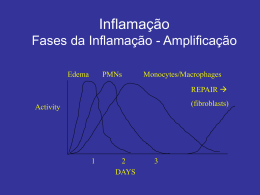
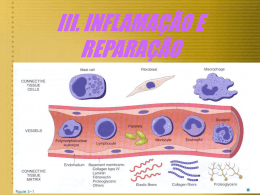
UNIVERSIDADE FEDERAL DE SANTA MARIA CENTRO DE CIÊNCIAS DA SAÚDE DEPARTAMENTO DE PATOLOGIA DISCIPLINA DE PATOLOGIA GERAL VETERINÁRIA (PTG 1002) Prof. Rafael Fighera Inflamação A inflamação é um processo ativo de resposta do organismo contra um agente nocivo, na tentativa de destruí-lo, diluí-lo ou encerrá-lo, ou seja, é uma situação salutar, mas que, em alguns casos, pode ser tão exacerbada a ponto de pôr em risco a vida do indivíduo. Logicamente que se não existisse processo inflamatório as infecções evoluiriam indefinidamente e levariam o individuo à morte. Entretanto, se esse processo for persistente e desimpedido, ele torna-se o próprio agente agressor. O entendimento desse conceito é fundamental para ressaltar que a inflamação tem função, embora isso possa parecer um paradoxo para aqueles indivíduos que padecem das agruras causadas por doenças que tem como base esse mecanismo e necessitam fazer uso constante de medicamentos antiinflamatórios. Durante séculos, muitos conceitos de inflamação foram utilizados, esses conceitos incluíram: 1) “inflamação é a reação dos tecidos a um irritante”; 2) “inflamação é um processo que tem início em seguida a agressão subletal a algum tecido”; 3) “inflamação é a resposta vascular e celular dos tecidos vivos à agressão”; e 4) ”inflamação é a reação dos vasos sanguíneos que leva ao acúmulo de líquidos e leucócitos nos tecidos extravasculares”. O termo inflamação, oriundo do verbo inflamar, provém da transliteração latina do grego inflammare, que significa atear fogo. A história da inflamação, assim como boa parte da história da cirurgia, está intimamente ligada à história das guerras, principalmente pela contaminação das feridas adquiridas nos campos de batalha. Embora haja fortes indícios de que a inflamação já fosse conhecida desde 3.000 a.C., foi apenas no século I que o escritor romano Cornelius Celsus (25 a.C.-50 d.C.) utilizou a expressão “rubor et tumor cum caore et dolore” para descrever o que ele chamou de sinais cardeais da inflamação. Essa expressão, que traduzida significa “rubor e tumor acompanhado de calor e dor”, denota com extrema acuracidade as principais características do processo inflamatório. No século XIX, o patologista alemão Rudolf Virchow (1821-1902) acrescentou a perda da função (function laesa) aos sinais cardeais da inflamação, embora o médico grego Claudio Galeno (129-216 d.C.) frequentemente receba o crédito por esse feito. Durante centenas de anos, desde que foi inicialmente descrita, a inflamação era considerada uma doença, até que em meados do século XVIII, o cirurgião e anatomista escocês John Hunter (1728-1793) cogitou pela primeira vez que a inflamação não só não era uma doença específica como tinha certa função para o organismo. Assim, independentemente do conceito adotado, o leitor deve ter em mente que inflamação é uma resposta protetora que tem como principal função livrar o organismo da causa da agressão (por exemplo, microorganismos) e de suas consequências (por exemplo, tecidos necróticos). O processo inflamatório pode ser dividido em agudo ou crônico, tendo em vista para isso a evolução do quadro. A inflamação aguda é curta, pois varia de horas a poucos dias, já o processo inflamatório crônico é longo, pois perdura de semanas a 1 meses. Alguns patologistas frequentemente utilizam o termo subagudo para descrever inflamações com evolução intermediária, entre uma e três semanas. A maioria dos casos de inflamação crônica evolui de inflamações agudas mal debeladas, o que é denominado cronificação da inflamação. Ocasionalmente, pacientes com inflamações crônicas podem demonstrar sinais clínicos, achados laboratoriais e histopatológicos de inflamação aguda, o que é descrito como agudização da inflamação ou inflamação crônico-ativa. Em relação ao infiltrado inflamatório, sabe-se que neutrófilos e monócitos começam a chegar ao tecido 4-6 e 12 horas após a injúria, respectivamente. Em média, os neutrófilos predominam no tecido durante 6-24 horas e são substituídos por macrófagos em 24-48 horas. O conhecimento básico sobre