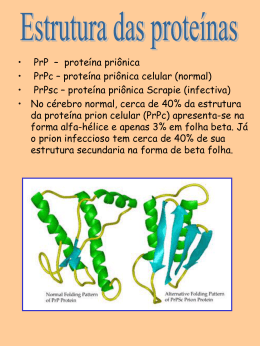
PROTEÔMICA 13/11/2007 O que é Proteômica? “Identificação, caracterização e quantificação de todas as proteínas envolvidas em uma cadeia particular, organela, célula, tecido, órgão ou organismo que pode ser estudado com o objetivo de fornecer dados confiáveis para o entendimento de determinado sistema.” A nomenclatura ômica… Genômica DNA (Gene) Transcrição Transcriptômica RNA Tradução Genômica Funcional Proteômica PROTEÍNA Reação enzimática Metabolômica METABOLITE Lições sobre os Projetos genômicos A complexidade evolucionária não é primariamente determinada pelo maior número de genes, mas som pela variação do número de proteínas sintetizadas. Isso é alcançado pela geração de múltiplas proteínas a partir de um único gene, como por exemplo: Diferentes combinações de exons por splicing alternativo Processamento pós-traducional (ex, clivagem de pró-peptídeos) Modificações pós-traducionais (ex acetilação, glicosilação) Modificações no dogma central: DNA --> RNA --> proteína(s) É importante então realisar análises a partir dos PRODUTOS do gene, e não tanto dele mesmo. Por que estudar a expressão de proteínas? mRNA inativo Controle Degradação RNA DNA Transcrito de RNA Primário mRNA mRNA Controle de Tradução Controle Transcricional Controle do Proces De RNA Controle Transport RNA proteína Proteína Controle Pós-Transducional Vantagens centrais da proteômica: Pesquisadores trabalham no nível de produtos gênicos, que são realmente expressos e levam a um produto real, não somente sendo expresso e tendo mRNA. Limitações centrais da proteômica: Geralmente, somente uma fração das proteínas sintetizadas pode ser detectada num experimento, enquanto a expressão de todos os genes pode ser monitorada num experimento de array. Pré-requisito básico da proteômica: O genoma sequenciado do organismo investigado ou pelo menos uma coleção de cDNAs. Background experimental Eletroforese 2D Método para separação e visualização de proteínas SEparação por carga e massa Espectometria de Massa Análise de alta confiabilidade e identificação de proteínas. Fragmentação de proteínas Análise de peptídeos 2D-SDS PAGE gel A primeira dimensão (separação por ponto isoelétrico) - Gel com um gradiente de pH imobilizado - Corrente elétrica leva aproteínas carregadas a mover até alcançar o ponto isoelétrico Ponto Isoelétrico (pI) Separação por carga: 4 Gradiente de pH estável 5 Baixo pH: Proteína Carregada + 6 7 8 Alto pH: 9 Proteína carregada 10 - No ponto isoelétrico, a proteína não tem carga e portanto não migra mais no campo elétrico. 2D-SDS PAGE gel A primeira dimensão (separação por ponto isoelétrico) - Gel com um gradiente de pH imobilizado - Corrente elétrica leva aproteínas carregadas a mover até alcançar o ponto isoelétrico A segunda dimensão (separação por massa) -pH gel strip e colocado em um gel SDS -SDS desnatura a proteína (movimento somente devido a massa) e elimina carga. 2D-SDS PAGE gel Exemplo de técnica de 2D-gel Vantagens vs Desvantagens Boa resolução para proteínas Detecção de modificações póstranscricionais Não funciona bem para proteínas altamente hidrofóbicas Limitado pelo range de pH Difícil detecção de proteínas pouco abundantes Análise e quantificação sáo difíceis Background Experimental Espectometria de Massas O que é Mass Spec? Ferramenta analítica para determinar a composição molecular de uma amostra ou os pesos moleculares Somente picomolares de concentrações são requeridas Acurácia de 0.01%, com somente 5 ppm de pequenas moléculas orgânicas Para 40 kDa, erro de 4 Da Pode-se identificar substituições de aminoácidos ou modificações pós-traducionais Que tipo de informação é gerada? Informação estrutural Tandem mass spectrometers – particularmente, uma sequencia definida Fragmentação de amostra – análise de produtos Sequenciamento de peptídeos Identificação de compostos individuais em misturas complexas Como funciona um espectômetro? 3 partes fundamentais: fonte de ionização, o analisador e o detector Amostras fáceis de manipular se ionizadas Separação no analisador de acordo com a razão massa-carga (m/z) Detecção de íons em separado e abundância relativa Sinais enviados ao sistema de dados e apresentados em um espectro m/z Esquematização O analisador, o detector e o ionizador estão em vácuo, para evitar movimento indesejado de íons A operação está sob completo controle de sistema Esquema típico de um TOF-MS Introdução da amostra e Ionização Ionização direta ou via cromatografia para separação de componentes (HPLC, GC, eletroforese capilar) Ionização pode resultar em íons positivamente carregados (proteínas) ou negativamente carregados (sacarídeos e oligonucleotídeos) Metodologias de Ionização Atmospheric Pressure Chemical Ionisation (APCI) Chemical Ionisation (CI) Electron Impact (EI) Electrospray Ionisation (ESI) Fast Atom Bombardment (FAB) Field Desorption / Field Ionisation (FD/FI) Matrix Assisted Laser Desorption Ionisation (MALDI) Thermospray Ionisation Detecção dos Íons O detector monitora o íon, amplifica seu sinal e o transmite para o sistema Detectores comuns: photomultiplier, electron multiplier, micro-channel plate Espectometria de massas é uma metodologia poderosa para analisar a estrutura de compostos orgânicos, mas tem algumas limitações: Nada pode ser caracterisado se não estiver em uma solução DEVIDAMENTE LIMPA A técnica não tem abilidade de apresentar uma análise seletiva de uma mistura complexa Para moléculas grandes, resultados são difíceis de interpretar. Tandem MS ou MS/MS tem 2 mass spec em série: O primeiro (MS1) é usado para selecionar, de uma fonte primária de íons, aqueles que possuem um m/z particular, o qual passa para a região de fragmentação, onde ocorrerá a dissociação. No segundo (MS2), haverá a análise dos íons gerados. DE verdade, MS1 é uma fonte de íons para MS2. MS1 MS2 Sequenciamento de peptídeos Peptídeos de 2.5 kDa ou menos dão melhores resultados. Proteínas retiradas de géis 2-D e digeridas Peptídeos fragmentados nas ligações peptídicas na Tandem mass spectrometry Alguns peptídeos geram informações para uma sequencia completa, enquanto a maioria gera sequencias parciais de 4-5 aa Geralmente essas “tag” sequences são suficientes para identificação por database Identificação de proteínas por MS Library Spot removed from gel Artificial spectra built Fragmented using trypsin Spectrum of fragments generated MATCH Artificially trypsinated Database of sequences (i.e. SwissProt) Como funciona o sequenciamento de proteínas Tandem MS: dois analisadores de massa em série com uma célula de colisão no meio Célula de colisão: um local aonde os íons colidem com um gás (He, Ne, Ar) resultando em fragmentação do íon Ser-Glu-Leu-Ile-Arg-Trp Collision Cell A fragmentação dos petídeos na células de colisão ocorre de maneira predizível, principalmente nas ligações peptídicas Ser-Glu-Leu-Ile-Arg Os íons resultantes tem massas que são consistentes com uma database de pesos moleculares de dipeptídeos, tripeptídeos, tetrapeptídeos... Ser-Glu-Leu Ser-Glu-Leu-Ile Etc… Vantagens vs. Desvantagens Determinação de peso molecular e sequencia de aa. Detecção de modificações póstranscricionais Alta confiabilidade Alto custo Requer sequencia de peptídeos em databse Após a proteômica….. Genômica funcional ProteinChipTM. Limitações da Proteômica Limitações experimentais: Análise em larga escala de proteínas pode ser difícil devido a alguns problemas, como: -Proteínas são frágeis -Podem existir em múltiplas isoformas -Não há equivalente para proteína de um PCR para amplificação em larga escala Limitações na análise de dados: -Dados as vezes contêm muito ruído, que podem ser difíceis de separar dos sinais específicos. Pode resultar em gasto de tempo desnecessário analisando espectros não importantes. -As análises de banco de dados para os espectros de mass spec são somente SCORES, deixando a análise para intervenção manual para eliminação de falsos positivos. Limitações biomédicas: -Na prática é muito difícil de traçar a completa progressão da doença. -As vezes, usar proteômica para monitorar a bioquímica de uma doença é como usar uma câmera fotográfica para filmar jogo de futebol Explains the whole process nicely -- article http://swehsc.pharmacy.arizona.edu/analysis/Proteomics_New s.htm Mascot Home page -- help section http://www.matrixscience.com/help_index.html Presentation about MS MS data http://sashimi.sourceforge.net/extra/oral.pdf http://www.genetik.unibielefeld.de/D1E33C76A7CCA010AAD3B435B51404EE/Geno me_Research_WS2002_03/stunde_ws0203_10.pd http://bmbus6.leeds.ac.uk/mres/5130/MassSpectrometry.ppt Some info about drug discovery/economic issues n such: http://monod.uwaterloo.ca/cs798/proteomics.pdf Paper on interpreting MSMS data http://chem-ncms.unl.edu/asms2003/kurt.pdf How to estimate correctness of MS MS prediction -- EM !!! http://www.proteomecenter.org/PDFs/Keller.AnalChem.02.pdf http://www.nature.com/cgitaf/DynaPage.taf?file=/nbt/journal/v21/n3/full/nbt0303-221.html http://www.esainc.com/MolecularProteomics/molecular_proteomic s.htm http://genome.ucsd.edu/classes/be202/ppt/11 Internet sites www.astbury.leeds.ac.uk/Facil/MStut/mstutorial.htm (Dr Alison E. Ashcroft at Leeds) www.asms.org (The American Society for Mass Spectroscopy) www.spectroscopynow.com (Base Peak) Mass Spec tools www.expasy.ch/tools/#proteome http://prowl.rockefeller.edu www.mann.embl-heidelberg.de Bibliography Internet sites : http://www.google.com • • • • • • http://www.bmss.org.uk/what_is/whatis.html http://www.duke.edu/~mdfeezor/NSHome/inform/msms1.html http://www.astbury.leeds.ac.uk/Facil/MStut/mstutorial.htm http://ms.mc.vanderbilt.edu/tutorials/ms/3.htm http://www.garvan.unsw.edu.au/public/corthals/book/IPMS.html http://www.micromass.co.uk/basics/Glossary.html Metodologias de Ionização Further Reading 1. MALDI beginner: http://www.srsmaldi.com/Maldi/Guide.html 2.MALDI lab user: http://www.srsmaldi.com/Maldi/Lab.html 3. MALDI tutorial: http://ms.mc.vanderbilt.edu/tutorials/maldi/maldi-ie_files/frame.htm 4. Ionization Methods 1: http://www.jeol.com/ms/docs/ionize.html 5. Ionization Methods 2: http://www.waters.com/Waters_Website/Applications/lcms/lcms_itq.htm
Baixar