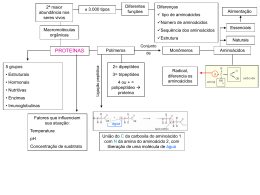
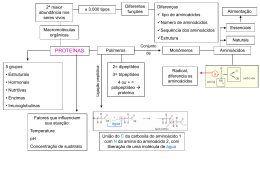
Degustação de trechos de livros sugeridos Determinação de estrutura de proteínas Fonte: Wilson & Walker. Principles and Techniques of Biochemistry and Molecular Biology, 7ed. Página 328Massa molecular relativa Existem três métodos disponíveis para a determinação da massa molecular relativa, Mr, frequentemente denominado de “peso molecular”. Os dois primeiros descritos abaixo são rápidos e simples e resultam em valores com precisão de 5-10 %. Dependendo do objetivo do experimento, necessita-se saber um valor estimado de tamanho da proteína e esses métodos em geral são suficientes para tal finalidade. O terceiro método baseado na determinação da massa por espectrometria de massa requer pessoas especializadas na utilização do instrumento e dão valores de massa com precisão de 0.001 %. Este tipo de precisão é essencialmente importante quando se quer determinar modificações póstraducionais. - Eletroforese em gel de SDS-poliacrilamida (SDS-PAGE, SDS-polyacrylamide gel electrophoresis) Neste tipo de eletroforese as proteínas são separadas com base no tamanho, o qual por sua vez está diretamente relacionado com a massa molecular. Para a determinação da massa da proteína, aplica-se uma amostra contendo uma mistura de proteínas com massas conhecidas (marcadores de peso molecular) no mesmo gel. Após a eletroforese, cora-se o gel e mede-se a distância percorrida por cada proteína marcadora e calcula-se a razão entre a distância percorrida pela proteína sobre a distância total da corrida de eletroforese. Com esses dados constrói-se uma curva de Log Mr (massa da proteína marcadora) vs migração relativa (da proteína marcadora). Determina-se a distância percorrida pela proteína e calcula-se a sua migração relativa. Com esse dado, determinase a massa molecular utilizando-se a equação da reta obtida na curva padrão. - Cromatografia de exclusão (filtração em gel) O volume de eluição da proteína na cromatografia de exclusão é determinado pelo tamanho da proteína. Esse volume de eluição tem relação direta com o logarítimo da massa molecular da proteína (Log Mr). Com a finalidade de determinar a massa de uma proteína, primeiro é necessário construir uma curva de calibração (curva padrão) utilizando várias proteínas de massa conhecida. Em geral faz-se a cromatografia de exclusão utilizando-se o equipamento de HPLC com colunas cujas dimensões variam de 1 a 30 cm e fluxos de 1 mL/min. Curvas de calibração lineares são obtidas plotando-se o Log Mr vs Kd (eluição relativa). O Kd é calculado seguindo a equação: Kd = (Ve-Vo) (Vt-Vo) Onde, Vo é o volume morto (volume necessário para que as moléculas totalmente excluídas da coluna saia, em geral é um volume muito pequeno), Vt é o volume necessário para que moléculas muito pequenas que entram em todos os poros saiam 1 (volume total da coluna) e Ve é o volume necessário para que a proteína de interesse saia. Esse método dá valores de massa com precisão de 10%. - Espetrometria de massas Usando métodos de ionizações brandas, como o electrospray (ESI) ou o MALDI (dessorção/ionização a laser assistida por matriz), pode-se produzir íons moleculares intactos para proteínas e, portanto pode-se determinar a sua massa molecular de forma precisa por espectrometria de massas. Em geral, o ESI produz íons de moléculas com massas de até 100 kDa, enquanto o MALDI produz íons de moléculas com massas de até 200 kDa. Em ambos, necessita-se de quantidades muito pequenas de amostra (pmols). A espectrometria de massa possibilita a determinação de massa com alta precisão para proteínas e peptídeos, e este tipo de dado pode ser utilizado para deduzir pequenas alterações na estrutura básica da proteína. Análise de aminoácidos A determinação da composição de aminoácidos e sua proporção relativa em uma proteína pode ser alcançada realizando-se uma hidrólise total da proteína e analisandose os aminoácidos livres por técnicas cromatográficas. A hidrólise pode ser feita aquecendo-se a proteína em HCl 6 M por 14 h a 110 C, no vácuo. Infelizmente a hidrólise ácida destrói ou modifica quimicamente aminoácidos como asparagina, glutamina e triptofano. Asparagina e glutamina são convertidas aos seus respectivos ácidos aspartato e glutamato e são analisados e quantificados como tal. Triptofano é completamente destruído e é melhor determinado espectrofotometricamente na proteína intacta. Os aminoácidos livres são separados por cromatografia. Atualmente, isto é feito derivatizando-se (reação em que o aminoácido é acoplado a um composto que absorve luz ou que fluoresce). Reagentes utilizados para tal finalidade incluem o-ftalaldeído e 6aminoquilil-N-hidroxisuccinimidyl carbamato (AQC), fenilisotiocianato, etc. Determinação da estrutura primária Por vários anos o sequenciamento de aminoácidos foi realizado utilizando-se proteínas isoladas (purificadas). Isso significa que a maioria dos dados de sequências disponíveis de proteínas eram limitadas a aquelas proteínas que podiam ser purificadas em quantidades suficientemente grandes para serem sequenciadas. O conhecimento da sequência de aminoácidos