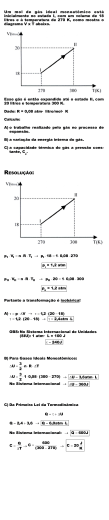
Termodinâmica Química – P1 Noturno Instruções: Gases e Primeira Lei da Termodinâmica 29.09.2006 Prof. Fabricio R. Sensato Nome:___________________________RA_________Eng. Materiais Dados T(K) = 273 + T(oC) R = 0,0820578 atmLmol-1K-1 R = 8,31447 JK-1mol-1 Equação dos gases ideais: pV = nRT 1m3=1000L 1Pa = 1kgm-1s-2; 1bar = 105 Pa; 1atm = 101 325 Pa; 1J=1kgm2s-2 dU = dq + dw CP,m – CV,m = R (gás ideal) dU = nCV,mdT; dH = nCP,mdT dw = -pextdV ∆H = ∆U + p∆V (p=cte) ∆rHo = Σ ν∆fHo(produtos) - Σ ν∆fHo(reagentes) Massas atômicas H: 1,0079 C: 12,0107 O: 15,9996 Ar: 39,95 • Reprodução do Conselho de Faculdade 002/04 (Comunicação entre alunos com objetivos ilícitos, ou cola, sob quais formas durante a realização de provas): Se a improbidade ocorrer em provas de aproveitamento anteriores à última (P1 e P2), atribuir nota zero à respectiva prova e suspender o aluno de prestar a prova seguinte da mesma disciplina. 1) (2,0 pontos) Seria possível que uma amostra de 25 g de argônio gasoso, Ar(g), num vaso de volume igual a 1,5 L, exercesse uma pressão de 1,97 atm, a 30 oC, se o seu comportamento fosse de um gás ideal? Em caso negativo, qual seria a pressão do gás? (b) Que pressão teria o argônio se ele fosse um gás de van der Waals? Para o argônio, a = 1,337 L2 atm mol-2 e b = 0,0320 L mol-1. Por que os valores de pressão calculados pelas equações do gás ideal e do gás de van der Waals são semelhantes (ou bem diferentes)? Dê uma explicação em termos das interações intermoleculares. 1 mol de Ar × 25 g Ar = 0,63 mol n(Ar) = 39,95 g Ar Gás ideal: 0,63 mol × 0,0820574atmLmol −1K −1 × 303K = 10 atm 1,5L R. Não seria possível que 25 g Ar (V=1,5L; T=303K) exercesse uma pressão de 1,97atm Gás de van der Waals p= p= 0,63 mol × 0,0820574atmLmol −1 K −1 × 303K 1,5L - 0,63 mol × 0,0320Lmol-1 2 0,63 mol − 1,337L2 atmmol − 2 × = 10 atm 1,5L Os valores de pressão calculados pela equação do gás ideal e pela equação de van der Waals são iguais (considerando o número de algarismos, de fato, significativos), pois no âmbito das variáveis de estado do problema (T, V e p) as interações intermoleculares podem ser negligenciadas. • A avaliação tem duração de 90 min. • Avaliação individual, sem consulta; • É permitido o uso de calculadora (mas não é permitido o uso de calculadoras contidas em celulares ou palmtops); • Telefones celulares devem permanecer desligados durante a realização da prova • O empréstimo de qualquer material não é permitido; • Todos os dados necessários para a resolução da prova figuram na folha de questões; • As questões devem ser resolvidas na própria folha de questões. Se necessário, utilize o verso da folha de questões; • Não desate o maço que lhe foi entregue; • Empregue o número correto de algarismos significativos; • Resolução e respostas podem ser dadas a lápis ou caneta. • Ao terminar a prova, deixe a sala sem qualquer alarde. 