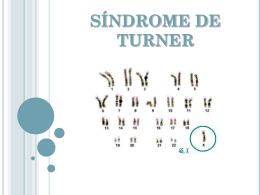
Revista Eletrônica Novo Enfoque, ano 2010, v. 11, n. 11, p. 34 – 42 ESTUDO CITOGENÉTICO DE 27 PACIENTES ENCAMINHADOS POR SUSPEITA DE sÍNDROME DE tURNER Costa, F.B.1,2; Aragão, H.L.P.2; Ramos, M.C.2; Bastos, E.F.2, 1Universidade Castelo Branco – Aluna do curso de Biomedicina 2Setor de Citogenética – Laboratórios Médicos Dr. Sérgio Franco RESUMO A síndrome de Turner (ST) é um distúrbio cromossômico caracterizado pela monossomia total ou parcial do cromossomo X cujas características fenotípicas incluem principalmente alterações no desenvolvimento sexual e um padrão específico de malformações congênitas como pescoço alado e “cubitus valgus”. Corresponde ao tipo de alteração cromossômica mais frequente em amostras de abortos espontâneos ocorrendo em aproximamente 1:3000 nativivos do sexo feminino. Diferentes cariótipos já foram descritos associados a ST com uma maior frequência do cariótipo clássico (45,X). O presente estudo tem por objetivo caracterizar citogeneticamente 27 casos de pacientes encaminhadas para avaliação citogenética por suspeita clínica de síndrome de Turner e correlacionar nossos achados laboratoriais com os dados da literatura realizando uma revisão atualizada dos diferentes tipos de cariótipos que podem ser encontrados nos pacientes com esta síndrome. Palavras-chave: Síndrome de Turner. Cariótipo. Bandeamento. Citogenética. INTRODUÇÃO A síndrome de Turner (ST), descrita em 1938 por Henry Turner, é um distúrbio genético de origem ainda desconhecida e sem qualquer afinidade hereditária, está associada à monossomia cromossômica e ocorre em aproximadamente 1:3000 nativivos do sexo feminino. É decorrente da presença de um cromossomo X e perda total ou parcial do segundo cromossomo sexual. (1-2) Geralmente não possuem cromatina sexual e muitas dessas concepções terminam em aborto, sendo provável que 97% desses conceptos sejam eliminados chegando a termo apenas 3%. (5) As características das recémnascidas portadoras podem ser semelhantes às crianças normais, exceto pelo baixo peso e, eventualmente, pelo linfedema das extremidades (pés e mãos). Dessa forma, a maioria dos diagnósticos ocorre por ocasião da primeira infância em razão da baixa estatura acentuada ou durante a puberdade, diante da ausência de desenvolvimento dos caracteres sexuais secundários e amenorreia primária ou secundária. Na idade adulta, essas mulheres podem apresentar todo o espectro fenotípico da síndrome, como a tireoidite autoimune e anomalias renais e cardíacas, dependendo da extensão e da localização da lesão cromossômica. O perfil hormonal é característico em razão da insuficiência ovariana, acarretando baixos níveis de estrógeno e altos níveis dos hormônios folículo estimulante e luteinizante (FSH e LH, respectivamente). A infertilidade apresenta-se na quase absoluta maioria como um quadro referente a alguns folículos. A surdez é uma complicação comum em todas as idades. Postula-se o irreversível. (2) Poucos são os casos relatados de Turner férteis, em cujos ovários certamente que as mulheres com ST têm risco elevado de apresentar baixa autoestima e, por conseguinte, qualidade de vida social comprometida. (3) É importante que o acompanhamento destas pacientes seja feito visando a antecipar, prevenir e tratar as complicações mais frequentes. O tratamento de reposição hormonal melhora bastante a qualidade de vida e deve ser oferecido a todas as pacientes. (4) Citogeneticamente, a Síndrome de Turner caracteriza-se pela presença de um cromossomo X normal e pela perda parcial ou total do outro cromossomo sexual, (7) apresentando um cariótipo do tipo 45,X, sendo os portadores fenotipicamente fêmeas. Os portadores apresentam estatura retardada, ovários rudimentares, não apresentam menstruação e sem as características sexuais secundárias. (6) Aproximadamente 50% da pacientes com Síndrome de Turner apresentam cariótipo 45,X e 25% apresentam mosaicismo 45,X/46,XX. Mas outras anomalias como isocromossomo de braço longo também podem estar presentes. (7) Outras alterações numéricas também podem estar presentes: 45,X/47,XXX e 45,X/46, XX/47, XXX, havendo pouca descrição