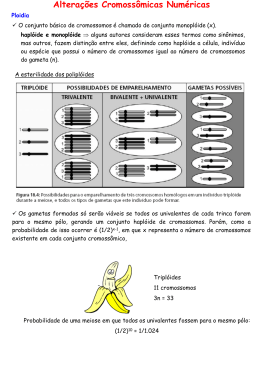
CITOGENÉTICA CLÍNICA CROMOSSOMOPATIAS DOS AUTOSSOMOS E DOS CROMOSSOMOS SEXUAIS Profa. Dra. Ana Elizabete Silva Genética Humana DOENÇAS CROMOSSÔMICAS Alteração cromossômica: numérica ou estrutural • > 100 síndromes • 1% nativivos • 1:160 neonatos • 50% dos abortos espontâneos Exemplo mais conhecido: Síndrome de Down ALTERAÇÕES NUMÉRICAS ou no número de cromossomos Tipos: Euploidias perda ou adição do lote haplóide -Triploidia (69,XXX) -Tetraploidia (92,XXXX) freqüentes em abortos espontâneos Aneuploidias perda ou adição de um ou + cromossomos EUPLOIDIAS Triploidia (69,XXX): apenas 1/10.000 zigotos triplóides nascem vivos, os que sobrevivem há suspeita de mosaicismo Tetraploidia (92,XXXX): incompatível com a vida recém-nascidos vivos são mosaicos triplóide/tetraplóides Freqüentes em abortos espontâneos TRIPLOIDIA http://www.assis.unesp.br/~egalhard/Numericas.htm ORIGEM DAS EUPLOIDIAS Triploidia: Diginia (erro materno) •óvulo 2n (erro na meiose I e II) + gameta n •fusão de ovócito e corpúsculo polar + gameta n Diandria (erro paterno) •óvulo n + dois gametas n (dispermia) •óvulo n + gameta 2n Tetraploidia erro na 1a. divisão mitótica no embrião (endorreduplicação) MIXOPLOIDIA MOSAICO E QUIMERA ABERRAÇÕES CROMOSSÔMICAS E ABORTOS ESPONTÂNEOS Incidência de abortos espontâneos: 15% a das gestações antes da 22 . semana Incidência de aberrações cromossômicas: 50% dos abortos entre 8a. e 15a. semanas apenas 5% dos abortos ocorrem durante as últimas semanas de gestação ABORTOS: causas imunológicas; anomalias uterinas; defeitos na fase luteal; diabetes; hipertireoidismo; anomalias cromossômicas DISTRIBUIÇÃO DOS TIPOS DE ANOMALIAS CROMOSSÔMICAS EM ABORTOS Difere daquelas dos recém-nascidos Exemplos: 1. monossomias X: 25% (presentes em recémnascidos e abortos) 2. trissomia 16: 32% (observada apenas em aborto) 3. outras trissomias: 21= 12% (60% são abortados) 15= 10% 22= 9,7% 4. 12,5% são poliplóides: triplóides: a maioria (17%), tetraplóides: ABORTO: 47,XX, +16 DIPLOIDIA UNIPARENTAL -Todos os cromossomos são derivados de apenas um genitor 1-Mola Hidantiforme: consiste num concepto anormal com constituição cromossômica 46,XX, todos cromossomos de origem paterna (homozigosidade para todos os loci) origem: degeneração do pró-núcleo feminino do ovo fertilizado e subsequente duplicação do DNA do pró-núcleo masculino. não há o desenvolvimento do embrião, apenas das membranas e placenta Frequência: Ocidente= 1/2000 Oriente (Taiwan)= 1/200 Mola parcial: são triplóides (2/3 dos casos: conjunto extra origem paterna 2-Teratoma Ovariano: consiste numa massa desorganizada de tecido embrionário com dois genomas maternos (46,XX), mas sem qualquer das membranas extraembrionárias e placenta. DISSOMIA UNIPARENTAL: ambos os homólogos derivados do mesmo genitor ANOMALIAS ESTRUTURAIS BALANCEADAS INVERSÃO INSERÇÃO TRANSLOCAÇÃO RECÍPROCA TRANSLOCAÇÃO ROBERTSONIANA Crossing na alça de pareamento em inversões paracêntrica e pericêntrica Inversão paracêntrica Inversão pericêntrica Translocação Recíproca Balanceada Translocação Robertsoniana ANEUPLOIDIAS Autossômicas 47, XX ou XY, + 21 (S. Down) 47, XX ou XY, + 18 (S. Edwards) 47, XX ou XY, + 13 (S. Patau) Dos cromossomos sexuais 45, X (S. Turner) 47, XXY (S. Klinefelter) 47, XXX (triplo X) 47, XYY (duplo Y) Tipos Freqüência Aberrações cromossômicas numéricas Autossomos 47,+21 1:800 47,+18 1:7500 47,+13 1:227000 Cromossomos sexuais 47,XYY 1:1000 RN do sexo masculinos 47,XXY 1:1000 RN do sexo masculinos 45,X 1:4000 RN do sexo feminino 47,XXX 1:900 Aberrações cromossômicas estruturais Rearranjos balanceados Robertsonianos 1:1100 Outros rearranjos balanceados 1:885 RN do sexo feminino Não-disjunção meiótica ANEUPLOIDIAS MÚLTIPLAS 48,XXY, +21 48,XXX, +21 46,x,+21 48,XXY,+18 48,XYY,+18 Hiopoulos et al. (2004): 1º. Caso de gêmeos 48,XXY,+21 (óbito) TRISSOMIA DO 21 - Síndrome de Down Incidência: 1/ 800 Características:hipotonia RM occipital plano pescoço curto ponte nasal baixa prega epicântica inclinação mongolóide dos olhos orelhas pequenas e malformadas língua