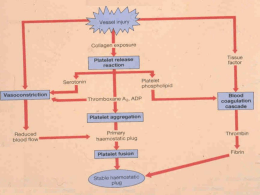
emergências oncológicas Manuseio de complicações hemorrágicas adquiridas no tratamento do paciente oncológico Divulgação Introdução Alexandre Mello de Azevedo * Médico hematologista do Centro de Tratamento Oncológico (CENTRON) Contato: omomom Daniel Tabak * Hematologista-Oncologista; diretor médico do Centro de Tratamento Oncológico (CENTRON); membro titular da Academia Nacional de Medicina Contato: [email protected] Simone Maradei Isabela Gonçalves A. Pereira Marcia Trindade Schramm Leonardo Javier Arcuri * Médicos hematologistas do Centro de Tratamento Oncológico (CENTRON) 18 abril/maio 2011 Onco& O sangramento é uma complicação frequente e potencialmente letal em pacientes com câncer. Pode estar diretamente relacionado à doença, ao tratamento antineoplásico ou, ainda, a fatores não relacionados à neoplasia em si. O termo hemostasia refere-se ao processo pelo qual o sangramento é controlado no local da lesão endotelial. É um mecanismo dinâmico, que inclui a ação do subendotélio, endotélio, plaquetas e proteínas plasmáticas. Didaticamente, é dividido em três fases: vascular, plaquetária e plasmática. Defeitos na hemostasia primária (plaquetas e fator de von Willebrand [FvW]) tipicamente resultam em sangramento mucocutâneo. Por outro lado, a sintomatologia de defeitos na cascata de coagulação (hemostasia secundária) é bem variável, podendo haver acometimento de tecidos profundos, levando a hematomas, sangramento retroperitoenal ou até no sistema nervoso central (SNC). Não importa quão grave seja a situação, devese sempre tentar obter uma história completa, que inclua sintomas associados, tempo de evolução e tratamentos/medicações recentemente realizados. Abordaremos neste texto o diagnóstico e o manejo de sangramentos em pacientes com câncer. Eventualmente, deparamo-nos com alterações em testes laboratorias de coagulação sem repercussões clínicas, e esse tema também será abordado. Avaliação laboratorial O sucesso da terapia baseia-se no diagnóstico correto. O primeiro passo na avaliação é obter um hemograma com contagem de plaquetas, tempo de protrombina (TP e INR), tempo de tromboplastina parcial ativada (TTPa) e fibrinogênio. A trombocitopenia isolada e grave (< 20.000/µl) normalmente é secundária a púrpura trombocitopênica idiopática (PTI), trombocitopenia induzida por drogas (TID), microangiopatia trombótica ou púrpura transfusional. Deve-se lembrar que a trombocitopenia deve ser sempre confirmada por hematoscopia, uma vez que o satelitismo (aderência a leucócitos) e agregados plaquetários podem causar trombocitopenia espúria, sem qualquer repercussão clínica. É importante notar que a trombocitose intensa (> 1.000.000/µl) também pode causar sangramento, principalmente por consumo de FvW pelo número aumentado de plaquetas. O TP e o TTPa avaliam a via extrínseca e a via intrínseca da coagulação, respectivamente. Essa divisão é de importância apenas diagnóstica e laboratorial. Alterações do fibrinogênio, protrombina, fatores V e X causam aumento tanto do TP quanto do TTPa. Alterações do fator VII aumentam apenas o TP, enquanto alterações do FvW, VIII, IX, XI e XII causam aumento do TTPa. As alterações dos fatores de coagulação identificadas pelos testes TP e TTPa podem ser causadas por consumo ou diminuição de produção, ou por produção de autoanticorpos (inibidores). Diferenciam-se esses dois grupos pelo “teste da mistura”, misturando-se plasma normal com o do paciente em iguais proporções (1:1). Quando a mistura não corrige o TP ou o TTPa, isso indica a presença de algum inibidor. Quando a mistura corrige o defeito, o mais provável é que haja deficiência quantitativa ou qualitativa de algum fator. Há, portanto, três padrões de alteração do TP e do TTPa, que indicam como deve prosseguir a investigação. Esses três padrões estão sumarizados na Tabela 1. Ensaios de TP e TTPa mais modernos não são alterados por anticoagulante lúpico. Se houver suspeita de anticorpo inibidor (o mais comum é contra o FVIII), a atividade de cada fator suspeito deverá ser testada separadamente. O padrão do anticorpo inibidor do FVIII, por exemplo, é uma baixa atividade do FVIII que não se corrige pela mistura 1:1 com plasma normal. Uma vez identificado, o anticorpo inibidor deve ser quantificado em sua atividade. A dosagem de fibrinogênio tem mais importância terapêutica do que diagnóstica. Níveis de fibrinogênio < 100 mg/dl devem ser tratados. O plasma fresco congelado (PFC) tem pouco fibrinogênio, e a reposição deve ser feita com crioprecipitado, transfundindo-se 1 U a cada 10 kg do paciente. Transfusão maciça O sangramento agudo em pacientes com câncer pode levar à transfusão de grandes quantidades de sangue em pouco tempo. A transfusão maciça leva à diluição de fatores de coagulação e plaquetas. O diagnóstico é feito pela história, associada a TP e TTPa aumentados. A transfusão de mais de 10 U de hemácias é um fator de risco para coagulopatia por transfusão. Deve-se repor PFC (15 ml/kg). Se houver CID associada, deve-se manter as plaquetas > 50.000/µl. Se o fibrinogênio