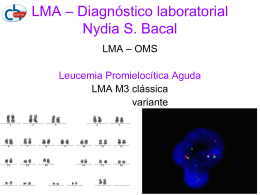
LEUCÓCITOS MORFOLOGIA, FUNÇÃO E ANOMALIAS LEUCOCITÁRIAS Profa. Dra. Larissa Gorayb F Mota Hematologia II – 8° Semestre/ Farmácia GRANULOPOESE GRANULÓCITOS GRANULÓCITOS - FUNÇÃO Células móveis: CS Parede dos vasos Partículas estranhas • ADESÃO: parede de vasos • DEFORMAÇÃO: atravessam pequenos pertuitos • QUIMIOTAXIA: encontram partículas estranhas ao meio CITOESQUELETO: proteínas contráteis formadas de microtúbulos e microfilamentos Movimentação Pseudópodos Material englobado Fagossoma enzimas MPO (atua na presença de H2O2) GRANULÓCITOS - FUNÇÃO • Relação entre enzimas, proteínas contráteis e membrana celular (receptores) Integridade do aparelho locomotor Presença de receptores Substâncias elaboradas ADESÃO QUIMIOTAXIA • Deficiência de proteínas de membrana: adesão imcompleta, falha na quimiotaxia e fagocitose ANOMALIAS LEUCOCITÁRIAS granulócitos • Alterações na forma e função dos leucócitos mononucleares • Células hipofuncionantes: manifestações clínicas - Anomalias leucocitárias propriamente ditas: alteração de forma função (variável) - Anomalias funcionais dos leucócitos ANOMALIAS LEUCOCITÁRIAS PROPRIAMENTE DITAS ANOMALIA DE PELGER-HÜET • Hereditária autossômica dominante • Anomalia na forma do núcleo dos neutrófilos – frequente - Falta de segmentação com assincronismo de maturação - Núcleos arredondados em bastão (lentes de óculos) - Citoplasma c/ coloração e granulações normais • Forma heterozigótica e homozigótica: sem deficiência funcional • Leucemias, metástases medulares, mielomas, malária NEUTRÓFILOS NORMAIS E PH ANOMALIA DE MAY-HEGGLIN • Hereditária autossômica dominante • Alteração no citoplasma de granulócitos e monócitos - Granulações grandes contendo RNA - Presença de plaquetas gigantes, deficientes (n° e forma) HIPERSEGMENTAÇÃO DOS NEUTRÓFILOS • Constitucional ou devida a def. vitamina B12 e folatos • Neutrófilos com > número de segmentos (+5/célula) • Rara GIGANTISMO DOS NEUTRÓFILOS • Hereditária autossômica dominante • Rara e sem repercussão para a função celular • Neutrófilos normais: 12 µm Gigantismo: 16 µm ANOMALIA DE ALDER-REILLY • Grânulos de linfócitos ou neutrófilos, monócitos • Granulações lisossomais acumuladas citoplasma • Comum em mucopolissacaridoses • Sem alteração de função c/ quadro variável: lesões oculares ósseas cardíacas neurológicas ANOMALIA DE ALDER-REILLY ANOMALIAS FUNCIONAIS DOS LEUCÓCITOS SÍNDROME DO LEUCÓCITO PREGUIÇOSO • Alteração na polimerização das proteínas contráteis • Motilidade dos neutrófilos: lentos e quimiotaxia deficiente • Rara. Capacidade de morte bacteriana comprometida SÍNDROME DE CHÉDIAK-HIGASHI-STEINBRINCK • Hereditária autossômica recessiva • Defeito genético na membrana lisossomal: grânulos gigantes def. quimiotaxia MPO anormal ↓defesa imunológica • Quadro clínico grave – homozigótica (incompatível c/ sobrevida) heterozigóticos (+ brando) • Albinismo total ou parcial, manifestações neurológicas adeno-hepato-esplenomegalia e DEFEITO NA GERAÇÃO DE H2O2 • Doença granulomatosa crônica: puramente funcional - Geração deficiente de H2O2 decorrente da ↓NADPH e NADH (precursores) • Deficiência de 6-GPD: indivíduos c/ def. nos eritrócitos podem ter def. nas células granulocíticas - Polimerização anormal de tubulina • Deficiência de MPO: sem quadro clínico severo desde que outros mecanismos estejam normais ANOMALIAS DEVIDAS A ALTERAÇÕES EXTRACELULARES • Deficiência de fatores quimiotáticos: Fagócitos (neutrófilos e monócitos) – normalidade intrínseca e produção de substâncias que atuam na quimiotaxia Agentes quimiotáticos importantes: natureza proteica frações do