protrusa prega palmar transversa única clinodactilia defeitos cardíacos Síndrome de Down diferentes etnias: indígenas, caucasóides, orientais e mulatos Incidência de SD na progênie de mães com idade avançada Idade materna (anos) Risco de SD em RN 15-19 1/1250 20-24 1/1400 25-29 1/1100 30 1/900 35 1/350 40 1/100 45 ou + 1/25 SÍNDROME DE DOWN Trissomia simples - 95% não disjunção meiose materna meiose I 75% meiose II 25% meiose paterna II 5% SÍNDROME DE DOWN Translocação robertsoniana 4-5% 46,XX ou XY,der(14;21)(q10;q10) 46,XX ou XY,der(21;21)(q10;q10) SÍNDROME DE DOWN Translocação 21q21q 1,6% Risco de afetados em portadores normais - 100% Mosaico (46, XX ou XY/47,XX ou XY,+21) 1-3% expressão clínica geralmente mais branda SÍNDROME DE DOWN Trissomia parcial do 21 “região crítica” - 21q22 • gene DYRK – retardo mental • gene APP (proteína precursora beta amilóide) • demência do tipo Alzheimer - CRB1 – neurodegenerações - CSTB – doenças neurodegenerativas - S100B – desenv. e maturação sistema nervoso - DYRK1A2 – prolif. céls. neurais, neurogênese, dif. neural -APP – D. Alzheimer (placas senis) - SOD – proteção contra deriv. oxigênio: envelhecimento - SIM2 – patogênese de RM - BHLB1 – deficiência de aprendizagem - DSCAM – candidato para cardiopatias - SH3BGR – doenças cardíacas congênitas - TIAM – desenvolvimento de leucemias - AML1 – hematopoiese normal (LLA e LMA infantil) REGIÃO CRÍTICA PARA SÍNDROME DE DOWN TRISSOMIA DO 18 (S. Edwards) Incidência : 1/7500 Características: hipertonia RM occipital proeminente orelhas de implantação baixa e malformadas micrognatia punhos cerrados esterno curto malformações cardíacas 47, XX ou XY, +18 95% mosaicismo trissomia 18q SÍNDROME DE EDWARDS TRISSOMIA DO 13 (S. Patau) Incidência: 1/22.700 Características: malformações SNC RM hipertonia microcefalia microftalmia orelhas malformadas fendas labial e palatina defeitos cardíacos e urogenitais 80% - 47, XX ou XY, +13 trissomia 13q SÍNDROME DE PATAU SÍNDROME DO CRI-DU-CHAT (5p-) (S. DO MIADO DO GATO) Incidência: 1/50.000 RM + Malformações Congênitas SÍNDROME CRI-DU-CHAT (5p-) Outras síndromes com deleção cromossômica: Síndromes do 4p-, 18q- e 13q- 4p18q- 13q- SÍNDROMES DE MICRODELEÇÃO Síndrome Deleção cromossômica Prader-Willi 15q11-13 Angelman 15q11-13 Langer-Giedion 8q24.11-q24.13 Miller-Dieker 17p13.3 Anomalia velo-cárdiofacial de DiGeorge 22q11 Smith-Manegis 17p11.2 Williams 7q11.23 *WAGR 11p13 Rubinstein-Taybi 16p13.3 Retinoblastoma 13q14 (*)Tumor de Wilms, Aniridia, Anomalias Genitourinárias, RM SÍNDROMES DE MICRODELEÇÃO 600Kb: alelos selvagens 460Kb: deleção de NF2 afetado Deleção do 22: FISH: centrômeros 14 e 22 gene NF2: 22q SÍNDROMES DOS CROMOSSOMOS SEXUAIS Aneuploidias dos cromossomos X e Y: 1:400-500 nascimentos Fenótipos menos graves Indicação clínica para cariótipo: Atraso do início da puberdade Amenorréia Infertilidade Genitália ambigua SÍNDROME DE TURNER (45,X) Incidência: 1/2500 sexo feminino Características: baixa estatura disgenesia gonadal pescoço alado implantação baixa dos cabelos tórax largo mamilos espaçados cúbito valgo defeitos renais e cardíacos SÍNDROME DE TURNER CARIÓTIPO: 45,X : 40-60% mosaicos 45,X/46,XX: 15% 46,X,i(Xq): 10% mosaicos 45,X/46,X,i(Xq): 8% deleções 46,XXq- ou 46,XXp- 6% outros mosaicos 45,X/? 8% Deleção do gene SHOX (Xp e Yp) = baixa estatura OUTRAS ALTERAÇÕES 5-10%: apresentam linhagem celular com cromossomo Y → gonadoblastoma → risco de malignização Análise de DNA: sondas do Y em pacientes com ST Cariótipo provável: 45,X/46,XY (comprovado por FISH ou PCR em diferentes tecidos: sangue, pele e músculo) TRIPLO X (47, XXX) Incidência: 1/1000 Características: estatura RM esterilidade irregularidade menstrual Formas variantes: 48, XXXX e 49,XXXXX SÍNDROME DE KLINEFELTER (47,XXY) Incidência: 1/1000 Características: estatura membros longos pênis e testículos pequenos esterilidade ginecomastia mosaicismo (15%) = 46,XY/47,XXY Formas variantes: 48, XXYY 48, XXXY 49, XXXXY SÍNDROME DO DUPLO Y (47,XYY) Incidência: 1/1000 Características: estatura problemas de comportamento inteligência normal
Baixar