ENZIMAS E CINÉTICA ENZÍMICA
ÌNDICE
1. Revisão dos conceitos de Keq e de QR................................................................................... 2
2. Revisão de conceitos de cinética química. ............................................................................. 3
3. O que são enzimas? ................................................................................................................ 5
4. O que é a cinética enzímica? .................................................................................................. 6
5. Como fazer um estudo cinético de uma enzima?................................................................... 7
6. Que tipo de resultados podem ser obtidos no estudo cinético de uma enzima?..................... 7
6.1. Noção de actividade enzímica ou actividade catalítica de uma enzima. ..................................................... 7
6.2. Noção de v0 ou velocidade inicial. .............................................................................................................. 8
6.3. Influência da quantidade de enzima na velocidade de conversão v0. .......................................................... 8
6.4. Influência da temperatura na actividade enzímica....................................................................................... 9
6.5. Influência do pH........................................................................................................................................ 10
6.6. Influência da concentração dos substratos na actividade enzímica e saturabilidade. ................................ 10
6.7. Influência da concentração do substrato em enzimas com “cinética de tipo michaeliano ou hiperbólico”.
.......................................................................................................................................................................... 11
6.8. Influência da concentração de substrato em enzimas “com cinética de tipo cooperativo ou sigmoide”. .. 15
6.9. Representações gráficas lineares nas enzimas “de cinética michaeliana” e de “cinética de tipo
cooperativo”. .................................................................................................................................................... 18
6.10. Modificadores da actividade enzímica: inibidores e activadores. ........................................................... 20
6.11. Inibidores competitivos. Sua representação gráfica linear. ..................................................................... 20
6.12. Inibidores não competitivos. Sua representação gráfica linear................................................................ 23
6.13. Efeitos alostéricos. .................................................................................................................................. 26
7. Para que pode servir o estudo cinético de uma enzima? ...................................................... 28
8. Nota final: a actividade das enzimas como factores de regulação do metabolismo. ........... 30
9. Bibliografia consultada: ....................................................................................................... 32
Este texto foi escrito por Rui Fontes em Janeiro de 1996 e corrigido em Dezembro de 2005. O
autor agradece a todos os que lerem este texto todas as críticas que entendam fazer.
1. Revisão dos conceitos de Keq e de QR.
Todas as reacções tendem a alcançar um equilíbrio, mas nem sempre isto é aparente. Se a
concentração de um dos reagentes está estequiometricamente em defeito em relação aos
outros e a reacção tem uma constante de equilíbrio de valor elevado pode parecer que, quando
macroscopicamente a reacção terminou, esse reagente foi completamente consumido. Para
efeitos práticos foi exactamente isso que ocorreu mas, em rigor, algumas moléculas desse
reagente permanecem no meio. Todos os sistemas que reagem alcançam um estado de
equilíbrio no qual permanecem pelo menos algumas moléculas dos reagentes.
São características dos estados de equilíbrio:
a) As propriedades macroscópicas do sistema mantêm-se constantes no tempo.
b) À escala microscópica, a reacção prossegue nos sentidos directo e inverso com velocidades
iguais.
c) Um mesmo estado de equilíbrio pode obter-se quer a partir dos reagentes quer dos produtos.
d) Um estado de equilíbrio químico só pode obter-se, no exacto sentido da palavra em sistemas fechados. Um ser vivo não é um sistema fechado e por isso, em sentido estrito, o
equilíbrio químico não existe nos seres vivos.
Associada a uma reacção química determinada (em condições de pressão, temperatura e
concentração dos reagentes e produtos determinadas) descrita pela equação:
aA+bB →pP+qQ
existem dois valores de constantes de equilíbrio:
a) uma constante de equilíbrio estequiométrica em que quer o numerador quer o denominador
são um produto de concentrações.
[P](eq) p [Q](eq) q
Keq = ⎯⎯⎯⎯⎯⎯⎯⎯
[A](eq) a [B](eq)b
(1.1)
Esta Keq não é verdadeiramente uma constante pois depende em certa medida da
grandeza dessas concentrações.
b) uma constante de equilíbrio termodinâmica em que quer o numerador quer o denominador
são um produto de actividades:
aP (eq) p aQ(eq) q
Keq = ⎯⎯⎯⎯⎯⎯⎯⎯
aA (eq) a aB (eq)b
(1.2)
Para a mesma reacção também se podem definir quocientes de reacção (QR) estequiométrico e
termodinâmico. As equações que definem QR têm um aspecto semelhante à das constantes de
equilíbrio mas em vez das concentrações (ou das actividades) de equilíbrio usam-se as
concentrações (ou as actividades) que efectivamente se observam num dado momento da
reacção.
Em reacções em meio aquoso é costume ignorar a concentração da água assim como a
concentração das substâncias que intervenham na reacção mas que não estejam dissolvidas na
água (por exemplo precipitados) quando se definem quer as constantes de equilíbrio quer os
Página 2 de 32
quocientes de reacção. Admitindo-se que estas “concentrações” não variam durante o
processo reactivo; quer à concentração da água quer à dessas substâncias na fase sólida se
atribui convencionalmente o valor 1.
Em investigação bioquímica laboratorial não é habitualmente necessário conhecer com rigor o
valor da constante de equilíbrio de uma reacção implicada no estudo que se está a realizar; ter
uma ideia da ordem de grandeza da constante de equilíbrio é, em geral, suficiente e por isso
indiferente usar uma constante termodinâmica ou estequiométrica.
No ser vivo, os compostos químicos reagem entre si respeitando a lei do equilíbrio químico.
Admitamos que num dado momento, no citoplasma da célula, os compostos A, B, P e Q,
todos hidrossolúveis, estão em presença uns dos outros em concentrações tais que o valor do
quociente de reacção (QR) é inferior ao da constante de equilíbrio (Keq): a reacção tem
tendência a processar-se no sentido da formação dos produtos P e Q com consumo de A e B.
No caso em que QR > Keq a reacção tem tendência a ocorrer no sentido inverso; quando QR =
Keq a reacção, macroscopicamente, não tem tendência a evoluir em nenhum dos sentidos.
2. Revisão de conceitos de cinética química.
Quando se diz que uma determinada reacção tem tendência a ocorrer (Keq > QR) não se quer
dizer com isso que ela ocorra de facto a uma velocidade apreciável num intervalo de tempo
determinado. A velocidade a que ocorre uma determinada reacção depende de vários factores
como a natureza dos reagentes e a sua concentração, a temperatura, e nalguns casos da
presença de radiações e de catalisadores.
As reacções químicas elementares de 1ª ordem (ou unimoleculares: A → P + ...) obedecem à
lei de velocidade expressa pela equação:
d [A]
- ⎯⎯⎯ = - k [A]
(2.1)
dt
A velocidade de reacção é, neste caso, proporcional à concentração do reagente A sendo k a
constante de proporcionalidade; porque relaciona a concentração de um reagente com a
velocidade da sua conversão em produtos diz que k é uma constante cinética.
Pode acontecer que uma determinada reacção (A + B → P + ...) seja elementar e de 1ª ordem
em relação a cada um dos dois reagentes A e B, sendo globalmente de 2ª ordem ou
bimolecular:
d [A]
- ⎯⎯⎯ = - k1 [A] [B]
dt
(2.2)
Os valores de k e k1 (constantes cinéticas) são independentes da concentração dos reagentes
mas variam com a natureza destes e com a temperatura, aumentando com esta de acordo com
a equação de Arrhenius.
Atentemos no esquema que se apresenta a seguir:
A+B
k1
P+Q
k2
Página 3 de 32
Se a transformação de A + B em P + Q é uma reacção química elementar o valor da velocidade de reacção que podemos observar experimentalmente na ausência de P e Q é igual a
k1 [A] [B]. Se a reacção inversa também é uma reacção química elementar então, na ausência
de A e de B, a velocidade que se pode observar experimentalmente nesta reacção é igual a k2
[P] [Q]. Qualquer que seja a concentração de A, B, P e Q a velocidade de formação de P (=
velocidade de formação de Q = velocidade de consumo de A ou de consumo de B) que
podemos observar experimentalmente, sem recurso a compostos marcados radioactivamente,
é dada pela equação:
v aparente = k1 [A] [B] - k2 [P] [Q]
(2.3)
De notar que a velocidade microscópica a que ocorrem as reacções elementares não é
influenciada pela presença de produtos e este facto pode ser comprovado experimentalmente
usando compostos marcados radioactivamente.
A equação anterior permite-nos relacionar a Keq com as constantes k1 e k2.. O sistema está em
equilíbrio quando as propriedades macroscópicas do sistema se mantém constantes no tempo
ou seja, quando v aparente = 0. Donde:
[P](eq) [Q](eq)
k1
Keq = ⎯⎯⎯⎯⎯⎯ = ⎯⎯
[A](eq) [B](eq)
k2
(2.4)
Deve notar-se que conhecer o valor da Keq não nos diz nada acerca do valor absoluto das
constantes k1 e k2 . Se, por exemplo, na reacção em análise o valor de Keq for 1 a única
informação que este valor fornece a um cinetista é que k1 = k2 . Assim, podemos dizer sem
contradição, que uma reacção se está a processar muito rapidamente (ou muito lentamente) e
que se encontra no estado de equilíbrio.
A maioria das reacções químicas e todas as reacções em que intervêm catalisadores são
reacções complexas e podem tentar interpretar-se como uma sequência de reacções
elementares em que ocorre a formação de compostos intermediários que não chegam a atingir
no meio reactivo uma concentração apreciável por métodos correntes de análise.
Na presença de um catalisador a velocidade das reacções modifica-se de forma marcada. Se
não se disser explicitamente que se trata de um catalisador negativo admite-se implicitamente
que estamos a falar de aumento da velocidade das reacções. Do ponto de vista de um cinetista
interessado no mecanismo das reacções químicas o catalisador é um novo reagente que foi
adicionado no meio reactivo e que tem a característica especial de ser regenerado no final do
processo reactivo microscópico em que intervém. A sua concentração total não varia durante
o processo reactivo.