for < 100 mg/dl, deve-se repor crioprecipitado (1 U/10 kg de peso). Os testes de coagulação devem ser repetidos após a reposição para guiar a terapia adicional. O objetivo é manter INR < 2, TTPa < 1,5 (relação paciente/ controle) e fibrinogênio > 100 mg/dl. Complicações da transfusão maciça A complicação mais comum é a hipotermia, que prejudica a função plaquetária e a eficiência das reações de coagulação, além de aumentar a fibrinólise. Alterações eletrolíticas são raras, e não se deve atribuir a acidose à transfusão maciça. Não se deve repor empiricamente cálcio (o citrato presente na bolsa de sangue é rapidamente metabolizado) nem bicarbonato. Sangramentos por problemas em fatores da coagulação Doença de von Willebrand adquirida A doença de von Willebrand (DvW) adquirida pode ocorrer nos linfomas, nas síndromes mieloproliferativas, no mieloma múltiplo (MM) e em outras gamopatias monoclonais. A fisiopatologia pode envolver um de vários mecanismos possíveis: anticorpos contra o FvW, proteólise do FvW, ligação anormal do FvW a células tumorais ou diminuição da síntese do FvW. Mais comumente, manifesta-se por sangramento difuso em feridas cirúrgicas, epistaxe ou sangramento do tubo digestivo em um paciente sem história pessoal ou familiar de sangramento. Os pacientes com DvW adquirida podem apresentar fenótipos diferentes, incluindo o tipo 1 (distúrbio quantitativo) ou o tipo 2 (distúrbio qualitativo). A resposta ao tratamento nos pacientes com DvW adquirida é variável. A desmopressina (DDAVP) é efetiva em muitos pacientes com DvW adquirida dos tipos 1 e 2; no entanto, uma vez que o mecanismo de destruição da molécula é mediado por anticorpos, a magnitude e a duração do efeito são frequentemente menores que o esperado. Em pacientes que apresentam sangramento ativo, indicam-se idealmente doses altas de concentrado de FvW, nem sempre disponíveis. Na indisponibilidade do produto purificado, pode-se recorrer à transfusão de crioprecipitado (80 a 100 U de FVIII por bolsa). O fator VIIa (FVIIa) pode ser indicado em pacientes com sangramentos graves (risco de vida ou lesões irreversíveis) e/ou portadores de inibidores fortes, nos quais os concentrados de FvW podem não ser eficazes. Inibidores adquiridos do fator VIII Em pacientes idosos com câncer, a deficiência de FVIII causada por autoanticorpos é a mais frequente complicação por deficiência adquirida de um fator da coagulação. As neoplasias linfoproliferativas são as mais frequentemente associadas. Diferentemente da hemofilia clássica, esses pacientes costumam apresentar equimoses extensas, além de possíveis hematomas em grupos musculares e tecidos moles. O diagnóstico é feito pelo prolongamento do TTPa, pelo teste positivo para a presença de um inibidor (teste da mistura) e pela dosagem subnormal de FVIII. O concentrado de FVIIa recombinante é o tratamento de escolha para pacientes com sangramentos graves e risco de vida. A dose é de 90 µg/kg, repetida a cada 2 ou 3 horas até a cessação do sangramento. Na indisponibilidade do FVIIa, pode-se recorrer a altas doses do Tabela 1: Diagnósticos diferenciais das alterações em TP e/ou TTPa TP aumentado 1. Deficiência de vitamina K 2. Terapia com warfarina 3. Doença hepática 4. Deficiência/Inibidor de fator VII TTPa aumentado 1. Heparina 2. Deficiência/Inibidor de FvW, VIII ou IX (risco alto de sangramento) 3. Deficiência/Inibidor de FXI ou XII (risco baixo ou ausente de sangramento) 4. Anticoagulante lúpico (risco de sangramento ausente) TP e TTPa aumentados 1. Heparina ou warfarina em altas doses 2. Coagulopatia por transfusão maciça (sem reposição adequada de plasma) 3. Deficiência de vitamina K 4. Doença hepática grave 5. Deficiência/Inibidor de FII (anticoagulante lúpico), V (estreptomicina) ou X (amiloidose) 6. Coagulação intravascular disseminada (CID) 7. Hipofibrinogenemia 8. Paraproteinemia 9. Leucemia promielocítica aguda Onco& abril/maio 2011 19 próprio FVIII recombinante e/ou ao tratamento imunossupressor, visando à redução da produção do inibidor. Coagulação intravascular disseminada (CID) A CID é a manifestação clínica da ativação descontrolada da trombina. A ativação da trombina leva a: (1) conversão de fibrinogênio em fibrina, (2) ativação e consumo de plaquetas, (3) ativação do FV e do FVIII, (4) ativação da proteína C (e a consequente degradação dos fatores Va e VIIIa), (5) ativação do endotélio vascular, e (6) fibrinólise. Descrevem-se quatro padrões clínicos na CID: 1. Forma assintomática ou “CID crônica” Pode haver evidência laboratorial de CID, mas sem sangramentos ou tromboses significativas. É uma situação em geral transitória, frequentemente encontrada em pacientes com tumores sólidos (ex: adenocarcinomas) ou hematológicos (ex: leucemias mielocíticas). Geralmente, caracteriza-se por um estado protrombótico compensado. A progressão da doença de base pode, no entanto, sobrepujar os mecanismos anticoagulantes naturais e precipitar os sintomas característicos. 