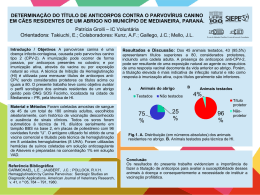
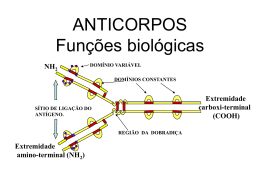
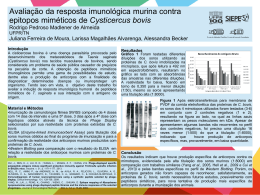
Ácido Fólico
O ácido fólico tem um papel importante na manutenção da hematopoiese, no
processo de síntese do DNA e em diferentes processos metabólicos. É absorvido no
intestino delgado e armazenado no fígado, mantendo uma reserva suficiente por
um período de 3 a 5 meses.
A deficiência está associada à escassez na dieta, a situações que levam ao aumento
da ingestão (medicamento), a fármacos, a patologias que interferem no
metabolismo e/ou na absorção e aos síndromes de má absorção.
A sua avaliação é útil na investigação de anemias, no acompanhamento da terapia
de reposição, na avaliação de alcoólicos crónicos, durante a gravidez, nos
diferentes síndromes de má absorção e nos distúrbios de absorção pós-cirurgia
para obesidade mórbida.
A anemia megaloblástica é a principal manifestação clínica da deficiência de ácido
fólico. Outras manifestações como doença depressiva, alterações neurológicas e
psiquiátricas e fetos com defeitos do tubo neural estão associadas também à sua
deficiência.
Níveis séricos diminuídos são encontrados também na gravidez (33%), em anemias
hemolíticas, em hepatopatias, em neoplasias, em pacientes hemodialisados, no
alcoolismo crónico, em alterações intestinais que levam a má absorção e no uso de
alguns medicamentos.
As drogas podem interferir tanto na sua absorção como no seu metabolismo. O uso
de anticonvulsivantes (fenitoína, fenobarbital e primidona), de contraceptivos orais
e, mais raramente, o uso de antiácidos e antagonistas do receptor H2 alteram a
absorção. O seu metabolismo pode ser alterado por uso de antimicrobianos que
levam à diminuíção das bactérias intestinais importantes no processo de
metabolização. Os quimioterápios, especialmente o metotrexate e o uso de álcool,
também interferem no seu metabolismo. O álcool interfere tanto no metabolismo
como na absorção.
O uso crónico de anticonvulsivantes leva quase sempre à macrocitose (aumento do
MCV), mesmo na ausência de anemia.
Ácido Lático
Em condições adequadas de oxigenação, a glicose é metabolizada por via aeróbica
para a produção de energia. Os produtos de seu metabolismo são convertidos em
piruvato, que por sua vez é metabolizado no ciclo de Krebs. Já em condições de
hipóxia tecidual grave, o metabolismo aeróbico torna-se incapaz de funcionar e,
assim, o piruvato é convertido em ácido láctico, utilizando o metabolismo
anaeróbico.
O ácido láctico é um ácido com pH 3,9, e, por isso, praticamente todo ele é
encontrado na circulação sob a forma de L-lactato. É derivado principalmente do
músculo-esquelético, cérebro, rins e eritrócitos. A sua concentração sanguínea
depende da taxa de produção nos tecidos e das taxas de metabolização hepática e
renal. Cerca de 65% do lactato produzido é usado pelo fígado, especialmente na
gliconeogénese. A remoção extra-hepática do lactato ocorre por oxidação na
musculatura esquelética e no córtex renal. Com a normalização dos níveis de
oxigenação, o lactato acumulado nas células é novamente convertido em piruvato,
retornando ao ciclo normal.
Os níveis séricos do ácido láctico estão relacionados com a disponibilidade de
oxigénio. Na avaliação da oxigenação, a concentração de pO2 indica a captação
alveolar pulmonar do O2. A medida da saturação de O2 determina o teor de
oxigenação arterial, enquanto o lactato é uma medida sensível e confiável de
hipóxia tecidual. Eleva-se precocemente antes que outras alterações clínicas ou
gasométricas possam ser detectadas, é por isso considerado um marcador precoce
de hipóxia tecidual.
SECUNDÁRIA À HIPÓXIA TECIDUAL
Choque, hipovolemia, insuficiência ventricular esquerda, infarte agudo do
miocárdio, edema pulmonar, estados per e pós- operatórios, cirurgia cardíaca,
circulação extracorpórea
DISTÚRBIOS METABÓLICOS
(PRODUÇÃO EXCESSIVA - DIIMINUIÇÃO DA REMOÇÃO HEPÁTICA)
Diabetes mellitus, insuficiência hepática, neoplasias, linfomas, leucemias,
intoxicações por drogas (acetaminofeno e salicilatos), etanol, metanol,
erros inatos do metabolismo, exercício excessivo e hiperventilação.
A acidose láctica pode ter origem na hipóxia tecidual ou pode ocorrer em
consequência de outras patologias que levem a alterações metabólicas. Em geral,
as etiologias desassociadas da hipóxia apresentam valores menos elevados.
Em pacientes com acidose metabólica, uma diferença aniónica elevada sugere o
diagnóstico de acidose láctica. É do que se deve suspeitar quando a soma dos
aniões menos a soma dos catiões ultrapassa 18 mEq/L. Isso, é claro, na ausência
de outras causas que possam levar a uma diferença aniónica, como insuficiência
renal, cetonémia importante e intoxicação por metanol.
[(Na+ + K+ ) - (Cl-+HCO3-)] > 18 mEq/L
Os seus níveis podem estar alterados noutros líquidos orgánicos como líquor e
líquidos pleural, ascítico e peritoneal, auxiliando no diagnóstico diferencial entre
infecções bacterianas e virícas.
Ácido Úrico
O ácido úrico é o maior produto do catabolismo das purinas. É armazenado no
organismo num pool de alto turnover, sendo oriundo do catabolismo das proteínas
da dieta e de fontes endógenas, concentrando-se principalmente no fígado. Cerca
de 60% desse é trocado diariamente por formação e excreção concomitantes.
O ácido úrico é excretado principalmente por via renal. Apenas uma pequena
parcela (1/3) é eliminada por via gastrointestinal. Não existe uma relação directa
entre os valores séricos e os valores urinários.
Os níveis séricos do ácido úrico são determinados pela relação entre a dieta, a
produção endógena e os mecanismos de reabsorção e de excreção. Os mecanismos
de reabsorção e de excreção renais são complexos, e podem ocorrer alterações na
filtração glomerular, na reabsorção tubular proximal, na secreção tubular e na
reabsorção após secreção.
Diversos factores como dieta, predisposição genética, sexo, idade, peso,
medicamentos, uso de álcool e associação com outras patologias como diabetes
mellitus e distúrbios lipídicos podem alterar os valores séricos e levar a um
desequilíbrio entre a absorção e a excreção de ácido úrico. Os seus valores sofrem
uma variação diurna, com valores mais elevados pela manhã e mais baixos à noite.
A hiperuricémia é a forma comum de se definir o aumento da concentração sérica
de ácido úrico que ultrapasse os valores de referência. Pode ocorrer por diferentes
mecanismos, associados com aumento da produção ou diminuição da excreção
renal. Ocorre nas dietas ricas em carnes, especialmente vísceras (fígado e rim),
vegetais leguminosos e trigo. Também é encontrada nas dislipidémias, nas anemias
hemolíticas, na anemia perniciosa e noutras situações em que há aumento do
turnover de ácidos nucleicos (excesso de destruição celular), como ocorre nas
neoplasias e no curso de quimioterapia e de radioterapia, especialmente no
tratamento de linfomas e de leucemias. A policitemia, o mieloma múltiplo e o
infarte agudo do miocárdio extenso podem também aumentar o metabolismo das
nucleoproteínas. Alterações da função renal, hipertensão arterial, hipotireoidismo,
hiperparatireoidismo, diabetes insipidus, diabetes mellitus, doença de Addison e
uso de drogas como salicilatos e alguns diuréticos podem induzir à diminuição da
velocidade de excreção de ácido úrico.
Portanto, os níveis séricos do ácido úrico podem apresentar-se alterados numa
gama de situações clínicas, incluindo a gota. A gota é responsável por apenas 10 a
15% das hiperuricemias. A maioria dos pacientes com gota sofre tanto de
superprodução como de hipoexcreção. A gota caracteriza-se clinicamente por
hiperuricémia, precipitação de urato monossódico em fluidos biológicos
supersaturados e depósito de urato por todo o corpo, com excepção do sistema
nervoso central, mas com maior predisposição para articulações, cartilagem
periarticular, ossos, bursa e tecidos moles subcutâneos. São comuns ataques
recorrentes de artrite, nefropatia e, frequentemente, nefrolitíase.
Os depósitos de uratos são responsáveis pelos sinais e sintomas da artrite gotosa,
pois levam a uma severa reacção inflamatória no local. Nos rins são descritos três
tipos distintos de lesões: a nefropatia gotosa com depósito de uratos no
parenquima, o depósito intratubular agudo de cristais de urato e a nefrolitíase.
Os homens respondem por cerca de 90% dos casos de gota. Normalmente, ela é
classificada como primária quando decorre de um erro metabólico, directamente
ligado ao aumento da produção ou à diminuição da excreção, e como secundária
quando decorre do aumento do ácido úrico em consequência de outras numerosas
etiologias.
AUMENTO DA FORMAÇÃO
Aumento da síntese de purinas
Desordens metabólicas hereditárias
Excesso de ingestão de purinas
Aumento do turnover de ácidos
nucléicos
Hipóxia tecidual
DIMINUIÇÃO DA SECREÇÃO
Idiopática
Insuficiência renal crónica
Aumento da reabsorção renal
Drogas (diuréticos e salicitatos)
Intoxicação por chumbo
Hipertensão arterial
Outras doenças endócrinas
A hipouricémia é rara, podendo ser secundária a diferentes situações como uma
doença hepatocelular grave, que leva à diminuição da síntese de purina, deficiência
da reabsorção tubular de ácido úrico congénita, como no síndrome de Fanconi, ou
adquirida, por supertratamento com drogas uricosúricas, na secreção inadequada
da hormona antidiurética, na doença de Wilson, na xantinúria, nas intoxicações por
metais pesados e nas dietas pobres em purina.
A quantidade de ácido úrico presente na urina varia de acordo com o pH: é tanto
menor quanto maior for o pH. A excreção urinária de ácido úrico aumentada pode
ocorrer isolada ou associada a outros distúrbios metabólicos (com aumento da
produção endógena), pelo aumento da ingestão de purinas e pelo uso de drogas
uricosúricas, principalmente na fase inicial do tratamento. A diminuição dos níveis
urinários de ácido úrico pode estar associada a gota crónica e a uma dieta pobre
em purinas.
Como já citado, não existe correlação directa entre os níveis séricos e urinários do
ácido úrico. A sua avaliação é útil na investigação das litíases renais. Os cristais de
ácido úrico são achados frequentes em crianças em fase de crescimento acelerado
e noutras situações de aumento do metabolismo de nucleoproteínas. Algumas
drogas, como antiinflamatórios, aspirina, vitamina C, além dos diuréticos, podem
alterar a sua excreção.
Ácido Valpróico
O ácido valpróico (valproato de sódio) é utilizado no tratamento das convulsões
tónico-clónicas, mioclónicas e atónicas e das ausências. Embora o seu mecanismo
de acção não seja totalmente conhecido, acredita-se que actue aumentando a
atividade do sistema inibidor, mediado pelo ácido gama-aminobutírico (GABA).
A absorção é rápida e completa, com picos séricos entre 1 a 4 horas após a
administração oral. As concentrações séricas terapêuticas usuais são de 50 a 100
mg/mL. A sua acção inicia-se cerca de 4 horas após a administração oral, e o
estado de equilíbrio normalmente é alcançado num período entre 1 a 4 dias.
Cerca de 90% da droga é metabolizada no fígado, e uma grande percentagem
(93%) está ligado a proteínas plasmáticas, principalmente à albumina. Por isso, em
situações com diminuição proteica, cirrose, uremia e uso de drogas que competem
com essa ligação, a taxa de ácido valpróico livre (fração biologicamente activa)
pode aumentar.
A semi-vida sérica no adulto é de 16 horas, podendo ser prolongada por uso de
bebidas alcoólicsa e diminuída pela associação com fenitoína, fenobarbital,
carbamazepina e primidona. Com o uso crónico, a semi-vida é reduzida para 12
horas. Em crianças, a semi-vida é de 12 horas. Em recém-nascidos e nas doenças
hepáticas em que o metabolismo é reduzido, a semi-vida é prolongada.
O ácido valpróico deve ser utilizado com cautela em gestantes, devido aos seus
prováveis efeitos teratogénicos. Os efeitos colaterais incluem sedação, distúrbios
gástricos, reacções hematológicas (a mais comum é a plaquetopenia), ataxia,
ganho de peso, sonolência e coma. Foram descritos casos raros de
hepatotoxicidade e pancreatite graves ou fatais.
A sua dosagem é de grande utilidade para avaliar os níveis de adesão ao
tratamento e detectar as concentrações em níveis tóxicos. Níveis baixos podem ser
encontrados por falta de adesão à terapia.
Normalmente, a colleita deve ser realizada em pacientes que estejam medicados
com o medicamento há pelo menos, 2 a 4 dias, e sempre antes da próxima dose.
Entretanto, nos casos de suspeita de intoxicação, a colheita pode ser realizada a
qualquer altura. Para facilitar a interpretação, é importante conhecer a hora em que
a última dose foi ingerida.
Agregação Plaquetária
Entre as propriedades das plaquetas estão a manutenção da homeostasia, a adesão
à superfície endotelial danificada, a agregação em resposta a uma variedade de
estímulos e a secreção de factores de coagulação, vasoconstritores e factores de
crescimento após a sua activação.
O processo de formação do trombo plaquetário inicia-se com a lesão endotelial.
Quando ocorre uma lesão vascular, a matriz colagénea e as proteínas
subendoteliais ficam expostas. É nesse local que os receptores de membrana das
plaquetas se ligam, resultando na adesão plaquetária, a primeira etapa do processo
de formação do trombo plaquetário. Múltiplos agonistas são gerados neste
momento. Eles induzem a activação plaquetária, ocasionando alterações nos
receptores da glicoproteína (GP) IIb/IIIa e levando a um estado de receptividade à
ligação do fibrinogénio. Nessa fase, as plaquetas encontram-se definitivamente
activadas. Em seguida, inicia-se o processo de agregação plaquetária, com a
ligação múltipla e cruzada do fibrinogénio aos receptores GP IIb/IIIa.
As causas de diminuição da agregação plaquetária podem ser congénitas ou
adquiridas. Entre as causas congénitas estão a doença de von Willebrand, a
trombastenia de Glanzmann e o síndrome de Bernard-Soulier. Todas estas
patologias estão relacionadas com defeitos na fase de adesão plaquetária.
As plaquetas aderem a superfícies estranhas por meio da ligação das glicoproteínas
da sua membrana, tendo como participante indispensável uma proteína plasmática,
na verdade um componente do complexo molecular do fator VIII da coagulação
chamado fator de von Willebrand. Esse mecanismo pode ser estudado em
laboratórios pelo tempo de sangria, teste de adesividade plaquetária, teste de
agregação plaquetária com ristocetina, dosagem do co-factor da ristocetina e
dosagem do fator VIII.
A doença de von Willebrand e a ausência congénita do factor de von Willebrand ou
do co-factor da ristocetina fazem com que a agregação seja anormal com todos os
estimulantes utilizados usualmente. Na doença de von Willebrand dos tipos IA e
IIA, a agregação com a ristocetina é geralmente anormal, mas está aumentada no
tipo IIB.
A trombastenia de Glanzmann, embora rara, é a principal doença autossómica
recessiva que afecta a interacção entre as plaquetas.
Caracteriza-se por um prolongamento do tempo de sangria, episódios recorrentes
de sangramentos mucocutâneos e ausência de resposta agregante aos estimulantes
normais com resultado positivo à ristocetina.
Na síndrome de Bernard-Soulier, ocorre ausência de resposta das plaquetas ao
factor de von Willebrand; as plaquetas respondem normalmente aos estimulantes
usuais, mas sem se agregarem em resposta à ristocetina.
Entre as condições adquiridas que causam diminuição da agregação plaquetária,
temos o uso de medicações inibidoras, doenças auto-imunes que produzem
anticorpos contra as plaquetas, desordens mieloproliferativas, uremia por
insuficiência renal, desordens adquiridas do armazenamento de ADP e produtos de
degradação da fibrina.
Algumas condições podem produzir aumento da agregação plaquetária, como
sejam, quadros de hipercoagulabilidade que indicam um risco de acidente vascular
cerebral, trombose venosa profunda e outras condições associadas à formação de
coágulo.
Na avaliação de pacientes com desordens plaquetárias qualitativas, deve ser
sempre considerado o estudo da agregação plaquetária, indicado também para os
pacientes com sangramento mucocutâneo de natureza prolongada e contagem
plaquetária normal.
O teste de agregação pode ser utilizado na monitorização de pacientes tratados
com antiagregantes plaquetários, como ácido acetilsalicílico e ficlodipina. O teste
com ácido araquidónico em vigência do uso dessas drogas resulta na agregação
diminuída ou ausente. O quadro a seguir actualiza os resultados da agregação
plaquetária nas diversas patologias e com os diferentes agentes agregantes.
ADP
1º
Fase
Glanzmann
A
Von Willebrand
N
Bernard- Soulier
N
Uso de AAs ou
N
similares
PATOLOGIA
Adrenalina Ristocetina Colagénio
Ácido
Araquidónico
2º
Fase
A
N
N
A
N
V
V
V
A
A
N
N
A
N
N
A
V
N
V
A
A=Anormal N=Normal V=Variáveis
O exame é realizado a partir do plasma do paciente em num instrumento fotóptico
denominado agregómetro. O plasma enriquecido em plaquetas é colocado em
contato com agentes agregantes. Ocorre, então, a formação crescente de grandes
agregados plaquetários, acompanhados de diminuição da turbidez da amostra. A
mudança na densidade óptica é transmitida pelo instrumento, em percentagem de
agregação. Os agentes agregantes normalmente utilizados no teste são ADP
(adenosina difosfato), colagénio, adrenalina, ácido araquidónico e ristocetina.
A informação clínica do paciente quanto ao uso de medicamentos de acção
plaquetária é importante para se ter certeza de que o resultado observado, caso
alterado, será devido a uma desordem qualitativa subjacente. A disfunção
plaquetária poderá ser observada nalgumas condições clínicas, como insuficiência
renal e desordens mieloproliferativas.
Albumina
De todas as proteínas séricas, a albumina é a que está presente em maior
concentração, correspondendo a cerca de 60% do total de proteínas. É sintetizada
exclusivamente pelo fígado, aparecendo primeiro no citoplasma dos hepatócitos
como um precursor chamado pré-albumina. A sua semi-vida biológica é de cerca de
3 semanas. Tem um papel muito importante em diversas funções do organismo,
como sejam a manutenção da pressão osmótica do plasma e o transporte de
substâncias. Por isso, está relacionada fisiopatologicamente às alterações do
equilíbrio hídrico e aos mecanismos de desentoxificação do organismo, já que tem a
capacidade de fixar substâncias, não só do tipo fisiológico, tais como bilirrubina,
magnésio, cálcio e ácido úrico, como também diversos medicamentos, como
penicilina e sulfa-penicilinas, entre outros.