Admitamos que a reacção de transformação de A em P+Q é, na ausência de catalisador, uma
reacção elementar de primeira ordem, e que v(sem catalisador) = k1 [A].
k1
A ⎯
⎯→ P + Q
Admitamos que na presença de catalisador o mecanismo reactivo é diferente. Cada vez que
uma molécula de A se encontra com uma molécula de catalisador C elas reagem para gerar
um complexo activado A•C e que este complexo se pode dissociar em C + P + Q. As
constantes de velocidade dependem, como dissemos, da natureza dos reagentes e portanto os
valores das constantes de velocidade associadas ao processo de formação de A•C e ao
Página 4 de 32
processo de dissociação deste complexo em P + Q + C podem ser muito maiores que o valor
de k1.
Admitindo que o catalisador C é um reagente temos também de admitir que é um produto.
A ⎯
⎯→ P + Q
C+A ⎯
⎯→ P + Q + C
[P](eq) [Q](eq)
Keq1 = ⎯⎯⎯⎯⎯⎯⎯
[A](eq)
[P](eq) [Q](eq) [C]
Keq2 = ⎯⎯⎯⎯⎯⎯⎯⎯⎯
[A](eq) [C]
(2.5)
(2.6)
As expressões Keq1 e Keq2 referem-se, respectivamente, às reacções não catalisada e
catalisada mas é óbvio que Keq1 = Keq2 = Keq. Os catalisadores modificam a velocidade das
reacções mas não modificam o valor da constante de equilíbrio nem o sentido em que a
reacção, macroscopicamente, vai ocorrer e que pode ser previsto pela relação Keq /QR. Se a
constante de equilíbrio não for demasiado alta impedindo-nos de observar a reacção inversa
(síntese de A a partir de P+Q) podemos constatar que esta reacção também é catalisada pelo
mesmo catalisador C.
3. O que são enzimas?
A palavra “enzima” (do Grego: en, na + zima, levedura) foi inventada em 1878 por Fredrich
Wilhelm Kühne. Na época, esta palavra não era inocente pois apoiava uma das teorias em
disputa acerca das causas das fermentações. Com esta palavra se apoiava a teoria de Justus
Liebig segunda a qual havia dentro da levedura substâncias químicas (fermentos = enzimas)
responsáveis pela transformação da glicose em dióxido de carbono e etanol contra a teoria
vitalista em moda na época, que defendia que as fermentações só podiam ocorrer na presença
da levedura (a célula viva).
Em 1897 os irmãos Buchner foram capazes de obter a partir da levedura um extracto isento de
células capaz de levar a cabo a fermentação da glicose. Desta forma se apoiava de forma
marcante a teoria de Liebig.
Com o desenvolvimento da ciência bioquímica se foi consolidando a ideia de que existiam
dentro dos seres vivos substâncias capazes de catalisar de modo muito específico
determinadas reacções químicas e que o conjunto sequenciado dessas reacções explicavam as
transformações químicas observadas nos seres vivos.
A natureza proteica das enzimas só foi definitivamente aceite nos anos 30 deste século, na
sequência dos trabalhos de James Summer (que purificou e cristalizou a urease do feijão) e de
John Northrop e Moses Kunitz (que demonstraram correlação directa entre a actividade
catalítica de preparações purificadas de enzimas digestivas e o seu conteúdo proteico).
Curiosamente, estudos da década de 80 de Thomas Cech num protozoário (Tetrahymena
thermophila) e de Sidney Altman em E. coli demonstraram actividade catalítica em certas
moléculas de RNA. Daqui surgiu o termo ribozimas: catalisadores biológicos de natureza não
proteica mas sim ácidos ribonucleicos.
Tal como acontece com todos os catalisadores quando se diz que uma enzima E catalisa a
transformação A→P está-se implicitamente a dizer que também catalisa a transformação
Página 5 de 32
inversa; a reacção vai progredir macroscopicamente no sentido A→P ou no sentido P→A
dependendo da relação Keq/QR.
Numa reacção enzímica chamam-se aos reagentes substratos da enzima.
Relativamente aos catalisadores não enzímicos as enzimas são em geral mais potentes,
actuam em condições “ pouco agressivas “ (pH próximo da neutralidade, temperatura <
100°C, etc.), têm uma enorme especificidade quer em relação aos substratos quer em relação
aos produtos da reacção que catalisam e a sua actividade pode ser, frequentemente, regulada
por substâncias diferentes dos substratos e dos produtos.
Sendo as enzimas moléculas proteicas o seu tamanho é, geralmente, muito grande relativamente ao tamanho das moléculas dos substratos. Este facto assim como a enorme
especificidade das enzimas relativamente aos substratos com que podem interagir levaram à
introdução do conceito de “sítio activo” (ou talvez mais correctamente “sítio catalítico”), um
local específico modelado de tal forma que permite a interacção específica com o substrato ou
substratos e onde ocorre a reacção química.
Fig.1: Modelos de chave-fechadura e de encaixe induzido para a interacção enzima-substrato. O primeiro
modelo (chave-fechadura) surgiu para explicar a alta especificidade das enzimas em relação aos substratos
nomeadamente a sua interacção com apenas um dos enantiómeros de uma mesma substância. O segundo modelo
(encaixe induzido) surgiu da necessidade de explicar certos casos de inibição competitiva (ver capítulo 6.11).
Em algumas enzimas estudos de difracção de raios X apoiam o segundo modelo (por exemplo, no caso da
transcarbamílase do aspartato).
Em 1894, Emil Fisher descobriu que as enzimas da via glicolítica podem distinguir enantiómeros (formas D de L) e esse facto levou-o a propor uma analogia com “a chave e a
fechadura” como modelo de interacção entre as enzimas e os seus substratos. Este modelo
admite a existência de locais preformados na enzima onde os substratos se podem ligar e
reagir. Koshland, em 1959, propôs um modelo diferente a que podemos chamar de “encaixe
induzido”; quando os substratos interagem com a enzima provocam nestas modificações
conformacionais que ocorrem não só no “sitio catalítico” mas também se podem repercutir
em toda a estrutura do proteído (ver Fig.1).
4. O que é a cinética enzímica?
A cinética enzímica é um ramo da bioquímica que estuda as enzimas em acção; a sua actividade catalítica. No entanto, pode também ser encarada como um ramo da cinética química
que estuda as propriedades catalíticas das enzimas.
O estudo cinético de uma enzima visa primariamente caracterizar, ou seja descrever, a
actividade dessa enzima. In vitro estuda-se a actividade da enzima procurando saber que tipo
de reacções pode catalisar, com que substratos pode interactuar e como se modifica essa
Página 6 de 32
actividade (qualitativa e/ou quantitativamente) quando se fazem variar as condições em que é
ensaiada. O valor de pH, a temperatura, o tempo de incubação, as concentrações dos
substratos, de cofactores ou de outras substâncias (inibidores ou activadores) são exemplos de
condições de ensaio que podem ser modificadas com o objectivo de observar como varia a
actividade da enzima.
5. Como fazer um estudo cinético de uma enzima?
O estudo cinético de uma enzima é feito in vitro. Para esses estudos podem ser usados, como
fonte de enzima, preparações que a contenham em estado mais ou menos purificado; quanto
mais purificada estiver a preparação enzímica mais fácil será o seu estudo cinético. De acordo
com os objectivos definidos não devem estar presentes na preparação enzimas que interfiram
no estudo que estamos a fazer.
Durante o complexo processo de purificação podem ocorrer alterações nas características
cinéticas da enzima de tal forma que as características observadas podem não ser as mesmas
da enzima no seu estado nativo; além disso as condições que usamos in vitro para estudar a
enzima são diferentes daquelas que a enzima tem no seu meio natural, a célula. Se o objectivo
dos estudos visa compreender o papel da enzima na célula os resultados experimentais devem
ser interpretados com especial prudência. Estes estudos podem servir de guia para planear e
interpretar experiências realizadas em condições menos artificiais.
Quando uma actividade enzímica é estudada escolhe-se um meio de ensaio apropriado para o
seu estudo; o objectivo pode ser estudar como varia a actividade catalítica da enzima quando
modificamos as características desse meio, mas numa fase precoce do estudo é necessário
buscar condições que permitam, no mínimo, reconhecer a existência dessa actividade.
Se a enzima catalisa a transformação reversível A + B
P + Q e a constante de
equilíbrio é muito superior à unidade as dificuldades do estudo podem ser minimizadas se
escolhermos estudar a transformação de A + B em P + Q e não o inverso. Assim
adicionaríamos no meio de ensaio os compostos A e B. Um amortecedor de pH com um pH
determinado assim como outras substâncias como, por exemplo, cofactores essenciais à
actividade da enzima estão, em geral, também presentes no meio de ensaio.
O meio de ensaio está a uma temperatura determinada e a reacção pode ser iniciada adicionando ao meio a preparação que contém a enzima. Para determinar a velocidade de
conversão de A+B em P+Q é indispensável termos uma maneira de seguir (de modo contínuo
ou descontínuo) como cresce a concentração de P ou Q (ou desce a concentração de A ou B)
em função do tempo.
6. Que tipo de resultados podem ser obtidos no estudo cinético de uma enzima?
6.1. Noção de actividade enzímica ou actividade catalítica de uma enzima.
A actividade enzímica é uma propriedade medida pelo aumento de velocidade de conversão
de uma reacção química que uma enzima produz num sistema de ensaio especificado.
Num meio de ensaio foram introduzidos os compostos A e B: a velocidade de conversão na
reacção A + B → P + Q pode ser expressa em µmol de P (ou Q) formados / min e esta
velocidade (v1) pode não ser nula na ausência de enzima. Se a reacção é catalisada pela
enzima a velocidade de conversão na presença desta pode ser v2. A quantidade ( v2 - v1 ) é
uma medida da actividade catalítica presente na preparação enzímica adicionada ao meio de
ensaio. Em geral o valor de v1 é tão pequeno comparado com v2 que se pode considerar v2 o
valor da actividade catalítica da enzima.
Página 7 de 32
É importante referir que a velocidade de conversão (e a actividade enzímica) é uma
quantidade extensiva e que portanto as suas dimensões são quantidade de substância / tempo
(n/t). A actividade enzímica de um dado volume de preparação pode ser expresso em UI
(Unidades Internacionais = µmoles de produto formado ou substrato consumido por min).