os tempos de chegada e permanência das células inflamatórias nos tecidos lesados é baseado em técnicas antigas de escarificação da pele, um método conhecido no passado como técnica da janela na pele de Rebuck. Baseado nesse antigo experimento, utiliza-se como regra geral que a inflamação aguda é mediada por neutrófilos e a inflamação crônica é mediada por macrófagos. Embora esse seja um conceito geral válido, é importante saber que existem inúmeras exceções. Por exemplo, em doenças virais, os linfócitos predominam desde o início; em doenças parasitárias, os eosinófilos são vistos do começo ao fim do processo e, em algumas doenças bacterianas, os neutrófilos podem predominar por semanas a meses. Vários tipos celulares podem constituir um quadro inflamatório, alguns deles encontram-se já no tecido afetado, como é o caso dos mastócitos, macrófagos residentes e fibroblastos; já outros adentram o tecido via sangue, são eles: neutrófilos, eosinófilos, basófilos, monócitos, linfócitos e plaquetas. Linfócitos são também frequentemente incluídos como células locais, já que ao realizarem “patrulhamento tecidual” migram constantemente da circulação para o tecido e vice-versa. É importante ressaltar que muitas proteínas e mediadores químicos auxiliam desde a saída das células do interior do vaso sanguíneo até a fagocitose do agente lesivo. Não se deve esquecer também que tanto o processo imune quanto a hemostasia estão intimamente ligados à inflamação, aumentando a complexidade das reações. Embora muitas possam ser as causas da inflamação, elas podem ser divididas em: aquelas incitadas por agentes vivos e por agentes não-vivos. Dentre os agentes vivos causadores de inflamação estão: vírus, bactérias, riquétsias, fungos, protozoários e metazoários. Dentre os agentes não-vivos destacam-se as agressões físicas (trauma, calor, frio e irradiação) e químicas. A inflamação ocorre por cinco mecanismos sequenciais: alteração no calibre vascular e consequentemente no fluxo vascular; aumento na permeabilidade vascular e consequentemente no extravasamento vascular; migração celular e consequentemente acúmulo de leucócitos no local da lesão; ativação celular e fagocitose (Esquema 1). Alteração no calibre vascular As alterações no calibre dos vasos e consequentemente no fluxo desses ocorrem logo após o desencadear da lesão e se caracterizam por uma vasoconstrição neurogênica reflexa seguida de vasodilatação, principalmente de arteríolas. Essa vasoconstrição neurogênica reflexa é a mesma que propicia o início da hemostasia primária. É importante ressaltar que a dilatação desses vasos leva a um maior afluxo sanguíneo, carreando células, proteínas e mediadores químicos não-protéicos até o local desejado. Essa vasodilatação é mantida durante toda a inflamação aguda através da liberação de óxido nítrico, prostaglandina D2 e leucotrieno B4. Essas alterações são responsáveis por 2 dois dos sinais cardeais clássicos da inflamação: rubor e calor. Atualmente, a palavra rubor é menos utilizada e tem sido gradativamente substituída por eritema. Embora esses sinais tenham sido descritos no século I, foi somente no século XIX que o patologista alemão Julius Cohnheim (1839-1884), um discípulo de Virchow, pôde explicá-los com maiores detalhes através da observação microscópica de membranas podálicas, mesentério e língua inflamados de sapos. Aumento da permeabilidade vascular Para que o processo inflamatório prossiga, é necessário que proteínas e mediadores químicos não-protéicos, trazidos pelo maior afluxo sanguíneo, saiam dos vasos e exerçam suas funções no interstício. A estimulação endotelial pelos mediadores químicos não-protéicos permite a passagem dos mediadores químicos protéicos do interior do vaso para o tecido. Com a perda protéica, a pressão oncótica do vaso diminui e a do tecido aumenta; esse fato, associado ao aumento da pressão hidrostática decorrente da vasodilatação, leva à passagem de grande quantidade de líquido rico em proteínas (exsudato) para o tecido (exsudação), ocasionando edema. Essas alterações são responsáveis por dois dos sinais cardeais clássicos da inflamação, tumor e dor, e foram