2. Forma predominantemente hemorrágica Os sangramentos costumam ter causa multifatorial: depleção de fatores procoagulantes, disfunção plaquetária, trombocitopenia e fibrinólise excessiva. Os sangramentos são geralmente difusos, em sítios múltiplos. 3. Forma predominantemente trombótica Apesar da ativação generalizada dos processos da coagulação, a trombose em vasos grandes é pouco frequente em pacientes com CID aguda, sendo observada principalmente na microcirculação, onde determina isquemia e pode precipitar a síndrome de disfunção de múltiplos órgãos (ex: fígado, rins). Nos pacientes com câncer, a trombose pode ser o principal fator complicador. A trombose é mais frequentemente venosa, mas há relatos de tromboses arteriais e de endocardite trombótica não bacteriana. 4. Purpura fulminans (PF) É a associação entre CID e equimoses e necrose simétrica da pele das extremidades. Dois modos de apresentação são descritos: A PF primária ocorre caracteristicamente após infecções virais (ex: varicela) em hospedeiros imunodeficientes. Nesses casos, a PF começa com eritema e dor em uma extremidade, progredindo rapidamente para necrose isquêmica e escurecimento da pele. A PF secundária é mais frequentemente associada à meningococcemia, mas pode ocorrer em qualquer tipo de infecção grave. Pode ocorrer em pacientes com câncer ou esplenectomizados com síndrome séptica. Em geral, o quadro clínico é de sépsis, e as lesões cutâneas frequentemente envolvem as extremidades, podendo levar à gangrena e à amputação. O tratamento deve ser primariamente dirigido à causa subjacente, 20 abril/maio 2011 Onco& o que nem sempre é possível. Inicialmente, medidas de ressuscitação e reposição volêmica devem ser observadas, uma vez que a má perfusão do fígado é um dos principais obstáculos à correção das anormalidades hemostáticas da CID. A reposição de plaquetas e fatores da coagulação (ex: fibrinogênio) através da transfusão de produtos como o crioprecipitado, o PFC e os concentrados de plaquetas não deve ser feita com o único objetivo de corrigir as anormalidades laboratoriais encontradas, mas sim para o controle de sangramentos clinicamente relevantes ou o preparo do paciente para a realização de procedimentos invasivos. O uso de heparina permanece polêmico, e deve se restringir aos pacientes que demonstram sinais claros de oclusão circulatória por deposição excessiva de fibrina (ex: insuficiência renal, isquemia de extremidades) sem evidência de hemorragia significativa concomitante, situação esta que é mais comum na CID crônica. É bom lembrar que a heparina exerce seu efeito pela potencialização da ação da antitrombina III (ATIII), e que esta última pode estar diminuída. Já existem concentrados de ATIII recombinante para reposição. A monitorização da anticoagulação pelo TTPa é pouco confiável na CID descontrolada, sendo mais indicados os níveis séricos de heparina. Em alguns pacientes sépticos com CID, a reposição com concentrado de proteína-C ativada recombinante tem mostrado bons resultados preliminares. Os antifibrinolíticos, como o ácido aminocaproico ou tranexâmico, podem ser utilizados nos casos que não respondem às medidas iniciais mas que trazem o risco de complicações trombóticas. Problemas de função e número de plaquetas Púrpura trombocitopênica idiopática (PTI) A PTI afeta 1:20.000 indivíduos, sendo mais comum em mulheres jovens. Pode estar associada a neoplasias, principalmente hematológicas, como a leucemia linfoide aguda (LLA) e linfomas. A fisiopatologia consiste na formação de autoanticorpos dirigidos contra glicoproteínas da membrana plaquetária, principalmente contra os complexos GPIIb-IIIa e GPIb-IX. A PTI tem chances de ocorrer em qualquer momento do curso de uma neoplasia, inclusive antecedendo o diagnóstico de uma eventual recidiva do câncer em pacientes que se encontram em remissão. De um modo geral, os pacientes são assintomáticos e a trombocitopenia pode ser um achado laboratorial. Os pacientes toleram bem contagens plaquetárias baixas, existindo um risco maior de sangramento com plaquetas abaixo de 5.000/µl. Não existe um teste laboratorial específico; é um diagnóstico de exclusão. O tratamento inicial consiste em pulsoterapia com dexametasona 40 mg/d durante 4 dias. Nos pacientes com trombocitopenia severa (< 10.000/µl) ou sangramento ativo, um ou mais tratamentos devem ser instituídos na tentativa de induzir uma resposta mais rápida. Tanto a imunoglobulina intravenosa (IgIV) na dose de 2 g/kg divididos em dois dias quanto o anticorpo anti-D na dose de 75 µg/kg em dose única podem induzir resposta em mais de 80% dos casos em 24 a 48 horas. Trombocitopenia induzida por droga (TID) Os pacientes com TID apresentam trombocitopenia de 1 a 3 semanas após o uso da medicação causadora. Vários medicamentos estão implicados no desenvolvimento da TID (Tabela 2). A terapia inicial consiste em suspender a medicação suspeita. Na presença de múltiplas medicações, deve-se suspender a que apresentar maior associação. O tratamento consiste em IgIV em altas doses (0,4 g/kg/d) durante 5 dias. O número de plaquetas costuma aumentar em 3 a 5 dias. Outra opção é a plasmaférese, porém com resultados inferiores. A transfusão de plaquetas, por