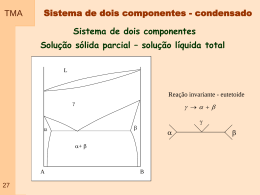
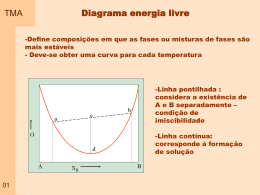
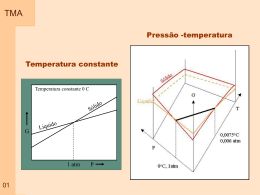
Misturas Simples TMA Diferenciação entre substâncias Puras e misturas simples Assume-se em geral que em misturas simples não existe reação Termodinamicamente Energia associada com a mistura apenas e com as interações entre as substâncias, que não envolvam reação Misturas de dois componentes A e B X A XB 1 Para gases ideais p A pB X A X B p p 01 Volume Parcial Molar TMA Adiciona 1 mol de H2O Adiciona 1 mol de H2O Volume aumenta de 18 cm3mol-1 Volume aumenta de 14 cm3mol-1 Volume Molar da H2O : 18 cm3mol-1 Volume Molar da H2O : 14 cm3mol-1 A diferença de aumento de volume depende do tipo de molécula que envolvem as moléculas de água adicionadas Definição : Volume molar da substância A em uma mistura é a variação de volume por mol de A adicionada a um grande volume de mistura. 02 Volume Parcial Molar – representação TMA T= 25 oC Volume Parcial Molar da água Etanol Água Percentual de etanol V V dV dnH 2O dne tanol nH O n e tanol p,T ,nH2O 2 p,T ,ne tan ol 03 Volume Parcial Molar do etanol VH 2O V nH O 2 p ,T ,ne tan ol V Ve tanol ne tanol p ,T ,nH2O V VJ nJ p,T ,n Onde n´ significa todos os outros componentes presentes TMA Volume Parcial Molar – cont. Definição : O volume parcial molar ( que pode ser positivo ou negativo) é o coeficiente angular da curva do volume total da amostra pelo numero de moles de um dos componentes, quando são mantidos constantes a Pressão; a temperatura; e a quantidades dos outros componentes Misturas de dois componentes A e B V VA n A p ,T ,nB Qual o significado do volume parcial molar negativo ? V V dV dnA dnB n n A p,T ,nB B p,T,nA O que acontece se a Temperatura variar? dV VAdnA VB dnB V VAnA VB nB 04 Quantidade de A, (nA) TMA Energia Livre de Gibbs Parcial Molar Definição : O conceito de quantidade parcial molar pode ser aplicada a qualquer variável extensiva, que seja função de estado. Ex. Energia livre de Gibbs parcial molar. Complemento: Para uma substância Pura o potencial químico representa a Energia Livre de Gibbs Molar. Para uma substância em uma mistura, o potencial químico se define como a Energia Livre de Gibbs Parcial Molar. G J nJ p ,T ,n Onde n´ significa todos os outros componentes presentes Definição: O potencial químico é o coeficiente angular da curva de Energia Livre de Gibbs em função do número de moles do componente J, quando são mantidos constantes a Pressão; a 05 temperatura; e a quantidades dos outros componentes. TMA Energia Livre de Gibbs Parcial Molar - cont Misturas de dois componentes A e B dG Vdp SdT G G dG dnA dnB n A p ,T ,nB nB p ,T ,n A Se não há variação de massa Se P e T são constantes com variação de massa (nJ) G G dG Vdp SdT dnA dnB n n A p ,T ,nB B p ,T ,n A 06 Geral – Equação fundamental geral da Termodinâmica Energia Livre de Gibbs Parcial Molar - cont TMA Definição : O potencial químico mostra como as grandezas termodinâmicas extensivas A, H, U e G dependem da composição G U H A J n n n n J p ,T ,n J S ,V ,n J S , p ,n J T ,V ,n ´ ´ ´ ´ G G dG Vdp SdT dnA dnB n A p ,T ,nB nB p ,T ,n A dG Vdp SdT AdnA B dnB Se P e T = Constante dG AdnA B dnB Equação de Gibbs-Duhem nAd A nB dB 0 d A 07 n d J J J nB d B nA 0 Função de estado dG AdnA B dnB nAd A nB dB ? G AnA B nB dG AdnA B dnB Energia Livre de Gibbs de misturas TMA Misturas de dois componentes A e B