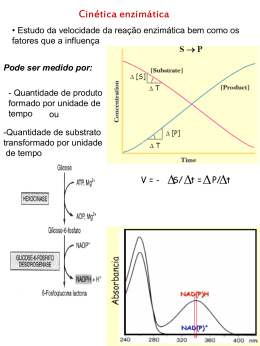
6.2. Noção de v0 ou velocidade inicial.
Fig.2: Quantidade de produto formado versus tempo. A actividade enzímica é uma quantidade extensiva (n/t) e
em cada momento t corresponde ao declive de uma curva como a desta figura. A velocidade de conversão A →
P diminui com o tempo de ensaio. v0 é o valor do declive no tempo zero e o valor da actividade catalítica para as
condições de ensaio nesse tempo zero. No caso em análise o resultado da operação de dividir a quantidade
(µmoles) de produto formado ao fim de 5 minutos por 5 min poderia ser uma boa estimativa de v0: a actividade
da enzima na preparação enzímica que foi adicionada neste meio de ensaio seria 1UI.
Quando se faz uma experiência visando desenhar um gráfico quantidade de P formado versus
tempo e se espera tempo suficiente, obtemos quase sistematicamente um gráfico com um
aspecto semelhante ao da Fig. 2. Num dado momento t a actividade catalítica é dada pelo
declive da curva nesse tempo t. À medida que o tempo de ensaio aumenta diminui a
velocidade e esse facto pode ser causado pela diminuição de concentração de reagentes (que
se vão consumindo), pelo aumento de concentração de produtos (que podem tornar
significativa a velocidade da reacção inversa1) ou/e simplesmente porque a enzima é instável
nas condições de ensaio. A esmagadora maioria dos estudos de cinética baseiam-se na
estimativa da velocidade para tempo zero (v0) ; nesse momento as características do meio de
ensaio, nomeadamente a concentração dos substratos, são aquelas que foram escolhidas para o
realizar.
6.3. Influência da quantidade de enzima na velocidade de conversão v0.
Mantendo invariantes todas as outras condições de ensaio a velocidade de conversão v0 é
directamente proporcional à quantidade de enzima adicionada no meio de ensaio. O gráfico v0
versus quantidade de enzima é uma linha recta de declive positivo que passa pela origem (ver
Fig. 3). Este facto permite-nos definir quantidades derivadas da actividade catalítica. Se
conhecermos o número de moles de enzima adicionada no meio de ensaio (o que só muito
1 Se, de modo contínuo, um dos produtos é eliminado do meio reactivo a reacção inversa não pode ter lugar. No
entanto se os produtos são mais que um, o produto ou produtos não eliminados podem ligar-se à enzima e
diminuir a velocidade de catálise; i.e. provocar inibição da actividade enzímica.
Página 8 de 32
Fig.3: Actividade enzímica versus quantidade de enzima. Os três gráficos desta figura representam o resultado
de uma mesma experiência. Em cada um de 4 tubos de ensaio contendo um meio especificado foram adicionadas
4 diluições de uma mesma preparação enzímica; 1E, 2E, 3E e 4E representam as distintas quantidades de enzima
adicionadas. No gráfico da esquerda estão representados os traçados obtidos (quantidade de produto formado
versus tempo) para cada um dos tubos e as rectas que estimam v0. O gráfica acima à direita representa v0 versus
quantidade de enzima. Os pontos do gráfico abaixo à direita representam a actividade específica obtida em cada
um dos quatro ensaios (neste caso em UI/mg proteído). A equação 6.7.7 mostra claramente que para
determinadas condições de ensaio fixadas v0 é proporcional a [Et], a concentração total de enzima no meio de
ensaio.
raramente acontece) podemos calcular a actividade específica e, por exemplo, exprimi-la em
moles de substrato convertido por segundo e por mole de enzima (s-1). Para permitir comparar
os resultados de experiências diferentes é costume exprimir a actividade específica em UI/mg
de proteína, UI/ml de preparação enzímica ou em UI/g de tecido donde essa enzima foi
extraída.
6.4. Influência da temperatura na actividade enzímica.
Quantidade de produto formado
30º
45º
60º
t1
tempo
t2
Fig.4: Quantidade de produto formado versus tempo a várias temperaturas. O uso de temperaturas elevadas pode
dificultar ou mesmo impedir uma boa estimativa de v0; assim é habitual usar temperaturas de ensaio em que a
enzima é estável. Esta figura mostra como pode variar a quantidade de produto ao longo do tempo para ensaios
de uma mesma enzima a várias temperaturas. Se usássemos um tempo de incubação suficientemente curto
poderíamos concluir que a enzima era mais activa a 60°C; nesse tempo muito curto a velocidade da reacção
enzímica foi alta e ainda não houve tempo suficiente para que a desnaturação se processasse em extensão
apreciável. Temperaturas mais baixas facilitam a estimativa de v0 pois a actividade mantém-se constante durante
mais tempo.
Página 9 de 32
Em todas as reacções químicas, incluindo as catalisadas por enzimas, a velocidade das
reacções aumenta com a temperatura, mas a velocidade com que um proteído sofre
desnaturação (e uma enzima se inactiva) também aumenta com a temperatura. O número de
enzimas capazes de resistir a temperaturas da ordem dos 100°C durante alguns segundos é
extremamente restrito. A temperaturas elevadas a velocidade de desnaturação da enzima é
muito alta podendo acontecer que a actividade enzímica se anule pouco tempo após o início
do ensaio (ver Fig. 4).
6.5. Influência do pH.
O pH a que uma reacção enzímica é estudada é normalmente imposto pelo experimentador
adicionando no meio de ensaio uma quantidade adequada de um determinado amortecedor de
pH. Um determinado amortecedor só funciona adequadamente como amortecedor de pH
numa faixa limitada de valores de pH e se o objectivo é estudar uma reacção enzímica numa
ampla gama de valores de pH temos de escolher amortecedores diferentes. Assim, um
determinado resultado experimental actividade versus pH pode reflectir apenas a influência
do amortecedor na actividade catalítica. Excluída esta possibilidade os gráficos experimentais
actividade versus pH podem traduzir que o estado ligado ou desligado a protões num
determinado local da enzima (ou nos substratos) se pode reflectir na actividade enzímica. Para
determinadas condições de ensaio existe um pH óptimo, o pH a que se obtém maior
actividade (ver Fig. 5).
2000
1500
25 mM
1000
500
750 µM
100 µM
0
8.5
9
9.5
10
10.5
pH
Fig.5: Esta figura foi recolhida e simplificada a partir de um artigo publicado no Biochemical Journal: R.K.
Morton, 1957, Biochemical Journal, 65: 674. Mostra um gráfico actividade versus pH para a actividade
hidrolítica da fosfátase alcalina de intestino de vitelo sobre o fenil-fosfato a distintas concentrações deste
composto.
6.6. Influência da concentração dos substratos na actividade enzímica e
saturabilidade.
As observações feitas em estudos sobre a influência da concentração de substratos, (assim
como a de inibidores e activadores) na velocidade das reacções enzímicas sempre foram
temas de especial interesse para os cinetistas.
Página 10 de 32
Quando se estuda a influência da concentração de um substrato na actividade duma enzima
começa por fixar-se a concentração dos outros substratos (se os houver) e das demais
condições de ensaio e estuda-se como varia a actividade versus concentração de um dos
substratos.
Em 1902 Adrian Brown estudando a reacção de hidrólise da sacarose [sacarose (+ H2O) →
glicose + frutose] e usando como catalisador sacarase (= invertase ou β-frutofuranosídase)
observou que para concentrações altas do substrato sacarose a ordem de reacção era zero em
relação à sacarose; ou seja, nessas concentrações a velocidade da reacção não variava quando
variava a concentração de substrato. Brown propôs um mecanismo que explicaria o
fenómeno:
E• sacarose ⎯
⎯→ E + glicose + frutose
E + sacarose
A enzima formaria com o substrato um complexo intermediário (mais tarde chamado
complexo de Michaelis); este complexo daria origem aos produtos regenerando-se a enzima e
a velocidade de reacção seria proporcional à concentração deste complexo. Se a concentração
de sacarose é muito alta relativamente à concentração de enzima, a enzima pode estar
saturada de substrato; aumentando a concentração de substrato a concentração do complexo
intermediário não pode aumentar mais e portanto não aumenta a velocidade de reacção. Ver
Fig. 6.
enzima s
enzima h
v0
[Substrato]
Fig. 6: Nalgumas enzimas o gráfico actividade versus concentração do substrato tem um aspecto semelhante a
enzima h (uma hipérbole rectangular passando pela origem); noutras um aspecto semelhante a enzima s (um
sigmoide). Em ambos os casos para altas concentrações de substrato a actividade é pouco sensível a variações na
concentração de substrato: as enzimas são saturáveis.
6.7. Influência da concentração do substrato em enzimas com “cinética de tipo
michaeliano ou hiperbólico”.
Para muitas enzimas o gráfico v0 versus concentração de um dos substratos, mantendo
constantes todas as outras condições do meio, é um ramo de hipérbole rectangular passando
pela origem (ver Fig. 6). Um tratamento matemático possível que explica este tipo de gráficos
em reacções enzímicas com apenas um substrato (ou mais de um mas os outros a
concentração fixada) foi proposto por Leonor Michaelis e Maude Menten em 1913.
Estes autores admitiram vários pressupostos:
Página 11 de 32
1) Um mecanismo representado pelo esquema seguinte:
k1
k3
E+S
E•S ⎯
⎯→ E + P +....
k2
A velocidade de formação do complexo E•S seria de 1ª ordem em relação a S (o substrato) e
a E (a enzima livre; não ligada a S) e a constante cinética associada ao processo seria k1. Quer
a velocidade de formação dos produtos quer a de dissociação do complexo E•S em S + E
seriam proporcionais à concentração deste complexo: ambos os processos seriam de 1ª ordem
em relação a E•S e as constantes cinéticas seriam respectivamente k3 e k2.
2) A velocidade de formação dos produtos a partir do complexo E•S era muito lenta
relativamente à velocidade de dissociação do complexo em E + S (k2>>k3) donde se poderia
admitir que a reacção de formação e dissociação do complexo E• S se encontrava num estado
muito próximo do equilíbrio químico.
3) Que a concentração de sítios catalíticos na enzima, [Et], era muito baixa relativamente à
concentração de [S] de maneira que para que esse estado de equilíbrio fosse atingido muito
poucas moléculas de S tinham que reagir com E; que esse estado de equilíbrio se atingia tão
rapidamente que na escala de tempo da transformação macroscópica de S em P se poderia
considerar instantâneo e se mantinha durante todo o tempo em que ocorria a transformação S
→ P+...