ser pouco eficaz, deve ser reservada apenas para as situações de sangramento grave e potencialmente fatal. Devem ser usadas plaquetas negativas para o antígeno HPA-1a. Púrpura pós-transfusional (PPT) É uma condição clínica grave que se caracteriza pela queda repentina do número de plaquetas, geralmente até níveis < 10.000/µl, e púrpura, 7 a 14 dias (em média, 9 dias) após a transfusão de componentes sanguíneos contendo plaquetas. Acomete principalmente mulheres multíparas, porém também é descrita em outras populações, como pacientes politransfundidos. É causada por aloanticorpos do receptor contra antígenos plaquetários do doador, ausentes no receptor. Essa destruição plaquetária ocorre, na maioria das vezes, pela presença de anticorpos contra o antígeno plaquetário HPA-1a, que está presente em 98% dos indivíduos, ocasionando uma destruição tanto das plaquetas infundidas quanto das plaquetas do próprio paciente. O quadro clínico é autolimitado e se resolve geralmente em três semanas. Porém, de 10% a 15% dos pacientes evoluem para óbito por sangramento no SNC. Refratariedade às plaquetas A refratariedade à transfusão de plaquetas caracterizase por um incremento plaquetário inadequado após a transfusão de concentrado de plaquetas. Esse mau aproveitamento se deve a inúmeras causas não imunes, como febre, infecção e grande esplenomegalia, bem como a fatores imunes, como a aloimunização contra antígenos do sistema HLA de classe I. Outras causas imunes importantes são: aloimunização contra antígenos plaquetários específicos e o uso de plaquetas ABO-incompatíveis. O diagnóstico é simples e consiste na ausência de incremento da contagem de plaquetas após três transfusões no período de duas semanas. O cálculo do incremento corrigido da contagem (ICC) após transfusão pode ser feito com o emprego de fórmula específica: ICC = IP x SC/dose (x 1011), no qual IP = incremento plaquetário desejado; SC = superfície corporal (m2). Se o ICC for menor que 7,5 a 10 x 109 em uma amostra colhida de 10 minutos a 1 hora após a transfusão, ou se o ICC for menor que 4,5 a 5 x 109 em uma amostra colhida 24 horas após a transfusão, pode-se definir como refratariedade à transfusão. Deve-se realizar, sempre que possível, a contagem plaquetária pré e pós-transfusional em pacientes politransfundidos. Uma vez constatada a refratariedade, devem ser utilizadas preferencialmente plaquetas ABO-idênticas. Em pacientes que estejam recebendo concomitantemente anfotericina B, deve-se fazer um intervalo de 2 horas entre a infusão do medicamento e a transfusão das plaquetas. Caso essas medidas não sejam eficazes, deve-se optar por transfundir apenas em caso de sangramento. Tabela 2: Drogas que comumente induzem trombocitopenia Drogas antiarrítmicas Procainamida, quinidina Agentes anti-GPIIb/IIIa Abciximab, eptifibatide, tirofiban Agentes antimicrobianos Anfotericina B, rifampicina, vancomicina, trimetoprim-sulfametoxazol Bloqueadores H2 Cimetidina, ranitidina Outras Acetaminofeno, amrinona, sais de ouro, heparina, quinine, efalizumab, carbamazepina, hidroclorotiazida, anti-inflamatórios não esteroides “O sangramento é uma complicação frequente e potencialmente letal em pacientes com câncer. Pode estar diretamente relacionado à doença, ao tratamento antineoplásico ou, ainda, a fatores não relacionados à neoplasia em si.” Trombocitopenia induzida por heparina (TIH) A TIH é uma síndrome imuno-hematológica mediada por um anticorpo que ocasiona ativação plaquetária na presença de heparina, induzindo à Onco& abril/maio 2011 21 agregação plaquetária e, consequentemente, a eventos trombóticos. A frequência de TIH em pacientes que recebem heparina não fracionada é maior quando comparada aos pacientes que recebem heparina de baixo peso molecular (HBPM). Os pacientes que apresentam uma queda de 30% a 50% na contagem de plaquetas durante o tratamento com heparina, mesmo que não apresentem trombocitopenia, também têm um risco aumentado de desenvolver eventos trombóticos. Há dois tipos distintos de TIH: tipo I e tipo II. O tipo I é a forma menos severa e mais frequente. Caracteriza-se por trombocitopenia leve, quase sempre > 100.000/µl, que se inicia precocemente após o uso da heparina. Entretanto, a TIH tipo II caracteriza-se por trombocitopenia mais severa, que geralmente surge de 4 a 14 dias após o início da administração da heparina. O diagnóstico clínico deve ser confirmado através de testes laboratoriais capazes de detectar anticorpos heparina-dependentes ou antígenos heparina-fator 4 plaquetário. Entre as medidas terapêuticas, a mais importante é a suspensão da droga envolvida. Devido à participação da trombina na patogênese da TIH, o tratamento primário deve incluir uma droga que reduza a geração de trombina, ou seja, inibidores de trombina. O argatroban é um inibidor sintético da trombina, de metabolização hepática. Sua atividade farmacológica faz com que ele atinja uma rápida eficácia terapêutica antitrombótica, com mínimo risco de sangramento e rápida restauração da hemostasia ao normal no momento da suspensão. A hirudina, mais