2) (2,0 pontos) A 25 oC, tem-se as seguintes entalpias de formação: ∆fHo do SO2(g) é igual a -296,81 kJ/mol, enquanto ∆fHo do H2O(l) é -285,83 kJ/mol. Para as reações a 25 oC: 2H2S(g) + Fe(s) → FeS2(s) +2H2(g) ∆rHo = -137,0 kJ/mol H2S(g) +3/2O2(g) → H2O(l) + SO2(g) ∆rHo = -562,0 kJ/mol Calcule a variação de entalpia de formação do H2S(g) e do FeS2(s) Da equação de oxidação do H2S(g), pode-se determinar sua entalpia padrão de formação: ∆rHo = Σ ν∆fHo(produtos) - Σ ν∆fHo(reagentes) ∆rHo = 1× ∆fHo[H2O(l)] + 1× ∆fHo[SO2(g)] - ∆fHo[H2S(g)] -562,0 = -285,83 kJ + (-296,81) – ∆fHo[H2S(g)] ∆fHo[H2S(g)] = -20,6 kJ Uma vez conhecido o valor de ∆fHo[H2S(g)], pode-se determinar ∆fHo[FeS2(s)] considerando-se equação termoquímica da reação entre o H2S(g) e o Fe(s) ∆rHo = 1× ∆fHo[FeS2(s)] - 2× ∆fHo[H2S(g)] -137,0 kJ = ∆fHo[FeS2(s)] - 2×(-20,6) ∆fHo[FeS2(s)] = -178,2 kJ 3) (2,0 pontos) Três mols de um gás ideal são comprimidos isotermicamente de 60L para 20L, usando-se uma pressão constante de 5 atm. Calcule q, w, ∆U e ∆H. Justifique qualquer consideração assumida. Processo isotérmico (gás ideal): ∆U = ∆H = 0 q = −w ∫ w = − pext dV ⇒ w = − pext × ∆V ⇒ w = −5atm × (20L − 60L) ⇒ w = 5 × 40atm L 1J w = 200 atm L × 2 -2 1kg m s q = -w = -20,3 kJ 101325 Pa 1kgm-1s -2 1 atm 1Pa 1m3 = 20,3kJ 1000L 4) (2,0 pontos) A 25 oC o coeficiente de expansão da água é α = 2,07×10-4 K-1 e a densidade é 0,9970 g/cm3. Se elevarmos 200 g de água de 25 oC para 50 oC, à pressão constante de 101 kPa, a) Calcule w b) Dado CP,m = 75,30 JK-1mol-1, calcule q, ∆U e ∆H α= 1 V ( ) ∂V ∂T p ∫ w = − pext dV ⇒ w = − pext × ∆V ∂V dV = dT ∂T p dV = αV dT ⇒ ∂V Como = α V, tem-se ∂T p V2 T2 V1 T1 ∫ dV = ∫ αV dT ⇒ ∆V = αV ∆T (assume-se, assim, que o volume é independente da temperatura) 1cm3 O volume pode ser determinado pela densidade: ×200g = 201 cm3 0,9970 g ∆V = 2,07×10-4 K -1× 201 cm3 × (25K) ⇒ ∆V = 1,04 cm3 5) (2,0 pontos) Um gás ideal, CV,m = (5/2)R, é expandido adiabaticamente contra uma pressão constante de 1 atm até que seu volume seja o dobro. Se a temperatura inicial é 25 oC e a pressão inicial 5 atm, calcule q, w, ∆U, T2, e ∆H. Justifique qualquer consideração assumida. R: processo adiabático: q = 0 e 1ª Lei da termodinâmica, torna-se ∆U = w ∫ w = − pext dV ⇒ w = − pext × ∆V ⇒ w = − pext × (2V1 − V1 ) → w = − pext × V1 w = − pext × 1mol×8,31447JK -1mol-1×298K nRT ⇒ w = −1 atm × ⇒ w = -0,5 kJ p1 5 atm Como ∆U = w e ∆U = nCV,m∆T ⇒ -0,5 kJ = 1mol×(5/2)R×(T2-T1) -500 J = 1mol×(5/2)8,31447 JK-1mol-1 × (T2-298K) ⇒ T2 = 274 K ∆H = nCP,m∆T; (como CP,m-CV,m = R; CP,m = (7/2)R) ∆H = nCP,m∆T ⇒ ∆H =1 mol (7/2) × 8,31447 JK-1mol-1 × (-24K) ⇒ ∆H = -0,7 kJ w = − pext × ∆V ⇒ w = −101kPa×1,04cm3 -1 -2 1000 Pa 1 kg m s w = −101kPa×1,04cm3 1 kPa 1 Pa 1J 1 kg m 2s -2 1 m 3 = −0,105J 100 cm ∫ ∆H = nCP,m dT ⇒ ∆H = nC P ,m ∆T 1 mol H 2 O nH 2 O = ×200 g = 11,1 mol de H 2 O 18,02 g H 2 O ∆H = nC P ,m ∆T ⇒ ∆H =11,1 mol ×75,30 J K -1mol-1 × 25K ⇒ ∆H = 20,9 kJ H = U + pV ⇒ ∆H = ∆U + p∆V (p = cte) ∆U = ∆H − p∆V ⇒ ∆U = 20,9 kJ - 0,105 × 10-3 kJ ⇒ ∆U = 20,9 kJ (∆H p∆V !!!!) Nota: Em geral é justificável ignorar a diferença entre a entalpia e a energia interna de fases condensadas, exceto em pressões muito elevadas, quando o produto pV não é desprezível 6) (questão bônus, até 2,0 pontos). Discorra conceitualmente sobre os principais tópicos por ora (até agora) estudados na disciplina de termodinâmica química. Sugestões de tópicos: i) gases ideais & gases reais; ii) equação de estado; iii) fator de compressibilidade; iv) primeira lei da termodinâmica; iv) processos reversíveis e irreversíveis; v) calorimetria e termoquímica; vi) Cp e Cv; vii) entalpia; viii) processos isotérmicos e adiabáticos; ix) entalpia de reação e temperatura; x) compressibilidade isotérmica, kT; x) pressão interna, πT; xi) coeficiente de expansão térmica, α; xii) Efeito Joule-Thomson. Use o verso da folha se necessário.
Baixar