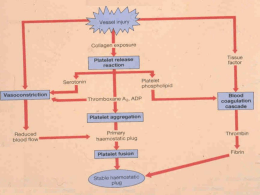
Faculdade de Medicina da Universidade de Coimbra Mestrado Integrado em Medicina Bioquímica I Seminário Orientado 6: Composição Bioquímica de fluidos biológicos: o plasma; a cascata de coagulação Trabalho realizado por: José Eduardo Mateus José Miguel Guerreiro José Pedro Leite Juliana Maciel O Sangue Plasma O Sangue é um tecido conjuntivo líquido que circula pelo sistema vascular sanguíneo dos animais vertebrados e que tem como função a manutenção da vida do organismo. Células Sanguíneas: • Eritrócitos • Leucócitos • Plaquetas Plasma • É o componente líquido do sangue, no qual as células sanguíneas estão suspensas. É um líquido de cor amarelada e é o maior componente único do sangue, compondo cerca de 55% do seu volume total. Constituição: • Água • Ureia • Proteínas • Ácido Úrico • Glucose • Creatina • Aminoácidos • Iões Inorgânicos • Lípidos • Hormonas Plasma (Constituição) Plasma = Soro • O soro é essencialmente o plasma desprovido de fibrinogénio. • O soro é preparado deixando-se o sangue coagular, enquanto que o plasma é preparado por centrifugação das células sem que tenha ocorrido a coagulação. Células Sanguíneas Como se formam? Hematopoiese Processo de formação, desenvolvimento e maturação dos elementos do sangue a partir de um precurssor celular comum e indiferenciado conhecido como célula mãe hematopoiética pluripotente, célula tronco ou hemocitoblasto. Células Sanguíneas • Eritrócitos: São as células que existem em maior quantidade no sangue e são responsáveis pela coloração avermelhada deste, devido à presença, no seu interior, de um pigmento denominado hemoglobina. Possuem a forma de um disco bicôncavo e são anucleados. • Leucócitos: Em menor quantidade, os leucócitos possuem núcleo e constituem a parte celular do sistema imunológico que actua contra substâncias estranhas e microorganismos patológicos (vírus, bactérias, fungos, etc). Também participam nas reações alérgicas, na produção de histamina. • PLAQUETAS Plaquetas / Trombócitos • Porção celular do sangue • Fragmentos de células poliplóides (Megacariócitos) • Pequenas e sem núcleo, mas com mitocôndrias REL •Têm uma fina camada de glicoproteínas • Estrutura activa e activável • Vida curta (10 dias) FUNÇÕES: Hemostase primária e secundária e preservação da integridade do endotélio vascular. Hemostase Capacidade de estancar uma hemorragia, isto é, processo de prevenção da perda de sangue quando um vaso é lesionado. Hemostase primária • Vasoconstrição Lúmen do vaso – – – – fluxo local de sangue pressão sanguínea Resulta da libertação de serotonina Actua num curto período de tempo Previne a perda acentuada de sangue É mais notória nos vasos de maior calibre devido à natureza muscular da sua parede ● As plaquetas ligam-se à superfície lesionada ● Adesão das plaquetas ao colagénio Factor Von Willebrand Proteína plasmática que se liga ao colagénio exposto dos tecidos. Serve de ponte entre os vasos danificados e as plaquetas. ● Quando as plaquetas aderem à parede vascular começam a activar-se. Activação das plaquetas • São activadas por exposição ao colagénio • Alteram a sua forma • Libertação de grânulos plaquetares ADP e Tromboxano A2 Activação das plaquetas vizinhas Adesão das plaquetas activadas Formação do tampão plaquetar Adesão e agregação das plaquetas Tampão plaquetário Paragem da Hemorragia Hemostase secundária A hemostase secundária consiste na coagulação sanguínea. Este processo pode ser visto como uma cascata de reacções enzimáticas que ocorrem por meio de activação proteolítica – através dos factores de coagulação: 1- são, por convenção, representados em numeração romana; 2- a maioria deles é sintetizada no fígado com intervenção da vitamina K. A coagulação ocorre através de duas vias Via Intrínseca Via extrínseca Todos os elementos necessários à sua activação estão presentes no plasma. Necessita de elementos exteriores ao plasma para se activar. É activada quando o sangue entra em contacto com a parede lesada do vaso. É activada quando o sangue entra em contacto com o factor tecidual (factor III – tromboplastina). Experiências feitas nas últimas três décadas vieram demonstrar que as duas vias não exibem funcionamento independente. A via extrínseca inicia e tem maior importância em todo o processo. O ponto comum entre as duas vias é a activação do factor X. A partir deste ponto, o processo tem o nome de via final comum. O factor Xa age, em presença de cálcio e do factor Va, sobre a protrombina (factor II), transformando-a em trombina. A trombina tem várias funções: 1- converte o fibrinogénio (factor I) em fibrina. 