potente, liga-se à trombina e forma um complexo não covalente irreversível, e com isso inibe todas as funções proteolíticas da trombina. Entretanto, a metabolização é renal, devendo a dose ser corrigida nos pacientes com função renal comprometida. Púrpura trombocitopênica trombótica (PTT) A PTT é uma microangiopatia trombótica caracterizada pela oclusão difusa de arteríolas terminais e de capilares por trombos ricos em plaquetas e em FvW. Na PTT adquirida, muitos pacientes não apresentam os cinco sinais e sintomas clássicos da doença, que são: trombocitopenia, anemia hemolítica microangiopática (AHMA), alterações neurológicas, comprometimento renal e febre. Porém, estão sempre presentes a AHMA, a trombocitopenia e as alterações neurológicas. Os níveis de lactato desidrogenase (LDH) estão sempre aumentados, constituindo um importante fator prognóstico na PTT. A mortalidade entre os pacientes não tratados é alta, em torno de 95% a 100%. Atualmente, a plasmaférese constitui a base do tratamento, podendo reduzir a mortalidade para menos de 20%. Por isso, é importante realizá-la o mais precocemente possível, tão logo se estabeleça o diagnóstico. Ela deve ser realizada diariamente, com trocas de 1 a 1,5 vez o volume de plasma do paciente, utilizando PFC como fluido de reposição. Não se sabe ao certo o número ideal de sessões, porém ela deve ser mantida até que se estabeleça uma remissão estável, que consiste na normalização do quadro neurológico, da contagem de plaque- 22 abril/maio 2011 Onco& tas e do nível de LDH, e no aumento da hemoglobina por pelo menos dois dias consecutivos. Síndrome hemolítico-urêmica (SHU) A SHU é uma microangiopatia trombótica caracterizada por anemia hemolítica e trombocitopenia, mas com predomínio de envolvimento da microcirculação renal. Ela pode estar associada ao uso de medicações imunossupressoras como a ciclosporina e o tacrolimo, bem como a agentes antineoplásicos como a mitomicina-C, a carboplatina e a gemcitabina. Além disso, a SHU pode ser uma complicação clínica secundária ao transplante alogeneico de medula óssea, com uma incidência em torno de 15%, podendo ocorrer também no contexto do transplante autólogo (5%). Em transplantados, as infecções pelo citomegalovírus também podem causar a SHU. O tratamento é incerto. Deve-se tratar ou remover, se possível, o fator desencadeante. Nesses casos, a plasmaférese não parece eficaz e o prognóstico é sombrio. Distúrbios hemorrágicos adquiridos associados a neoplasias hematológicas Leucemia promielocítica aguda (LPA) A LPA caracteriza-se pela presença em número aumentado de promielócitos anormais. Pode cursar com grave coagulopatia e deve ser encarada como uma emergência médica, requerendo uma série de medidas de suporte que devem ser iniciadas rapidamente e de forma simultânea. Uma fração significativa desses pacientes desenvolve hemorragia fatal durante a avaliação diagnóstica, antes ou durante os primeiros dias do tratamento de indução. A fisiopatologia dessa coagulopatia é complexa, e os defeitos hemostáticos são múltiplos. O mecanismo patogênico mais convincente aponta para propriedades da célula leucêmica, que por meio da liberação de uma série de mediadores ativa a coagulação através de três mecanismos principais: CID, fibrinólise e liberação de enzimas procoagulantes. Esse processo é intensificado pela trombocitopenia e rápida liberação celular de produtos tumorais induzidos pela quimioterapia. A terapêutica apropriada para a LPA consiste no tratamento simultâneo da coagulopatia e da leucemia. O tratamento de escolha atualmente consiste no uso do ácido transretinoico (ATRA) em combinação com quimioterapia à base de antracíclicos. O ATRA atua beneficamente sobre a coagulopatia de forma precoce, e deve ser iniciado nos casos suspeitos, mesmo antes da comprovação citogenética e/ou molecular da LPA. O tratamento de suporte consiste em transfusão vigorosa de PFC, fibrinogênio e/ou crioprecipitado e plaquetas. Deve-se almejar um fibrinogênio > 100 a 150 mg/dl e plaquetas > 30 a 50 x 109/L. A terapia de reposição deve continuar até o desaparecimento total de sinais clínicos ou laboratoriais de coagulopatia, e deve ser intensificada em pacientes que têm fatores de risco adicionais (idade avançada, hiperleucocitose, creatinina sérica aumentada, sangramento ativo). A melhora nos níveis de fibrinogênio é um bom marcador de resposta ao tratamento. O papel do FVIIa recombinante ou do complexo protrombínico para o tratamento de hemorragias graves na LPA permanece incerto, e recomenda-se que o uso de agentes procoagulantes nesse contexto deva se restringir ainda a estudos clínicos, tendo em vista o risco de complicações trombóticas. Deve-se ainda evitar cateterização de acesso venoso central, punção lombar ou outros procedimentos invasivos antes e durante a terapia de indução, enquanto a coagulopatia estiver presente, clinica e/ou laboratorialmente. Outras leucemias e síndrome mielodisplásica (SMD) A trombocitopenia é a causa mais comum de sangramento relacionado a essas condições. Entretanto, nos pacientes com contagens de