Para a reacção de dissociação do complexo E•S definiram a constante de equilíbrio Ks (s de
substrato).
[S] [E]
Ks = ⎯⎯⎯⎯⎯⎯ =
[E•S]
k2
⎯⎯
k1
(6.7.1)
Com base nestes pressupostos definiram v0 como a velocidade da reacção para concentração
nula de produtos:
v0 = k3 [E•S]
(6.7.2)
k3 é a constante de velocidade para a reacção elementar e unimolecular E•S → E + P +...
A concentração total de enzima ([Et]) é igual à soma das concentrações de enzima livre ([E])
mais enzima complexada ([E•S]) quaisquer que sejam as unidades em que estas se exprimam:
[Et] = [E] + [E•S]
(6.7.3)
As equações 6.7.1, 6.7.2 e 6.7.3 constituem um sistema que pode ser desenvolvido de forma a
obter v0 em função de [Et], Ks e [S].
Ks [E•S]
[E] = ⎯⎯⎯⎯⎯
[S]
(6.7.4)
Página 12 de 32
Ks [E•S]
Ks
[Et] = ⎯⎯⎯⎯ + [E•S] = ( ⎯⎯⎯ + 1) [E•S]
[S]
[S]
(6.7.5)
[Et]
[Et] [S]
[E• S] = ⎯⎯⎯⎯⎯⎯ = ⎯⎯⎯⎯
(Ks / [S] + 1 )
Ks + [S]
(6.7.6)
k3 [Et] [S]
v0 = ⎯⎯⎯⎯⎯⎯⎯
Ks + [S]
(6.7.7)
Um gráfico v0 versus [S] é um ramo de hipérbole rectangular passando pela origem tendo
uma assímptota horizontal (y = k3 [Et]) e uma assimptota vertical (x= -Ks).
Quando [S] >> Ks a equação de velocidade simplifica: v0= k3 [E]; nestas circunstâncias
praticamente toda a enzima está complexada com o substrato, está saturada ([Et] ≈ [E•S]), e
nas condições do ensaio não seria possível aumentar a velocidade aumentando a concentração
de substrato. Assim podemos definir o conceito de velocidade máxima como a actividade
enzímica para concentração saturante de substrato: Vmax = k3 ⏐Et ⏐ e nestas circunstâncias v0 =
Vmax. É de notar que o valor de Vmax obtido em determinadas condições de temperatura e pH
pode ser diferente daquele que é obtido noutras condições de temperatura e pH. Vmax apenas
significa v0 para concentrações saturantes de substrato.
Assim podemos escrever a equação de Michaelis-Menten como se segue:
Vmax [S]
v0 = ⎯⎯⎯⎯⎯⎯⎯
Ks + [S]
(6.7.8)
Quando [S]<<Ks a equação de velocidade simplifica: v0 = Vmax [S]/Ks; para muito baixas
concentrações de substrato praticamente toda a enzima está na forma não complexada ([Et]
≈[E]) e a velocidade é directamente proporcional à concentração de substrato. Quando [S] =
Ks então [E] = [E•S] e a enzima está igualmente distribuída entre as formas complexada e
não complexada: v0 = Vmax / 2. Ver Fig.7.
Página 13 de 32
Fig. 7: Distribuição das formas enzímicas complexada e não complexada com o substrato para três
concentrações de substrato. Para valores de concentração de substrato muito superiores a Ks a soma no
denominador da equação 6.7.8 pode simplificar para [S]; a enzima está saturada, v0 ≈ Vmax e a hipérbole, para
estas concentrações de substrato, “tende” para uma recta de declive nulo. Para valores de concentração de
substrato muito inferiores a Ks a soma no denominador da equação 6.7.8 pode simplificar para Ks; v0 é
proporcional a [S] (e a constante de proporcionalidade = Vmax/Ks) e a hipérbole, para estas concentrações de
substrato, “tende” para uma recta de declive = Vmax/Ks. Quando a concentração de substrato é igual a Ks a enzima
está hemisaturada e na equação 6.7.8 o denominador pode simplificar para 2*[S]; v0 = Vmax/2. Ks é a
concentração de substrato para a qual v0 é igual a metade de Vmax. Na equação 6.7.9 Ks será substituído por Km e
todas estas afirmações continuam a fazer sentido.
Um dos pressupostos de Michaelis e Menten, concretamente a imposição de k3<<k2 e a
consequente a necessidade de equilíbrio químico na formação e dissociação de E•S, foi
questionados por Briggs e James Haldane em 1925. Estes autores resolveram
matematicamente o problema que o mecanismo aceite por Michaelis e Menten coloca,
admitindo que a concentração do complexo E•S se mantém constante (estacionária) durante
praticamente todo o processo catalítico; ou seja que a soma das velocidades de desagregação
do complexo E•S (k2 [E•S] + k3 [E•S]) iguala a velocidade da sua formação (k1 [S] [E]).
O desenvolvimento matemático com base neste pressuposto leva a uma equação semelhante à
proposta por Michaelis e Menten mas em que a constante de equilíbrio Ks é substituída por
uma outra Km sendo que Km = (k2 + k3 )/ k1.
Vmax [S]
v0 = ⎯⎯⎯⎯⎯⎯⎯
Km + [S]
(6.7.9)
Km , a constante de Michaelis (em homenagem a Leonor Michaelis) poderia assim ser definida
como o valor da concentração de substrato quando a velocidade de reacção é metade de Vmax
(ver Fig. 8).
Página 14 de 32
Fig.8: Actividade versus concentração de substrato em enzimas com cinética de tipo hiperbólico. O Vmax é o
limite para que tende v0 quando a concentração do substrato S tende para infinito. O Km é a concentração de
substrato para o qual o valor de v0 é metade do valor de Vmax. A equação v0= Vmax [S]/ (Km + [S] ) descreve uma
hipérbole rectangular passando pela origem, com uma assímptota horizontal (y= Vmax) e outra vertical (x=- Km).
Aceitando como Michaelis que k3<<k2 a constante de Michaelis será a constante de
dissociação do complexo E•S em S + E (enzima livre). Se aceitarmos tal como Briggs e
Haldane o valor do Km (Km = (k2 + k3 )/ k1) será tanto maior quanto maiores forem os valores
das constantes cinéticas associadas à dissociação do complexo E•S (em E+S no caso de k1;
em E+P no caso de k3) e tanto menor quanto maior for o valor da constante cinética associada
à formação do complexo E•S. Em ambos os casos o valor do Km é uma medida da afinidade
do substrato em relação à enzima (quanto maior for o seu valor menor será a afinidade).
Como Briggs e Haldane podemos aceitar que Km = Ks + k3/k1; nesse caso Km pode ter um
valor da mesma ordem de grandeza de Ks se admitirmos que o valor de k3/k1 não é muito
grande relativamente ao valor de Ks.
Para determinadas condições de ensaio os valores de k1, k2 e k3 são constantes e portanto
constante o valor de Km; este valor não varia com a quantidade de enzima e é portanto uma
característica da actividade dessa enzima.
6.8. Influência da concentração de substrato em enzimas “com cinética de tipo
cooperativo ou sigmoide”.
a) Quando se estuda a influência da concentração de alguns substratos na actividade de
algumas enzimas podem obter-se gráficos v0 versus concentração de substrato com a forma de
um sigmoide (ver Fig. 6). Este fenómeno ocorre por exemplo nos casos da glicocínase, da
fosfofrutocínase-1, da cínase do piruvato, da desidrogénase do glutamato, da
amidofosforibosil-transferase e da transcarbamílase do aspartato.
Para baixas concentrações de substrato a actividade cresce de forma exponencial com a
concentração deste. Para altas concentrações de substrato manifesta-se o fenómeno da
saturação; o facto de existir uma quantidade limitada de enzima no meio de ensaio implica
que, quando a enzima está próxima da saturação, as variações da concentração de substrato
provocam modificações mínimas na actividade.
Página 15 de 32
b) Às cinéticas de tipo sigmoide chamam-se também de tipo cooperativo ou de
cooperatividade positiva. A palavra cooperatividade surgiu na sequência das teorias que
foram formuladas para explicar o traçado sigmoide nos gráficos que relacionam a quantidade
de oxihemoglobina (hemoglobina•O2) com a pressão parcial de oxigénio (pO2); a chamada
curva de saturação da hemoglobina.
A hemoglobina não é uma enzima mas sim um proteído tetramérico, em que cada um dos
quatro monómeros (muito semelhantes entre si) pode ligar uma molécula de O2. Um outro
proteído capaz de ligar o O2 é a mioglobina mas é monomérica e apenas pode ligar uma
molécula de O2 por molécula. Em contraste com a hemoglobina a curva de saturação da
mioglobina é uma hipérbole (como a das enzimas de tipo michaeliano).
Estes factos induziram Jacques Monot, Wyman e Changeux a propor em 1965 um modelo
teórico que explica a sigmoidicidade da curva de saturação da hemoglobina. Resumidamente
o modelo de Monot, Wyman e Changeux admite a existência de dois estados conformacionais
possíveis para a molécula tetramérica da hemoglobina: R e T; estas duas conformações
estariam em equilíbrio químico e, na ausência de O2 a forma T seria largamente
predominante. Admite ainda que a afinidade do O2 para a forma R é muito superior à sua
afinidade para a forma T; a introdução e o enriquecimento em O2 num meio contendo
hemoglobina aumentaria a razão entre o número de tetrameros na forma R e T.
Assim a ligação de uma molécula de O2 à hemoglobina faz aumentar a concentração da forma
R (a forma que melhor liga o O2) facilitando a ligação de outras moléculas de O2. As
moléculas de O2 já ligadas na hemoglobina cooperam na ligação das que podem ser ligadas se
a pO2 for aumentada.
Um modelo alternativo foi proposto por Koshland em 1966 desenvolvendo para o caso de
proteídos (ou enzimas) com mais que um local de ligação para o ligando (ou o substrato) um
modelo por si formulado em 1959 (o modelo de encaixe induzido). Este modelo, na sua
formulação mais simples, admite que a ligação de uma molécula de O2 num dos monómeros
da hemoglobina induz uma modificação conformacional na estrutura desse monómero; esse
monómero modificado pode influenciar a conformação dos outros protómeros de um mesmo
tetramero. No caso da hemoglobina a ligação do oxigénio induziria uma forma
conformacional com maior afinidade para o oxigénio.