também descritas pelo patologista alemão Julius Cohnheim (1839-1884). Atualmente, a palavra tumor é pouco utilizada para descrever esse tipo de alterações, pois foi gradativamente substituída por edema e tumefação. Baseado nesses aspectos, para que ocorra edema inflamatório, desencadeia-se um mecanismo de extravasamento vascular decorrente da permeabilidade vascular aumentada. Vários desses mecanismos são propostos para explicar esse fenômeno, são eles: formação de lacunas intercelulares, transcitose aumentada, reorganização citoesquelética, lesão endotelial direta, lesão endotelial mediada por leucócitos, extravasamento prolongado tardio e extravasamento de novos vasos sanguíneos. Tais mecanismos foram inicialmente estudados por Sir Thomas Lewis (1881-1945), cardiologista e cientista clínico gaulês, que em 1927 estabeleceu o conceito corrente de que substâncias químicas, como a histamina, induzidas localmente pela lesão medeiam as alterações vasculares da inflamação. A formação de lacunas intercelulares por onde passam proteínas plasmáticas e mediadores químicos é, com certeza, o mecanismo mais óbvio e fácil de ser compreendido. Esse processo é mediado pela histamina, bradicinina, leucotrienos e substância P, que causam contração das células endoteliais de vênulas com diâmetro entre 20 e 60 m. Acredita-se que o motivo pelo qual esse mecanismo afeta apenas vênulas esteja relacionado ao fato desses vasos terem um maior número de receptores para histamina e substância P. A transcitose aumentada é outro mecanismo descrito apenas para vênulas, onde através de “falsos canais” as proteínas plasmáticas podem passar pelo interior das células endoteliais. Esses “falsos canais” são formados por vesículas e vacúolos (organelas vesiculovacuolares) estimulados pelo fator de crescimento endotelial vascular (VEGF). Para alguns autores, a transcitose aumentada pode ser um mecanismo alternativo de formação de lacunas intercelulares e, assim, também estar relacionada à estimulação endotelial pela histamina. A reorganização citoesquelética ou retração endotelial é o processo pelo qual as células endoteliais separam-se uma das outras, quando estimuladas pela interleucina-1, fator de necrose tumoral- (TNF-) e interferon- (IFN-). Assim como no fenômeno anterior, as vênulas são as estruturas vasculares afetadas. 3 A lesão endotelial direta a qualquer tipo de pequeno vaso sanguíneo (vênulas, capilares e arteríolas) causa além de hemorragia, aumento da permeabilidade vascular. Esse é o mecanismo típico de situações em que há necrose endotelial, como, por exemplo, nas queimaduras, mantendo-se até o momento da reparação ou trombose do vaso. A lesão endotelial mediada por leucócitos é outro mecanismo basicamente venular decorrente da liberação de substâncias tóxicas pelos leucócitos, quando do contato com o endotélio, no momento da aderência. Essa forma de aumento da permeabilidade vascular é fundamentalmente importante na patogênese de várias doenças glomerulares e pulmonares, principalmente glomerulonefrites por imunocomplexos e síndrome da angústia respiratória adquirida (SARA), respectivamente. O extravasamento prolongado tardio é uma forma de aumento da permeabilidade vascular que ocorre em vênulas e capilares, algumas horas após a exposição a mediadores químicos. Acredita-se que o próprio efeito direto do agente nocivo ou a liberação de substâncias ainda desconhecidas ocasione essa forma de extravasamento vascular. Um exemplo típico desse tipo de processo é a lesão induzida por radiação ultravioleta e raio x. O extravasamento de novos vasos sanguíneos, formados através da angiogênese, decorre da permeabilidade aumentada desses vasos neoformados, mediada principalmente pelo VEGF. Além disso, sabe-se que as células endoteliais dos vasos neoformados possuem uma densidade de receptores para histamina. Esse tipo de aumento da permeabilidade vascular ocorre tanto em vênulas como em capilares e arteríolas. Migração celular O próximo passo na resposta inflamatória é permitir o extravasamento de leucócitos, a fim de que esses possam chegar ao tecido afetado e assim exercerem suas funções. Basicamente