A B G AnA B nB Se P e T = Constante Exemplo clássico : Mistura de dois gases ideais A 08 Inicial (gases separados) Final (Mistura de gases) (Sem reação) Gi Gf T, P constante B G=Gf- Gi TMA misG nRTXA lnXA XB lnXB 09 mis H 0 G misS mis T P,nA ,nB misS nRXA lnXA XB lnXB Potencial Químico de líquidos TMA pA A RT Ln p A Potencial químico gás puro Para falarmos em fases condensadas e Importante a relação entre fase condensada e pressão de vapor T re 10 Átomos de A líquido rc Na condição de equilíbrio existe um equilíbrio entre evaporar e condensar. Existe um equilíbrio entre a taxa de evaporação e taxa de condensação No equilíbrio define-se a pressão de vapor TMA Taxa de evaporação – É função da força de ligação entre átomos. Quanto maior a força de ligação menor a taxa de evaporação. Taxa de condensação – depende da concentração de átomos de A no estado vapor que se chocam com a superfície do líquido por unidade de tempo. Esta taxa de condensação é proporcional à pressão parcial T T re rc Átomos de A líquido 11 re rc Átomos de B líquido T re rc O que acontece se átomos de A e B forem misturados? Potencial Químico de líquidos TMA A A RT n pA A RT n p A 1 O potencial químico de um líquido puro será dado por * p *A A RT n A 1 A RT n p *A O potencial químico do liquido A em uma mistura de dois líquidos A e B pA A RT n * pA * A 12 Potencial químico gás puro Lei de Raoult TMA François Raoult – Químico Frances pA A RT Ln * pA *A RT LnX A * A 13 Pressão Total Pressão Para misturas de líquidos similares a relação da pressão parcial de vapor de cada componente da mistura em relação à pressão de vapor do componente puro, é aproximadamente igual à fração molar do liquido presente na mistura. pA XA * pA Pressão Parcial de A Pressão Parcial de B Fração Molar de A Lei de Raoult - interpretação TMA -Considerando a taxa de evaporação das moléculas -A presença de um segundo componente interfere na remoção de moléculas do meio líquido mas não impedem a condensação. -Taxa de vaporização T axade vaporização kXA -Taxa de condensação T axade condensação k´pA -No equilíbrio T axade condensação T axade evaporação kX A k ´ pA 14 kX A pA ´ k Para um líquido puro XA 1 k p ´ k * A pA XA * pA TMA Lei de Raoult – Afastamento do comportamento Comportamento Ideal 15 Comportamento Real TMA Lei de Raoult – Afastamento do comportamento Característico de misturas de líquidos que apresentam diferenças químicas significativas Quando a solução é muito diluída o solvente comporta-se como ideal Quando em soluções diluídas o soluto apresenta pressão de vapor definida por uma constante de proporcionalidade Lei de Henry 16 Lei de Henry TMA Soluções reais, com baixas concentrações, a pressão de vapor do soluto é proporcional a fração molar, mas a constante de proporcionalidade não é a pressão de vapor do componente puro. pA XA * pA 17 Pressão William Henry – Químico Inglês pA XA kB São conhecidas como soluções ideais diluídas onde solvente é um líquido puro, ligeiramente modificado. O soluto presente em baixas concentrações Fração Molar de A TMA - Se as ligações A-B forem mais fortes que A-A e B-B T re re Numero de ligaçoes A-A diminuem Numero de ligações A-B aumentam Taxa de evaporação de A diminui se XA diminui T re Numero de ligaçoes B-B diminuem Numero de ligações A-B aumentam Taxa de evaporação de B diminui se XB diminui Lei de Henry – define a dependência entre taxa de evaporação