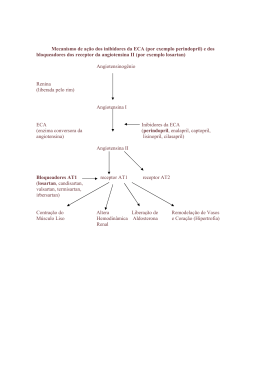
O SISTEMA RENINA-ANGIOTENSINA-ALDOSTERONA Classicamente, o sistema renina-angiotensina-aldosterona (S-RAA) é descrito como um eixo endócrino no qual cada componente de uma cascata é produzido por diferentes órgãos, um arranjo que é exemplo de interação de vários sistemas orgânicos, engajados todos na luta para manter a estabilidade hemodinâmica. Teleologicamente, pode-se afirmar que a ativação do S-RAA é atitude de grande responsabilidade, em função das conseqüências que sua atuação determinam, daí a necessidade de que muitos órgãos participem desse processo. Estão identificados no corpo humano dois diferentes tipos de sistemas renina-angiotensina-aldosterona: o circulante, descrito há já bastante tempo, e o local, descrito mais recentemente, que parece desempenhar papel crucial na manutenção da homeostase circulatória. No S-RAA circulante, o angiotensinogênio é produzido pelo fígado, que requer glicorticóides do córtex adrenal e estrógeno das gônadas; a renina é liberada pelos rins, enquanto que a enzima de conversão de angiotensina I em angiotensina II (ECA) é encontrada no endotélio vascular de vários órgãos. Uma vez ativada a cascata, surgem a angiotensina I e a angiotensina II, que circulam pelo sangue ativando suas estruturas-alvo: vasos sangüíneos (sobretudo arteríolas e veias sistêmicas), rins, coração, adrenais e o sistema nervoso simpático. A lógica fundamental que preside o funcionamento do sistema é responder a uma instabilidade hemodinâmica e evitar redução na perfusão tecidual sistêmica. Atua de modo a reverter a tendência à hipotensão arterial através indução de vasoconstricção arteriolar periférica e aumento na volemia por meio de retenção renal de sódio (através da aldosterona) e água (através da liberação de ADH-vasopressina). Portanto, o sistema renina-angiotensina-aldosterona se soma ao sistema simpático e ao ADH, compondo o trio de sistemas neuro-hormonais de compensação cardiovascular. Copyright © 2001 Fisiopatologia Aplicada Mecanismos de Ativação do Sistema RAA São cinco as condições principais nas quais o S-RAA é ativado: (1) Insuficiência cardíaca; (2) Restrição de sódio; (3) Contração do compartimento intravascular (desidratação, hemorragia, diarréia); (4) Aumento do tônus simpático (5) Hipotensão arterial O agente central do S-RAA é a angiotensina II, que tem receptores nos seguintes órgãos-alvo: rins, coração, cérebro e adrenais, além dos vasos sangüíneos. A cascata do sistema renina-angiotensina-aldosterona São cinco os elementos da cascata: (1) Angiotensinogênio (2) Renina (3) Angiotensina I (4) Angiotensina II (5) Angiotensina III Angiotensinogênio É uma alfa-2 globulina produzida pelo fígado em presença de glicocorticóides e estrógenos, que circula no plasma como um peptídeo biologicamente inativo, sobre o qual irá atual a renina, gerando uma seqüência de substâncias ativas. Em condições como síndrome de Cushing e durante o uso de anticoncepcionais orais, a ocorrência de hipertensão arterial sistêmica volumedependente é possível por um efeito de ação de massas: como nos ensina as leis do equilíbrio químico, o aumento na produção de angiotensinogênio (substrato) faz Copyright © 2001 Fisiopatologia Aplicada com que os níveis de angiotensinas sejam elevados para uma mesma quantidade de enzima renina. Renina É a enzima proteolítica que converte angiotensinogênio em angiotensina I (convertendo uma substância de 411 aminoácidos em outra de 10 aminoácidos), sendo assim responsável pela etapa limitante da síntese de angiotensinas. A prórenina é o precursor inativo da renina, que usualmente é encontrado em baixas concentrações plasmáticas mas, em diabéticos (tipo I e II) têm sido encontrados níveis elevados a ponto de ter sido proposto como marcador de nefropatia diabética. Convém lembrar que a nefropatia diabética é um exemplo de hiperaldosteronismo hiporeninêmico, tendo sido sugerido que os diabéticos com renina baixa seriam os casos de extrema capacidade de converter a pró-renina em re-nina. Por outro lado, o aparelho reprodutor feminino é outra fonte importante de pró-renina, sendo sua produção regulada pelo nível de gonadotropinas circulantes. Por tudo isso, aceita-se que níveis plasmáticos elevados de pró-renina podem ser marcadores de anormalidades vasculares em diabéticos, de modificações fisiológicas durante o ciclo menstrual e de tumores produtores de renina, que são raros mas foram descritos. A liberação de renina é cuidadosamente controlada pelo aparelho justaglomerular, composto de mácula densa da primeira porção