fenotípica por se apresentarem poucos casos descritos. (7) O cariótipo 45,X/46,XX, geralmente apresenta menos anomalias, mas é necessário levar em consideração a proporção de cada linhagem celular. (7) Já o cariótipo 45,X é conhecido como clássico da Síndrome de Turner e apresenta o fenótipo de baixa estatura, disgenesia gonadal e as alterações descritas anteriormente. Mas não só apenas alterações numéricas que caracterizam a Síndrome de Turner. Alterações estruturais também se fazem presente. O isocromossomo do braço longo do X pode estar presente em 46, X,i (Xq) ou 45,X,i (Xq). Deleções também podem estar presentes como: 46, X, del (Xp) ou 45,X/46,X, del (Xp) e parecem ser menos afetadas do que 45,X. Se a deleção comprometer apenas a extremidade distal do braço curto do X, apresenta poucos sinais clínicos. Mas se compromete a maior parte ou todo o braço curto, o diagnóstico pode se confundir com o cariótipo 45, X, pois apenas metade dessas pacientes apresenta amenorreia primária. (7) Já quando o cariótipo se apresenta como 46, X, del (Xq) ou 45,X/46,X, del (Xq), são vistos poucos sinais fenotípicos, pois essas pacientes apresentam estatura normal e quase sempre são detectadas pela amenorreia primária ou secundária. (7) Alterações como cromossomo X em anel (r), translocações X/X e X/autossomo são as mesmas das descritas nas deleções dos braços curto e longo. (7) Estudos mostram que cerca de 78% de casos de monossomia do cromossomo X o gameta sem o cromossomo sexual têm origem paterna. 35 Outros 22% têm origem materna. Essa não disjunção paterna pode ser explicada pela não disjunção XY ter uma maior tendência de ocorrer, pois no caso do XY, o pareamento é feito pela ponta, tornando este par mais suscetível à não-disjunção que os demais. (7) No caso do cariótipo 46, X,i (Xq), o gameta que contém o isocromossomo pode ter se originado de um erro na duplicação do centrômero na meiose materna ou paterna (sendo de probabilidade igual) ou pode haver uma translocação entre os dois braços longos de cromossomos X homólogos na linhagem germinativa. (7) Cariótipos 45,X/46,XX podem ter origem na perda de um dos cromossomos X em uma das divisões iniciais do embrião, caracterizando assim um mosaicismo. Linhagens celulares aneuploides como 45X/46,XX/47,XXX podem ter origem de uma não disjunção de um dos cromossomos X em uma das divisões iniciais do embrião. (7) Algumas deleções de regiões do X podem ter origem em translocações presentes em estado equilibrado nos pais ou podem ter ocorrido em sua linhagem germinativa. (7) Cromossomo X anormal pode ser encontrado em mosaico com linhagem 45,X. Também pode ocorrer a perda do cromossomo anormal na linhagem 45,X. Ambas as linhagens também podem ter se originado simultaneamente, como resultado da segregação mitótica após troca anormal entre cromátides-irmãs. (7) Assim sendo, o diagnóstico da síndrome de Turner é feito através da realização de cariótipo e deve ser suspeitado em pacientes do sexo feminino com quadro clínico sugestivo, onde meninas com estatura abaixo do alvo genético para a idade devem ser investigadas, pois este pode ser o único achado em alguns casos. (4) Este estudo visa a caracterizar citogeneticamente 27 pacientes encaminhadas por suspeita clínica de síndrome de Turner; correlacionar nossos achados laboratoriais com os dados da literatura além de fazer uma revisão atualizada dos diferentes tipos de cariótipo que podem ser encontrados nos pacientes com esta síndrome. Método Foram selecionados 27 casos, entre janeiro de 2009 e março de 2010, encaminhados por suspeita clínica de ST ao Setor de Citogenética do Laboratório Sérgio Franco Medicina Diagnóstica. A análise citogenética foi realizada em cromossomos metafásicos corados por bandeamento GTG e classificados segundo o Sistema Internacional de Nomenclatura Citogenética (ISCN, 2009) (10). Para a obtenção dos cromossomos (prometafásicos e metafásicos) utilizou se a técnica descrita por Moorhead e cols., (1960) (11). Foram colhidos 5ml de sangue periférico dos pacientes com seringa descartável heparinizadas. Foram implantadas 12 gotas de sangue total em 6,0 mL de meio de cultura RPMI – 1640, contendo