cinco são as etapas para que essa perfeita sequência de eventos denominada migração celular ou extravasamento celular concretize-se, são elas: marginação, rolagem, aderência, transmigração e quimiotaxia. Os mecanismos descritos a seguir são semelhantes para todos os tipos de leucócitos, entretanto, são pequenas diferenças durante a migração que justificam a predominância de cada tipo celular específico em relação à etapa do processo e ao estímulo agressor. A alteração no calibre vascular, mais especificamente a vasodilatação, associada ao aumento da permeabilidade vascular leva ao alentecimento do fluxo sanguíneo e ao aumento relativo na concentração de eritrócitos nos pequenos vasos sanguíneos regionais e, consequentemente, ao aumento da viscosidade sanguínea, um fenômeno denominado estase sanguínea. Essa estase sanguínea, evidente em 15-30 minutos após o desencadear do processo inflamatório, é fundamental para o início da migração celular, pois o sangue que flui dentro do vaso em uma coluna axial central, ou seja, centripetamente, passa para uma situação inversa quando o fluxo sanguíneo diminui, fenômeno denominado efeito centrífugo ou quebra do fluxo axial. Esse efeito leva ao acúmulo de leucócitos na periferia das vênulas, fenômeno denominado marginação, a primeira fase da migração celular. Após a marginação, os leucócitos se aderem transitoriamente ao endotélio e giram sobre seus eixos, situação descrita como rolagem. Em um determinado ponto dessa rolagem, os leucócitos repousam e assim aderem-se firmemente ao endotélio, essa situação é melhor entendida quando comparada a “bolas de gude sobre as quais um riacho corre sem perturbá-las”, conforme mencionado por Tucker Collins, no capítulo 4 sobre inflamação do clássico livro de Robbins. Na histologia, vênulas repletas de leucócitos marginados, aderidos ou não-aderidos ao endotélio, podem ser vistas em áreas muito iniciais de inflamação, quando há apenas edema intersticial, pois os leucócitos ainda não transmigraram. Essa lesão tem sido denominada por alguns patologistas como leucocitostase. Nos casos em que há adesão de muitos leucócitos ao endotélio, ao ponto de dar um aspecto de revestimento interno ao vaso sanguíneo, o fenômeno é denominado pavimentação. Os fenômenos de aderência e rolagem embora pareçam bastante simples são na verdade mecanismos mediados por várias moléculas que atuam ligando os leucócitos ao endotélio vascular, essas moléculas pertencem à quatro famílias: selectinas, imunoglobulinas, integrinas e glicoproteínas semelhantes à mucina (Quadros 1, 2 e 3). A transmigração intercelular ou diapedese é o mecanismo pelo qual o leucócito deixa o vaso sanguíneo, basicamente as vênulas, e chega ao tecido, esse processo é complexo e depende das moléculas de aderência encontradas nas junções entre duas células endoteliais, uma dessas proteínas é a chamada molécula de aderência plaquetária à célula endotelial (PECAM-1 ou CD31), uma imunoglobulina. Após passar pelas junções interendoteliais, os leucócitos adentram o tecido degradando a membrana basal através da secreção de colagenases. Após saírem do vaso, os leucócitos emigram nos tecidos em direção ao local da lesão, por um processo conhecido como quimiotaxia. Nesse ponto, uma pergunta é necessária: como os leucócitos sabem qual caminho seguir, ou seja, como se guiam até o local da injúria se eles não “enxergam”, “escutam” ou “farejam”? A resposta para essa pergunta está na quimiotaxia, um mecanismo pelo qual os leucócitos movimentam-se guiados por um gradiente químico (substâncias quimiotáticas). Basicamente todos os tipos de leucócitos utilizam substâncias quimiotáticas para se orientar, mas os neutrófilos e os monócitos fazem isso com mais eficiência. Várias substâncias encontram-se no grande grupo dos agentes quimiotáticos para neutrófilos, dentre elas: produtos bacterianos (peptídeos com aminoácido terminal n-formilmetionina - FMLP), membros do sistema complemento (C5a), produtos da via lipoxigenase (leucotrieno B4), metabólitos lipídicos (PAF) e quimiocinas (interleucina8). Além de serem auxiliados por substâncias quimiotáticas, os neutrófilos podem ser