A hiperalbuminémia é rara e na maioria dos casos indica situações clínicas de
desidratação ou hiperinfusão com albumina.
Já a hipoalbuminémia é frequente, e pode apresentar-se em consequência de
diferentes mecanismos, como diminuição da síntese por lesões hepáticas, má
nutrição e síndromes de má absorção, aumento do catabolismo, como na fase de
resposta aguda, perdas excessivas, como no síndrome nefrótico, outras lesões
renais com perda protéica, disfunção da tiróide, úlcera péptica, alcoolismo crónico,
gravidez, hemorragias, queimaduras, perdas intestinais e perdas para espaço
interestecial, como ascites e outros derrames volumosos.
Nos casos de doenças crónicas como tuberculose e neoplasias, a diminuição ocorre
tanto por alteração da síntese como pelo aumento do catabolismo. A queda de
albumina pode também estar relacionada a uma rápida hidratação, superhidratação, necrose grave difusa do fígado, hepatite crónica activa e neoplasias.
Portanto, essa avaliação é clinicamente importante na verificação das condições
nutricionais, no acompanhamento da síntese, do catabolismo e das perdas
protéicas. Níveis séricos entre 2,0 e 2,5 g/dL correlacionam-se com a manifestação
de edema.
Consultar a electroforese das proteínas
Aldolase
É uma enzima presente em quase todos os tecidos. É composta pelas subunidades
A, B e C, que formam quatro isoenzimas, sendo que a subunidade A responde pela
maior parcela da aldolase sérica.
AAAA
BBBB
CCCC
AAAC
predomina nos músculos esqueléticos
predomina no fígado
predomina no cérebro e noutros tecidos
presente nos tecidos, porém em baixa concentração
A determinação sérica da aldolase é de grande importância clínica na avaliação das
doenças primárias da musculatura esquelética. As maiores elevações são
encontradas na distrofia muscular progressiva de Duchenne. Níveis alterados
também podem ser observados nas miosites e dermatomiosites. Os seus níveis
correlacionam-se com a massa muscular. A perda muscular progressiva faz
diminuir os valores por perda da capacidade de síntese; portanto, só se encontram
valores alterados nas fases precoces das distrofias, quando a massa muscular se
encontra relativamente preservada. Valores normais são encontrados nas
patologias musculares de causa neurológica como esclerose múltipla, poliomielite e
miastenia gravis.
A sua elevação não é específica para doenças musculares. O aumento da aldolase
pode ser encontrado no infarte do miocárdio (pico em 24 a 48 horas), hepatites
virícas agudas, pancreatite hemorrágica, gangrena, tumores da próstata,
neoplasias hepáticas primárias e secundárias, anemia megaloblástica, leucemia
granulocítica e em pacientes medicados com fenotiazida.
Alfa-1-Antitripsina
É uma das proteínas de fase aguda e, como tal, apresenta-se aumentada nas
doenças inflamatórias agudas e crónicas, neoplasias, pós-traumas ou cirurgias e
durante a gravidez ou terapia com estrogénio.
Níveis diminuídos estão associados a deficiência congênita. Na deficiência
homozigota, cerca de 10% das crianças desenvolvem hepatopatias importantes,
inclusive hepatite neonatal e cirrose. Os adultos tendem a apresentar quadro de
enfisema na terceira ou quarta década de vida e quadros de cirrose assintomática,
hepatite crónica activa e carcinoma hepatocelular.
A doença é sugerida pela ausência da banda alfa-1 na eletroforese (a alfa-1antitripsina representa 90% dessa banda) e confirmada pela dosagem sérica da
alfa-1-antitripsina. Nos homozigotos, os níveis séricos encontram-se
acentuadamente diminuídos ou, mais raramente, ausentes. Nos heterozigotos, os
níveis séricos apresentam-se diminuídos em níveis moderados.
A alfa-1-antitripsina deve ser investigada em todos os lactentes com complicações
hepáticas.
Alfa-1-Glicoproteína Ácida
Proteína também conhecida como orosomucóide, é sintetizada basicamente no
fígado, podendo ser sintetizada, ainda, por leucócitos e por células tumorais. É
filtrada em grande quantidade pelo glomérulo, o que acarreta uma semi-vida curta,
de cerca de 5 dias.
Como uma proteína de fase aguda, grandes concentrações séricas podem ser
observadas em processos infecciosos e inflamatórios agudos, durante a gravidez,
infarte agudo do miocárdio, mielomas, doença de Hodgkin, neoplasias, traumas,
queimaduras e colagenoses. Níveis séricos diminuídos podem ser encontrados em
estados de desnutrição, lesões hepáticas graves e patologias com grande perda
protéica.
Os níveis encontrados nos derrames cavitários podem ser de ajuda no diagnóstico
diferencial entre transudados e exsudados. São encontrados níveis baixos nos
transudados, elevados nos exsudados e extremamente elevados nos exsudados de
origem neoplásica.
A sua função biológica ainda não foi bem definida. No entanto, alguns trabalhos
indicam uma participação importante no transporte de hormonas e drogas. É um
dos melhores marcadores de actividade inflamatória, já que retorna a níveis
normais em menos tempo do que as outras proteínas de fase aguda.
Alfa-Fetoproteína
A alfa-fetoproteína (AFP) é a principal glicoproteína plasmática precoce do feto
humano. É sintetizada pelo fígado fetal, os seus níveis elevam-se durante a 14ª
semana de gestação, atingindo os índices normais do adulto em 6 a 10 meses após
o nascimento. Nos adultos, está presente em níveis baixos, em homens e em
mulheres não-grávidas saudáveis.
Em pacientes com carcinomas hepatocelulares e tumores testiculares (nãoseminomas), encontram-se níveis elevados. Valores elevados também podem ser
encontrados em cerca de 20% dos carcinomas gástricos e pancreáticos e numa
pequena percentagem de carcinomas do pulmão e do cólon.
Nem sempre as elevações de AFP estão associadas a malignidade. Os níveis podem
estar elevados em doenças inflamatórias do fígado, como hepatite viríca, hepatite
crónica e cirrose hepática. Níveis altos de AFP também podem estar presentes em
doenças inflamatórias intestinais, como a doença de Crohn, e na colite ulcerosa,
que também produzem elevações do antígeno carcinoembrionário (CEA).
A maior indicação da determinação de AFP é a monitorização do tratamento de
carcinomas hepatocelulares e de tumores testiculares (não-seminomas) e das suas
recidivas. Após o tratamento, os valores retornam ao normal dentro de 4 a 6
semanas. O aumento após esse período ou a permanência de valores elevados
indicam, respectivamente, recidiva e persistência da doença.
Os níveis de AFP estão correlacionados ao tamanho do tumor. Nos tumores
testiculares, é utilizada em associação com a beta-gonadotrofina coriónica (bHCG),
sendo extremamente útil no diagnóstico diferencial. Os seminomas cursam com
AFP negativa e bHCG elevada; nos não-seminomas, ambas se elevam.
É inútil a sua utilização como rastreamento na população, por causa de
significativas elevações em diferentes patologias, benignas e malignas.
Consultar Gonadotrofina Coriônica.
Alfa-Fetoproteína na Gravidez
A alfa-fetoproteína (AFP) produzida pelo feto é transferida para o líquido amniótico
por meio da urina fetal. Os níveis no soro fetal estão em concentrações
aproximadamente 150 vezes mais baixas do que no líquido amniótico. A AFP
também aparece no soro materno por transferência pela placenta ou por difusão
através das membranas fetais. Picos acontecem em torno da 32a semana de
gestação.
Em contraste com os níveis típicos do segundo trimestre, de 10 a 12 mg/mL no
líquido amniótico, os níveis no soro materno durante esse mesmo período são de
apenas 30 a 35 ng/mL. No segundo trimestre, os níveis de AFP no soro materno
aumentam cerca de 15% por semana, enquanto os níveis no líquido amniótico
declinam aproximadamente 13% por semana. Em lesões fetais abertas, como
espinha bífida e anencefalia, ocorre perda da AFP para o líquido amniótico,
elevando os níveis de AFP.
No soro materno, a AFP é um teste de triagem para defeitos congênitos fetais como
os do tubo neural, espinha bífida, anencefalia e também para a síndrome de Down.
A triagem adequada para essas alterações requer um protocolo integrado que
envolve exames laboratoriais, ultra-sonografias, médicos obstetras e
aconselhamento genético.
Uma triagem falso-positiva pode ser provocada por subestimação da idade
gestacional, gestação múltipla inesperada ou hemorragia de placenta. As duas
razões anteriores podem ser excluídas por meio do exame de ultra-som e pela
repetição da avaliação, 1 semana depois. Se tais condições não estiverem
presentes, um teste positivo alto indica risco, com probabilidade,
aproximadamente, de 1 em 50 de uma anomalia fetal.
A combinação da idade materna na data da coleta do sangue e os níveis de AFP irá
predizer o fator de risco para a síndrome de Down. O HCG e o estriol são usados
freqüentemente para um aumento adicional da precisão da avaliação de risco da
síndrome de Down (teste triplo).
Consultar Teste Triplo.
Amilase
As amilases são enzimas que catalisam a hidrólise da amilopectina, da amilose e do
glicogênio. A amilase presente no sangue e na urina de indivíduos normais é de
origem pancreática (predominantemente forma P) e das glândulas salivares (forma
S).
A avaliação dos níveis séricos da amilase tem grande utilidade clínica no diagnóstico
das doenças do pâncreas e na investigação da função pancreática. Na pancreatite
aguda, os níveis de amilase podem alcançar valores de quatro a seis vezes o limite
superior de referência, elevando-se em 2 a 12 horas e retornando a níveis normais
em 3 a 4 dias. A magnitude da elevação da amilase não se correlaciona com a
gravidade da lesão pancreática. Cerca de 20% dos casos de pancreatite aguda
podem cursar com valores normais de amilase. Por isso, a dosagem concomitante
dos níveis de lipase é importante, permitindo o diagnóstico desses casos. Nos casos
que evoluem com formação de pseudocistos, os níveis de amilase continuam
elevados por mais tempo. Os abcessos pancreáticos também podem aumentar os
níveis séricos da amilase. As pancreatites crónicas cursam com níveis normais ou
pouco elevados de amilase.
O carcinoma pancreático cursa com níveis normais; a elevação aparece em menos
de 5% dos pacientes. Na maior parte dos casos, os níveis de amilase só se elevam
quando o tumor provoca a obstrução do ducto pancreático principal.
As causas não-pancreáticas de aumento da amilase incluem lesões inflamatórias
das glândulas salivares, como parotidite, apendicite aguda, gravidez tubárica rota,
úlcera péptica perfurada, trauma pancreático, obstrução intestinal, aneurisma
dissecante da aorta, pós-operatório de cirurgias toráxicas e abdominais,
queimaduras, doenças do trato biliar, traumas e uso de um grande número de
drogas como morfina e derivados. A amilase pode estar elevada também em
neoplasias como as pulmonares e as ováricas, e estudos apontam que a elevação
se dá à custa da amilase do tipo S.
Os níveis urinários de amilase permanecem alterados por períodos mais longos que
os séricos. Nos casos de complicação com pseudocisto de pâncreas, a amilase
urinária pode permanecer elevada por semanas após os níveis séricos terem
retornado ao normal. Nos indivíduos com função renal normal, a proporção entre a
clearance de amilase e a creatinina é constante, com valores de referência usuais
de 2 a 5%. Na pancreatite, a clearance da amilase está aumentado, e, portanto, a
proporção entre a clearance de amilase/creatinina está elevada. Valores acima de
8% são comuns na pancreatite aguda. Valores elevados podem ser encontrados
também em queimados, na insuficiência renal e no mieloma múltiplo.
Na macroamilasemia, a amilase encontra-se ligada a uma imunoglobulina, e o
complexo formado é muito grande para ser filtrado pelos glomérulos, o que leva a
uma hiperamilasemia aparente que não indica doença; os valores séricos são muito
altos, e os valores urinários, normais. Nesses casos, utilizam-se os resultados da
relação entre a clearance de amilase/creatinina para se fazer o diagnóstico
diferencial. Isso porque, na macroamilasemia, a relação é muito baixa, ao contrário
da relação encontrada na pancreatite aguda.
ANCA
Os anticorpos anticitoplasma de neutrófilos (ANCA) são auto-anticorpos contra uma
serina protease, a proteinase 3. Os ANCA estão associados primariamente à
granulomatose de Wegener. São muitas vezes os únicos anticorpos presentes nessa
patologia. São encontrados também em quadros de vasculite, poliartrite nodosa
microangiopática, poliangeíte microscópica, glomerulonefrite necrotizante
microscópica e síndrome de Churg-Strauss. Também podem estar presentes em
diversas patologias gastrointestinais e reumáticas, ou após o uso de certas drogas.
Quando esses anticorpos são incubados com neutrófilos normais fixados em etanol,
podem ser observados dois padrões de imunofluorescência: o citoplasmático (cANCA) e o perinuclear (p-ANCA).
O padrão c-ANCA é encontrado em cerca de 85% dos casos de granulomatose de
Wegener, e os seus títulos correlacionam-se com a atividade da doença. Em
pacientes com vasculite, esses anticorpos são geralmente dirigidos contra a
mieloperoxidase, o que leva à visualização do padrão perinuclear (p-ANCA). Esse
padrão também pode ser observado na poliartrite nodosa microangiopática, na
poliangeíte microscópica, na glomerulonefrite necrotizante microscópica e na
síndrome de Churg-Strauss.
O padrão p-ANCA deve ser distinguido dos anticorpos antinucleares (ANA). A sua
presença pode ser encontrada em doenças intestinais e na artrite reumatóide.
Técnicas de imunoblotting ou de ELISA são melhores do que a imunofluorescência
por apresentarem maior precisão e por permitirem a identificação dos antígenos
alvos associados a esses anticorpos.
Enzima Conversora da Angiotensina (ECA)
A enzima conversora da angiotensina (ECA) actua na conversão de angiotensina I
em angiotensina II, representando um papel importante na homeostase da pressão
arterial. É produzida principalmente no pulmão.
Embora, a sua medida tenha pouca utilidade no diagnóstico e tratamento da
hipertensão arterial, entre as alterações genéticas consideradas como possíveis
etiologias das doenças cardiovasculares, a que tem despertado maior atenção é o
polimorfismo genético do gene que codifica a ECA.
Num grande número de indivíduos, observaram-se diferenças marcantes nos níveis
plasmáticos da ECA. Essa variabilidade é efeito de um polimorfismo genético no
gene codificante para a enzima. A análise dos níveis de ECA no plasma circulante
demonstrou uma associação directa entre os genótipos relativos ao polimorfismo e
os níveis da proteína. Dessa forma, indivíduos que apresentam o genótipo DD
possuem aproximadamente o dobro da concentração de ECA circulante, em relação
a aqueles com genótipo II, enquanto o genótipo DI apresenta níveis intermediários
de ECA. O polimorfismo D/I encontra-se correlacionado com 47% da variação total
do nível de ECA circulante no plasma.
Estudos realizados demonstraram que o genótipo DD está associado à alta
incidência de EAM, consistindo num factor de risco independente, aumentando em
2,7 vezes o risco relativo de desenvolver tal patologia. O efeito deletério do
genótipo DD pode resultar de uma super expressão da ECA, levando a um aumento
local de angiotensina II em regiões vasculares como a circulação coronária,
podendo ser considerado como um novo e potente factor de risco para o EAM.
A utilização mais frequente do doseamento dos níveis de ECA é no diagnóstico e
acompanhamento da evolução da sarcoidose. Os seus níveis, encontram-se
elevados em 40-85% dos pacientes adultos com sarcoidose activa e diminuem em
resposta à terapia com corticosteróides. Na sarcoidose pulmonar pode estar
aumentada no soro e no lavado bronco-alveolar.
Não é um marcador específico de sarcoidose, visto que concentrações elevadas
podem também ser encontradas noutras patologias pulmonares como enfisema,
asma, carcinoma de pequenas células, carcinoma de células escamosas,
tuberculose, silicose, abestose e pneumonias.
Está elevada, porém, em menor frequência, noutras doenças granulomatosas como
a lepra e a tuberculose e noutras patologias como esclerose múltipla, doença de
Addison, hepatopatias, hipertiroidismo, doença de Gaucher e ocasionalmente em
linfomas não-Hodgkin e no linfoma de Lennert.
Níveis séricos diminuídos podem ser encontrados no hipotiroidismo, anorexia
nervosa, doença hepática crónica e em diferentes patologias malignas.
A determinação da ECA no LCR (líquido cefalo raquidiano) tem particular interesse
no diagnóstico da neurosarcoidoise que ocorre em 5% dos pacientes com
sarcoidose. Entretanto, a sua utilização é controversa, visto que, níveis elevados
podem também ser encontrados na esclerose múltipla, neuro-sífilis e em encefalites
virícas.
Anticardiolipina
A cardiolipina é um fosfolipído aniónico. A maioria dos anticorpos anticardiolipina
(ACA) reage cruzadamente com outros fosfolipídos. Devido a esta característica e
pela sua maior facilidade de detecção, são utilizados na investigação da presença
de anticorpos antifosfolipídos.
Assim como os anticorpos anticardiolipina, os anticoagulantes lúpicos (ACL) são
também anticorpos antifosfolipídos. Interferem nos procedimentos de screening de
coagulação, sendo uma causa comum de prolongamento do aPTT (tempo de
tromboplastina parcial ativado).
Existem algumas características clínicas comuns aos pacientes com a síndrome de
antifosfolipídio (AFL): trombose venosa, trombose arterial, abortos recorrentes e
trombocitopenia. Esses anticorpos são hoje reconhecidos como uma das causas
mais importantes de hipercoagulabilidade e trombose.
Aproximadamente 50% dos pacientes apresentam síndrome AFL primária, isto é,
não-associada a doenças sistémicas, enquanto os restantes apresentam a forma
secundária.
A trombose é a apresentação mais comum da síndrome de AFL, e o local da
trombose (arterial ou venosa) pode definir síndromes com diferentes características
clínicas e laboratoriais: anticardiolipina (AFL-ACA) - positivo e anticoagulante lúpico
(AFL-ACL) - positivo.
A prevalência, a etiologia e a conduta desses síndromes, relacionadas porém
distintas, são diferentes. A apresentação AFL-ACA é mais comum do que a AFLACL. A relação de prevalência é de 5 para 1. Ambas estão associadas a tromboses,
abortos e trombocitopenia.
A AFL-ACA está associada a trombose arterial e venosa, inclusive com quadros de
trombose venosa profunda e embolia pulmonar, doenças coronárias e
cerebrovasculares prematuras. Em contrapartida, o síndrome AFL-ACL é associado
mais frequentemente a trombose venosa, que envolve os quadros de trombose
venosa periférica, mesentérica, renal, hepática, sistema porta e veia cava. A
trombose arterial raramente acontece na síndrome AFL-ACL. A atividade dos ACA
em níveis elevados, na presença de ACL, é um fator de risco importante para
trombose arterial.
Os abortos normalmente acontecem no terceiro trimestre da gravidez e são
associados a trombose da placenta. Embora as mulheres com abortos espontâneos
periódicos no primeiro trimestre possam ter AFL, outras causas de perda fetal no
primeiro trimestre são mais comuns.
