARS Doença autossômica recessiva Trombocitopenia: grave apenas no primeiro ano de vida, a partir de quando verifica-se o aumento progressivo do número de plaquetas Anomalias dos membros: ausência ou hipoplasia bilateral do rádio em 100% dos casos, podendo associar-se a hipoplasia do cúbito, defeitos das mãos e membros inferiores Achados ocasionais: tetralogia de Fallot, defeitos do septo atrial TARS TARS Prognóstico: depende da trombocitopenia nos primeiros meses de vida (alto risco de hemorragia gastrointestinal ou cerebral) Atraso do desenvolvimento em 7% dos casos (hemorragia intracraniana) Diagnóstico pré-natal: ecografia evidência aplasia bilateral do rádio e trombocitopenia estabelecida por cordocentese CAMT Síndrome autossômica recessiva Trombocitopenia grave que pode evoluir para pancitopenia devido a redução dos precursores eritróides e mielóides Cerca de 10 a 30% dos casos apresentam anomalias ortopédicas ou neurológicas Sangramento grave, hemorragia intracraniana, gastrointestinal e pulmonar Conclusão Tais alterações plaquetárias são raras e a classificação destes distúrbios é uma tentativa de promover a compreensão dos transtornos e fornecer uma análise abrangente sobre esse tópico Doenças adquiridas devem ser cuidadosamente excluídas do processo de investigação Como terapia, em caso de episódios de pequenos sangramentos, nenhum tratamento é necessário. No entanto, para episódios de hemorragias mais graves, a transfusão plaquetária é o tratamento de escolha. Obrigada!
Baixar