e composição da mistura 18 PAo 0 19 Pressão de vapor Pressão de vapor TMA Xa 0 1 Se A-B maior que A-A e B-B re´(A)< re(A) Linha de Henry acima da Raoult PAo de Xa 1 Se A-B menor que A-A e B-B re´(A)> re(A) Linha de Henry abaixo da de Raoult TMA Energia Livre de Gibbs de misturas Misturas de dois componentes A e B Solução ideais – que segue a lei de Raoult 20 TMA Energia Livre de Gibbs soluções reais O que foi considerado até agora: -Mistura de gases ideais : não existe interação entre componentes -mistura de líquidos ideal : existe interação entre moléculas mas, em média as forças de interação entre A e B são semelhantes às interações A-A e B-B. -Soluções reais: as interações entre A-A, A-B e B-B são diferentes O tipo de interações em soluções reais definem se a mistura resultará em: - miscibilidade total - Imiscibilidade total - miscibilidade parcial 21 TMA Energia Livre de Gibbs soluções reais Para soluções reais usa-se a função excesso XE Exemplo: SE é a diferença entre a variação de entropia observada e a variação de entropia se a solução fosse ideal 22 TMA Energia Livre de Gibbs soluções reais Exemplo: Função modelo para entalpia H E mis H mis H ideal H E nRTX A X B nwX A X B Medida da força de interação A-B Endotérmico Exotérmico 23 TMA Energia Livre de Gibbs soluções reais Exemplo: Função para Energia Livre de Gibbs mix G nRT X A ln aA X B ln aB mixG nRT X A ln X A X B ln X B X A X B Para > 2 pode gerar separação de fase 24 ? Atividade em solução A atividade de um componente em qualquer estado em uma temperatura T é definido como a relação entre a fugacidade da substancia do estado e a fugacidade no estado de padrão. Atividadede A a i G RTXA ln XA XB ln XB G RTXA ln XA XB ln XB G RTXA ln a A XB ln a B fi f io Solução de gases ideais Solução de fases condensadas ideais Solução de fases condensadas reais Para uma solução real: •a atividade de A na solução não pode ser considerada igual a fração molar •Existe um fator de aproximação chamado de coeficiente de atividade •Este coeficiente é a relação entre a atividade e a fração molar G RTXA ln a A XB ln a B • Pode ser maior ou menor que 1. •Se for maior que 1 diz-se que a aA XA Determinar o coeficiente de atividade é importante para determinar a variação da energia livre solução apresenta deslocamento positivo em relação à lei de Raoult. •Se for igual a 1 a solução comporta-se como solução ideal •Se for menor que 1 diz-se que a solução apresenta deslocamento negativo em relação à lei de Raoult. •H de mistura não é zero •O aumento da temperatura tende a aproximar o comportamento de misturas do comportamento ideal. •Se >1 o aumento da temperatura tende a diminuir o valor de •Se <1 o aumento da temperatura tende a aumentar o valor de •Se H de mistura é negativo – processo exotérmico – tendência para formação de compostos •Se H de mistura é positivo – processo endotérmico – tendência para formação de separação de fases Variação de energia livre – Diagrama de fases -Sistema isobárico -Temperatura e composição variando -A fase existente será aquela que apresenta menor energia livre -Existe uma relação quantitativa de fases que , para uma determinada temperatura e composição correspondem a mínima energia c a b G d A XB B o a p n G G b r m A s q XB B Ni Cu Ni Cu Solução - Líquido Solução Sólida Cu Zn Solução sólida Zn Cu Composto de Cu Zn O que define a solubilidade Solução em cobre
Baixar