A contagem de plaquetas pode ser normal ou ligeiramente baixa. Aproximadamente
30% dos pacientes têm contagem de plaquetas abaixo de 100.000/mL durante o
curso da doença. Paradoxalmente, esses pacientes ainda apresentam risco para
trombose. O tratamento é iniciado quando a contagem de plaquetas se encontra
abaixo de 50.000/mL (especialmente quando abaixo de 35.000/mL), por causa do
risco aumentado de hemorragia.
Os anticorpos anticardiolipina são medidos por testes imunoenzimáticos. A
utilização combinada da pesquisa desses anticorpos com o anticoagulante lúpico
(ACL) melhora a sensibilidade para a descoberta de anticorpos antifosfolipído.
Aproximadamente 60% dos pacientes são positivos tanto para ACL quanto para
ACA, e 40% são positivos somente para ACA ou ACL.
Podem ser encontrados associados a doenças como lúpus eritematoso sistémico
(LES), outras doenças do tecido conjuntivo, infecção pelo HIV e por outros
microrganismos, neoplasias e um quadro induzido por drogas. São encontrados
numa pequena parcela da população saudável.
No LES, a presença do anticorpo anticoagulante lúpico ocorre em cerca de 70% dos
casos . Na pesquisa de sífilis, pode levar a uma reação de VDRL falsamente
positiva.
O diagnóstico de síndrome antifosfolipídico exige a constatação da presença de uma
das características clínicas citadas e um teste positivo. Os testes podem ser o do
anticoagulante lúpico, os de anticorpos anticardiolipina ou ambos.
Consultar Anticoagulante Lúpico.
Anticorpos Anti-célula Parietal
A pesquisa de anticorpos anti-célula parietal é importante para auxiliar o
diagnóstico diferencial da anemia perniciosa e da atrofia gástrica. Anticorpos anti-
célula parietal são encontrados em cerca de 90% dos pacientes com anemia
perniciosa e em 60% dos casos de atrofia gástrica. A sua presença não se
correlaciona com a má absorção de vitamina B12, mas sim com o nível de
destruição das células parietais.
Não são anticorpos específicos, sendo encontrados em 20 a 30% dos pacientes com
doenças auto-imunes e em 16% de pacientes assintomáticos com mais de 60 anos.
Podem estar presentes em pacientes com úlcera ou carcinoma gástrico.
É possível ocorrer reactividade cruzada entre os anticorpos anti-célula parietal e
antitiroideus em pacientes que apresentem concomitância das patologias (tiroidite e
anemia perniciosa).
Nalguns casos, pode ocorrer um decréscimo nos níveis de anticorpos, decorrente,
provavelmente, da diminuição do número de células parietais.
Anticorpos anti-célula parietal (ACP) são encontrados em aproximadamente 5% da
população saudável. Alguns estudos referem uma alta frequência (90-100%) de
ACP na anemia perniciosa. A relação de ACP com as gastrites tipo A (caracterizadas
por atrofia da mucosa do fundo gástrico, acloridria, tendência a evoluir para anemia
perniciosa e associação com doença endócrina auto-imune) é bem conhecida.
O papel patológico dos ACP na anemia perniciosa ainda é obscuro. Nas doenças
hepáticas auto-imunes, são encontrados ACP em 100% dos pacientes com cirrose
biliar primária, em 75% das hepatites auto-imunes e em 29% dos pacientes com
doença hepática crónica.
Anticorpos Anticoagulante Lúpico
Os anticorpos anticoagulantes lúpicos (ACL) são imunoglobulinas da classe IgG ou
IgM. Assim como os anticorpos anticardiolipina (ACA), os anticoagulantes lúpicos
fazem parte da família dos antifosfolipídos e interferem nos procedimentos de
screening de coagulação que dependem da presença de fosfolipídios. Constituem
uma causa comum do prolongamento do tempo de tromboplastina parcial ativado
(APTT).
Os ACL são espécie-específicos e são neutralizados pela adição de fosfolipídos
(plasma rico em plaquetas). São anticorpos muito heterogéneos no que diz respeito
às suas características imunológicas e à variação de complexos fosfolipídicos e
protéicos que actuam como seu alvo antigénico.
Dados recentes sugerem que outras proteínas, como a proteína C, a proteína S e a
trombomodulina, são também alvos para ACL.
Apesar da sua actividade anticoagulante in vitro, na prática os anticoagulantes
lúpicos estão relacionados com manifestações tromboembólicas recorrentes
arteriais (menos frequentemente) e venosas, abortos repetidos, e, em certos casos,
são encontrados em pacientes hígidos, assim como em diferentes situações clínicas,
como doenças auto-imunes, neoplasias, quadros infecciosos virais, bacterianos e
parasitários, distúrbios neurológicos e uso de alguns medicamentos.
A detecção laboratorial de ACL não deve ser baseada num único teste. Deve-se
realizar uma combinação de testes de screening com ensaios para excluir
deficiências de factor de coagulação ou a presença de um inibidor de factor, os
quais podem dar origem a resultados falso-positivos para ACL. Ou seja, a detecção
deve ser realizada em etapas: screening para identificação da alteração; exclusão
de déficit de factor, confirmando assim a presença de um inibidor e a
caracterização do tipo de inibidor.
É importante também a interferência da heparina e dos anticoagulantes orais nos
resultados, determinando, portanto, que o teste seja realizado apenas após 2
semanas da suspensão dos anticoagulantes orais e 48 horas após a última dose de
heparina.
Na avaliação do síndrome antifosfolipídico, alguns dados indicam que os ensaios de
ACL predizem com mais segurança trombose, perda fetal recorrente e
trombocitopenia do que os ensaios para ACA. Entretanto, aproximadamente 60%
dos pacientes são positivos tanto para ACL quanto para ACA, enquanto os 40%
restantes são positivos apenas para ACA ou para ACL.
Consultar Anticardiolipina.
Anticorpos Anti-GAD
Os anticorpos antidescarboxilase do ácido glutâmico (anti-GAD), anti-insulina (antiIN) e anti-ilhota (anti-IL) são evidências auto-imunes que predizem o aparecimento
de diabetes mellitus insulino-dependente (DMID).
Esses anticorpos estão presentes na maioria dos pacientes recentemente
diagnosticados como portadores de DMID e em 80% daqueles que irão progredir
para esse quadro. Diferenças marcantes são encontradas de acordo com o sexo do
indivíduo.
Nas mulheres portadoras de DMID, independentemente da idade, o teste
apresenta-se positivo em mais de 80%, enquanto nos meninos menores de 10 anos
essa taxa diminui para cerca de 50%, atingindo os valores da população diabética
feminina nos homens adultos.
Resumidamente, a diabetes mellitus permanece sendo diagnosticado através de
critérios clínicos e da determinação sérica de glicose. Os marcadores imunológicos
ainda não têm um papel diagnóstico definido.
O valor preditivo positivo de um teste isoladamente é baixo na população em geral,
mas a combinação de vários marcadores aumenta esse valor, alcançando níveis
entre 50 e 60% nalguns estudos.
Anticorpos Anti-ilhotas Pancreáticas
Anticorpos anti-ilhota (anti-IL), anticorpos anti-insulina (anti-IN) e anticorpos
antidescarboxilase do ácido glutâmico (anti-GAD) são evidências auto-imunes que
predizem o aparecimento de diabetes mellitus insulino-dependente (DMID).
Anticorpos anti-ilhotas demonstram uma reação imunológica contra as células
productoras de insulina. Esse teste tem sido utilizado para se prever o
aparecimento de diabetes mellitus insulino-dependente (DMID).
Chegou-se a essas conclusões após estudarem três grupos distintos de indivíduos:
pacientes com DM auto-imune, os seus parentes de primeiro grau e indivíduos
normais. Entre 80 a 85% dos pacientes com DMID e 3 a 4% dos parentes de
primeiro grau de pacientes portadores de DMID apresentam anticorpos anti-ilhotas,
mesmo antes de se mostrarem com aumento da glicemia.
A especificidade desse teste para DMID encontra-se entre 96 e 99%. Um resultado
positivo em indivíduos normais pode representar um aumento transitório
inespecífico ou uma progressão para DMID. Como principal indicação clínica, temos
o parentesco em primeiro grau com indivíduos portadores de DMID, para que se
possa ter um marcador preditivo do aparecimento da doença. O método usual para
se detectar anti-ilhotas é a imunofluorescência indireta.
A classe de anticorpos IgG antigliadina (AGA IgG) foi o primeiro marcador
sorológico para essa doença. É mais sensível que a classe IgA (AGA IgA), embora
seja menos específica. Apesar de o AGA IgA ser mais específico para a DC, 2% dos
pacientes com essa patologia têm deficiência selectiva dessa classe de anticorpos.
Resultados positivos de AGA IgG e IgA têm sensibilidade de 96% a 100% e
especificidade de 96% a 97%. Esses anticorpos também são utilizados na
monitorização do tratamento da DC e tendem a diminuir ou a desaparecer com
uma dieta livre de glúten.
Anticorpos Antiinsulina
Os anticorpos anti-insulina (anti-IN), anti-ilhota (anti-IL) e os anticorpos
antidescarboxilase do ácido glutâmico (anti-GAD) são evidências auto-imunes que
predizem o aparecimento de diabetes mellitus insulino-dependente (DMID).
Os anticorpos anti-insulina estão presentes em 50% dos pacientes com DMID no
momento do diagnóstico, antes do início da insulinoterapia. A sensibilidade do teste
é de 50 a 70%, e ele é mais vezes positivo em crianças e adultos jovens.
O doseamento de insulina em presença desses anticorpos é prejudicado, tornandose impreciso para o acompanhamento clínico.
Anticorpos Antinucleares
Os anticorpos antinucleares (ANA) são encontrados frequentemente numa
variedade de doenças reumáticas auto-imunes. São úteis na investigação dessas
doenças, auxiliando no diagnóstico de patologias como lúpus eritematoso sistémico
(LES), esclerose sistémica progressiva (ESP), doença mista do tecido conjuntivo
(DMTC), síndrome de Sjögren (SS), poliomiosite e dermatomiosite.
Os ANA podem estar presentes numa pequena percentagem da população
aparentemente sã; esta frequência é maior nas mulheres e aumenta com a idade.
Segundo a literatura, a positividade entre 20 a 60 anos varia entre percentagens
mais altas (13,3%) para títulos baixos e mais baixas (3,3%) para títulos mais
elevados.
A pesquisa dos anticorpos antinucleares pode ser realizada por diferentes técnicas;
a imunofluorescência indireta é a mais utilizada, devido à sua maior sensibilidade,
reprodutibilidade e facilidade de execução. A sensibilidade aumenta
significativamente quando se utiliza como substrato láminas com células Hep 2
(células de carcinoma de laringe humana), por causa da maior expressão de
antígenios na superfície dessas células comparados aos expressos nas células de
fígado ou de rins de camundongos, que se utilizavam anteriormente. Em geral, é
possível detectar melhor a presença de ANA por associação de três anti-soros
isotipos específicos (anti-IgG, anti-IgA e anti-IgM). A pesquisa realizada por essa
técnica serve de triagem inicial para a presença de ANA. É normalmente
denominada pesquisa de factor antinuclear (FAN) ou ANA (antinuclear antibody).
A realização de outras metodologias, tais como as imunoenzimáticas, permite uma
avaliação isolada dos diferentes auto-anticorpos, sugeridos pelos padrões de
fluorescência encontrados no FAN. A identificação desses ANA permite um
diagnóstico mais preciso, e eles podem ser utilizados, em muitos casos, como
marcador para uma das diferentes doenças reumáticas auto-imunes. Muitas dessas
patologias apresentam um perfil distinto de auto-anticorpos que auxilia o
diagnóstico.
Fator Antinuclear - FAN
Nas pesquisas realizadas por imunofluorescência indirecta, os resultados são
expressos em títulos, segundo as diluições empregues. É possível trabalhar
utilizando-se como limite títulos de 1/40, que permitem maior sensibilidade, ou de
1/80, que permitem maior especificidade, ou seja, um menor número de falsopositivos.
Na prática clínica, são considerados significativos títulos superiores a 1/160. Os
resultados com títulos positivos são acompanhados pela descrição do padrão de
fluorescência encontrado, que serve como orientação da presença de um antígeno
específico e, em alguns casos, como padrão diagnóstico. Em determinadas
situações, em especial no LES em actividade, pode ocorrer a observação de mais de
um padrão de fluorescência.
PADRÕES DE
FLUORECÊNCIA
PERIFÉRICO
HOMOGÊNIO
PERIFÉRICO/
HOMOGÊNEO
PONTILHADO
GROSSO
PONTILHADO
FINO NUCLEAR
NUCLEOLAR
CENTROMÉRICO
ANTÍGENOS
PATOLOGIA
RELACIONADAS
DNA de dupla hélice ou de hélice
LES
única, histona e RNA
DNA de dupla hélice ou de hélice LES, AR, lúpus induzido por
única, histona e RNA
drogas
DNA de dupla hélice ou de hélice
LES
única, histona e RNA
SM, RNP
LES, DMTC
SS-A (RO) SS-B (La)
SS, LES
Proteínas Nucleolares
Proteínas de centrómeros
ESP
Síndrome de CREST
O padrão pontilhado, pode também ser referido como padrão salpicado.
Outros padrões de fluorescência podem ser encontrados em menor frequência. O
pontilhado citoplasmático grosso ou reticular, associado à presença de anticorpos
antimitocôndrias em pacientes com cirrose biliar primária; o citoplasmático
homogêneo, relacionado a proteínas ribossômicas em pacientes com LES; e o
padrão pontilhado fino citoplasmático, que pode estar associado à presença de
anticorpos anti-JO-1, anti-PCNA e proteínas da membrana celular, em pacientes
com LES, outras patologias do tecido conjuntivo e hepatopatias auto-imunes.
Anticorpos Anti-DNA Nativo ou de Dupla Hélice
A pesquisa desse anticorpo, realizada por imunofluorescência indirecta, utilizando
como substrato a Crithidia luciliae, apresenta boa sensibilidade e alta
especificidade. A técnica imunoenzimática, apesar de mais sensível, é menos
específica, levando a riscos de resultados falso-positivos (identificação de DNA de
hélice única).
A sua presença é considerada um marcador para o diagnóstico de lúpus
eritematoso sistémico, sendo detectado em 50 a 70% dos casos em actividade. Ao
contrário de outros auto-anticorpos, os seus títulos apresentam relação directa com
a actividade da doença, sendo extremamente úteis para o acompanhamento da
terapia e da reactivação da doença. Quanto maior o título, maior a actividade da
doença.
Nalguns casos (cerca de 30%), apesar de as evidências clínicas e laboratoriais
indicarem lúpus eritematoso sistémico, o anticorpo anti-DNA nativo pode não ser
detectado. Acredita-se que, nesses casos, os auto-anticorpos estejam sob a forma
de imunocomplexos.
Anti-ENA - Auto-anticorpos contra Antígenos Extraídos do
Núcleo
AUTO-ANTICORPOS CONTRA ANTÍGENOS EXTRAÍDOS DO NÚCLEO
SM
LES
RNP
DMTC, LES, ESP, AR e Polimiosite
SS-A (Ro)
SS, LES, lúpus neonatal e forma cutânea subaguda
SS-B (La)
SS, LES, lúpus neonatal e forma cutânea subaguda
Scl 70
ESP
Jo -1
Polimiosite e dermatomiosite
O padrão pontilhado, pode também ser referido como padrão salpicado.
Outros padrões de fluorescência podem ser encontrados em menor frequência. O
pontilhado citoplasmático grosso ou reticular, associado à presença de anticorpos
antimitocôndrias em pacientes com cirrose biliar primária; o citoplasmático
homogêneo, relacionado a proteínas ribossómicas em pacientes com LES; e o
padrão pontilhado fino citoplasmático, que pode estar associado à presença de
anticorpos anti-JO-1, anti-PCNA e proteínas da membrana celular, em pacientes
com LES, outras patologias do tecido conjuntivo e hepatopatias auto-imunes.
Sm
Os auto-anticorpos anti-Sm ("Smith") são dirigidos contra ribonucleoproteínas de
baixo peso molecular. Estão presentes em 20 a 30% de pacientes de LES,
geralmente acompanhados de positividade para outros anticorpos, entre eles o
anti-RNP. Em contraste com os auto-anticorpos anti-DNA de dupla hélice, que
tendem a apresentar níveis flutuantes em paralelo à actividade da doença, os autoanticorpos anti-Sm não variam muito com o passar do tempo. A sua presença
correlaciona-se com o padrão pontilhado grosso na pesquisa de ANA por
imunofluorescência indirecta. Podem ser encontrados também, embora mais
raramente, noutras doenças do tecido conjuntivo, como a DMTC. É considerado um
marcador diagnóstico do LES.
A sua positividade noutras patologias que não as do tecido conjuntivo tem sido
descrita nalguns estudos que relatam a sua presença em gamapatias monoclonais e
uveítes.
RNP
Os auto-anticorpos anti-RNP são encontrados em altos títulos na doença mista do
tecido conjuntivo (DMTC). São úteis como marcadores para a DMTC, já que são
detectados em altos títulos em 90% dos pacientes com essa patologia. Podem
apresentar-se positivos também, porém com menos frequência, no LES, ESP, SS,
artrite reumatóide e polimiosite.
Nos pacientes com LES, os auto-anticorpos anti-RNP apresentam-se positivos em
cerca de 26 a 40% dos casos, geralmente com outros anticorpos antinucleares
também positivos. A sua presença no LES é considerada indício de bom
prognóstico, pois indica menor probabilidade de lesão renal.
SS-A/Ro - SS-B/La
São anticorpos relacionados com uma classe de partículas de ribonucleoproteínas
que são compostas por pequenos ácidos ribonucleicos.
A detecção de auto-anticorpos SS-A/Ro é possível em aproximadamente 60 a 70%
dos pacientes com síndrome de Sjögren, e com menor freqüência, na artrite
reumatóide, nas vasculites associadas ao síndrome de Sjögren, noutras doenças do
colágeno e em cerca de 5% da população hígida. Estão presentes em 40 a 50% de
LES, em quase 100% dos casos de mães com filhos com lúpus neonatal e também
nos casos da forma cutânea subaguda do lúpus.
A presença de anti-SS-B/La dá-se em 40 a 50% dos casos de síndrome de Sjögren
e em 6 a 15% dos casos de LES. No lúpus neonatal, quase sempre são detectados
auto-anticorpos SS-A/Ro na mãe. Cerca de 2% de crianças nascidas de mães com
auto-anticorpos SS-A/Ro apresentam a patologia.
Na maioria das patologias em que se encontram presentes, os anticorpos antiSSA/Ro são identificados com maior freqüência do que os anti-SSB/La. No
síndrome de Sjögren, em cerca de 50% dos casos os auto-anticorpos SS-A/Ro
estão acompanhados da presença de anti-SS-B/La.
Scl 70
Na literatura actual, os anticorpos anti-Scl 70 são identificados como antitopoisomerase I. Estão presentes em 64 a 75% de pacientes com ESP, mas só são
vistos, raramente, na síndrome de CREST (calcinoses, dismotilidade esofagiana,
fenómeno de Raynaud, esclerodactilia, telangicectasia), em poucos casos de
fenómeno de Raynaud primário (10%), na síndrome de Sjögren (5%) e em cerca
de 1% dos casos de DMTC e LES. É considerado marcador diagnóstico da ESP.