plaquetas normais ou elevadas, complicações hemorrágicas podem estar associadas a disfunção plaquetária adquirida ou até mesmo a morfologia alterada dos megacariócitos. Defeitos adquiridos de plaquetas relacionados a manifestações hemorrágicas são mais comuns nas leucemias mieloides agudas (LMA), mas têm sido descritos, também, em leucemias linfoblásticas e mielomonoblásticas, tricoleucemia e síndromes mielodisplásicas (SMD). Outros fatores de risco, como hipertensão, anormalidades intravasculares, sépsis, CID, alterações dos fatores de coagulação e hiperleucocitose podem contribuir para hemorragias. A infiltração hepática por células leucêmicas pode comprometer a produção de fatores de coagulação. A coagulopatia mais comumente encontrada em LLA está relacionada ao uso da L-asparaginase, que diminui a síntese hepática de certas proteínas, alterando a produção de fatores da coagulação. Felizmente, apesar dos baixos níveis de fibrinogênio nesses casos, o sangramento é raro. Os sintomas dependem da gravidade da tendência hemorrágica e de sua localização, e consistem em epistaxe, hematúria, sangramento gastrintestinal, petéquias e até mesmo sangramento intracraniano, cursando, por exemplo, com cefaleia e turvação visual. A correção da causa subjacente, quando possível, é o melhor tratamento. Transfusão profilática de plaquetas tem sido universalmente aplicada como terapia de suporte em pacientes portadores de leucemias agudas e está indicada nos pacientes com plaquetas < 10.000/µl, sistematicamente, ou naqueles com > 10.000/µl e sangramento ativo. O tratamento para a coagulopatia consiste na infusão de PFC, complexo protrombínico, ATIII e concentrado de fibrinogênio, quando indicados. O ácido tranexâmico e similares podem ser considerados em estados hiperfibrinolíticos, sendo contraindicados em sangramentos de trato urinário. Medidas locais como a infiltração de vasoconstritores, a embolização arterial ou até mesmo a intervenção cirúrgica podem ser consideradas. Radioterapia pode ser usada com efeito hemostático em alguns casos de sangramento genital ou pulmonar. Na SMD, múltiplos defeitos funcionais são encontrados nas plaquetas. Isso inclui redução na agregação plaquetária e diminuição dos estoques de plaquetas, do FvW e do fibrinogênio. Nesses pacientes, as hemorragias podem acontecer mesmo na vigência de plaquetas > 50.000/µl e suporte transfusional. Síndromes mieloproliferativas (SMP) Tendência a sangramento e defeitos qualitativos de plaquetas são comumente encontrados nas SMP. Essas anormalidades refletem características do clone leucêmico, assim como alterações adquiridas nas plaquetas circulantes. Descrevem-se a diminuição do receptor alfa-2 adrenérgico plaquetário e da produção de tromboxano-A2 e anormalidades da expressão dos complexos GPIIb-IIIa, GPIb e GPIa-IIa. Anormalidades adquiridas do FvW plasmático têm sido documentadas em pacientes com SMP e elevadas contagens plaquetárias, e estão relacionadas a hemorragias. O FvW plasmático (particularmente os grandes multímeros) está diminuído, tendendo a se normalizar após o tratamento citorredutor. “Não importa quão grave seja a situação, deve-se sempre tentar obter uma história completa, que inclua sintomas associados, tempo de evolução e tratamentos/medicações recentemente realizados.” Disproteinemias A disproteinemia consiste na produção anormal de imunoglobulinas, que podem interferir nas várias fases da coagulação e acarretar hemorragias. Interações fisiopatológicas entre paraproteínas e fatores de coagulação, plaquetas e vasos sanguíneos podem produzir anormalidades hemostáticas. Coagulopatias têm sido descritas em pacientes com amiloidose primária (AL), macroglobulinemia de Waldenström (MW), mieloma múltiplo (MM) e gamopatia monoclonal de significado indeterminado (GMSI). Piora Onco& abril/maio 2011 23 na função plaquetária, demonstrada por aumento do tempo de sangramento ou alterações da curva de agregação plaquetária, tem sido associada a hemorragias clinicamente significativas relacionadas a altos níveis de imunoglobulina, principalmente de IgM. O mecanismo fisiopatológico seria a infiltração das plaquetas por paraproteínas. A correção dessas alterações pela plasmaférese sugere que também a hiperviscosidade esteja relacionada. A DvW adquirida pode complicar o curso de neoplasias, particularmente os linfomas. A deficiência do FX, isolada ou não, é a coagulopatia mais comumente descrita na amiloidose, e o grau de deficiência do FX não se correlaciona com a gravidade do sangramento. A reposição do FX com PFC ou complexo protrombínico corrige essa deficiência adquirida. Opções adicionais incluem esplenectomia e FVIIa. A resposta à quimioterapia é geralmente insatisfatória. Complicações do tratamento antitrombótico nas neoplasias A principal complicação do tratamento antitrombótico são as hemorragias e, menos frequentemente (< 1%), a necrose de pele ou de tecidos. Os sinais, os sintomas e o grau de severidade variam de acordo com a localização, o grau e a extensão do sangramento. Além disso, reações de hipersensibilidade também podem