estimulados a movimentarem-se até o local da injúria por citotaxígenos, que são substâncias capazes de gerar um fator quimiotático após interação com o soro, plasma, componentes do complemento, mastócitos ou linfócitos sensibilizados. Exemplos de citotaxígenos incluem complexos antígeno-anticorpo, endotoxinas, bactérias e diversas enzimas que reagem com membros do sistema complemento ou com constituíntes dos lisossomos. Substâncias quimiotáticas para monócitos incluem: substâncias liberadas por neutrófilos mortos, fragmentos de degradação de colágeno e fibronectina, fator de crescimento derivado de plaquetas (PDGF), membros do sistema complemento (C5a) e quimiocinas, como a proteína inflamatória de macrófagos-1 (MIP-1) e a fractalcina. Fagocitose A palavra fagocitose vem da transliteração latina do grego, em que phagein significa comer, kytos significa vaso oco e refere-se à célula e osis significa doença, processo patológico ou aumento, ou seja, “fagocitose é o ato de comer realizado pela célula”. O termo fagocitose foi empregado pela primeira vez pelo biólogo russo Elie Metchnikoff (1845-1916) em 1882, após observar a ingestão de espinhos da rosa por amebócitos de larvas de estrela-do-mar e de bactérias por leucócitos de mamíferos, fato que lhe valeria compartilhar o prêmio Nobel de Medicina em 1908 com Paul Ehrlich. A fagocitose e a 5 liberação de enzimas pelos fagócitos (leucócitos que realizam fagocitose) se constituem na principal função dessas células. Múltiplas células no organismo possuem a capacidade de realizar fagocitose; em relação a leucócitos, os neutrófilos e os macrófagos são os que realizam essa função com maior eficiência. No entanto, eosinófilos e basófilos também têm capacidade de englobar partículas sólidas. A fagocitose pode ser dividida em três processos, a saber: reconhecimento e fixação, engolfamento e destruição (degradação) do material ingerido. Embora tanto os neutrófilos como os macrófagos possam fagocitar bactérias na ausência de opsoninas (da transliteração latina do grego opson, que significa molho), essas substâncias quimioatraentes aumentam a eficiência da fagocitose, pois partículas suspensas em líquidos possuem cargas elétricas negativas e assim se repelem, esse fenômeno é conhecido como potencial zeta. O conceito de que existiam substâncias na circulação capazes de “encapar” bactérias é datado de 1900. As principais opsoninas são o fragmento Fc da imunoglobulina G (IgG), os fragmentos opsônicos de C3 (C3b e C3bi) e as proteínas de ligação aos carboidratos (colectinas). Essas moléculas possuem receptores específicos na membrana dos leucócitos e assim facilitam a fagocitose. Estes receptores são: FcRI, II e III (CD64, CD32 e CD16, respectivamente) para os fragmentos Fc da IgG; receptores 1, 2 e 3 (CD35, CD21 e CD18, respectivamente) para C3b e C3bi; e receptores C1q para as colectinas. O receptor 3 para C3bi é a mesma molécula de aderência descrita pelo nome MAC-1. A fagocitose que ocorre na ausência de opsoninas é denominada fagocitose de superfície, esse mecanismo foi descrito na década de 1940 por Wood e Irons. O engolfamento consiste na forma mais conhecida da fagocitose, ou seja, na extensão de um pseudópode por sobre o objeto a ser engolfado. Esse processo é estimulado pela ligação da opsonina ou do microorganismo ao receptor específico, ocorrendo semelhantemente ao quimiotaxismo. Durante a ingestão, a partícula circundada pelo pseudópode é invaginada juntamente com a membrana plasmática. Atualmente, diversas teorias sobre processos de invaginação de partículas têm sido propostas, em uma delas relata-se a utilização da membrana do retículo endoplasmático ao invés da membrana plasmática. Após o encerramento da partícula, o fagossoma formado se funde ao lisossomo (grânulo lisossômico, lisossomo primário ou grânulo azurofílico), formando o fagolisossomo. É no fagolisossomo que ocorre a liberação das enzimas lisossômicas que irão degradar o que foi fagocitado. Atualmente acredita-se que grânulos secundários (grânulos específicos) também possam esvaziar-se em vacúolos fagocíticos. Devemos ressaltar que a ligação da célula com a