A presença desses anticorpos aumenta em cerca de 17 vezes o risco de
desenvolvimento de fibrose pulmonar em pacientes com quadro de esclerodermia.
Esse risco implica a necessidade de monitorização mais frequente da função
pulmonar.
A positividade dos anticorpos anti-Scl-70 não tem valor preditivo para a
possibilidade de envolvimento cardíaco, renal ou sobrevida. Já a persistência de
auto-anticorpos anti-Scl-70 em pacientes com doença de Raynaud é indicativa da
possibilidade de desenvolvimento de esclerose sistémica.
JO-1
Anticorpos contra o antigénio Jo-1 (sintetase-1,2-de histidil-tRNA) são encontrados
em aproximadamente 30 a 50% dos pacientes adultos com miosites, incluindo
polimiosite, dermatomiosite e as síndromes superpostas. Aparecem em cerca de
60% dos casos de miosite e de doença intersticial pulmonar. A presença de
anticorpos anti-Jo-1 em pacientes com polimiosite idiopática normalmente é
acompanhada de doença grave, tendência a recidiva e mau prognóstico. Na miosite
juvenil com presença de anti-Jo-1, as manifestações clínicas e o curso de doença e
resposta à terapia são semelhantes à evolução nos adultos. É considerado
marcador diagnóstico de polimiosite.
Anticentrómero
Os anticorpos anticentrómero (AAC) são um subgrupo distinto dos anticorpos
antinucleares detectáveis, utilizando como substrato células de cultura de tecidos.
Os pacientes com anticentrómero-positivos não apresentam as características mais
graves da esclerose sistémica progressiva, como a doença renal. Por isso, a
positividade parece indicar um prognóstico mais favorável.
São encontrados em cerca de 22% dos pacientes com esclerose sistémica
progressiva e estão positivos em cerca de 70% dos casos de síndrome de CREST e
em cerca de 12% dos pacientes com cirrose biliar primária, dos quais
aproximadamente 50% apresentam características de esclerodermia. Por outro
lado, a esclerodermia, sob a forma da síndrome de CREST, está presente em cerca
de 4% dos casos de cirrose biliar primária.
Os casos de esclerodermia limitados ao síndrome de CREST que cursam com AAC
positivo apresentam, com mais frequência, calcinose e telangiectasias. Nestes
casos, é menor a ocorrência de fibrose pulmonar intersticial.
Os pacientes que apresentam fenómeno de Raynaud e AAC positivo sugerem a
possibilidade de desenvolvimento do síndrome de CREST.
Anticorpos Antitiroideus
As doenças da tiróide de etiologia auto-imune são acompanhadas da presença de
auto-anticorpos. A sua determinação está indicada na investigação diagnóstica das
tiróidites auto-imunes, principalmente a doença de Hashimoto, doença de Graves,
tiróidite pós-parto e mixedema idiopático.
Na investigação diagnóstica, recomenda-se a utilização da pesquisa associada de
mais de um anticorpo para avaliar, dessa forma, a presença de anticorpos contra os
diferentes antígenos. Os anticorpos mais pesquisados são antitireoperoxidase (antiTPO), antitireoglobulina (anti-TRG) e os anticorpos anti-receptores de TSH (TRAB).
Os anticorpos antimicrossomais foram durante muito tempo, em associação com os
doseamentos das hormonas tiróideias, considerados o padrão para o diagnóstico
das tiroidites auto-imunes. Hoje, podem ser substituídos pela pesquisa dos
anticorpos antiperoxidase tiroideu. A tireoperoxidase (TPO), principal enzima
envolvida no procedimento de síntese de hormona da tiróide, é uma glicoproteína,
expressa apenas em células foliculares da tiróide. Ela é o principal antigénio na
partícula microssomal da tiróide.
Os anticorpos antitireoperoxidase estão presentes em 4 a 9% dos adultos normais,
em 57-74% dos pacientes com doença de Graves, em 99-100% dos pacientes com
doença de Hashimoto ou mixedema idiopático, em 19% dos casos de tumores
diferenciados de tiróide e, raramente, em pacientes com tiróidite subaguda. A
prevalência de positividade em pacientes idosos (80 anos) é mais alta nas mulheres
(10%) comparadas aos homens (2%).
Os anticorpos antitireoglobulina estão presentes em títulos elevados em cerca de
25% dos casos de doença de Graves e em 55% dos pacientes com tiróidite de
Hashimoto. Raramente se observam casos em que os anticorpos estão ausentes
devido à produção de anticorpos restrita aos linfócitos intratiroideus. Eles podem
ser detectados, com menor frequência, noutras patologias auto-imunes, em
pacientes com carcinoma de tiróide e em percentagens ainda menores em
mulheres e indivíduos idosos hígidos.
Geralmente, as amostras apresentam-se positivas para anti -TRG e anti-TPO. A
positividade isolada para os anticorpos antitireoglobulina é menos frequente que o
achado isolado dos anticorpos antitireoperoxidase.
Durante a gravidez, a presença de anticorpos anti-TPO e TRAB pode ser
considerada fator preditivo de tiróidite pós-parto.
Durante as fases precoces da tiróidite de Hashimoto, encontramos anticorpos
antitireoglobulina significativamente elevados e níveis menos elevados de anti-TPO.
Tardiamente, os anticorpos anti-TRG podem desaparecer, enquanto os anti-TPO
permanecem presentes por muitos anos.
Pacientes que apresentam outras desordens auto-imunes, como síndrome de
Sjögren, lúpus eritematoso sistémico, artrite reumatóide e anemia perniciosa,
podem apresentar anticorpos antitireoglobulina positivos.
Outro anticorpo utilizado na investigação clínica das patologias tiroideias são os
anticorpos anti-receptores de TSH (TRAB), que podem actuar tanto estimulando
como inibindo as funções tiroideias, ligando-se a diferentes epitopos. Encontram-se
positivos na doença de Graves e nas tiroidites subagudas. Devido à sua capacidade
de atravessar a barreira placentária, podem levar à doença de Graves neonatal por
transferência passiva.
A positividade do TRAB no soro sugere doença auto-imune em actividade, mas não
define o estado funcional da glândula. O ensaio baseia-se na capacidade do soro
que contém TRAB de impedir a ligação do TSH. Esse teste também é útil na
avaliação de remissões ou recidivas da doença.
Antiestreptolisina O
Os Streptococcus beta-hemolíticos do grupo A causam infecções clínicas
especialmente de orofaringe e pele, endocardites e quadros não-supurativos, como
febre reumática e glomerulonefrite aguda.
A estreptolisina O é uma das toxinas extracelulares libertadas pelo Streptococcus
beta-hemolítico do grupo A. Ela é capaz de induzir o síntese de anticorpos
específicos, os anticorpos antiestreptolisina O (ASO), em cerca de 80% das
infecções. O uso de antibióticos, corticóides e drogas imunossupressoras pode inibir
a produção de ASO.
A ASO eleva-se na primeira semana, atingindo valores máximos em 2 a 4 semanas
após a infecção estreptocócica e retornando aos valores normais após 6 a 12
meses.
A manutenção de títulos de ASO elevados de anticorpos ou a sua elevação em
amostras seguidas são indicativas de infecção aguda, reinfecção ou lesões pósestreptocócicas.
Apesar de níveis séricos de ASO serem encontrados na maioria dos pacientes com
febre reumática e glomerulonefrite pós-estreptocócica, a sua maior indicação é no
seguimento de pacientes com febre reumática, já que nesses casos os títulos de
ASO se correlacionam melhor com a atividade da doença. Cerca de 80 a 85% dos
casos de febre reumática cursam com títulos elevados.
Antígeno Carcioembrionário (CEA)
O antígeno carcioembrionário (CEA) é normalmente sintetizado e secretado pelas
células que revestem o trato gastrointestinal do feto e em pequenas quantidades no
adulto. Em situações normais, é eliminado pelo intestino. Em desordens benignas e
malignas do trato gastrointestinal, pode ser detectado no sangue circulante.
Não é um antigénio órgão-especifico. A detecção no soro associa-se a várias
malignidades como as de estômago, pâncreas, gastrointestinais, mama, pulmão e
ovário.
O antigénio carcioembrionário está presente em doenças benignas como
hepatopatias, pancreatites, enfisema pulmonar, doenças benignas da mama, colite
ulcerosa, doença de Crohn e pólipo rectal. Níveis mais elevados são encontrados
em tabagistas.
O CEA é usado na monitorização de tumores gastrointestinais, particularmente no
carcinoma colorrectal. Segundo o The National Cancer Institute, o CEA é o melhor
método não-invasivo para acompanhamento do carcinoma colorrectal.
Níveis pré-operatórios muito altos são prognósticos de altas taxas de recidiva e de
baixas taxas de sobrevida. O CEA pode ser usado para monitorizar a eficácia do
tratamento, a recidiva da doença local ou metastática. Elevações dos níveis de CEA
são maiores nas metástases e menores na recidiva local. Os níveis mais altos são
encontrados nas metástases ósseas e hepáticas.
No decurso de quimioterapia ou de radioterapia, os níveis séricos podem elevar-se
temporariamente. A avaliação pós-operatória deve ocorrer em torno da 4ª semana.
A sua avaliação não é recomendada como screening, devido à incidência de
elevação de CEA noutras patologias.
Cerca de 63% dos pacientes com carcinoma colorretal têm elevações de CEA. As
alterações correlacionam-se com o estadiamento do tumor. Apresenta-se alterado
em 20% dos pacientes em Duke A, 58% em Duke B e 68% dos casos em Duke C.
A avaliação bimensal é recomendada durante os primeiros 2 anos após a cirurgia;
nos anos subsequentes, as avaliações podem ser mais espaçadas, até completar 5
anos.
Noutros tipos de tumor em que o CEA é detectado, os níveis no pós-operatório
podem ser úteis no acompanhamento da terapia e na identificação do aparecimento
da doença metastática, especialmente nos carcinomas de mama associados com CA
15-3 e os de pâncreas associados ao CA 19-9.
Antimitocôndria
A avaliação dos anticorpos antimitocondriais é importante na investigação
diagnóstica das doenças hepáticas crónicas, em que está presente em 90% dos
casos de cirrose biliar primária, especialmente no diagnóstico diferencial com outras
colangites.
Os anticorpos antimitocondriais (AMA) podem também estar presentes em 25% a
30% dos casos de hepatite crónica activa e mais raramente noutras doenças
hepáticas, como obstrução biliar extra-hepática, hepatite vírica, cirrose de
diferentes etiologias, hepatite induzida por drogas e neoplasia hepática. São
encontrados em baixos títulos em cerca de 1% de adultos saudáveis e em doenças
auto-imunes.
Os níveis de anticorpos não se correlacionam com a gravidade e o tempo de
evolução da doença, nem definem o prognóstico. Os AMA desaparecem
aproximadamente 1 mês após o transplante hepático e diminuem com o uso de
ciclosporina como tratamento.
O diagnóstico da cirrose biliar primária deve-se basear nos achados clínicos,
laboratoriais e histológicos.
Aproximadamente 4% de pacientes com cirrose biliar primária têm esclerodermia,
frequentemente como síndrome de CREST. Além disso, às vezes é encontrada
reactividade para AMA nalguns pacientes com síndrome de CREST ou esclerodermia
difusa, na ausência de doença hepática.
Diferentes antigénios são encontrados na membrana mitocondrial, fazendo com que
existam diferentes tipos de anticorpos antimitocondriais. A identificação dos
diferentes tipos (M1, M2, M3, M4, M5, M6, M7, M8 e M9) é possível com o uso de
antigénios recombinantes em ensaio imunoenzimático e permite um diagnóstico
específico dos AMA. A cirrose biliar primária está relacionada a M2, e o tipo M9
pode ser encontrado em membros saudáveis da família. Os dos diferentes tipos
restantes correlacionam-se com outras patologias, como doenças do colágeno (M5)
e hepatite induzida por drogas (M6).
A imunofluorescência indirecta usando células HEp-2 é 100% sensível e específica
para identificação de AMA total.
Antimúsculo Liso
Os anticorpos antimúsculo liso (AML), ou (f-actina) são encontrados na maioria dos
pacientes com hepatite crónica auto-imune. Em geral, a ausência de AML e
anticorpos antinucleares são argumentos para o diagnóstico de formas não autoimunes de hepatites crónicas. Nas hepatites víricas crónicas, eles podem ser
detectados em títulos mais baixos em cerca de 10% dos casos. Podem ser
encontrados em aproximadamente 2% dos adultos normais, porém sempre em
baixos títulos. A presença de anticorpos antinucleares pode interferir, levando à
positividade com títulos baixos.
Títulos elevados são característicos da hepatite crónica auto-imune. Quando
presentes noutras patologias, normalmente apresentam-se com títulos baixos,
<1/80. Cerca de 50% dos pacientes com cirrose biliar primária apresentam AML.
Com menor frequência são encontrados nas uveítes, nas hepatites induzidas por
drogas, na alopécia, na doença hepática alcoólica, nas neoplasias, na hipertensão
pulmonar primária e transitoriamente em hepatites agudas e outras infecções
víricas, incluindo mononucleose infecciosa.
A sua presença não tem valor preditivo para o desenvolvimento de doença
hepática. O diagnóstico deve ser sempre acompanhado da avaliação de outros
anticorpos como os anticorpos antinucleares e do perfil hepático. O achado isolado
de AML não tem significado clínico.
Apoliproteína A-I
A apolipoproteína A (ApoA) é um dos principais componentes da lipoproteínas de
alta densidade (HDL). Os seus dois componentes principais são a Apo-A-I e a ApoA-II.
A Apo-A-I é constituída por 245 aminoácidos e representa cerca de 75% da Apo-A
nas HDL. É sintetizada no fígado e no intestino e responsável pela activação de uma
enzima envolvida na esterificação do colesterol no plasma. O colesterol esterificado
pode então ser removido dos tecidos extra-hepáticos e células periféricas e
transportado até o fígado, para então ser metabolizado e excretado. A Apo-A-II
representa cerca de 20% da Apo-A nas HDL. É constituída por 154 aminoácidos, e o
seu papel fisiológico ainda é pouco conhecido.
Valores da Apo-A-I elevam-se durante a gravidez, na doença hepática e na
administração de estrogénios. Valores diminuídos são encontrados na colestase, na
septicémia, na cirrose hepática aguda, em pacientes que consomem insulina, na
aterosclerose e nos casos de deficiências hereditárias.
A avaliação da Apo-A-I tem sido apontada como um bom parâmetro preditivo de
risco de doença coronária. Os seus níveis são inversamente proporcionais ao risco
de doença coronária prematura. Valores elevados de Apo-A-I (HDL) e baixos de
Apo-B (LDL) correlacionam-se com baixo risco aterogénico.
Avaliação da Função Gonadal
DISTÚRBIOS DA REPRODUÇÃO FEMININA
Puberdade Precoce
A puberdade precoce caracteriza-se por aceleração da velocidade de crescimento,
desenvolvimento das mamas e crescimento de pelos pubianos. Pode ser classificada
como puberdade precoce verdadeira (dependente de GnRH), indicando activação
precoce do eixo hipotalámico-hipofisário-gonadal, ou pseudopuberdade precoce
(independente de GnRH), quando o aumento das hormonas esteróides ocorre sem
o estímulo das gonadotrofinas. Ambas as condições podem estar relacionadas a
doenças graves, sendo necessária cuidadosa anamnese, exame clínico e métodos
de diagnóstico por imagem para descartar doenças do SNC, como tumores,
traumatismo craniano ou encefalite, ou mesmo massa pélvica ou abdominal.
A avaliação hormonal deve incluir função tiroideia (TSH, T4L), esteróides (estradiol,
testosterona, progesterona, sulfato de deidroepiandrosterona (S-DHEA),
androstenediona e 17 hidroxiprogesterona), FSH, LH, HCG e teste LH-RH.
Na puberdade precoce verdadeira, os valores de LH, FSH e estrógenos são
compatíveis com maturidade sexual, enquanto valores elevados de estrogénios e
baixos de FSH e LH são geralmente observados na presença de tumores secretores
de estrogénios. Níveis elevados de HCG podem ser indicativos de teratoma ou de
disgerminoma do ovário.
Níveis elevados de 17-hidroxicorticosteróides, progesterona e androgénios adrenais
confirmam o diagnóstico de hiperplasia adrenal por deficiência de 21-hidroxilase,
enquanto o aumento do 11-desoxicortisol indica hiperplasia adrenal por deficiência
de 11-hidroxilase.
Suspeita-se de adenoma ou carcinoma do córtex adrenal quando o S-DHEA e/ou a
androstenediona estão aumentados. Um aumento na testosterona sugere
precocidade de origem ovariana.
Outros testes úteis no diagnóstico da precocidade sexual são a ultra-sonografia dos
ovários, a ressonância magnética (RM), a tomografia computadorizada (TC) ou a
ultra-sonografia das adrenais, a determinação da idade óssea e RM ou TC de
crânio.
Amenorréia
É definida como a ausência de menstruação por 3 meses ou mais em mulheres que
já menstruaram anteriormente. Podem também ser consideradas amenorréicas as
mulheres nas quais a menarca não ocorreu até os 16 anos, independentemente da
presença ou ausência de caracteres sexuais secundários.
Segundo Speroff, as causas de amenorréia podem ser classificadas em quatro
grupos (compartimentos):
CLASSIFICAÇÃO ETIOLÓGICA DA AMENORRÉIA
AMBIENTE
GRUPO IV
SNC - HIPOTÁLAMO
*GnRH-TRH
GRUPO III
HIPÓFISE ANTERIOR
*TSH, FSH, LH, PROLACTINA
GRUPO II
OVÁRIO
*ESTROGÊNIOS
*PROGESTERONA
*ANDROGÊNIOS
GRUPO I
ÚTERO, CERVICE E VAGINA
Stress
Aumento de Peso
Distúrbios Psicológicos
Exercícios físicos
Tumor Hipofisário
Insuficiência hipofisária
Síndrome de Sheeham
Perimenopausa
Síndrome de Resistência
Ovárica
Insificiência Ovárica
Anormalidades Cromossómicas
Anomalia do Ducto de Müller
Testículo Feminilizante
Síndrome de Asheeman
Amenorréia e infertilidade podem ser os primeiros sintomas de disfunção tiroidéia.
Em casos de hipotiróidismo, o aumento de TSH induz a hiperprolactinémia.
Infertilidade
Ocorre em 15% dos casais, e 50% dos casos devem-se a problemas na mulher.
Dessas, 40% não ovulam, 10% têm fase lútea inadequada, 40% têm anomalias
tubáricas e 10% têm factores cervicais ou tiroideios. Embora a causa da
insuficiência não seja encontrada na maioria das mulheres, ela pode resultar de
anomalias dos eixos hipotalámico-hipofisário ovárico, tiroideio ou adrenal.
A avaliação laboratorial de não ovulação deve incluir FSH, LH, prolactina e
progesterona. Em presença de sinais e sintomas de disfunção tiroideia ou de
hiperandrogenismo, deve-se fazer avaliação de hormonas tiroideias, do S-DHEA e
da testosterona.
