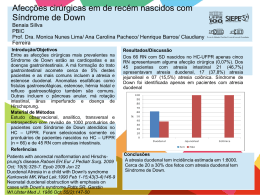
ATRESIA AURAL PIBIC - Programa Institucional de Bolsas de Iniciação Científica Mariana de Faria Navarrete¹ Daniel Mochida Okada² Fábio Akira Suzuki³ 1. Acadêmicos do curso de Medicina da Universidade Cidade de São Paulo, internos do Hospital do Servido Público Estadual. 2. Médico assistente do Instituto de Assistência Médica ao Servidor Público Estadual, professor da disciplina de Otorrinolaringologia do curso de Medicina da Universidade Cidade de São Paulo. 3. Diretor do Centro de Desenvolvimento e Pesquisa do Hospital do Servidor Público Estadual, professor da disciplina de Otorrinolaringologia do curso de Medicina da Universidade Cidade de São Paulo. Introdução Atresia aural congênita refere-se a um espectro de deformidades da orelha presentes ao nascimento que envolve algum grau de insuficiência do desenvolvimento do canal auditivo externo e, muitas vezes, da membrana timpânica e dos ossículos da orelha média. Epidemiologia A incidência estimada está entre 1:9000 e 1:22000 nascidos vivos. O sexo masculino é mais afetado que o feminino numa relação de 2,5:1. A maioria dos casos é unilateral. Etiologia Este tipo de malformação pode estar relacionado a síndromes ou ocorrer isoladamente. Acarreta perda auditiva devido à alteração na condução do som do ambiente até a cóclea, por isso requer diagnóstico e avaliação precoce a fim de evitar seqüelas tardias da perda auditiva como problemas de aquisição de linguagem e aprendizado e problemas da fala. Objetivo do trabalho Este estudo tem por objetivo revisar a literatura nacional, internacional e dos bancos de dados eletrônicos e caracterizar os casos de atresia aural dos pacientes nascidos no Hospital do Servidor Público Estadual (HSPE) entre Janeiro/2006 a Agosto/2010. Metodologia Este trabalho baseou-se na pesquisa transversal de dados estatísticos do arquivo médico do HSPE e na revisão assistemática da literatura nacional, internacional e bancos de dados eletrônicos. Foram analisados os prontuários de todos os pacientes nascidos vivos no período de Janeiro de 2006 a Agosto de 2010. Resultados No período de Janeiro de 2006 a Agosto de 2010 foram 5033 nascidos vivos no Hospital do Servidor Público Estadual. Desses, apenas três casos apresentaram alguma malformação auricular: Fevereiro/2007: recém-nascido do sexo masculino que apresentava um apêndice auricular acessório esquerdo como um fator isolado. Abril/2008: recém-nascido masculino que apresentava agenesia auricular unilateral acompanhada de malformações cardíacas (tetralogia de Fallot), diagnosticando-se síndrome de Goldenhar. Setembro/2010: recém-nascido do sexo masculino que apresentou ao nascimento síndrome Charge. Síndrome de Goldenhar Anomalia congênita rara. Herança autossômica dominante ou recessiva. Acomete predominantemente o sexo masculino. Sua incidência varia de 1:3500 a 1:5600 nascidos vivos. Caracteriza-se por alterações oculares, auriculares e vertebrais, além de outras manifestações decorrentes de erros na morfogênese do 1º e 2º arcos branquiais no período de organogênese. Síndrome de Goldenhar Síndrome de CHARGE Acomete cerca de 1:9000 nascidos vivos. Há relatos na literatura que sugerem tanto a herança autossômica dominante como a autossômica recessiva. Caracteriza-se por um conjunto específico de malformações congênitas: C - Coloboma de íris H - Cardiopatia congênita A - Atresia de coanas R - Retardo de crescimento e desenvolvimento G - Anomalias genitais E - Anomalias do pavilhão auricular e/ou surdez Síndrome brânquio-otorrenal Doença autossômica dominante, mutações no gene EYA 1. Sua incidência é de 1:40000 recém-nascidos vivos. As manifestações clínicas da SBOR decorrem de transtorno genético e envolvem perda auditiva de condução, neurossensorial ou mista, malformações da orelha externa, média e interna, cistos ou fístulas branquiais e anomalias renais que podem variar de hipoplasia a agenesia renal. provocada por Síndrome brânquio-otorrenal Síndrome de Treacher Collins Deformidade rara. Ocorre vivos. Apresenta grande variedade de alterações, sendo que as manifestações clínicas mais freqüentes são: obliqüidade antimongolóide das fendas palpebrais, hipoplasia malar, hipoplasia mandibular, malformação dos pavilhões auriculares e coloboma palpebral inferior. em aproximadamente 1:50000 nascidos Síndrome de Treacher Collins Conclusão Observa-se que na maioria dos casos as deformidades da orelha presentes ao nascimento são decorrentes de alterações genéticas que cursam com outras malformações e predominam no sexo masculino. A ampla variabilidade clínica das malformações auriculares exige atenção e perícia. Embora seja baixa a prevalência, o diagnóstico e tratamento precoces visam melhorar a qualidade de vida do paciente reduzindo danos e complicações. O otorrinolaringologista e o pediatra devem estar capacitados para reconhecer seus sinais e sintomas característicos. Referências Bibliográficas 1. Bento RF, Miniti A, Butugan O. Doenças congênitas do ouvido. Tratado de Otologia, 1ª edição; 1998. 2. Gates GA, Valente M. Fitting strategies for patients with conductive hearing loss. New York: Thieme Medical Publishers; 1994. 3. De la Cruz A, Chandraseckhar SS. Congenital Malformation of the Temporal Bone. Otologic Surgery. 1994. 4. Kamerer DB. Congenital and Acquired Atresia of the External Auditory Canal. Operative Otolaryngology, Head and Neck Surgery; 1997. 5. Moore LK, Persaud TVN. Embriologia Clínica, 7ª edição; 2008. 6. Davenport SLH, Hefner MA, Mitchell JA. The spectrum of clinical features in CHARGE syndrome. Clin. Genet., v. 29, p. 298310; 1986. 7. Terhaar B. Oculo-auriculo-vertebral dysplasia (Goldenhar syndrome) concordant in identical twins. Acta Genet Med. Gemellol; 21:116-124; 1972. 8. Ermeso GA, Lugo NT, Ceballos MF. Síndrome branquiootorrenal. Rev. Cubana Pediatria; 77(2); 2005. 9. Marres HAM, Cremers CWRJ, Dixon MJ, Huygen PLM, Joosten FBM. The Treacher Collins syndrome: a clinical, radiological, and genetic linkage study on two pedigrees. Arch Otolaryngol Head Neck Surg; 121(5):509-514; 1995.
Baixar