antibióticos (penicilina – 5.000.000 U) e estreptomicina (0,0004 g/mL (Cultilab). Após 71 horas de incubação, a cultura foi interrompida adicionando-se 0,2mL de colchicina (Colcemid – Cultilab) numa concentração de 10mg/mL. Após 1 hora, o material foi centrifugado, desprezando-se o sobrenadante e ao pellet celular foram adicionados 6,0 mL de solução hipotônica (KCL 0,075M), permanecendo por 20 minutos na estufa a 37°C. Após sucessivas fixações com metanol/ácido acético (3:1), as células foram 36 ressuspensas em um volume menor de fixador para posterior preparo das lâminas. Para o bandeamento GTG (Seabright, 1971) (9), as lâminas com cerca de 5 dias de preparo foram tratadas com uma solução de tripsina e coradas Giemsa a 3% segundo protocolo apropriado. Os cromossomos foram analisados em microscopia ótica e classificados de acordo com o Sistema Internacional de Nomenclatura para a Citogenética Humana (ISCN -2009); e o cariótipo montado utilizando um programa de cariotipagem Cytovision. (10). Fig. 1 - Cariótipo 45,X observado na maioria dos casos investigados. RESULTADOS Na tabela 1 mostra a relação de encontrados. A idade das pacientes doa casos investigados com a suspeita variou entre 0 e 36 anos. Clínica e os resultados citogenéticos INDICAÇÃO CLÍNICA IDADE CARIÓTIPO Síndrome de Turner? 0 45,X [20] Síndrome de Turner? 0 45,X[20] Síndrome de Turner? 6 45,X[20] Síndrome de Turner? 6 45,X[20] Hipogonadismo, hipogonadotrófico 13 45,X [20] Hipogonadismo 15 45,X[20] 37 Síndrome de Turner, hipogonadismo, hipogonadotrófico 15 45,X[20] Síndrome de Turner? 15 45,X [20] Síndrome de Turner? 16 45,X [20] Síndrome de Turner? 16 45,X [20] Disgenesia gonadal, síndrome de Turner? 18 45,X [20] Hipertireoidismo 36 45,X [20] Amenorreia 14 45,X[16]/46,XX[4] Atraso no desenvolvimento 1 45,X [4]/46,X,r (?) [16] Síndrome de Turner? 11 45,X [16]/46,X,r (?) [4} Baixa estatura, osteopenia difusa 11 45,X [12]/46,X.r (?) [8] Baixa estatura, microcefalia, micromelia, deformidade de Maderling, antecedente de PIG, atraso do desenvolvimento cognitivo 13 45,X [15]/46,X,r (?) [5] Atraso no crescimento, amenorreia 14 45,X [8]/46,X,r (?) [12] Síndrome de Turner? 3 45,X [2]/46,X,i (X)(q10)[18] Síndrome de Turner? 3 45,X [2]/46,X,i (X)(q10)[16] Síndrome de Turner? 24 45,X [4]/46,X,i (X)(q10)[16] Menopausa precoce 29 45,X [12]/47,XXX [8] Síndrome de Turner? 10 46,XX,inv per (9) [20] Síndrome de Turner, baixa estatura 10 46,XX[30] Síndrome de Turner, baixa estatura 12 46,XX[30] 38 Síndrome de Turner? 12 46,XX[30] Síndrome de Turner? 14 46,XX[30] Tabela 1: Relação de casos com indicação clínica, idade e resultado citogenético. Fig. 2 – Anel de cromossomo X (seta) em paciente com cariótipo 45,X/46,X,r (?). Fig 3 – Isocromossomo de Xq (seta) observado em paciente com cariótipo 45,X/46,X,i (X)(q10). 39 Fig 4 - Cariótipo 47,XXX em paciente em mosaico. CONCLUSÃO A suspeita diagnóstica da síndrome pode ser feita em neonatos do sexo feminino pela observação do pescoço alado e do linfedema. Se a ST não for diagnosticada na lactância ou infância, geralmente será diagnosticada tardiamente na fase pós-púbere devido à ocorrência de baixa estatura e/ou amenorreia, sendo necessária, em ambos os casos, uma confirmação citogenética (8). Todos os casos avaliados neste estudo possuíam indicação clínica de ST ou uma ou mais características incluídas nos “estigmas” de Turner, como: baixa estatura, hipogonadismo etc. A baixa estatura está relacionada à ausência de um gene do cromossomo X que, em mulheres saudáveis, a sua duplicidade determina estatura normal. No caso da ST devido à monossomia do X, esse gene perde seu homólogo determinando assim anormalidade na estatura (8). A hipótese que explica essa diferença se faz porque a inativação do X em mulheres normais não é completa, existindo vários locos que podem escapar a essa inativação. No braço curto dos cromossomos X, há necessidade de um gene aparecer em dose dupla para o desenvolvimento gonadal normal: o gene ZFX. Por isso, mulheres com monossomia de X, isocromossomo de braço longo do X e deleções de braço curto, apresentam não só disgenesia gonadal como também os estigmas somáticos