As disfunções da fase lútea podem ser avaliadas pelo doseamento de progesterona,
cujos níveis de 10.000 pg/mL ou mais, 5 a 10 dias antes do período menstrual
seguinte, descartam uma fase lútea inadequada. A biópsia de endométrio realizada
2 a 3 dias antes da menstruação é útil no diagnóstico.
Hirsutismo
O aumento do crescimento de pelos (hirsutismo) ocorre por aumento da produção
de androgénios (ováricos ou adrenais) ou aumento da sensibilidade da pele aos
andrógenios circulantes. Mais de 90% das pacientes têm ovários poliquísticos ou
hirsutismo idiopático.
A avaliação hormonal deve incluir testosterona, androstenediona e S-DHEA. A
sensibilidade da pele aos androgénios é modulada pela enzima 5-alfa-redutase, que
converte a testosterona em deidrotestosterona, a hormona mais activa no
crescimento dos pelos.
O síndrome do ovário poliquístico envolve o eixo hipotálamo-hipófise-ovário e
adrenal. Esses pacientes cursam com o aumento da produção de estrógenios e
androgénios, evidenciado pelo aumento dos níveis circulantes de testosterona,
androstenediona, deidroepiandrosterona, S-DHEA, 17-hidroxiprogesterona e
estrona. Deve-se avaliar ainda FSH, LH e hormonas tiroideios. A prolactina tende a
estar ligeiramente elevada.
DISTÚRBIOS DA REPRODUÇÃO MASCULINA
Hipogonadismo/ Infertilidade
O hipogonadismo masculino ou infertilidade pode ser classificado como prétesticular, testicular ou pós-testicular.
As causas pré-testiculares (hipogonadismo secundário) são geralmente
hipotalámicas ou hipofisárias, podendo ser congénitas ou adquiridas. As alterações
hipotalámicas congénitas são mais frequentes do que as hipofisárias, e podem
causar apenas puberdade atrasada ou hipogonadismo hipogonadotrófico idiopático.
No geral, o crescimento progride sem sinais puberais e com testículos muito
pequenos.
A deficiência gonadotrófica hipofisária adquirida é mais frequente que a
hipotalámica e, geralmente, causada por tumores. A disfunção eréctil ou impotência
pode ser uma manifestação precoce, bem como a deficiência da hormona de
crescimento (HGC).
Tumores secretores de prolactina também podem causar sintomas de disfunção
eréctil e infertilidade, uma vez que o aumento de prolactina suprime a produção de
GnRH. Outras causas de hipogonadismo pré-testicular são: hipotiroidismo,
síndrome de Cushing e cirrose alcoólica.
As causas testiculares (hipogonadismo primário) podem ser congénitas ou
adquiridas. As causas congénitas incluem síndrome de Klinefelter, criptorquidismo e
hipoespermatogénese idiopática. As causas de insuficiência testicular adquiridas
mais frequentes são orquite (que pode ser causada pela parotidite) e traumatismo
físico. Outras causas são radiação e agentes antineoplásicos.
As causas pós-testiculares devem-se geralmente ao bloqueio funcional ou à
obstrução mecânica do transporte do esperma ou alterações funcionais do
esperma.
As manifestações clínicas incluem distribuição anormal de pelos faciais e corporais,
distribuição anormal e diminuição da massa muscular, distribuição anormal da
gordura corporal, aumento da proporção envergadura/altura ou diminuição da
proporção tronco/membros inferiores, diminuição do volume testicular, diminuição
ou ausência de olfacto e ginecomastia.
É necessário investigar o desenvolvimento físico e a idade da puberdade, as
alterações de líbido, disfunção eréctil, infertilidade e dificuldades conjugais
relacionadas com o desempenho sexual.
A síndrome de Kallmann caracteriza-se por ausência ou diminuição de olfacto,
cegueira para cores, agenesia renal, surdez neurossensorial, fenda labial ou
palatina, diminuição ou ausência das características sexuais secundárias em
homens e testículos pequenos.
No hipogonadismo devido à hiperprolactinemia, os pacientes podem apresentar
galactorréia, disfunção eréctil e diminuição da líbido.
A síndrome de Klinefelter após a puberdade caracteriza-se por aparência tipo
eunuco, testículos firmes e pequenos, ginecomastia, azoospermia e por vezes
atraso mental.
A avaliação laboratorial inclui os doeamentos séricas de testosterona, LH, FSH,
prolactina e SHBG.
No hipogonadismo primário, a testosterona sérica encontra-se baixa, com altos
níveis de LH e FSH. No hipogonadismo secundário, a LH e a FSH encontram-se
também diminuídos. A patologia hipofisária pode ser distinguida da patologia
hipotalámica por meio de testes dinâmicos da função endócrina. O teste de
estímulo com GnRH avalia a reserva de gonadotrofinas. O aumento da FSH e da LH
após o teste indica patologia hipotalámica.
No hipogonadismo primário (LH e FSH elevadas), um cariótipo alterado com
genótipo masculino e azoospermia sugere síndrome de Klinefelter.
O hipotiroidismo e a hiperprolactinemia podem ser excluídos com dosagens séricas
de TSH e de prolactina, respectivamente. A pesquisa de tumores deve ser realizada
com tomografia computadorizada e ressonância magnética.
Infertilidade
Geralmente, o único sinal de infertilidade é a incapacidade de engravidar a parceira.
O paciente pode ter história de criptorquidismo e infecção do trato genito-urinário.
O espermograma continua a ser o teste mais valioso na investigação da
infertilidade masculina.Testes especiais como anticorpos antiespermatozóides,
interação com o muco cervical e testes de penetração do esperma só devem ser
realizados após os exames de triagem.
A azoospermia pode ocorrer devido a obstrução bilateral, ejaculação retrógrada ou
ausência congénita dos vasos deferentes ou das vesículas seminais. Exames
radiológicos dessas estruturas auxiliam o diagnóstico.
Disfunção Eréctil
A disfunção eréctil é um síndrome complexo que envolve factores arteriais,
venosos, sinusoidais, neurológicos, hormonais, bioquímicos, sociais e psicológicos.
É definida como a incapacidade de obter uma erecção longa durante o tempo
suficiente para uma relação plenamente satisfatória. Geralmente, ocorre uma
disfunção eréctil parcial, mais frequentemente do que uma falta completa de rigidez
do pénis. A perda de erecção antes da ejaculação é a queixa mais comum.
A disfunção eréctil pode ser classificada como fisiológica ou funcional. Uma
disfunção neurogénica envolve incapacidade de iniciar a erecção devido à ausência
dos neurotransmissores endógenos, e pode ser causada por diabetes, doenças da
medula espinal, lesões da cauda da medula espinal, polineuropatia, mielopatia e
esclerose múltipla, entre outras. Determinados medicamentos, estados de
ansiedade e o uso de bebidas alcoólicas também podem acarretar problemas.
Drogas que atuam no SNC ou no pico adrenérgico da ansiedade podem causar
diminuição do fluxo sanguíneo do pénis, assim como drogas anti-hipertensivas
(betabloqueadores, tiazidas, metildopa), cimetidina e alguns antilipémicos. O
alcoolismo de longa duração e a cirrose hepática podem prejudicar os mecanismos
de condução nervosa intrapenianos e acarretar atrofia testicular ou ginecomastia.
A segunda classificação é baseada nas desordens vasculares. As principais causas
são arteriosclerose, síndrome de Leriche, outras obstruções vasculares,
microangiopatia, síndromes anginosas, insuficiência cardíaca congestiva e
priapismo. A disfunção vascular pode ser arterial, venosa ou mista. Na disfunção
arterial, não ocorre o enchimento do corpo cavernoso. Estes casos geralmente são
provocados por hipertensão, diabetes, dislipidemias, angina e tabagismo. Na
disfunção venosa, o corpo cavernoso não consegue reter o sangue, e isso deve-se,
normalmente, a traumas ou à idade.
As causas endócrinas da disfunção eréctil incluem a deficiência de testosterona por
doença hipofisária ou testicular, o excesso de estrogénio e a hiperprolactinemia.
Outras causas são o hipertiroidismo, o hipotiroidismo e o síndrome de Cushing.
A avaliação laboratorial, portanto, deve incluir glicemia de jejum, perfil lipídico,
avaliação hepática, renal e da tiróide, bem como doseamentos de prolactina,
testosterona livre, LH e FSH.
Prostatite
O termo prostatite inclui inúmeras condições como prostatites bacterianas agudas e
crónicas, não-bacterianas e prostatodinia.
Nas prostatites bacterianas agudas, o início é abrupto, com febre e infecção do
trato urinário, sintomas irritativos e obstrutivos, dor lombar baixa ou no períneo,
astenia, mialgia e artralgia.
Geralmente, ocorre em pacientes jovens por bacilos Gram-negativos. O exame
simples de urina geralmente revela piúria, indicativa de infecção urinária, e, à
cultura, o Gram-negativo encontrado com mais frequência é Escherichia coli.
A prostatite bacteriana crónica ocorre em pacientes mais velhos, com infecção
urinária de repetição, geralmente por Gram-negativos. Muitos desses pacientes
apresentam cálculos na próstata, o que facilita a cronicidade da infecção. A coleta
após massagem prostática é aconselhável nesses casos.
O tipo mais comum de prostatite é a não-bacteriana, geralmente causada por
Chlamydia trachomatis, Ureaplasma urealyticum e Trichomonas vaginalis.
Neoplasias
Os tumores testiculares são raros e representam menos de 1% das mortes por
cancro nos homens. Apesar da baixa prevalência, em alguns países é a neoplasia
mais comum em homens entre os 29 e os 40 anos de idade. A maioria dos tumores
testiculares (95%) é formada por células germinativas e divide-se em seminomas e
não-seminomas.
Entre os tumores não-seminomatosos estão o carcinoma de células embrionárias
coriocarcinoma e o teratoma, que normalmente metastatizam por vias linfática e
hemática, além de serem resistentes à radioterapia. As metástases dos tumores
seminomatosos são, normalmente, apenas por via linfática, para linfonodos
regionais, retroperitoneais, mediastinicos e supraclaviculares, e são sensíveis à
radioterapia. Quarenta por cento dos tumores de células germinativas são formados
por componentes seminomatosos e não-seminomatosos, mas a presença de
qualquer componente não-seminomatoso classifica-o como tal. O sinal clínico mais
comum da apresentação do tumor de testículo é o intumescimento indolor do
órgão, que pode tornar-se doloroso por hemorragias no interior da neoplasia ou
após traumatismos. Outros sintomas que podem ser encontrados são disúria, perda
de peso, feminilização, ginecomastia e diminuição da libido.
Paralelamente à realização de exames de imagem, os marcadores tumorais são de
grande auxílio no diagnóstico desses tipos de cancro. A determinação sérica da
hormona gonadotrófica coriónica (HCG) e de alfafetoproteína (AFP) é importante na
investigação diagnóstica, especialmente se o método utilizado dosear o HCG total e
a fracção beta livre.
A presença de níveis aumentados de AFP indica a existência de um componente
não-seminomatoso no tumor de testículo. Num estudo de pacientes com tumores
não-seminomatosos, os níveis séricos de HCG e da fração beta livre estavam
aumentados em 60% e em 40-70% dos casos, respectivamente. Apesar de poucos
tumores seminomatosos estarem associados ao aumento de HCG, níveis elevados
(acima de 5.000 UI/L) estão ligados à presença de tumor não-seminomatoso.
A avaliação conjunta dos dois analítos aumenta o valor preditivo dos testes em
relação à dosagem isolada, pois permite a detecção de aproximadamente 90% dos
tumores não-seminomatosos. Na investigação de seminoma, os níveis séricos de
HCG estão aumentados somente em 7 a 16% dos pacientes, e os valores são
inferiores a 200 UI/L. Porém, quando se utiliza a dosagem da fração beta livre, 20 a
50% dos pacientes com seminoma apresentam níveis séricos aumentados,
mostrando a importância de se dosear essa fracção. Nesses casos, é necessário
realizar o exame histológico do material de punção testicular para diferenciar
seminomas de não-seminomas.
Existem ainda os tumores testiculares de células de Sertoli ou células de Leydig,
que geralmente são benignos.
Não existe um consenso entre os mais de 40 ensaios existentes hoje no mercado
para as determinações de HCG ou das moléculas relacionadas. Variações entre os
resultados encontrados entre ensaios são comuns, especialmente se a dosagem de
HCG é para a investigação de gestações complicadas com aborto espontâneo, préeclâmpsia, doença trofoblástica, síndrome de Down ou neoplasias do testículo, da
bexiga ou do ovário.
Essas variações entre os resultados de HCG devem-se ao facto de que várias
formas da molécula de HCG podem estar presentes no sangue e na urina de
gestantes e de indivíduos com tumores: molécula de HCG intacta, fraccionada,
hiperglicosilada, hipoglicosilada, sem a extensão C-terminal, sub-unidade livre, subunidade grande, sub-unidade beta livre, sub-unidade beta livre fraccionada e a
fracção beta core. Muitas dessas sub-unidades são produtos de diferentes etapas
de degradação do HCG que fazem parte de um processo de desativação da
molécula.
No início de uma gestação normal, a principal forma de HCG encontrada no sangue
é a não-fraccionada, que aumenta exponencialmente no primeiro trimestre de
gravidez. Em pacientes com cancro de testículo ou doença trofoblástica, as
principais formas de HCG encontradas têm sido o HCG fraccionado e a fracção beta
livre, que é indicada como o melhor marcador tumoral para o cancro de testículo.
A metodologia que utiliza a associação HCG fraccionada, HCG não-fraccionada e
fracção beta livre é a que melhor se aplica para a dosagem de HCG, seja na
investigação de gravidez ou de cancro.
A maioria das neoplasias de próstata é de adenocarcinomas, detectados quando se
acha um nódulo de consistência difusa, por meio do toque retal. Em geral, o
paciente não apresenta, até então, queixas clínicas. Alguns pacientes manifestam
obstrução do trato urinário ou disfunção eréctil. O screening com dosagem do
antigénio específico da prostata (PSA) tem facilitado, juntamente com o toque retal,
o diagnóstico mais precoce. Perante as alterações desses parâmetros, exames
complementares, como o ultra-som trans-rectal e a biópsia da próstata, tornam-se
necessários para o diagnóstico.
AVALIAÇÃO LABORATORIAL
Hormona Folículo-Estimulante (FSH)
É uma hormona glicoproteica produzida pela hipófise e secretada de forma pulsátil,
podendo ser doseada em pool. Eleva-se nas deficiências do ovário e testicular
(hipogonadismo primário). É indicado no diagnóstico da menopausa. Encontra-se
diminuída nas doenças hipotalámicas e hipofisárias, excepto em casos de tumores
produtores de gonadotrofinas, quando se encontra elevada. Pode elevar-se também
em casos de comprometimento da espermatogénese. A relação LH/FSH maior do
que 2 pode sugerir o diagnóstico de ovário poliquístico. É útil também na avaliação
da puberdade precoce.
Hormona Luteínica (LH)
É uma hormona glicoproteica produzida pela hipófise e secretada de forma pulsátil,
podendo ser doseada em pool. O doseamento seriado é indicado no diagnóstico da
ovulação. Eleva-se no hipogonadismo primário e encontra-se diminuída nas
doenças hipotalámicas e hipofisárias, excepto em casos de tumores produtores de
gonadotrofinas, quando se apresenta elevada. A sua relação com FSH é valorizada
no diagnóstico do síndrome do ovário poliquístico. É útil também na avaliação da
puberdade precoce.
Testosterona
É uma hormona esteróide androgénica secretada pelos ovários e gandulas suprarenais na mulher e pelos testículos nos homens. É útil no diagnóstico do
hipogonadismo masculino, bem como na avaliação da puberdade. Na mulher, é
indicado na avaliação do hirsutismo e da virilização. A testosterona circula ligada à
SHBG (globulina ligada às hormonas sexuais), com uma pequena fracção que
circula na forma livre.
3-Alfa-Androstenediol Glicuronídeo
(3-Alfa-Diol)
É o principal metabolito da deidrotestosterona. Nos casos de aumento da actividade
da 5-alfa-reductase, ocorre aumento tanto da DHT quanto dos seus metabolitos. É
importante no diagnóstico do hirsutismo idiopático, encontrando-se aumentado
mesmo com níveis normais de testosterona.
Estrona
Provém principalmente da conversão periférica da androstenediona. É útil na
avaliação dos tumores feminilizantes e da menopausa, bem como na avaliação da
puberdade e do síndrome do ovário poliquístico.
Estradiol (17-Beta-estradiol)
É o estrogénio mais potente produzido pelas gonadas. Na mulher, é produzido pelos
folículos ováricos, e nos homens, pelas células de Leydig e por conversão periférica
da testosterona.
É útil no diagnóstico da puberdade precoce e dos tumores feminilizantes no
homem. No hipogonadismo feminino, encontra-se em níveis baixos, apesar de
grande superposição com valores normais. No hipogonadismo masculino,
dependendo da etiologia, os valores podem estar normais ou elevados. Nos
tumores feminilizantes no homem (supra-renais ou testiculares) produtores de
estrogénio, o estradiol estará elevado.
Estriol
O estriol é produzido pela placenta a partir da conversão do sulfato de deidroepiandrosterona produzido pela supra-renal fetal. Por isso, o seu doseamento é útil
na avaliação da integridade feto-placentária. Os seus valores elevam-se
gradualmente durante a gestação normal. O seu doseamento seriado é utilizado
como um marcador no acompanhamento das gestações de alto risco, visto que sua
queda indica sofrimento fetal.
Progesterona
É um esteróide secretado pelas gonadas e glândulas supra-renais. É o principal
marcador da ovulação. Para essa avaliação, devem ser colhidas amostras seriadas.
A principal indicação desse doseamento é o diagnóstico de ciclos anovulatórios e a
disfunção da fase lúctea.
Testosterona Livre
A testosterona livre é responsável pelo efeito metabólico da testosterona, uma vez
que não sofre a influência dos níveis de SHBG. É importante no diagnóstico do
hipogonadismo masculino e do hirsutismo feminino.
Androstenediona
É um androgénio produzido pelas supra-renais e gónadas. Útil na avaliação do
hiperandrogenismo (hirsutismo, acne), no acompanhamento da hiperplasia
congénita de supra-renal, tumores virilizantes de supra-renal e ovário.
Deidrotestosterona (DHT)
Provém da transformação periférica de testosterona no homem e da
androstenediona na mulher, pela ação da 5-alfa-redutase. É útil na avaliação do
hirsutismo feminino, sugerindo maior conversão periférica. Também é útil nas
avaliações do defeito da 5-alfa-redutase, nos casos de pseudo-hermafroditismo
masculino, quando as concentrações de DHT são desproporcionalmente baixas em
relação às de testosterona.
Eleva-se também na hiperplasia congénita da supra-renal e nos carcinomas do
ovário e adrenais. Durante a gestação, grandes quantidades são produzidas pela
placenta.
Globulina Ligada às Hormonas Sexuais (SHBG)
É uma glicoproteína sintetizada pelo fígado. A sua função é transportar as
hormonas sexuais. É útil na avaliação complementar do hiperandrogenismo, uma
vez que a sua elevação pode levar ao aumento dos níveis de testosterona total mas
não dos de testosterona livre. Várias circunstâncias alteram seus valores, como
estrógenios e hormonas tiroidéias (hipertiroidismo). Tamoxifen, fenitoína e cirrose
hepática elevam os níveis de SHBG. Já os androgénios, os glicocorticóides, a
hormona de crescimento, o hipotiroidismo, a obesidade e a acromegalia cursam
com níveis diminuídos de SHBG.