Em ambas as teorias a ligação de uma molécula de ligando (o oxigénio ou um substrato) a
um dos monómeros da proteína em análise influência positivamente a ligação de uma segunda
molécula de ligando aos outros monómeros.
Os modelos matemáticos e as equações associadas a qualquer destas teorias podem
(escolhidos valores adequados para os parâmetros dessas equações) adequar-se de forma
perfeita aos dados experimentais da curva de saturação da hemoglobina.
Essas equações e essas teorias podem também explicar e descrever adequadamente as curvas
sigmoides nos gráficos actividade versus concentração de substrato de todas as enzimas
oligoméricas com mais de um local de ligação para o substrato. No entanto, deixam sem
explicação alguns casos de enzimas monoméricas que apresentam cinéticas de tipo sigmoide,
como por exemplo a glicocínase. Foram propostas teorias que explicam estas situações; em
muitas delas se admite a existência de mais de uma forma conformacional para a enzima.
É de notar que nem todas as enzimas oligoméricas têm cinéticas de tipo cooperativo; por
exemplo os dados experimentais obtidos com a desidrogénase láctica (uma enzima
tetramérica) ajustam-se a uma cinética de tipo michaeliano.
c) As equações resultantes do tratamento matemáticos dos modelos de Monot, Wyman e
Changeux e de Koshland são extremamente complexas; na prática só muito raramente se
usam essas equações para estudar e descrever os resultados experimentais obtidos com
enzimas com cinéticas de tipo cooperativo.
Página 16 de 32
Uma forma mais simples e muito mais comum é usar (para a descrição paramétrica dessas
sigmoides) de forma pragmática uma equação proposta por Hill em 1913 para descrever a
curva de saturação da hemoglobina.
A equação de Hill pode escrever-se da seguinte maneira:
Vmax [S]h
v0 = ⎯⎯⎯⎯⎯⎯⎯
(6.8.1)
(S50)h + [S]h
[S] é a concentração de substrato. S50 é a concentração de substrato para a qual a enzima está
hemisaturada e em que a velocidade é metade de Vmax. A expressão “constante de Michaelis”
(Km) deve ser reservada para as cinéticas hiperbólicas. h é o coeficiente de Hill e é uma
medida do grau de cooperatividade (ou sigmoidicidade): quando h=1 a equação acima
simplifica e é igual à equação de Michaelis-Menten (ausência de cooperatividade), quando
h>1 dizemos que a cooperatividade é positiva e o gráfico v0 versus ⏐S⏐ é um sigmoide (ver
Fig. 9).
No caso da hemoglobina a equação de Hill teria de ser reescrita:
Y= pO2h / ((p50)h + pO2h)
(6.8.2)
Y é a fracção de hemoglobina saturada (o número de protómeros ligados ao O2 / número total
de protómeros), p50 a pressão de oxigénio correspondente a hemisaturação e pO2 a pressão de
oxigénio. Para a hemoglobina, Hill determinou experimentalmente o valor h como sendo 2,8.
Koshland propôs um outro parâmetro (RS = índice de cooperatividade) como forma de medir
a sigmoidicidade das curvas em análise. RS seria a razão entre as concentrações de substrato
(ou de ligando) requeridas para obter para obter um v0 de 90% de Vmax (ou 90% de saturação)
e um v0 de 10% de Vmax (ou 10% de saturação). Rs é igual a 81 nos casos de cinética
michaeliana e tem um valor inferior a este nos casos de enzimas com cinéticas de tipo
cooperativo.
d) Por razões que serão discutidas à frente é frequente chamar às enzimas que exibem uma
cinética de tipo cooperativo enzimas alostéricas. No entanto estas duas expressões não são
sinónimas: existem muitas enzimas sensíveis a efectores alostéricos e que podem por isso
chamar-se também de alostéricas que não apresentam cinéticas de tipo cooperativo.
10
Vmax
h=4
h=2
h=1
Vo
Vmax/2
5
0
0
5
10
S50
15
[Substrato]
Fig.9: Gráficos v0 versus [S] usando a equação 6.8.1 para diferentes valores de h; S50=5 e Vmax = 10 para todos
os casos. S50 é a concentração de substrato para v0= Vmax/2.
Página 17 de 32
6.9. Representações gráficas lineares nas enzimas “de cinética michaeliana” e de
“cinética de tipo cooperativo”.
a) Os resultados experimentais do estudo da influência da concentração de substrato na
actividade enzímica adaptam-se bem à equação de Michaelis-Menten em muitas enzimas
(enzimas com cinética michaeliana). Nestes casos os valores de Km e de Vmax descrevem de
modo adequado o resultado da experiência. Embora actualmente existam métodos estatísticos
sofisticados e inclusive programas informáticos para calcular a partir dos dados experimentais
os valores destes dois parâmetros continuam a ser usados métodos de representação gráfica
para o seu cálculo.
A representação gráfica de Lineweaver-Burk ou de dupla inversão é sem dúvida a mais
popular e merece uma referência neste texto.
Invertendo ambos os termos na equação de Michaelis-Menten (ver equação 6.7.9) obtemos:
1
Km
⎯⎯ = ⎯⎯⎯
v0
Vmax
1
⎯⎯
[S]
+
1
⎯⎯⎯
Vmax
(6.9.1)
O gráfico 1/v0 versus 1/[S] é uma recta de declive positivo (Km / Vmax) que intercepta o eixo
das ordenada num ponto cujas coordenadas são (0,1/ Vmax) e o eixo das abcissas noutro ponto
de coordenadas (-1/ Km, 0). Ver Fig. 10.
Assim um método simples de calcular o Km e o Vmax a partir de resultados experimentais é
começar por calcular os inversos 1/ v0 e 1/[S], desenhar os pontos correspondentes às
coordenadas (1/[S]i , 1/v0 i ) e desenhar uma recta que una esses pontos. O inverso do valor da
ordenada na origem será o Vmax; o simétrico do inverso do valor de x onde a recta cruza o eixo
das abcissas é o Km.
É de notar que num gráfico de Lineweaver-Burk os pontos mais próximos dos eixo das
ordenadas são os que representam as concentrações de substrato mais elevadas. Assim o
ponto de abcissa zero representa uma concentração infinita de substrato; de facto o Vmax é um
parâmetro e raramente um resultado experimental que se obtenha directamente. Um cálculo
simples com a equação de Michaelis-Menten leva-nos a concluir que quando a concentração
de substrato é 10 vezes superior ao Km, v0 será 90,9 % do Vmax.
Página 18 de 32
1/vo
declive = Km / Vmax
1 / Vmax
1/[S]
-1/Km
Fig.10: Gráfico de Lineweaver-Burk ou de dupla inversão em enzimas com cinética de tipo hiperbólico. A
equação 6.9.1 mostra que neste tipo de cinética o gráfico 1/v0 versus 1/[S] é uma recta de declive positivo. Os
pontos da recta à esquerda do eixo das ordenadas não têm significado experimental pois não existem
concentrações negativas de substrato. No entanto podem ter um significado cinético: o valor no eixo das abcissas
para o ponto de intercepção da recta com o eixo das abcissas é -1/Km. Quando associado a duas enzimas (ou a
uma mesma enzima em condições de ensaio distintas) existem dois valores de Km distintos o valor mais alto
corresponde com um ponto de intercepção mais próximo do ponto 0,0. O valor no eixo das ordenadas para o
ponto de intercepção da recta com o eixo das ordenadas é 1/Vmax. Quando associado a duas enzimas (ou a uma
mesma enzima em condições de ensaio distintas) existem dois valores de Vmax distintos o valor mais alto
corresponde com um ponto de intercepção mais próximo do ponto 0,0.
b) A equação de Hill também é linearizavel:
v0= Vmax [S]h / ((S50)h + [S]h)
(6.9.2)
Vmax - v0 = Vmax - Vmax [S]h / ((S50)h + [S]h)
(6.9.3)
Vmax - v0 = Vmax (S50)h / ((S50)h + [S]h)
(6.9.4)
v0
⎯⎯⎯⎯
Vmax - v0
=
Vmax [S]h /((S50)h + [S]h)
[S]h
⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯ = ⎯⎯⎯⎯
Vmax (S50)h / ((S50)h + [S]h)
(S50)h
log
v0
⎯⎯⎯⎯
Vmax - v0
[S]h
[S]
= log ⎯⎯⎯⎯ = h log ⎯⎯⎯
(S50)h
(S50)
log
v0
⎯⎯⎯⎯
Vmax - v0
= h log [S] - h log (S50)
(6.9.5)
(6.9.6)
(6.9.7)
O gráfico log [v0 / (Vmax - v0) ] versus log [S] é uma recta cujo declive é h e a ordenada na
origem é o simétrico de (h log (S50)). A aplicação desta equação a resultados experimentais
pode permitir determinar graficamente os valores de h e de S50 se previamente o valor de Vmax
tiver sido estimado. Ver Fig. 11.
Página 19 de 32
vo
log
declive = h
Vmax-vo
log [S]
-h log S50
log S50
Fig.11: Gráfico linear obtido a partir do desenvolvimento matemático da equação de Hill. A equação 6.9.7
mostra que o gráfico log (v0/( Vmax- v0)) versus log ⏐S⏐ é uma recta de declive positivo h. Quando v0 = Vmax/2 a
concentração de substrato é S50, a fracção v0/( Vmax- v0) toma o valor 1 e o log desta mesma fracção é zero.
6.10. Modificadores da actividade enzímica: inibidores e activadores.
Quando comparamos um par de ensaios enzímicos feitos nas mesmas condições (inclusive a
concentração de substrato), excepto a presença e a ausência de uma substância M, pode
acontecer que as actividades sejam significativamente diferentes. Nestes casos pode ser útil
usar o conceito de grau de modificação (inibição ou activação) induzido por M (inibidor ou
activador). O grau de inibição pode ser definido como a variação de actividade provocada
pelo inibidor relativamente ao ensaio em que ele estava ausente: = (v0 - v0inibidor) /v0. No caso
de activação o grau de activação seria: = (v0activador - v0) /v0. Ver Fig. 12.
140
80
}
∆v
{
o
o
60
40
20
+inibidor
Actividade enzímica
100
+activador
∆v
120
0
Fig. 12: Pela adição de determinadas substâncias ao meio de ensaio podemos eventualmente aumentar ou
diminuir a actividade catalítica da enzima. Quando em determinadas condições de ensaio uma substância pode
aumentar a actividade enzímica diz-se um activador; se diminui a actividade catalítica diz-se um inibidor. O grau
de activação ou de inibição pode ser expresso em valor percentual e neste caso o seu valor será 100*∆v0 /(v0 na
ausência de modificador).