do túbulo contorcido distal, de células contíguas do mesângio e de células especializadas que fazem parte da parede da arteríola aferente. Além de monitorizar o teor de sódio no sangue que penetra no glomérulo via arteríola aferente, as células intraglomerulares são mecanorreceptores sensíveis à distensão, de modo que deflagram a liberação de renina sempre que a pressão sangüínea ou natremia estiver baixa. Já as células que compõem a mácula densa são osmoreceptores, que reagem a um aumento no teor de sódio presente no ultrafiltrado que segue em direção ao túbulo distal; de fato, a quantidade de sódio que passa pela mácula Copyright © 2001 Fisiopatologia Aplicada densa sob condições fisiológicas é muito pequena, pois no segmento imediatamente anterior (alça ascendente de Henle) há expulsão ativa de NaCl, persistindo pouco sódio no líquido tubular. A inibição da bomba Na+K+/Cl (dita "bomba de Cloreto")m provocada pelos poderosos diuréticos de alça (furosemida, bumetamida) sobre a alça de Henle, interfere com o mecanismo multiplicador de contracorrente renal, responsável pela hipertonicidade medular. A isso se soma a oferta de quantidades muito elevadas de NaCl ao túbulo distal, superando sua capacidade de reabsorção, a despeito de intensa troca de Na+ por H+ e K+, resultando em diurese volumosa, natriurese, e intensa secreção de potássio e hidrogenionte. O resultado final do uso dessas drogas é diurese intensa com espoliação de potássio e indução de alcalose metabólica. Assim, a ativação do sistema RAA intra-renal, pelo excessivo aporte de sódio à mácula densa, induz produção local de renina. As prostaglandinas renais também podem participar desse processo, através da indução de alterações no nível da filtração glomerular. A Enzima de Conversão de Angiotensina (ECA) A angiotensina I (de 10 aminoácidos) é convertida em angiotensina II (de 8 aminoácidos) pela enzima de conversão (ECA), uma dipeptidil-carboxilase. Descrita inicialmente em 1956 (1), a ECA passou a se procurada como uma enzima capaz de eliminar do sangue tanto a angiotensina I quanto a bradicinina e a serotonina. No Brasil, foi possível bloquear a conversão de angiotensina I e a eliminação de bradicinina empregando-se veneno da cobra jararaca, logo a seguir, foi demonstrado que a cininase II (enzima que inativa a bradicinina) e a enzima de conversão (ECA) são uma mesma substância. A enzima de conversão de angiotensina I tem sido encontrada no endotélio vascular pulmonar e sistêmico, que assim se mostra capaz de inativar um vasodilatador (a bradicinina) e ativar um vasoconstrictor (angiotensina II). O uso de inibidores da enzima de conversão da angiotensina I (captopril, enalapril, Copyright © 2001 Fisiopatologia Aplicada lisinopril) não apenas bloqueia os efeitos vasoconstrictores (e hipervolêmicos) da angiotensina, mas também potencializa a vasodilatação induzida pela bradicinina. Embora tenha efeito vasoconstrictor menos prolongado que o obtido com doses equivalentes de endotelina e de tromboxane A2, a angiotensina II é considerada a mais poderosa substância vasoconstrictora sistêmica, enquanto aqueles exercem seus efeitos mais intensamente nos próprios locais onde são formados. A angiotensina II exerce várias ações e efeitos atuando principalmente em nível de: (1) vasos sangüíneos; (2) coração; (3) rins; (4) sistema endócrino. Sobre os vasos A este nível, a angiotensina determina contração das artérias como aorta, coronárias, femoral e carotídeas, através tanto do aumento de AMP cíclico quanto ativação da fosfolipase C, com geração de inositol-trifosfato (IP3). Participa, também do processo que resulta em hipertrofia vascular. Sobre o coração Atuando sobre seus receptores de superfície, a angiotensina II causa um aumento dose-dependente da força de contração miocárdica, de modo que independe de seus efeitos hemodinâmicos sobre as condições de carga imposta ao coração; esta melhoria no estado inotrópico também é independente da potencialização adrenérgica. A maior contratilidade decorre do aumento na intensidade da corrente lenta de cálcio. A despeito desse efeito inotropo positivo, a angiotensina II não altera o Copyright © 2001 Fisiopatologia Aplicada cronotropismo cardíaco, ou seja, não determina taquicardia. Por outro lado, como o efeito vasoconstrictor coronariano supera o estímulo vasodilatador (visto em todo e qualquer aumento da força de contração miocárdica), o fluxo sangüíneo coronariano é relativamente reduzido. Do mesmo modo que no músculo liso vascular, tudo indica que a angiotensina II participa do processo de