Avaliação Dinâmica da Função Endócrina
Muitas patologias endócrinas podem ser diagnosticadas por meio dos doseamentos
dos níveis hormonais sérios ou urinários, em condições basais. Contudo, o
doseamento isolado das hormonas nem sempre permite a distinção entre o normal
e o patológico.
Os valores de normalidade tão alargados para concentrações séricas de algumas
hormonas tornam imprecisa a interpretação de valores individuais se o valor normal
prévio para determinado paciente for desconhecido. Por exemplo, uma
concentração sérica de tiroxina (T4) no limite máximo para a população em geral
pode estar associada a hipertiroidismo num paciente cuja concentração,
usualmente, estaria no limite mínimo da faixa de normalidade.
Além disso, há pequenos níveis de disfunção endócrina que podem estar
compensados em condições basais. Portanto, os níveis séricos de cortisol podem
estar normais em pacientes com insuficiência adrenocortical parcial devido ao
aumento de secreção de corticotrofina (ACTH).
No diagnóstico de alterações parciais dos mecanismos de controlo endócrinos,
podemos utilizar três tipos de testes funcionais: doseamentos hormonais seriados,
doseamentos de várias hormonas e testes de reserva endócrina e retro-controlo
endócrino. Todos os três são da máxima importância no diagnóstico, mas podem
sofrer a influência de inúmeros factores que tornariam sua interpretação bastante
complexa.
Doseamentos Hormonais Seriados
Nalgumas circunstâncias, a variação de secreção hormonal reflecte processos pouco
compreendidos de exacerbação ou de remissão de patologias. Dessa forma,
doseamentos seriados dos níveis de cálcio e PTH (paratormona) por períodos
prolongados podem ser necessárias para diagnóstico de hiperparatiroidismo. Outras
condições clínicas, como o síndrome de Cushing, podem apresentar o mesmo
padrão de exacerbação/remissão. Noutras circunstâncias, a variação na secreção
hormonal endócrina resulta de alterações rítmicas como uma secreção pulsátil ou
um ritmo circadiano básico. A perda do ritmo circadiano de secreção de cortisol
pode ser um sinal precoce de síndrome de Cushing. O ritmo circadiano da secreção
hormonal pode ser alterado por inúmeros factores, como distúrbios do sono,
drogas, doenças psiquiátricas e stres. A demonstração de uma variação diurna
normal pode ser uma boa evidência de normalidade na sua função. No entanto, a
sua ausência não indica necessariamente uma doença endócrina primária. Na
verdade, alterações desse ritmo circadiano indicam a necessidade de testes
diagnósticos adicionais.
Doseamento de várias Hormonas
Como o sistema endócrino funciona basicamente sob retro-regulação, o
doseamento de várias hormonas (T4 e TSH, cálcio e PTH, testosterona e LH )
permite uma avaliação dos níveis individuais. Por exemplo, uma vez que a faixa de
normalidade do T4 é ampla, os níveis de determinado paciente poderiam cair para
a metade e ainda permanecer na faixa normal. Nesse caso, contudo, uma
concentração de T4 próxima aos limites mínimos normais, associada a elevados
níveis de TSH, indica o estadio inicial de uma falência tiroidiana compensada.
Conforme observamos na tabela a seguir, baixos níveis de ambos os pares
hormonais apontam deficiência da hormona trófica (ex: insuficiência hipofisária no
caso de TSH e T4). Altos níveis da hormona-alvo com baixos níveis da hormona
trófica sugerem secreção autónoma do órgão-alvo (ex: adenomas adrenais
hiperfuncionantes inibindo a secreção de ACTH).
ALTO
NÍVEL DA HORMONA DA GLÂNDULA
BAIXO
NORMAL
ALTO
Secreção autónoma
de hormonas tróficas
ou resistência à
acção hormonal da
glândula alvo
NORMAL
BAIXO
Normal
Insuficiência
Hipofisária
Secreção autónoma
do órgão alvo
Elevados níveis dos pares hormonais são compatíveis com os mecanismos de várias
doenças. A secreção autónoma de uma hormona trófica pode ser tópica ou
ectópica. Por exemplo, o síndrome de Cushing pode resultar da secreção hipofisária
de ACTH ou da secreção de ACTH por tumores pulmonares. Outra possibilidade é a
secreção de factores liberados a partir de tumores em órgãos periféricos, causando
hipersecreção de hormonas hipofisárias, como por exemplo a acromegalia
resultante da secreção ectópica de fatores liberadores da hormona de crescimento.
Por outro lado, essa elevação combinada da hormona trófica e da hormona da
glândula-alvo pode dever-se à resistência da acção da hormona da glândula-alvo.
Essa resistência pode ser herdada (como nos casos de defeito de receptor de
androgénios que causam resistência à acção da hormona e resultam em níveis
elevados de LH e de testosterona), ou adquirida (como no caso de resistência
insulínica da obesidade, que pode levar a hiperinsulinismo e hiperglicemia).
Elevação de TSH e T4 pode indicar tanto secreção autónoma de TSH quanto à
resistência da acção da T4.
Contudo, doenças em estadio inicial podem tornar necessárias informações
adicionais por meio de testes dinâmicos.
Testes da Função Endócrina
Os testes dinâmicos baseiam-se em estímulo ou em supressão da produção
hormonal.
Testes de Estimulação
São utilizados na suspeita de hipofunção endócrina para avaliar a capacidade de
reserva de síntese e secreção hormonal. Esses testes são realizados de duas
maneiras:
1) Administração de uma hormona trófica para testar a capacidade do órgão-alvo
de aumentar a produção hormonal. Essa hormona pode ser um factor liberador
hipotalámico, como o TRH, ou uma hormona hipofisária, como o ACTH. Nesses
casos, a capacidade do órgão-alvo é avaliada pelo doseamento dos níveis
hormonais séricos - nos exemplos citados, o TSH e o cortisol.
2) Estimulação da secreção de uma hormona trófica endógena ou factor
estimulador e medição do efeito desses estímulos antiestrogénicos em nível
hipotalámico, diminuindo o retro-controlo negativo e causando um aumento na
secreção de gonadotrofinas, que pode ser seguido por ovulação e/ou aumento na
formação de esteróides gonodais.
Testes de Estimulção mais Frequentes
ÓRGÃO/ SISTEMA
Hipotálamo-hipófise
Hipófise
Tiróide
Supra Renal
Gónadas
Ilhotas Pancreáticas
Paratiróide
Balanço Hídrico
Metabolismo do
Cálcio
ESTÍMULO
Hipoglicemia
Metirapona
Levodopa
Anginina
Citrato de clomifeno
Exercício
Restrição hídrica
RTH
LHRH
CRF
GHRH
TSH
Pentagastrina
Cálcio
Certrosina
Metirapona
Mudança de postura
HCG
Glicose
PTH
Vasopresina
Sobrecarga de Cálcio
RESPOSTA
GH e ACTH (cortisol)
ACTH (cortisol e 11-desoxicortisol)
GH
GH
Gonodotrofinas
GH
Vasopresina
TSH e prolactina
Gonodotrofinas
ACTH (cortisol)
GH
Captação de iodo radiotaivo
Calcitonina
Calcitonina
Cortisol
Cortisol e 11 desoxicortisol
Renina e aldosterona
Testosterona
Tolerância a glicose e libertação de
insulina
AMPc e fósfatos urinários
Concentração urinária
Cálcio Urinário
Testes de Supressão
São utilizados em casos de suspeita de hiperfunção endócrina. Como nos testes de
estimulação, são utilizadas hormonas exógenas ou factores reguladores conhecidos
para avaliar a inibição da produção hormonal endógena, por exemplo, a
administração de glicocorticóide (dexametasona) a pacientes com suspeita de
síndrome de Cushing para avaliar a capacidade de inibição da secreção do ACTH e
portanto da síntese adrenal de cortisol. A falência da supressão nesses testes indica
a presença de secreção autónoma da hormona da glândula-alvo ou de hormonas
tróficas (hipofisárias ou ectópicas) que não estão sob retro-regulação normal.
Testes de Supressão Mais Frequentes
ÓRGÃO/ SISTEMA
Hipotálamo-hipófise
Tiróide
Supra-renal
Ilhotas Pancreáticas
ESTÍMULO
Glicose
Dexametasona
T4
Dexametasona
Salina
Clonidina
Jejum
RESPOSTA
GH
ACTH (cortisol)
Captação de iodo radiotaivo
Cortisol
Renina e aldosterona
Noradrenalina plasmática
Glicose e insulina
Interpretação dos Testes Funcionais
São a melhor avaliação dos distúrbios endócrinos leves, como por exemplo o teste
de estímulo com cortrosina para diagnosticar a insuficiência adrenocortical parcial
com secreção de cortisol basal normal. Os testes dinâmicos também são úteis na
determinação do defeito patogénico, tal como o estímulo com LHRH para
diagnosticar se o hipogonadismo hipogonadotrófico ocorre em consequência de
insuficiência hipofisária ou hipotalámica. Em pacientes com síndrome de Cushing, a
supressão da produção de cortisol em resposta a altas doses de dexametasona
sugere hipersecreção hipofisária de ACTH, uma vez que tumores adrenais ou
secreção ectópica de ACTH não respondem ao teste de supressão.
A principal dificuldade na interpretação dos testes é a definição adequada do que
seja resposta normal em indivíduos normais que apresentam outras patologias. Por
exemplo, os atletas têm uma resposta aumentada do ACTH e do cortisol ao
estímulo com cortrosina. Além disso, inúmeros factores influenciam a resposta de
um teste funcional, como idade, sexo, fumo, uso de determinadas drogas,
obesidade, desnutrição, insuficiência renal crónica, cirrose hepática e outras
patologias associadas. Por exemplo, a resposta do TSH ao estímulo com TRH
diminui em homens com mais de 60 anos. Citamos ainda a resposta sub-normal do
GH ao estímulo de hipoglicemia induzida por insulina e a administração de arginina
e de levodopa em pacientes obesos, que normalizam após o retorno ao peso ideal.
Por outro lado, pacientes com desnutrição grave, insuficiência renal crónica ou
cirrose hepática costumam ter níveis de GH basal elevados, que não respondem à
supressão ou respondem com um aumento paradoxal após sobrecarga de glicose.
Na avaliação da baixa estatura, a realização de dois testes aumenta a sensibilidade
de 80 para mais de 90%.
Várias doenças psiquiátricas estão associadas a testes dinâmicos da função
endócrina, alterados na ausência de patologia endócrina específica. A mais
frequente é a depressão. Pacientes com depressão primária grave não apresentam
supressão adequada do cortisol, após administração de dexametasona,
normalizando essa resposta após o tratamento da depressão. Em 20% dos
pacientes com doença psiquiátrica aguda, são encontradas concentrações elevadas
de T4 e T4 livre sem evidência clínica de tireotoxicose. A resposta do TSH ao TRH
apresenta-se subnormal ou ausente em cerca de 25% dos pacientes psiquiátricos
sem doença tiroidéia, principalmente em casos de depressão unipolar ou bipolar.
O uso de drogas também interfere nos testes dinâmicos. Glicocorticóides em doses
farmacológicas, progestagénios, teofilina e cloropromazina bloqueiam a resposta do
GH aos estímulos usuais. A ingestão crónica e excessiva de álcool diminui a
resposta da prova de supressão do cortisol à dexametasona. O uso de levodopa, de
dopamina e de aspirina em altas doses bloqueia a resposta do TSH ao TRH. A
fenitoína aumenta a captação celular e o metabolismo do T4, levando a baixos
níveis de T4 livre, além de diminuir em 50% a resposta do TSH ao TRH. Portanto, a
avaliação da função tiridéia é prejudicada principalmente quando nos encontramos
diante de um paciente com uso dessa droga com níveis baixos de T4 e TSH normal.
Deve ser considerada, ainda, a possível necessidade de estímulos repetidos para a
obtenção de uma resposta normal. Por exemplo, pacientes com hipogonadismo
hipogonadotrófico por doença hipotalámica com resposta subnormal do LH ao
estímulo com LHRH podem normalizar a resposta ao teste após 1 semana de
administração diária de LHRH. Esse protocolo permite a distinção entre
hipogonadismo hipotalámico e hipofisário.
Para melhor compreensão das provas funcionais, a figura acima esquematiza a
regulação da secreção das hormonas hipofisárias por intermédio de hormonas
hipotalámicas estimulantes e inibidores.
Os Testes Mais Frequentes
Estimulação da Hormona de Crescimento
A maioria dos autores considera normal uma resposta superior a 7 ng/mL, embora
outras escolas admitam valores > 10 ng/mL.
- Após Exercício
Colher a amostra basal em repouso, submeter o paciente a 20 minutos de
exercício. Colocá-lo em repouso durante 10 minutos antes da colheita da amostra,
após esforço. Aproximadamente 10% dos pacientes normais deixam de responder a
esse estímulo. Portanto, a baixa resposta não faz o diagnóstico de deficiência de
GH. A sua realização está contra-indicada nas doenças sistémicas nas quais o
esforço possa desencadear ou agravar o quadro.
- Após Clonidina
Colher amostras nos tempos: basal, 60, 90 e 120 minutos. O paciente deverá
permanecer deitado durante toda a prova e por mais 1 hora após a colheita da
amostra de 120 minutos, prevenindo os efeitos da hipotensão causada pela droga.
A pressão arterial deve ser monitorizada durante toda a prova.
- Após Insulina
São realizadas os doseamentos da GH e da glicemia nos tempos: basal, 30, 60, 90
e 120 minutos. É indispensável a monitorização médica contínua devido ao risco de
hipoglicemia. A informação obtida nessa prova não parece superior à obtida com
outros testes de libertação de GH. A sua realização é contra-indicada em pacientes
com história de crises convulsivas, cardiopatias, insuficiência renal ou hepática,
hipocortisolismo, em gestantes ou no uso concomitante de algumas drogas como
hipoglicémicos orais e betabloqueadores. Para que o teste seja considerado válido,
deve ocorrer queda da glicemia a valores abaixo de 40 mg/dL ou aparecimento de
sinais clínicos de hipoglicemia.
- Após Piridostigmina (Mestinon)
Colher amostras para doseamento de GH nos tempos: basal, 60, 90 e 120 minutos.
A sua realização é contra-indicada em pacientes com história de asma brônquica,
diarréia e cólica intestinal.
- Após TRH
Colher amostras para doseamento de GH nos tempos: basal, 30 e 60 minutos. Em
indivíduos normais, não ocorre aumento dos níveis de GH, enquanto em muitos
acromegálicos pode ocorrer um efeito paradoxal da droga com aumento da
secreção de GH. Essa resposta não apresenta a mesma uniformidade que a
resposta anormal à administração de glicose por via oral. A sua realização é contraindicada em pacientes com história de hipertensão arterial, doenças
cerebrovasculares e glaucoma.
Supressão da Hormona de Crescimento
- Após Glicose
São realizados os doseamentos de GH e de glicemia nos tempos: basal, 30, 60, 90,
120 e 180 minutos. Nos indivíduos normais, ocorre diminuição dos níveis de GH até
valores inferiores a 2 ng/mL. Nos acromegálicos, pode ocorrer uma resposta
paradoxal, com elevação dos níveis de GH.
- Após Parlodel
São realizados os doseamentos de GH nos tempos: basal, 60, 120, 180, 240 e 360
minutos. Indicado para determinar a possível resposta dos pacientes portadores de
tumores produtores de GH à terapêutica medicamentosa com bromoergocriptina,
devendo ocorrer queda expressiva (50% dos níveis basais).
Estimulação da Prolactina
- Após TRH
São realizados os doseamentos de prolactina nos tempos: basal, 30 e 60 minutos.
A pressão arterial deve ser monitorizada, pois pode ocorrer elevação dos níveis da
PA. Em mulheres normais, deve ocorrer aumento de pelo menos três vezes o valor
basal, enquanto nos homens se espera um aumento de pelo menos duas vezes o
valor basal.
Níveis elevados no doseamento basal e uma resposta reduzida ou ausente sugerem
a presença de tumor, embora esse padrão de resposta não seja suficiente para
diagnosticar ou excluir a possibilidade de prolactinoma. Níveis diminuídos no
doseamento basal, com resposta baixa, são um achado raro, excepto quando
associados à falência precoce da lactação no síndrome de Sheeham.
Supressão da Prolactina
- Após Parlodel
Dosear a prolactina basal (8:00) e a cada 2 horas em quatro amostras (10,12,14 e
16 horas). Indicado para determinar a possível resposta dos pacientes portadores
de tumor produtor de prolactina à terapêutica medicamentosa. Espera-se uma
queda do nível basal de mais de 50% após 4 horas.
Estimulação da Hormona Tireotrófica
- Após TRH
É realizado o doseamento do TSH nos tempos: basal, 30 e 60 minutos, e se
necessário, 90 e 120 minutos. Normalmente ocorre um pico de TSH entre 20 a 30
minutos após a administração do TRH. O incremento de TSH é de cinco a quinze
vezes o nível basal, variando com o sexo (maior em mulheres) e com a idade
(menor em idades avançadas).
Em pacientes com hipotiroidismo primário, o TSH parte de valores basais altos,
eleva-se mais ainda e não se normaliza aos 60 minutos. Já no hipotiroidismo
secundário por deficiência hipofisária, o TSH encontra-se em níveis inferiores no
doseamento basal e não responde ao TRH.
No hipotiroidismo hipotalámico, a resposta do TSH ao TRH é tardia, registando-se
elevação gradual do TSH até 60 minutos.
Nos pacientes com hipertiroidismo, a secreção do TSH encontra-se bloqueada pelo
excesso de T3 eT4, não havendo resposta ao estímulo com o TRH.
Estimulação das Gonadotrofinas Hipofisárias
- Após LHRH
São realizados os doseamentos de LH e de FSH nos tempos: basal, 15, 30 e 60
minutos. Em homens, ocorre um aumento de LH de duas a dez vezes o nível basal,
e o aumento de FSH é de metade a duas vezes esse nível. Nas mulheres, o
aumento de LH é de três a quatro vezes o nível basal na fase folicular e mais
acentuado na fase lúctea, enquanto o aumento de FSH é de metade a duas vezes o
basal.
Estimulação Hipotálamo-Adreno-Hipofisária
- Após Insulina
Esse teste avalia o estímulo de ACTH e de cortisol. São realizados doseamentos de
ACTH e de cortisol nos tempos: basal, 30, 60 , 90 e 120 minutos. Para que o teste
seja considerado válido, devem ocorrer queda da glicemia para valores abaixo de
40 mg/dL ou o aparecimento de sinais clínicos de hipoglicemia. É indispensável a
monitorização médica contínua, devido ao risco de hipoglicemia.
Esta prova deve ser precedida da prova de estímulo com ACTH, e só devem ser
submetidos a ela os pacientes com estímulo positivo.
Normalmente, o ACTH aumenta três vezes ou mais em relação ao nível basal,
devendo exceder 150 ng/mL. O cortisol eleva-se a valores superiores a 200 ng/mL.