6.11. Inibidores competitivos. Sua representação gráfica linear.
Quando se estuda o efeito da concentração de substrato na actividade podemos, multiplicando
o número de tubos, fazê-lo também na presença de várias concentrações de uma substância I:
um inibidor da actividade da enzima.
Algumas vezes observa-se que o inibidor não altera o Vmax mas que é o valor de Km que
cresce linearmente com o valor de I. O grau de inibição é marcado para baixas concentrações
de substrato diminuindo ou podendo mesmo deixar de observar-se inibição para altas
concentrações de substrato. Ver Fig. 13.
Página 20 de 32
Nos casos em que a cinética é hiperbólica um mecanismo enzímico como o que é apresentado
a seguir pode explicar o fenómeno:
k1
E+S
+
I
k5
k3
E•S ⎯
⎯→ E + P +....
k2
k6
E•I
O esquema acima representa um mecanismo em que E, I e E•I estão em equilíbrio químico
durante todo o processo catalítico; existe assim uma constante Ki (constante de inibição) que
relaciona as concentrações de equilíbrio destas três substâncias e que pode ser definida como
a constante de dissociação do complexo E•I.
[I] [E]
Ki= ⎯⎯⎯⎯⎯
[E•I]
k6
= ⎯⎯
k5
(6.11.1)
O desenvolvimento matemático deste modelo conduz à seguinte equação:
Vmax [S]
v0 = ⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯
Km (1 + [I] / Ki ) + [S]
(6.11.2)
Esta equação mostra que o valor da constante de Michaelis para um determinado valor de
[I], a constante de Michaelis aparente (=observável) nessas condições pode ser dada pela
expressão: Km (aparente) = Km (1 + [I] / Ki ) = Km + ( Km / Ki ) [I]. A constante Ki pode ser
redefinida como a concentração de inibidor que dobra o valor de Km; quanto menor for o seu
valor maior será a capacidade inibidora da substância I.
Ambos os termos da equação 6.11.2 podem ser invertidos dando origem à equação de
Lineweaver-Burk:
1
Km
1
⎯⎯ = ⎯⎯⎯ ( 1 + [I]/ Ki ) ⎯⎯
v0
Vmax
[S]
+
1
⎯⎯⎯
Vmax
Página 21 de 32
(6.11.3)
Vmax
10
0.9
9
S
vo
0.8
1/vo
8
0.7
7
6
S+I
0.6
S+I
0.5
5
0.4
4
0.3
3
S
0.2
2
1/Vmax
1
0
0.1
0
0
10
Km Km'
20
30
40
-0.3
0.2
0.7
1/[S]
[S]
-1/Km -1/Km'
Fig.13: Os inibidores competitivos actuam elevando o Km aparente. Do lado esquerdo desta figura a hipérbole S
representa v0 versus ⏐S⏐na ausência de inibidor e a hipérbole S+I v0 versus ⏐S⏐na presença de uma determinada
concentração do inibidor I. Do lado direito estão os gráficos de Lineweaver-Burk correspondentes. Km e Km
representam respectivamente a constante de Michaelis observável na ausência e presença do inibidor. Na
presença do inibidor o valor de Km aumenta mas o valor de Vmax não se modifica: o efeito inibidor anula-se para
concentração “infinita” de substrato e as rectas de Lineweaver-Burk cruzam-se sobre o eixo das ordenadas.
Na posse de dados experimentais podemos num único gráfico desenhar uma família de rectas
(1/v0 versus 1/[S]) cada uma delas correspondendo a ensaios com uma determinada
concentração de inibidor. No tipo de inibição em discussão as rectas cruzam-se entre si sobre
o eixo das ordenadas. Concentrações crescentes de I correspondem-se com rectas com declive
crescente. Ver Fig. 13.
Este tipo de inibição diz-se competitiva pois que, para uma dada concentração de inibidor o
grau de inibição é sempre mais marcado em ensaios com baixas concentrações de substrato
que com altas concentrações de substrato. Aumentando a concentração de substrato pode ser
possível anular (Vmax é invariante) ou pelo menos diminuir o grau de inibição: o substrato e o
inibidor parecem ter afinidade para um mesmo “sítio activo” na enzima e competirem na
ligação a esse “sítio activo”. Se o substrato e I forem estruturalmente semelhantes esta ideia
fica reforçada. Ver Fig. 14.
Algumas vezes uma mesma enzima pode ligar-se a dois substratos diferentes (embora com
estruturas relacionadas) catalisando duas reacções diferentes: ligando A pode catalisar a
transformação A→P, ligando B pode catalisar a transformação B→Q. Neste caso A é um
inibidor competitivo da reacção B→Q e B um inibidor competitivo da reacção A→P. O valor
do Ki A (constante de inibição associada a A na reacção B→Q) é igual a KmA (constante de
Michaelis associada a A na reacção A→P); também KiB = KmB.
Página 22 de 32
Fig. 14: Modelo interpretativo na inibição de tipo competitivo. Os factos observados nas experiências de cinética
enzímica usando inibidores competitivos podem ser interpretados da seguinte maneira: no sítio activo da enzima
podem ligar-se de forma reversível quer o substrato quer o inibidor; substrato e inibidor competem por esse
local de ligação mas apenas se geram os produtos da actividade enzímica em estudo quando a enzima está ligada
ao substrato.
6.12. Inibidores não competitivos. Sua representação gráfica linear.
Em situações experimentais semelhantes àquelas que foram descritas no capítulo 6.11 às
vezes observa-se que o inibidor I provoca uma diminuição no Vmax sem provocar modificação
no valor de Km. Se a enzima tem uma cinética de tipo michaeliano dizemos que I é um
inibidor não competitivo. Ver Fig. 15.
Um mecanismo compatível com esta situação seria o representado no esquema a seguir:
k1
E+S
+
I
k5
k3
E• S ⎯
⎯→ E + P +....
+
I
k2
k6
k9
k10
k7
E•I + S
E•I•S
k8
Para que este modelo responda de forma adequada ao problema colocado, nomeadamente que
o valor da constante de Michaelis não seja modificado pela presença de I, teremos ainda de
impor que as constantes de dissociação dos complexos E•I (=Ki1) e E•I•S (=Ki2)
relativamente a I tenham o mesmo valor: Ki1 = Ki2.
Página 23 de 32
[I] [E]
Ki1= ⎯⎯⎯⎯⎯
[E•I]
k6
= ⎯⎯
k5
(6.12.1)
[I] [E•S]
Ki2= ⎯⎯⎯⎯⎯
[E•I•S]
k10
= ⎯⎯
k9
(6.12.2)
O desenvolvimento matemático deste modelo conduz à seguinte equação:
Vmax [Ki / (Ki + [I]) ] [S]
v0 = ⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯
Km + [S]
(6.12.3)
Esta equação mostra que o valor de Vmax para um determinado valor de [I], a velocidade
máxima aparente nessas condições pode ser dada pela expressão: Vmax (aparente) = Vmax [ Ki / (Ki
+ [I])]. A constante Ki pode ser redefinida como a concentração de inibidor que reduz a
metade o valor da Vmax.
Ambos os termos da equação 6.12.3 podem ser invertidos dando origem à equação de
Lineweaver-Burk:
1
Km
1
⎯⎯ = ⎯⎯⎯ ( 1 + [I]/ Ki ) ⎯⎯
v0
Vmax
[S]
+
1
⎯⎯⎯
Vmax
( 1 + [I]/ Ki )
(6.12.4)
Na posse de dados experimentais podemos num único gráfico desenhar uma família de rectas
(1/v0 versus 1/[S]) cada uma delas correspondendo a ensaios com uma determinada
concentração de inibidor. No tipo de inibição em discussão as rectas cruzam-se entre si sobre
o eixo das abcissas. Concentrações crescentes de I correspondem-se com rectas com declive
crescente. Ver Fig. 15.
O modelo pode ser descrito da seguinte maneira: quer a enzima livre E, quer o complexo E•S
podem ligar I com uma afinidade idêntica; I ligado à enzima não perturba a ligação de S à
enzima mas impede que a forma ligada a S seja capaz de gerar P. O inibidor I actua como se
eliminasse enzima activa da solução; de facto parte da enzima fica excluída do processo
catalítico. O modelo é compatível com a existência na enzima de um local distinto do “sítio
activo” onde I pudesse ligar-se; a ligação de I nesse local poderia provocar uma modificação
conformacional na enzima; na presença de I a enzima podia ligar o substrato mas não podiam
desencadear-se as reacções necessárias para a formação dos produtos. Se a estrutura de I for
muito diferente de S esta hipótese aparece reforçada. Ver Fig. 16.
Estamos assim a colocar a hipótese de que I se liga à enzima um sítio diferente do sítio activo,
um sítio alostérico (do Grego: allos, outro + stereos, espaço). Dixon e Webb num célebre
livro chamado “Enzymes” assim como Sols nos seus últimos escritos (ver referências)
comentam acerca da confusão de conceitos existente na literatura científica a propósito do
termo alostérico. Segundo estes autores alostérico deve ser um adjectivo que classifica um
activador ou um inibidor que actua ligando-se à enzima num local distinto do(s) substrato(s),
o seu efeito (o efeito alostérico), o local onde se ligam (o sítio alostérico) ou uma enzima
(enzima alostérica) onde ocorrem efeitos alostéricos. É frequente encontrar na literatura
científica a expressão enzima alostérica como sinónimo de enzima com cinética de tipo
cooperativo. Compreende-se o uso dessa expressão porque nestas enzimas é extremamente
Página 24 de 32
frequente observar efeitos comprovadamente alostéricos e no caso de poderem ligar mais de
uma molécula de substrato em distintos monómeros pode considerar-se que o local de ligação
de uma molécula de substrato é um sítio alostérico em relação ao local de ligação da segunda
molécula de substrato. Neste caso o substrato ligado num monómero pode ser designado de
modificador alostérico homotrópico usando-se a expressão heterotrópico para classificar os
modificadores alostéricos que não são substratos.