ocorrer. O risco de hemorragia está relacionado com a intensidade e a duração da terapia e pode resultar em morte ou deficiência permanente. A necrose está associada a trombose local, e usualmente surge cinco dias após o início do tratamento. A amputação do órgão acometido e o debridamento fazem parte do tratamento. O tratamento antitrombótico também pode levar à liberação de placas ateromatosas e desencadear a “síndrome dos dedos roxos”. Outros fatores, como viagens, mudanças na dieta, fatores ambientais e físicos, doenças associadas e uso concomitante de medicações, podem influenciar na resposta ao tratamento e aumentar os riscos de complicações. Antagonistas da vitamina K Os antagonistas da vitamina K (warfarina, coumadin) inibem os fatores de coagulação dependentes da vitamina K (II, VII, IX e X) e as proteínas C e S, anticoagulantes naturais. Eles não têm efeito direto em um trombo já estabelecido e também não revertem o dano tecidual isquêmico. Entretanto, uma vez que o trombo tenha se instalado, o objetivo é prevenir a sua extensão e as complicações tromboembólicas secundárias que podem resultar em sequelas fatais. Os principais determinantes do risco de sangramento são a intensidade do efeito anticoagulante, as características do paciente (idosos e comorbidades associadas), o uso concomitante de drogas que interferem na hemostasia (aspirina, acetaminofeno e anti-inflamatórios não esteroidais) e a duração do tratamento. A intensidade do efeito anticoagulante é o fator de risco mais importante para a hemorragia intracraniana, independentemente da indicação. O risco dobra a cada 1,0 ponto a mais no INR. A medicação deve ser descontinuada e a vi- 24 abril/maio 2011 Onco& tamina K oral ou intravenosa administrada, dependendo da intensidade do sangramento, pois oferece vantagens sobre a vitamina K subcutânea e sobre o plasma. O INR começa a diminuir somente de 12 a 36 horas após a suspensão do anticoagulante. Para os pacientes sem sangramentos e com INR alto mas < 5, a vitamina K pode ser omitida ou administrada com dose reduzida. Para os pacientes com INR entre 5 e 10, deve-se administrar de 1 a 2,5 mg de vitamina K por via oral ou, dependendo da urgência, por via intravenosa, juntamente com o plasma. Quando o INR for maior que 10, a dose deve ser de 2,5 a 5 mg. Nos pacientes com risco de hemorragia intracraniana, deve-se administrar 10 mg de vitamina K intravenosa, e também considerar o plasma e o complexo protrombínico para uma correção mais rápida. Agentes antiagregantes plaquetários O ácido acetilsalicílico (AAS) inibe a via da ciclo-oxigenase através da acetilação de aminoácidos. As principais complicações decorrentes do seu uso incluem sangramentos, reações alérgicas (broncoespasmo), úlceras gástricas, constipação e insuficiência renal. O risco de acidente vascular cerebral hemorrágico (AVCh) é de aproximadamente 1 em 2.500 pacientes/ano. As complicações podem ser resolvidas ou diminuídas com a redução ou suspensão da dose, a associação de protetores gástricos, a troca por outro agente antitrombótico ou até mesmo com a transfusão de plaquetas ou com a desmopressina, em casos emergenciais. Há pouca informação sobre o manuseio das complicações hemorrágicas que envolvem outros agentes, como ticlopidina, clopidogrel, dipiridamol, prasugrel ou abciximab. No entanto, a transfusão de plaquetas também está indicada nesses casos. Alguns estudos in vitro sugerem que a adição de crioprecipitado pode ajudar a restaurar a função plaquetária em pacientes que usam o tirofiban, o eptifibatide ou outros agentes antiagregantes que inibem os receptores plaquetários para o fibrinogênio. Heparina A heparina é utilizada para a profilaxia ou o tratamento da trombose. Ela aumenta a velocidade da ligação entre antitrombina e trombina e age como catalisador na reação de inativação da trombina. Inibe a coagulação através de alterações na função plaquetária e na permeabilidade capilar e tem o potencial de causar sangramento em menos de 3% dos pacientes, que pode ser revertido com o uso de sulfato de protamina (na maioria das situações, desnecessário, pois a heparina padrão tem meia-vida curta, de 30 a 60 minutos). A HBPM tem meia-vida mais longa e requer o uso de protamina. A dose varia de 0,5 a 1 mg para cada 100 U de heparina administrada. Ela não reverte completamente o efeito da HBPM, mas pode neutralizar o efeito antitrombótico. A heparina também pode induzir trombocitopenia, mas raramente é uma causa importante de sangramento. Ocorre em 1% a 3% dos pacientes com trombose venosa profunda que recebem heparina não fracionada e em menos de 1% com HBPM. A trombocitopenia aparece de 5 a 10 dias após o início da terapia, mas também pode ocorrer mais cedo em pacientes que fizeram tratamento recente com heparina. É mais comum em pacientes com câncer e apresenta taxas altas de complicações trombóticas. É uma condição pró-trombótica associada a um aumento da trombina, evidenciada pela presença de níveis elevados de complexos trombina-antitrombina. É considerada uma síndrome de hipercoagulabilidade adquirida importante. O