- Após Cortrosina
O teste de estimulação do cortisol - estimulação rápida com cortrosina simples. São
realizadas os doseamentos de cortisol nos tempos: basal, 30, 60 e 90 minutos. A
ausência de resposta do cortisol ao estímulo com cortrosina estabelece o
diagnóstico de insuficiência adrenocortical parcial em pacientes com secreção de
cortisol basal normal. Níveis de cortisol iguais ou superiores a 200 ng/mL, em
qualquer ponto da prova, indicam função adrenal normal.
Avaliação Hipofisária Total
São realizados doseamentos de TSH, prolactina, LH, FSH, cortisol, GH e glicose nos
tempos: basal, 30, 60 minutos. Os doseamentos de FSH, GH, cortisol e glicose
devem ser realizados também aos 90 minutos.
A resposta de cada sector (tireotrófico, gonadotrófico, corticotrófico ou lactotrófico),
bem como as contra-indicações, devem ser avaliadas isoladamente, como já foi
descrito anteriormente.
Betacaroteno
O caroteno é um precursor lipossolúvel da vitamina A encontrado em gorduras,
folhas e vegetais amarelos. Uma pequena porção do caroteno é absorvida pelo
intestino, contribuindo para a cor amarela do soro. Com a participação das
gorduras e dos sais biliares, a maior parte do caroteno é normalmente convertida
em retinol no trato intestinal.
A hipercarotenemia, isto é, a elevação do nível de caroteno, caracteriza-se por
pigmentação amarela da pele, sem nenhuma mudança na cor da esclerótica,
causada por ingestão excessiva de alimentos ou de complexos vitamínicos ricos em
caroteno. O indivíduo também pode apresentar prurido e perda de peso. A doença
é geralmente benigna e é tratada com mudanças na dieta.
Valores diminuídos indicam ingestão insuficiente ou má absorção. O doseamento de
caroteno é mais frequentemente utilizado como prova de triagem para a síndrome
de má absorção e no diagnóstico diferencial de hiperbilirrubinemia
Beta-2-Microglobulina
A beta-2-microglobulina é uma pequena molécula presente na superfície das células
nucleadas, principalmente linfócitos, que está relacionada com HLA (Human
Leucocyte Antigen). A sua presença é necessária para a inserção da molécula de
HLA na membrana celular, além de estabilizar a cadeia pesada do HLA. Ela parece
actuar na regulação da função dos leucócitos, mas isso ainda não foi
suficientemente definido.
Está, normalmente, presente no plasma, urina e líquor. É libertada durante o
processo de renovação das membranas celulares. Por ser uma proteína pequena, é
filtrada pelos glomérulos. No entanto, a maior parte é reabsorvida e degradada
pelas células epiteliais tubulares dos túbulos proximais. Por isso, a concentração
plasmática de beta-2-microglobulina é um bom índice da taxa de filtração
glomerular. É descrito um aumento progressivo dos valores normais acompanhando
a idade.
Níveis séricos elevados podem ocorrer em diversas patologias inflamatórias, como
hepatites, artrite reumatóide, lúpus eritematoso sistémico, SIDA, sarcoidose e em
pacientes com leucemias, linfomas e alguns tumores sólidos e patologias que
cursam com a diminuição da filtração glomerular.
Níveis urinários elevados podem ser encontrados em pacientes com desordens
renais tubulo-intersticiais, como intoxicação por metais pesados, drogas
quimioterápias, aminoglicosídeos, infecções urinárias altas e rejeição a
transplantes. Relatos recentes apontam a beta-2-microglobulina como o melhor
marcador de prognóstico no mieloma múltiplo.
Bilirrubina
A bilirrubina é o principal produto do metabolismo do heme da hemoglobina. Cerca
de 70% da bilirrubina são provenientes da destruição de eritrócitos velhos, 15%
provêm de fontes hepáticas, e o restante é proveniente da destruição de células
vermelhas defeituosas na medula óssea e nos citocromos.
A hemoglobina é metabolizada no baço e no sistema reticuloendotelial, dando
origem à biliverdina, que é reduzida a bilirrubina pela enzima biliverdina redutase.
Essa bilirrubina recém-formada circula no sangue ligada à albumina sérica (forma
não-conjugada). É transportada pelo sistema porta até o fígado, onde penetra no
hepatócito por dois mecanismos distintos: difusão passiva e endocitose.
Uma vez dentro do hepatócito, a bilirrubina desliga-se da albumina e forma um
complexo protéico com as chamadas proteínas Y e Z. Logo depois, liga-se a um
outro complexo chamado ligandina. É então transportada para o retículo
endoplasmático liso, onde se forma um substrato da enzima glicuronil transferase,
dando origem a um diglicuronídeo conjugado (mono- e triglicuronídeos também são
formados). A bilirrubina, agora já conjugada, é transportada até a membrana
celular. Na face oposta aos sinusóides e próxima aos canalículos biliares, ela é
excretada directamente. Só consegue ultrapassar a membrana quando conjugada.
Através dos canalículos biliares, alcança o trato intestinal, onde é metabolizada
pelas bactérias da flora intestinal, formando os urobilinogénios. A maior parte dos
urobilinogénios é absorvida e novamente excretada pelo fígado, e uma pequena
fracção é excretada pelos rins.
Existem portanto dois tipos de bilirrubina circulantes - a conjugada (bilirrubina
directa) e a não-conjugada (bilirrubina indirecta). No entanto, existe um terceiro
tipo de bilirrubina, chamada de bilirrubina delta, do tipo conjugada de reacção
rápida e ligada à albumina permanentemente por uma reacção covalente. Pelas
técnicas tradicionais, a bilirrubina delta era incluída nos resultados da bilirrubina
directa (conjugada) e na bilirrubina total.
Por estar fortemente ligada à albumina, a bilirrubina delta não é excretada pelos
rins e permanece elevada por muitor tempo, na verdade, por períodos
correspondentes à semi-vida da albumina (cerca de 19 dias), mesmo após a
resolução da obstrução ou do período agudo da lesão hepática. Isso pode levar a
falsas interpretações. Entretanto, actualmente, os métodos automatizados de
última geração, especialmente a tecnologia de química seca, já separam a fracção
delta, que não é incluída em nenhuma das demais frações, nem mesmo no valor da
bilirrubina total.
Acompanhando os mecanismos envolvidos no metabolismo da bilirrubina, é possível
correlacionar o aumento de seus níveis séricos com alterações de uma dessas
etapas.
Os níveis séricos da bilirrubina não-conjugada (bilirrubina indirecta) são
determinados pela velocidade de produção e pela velocidade de remoção dessa
bilirrubina da circulação. Os distúrbios que alteram a capacidade de depuração do
fígado estão ligados à captação e/ou conjugação hepática. Os aumentos de
bilirrubina indirecta não levam ao aumento da bilirrubina na urina.
Os níveis séricos da bilirrubina conjugada (bilirrubina directa) são determinados
pela capacidade de excreção da bilirrubina pelo fígado, ou seja, pela integridade
fisiológica do hepatócito e da permeabilidade das vias biliares intra e extrahepáticas. Patologias que alterem essas funções cursam com aumento da
bilirrubina directa, e muitas vezes da bilirrubina indirecta, e com a presença de
bilirrubina na urina.
Anemias hemolíticas (hereditárias e adquiridas,
reabsorção extravascular, eritropoiese ineficaz.
Alterações da
Síndrome de Gilbert, drogas, acidose metabólica e
Captação
estados carenciados com hipoalbuminémia.
Alteração da
Síndrome de Cligler-Najjar (tipos 1 e 2: deficiência
Conjugação
da actividade da glicuronil transferase total e
parcial, respectivamente). Inibição da glicuronil
transferase por fármacos, especialmente em recém
nascidos.
Alterações da
Síndrome de Dubin-Johnson, síndrome de Rotor,
excreção
colestase intra-hepática recorrente benigna,
icterícia recorrente da gravidez, colestase por
fármacos, icterícia pós-operatória, quadros de
comprometimento da função hepática, como
hepatites e cirrose.
Obstruções das vias Lesões parciais ou completas dos ductos biliares por
biliares
estenose, cálculos ou tumores.
Produção excessiva
Brucelose
A brucelose é uma zoonose, e a forma humana é causada por uma das quatro
espécies: Brucella melitensis, a causa mais comum em todo o mundo, adquirida
pelo contacto com cabras, carneiros e camelos; Brucella abortus, adquirida por
contacto com bois; Brucella suis, adquirida por contacto com porcos; e Brucella
canis, adquirida por contacto com cães.
O contágio dá-se pelo contacto com a pele ou por ingestão de secreções
contaminadas por esses animais. Tais bactérias mantêm-se viáveis em solo seco
por cerca de 40 dias, e no caso de solo húmido esse prazo é ainda maior. São
destruídas pela pasteurização e fervura, mas resistem ao congelamento.
A contaminação relacionada com a ocupação profissional, como é o caso de
agricultores, veterinários, processadores de carne, são a fonte mais frequente. Na
população em geral, a fonte mais comum de contaminação é a ingestão de leite e
derivados não-pasteurizados e o consumo de carne crua. Pode ser transmitida de
pessoa a pessoa, pela placenta e durante a amamentação, e são citados casos
raros de contaminação por actividade sexual.
A infecção pode distribuir-se amplamente pelo organismo, causando lesões
praticamente em qualquer órgão, com mais frequência no coração, ossos e
articulações, aparelhos respiratório, gastrointestinal e geniturinário, globo ocular,
pele, sistema nervoso central e sistema endócrino.
O quadro clínico inicial é comum a outras doenças febris. O período de incubação
dura em média de 1 a 3 semanas, podendo, em alguns casos, durar vários meses.
A multiplicação intracelular do microrganismo ocorre nos gânglios linfáticos e
sistema reticuloendotelial. A gravidade do quadro é variável, podendo apresentarse desde uma forma leve a grave. O quadro apresenta sintomas comuns como
febre, mialgia, cefaléia, anorexia, artralgia e lombalgia. O exame clínico pode ser
pouco expressivo ou apresentar linfadenopatia, hepatoesplenomegalia, dor à palpação da coluna vertebral, dor abdominal e outras manifestações.
O diagnóstico é feito mediante a associação de história compatível com
probabilidade de infecção, sinais clínicos e deteccção de anticorpos por reacções de
aglutinação (com ou sem cultura positiva de sangue e tecidos). O diagnóstico é de
grande importância no período pré-natal, pois pode levar à morte fetal.
Os antigénios bacterianos têm a capacidade de induzir a formação de anticorpos
específicos, inicialmente da classe IgM e logo após das classes IgG e IgA . Esses
anticorpos aparecem a partir da segunda semana da doença, com picos entre a
terceira e sexta semanas. Títulos maiores ou iguais a 1/160 são considerados
significativos quando encontrados numa região não-endémica. Em áreas endémicas
e em profissionais de alto risco de contaminação, são considerados significativos
títulos iguais ou acima de 1/320.
Títulos altos de IgM indicam infecção aguda; altos títulos de IgG, infecção em
actividade; quando mais baixos, podem significar infecção antiga. Recomenda-se a
análise com intervalo de 2 semanas na avaliação dos casos duvidosos: variações de
quatro vezes o título anterior são sugestivas de infecção aguda. Reacções com
títulos baixos podem ser encontradas em pacientes vacinados contra a febre tifóide.
Podem ser encontradas reações cruzadas por infecção por outras bactérias e após
reacções intradérmicas, com antigénios de Brucella
BTA
O carcinoma da bexiga é um dos tipos de tumor de maior incidência na Europa.
Afecta principalmente homens entre 50 e 70 anos de idade, fumadores e pessoas
expostas a substâncias químicas, como tintas, couro e borracha.
Segundo as estatísticas, 1 em cada 5 pacientes diagnosticados com este tipo de
carcinoma tem expectativa de vida de cerca de 5 anos. No entanto, se o
diagnóstico for precoce, a taxa de sobrevida nesse mesmo período sobe de 20 para
94%.
Entre os sinais clínicos de carcinoma da bexiga, o mais frequente é a hematúria,
mas outros sintomas - como aumento da frequência urinária, dor pélvica e
suprapúbica, obstrução urinária e micro-hematúria, podem também manifestar-se.
Aproximadamente 80% dos tumores da bexiga são de células de transicão e estão
restritos, na sua maioria, à superfície mucosa, podendo ser resolvidos por meio de
ressecções trans-uretrais.
A detecção precoce do carcinoma da bexiga é essencial para se aumentar a
sobrevida de pacientes portadores da entidade mórbida. A recorrencia desse tumor
pode dar-se em cerca de 50% dos pacientes tratados, o que torna essencial a
monitorização a longo prazo.
A observação da recorrência do tumor em pacientes com diagnóstico prévio de
carcinoma da bexiga tem sido feita por meio de cistoscopia, podendo ser
suplementada, nalguns casos, pela citologia de amostra da urina ou do raspado da
bexiga. Entretanto, no que diz respeito à monitorização, a citologia é um exame
sensível para os tumores pouco diferenciados (alto grau) e pouco sensível para os
bem diferenciados (baixo grau). Por esse motivo, têm-se procurado outros testes
de maior sensibilidade que possam suplementar ou mesmo substituir os exames
existentes.
O teste ideal para o diagnóstico e a monitorização de pacientes com carcinoma da
bexiga deve ser rápido, de fácil realização, além de possuir sensibilidade e
especificidade altas. Estudos recentes mostraram que a detecção do BTA (bladder
tissue antigen) é significativamente mais sensível do que a citologia, não
importando o estadio ou o grau do tumor da bexiga.
O antigénio do tumor da bexiga reconhecido pelos anticorpos no teste BTA foi
inicialmente isolado da urina de pacientes com neoplasia da bexiga confirmada
histologicamente. Não é reconhecido na maioria dos indivíduos normais e em
pessoas com outras patologias do aparelho urinário. A especificidade do teste
mostrou-se equivalente à da citologia em indivíduos normais. Além de ser um teste
não invasivo que utiliza como material apenas uma pequena amostra isolada de
urina, estudos comprovaram que o BTA é significativamente mais sensível do que a
citologia, sejam quais forem o estadio ou o grau do tumor.
CA 125
O CA 125 é uma glicoproteína de alto peso molecular (>200 kD), de papel
fisiológico ainda desconhecido, presente em diferentes condições benignas e
malignas, sendo útil especialmente no acompanhamento dos carcinomas do ovário.
A concentração sérica do CA 125 é superior a 35 U/mL em aproximadamente 80%
das mulheres com carcinoma do ovário, 26% das mulheres com tumores benignos
de ovário e em 66% de pacientes em condições não-neoplásicas, inclusive o
primeiro trimestre da gravidez, fase folicular do ciclo menstrual, endometrioses,
miomas uterinos, salpingites agudas, tuberculose pélvico-peritoneal, cirrose
hepática, pancreatites e inflamações do peritoneu, do pericárdio e da pleura.
Outros carcinomas não-ováricos podem elevar os níveis de CA 125 como os de
endométrio, pâncreas, pulmão, mama e gastrointestinais.
Sómente cerca de 3% das mulheres saudáveis têm concentração sérica de CA 125
acima de 35 U/mL, e apenas 0,8%, acima de 65 U/mL.
Os níveis séricos correlacionam-se com o tamanho e o estadiamento do tumor. Em
pacientes com carcinoma limitado ao ovário (estadio I), apenas 50% se
apresentam positivas. Já 90% dos pacientes com carcinoma de ovário disseminado
(estadios II, III e IV) apresentam concentrações séricas acima de 35 U/mL.
A combinação da avaliação do CA 125 com o exame pélvico e a avaliação por ultrasom transvaginal aumenta a sensibilidade e a especificidade do exame como teste
de triagem para o carcinoma do ovário.
É útil também no diagnóstico diferencial de massas do ovário palpáveis. Quanto
mais altos os níveis de CA 125, maior a probabilidade da sua associação ao
carcinoma do foro ginecológico.
O CA 125 é usado no acompanhamento dos carcinomas do ovário, na avaliação de
lesões residuais, monitorização de recidivas e resposta à terapia. Na avaliação do
aparecimento de metástases, tem a capacidade de detectar 75% dos casos. É útil
também como marcador prognóstico. Um resultado negativo não exclui a presença
da doença.
Os valores diminuem em torno de 3 semanas após a conduta terapêutica. Os níveis
de CA 125 após a primeira etapa de quimioterapia ajudam a formulação do
prognóstico. Pacientes com diminuição acima de 50% nas concentrações de CA 125
têm sobrevida de cerca de 2 anos em 45% dos casos, comparada com a previsão
de 22% nos pacientes que apresentam diminuição do CA 125 inferior a 50%.
Elevações acima de 60 U/mL, após o primeiro ciclo de quimioterapia, sugerem
reincidência do tumor.
CA 15-3
O antigénio CA 15-3 é uma glicoproteína presente no epitélio mamário, detectada
em análises imunoenzimáticas utilizando-se anticorpos monoclonais específicos.
Elevações das concentrações séricas do CA 15-3 podem ser encontradas em creca
de 23% dos pacientes com carcinoma primário da mama e em cerca de 40 a 50%
dos pacientes com metástases. Apenas 23% dos pacientes com doença precoce
apresentam elevação da concentração do CA 15-3, e cerca de 16% dos pacientes
com doença benigna da mama apresentam valores alterados.
A sua utilização está indicada no acompanhamento da recorrência do carcinoma da
mama, monitorização de metástases e também para monitorização da resposta à
terapia.
Os valores do CA 15-3 correlacionam-se com a extensão e o estágio da lesão.
Entretanto, na prática, essa correlação é melhor observada na progressão do que
na regressão. São encontrados percentadens de positividade com valores acima de
25 U/mL em 5% dos pacientes no estadio I, 29% no estadio II, 32% no estadio III
e 95% no estadio IV.
As pacientes que desenvolvem doença metastática têm aumento de CA 15-3, que
pode preceder de 2 a 9 meses os sinais clínicos. As probabilidades de progressão
são respectivamente 52%, 85% e 96% para 1, 3 e 6 meses após um teste positivo.
A probabilidade de não-progressão após um teste negativo é de 91%. O CA 15-3 é
mais sensível do que o CEA para monitorização de recidiva, e os dois marcadores
devem ser utilizados em conjunto.
Um aumento de 25% nos níveis séricos do CA 15-3 indica progressão do carcinoma
em 95% dos pacientes. Uma diminuição de 25% dos níveis séricos do CA 15-3 está
associada a resposta à terapia. Variações menores do que 25% são,
frequentemente, associadas a doença estável. Uma elevação nos níveis de CA 15-3
pode acontecer nas primeiras semanas de tratamento e não deve ser confundida
com falha terapêutica.
Valores de CA 15-3 elevados correlacionam-se com a presença de metástase óssea,
mas valores normais não excluem a possibilidade de sua existência. Utilizando-se
como limite (cut-off) 26 U/mL, o CA 15-3 prediz recidivas em menor período de
tempo do que os métodos radiológicos e os critérios clínicos.
Como a maioria dos marcadores tumorais disponíveis, o CA 15-3 não pode ser
utilizado como teste de rastreio na população em geral. Pode estar presente
noutras patologias malignas do pâncreas, pulmão, ovário e fígado. Valores
alterados podem ser encontrados numa pequena parcela da população hígida (2 a
5%), geralmente em concentrações baixas, bem próximas aos valores superiores
de referência. Pacientes com condições benignas como hepatites crónicas, cirrose
hepática, sarcoidose, tuberculose e lúpus eritematoso sistêmico podem apresentar
concentrações séricas acima de 40 U/mL.