Como já referido as expressões enzima alostérica e enzima com cinética de tipo cooperativo
(sigmoide) não são sinónimas porque a existência de efeitos alostéricos não é exclusivo das
enzimas com gráficos v0 versus concentração de substrato de tipo sigmoide. O antónimo de
alostérico é isostérico e um efeito inibidor de tipo competitivo baseado num mecanismo de
competição com o substrato em relação ao sítio activo da enzima podia classificar-se desta
maneira. A possibilidade de um inibidor se ligar no sítio activo (inibidor isostérico) e ser um
inibidor não competitivo também existe. Se a ligação do inibidor ao centro activo é de tipo
covalente e estável (a velocidade com que se pode dissociar do centro activo é muito baixa ou
nula) e impede a ligação do substrato as moléculas de enzima ligadas ao inibidor estão
excluídas do processo catalítico.
Fig. 15: Os inibidores não competitivos actuam diminuindo o Vmax aparente e o grau de inibição é invariante
com a concentração de substrato. Do lado esquerdo desta figura a hipérbole S representa v0 versus [S] na
ausência de inibidor e a hipérbole S+I v0 versus [S] na presença de uma determinada concentração do inibidor I.
Do lado direito estão os gráficos de Lineweaver-Burk correspondentes. Vmax e Vmax’ representam
respectivamente o Vmax observável na ausência e presença do inibidor. Na presença do inibidor o valor de
Vmax diminui mas o valor de Km não se modifica: as rectas de Lineweaver-Burk cruzam-se sobre o eixo das
abcissas.
Página 25 de 32
Fig. 16: Modelo interpretativo na inibição de tipo não competitivo. Os factos observados nas experiências de
cinética enzímica usando inibidores não competitivos podem ser interpretados da maneira que a seguir se
descreve. Na enzima, para além do sítio activo onde o substrato pode ligar-se, existe um outro local onde, de
forma reversível, se pode ligar o inibidor. A enzima ligada ao inibidor pode ligar o substrato mas não é capaz de
gerar produtos. Para que de facto o valor do Km não seja modificado pela presença de inibidor temos de admitir
que a afinidade das formas enzímicas E e E•S pelo inibidor são iguais, i.e. Ki1 e Ki2 têm o mesmo valor.
6.13. Efeitos alostéricos.
a) Em enzimas com cinéticas de tipo cooperativo (mas não exclusivamente nestas como já
referido) é frequente observar efeitos activadores ou/e inibidores que foram interpretados
como estando relacionados com a interacção reversível de efectores (substâncias activadoras
ou inibidoras), com um ou mais sítios alostéricos existentes na enzima. Frequentemente, estes
efeitos, observados in vitro, foram interpretados como tendo importantes implicações
metabólicas na fisiologia química da célula. A ligação do efector (um ligando L) à enzima
provocaria uma alteração na conformação da enzima que se reflectiria numa modificação na
sua actividade catalítica. Outro possível mecanismo seria a existência prévia de duas formas
enzímicas (duas conformações) em equilíbrio químico com actividades distintas e que L teria
maior afinidade para uma das formas. Por exemplo, se L tem maior afinidade para a forma
enzímica com menor actividade aumentar a concentração de L no meio de ensaio corresponde
a aumentar a concentração da forma menos activa da enzima à custa da diminuição da forma
mais activa: L seria, neste caso, um inibidor.
Em alguns casos o efeito activador ou inibidor relaciona-se com uma alteração no valor do
Vmax relativamente a um ou mais dos substratos da enzima. Outras vezes o efeito activador ou
inibidor relaciona-se com uma alteração no valor do Km (ou do S50) relativamente a um ou
mais dos substratos da enzima. No primeiro caso diz-se que há um efeito de tipo V e no
segundo um efeito de tipo K e uma combinação dos dois também é possível. De notar que um
efeito de tipo K activador significa um diminuição no valor de Km (ou de S50), ou seja um
aumento da afinidade da enzima para o substrato; um efeito de tipo K inibidor significa um
aumento no valor de Km (ou de S50), ou seja uma diminuição da afinidade da enzima para o
substrato. Ver Fig. 17.
Em enzimas com cinéticas de tipo cooperativo em relação com um substrato S no qual se
pode observar um efeito K activador é frequente observar-se paralelamente com uma
diminuição do valor de S50 uma diminuição da sigmoidicidade da curva que representa a
Página 26 de 32
função v= ƒ ([S]); para valores altos de concentração de activador o aspecto sigmoide da
curva pode desaparecer e surgir um aspecto hiperbólico. Para o caso de inibidores de tipo K é
frequente observar-se o oposto: um aumento do valor do S50 e, paralelamente, um aumento
da sigmoidicidade da curva em análise. Ver Fig. 18.
Em algumas enzimas, como no caso da redútase dos nucleotídeos, podem observar-se efeitos
alostérico que se reflectem em alterações na especificidade em relação com determinados
substratos.
Em algumas enzimas oligoméricas compostas por subunidades diferentes observou-se que os
efeitos catalíticos da enzima estavam relacionados com um tipo de subunidades e que os
efeitos alostéricos com um segundo tipo: ou seja que o(s) centro(s) activo(s) e o(s) centro(s)
alostérico(s) se situavam em diferentes monómeros da enzima.
b) Em geral quando se fala de efeitos alostéricos pensa-se num local da enzima diferente do
sítio activo no qual o ligando L pode ligar-se de forma não covalente e que a velocidade com
que se atinge o estado de equilíbrio químico nessa ligação é extraordinariamente rápida e não
catalisada enzimicamente. Algumas enzimas podem apresentar conformações e características
cinéticas muito distintas consoante estão numa forma desligada ou ligada a um ou mais
grupos fosfato (ligação covalente de tipo fosfoester num resíduo aminoacídico hidroxilado
distante do sítio activo) sendo a transformação reversível entre as duas formas catalisada
enzimicamente por outras enzimas. Neste caso não é costume falar de efeito alostérico
embora de facto o local de ligação do fosfato seja distante do sítio activo.
Vmax
+Activador
14
+Activador
vo 12
Vmax
Vmax 10
9
8
10
vo
8
Vmax
+Inibidor
7
6
5
6
4
+Inibidor
4
3
2
2
1
0
0
0
10
20
30
40
[S]
S50
0
10
20
30
40
[S]
Km Km Km
Fig. 17: Efeitos alostéricos activadores e inibidores de tipo V (à esquerda) e de tipo K (à direita) em enzimas
com cinéticas de tipo sigmoide. Existe um efeito activador ou inibidor de tipo V quando, respectivamente o Vmax
aumenta ou diminui por acção do efector. No caso de efeitos de tipo K um efeito activador significa uma
diminuição no valor do S50 e um efeito inibidor um aumento no valor de S50.
Página 27 de 32
sem efector alostérico
vo
+ATP
+CTP
0
10
20
30
40
Aspartato (m M)
Fig. 18: Efeito de tipo K inibidor (CTP) e activador (ATP) sobre a actividade catalítica da transcarbamilase do
aspartato. A transcarbamilase do aspartato catalisa a seguinte reacção: fosfato de carbamilo + aspartato → Pi +
carbamil-aspartato; esta reacção integra-se na via metabólica da síntese dos nucleotídeos pirimidínicos. Na
presença de CTP o valor de S50 para o substrato aspartato aumenta e paralelamente aumenta a sigmoidicidade da
curva v0 versus ⏐aspartato⏐. O contrário ocorre na presença do ATP.
7. Para que pode servir o estudo cinético de uma enzima?
O estudo cinético de uma enzima informa-nos acerca do modo como a actividade da enzima
se modifica quando se alteram as condições do meio em que esta está a ser ensaiada.
a) Essas observações levantam uma pergunta: porquê? Como se poderia explicar este
comportamento cinético?
A tentativa de responder a esta pergunta levou à elaboração de modelos teóricos e matemáticos que visam fornecer uma explicação para os factos observados. Em grande medida
essas teorias limitam-se a propor um ou mais mecanismos de reacção compatíveis com as
observações feitas. Os mecanismos compatíveis com os dados experimentais podem
eventualmente ser testados por novas experiências de cinética enzímica mas o mais frequente
é que estudos químicos e estruturais da enzima em análise, dos substratos ou de enzimas com
actividades semelhantes forneçam dados decisivos para uma melhor compreensão da maneira
como esta funciona.
b) Outra pergunta pode ser colocada, e não é seguramente menos importante: para quê? Para
que podem servir os dados de observação das experiências de cinética enzímica?
b.1) Como já referido, o valor da actividade catalítica de uma determinada enzima num meio
especificado é directamente proporcional à quantidade de enzima presente nesse meio. Esta
observação assim como as características de especificidade (de substrato e tipo de reacção),
actividade versus temperatura e pH e a acção de modificadores da actividade de uma enzima
podem permitir, com base em estudos de cinética, o desenho de condições de ensaio
adequadas ao doseamento de uma enzima numa preparação biológica complexa. É importante
escolher condições de ensaio em que outras enzimas possíveis interferentes tenham baixa
actividade e em que a enzima em estudo tenha uma alta actividade. É frequente a escolha de
concentrações de substratos bastante mais altas que os Km (ou S50): nestas condições a
actividade enzímica é pouco sensível à diminuição da concentração de substrato durante o
ensaio tornando mais fácil estimar v0.
Página 28 de 32
O interesse desses métodos de doseamento é enorme e alguns exemplos podem ser referidos:
a) Em certas doenças, como por exemplo a pancreatite aguda, certas enzimas que estão
normalmente presentes no plasma sanguíneo em quantidades vertigiais, aumentam a sua
concentração no plasma sanguíneo. Nesta situação patológica o conteúdo enzímico das
células pancreáticas exócrinas é vertido no plasma. Uma actividade enzímica específica pode
ser avaliada através de um ensaio enzímico e é uma medida da quantidade de enzima presente
no plasma. O doseamento da actividade amilolítica no soro sanguíneo é um ensaio
laboratorial muito importante no diagnóstico da pancreatite aguda.
b) O efeito de hormonas e de condições fisiológicas (jejum, por exemplo) ou patológicas
(doenças genéticas em que uma enzima não é sintetizada ou é anormal sob o ponto de vista
funcional, por exemplo) nos níveis tecidulares de uma determinada enzima pode ser estudado
avaliando a actividade dessa enzima no tecido em estudo.
b.2) As características cinéticas de uma enzima, nomeadamente a especificidade em relação a
substratos, a inibidores, a activadores e ao tipo de reacção podem também ser usadas no
doseamento e na síntese de compostos químicos. O contributo da cinética enzímica para o
desenvolvimento de métodos de análise em contextos médicos (análises clínicas), científicos,
industriais ou de investigação criminal não pode deixar de ser realçado. Um exemplo é o uso
comum de uma enzima de origem bacteriana altamente específica, a glicose oxidase, no
doseamento da glicose.
b.3) O estudo cinético de uma enzima é importante para caracterizar essa enzima, desta maneira distinguindo-a funcionalmente das demais enzimas inclusive das que podem catalisar a
mesma reacção.