de uma proteína era (ainda é) um pré-requisito para a determinação da estrutura tridimensional da proteína e o conhecimento da função proteica. No entanto, atualmente os bioquímicos que trabalham com proteínas ficam normalmente satisfeitos com dados de algumas sequências curtas obtidas da região amino-terminal ou de uma sequência de um peptídeo interno, produzido pela clivagem parcial da proteína por proteases. Os dados do sequenciamento são em geral utilizados para: - Pesquisar sequências em banco de dados para verificar se a proteína de interesse já foi isolada e, portanto pode ser identificada. Para este tipo de busca, sequências muito curtas (3-5 resíduos), conhecidos como “sequence tags” são utilizadas. - Pesquisar sequências homólogas usando bancos de dados para identificar função de proteínas. Por exemplo, a busca pode mostrar sequências com identidade significante 2 com sequências de proteínas quinases conhecidas, sugerindo fortemente que a proteína em estudo seja também uma proteína quinase. - A sequência pode ser utilizada para desenhar probes de oligonucleotídeos a serem utilizados para selecionar clones a partir de bibliotecas de DNA complementar. Desta forma, o DNA codificante para a proteína pode ser isolado e a sequência do DNA, e portanto a sequência da proteína podem ser determinadas. A obtenção da sequência da proteína desta forma é mais trabalhosa e mais demorada do que realizar o sequenciamento direto da proteína. Um outro uso da sequência da proteína é para o controle de qualidade em indústrias biofarmacêuticas. Várias companias farmacêuticas produzem produtos que são proteínas, por exemplo, hormônios peptídicos, anticorpos, enzimas terapêuticas, peptídeos sintéticos. A análise da sequência, especialmente para determinar sítios e natureza das modificações pós-traducionais, como glicosilação, é necessário para garantir a integridade estrutural destas proteínas. - Degradação de Edman Em 1950, Per Edman publicou um método químico para a remoção sucessiva de resíduos de aminoácidos a partir da extremidade amino-terminal da proteína ou peptídeo. Estas séries de reações foram denominadas de degradação de Edman e o método continua sendo o meio químico mais efetivo de remover resíduos de aminoácidos de maneira sucessiva a partir de uma cadeia polipeptídica e então determinar a ordem em que os aminoácidos estão ligados à partir da extremidade amino-terminal de uma proteína ou peptídeo. No entanto, o método vem sendo cada vez menos utilizado atualmente e nãos será descrito em detalhes aqui. O desenvolvimento da espectrometria de massas nos últimos 20 anos tornou esta técnica o método de escolha para a determinação da sequência de proteínas. - Clivagem da proteína e produção de peptídeos Quando se estuda uma proteína existem várias ocasiões em que sua clivagem em fragmentos peptídicos menores é necessária. Peptídeos podem ser produzidos por clivagem química ou enzimática. Os métodos químicos tendem a produzir fragmentos peptídicos muito grandes, pois em geral as reações clivam em aminoácidos menos comuns, produzindo 2 ou 3 peptídeos grandes. Métodos enzimáticos tendem a clivar ligações peptídicas adjacentes a aminoácidos que são comuns em proteínas (ex. a tripsina cliva a cadeia onde tiver lisina ou arginina) produzindo 50 ou mais peptídeos. - Espectrometria de massas (MS) Devido ao absoluto requerimento da produção de íons em fase gasosa a técnica de espectrometria de massas por muitos anos ficou restrita à análise de compostos pequenos e apolares com massas menores que 500 Da. No entanto, no início da década de 80 introduziram-se técnicas de ionização como o electrospray e o MALDI que possibilitaram a análise de proteínas por espectrometria de massas. Embora a degradação de Edman ainda seja utilizada ocasionalmente, a espectrometria de massas é atualmente o método de escolha para a determinação da sequência de aminoácidos. Quando peptídeos são fragmentados no espectrômetro de massas, estes são fragmentados predominantemente na ligação peptídica (embora outras fragmentações 3 também ocorram, complicando a interpretação do espectro de massas). Isto significa que os fragmentos peptídicos gerados diferem sequencialmente da massa de um resíduo de aminoácido, podendo a sequência ser deduzida facilmente. Em particular, se há uma modificação, ela pode ser observada pelo acréscimo de massa correspondente. O uso da espectrometria de massas para a obtenção da sequência de aminoácidos e peptídeos está descrito em mais detalhes na Seção 9.5 (Ver Degustação 7). A espectrometria de massas em tandem (MS/MS ou MS2) é cada vez mais utilizado para a obtenção de dados de sequência. A proteína digerida (em geral utilizando-se tripsina) é introduzida no espectrômetro de massas (com ou sem separação prévia da mistura de peptídeos). O íon correspondente a um peptídeo é selecionado no primeiro analisador e fragmentado na câmara de colisão e os fragmentos gerados analisados em um segundo analisador de massas. O espectro de massa desses fragmentos fornece as informações necessárias para a obtenção da sequência daquele peptídeo (Detalhes na Seção 9.5). 4
Baixar