característicos da Síndrome, existindo evidências de que os genes do braço curto do X situados à extremidade distal e ao centrômero não se inativem quando em hemizigose tornando-se responsáveis pela baixa estatura. (7) Assim como descrito na literatura, nossos resultados mostram uma maior frequência de pacientes com cariótipo 45,X (Fig.1) que correspondeu 44,44% do total de casos investigados. A segunda anormalidade cromossômica mais frequente em nosso estudo foi à presença de uma alteração estrutural, cromossomo em anel (Fig.2) no braço curto de um dos cromossomos X. Todas as nossas amostras com deleção de X apareciam em mosaico com a presença simultânea de uma linhagem cromossômica X0. Este achado está, provavelmente, relacionado à instabilidade do cromossomo com alteração estrutural que pode ser perdido durante as sucessivas divisões celulares do desenvolvimento. 40 Outra alteração estrutural bastante encontrada em nosso estudo foi a presença de um isocromossomo de X (Fig. 3), cromossomo este constituído da perda do braço curto com duplicação do braço longo de X. Esta alteração também apareceu em mosaico, possivelmente, pela mesma origem descrita acima. Em um caso, encontramos o cariótipo 45,X/47,XXX (Fig. 4) correspondendo a uma alteração numérica originada, possivelmente, de uma não disjunção mitótica. Esta paciente foi encaminhada por apresentar menopausa precoce. Através deste estudo, também demonstramos que em 85,18% dos casos encaminhados para avaliação, o diagnóstico citogenético da síndrome de Turner foi confirmado, evidenciando mais uma vez a importância do estudo citogenético para o fechamento do diagnóstico clínico. Nos casos cuja citogenética não confirmou a síndrome, não podemos afastar a possibilidade de alteração a nível molecular ou a presença de um número muito pequeno de células monossômicas que pode não ter sido alcançado através da quantidade de células avaliadas. Assim sendo, a caracterização da Síndrome de Turner se dá através de diagnóstico clínico, mas também com a comprovação pelo cariótipo o que deveria ser feito logo nos primeiros meses de vida, em pacientes do sexo feminino com quadro clínico sugestivo, e em meninas com estatura abaixo da média para a idade, com a finalidade de proporcionar um direcionamento mais eficaz no tratamento e possibilitar uma qualidade de vida melhor para as portadoras da síndrome. REFERÊNCIAS 1-SURIZAN, Lígia Z C; SILVA, Roberto B P; MARINI, Sofia H V L; ET AL. A percepção da doença em portadoras da síndrome de Turner. Jornal de Pediatria Copyright © 2004 by Sociedade Brasileira de Pediatria. http://www.scielo.br/pdf/jped/v80n4/v80n4a11.pdf ) 2-CHVATAL, Vera Lúcia Soares; BÖTTCHER-LUIZ, Fátima; TURATO, Egberto Ribeiro. Respostas ao adoecimento: mecanismos de defesa utilizados por mulheres com síndrome de Turner e variantes. The defenses employed by women with Turner syndrome: dealing with the disease. www.scielo.com.br 3-JUNG, Monica de Paula; CARDOSO, Maria Helena Cabral de Almeida; VILLAR, Maria Auxiliadora Monteiro; LERENA JR, Juan Clinton. Revisitando o desvendamento da etiologia da síndrome de Turner. www.scielo.com.br 4-FONSECA,Krukemberghe. Síndrome de Turner. Acesso em 15/11/2009. http://www.brasilescola.com/biologia/sindrome-de-turner.htm. 5- http://www.portalsaofrancisco.com.br/alfa/sindrome-de-turner/. Acesso em 20/11/2009. 6- http://www.crazymania.com.br/biblioteca/citogenética_humana 7- OTTO, Priscila G; OTTO, Paulo Alberto; FROTA, Oswaldo. Genética Humana e clínica. 2 ed. São Paulo: Rocca, 2004. 8- SÍNDROME DE TURNER. Acesso em 12/11/2009. http://www.sindromes.org/sindromedeturner/ 9- SEABRIGTH, M. A rapid banding technique for human chromosomes. Lancet, 2: 971-972, 1971. 41 10- ISCN 2009: An International System for Human Cytogenetics Nomenclature, Recommendations of the International Standing Committee on Human Cytogenetic Nomenclature. Published in collaboration with 'Cytogenetic and Genome Research' Editor(s): Shaffer, L.G.; Slovak ML, Campbell LJ. 11- MOOREHEAD, P. S; NOWELL, P.C.; HELLMANS, W. J. et al. Chromosomes preparations of leucocytes cultures from peripheral blood. Exp. Cell Res, 20:613-16,1960. 42
Baixar