Dentro da mesma espécie biológica, duas enzimas estruturalmente distintas podem catalisar a
mesma reacção. Chamam-se a estas enzimas isoenzimas. Pode acontecer que estas enzimas
sejam funcionalmente indistinguíveis mas em alguns casos um estudo cinético sistemático
pode mostrar algumas diferenças como por exemplo no valor do Km (ou do S50) em relação a
um ou mais substratos, na especificidade, activação ou inibição por certos efectores, nos
valores de pH óptimo, etc. Exemplos são a hexocínase cerebral e a glicocínase hepática: para
além de especificidades diferentes tem valores de Km muito distintos em relação ao substrato
glicose e apenas a primeira é inibida pela glicose-6-P.
b.4) Os estudos in vitro da actividade das enzimas foram e são de extraordinária relevância no
esclarecimento das vias metabólicas dos seres vivos e da sua regulação.
O reconhecimento da existência (identificação) de cada uma das enzimas, das reacções por
elas catalisados (identificação de seus substratos e produtos) num determinado organelo
celular, célula, tecido ou organismo são aspectos chave na compreensão do metabolismo em
geral e das vias metabólicas em particular. O reconhecimento in vitro da possibilidade de
inibir uma enzima purificada com um determinado inibidor pode servir de base para
experimentar esse inibidor em sistemas menos artificiais como organelos celulares, células ou
animal de experiência. Os resultados dessas experiências podem fornecer dados importantes
para a compreensão da via metabólica em que essa enzima está integrada.
O efeito da concentração de substratos ou de outros factores como, por exemplo, substâncias
que adicionadas no meio de ensaio modificam a actividade de uma enzima pode fornecer
pistas para a descoberta da forma como na célula a actividade da enzima é regulada e orientar
a investigação futura quer de cinética enzímica quer por outros métodos. Normalmente é
muito difícil conhecer com precisão as concentrações estacionárias celulares dos
intermediários metabólicos quer porque estão habitualmente em concentrações muito baixas
quer porque no momento em que a célula é destruída para se fazer a análise esta concentração
Página 29 de 32
pode alterar-se dramaticamente. Apesar disso a estimativa das concentrações de substratos ou
de outras substâncias (como efectores da actividade enzímica) e a sua comparação com as
suas constantes de afinidade em relação a uma enzima podem ser decisivos para compreender
a regulação dessa enzima a nível celular.
Pode acontecer, como por exemplo no caso da glicocínase hepática, que o valor do Km para a
glicose seja mais alto que a concentração habitual de glicose dentro do hepatócito: a enzima
funciona in vivo em condições de sub-saturação no que respeita à glicose. Por isso, flutuações
na concentração estacionária da glicose resultantes, por exemplo, de situações pós-pandriais
ou de jejum, refletem-se em alterações na actividade desta enzima. Outro exemplo:
demonstrou-se que a fosfofrutocínase-1 é inibida in vitro de forma alostérica por
concentrações de ATP e de citrato da ordem de grandeza das encontradas na célula;
flutuações nessas concentrações são considerados factores importantes na regulação da
actividade dessa enzima a nível celular.
b.5) Muitos dos medicamentos usados no tratamento de doentes são inibidores específicos da
actividade de determinadas enzimas. Daremos três exemplos:
i) As sulfamidas são antibióticos que exercem a sua acção numa enzima bacteriana da via da
síntese do ácido fólico competindo com o seu substrato natural (são inibidores competitivos
isostéricos). Essa enzima não existe no homem (o ácido fólico é, para o homem, uma
vitamina) e portanto em doses adequadas as sulfamidas podem matar as bactérias sem
provocar efeitos graves no metabolismo humano.
ii) O metotrexato é usado no tratamento de doenças neoplásicas inibindo por mecanismo
competitivo isostérico a dihidrofolato redútase, uma enzima que catalisa a conversão do
dihidrofolato em tetrahidrofolato. As células neoplásicas reproduzem-se rapidamente e nestas
a síntese de ADN e dos nucleotídeos percursores é extremamente rápida. Para que a síntese de
um destes precursores (o TTP) possa ocorrer é necessária a síntese de tetrahidrofolato que está
dependente da actividade da dihidrofolato redútase. Assim em doses adequadas o metotrexato
pode impedir a multiplicação das células neoplásicas embora também exerça efeitos menos
desejados em células que, fisiologicamente, se multiplicam rapidamente.
iii) O alopurinol é um medicamento usado no tratamento da gota e exerce a sua acção por ser
um inibidor potente da xantina oxídase, uma enzima da via metabólica onde se sintetiza o
ácido úrico. A gota é uma doença em que as concentrações de ácido úrico estão muito
aumentadas no meio interno e estas altas concentrações, independentemente das causas que as
provocam, são nocivas para o organismo.
8. Nota final: a actividade das enzimas como factores de regulação do metabolismo.
Na célula a actividade de uma enzima pode depender de múltiplos factores.
Se, no citoplasma da célula ou num organelo celular, a razão entre as concentrações estacionárias de produtos e reagentes de uma reacção catalisada por uma determinada enzima
(seja A+B → P+Q) não se afasta muito da Keq podemos deste facto deduzir duas
consequências: i) que flutuações nessas concentrações estacionárias podem fazer com que na
reacção enzímica celular umas vezes predomine a reacção em sentido directo (A+B →P+Q) e
noutras a reacção em sentido inverso (P+Q →A+B) e ii) que a actividade dessa enzima é
suficientemente alta a nível celular para não ser um passo limitante da velocidade da via
metabólica em que está inserida. A regulação da actividade deste tipo de enzimas depende
basicamente da concentração de reagentes e produtos e da sua relação com os Km (ou S50) a
eles associados.
Noutras reacções a actividade da enzima a nível celular é relativamente baixa e o valor da QR
(tendo em conta concentrações estacionárias de reagentes e produtos) é muito mais baixo,
Página 30 de 32
pelo menos em condições fisiológicas, que o da Keq; neste caso a reacção progride na célula,
macroscopicamente, apenas em um dos sentidos (seja o sentido A+B→P+Q). Neste tipo de
enzimas é frequente observar-se que a sua actividade pode variar em função de outros factores
e mecanismos para além da concentração de substratos ou produtos (de acordo com as
premissas formuladas P e Q, embora não sejam, pelo menos na célula, substratos da enzima,
podem funcionar a nível celular como inibidores da transformação A+B→P+Q). Entre esses
factores ou mecanismos possíveis podem destacar-se:
i) Efeitos alostéricos inibidores exercidos por efectores que são produtos finais da via
metabólica em que a enzima se insere (inibição de tipo feed-back).
ii) Efeitos alostéricos activadores exercidos por efectores que são substratos da via metabólica
em que a enzima se insere (não necessariamente substratos da enzima), por efectores cuja
concentração está sujeita a regulação hormonal (caso da frutose-2,6-bisfosfato) ou por
substâncias indicadoras de déficit relativo de um produto dessa via metabólica (caso do AMP
e sua acção activadora na cínase-1 da frutose-6-P).
iii) Efeitos inibidores ou activadores resultantes da fosforilação covalente ou desfosforilação
da própria enzima através da acção catalítica de outras enzimas também estas reguladas por
mecanismos mais ou menos complexos.
iv) Alteração da quantidade de enzima na célula mediada através de efeitos positivos ou
negativos nos seus mecanismos de síntese ou de degradação.
De notar que as hormonas e os neurotransmissores podem exercer um papel determinante na
regulação das vias metabólicas através de vários mecanismos nos quais a regulação da
actividade enzímica é a chave do processo. Entre esses mecanismos podemos destacar:
i) Promoção da síntese de enzimas específicas.
ii) Promoção do transporte de substâncias através das membranas desta forma possibilitando
o acesso de substratos ou de reguladores da actividade enzímica (nomeadamente iões como o
cálcio), às enzimas.
iii) Promoção da síntese ou da degradação de efectores da actividade enzímica.
Página 31 de 32
9. Bibliografia consultada:
Cornish-Bowden A., Cárdenas M.L, 1987, Mini Review. Co-operativity in Monomeric Enzymes. J.
Theor. Biol. , 124: 1-23.
Dixon M & Webb E.C., 1979, Longman Group Ltd. Enzymes. 3rd edition. Great Britain.
Engel P.C.. 1981, Brammer WJ & Edidin M. eds. Enzyme Kinetics. A Steady-state Approach. 2nd
edition. London: Chapman and Hall.
Macarulla J.M., Marino A., Macarulla A., 1992. Editorial Reverté, S.A. Bioquímica Cuantitativa.
Barcelona
McGilvery R. W., 1983, W. B. Saunders Company. Biochemistry . A Functional Approach. 3rd
edition. Japan.
Murray R.K., Granner D.K., Mayes P.A. & Rodwell, 1990, Appleton & Lange. Harper’s
Biochemistry. 22nd edition. USA.
Nomenclature Committee of the International Union of Biochemistry. 1982. Symbolism and
Terminology in Enzyme Kinetics-recommendations 1981. Eur. J. Biochem: 128: 281-291.
Sols A. 1981. Multimodulation of Enzyme Activity. in Current Topics in Cellular Regulation. Vol. 19.
Academic Press, Inc., New York. Pag 77-101.
Sols A. 1990. Enzyme Regulation: from Allosteric Sites to Intracellular Behavior. in Selected Topics
in the History of Biochemistry: Personal Recollections, III. Vol. 37. Elsevier Sciences Publishers., Pag
177-199.
Voet D. & Voet J. G., 1990, John Wiley & Sons, Inc. Biochemistry. USA.
Página 32 de 32
Baixar




![+ [E] [S] - Sistemas EEL - Universidade de São Paulo](http://s1.livrozilla.com/store/data/000150897_1-f33431079ef06af434a6636d8f76dc77-260x520.png)