2- activa o factor XIII, responsável pela polimerização da fibrina. 3- quando gerada em excesso actua como anti-coagulante, através da activação da proteína C. O factor XIII polimeriza a fibrina. Forma ligações covalentes entre diferentes moléculas solúveis. Estabelecem-se ligações cruzadas entre os monómeros tornando-os numa teia insolúvel de polímero – o trombo ou coágulo. Mecanismos Anticoagulantes A tendência do sangue a coagular é equilibrada in vivo por reacções limitantes que tendem a impedir a coagulação no interior dos vasos sanguíneos e a degradar qualquer coágulo que se possa formar. • Estas reacções incluem a interacção entre: Tromboxano A2 (produzido pelas Plaquetas): promove a agregação plaquetária. Prostaciclina (produzida pelas células endoteliais): Inibe a agregação plaquetária. Induz a formação de coágulos no local de lesão do vaso sanguíneo, deixando o lúmen do vaso livre de coágulos. Três Mecanismos que se opõem à formação do Coágulo: 1. Actua na fase inicial da coagulação e utiliza uma proteína do plasma libertada pelas células endoteliais designada factor inibidor tecidual (Tissue Factor Pathway InhibitorTFPI) que se liga ao factor tecidual (factor VIIa), destruindo a capacidade destes em estimular a formação do factor Xa. 2. É desencadeado pela Trombina. Esta liga-se a um receptor da célula endotelial designado trombomodulina transformando-se num anticoagulante, visto que o complexo trombomodulinatrombina activa a proteina C que com a proteina S (seu cofactor) inactiva os factores V e VIII e o Inibidor do activador de Plasminogénio tecidual, aumentando a formação de Plasmina. 3. Proteína chamada Antitrombina III que inactiva a trombina e outros factores da coagulação. Para o fazer, a antitrombina circulante deve ela própria ser activada, o que acontece quando esta se liga à Heparina (substância presente na superfície das células endoteliais). A Antitrombina III previne assim a expansão do coágulo, inactivando rapidamente factores coagulantes trazidos ao local pelo fluxo sanguíneo. Sistema Fibrinolítico • TFPI, Proteína C e Antitrombina III funcionam em conjunto para limitar a formação do coágulo. O Sistema Fibrinolitico dissolve o coágulo após a sua formação. • Este sistema consiste numa proenzima do plasma, o plasminogénio, que pode ser activado em plasmina pelo activador do plasminogénio tecidual(t-PA) libertado pelas células endoteliais. Uma vez formada, a plasmina digere a fibrina, dissolvendo assim o coágulo. Activador do Plasmogénio tecidual Plasmina Plasminogénio Fibrina Fragmentos solúveis de Fibrina Anormalidades da Hemostasia: Doenças Hemorrágicas Existem doenças que afectam os sistemas da hemostasia e predispõem os seus portadores a fenómenos hemorrágicos espontâneos ou desencadeados por procedimentos cirúrgicos. • Alterações do fígado podem produzir deficiência de vitamina K e também de protrombina e dos factores VII, IX e X, levando a hemorragias. • As principais alterações da coagulaçao são a deficiência de Vitamina K, a Hemofilia e a Trombocitopenia. Hemofilia Algumas doenças hereditárias de coagulação que causam sangramento excessivo são as hemofilias: • Hemofilia A: deficiência do factor VIII. • Hemofilia B (Doença de Christmas): deficiência do factor IX. São ambas doenças recessivas ligadas ao sexo (cromossoma X). • Hemofilia C: Este tipo de hemofilia é determinada por um gene autossómico dominante não relacionado com o sexo e caracteriza-se pela ausência de um factor denominado PTA. Trombocitopenia • Há uma redução do número de plaquetas em circulação no sangue, pelo que predispõem os indivíduos a pequenas hemorragias em todos os tecidos. • A causa mais comum é a auto-imunidade, que promove o desenvolvimento de anticorpos contra as próprias plaquetas. • Em alguns casos os anticorpos aparecem em consequência de transfusões de sangue. • A irradiaçao da medula óssea também produz diminuição no número de plaquetas, com alterações da hemostasia. Fim
Baixar