diagnóstico é clínico e laboratorial (teste de agregação com heparina e ELISA para a detecção de anticorpos antiplaquetários). A frequência da síndrome em pacientes tratados com heparina é altamente variável e influenciada pela preparação da heparina (heparina fracionada bovina > heparina fracionada porcina > HBPM) e pelo perfil dos pacientes que recebem o tratamento (cirurgias, gravidez). Ocorre queda inexplicada e repentina das plaquetas, em mais de 50%, já nas primeiras 24 horas e até 4 dias após o início da aplicação. Raramente podem surgir lesões cutâneas ou reações sistêmicas agudas após a administração intravenosa. A heparina deve ser suspensa e recomenda-se a substituição por anticoagulantes como a lepirudina e o argatroban, a bivalirudina ou danaparoide, ou por inibidores diretos da trombina, como a hirudina ou o ximelagatran. Usualmente eles são bem tolerados e apresentam risco mínimo de sangramentos. 1. Estreptoquinase (SK) É obtida a partir de culturas de estreptococos-hemolíticos e, por ser um antígeno, pode causar reações alérgicas. Raramente, anafilaxia (0,5%), mas tremores, rash cutâneo ou febre podem ocorrer em até 10% dos pa- Referências bibliográficas 1. Craig S. Kitchens, Barbara M. Alving, Craig M. Kessler. Consultative hemostasis and thrombosis. 2a edição. Editora Saunders, 2007. 2. Eby C. Pathogenesis and management of bleeding and thrombosis in plasma cell dyscrasias. Br J Haematol. 2009;145(2):151-63. 3. Falanga A, Rickles FR. Management of Thrombohemorrhagic Syndromes (THS) in hematologic malignancies. Hematology Am Soc Hematol Educ Program. 2007:165-71. 4. Hook KM, Abrams CS. Treatment options in heparin-induced thrombocytopenia. Curr Opin Hematol. 2010;17(5):424-31. 5. Levine MN, Raskob G, Beyth RJ, Kearon C, Schulman S. Hemorrhagic complications of anticoagulant treatment: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126(3 Suppl):287S-310S. 6. Marcel M. Levi, Alvin H. Schmaier. Disseminated intravascular coagulation. Emedicine. Versão atualizada em 04/10/2009. Capturado online em 25/02/2001. Disponível em http://emedicine.medscape.com/article/199627-overview. 7. Michiels JJ, Berneman Z, Schroyens W, Finazzi G, Budde U, van Vliet HH. The paradox of platelet activation and impaired function: platelet-von Willebrand factor interactions, and the etiology of thrombotic and hemorrhagic manifestations in essential thrombocythemia and polycythemia vera. Semin Thromb Hemost. 2006;32(6):589-604. cientes tratados. A eficácia da estreptoquinase não é reduzida pela reação alérgica, mas deve ser evitada a sua reutilização por um período de um a dois anos. A hipotensão pode ocorrer durante administração rápida da solução (acima de 500 U/kg/min), mas geralmente responde a líquidos, dopamina, diminuição da velocidade de infusão, e raramente é necessária a interrupção. O pequeno sangramento é a complicação mais comum, ocorrendo em 3% a 4% dos pacientes, geralmente nos locais de punção. Os grandes sangramentos, por definição os que necessitam de transfusão de sangue, são menos comuns. O risco de AVCh é menor que 1% em todos os pacientes e, em pacientes acima dos 70 anos, é de 1,6%. 2. Ativador tecidual do plasminogênio (t-PA) A principal complicação do tratamento com t-PA é o sangramento. A incidência de AVCh em pacientes com mais de 70 anos é 1% maior do que nos tratados com estreptoquinase. Esse risco pode aumentar em pacientes com peso < 70 kg, pressão arterial > 170/95 mmHg e uma heparinização agressiva. 3. Anistreplase (APSAC) As complicações são semelhantes às da estreptoquinase. Por ser um antígeno, deve-se evitar reutilizá-la pelo período de um ano. 4. Uroquinase É frequentemente utilizado para trombólise nas salas de hemodinâmica e para tratamento da embolia pulmonar grave. 8. Sanz MA, Grimwade D, Tallman MS, Lowenberg B, Fenaux P, Estey EH, Naoe T, Lengfelder E, Büchner T, Döhner H, Burnett AK, Lo-Coco F. Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2009;113(9):1875-91. 9. Sanz MA, Montesinos P. Open issues on bleeding and thrombosis in acute promyelocytic leukemia. Thromb Res. 2010;125 Suppl 2:S51-4. 10. Sanz MA, Tallman MS, Lo-Coco F. Tricks of the trade for the appropriate management of newly diagnosed acute promyelocytic leukemia. Blood. 2005;105(8):3019-25. 11. Schünemann HJ, Cook D, Grimshaw J, Liberati A, Heffner J, Tapson V, Guyatt G.Antithrombotic and thrombolytic therapy: from evidence to application: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126(3 Suppl):688S-696S. 12. Tallman MS, Kwaan HC. Intravascular clotting activation and bleeding in patients with hematologic malignancies. Rev Clin Exp Hematol. 2004;8(1):E1. 13. Thomas G. DeLoughery. Management of acquired bleeding problems in cancer patients. Emerg Med Clin N Am. 2009;27(3):423-44. 14. Warkentin TE, Greinacher A. Heparin-induced thrombocytopenia: recognition, treatment, and prevention: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126(3 Suppl):311S-337S. 15. Zojer N, Ludwig H. Hematological emergencies. Ann Oncol. 2007;18 Suppl 1:i45-i48. Onco& abril/maio 2011 25
Baixar