opsonina Fc da IgG é suficiente para iniciar o engolfamento, já no caso da ligação com C3b e C3bi é necessário a ativação pela laminina e pela fibronectina extracelulares ou por certas citocinas. É importante frisar que no processo de fagocitose, assim como em qualquer outro tipo de endocitose, o material levado para o interior da célula em momento algum permanece livre no citoplasma. A destruição do organismo fagocitado depende intimamente da chamada explosão respiratória, mas mecanismos independentes de oxigênio também auxiliam nessa degradação, dentre eles a ação de substâncias dos grânulos, como: proteína bactericida por aumento da permeabilidade (BPI), lisozima, mieloperoxidase, lactoferina, proteína básica principal, defensinas, elastase e citocromo b. É importante ressaltar que as bactérias gram-positivas são rapidamente destruídas, enquanto as gram-negativas são mais resistentes, pois possuem uma parede celular menos digerível. Algumas bactérias específicas como Brucella spp. e Listeria monocytogenes são tão resistentes que se multiplicam no interior dos fagócitos. 6 Quando uma partícula é fagocitada por um leucócito, um processo denominado explosão respiratória (surto oxidativo) é desencadeado, esse fenômeno causa várias alterações celulares, como: aumento no consumo de oxigênio (duas a três vezes), aumento na utilização da glicose através da derivação da hexose monofosfato (via pentose fosfato) e da glicólise anaeróbica (via Embden-Meyerhof), aumento na oxidação do fosfato de nicotinamida-adenina-dinucleotídeo (NADPH) (cinco vezes) e da nicotinamida-adenina-dinucleotídeo (NADH) (30 vezes), aumento na síntese de lipídios de membrana, oxidação dos íons cloreto e iodeto, aumento na produção de metabólitos reativos do oxigênio e incremento na glicogenólise. A explosão respiratória foi descrita inicialmente por Baldridge e Gerard no ano de 1933. Nos últimos anos, o número de informações sobre a explosão respiratória aumentou demasiadamente, isso se deve ao fato de que muito tem se pesquisado sobre o assunto, tendo como base a doença granulomatosa crônica em humanos, um defeito congênito no metabolismo desse importante mecanismo neutrofílico. É interessante se perguntar como que os neutrófilos carregados de radicais livres extremamente tóxicos não danificam o tecido, já que eles são células lábeis, que morrem após a fagocitose. Embora a resposta para essa pergunta não seja uma descoberta nova, seu mecanismo foi descrito apenas recentemente. A morte dos neutrófilos ocorre por apoptose, o que permite a não liberação dos radicais livres no tecido; os macrófagos locais são responsáveis por eliminar essas células do “local da batalha”. Resultados da inflamação aguda Após esta breve revisão sobre os mecanismos inflamatórios, suas células e seus mediadores químicos, é importante lembrar que, na maior parte das vezes, a inflamação aguda resulta na cura com subsequente reconstituição do tecido danificado, seja por resolução completa, através da reparação tecidual por regeneração, por resolução por fibrose, através da reparação tecidual por substituição, ou, mais frequentemente, por uma associação de ambos esses processos. Logicamente que o que se espera de um processo inflamatório agudo é a resolução perfeita, sem sequelas importantes, entretanto, isso nem sempre ocorre e, em alguns casos, o organismo abre mão de outros mecanismos para debelar o problema. Um desses mecanismos é o encapsulamento do agente agressor junto a leucócitos viáveis e degenerados ali presentes (formação de abscesso). Outro mecanismo é a progressão da inflamação aguda para inflamação crônica. Infamação crônica A inflamação crônica é o resultado, na maior parte das vezes, de um processo agudo não-debelado ou ainda, decorrente de uma resposta primária de baixo grau frente a agentes infecciosos ou toxinas. As infecções persistentes que levam às reações imunológicas tardias (hipersensibilidade tipo IV) são bons exemplos de inflamação crônica. A exposição prolongada a agentes tóxicos e a auto-imunidade que ocorre em enfermidades específicas são também exemplos já consagrados de resposta crônica. Os monócitos