complemento C3 e C5 síntese e taxas - Defeito na síntese proteica: inibidores ou inativadores das frações do SC alteração no mecanismo inflamatório - Alguns exemplos: deficiência de quimiotaxia em diabéticos e artrite reumatóide drogas antiinflamatórias: fenilbutazona, corticóides ANOMALIAS DEVIDAS A ALTERAÇÕES EXTRACELULARES • Deficiência de opsoninas: substâncias que atuam na fagocitose por neutrófilos e monócitos (IgG, IgM, fatores de complemento) ATIVIDADE OPSONIZANTE - Defeito na síntese de proteínas: falha na opsonização e fagocitose > suscetibilidade à infecções DOENÇAS PROLIFERATIVAS DA LINHAGEM MIELÓIDE DOENÇAS PROLIFERATIVAS DA LINHAGEM MIELÓIDE • Diferentes enfermidades: proliferação anormal das células mielóides granulocíticas • Manifestação evidente no início da doença: ↑ células indiferenciadas, (leucemia mielóide aguda) jovens: blastos mielóides • Proliferação granulocítica evidente: células maduras com raras (leucemia mielóide crônica) formas blásticas • Síndrome mieloproliferativa • Síndrome mielodisplásica LEUCEMIA MIELÓIDE AGUDA (LMA) • Natureza malígna: proliferação anômala dos precursores granulocíticos da MO STEM CELL PARADA OU DIFICULDADE DE MATURAÇÃO • Qualquer etapa da granulocitogêse: formas indiferenciadas ou formas diferenciadas classificação LMA LMA - ETIOPATOGENIA • Hemopatias malígnas: fatores ambientais, herança e fatores individuais - Fatores ambientais: radiações (γ ou β) infecções (virais) condições sócio-econômicas - Herança genética: doenças constitucionais - Fatores individuais: tabagismo stress ↓ resistência física atividade profissional LMA - ETIOPATOGENIA • Doenças proliferativas: descontrole no mecanismo regulador de crescimento celular Sequência de ác. nucleicos c/ informações definidas GENES estimuladores ou inibidores (equilíbrio de função) • Fatores externos causam lesões cromossômicas: quebras, translocações, inversões, perdas, alteração de sequência aa mutação genética • Locais das alterações: genes de regulação de crescimento celular ficam com função modificada (oncogenes) LMA - INCIDÊNCIA • LMA : indivíduos de todas as idades, raça e sexo • OMS: final da 2ª década de vida (+50% dos casos) sexo masculino (60%) LMA - CLASSIFICAÇÃO • Aspecto morfológico: células predominantes • Padrão universal: FAB (grupo franco-americano-britânico) análise de sg periférico e MO por esfregaço % MIELOBLASTOS associada a % outros blastos M1, M2, M3, M4, M5a, M5b, M6 e M7 • Classificação MIC: morfológica, imunológica e citogenética reavaliação de M1 a M7: biologia da cél. leucêmica LMA - CLASSIFICAÇÃO MIC • Citoquímica e imunofenotipagem: origem das leucemias desde células mais indiferenciadas • Citogenética: correlação entre os tipos FAB c/ translocação (M2, M3, M5a) eosinofilia e inversão (M4) cultivo, coloração, visualização em microscópio • Imunofenotipagem: presença de marcadores imunológicos de 2 linhagens ex.: céls. leucêmicas c/ marcadores CD7 e CD13 formas híbridas, leucemias bifenotípicas Fase precoce no desenvolvimento celular citometria de fluxo, biologia molecular DIAGNÓSTICO CLÍNICO E DIFERENCIAL - LMA • Sintomas: palidez cutâneo-mucosa, febre e hemorragias, hepatomegalia, esplenomegalia, alterações da pele, sintomas neurológicos, dores ósseas, sinais de insuficiência respiratória, infiltrados tumorais células leucêmicas descontrole proliferação/maturação • Predileção à tecidos ↑ vascularizados e fç hemoformadora: CS hepato e esplenomegalia SNC, globo ocular • Diagnóstico diferencial: esfregaços de sg e MO com ↑ % de blastos hemograma c/ plaquetopenia, leucocitose (variável para formas subagudas e crônicas - anemia) pré-leucemia: anemia s/ alterações leucocitárias e plaquetárias - acompanhamento DIAGNÓSTICO DIFERENCIAL - LMA • Quando a anemia é presente: macrocítica/megaloblástica • Diagnóstico diferencial entre LMA-M6 e an. Megaloblástica DOSAGEM DE VITAMINA B12 E FOLATOS DIAGNÓSTICO LABORATORIAL - LMA • Hemograma e mielograma (colorações citoquímicas: peroxidase, sudan black) • Diagnóstico difícil: biópsia óssea (condições do tec. medular) fibrose, infiltração leucêmica, edema, necrose mau prognóstico • Radiografias e ultrassom: infiltrações localizadas • Exames de líquor, fezes, urina; culturas, reações sorológicas • Dosagens bioquímicas • Fundo de olho; coagulograma • citoquímicos; imunofenotipagem CLASSIFICAÇÃO FAB DAS LMA BLASTOS - LMA TRATAMENTO • Medidas de Suporte: melhora das condições gerais, ↓ riscos transfusões He/plaquetas; desinfecção (ATBterapia); ↓ingestão verduras cruas; ↓ hiperuricemia (alopurinol) • Tratamento Específico: Uso de quimioterápicos em combinação drogas quimioterápicas: daunomicina (DRM), citarabine (Ara-C), tioguanina (6TG) V.O: Protocolo DAT VP 16 (epipodofilotoxina) – M4 ou M5 - Tratamento de indução da remissão - Tratamento pós-indução TRATAMENTO QUIMIOTERÁPICO (Qt) - FASES • Indução da remissão: Remissão total ou completa (RC) da LMA RC: ausência de blastos no sg e 5% na MO qdo há remissão com blastos >5%: RP - parcial - RC: esquemas de Qt em porcentagem variável idade do paciente, grau de infitração leucêmica, condições físicas mais atingida após 1ª indução • Tratamento pós-indução: Manutenção da RC c/ terapêutica < agressiva Consolidação e manutenção da RC Qt: PÓS-INDUÇÃO • Consolidação da RC: 2-6 ciclos de Qt adicionais, semelhantes aos da indução c/ intervalos (2 semanas) • Manutenção da RC: doses < mesmas drogas antileucêmicas ciclos mensais (2-3 anos) – INSTITUÍDA OU NÃO • Intensificação tardia: mesma terapêutica da consolidação c/ intervalos de 4 semanas, fase tardia prevenção da recidiva (céls leucêmicas residuais) - Controle da Qt: mielograma semanal e biópsia MO - 2 semanas após RC início da indução LEUCEMIA MIELÓIDE AGUDA RESISTENTE OU RECIDIVADA • LMA resistentes à Qt clássica: alcançam RC c/ drogas de 2ª linha chances < qdo falha de protocolo • LMA pouco diferenciada: blastos indiferenciados c/ positividade p/ CD34 CD13 e CD33 positivos - Agentes diferenciadores: vit. D3, ác. retinóico, Ara-C ↓ doses (subcut.) - Fatores de crescimento: G-CSF, GM-CSF (granulócitos e monócitos) - Imunomodulação c/ Interleucina 2: ativa linfócitos NK – lise das céls resid. TRANSPLANTE DE MEDULA ÓSSEA (TMO) • Pacientes jovens <30 anos c/ doador compatível (HLA) • Singênico: irmão gêmeo • Alogênico: irmão ou outro doador compatível (HLA não idêntico) • Autólogo: próprio paciente c/ material de fase de RC • FASE CRÍTICA: Mielodepressão e Imunossupressão medidas de suporte: transfusões, fat. crescimento, Interleucinas TMO – CUIDADOS • Qdo transplante autólogo: material medular submetido à Qt • Decisão p/ TMO: idade, disponibilidade de doador, tipo de LMA > 60 anos: recidiva frequente (TMO qdo RC) • Falhas: esterilização por Qt, reação enxerto vs hospedeiro, lesão do estroma medular por Qt agressivas PROGNÓSTICO E EVOLUÇÃO • Qt intensivas: RC em até 85% (c/ fatores influenciadores) • Bom prognóstico: pouca idade, sexo feminino, < céls blásticas, plaquetas (nl), tipos: M1, M2 ou M3, ausência alterações cromossômicas, ausência de infiltração SNC, resposta rápida aos Qt (c/ < ciclos), ausência de hepato-esplenomegalia e de infecções • Uso de TMO alogênico: ↑ possibilidade de tratamento p/ > pessoas • Uso de TMO autólogo: pacientes idosos recebem TMO
Download