hipertrofia cardíaca. Sobre os rins A angiotensina II exerce efeitos importantes na hemodinâmica intra-renal e na homeostase hidrossalina. A vasoconstricção renal, predominantemente exercida sobre a arteríola aferente, aumenta a pressão de filtração glomerular mesmo com grande decréscimo na perfusão renal. Ao nível do túbulo distal, a angiotensina ativa diretamente a bomba eletrogênica de prótons, que troca Na+ tubular por H+ da célula, e indiretamente (via aldosterona) ativa a bomba Na+/K+. A somatória dos efeitos hemodinâmicos e sobre a membrana basal glomerular resulta em efeito proteinúrico, o qual pode ser bloqueado por inibição da enzima de conversão. O Kf (coeficiente de ultrafiltração glomerular) aumenta, enquanto que a área de superfície disponível para a filtração glomerular é reduzida, mediante contração das células mesangiais. Quando a vasoconstricção induzida pela angiotensina II sobre a arteríola aferente é exagerada, a nutrição dos túbulos renais fica prejudicada, uma vez que os capilares peritubulares são oriundos do sistema porta renal, especificamente, da arteríola aferente. Para evitar a necrose tubular aguda, que seria inevitável toda vez que os níveis de angiotensina II atingissem um valor crítico, os rins passam a produzir vasodilatadores locais (prostaglandinas) que determinam insensibilidade parcial aos vasoconstrictores sistêmicos, de modo a adequar a perfusão renal com a sobrevivência tubular. Angiotensina III Copyright © 2001 Fisiopatologia Aplicada Componente do Sistema RAA circulante, angiotensina III é poderoso indutor da secreção de arginina-vasopressina-ADH, potencializadora da atividade simpática e indutora da secreção de aldosterona, além do efeito vasoconstrictor similar ao da angiotensina II. A angiotensina III ocorre fisiologicamente no plasma em baixas concentrações. Ela é oriunda de vários órgãos, sobretudo cérebro, rins e coração. A proporção dos componentes do Sistema RAA é: Angiotensina I: 67% Angiotensina II: 30% Angiotensina III: 3% Isso equivale dizer que o nível de angiotensina II corresponde a 45% do nível de angiotensina I e o de angiotensina III corresponde a 4% do nível de angiotensina II circulante. Os estímulos para a formação de angiotensina II são os mesmos que para angiotensina I e II. Angiotensina III pode ser formada a partir da angiotensina I por ação de peptidases plasmáticas. Sua importância fisiológica reside em seu papel modulador da função autonômica, da secreção de ADH e secreção de aldosterona. Além disso, ela atua em nível do complexo vagal/gânglio solitário induzindo modificações na sensibilidade de reflexos baroceptores de modo eqüipotente ao obtido com angiotensina II. O fato interessante é que a produção de angiotensina III não é totalmente bloqueada com o uso de inibidores da enzima de conversão (ECA), uma vez que há outras vias metabólicas através das quais a angiotensina III pode ser produzida. O sistema renina-angiotensina-aldosterona local Ao que parece, o S-RAA circulante até aqui descrito serve primariamente para manter a estabilidade hemodinâmica diante de um estresse imediato como a queda súbita no débito cardíaco; o sistema atua regulando simultaneamente o débito (ao otimizar a pré-carga através da indução de hipervolemia) e a RPT (via arteríolo-constricção). Copyright © 2001 Fisiopatologia Aplicada Logo, a atividade do S-RAA retorna ao normal durante o estado latente ou compensado da ICC, porque a normalização do volume sistólico no estado de hipervolemia bloqueia a ativação sustentada do sistema. Diz-se, então que a resposta temporal do S-RAA frente a uma estabilidade hemodinâmica é tal que, após a ativação aguda durante a descompensação, há normalização ao longo da fase crônica compensada. Entretanto, o S-RAA local vai se tornando progressivamente mais ativo nessa fase compensada; quando se exaure esse recurso, sobrevém reativação sustentada do S-RAA circulante, com manutenção dessa vez transitória da estabilidade hemodinâmica, quando então se estabelece a descompensação típica da ICC. A ativação do sistema renina-angiotensina-aldosterona local permite a obtenção dos efeitos benéficos (aumento da contratilidade miocárdica, redistribuição do fluxo sangüíneo, hipervolemia) sem os desfavoráveis efeitos resultantes da ativação do sistema RAA sistêmico, os quais, em última análise, são os responsáveis diretos pela sombria evolução natural dos pacientes com ICC. Pelo menos quatro sistemas S-RAA foram identificados até o momento: 1) Vascular 2) Cardíaco 3) Renal 4) Encefálico Copyright © 2001 Fisiopatologia Aplicada
Baixar