circulantes constituem a principal fonte celular da inflamação crônica; esses monócitos chegam ao tecido e transforma-se em células ativas (macrófagos), com capacidade ilimitada de fagocitose. Além disso, as células ativadas possuem capacidade de desgranular, liberando mediadores químicos locais e causando grave destruição tecidual. Os mecanismos pelos quais os monócitos saem dos vasos e 7 vão até o tecido lesado são similares aos descritos na inflamação aguda para os neutrófilos. O acúmulo de macrófagos no local da lesão é decorrente basicamente da chegada de monócitos sanguíneos através da liberação de substâncias quimiotáticas, entretanto, outras formas de aumento no número de macrófagos locais incluem a imobilização dessas células no local da inflamação por parte de certas citocinas (por exemplo, o fator inibidor de macrófagos) e sua proliferação no tecido afetado, ou seja, é possível que ocorra mitose de macrófagos no tecido. As principais substâncias quimiotáticas para macrófagos são: C5a, quimiocinas (MIP-1 e fractalcina), PDGF, fragmentos de degradação do colágeno e fibronectina. A morte dos neutrófilos é o sinal chave para a quimiotaxia dos macrófagos. Alguns autores afirmam inclusive que substâncias liberadas dos neutrófilos mortos constituem-se em possíveis quimiotáticos para monócitos. A transmigração dos monócitos, sua diferenciação em macrófagos e sua migração para o local da lesão correspondem ao início do fenômeno que é classicamente definido como inflamação crônica. Os processos pelos quais os monócitos saem do vaso e chegam ao tecido são idênticos aos descritos para os neutrófilos e por isso não serão repetidos. A pergunta básica a ser feita é: como que, se essas células utilizam o mesmo mecanismo de migração, elas chegam ao tecido em momentos diferentes? Acredita-se que o principal motivo pelo qual os monócitos chegam depois ao tecido esteja relacionado com o tipo de substância quimiotática liberada pelo foco da lesão. Muitas substâncias que são atrativas de neutrófilos e que têm capacidade de estimular suas moléculas de aderência, não o são para monócitos. Como já descrito anteriormente, os macrófagos atuam de duas formas básicas no tecido, fagocitando agentes infecciosos e restos celulares ou desgranulando e assim liberando mediadores químicos no local da lesão. Esses produtos incitam lesão tecidual e fibrose, são eles: proteases neutras, metabólitos do ácido araquidônico, metabólitos tóxicos do oxigênio, óxido nítrico, elastase, colagenase, hidrolases ácidas, lipases, fosfatases, componentes do sistema complemento, citocinas fibrogênicas, ativadores do plasminogênio e fatores de crescimento, como o PDGF, fator de crescimento de fibroblastos (FGF), fator de crescimento transformador- (TGF-) e fator de angiogênese (FGF). Prof. Rafael Fighera, Méd. Vet., Me., Dr., Membro CBPA Laboratório de Patologia Veterinária Departamento de Patologia Centro de Ciências da Saúde Universidade Federal de Santa Maria 8 Quadro 1 - Relação das moléculas de aderência endoteliais e neutrofílicas. Moléculas de aderência neutrofílica 2 Integrinas LFA-1 MAC-1 Moléculas de aderência endotelial Imunoglobulinas ICAM-1, ICAM-2 e ICAM-3 ICAM-1 Glicoproteínas semelhantes à mucina Selectinas PSGL-1* P-selectina e E-selectina ESL-1* E-selectina *Glicoproteínas auxiliadas pelo sialil-Lewis X. Quadro 2 - Relação das moléculas de aderência endoteliais e monocíticas. Moléculas de aderência monocítica 1 Integrinas VLA-4 Moléculas de aderência endotelial Imunoglobulina VCAM-1 2 Integrinas LFA-1 Imunoglobulinas ICAM-1 e ICAM-2 Glicoproteínas semelhantes à mucina Selectinas PSGL-1* P-selectina e E-selectina ESL-1* E-selectina *Glicoproteínas auxiliadas pelo sialil-Lewis X. Quadro 3 - Relação das moléculas de aderência endoteliais e linfocíticas. Moléculas de aderência linfocítica 2 Integrinas LFA-1 MAC-1 Moléculas de aderência endotelial Imunoglobulinas ICAM-1 e ICAM-2 ICAM-1 Glicoproteínas semelhantes à mucina PSGL-1* ESL-1* Selectinas P-selectina e E-selectina E-selectina Selectinas Glicoproteína semelhante à mucina L-selectina GlyCAM-1 *Glicoproteínas auxiliadas pelo sialil-Lewis X. 9
Baixar