CA 19-9
O CA19-9, assim como o CA 242 e o CA 50, são marcadores tumorais de carcinoma
colorrectal e pancreático. Estes três marcadores possuem um epítopo em comum,
que os caracteriza como marcadores de carcinoma gastro-intestinal.
O CA 19-9 (antígeno carboidrato 19-9) é uma glicoproteína do tipo mucina, de alto
peso molecular (> 400 kD), que possui uma estrutura siálica originada do antigénio
do grupo sanguíneo Lewis. Consequentemente, não se expressa nos indivíduos Le
(a-, b-), levando a resultados falso-negativos em pessoas com esse perfil,
aproximadamente 6% da população.
É sintetizado normalmente pelas células dos ductos do pâncreas e da vesícula biliar
humana e pelo epitélio gástrico, do cólon, endometrial e salivar.
A concentração sérica do CA 19-9 encontra-se elevada em pacientes com
carcinoma de pâncreas (80%), carcinoma hepatocelular (67%), carcinoma gástrico
(40 a 50%) colorretal (30%) e alguns pacientes com carcinoma de mama (15%).
Condições benignas como pancreatite e icterícia podem levar ao aumento dos níveis
séricos (18%). A maioria dos pacientes com pancreatite tem valores séricos abaixo
de 75 U/mL (96%). Os níveis séricos podem apresentar-se alterados também em
pacientes com doenças como cirrose hepática, doenças intestinais inflamatórias e
em condições auto-imunes como artrite reumatóide (33%), lúpus eritematoso
sistémico (32%) e esclerodermia (33%).
A baixa sensibilidade e especificidade e a falta de opções de tratamento impedem o
uso de CA 19-9 como teste de triagem para carcinoma do pâncreas. Os níveis do
CA 19-9 são úteis para avaliar a recidiva do tumor e a presença de metástases. A
principal indicação para a utilização do CA 19-9 está na preparação para a conduta
cirúrgica.
Basicamente, todos os pacientes que apresentam valores séricos maiores do que
1.000 U/mL têm um tumor maior que 5 centímetros de diâmetro, e apenas 5% dos
pacientes desse grupo apresentam tumores ressecáveis. Porém, 50% dos pacientes
pré-tratados com CA 19-9 abaixo de 1.000 U/mL apresentam tumores ressecáveis.
Outros estudos têm mostrado que a maioria dos pacientes com concentração maior
do que 300 U/mL apresenta tumores irressecáveis.
Apesar da sensibilidade diagnóstica do CA 19-9 para tumores do pâncreas ser
maior do que a de outros marcadores, o diagnóstico precoce realizado por meio do
seu uso tem sido bem limitado. Ou seja, a capacidade diagnóstica em casos iniciais
é muito baixa. O CA-19-9 é utilizado na monitorização de carcinomas
gastrointestinais, especialmente do pâncreas. É descrito como menos eficiente do
que o CEA como marcador de carcinomas colorretais.
O doseamento do CA 242 está indicada no acompanhamento de tumores
pancreáticos e colorretais. Valores elevados de CA 242 são encontrados em 5 a
33% dos pacientes com doença benigna do cólon, estômago, fígado, pâncreas e
vesícula biliar; 68 a 79% de pacientes com carcinoma do pâncreas; 55 a 85% de
pacientes com carcinoma colorretal e 44% dos pacientes com carcinoma gástrico. O
CA 242 é supostamente menos eficiente do que o CA 19-9 e o CA 50 na detecção
do carcinoma pancreático.
Níveis elevados de CA 50 são encontrados em até 46% das doenças benignas do
pâncreas, em 35 a 38% das patologias benignas do trato biliar e em 22 a 59% das
patologias do fígado. Apresenta-se elevado entre 80 a 97% dos pacientes com
carcinoma do pâncreas. No carcinoma do cólon, a frequência varia de 19% nos
estadios mais iniciais até 73% nos estadios mais avançados.
Este marcador encontra-se elevado no carcinoma de esófago (41 a 71%),
estômago (41 a 78%), trato biliar (58 a 70%) e hepatocelular (14 a 78%).
Cálcio
No adulto, cerca de 98% do cálcio está localizado nos ossos, principalmente sob a
forma de hidroxiapatita, uma rede composta por cálcio e fósforo. O restante, cerca
de 2%, encontra-se no fluído extracelular e noutros tecidos, principalmente no
músculo esquelético.
O ião cálcio está entre os principais componentes minerais do organismo e
desempenha um papel fundamental na mineralização óssea. Tem importância vital
em vários processos fisiológicos, como o da coagulação sanguínea, a transmissão
dos impulsos nervosos, a manutenção do mecanismo de contracção e relaxamento
muscular esquelético e cardíaco, as activações enzimáticas, a regulação das
glândulas endócrinas e exócrinas e a manutenção da integridade e da
permeabilidade da membrana celular, principalmente em relação ao mecanismo de
troca sódio /potássio.
É absorvido de forma activa no duodeno e no jejuno superior. Cerca de 10 a 20%
do cálcio consumido é absorvido. A absorção é estimulada principalmente pela
vitamina D (estimula a absorção intestinal e a mineralização óssea), pelo meio
ácido, pela hormona de crescimento e por uma dieta rica em proteínas. A relação
cálcio/fósforo também é um factor importante na regulação da absorção.
Coeficientes elevados levam à formação de complexos de fosfato de cálcio
insolúveis. O pH alcalino, o teor de gordura do bolo alimentar e o cortisol inibem a
absorção.
A manutenção do equilíbrio dos níveis de cálcio no organismo envolve diferentes
órgãos: intestino delgado, rins e esqueleto. Os seus níveis são regulados por
diferentes hormonas: pela paratormona (que aumenta a reabsorção tubular e a
mobilização do cálcio ósseo), por hormonas derivados do metabolismo da vitamina
D3 e pela calcitonina (que actuam sobre a reabsorção óssea e nos túbulos renais).
Outras hormonas que afectam o metabolismo do cálcio, mas que não têm a sua
secreção alterada pelas alterações das concentracções do ião, são as hormonas
tireoidéias, do crescimento, glicocorticóides adrenais e esteróides.
Basicamente, os valores do cálcio dependem das taxas de absorção intestinal,
reabsorção óssea e perda renal. Concentrações anormais do cálcio ósseo indicam
distúrbios na função das glândulas paratiróides, doenças ósseas, neoplasias,
síndrome de má absorção ou desnutrição, deficiência de vitamina D, excesso de
antiácidos contendo cálcio e doenças renais. Actualmente, grande relevo tem sido
dado à complementação de cálcio, especialmente nas mulheres após a menopausa,
como meio de retardar ou evitar as manifestações de osteoporose.
A excreção do cálcio dá-se principalmente por via urinária, e uma pequena parcela
pela transpiração. A determinação do cálcio urinário é útil para o acompanhamento
das terapias de reposição, nas investigações de litíase renal e no seguimento de
patologias que envolvem o metabolismo do cálcio, como nas doenças ósseas
primárias e metastáticas, intoxicação por vitamina D, hipercalciúrias idiopáticas,
sarcoidoise e doenças da paratiróide.
O cálcio encontra-se no soro ou no plasma de três formas diferentes:
1. A forma fisiologicamente activa de cálcio livre e ionizado, que corresponde a
50% do cálcio total (difusível).
2. Em forma de complexos com outros iões, que correspondem a 5% do cálcio total
(difusível).
3. Os 45% restantes, que se apresentam ligados a proteínas plasmáticas,
especialmente à albumina (não-difusível).
A distribuição dessas formas está directamente ligada ao pH do fluido extracelular e
à concentração de proteínas. A acidose aumenta o cálcio ionizado, e a alcalose
diminui a sua concentração. Já o aumento de proteínas leva a um aumento do
cálcio ligado e também do cálcio total. O cálcio ionizado mantém os seus níveis
fisiológicos, ou seja, a sua concentração é constante e independente do cálcio
ligado.
Portanto, a avaliação do cálcio ionizado é útil nalgumas situações clínicas, como
distúrbios ácido-base, nos quais o cálcio ionizado está alterado sem que haja
alteração do cálcio total. Esta situação pode ocorrer na hemodiálise, nas
septicémias, mielomas, cirrose, transfusões, plasmaférese, distúrbios
cardiovasculares, insuficiência renal e no diagnóstico do hiperparatiróidismo.
Consultar Cálcio Ionizado.
Cálcio Ionizado
O cálcio é o quinto elemento mais comum no corpo humano. A distribuição desse
elemento no corpo abrange três compartimentos: esqueleto (98%), tecidos moles e
sangue (2%).
No plasma, o cálcio está presente de três formas distintas: ligado à proteína, livre
ou ionizado e formando complexos (principalmente com fosfato, bicarbonato e
citrato). Aproximadamente 40% do cálcio plasmático está ligado de forma
reversível às proteínas, e qualquer alteração na quantidade de proteína que se liga
ao cálcio altera a quantidade de cálcio total.
Cerca de 20% do cálcio ligado à proteína está ligado também à globulina. Em
pacientes com mieloma múltiplo, a globulina em alta concentração pode ligar-se a
uma grande quantidade de cálcio, provocando um aumento da quantidade de cálcio
total. A quantidade de cálcio ionizado presente no plasma é de 50% do cálcio total.
Apenas 10% do cálcio formam complexo com aniões.
Além da concentração de proteína, o balanço ácido-base também pode afetar a
fração ionizada. A alcalose causa redução na fracção de cálcio iónico circulante, e a
acidose, aumento. Num estado de acidose crónica, por exemplo, os níveis séricos
normais de cálcio total podem ocultar um aumento na concentração de cálcio
ionizado. Portanto, os níveis séricos normais de cálcio ionizado são importantes na
manutenção do metabolismo dependente de cálcio.
O nível sérico de cálcio ionizado controla, por exemplo, a secreção de hormonas das
paratiróides. Níveis baixos de cálcio ionizado estimulam a secreção de hormonas da
paratiróide, e os níveis altos suprimem essa secreção.
Finalmente, a determinação de cálcio ionizado é um indicador melhor do estado de
cálcio plasmático do que o cálcio total, por ser metabolicamente activo e melhor
regulado pelas hormonas reguladoras de cálcio
Cálculo Renal
A análise físico-química do cálculo renal é de grande importância clínica na
orientação preventiva da calculose. São avaliados os aspectos macroscópicos, como
tamanho, cor, forma, consistência, além da análise bioquímica, na qual são
identificados os elementos presentes. Os elementos frequentemente encontrados
são oxalato de cálcio, ácido úrico, fosfato de magnésio e, mais raramente, cistina.
TIPO DE CÁLCULO
ÁCIDO ÚRICO
CISTINA
FOSFATO de MAGNÉSIO
OXALATO
OXALATO OU FOSFAFTO DE CALCIO
CONDIÇÃO
Hiperglicemia, Hiperparatiroidismo, Hipertiroidismo,
Acidose tubular renal, etiologia desconhecida (comum),
intoxicação por vitamina D
Deficiência tubular renal
Infecção por Proteus ssp e outras infecções das vias
urinárias
Anestesia com metaxiflurano, deficiência de vitamina B6
Gota, distúrbios linfoprolifarativos
Capacidade Total de Fixação de Ferro
A capacidade total de fixação do ferro sérico ou TIBC (Total Iron-Binding Capacity)
representa uma estimativa aproximada de todas as proteínas transportadoras de
ferro, em especial da transferrina. O teste consiste na adição de ferro em excesso
ao soro, tentando a saturação das proteínas. A seguir, todo ferro excedente, nãoligado às proteínas é retirado, e logo após o ferro sérico é avaliado.
Como a transferrina não é a única proteína fixadora de ferro, a avaliação da
capacidade de fixação do ferro não representa exactamente a capacidade de
fixação da transferrina. Entretanto, por ser a transferrina a maior proteína
transportadora do ferro, a avaliação da capacidade total de fixação do ferro no soro
representa de forma significativa a sua capacidade de ligação ao ferro.
A anemia por deficiência de ferro é caracterizada por queda na concentração de
ferro, aumento do TIBC e diminuição da saturação de transferrina. O TIBC do soro
está aumentado na deficiência de ferro e diminuído na anemia por doenças
crónicas.
A redução da capacidade de fixação está associada à diminuição dos níveis séricos
da transferrina. O inverso também acontece: níveis elevados de transferrina são
seguidos pela elevação da capacidade de fixação do ferro. Na hepatite, ocorre um
aumento da libertação da transferrina pela necrose do hepatócito, levando a um
aumento da capacidade de combinação do ferro.
CAPACIDADE TOTAL DE FIXAÇÃO DO FERRO
AUMENTADA
DIMINUÍDA
Anemia ferropriva
Síntese protéica diminuída
Período da Infância
Desnutrição grave
Gravidez
Neoplasias
Hepatites e Insuficiência hepática
Hemocromatose
Anticontrceptivos orais
Nefropatias
Carbamazepina
É um fármaco antiepiléptico importante, utilizado no tratamento de crises
convulsivas simples e complexas, parciais e generalizadas no adulto, e também no
tratamento da nevralgia do trigémio, nevralgia glossofaringea e outras nevralgias
como as neuropatias diabéticas.
A carbamazepina é um composto tricíclico relacionado quimicamente com a
imipramina (antidepressivo tricíclico). A acção antiepiléptica é diminuir o fluxo de
sódio e de cálcio nos neurónios hiperexcitáveis. Uma redução da transmissão
sináptica excitatória explicaria a sua acção sobre as nevralgias.
A doseamento é importante para avaliar a eficácia do tratamento e impedir os
efeitos tóxicos por meio da manutenção dos níveis séricos do range terapêutico.
A concentração sérica terapêutica usual é de 4 a 12 mg/mL. Normalmente, alcançase um estado de estabilidade em 3 a 4 dias. Após a administração oral, a absorção
é lenta e apresenta grandes variações individuais, atingindo o pico sérico em horas.
A metabolização faz-se por via hepática, apresentando uma farmacocinética
própria, com a capacidade de induzir as enzimas hepáticas responsáveis pela sua
clearance (auto-indução).
A clearance da carbamazepina aumenta com o tempo, e as enzimas estão
completamente induzidas em cerca de 4 a 6 semanas, ocorrendo então o aumento
da clearance e a consequente diminuição da semi-vida do fármaco, que cai de 25 a
40 horas para 15 a 25 horas, depois da auto-indução. Como o metabolismo é
hepático, qualquer alteração da função hepática leva ao aumento da concentração
sérica do fármaco.
Cerca de 85% da carbamazepina está ligada às proteínas. A carbamazepina
apresenta um metabolito activo que é potencialmente tóxico - a carbamazepina 10,11 epóxido, que deve ser avaliado nos pacientes com sinais de intoxicação que
apresentem níveis séricos normais da carbamazepina.
Há a possibilidade de ocorrência de efeitos colaterais, como nistagmo, cefaléia,
ataxia, visão dupla e, menos frequentemente, rash (erupção) cutâneo. Podem
também ocorrer reacções hematológicas, sendo a mais comum a leucopenia. Em
casos raros, regista-se a ocorrência de formas graves, como anemia aplástica,
trombocitopenia e agranulocitose. Também podem surgir reacções de
hipersensibilidade, incluindo síndrome de Steven Johnson, hiponatremia,
especialmente em idosos, osteomalácia e efeitos sobre a condução cardíaca.
O uso em mulheres no primeiro mês de gestação aumenta o risco de defeitos na
formação do tubo neural.
A carbamazepina aumenta o metabolismo do ácido valpróico, clonazepam, teofilina
e warfarina. A sua concentração pode ser aumentada pelo uso concomitante de
cimetidina, isoniazida, eritromicina, lítio, fluoxetina e ácido valpróico. Possui ainda
um efeito antidiurético, reduzindo as concentrações da hormona antidiurética.
Podem ser encontrados frequentemente níveis baixos, por falta de adesão ao
tratamento. O uso de outras drogas anticonvulsivantes (politerapia) como
fenobarbital, fenitoína e primidona pode diminuir a concentração sérica da
carbamazepina sem levar a crises convulsivas.
Normalmente, a colheita deve ser realizada em pacientes que estejam a fazer o
medicamento há pelo menos 2 dias, e sempre cerca de 1 hora antes da próxima
dose. Entretanto, nos casos de suspeita de intoxicação, pode ser realizada em
qualquer momento. Para facilitar a interpretação, é importante conhecer o horário
da última dose ingerida.
Ceruloplasmina
A ceruloplasmina é uma glicoproteína produzida pelo fígado, responsável pelo
transporte de 80 a 95% do cobre plasmático. É uma proteína de resposta de fase
aguda, migrando na região alfa-2-globulina na electroforese das proteínas.
Normalmente não é visível, mas apenas nas situações que levam a grandes
elevações séricas.
A sua principal utilização clínica é no diagnóstico diferencial entre várias doenças
hepáticas e na doença de Wilson, que é classicamente acompanhada pela
diminuição dos níveis séricos da ceruloplasmina. É uma doença autossómica
recessiva, em que ocorre diminuição da capacidade de ligação do cobre à
ceruloplasmina, resultando no aumento dos níveis de cobre livre no plasma e nos
tecidos, especialmente fígado e cérebro.
Outras patologias que levam a lesão hepática grave, desnutrição, síndrome
nefrótica e enteropatias com perda de proteínas podem cursar com níveis
diminuídos de ceruloplasmina.
Níveis excessivos de zinco na dieta podem levar a um bloqueio da absorção
intestinal de cobre, levando inicialmente a uma diminuição dos níveis de
ceruloplasmina. O período necessário à eliminação do zinco supera o bloqueio à
absorção do cobre, seguindo-se a um aumento do cobre sérico e da ceruloplasmina.
Apresenta-se elevada em diferentes distúrbios que levam à reacção da fase aguda,
como: carcinomas, leucemias, doença de Hodgkin, traumas, obstrução biliar,
gravidez, intoxicação por cobre, uso de contraceptivos orais, fenitoína e terapia
com estrogénios.
Chlamydia
O género Chlamydia compreende bactérias Gram-negativas desprovidas de
motilidade, que se classificam nas espécies trachomatis, pneumoniae e psitacci.
Incapazes de sintetizar o seu próprio DNA, são parasitas intracelulares obrigatórios
de células eucarióticas, completando o seu ciclo de multiplicação em 48 a 72 horas.
As espécies trachomatis e pneumoniae são reconhecidas como patogénios
humanos, e a psitacci é reconhecida primariamente como patogénio animal. A
psitacci infecta aves, bovinos e ovinos e pode desencadear em humanos uma
doença respiratória por exposição ao material infeccioso (geralmente fezes de aves
infectadas).
A Chlamydia trachomatis possui 20 sorotipos diferentes já identificados. Os
sorotipos L1,L2e L3 são responsáveis pelo síndrome linfogranulomatoso venéreo;
os A, B, Ba e C são frequentemente associados ao tracoma, e os de D a K estão
ligados a outras manifestações sexualmente transmitidas, sendo que os sorotipos
D, E e F são os mais frequentes.
A infecção por C. trachomatis pode perdurar por muitos anos de forma
assintomática nos indivíduos infectados não-tratados. As infecções causadas por
Chlamydia trachomatis incluem:
o Linfogranuloma venéreo - doença sistémica, sexualmente transmissível, causada
por determinados serotipos da espécie;
- o Tracoma - traduz-se por uma conjuntivite crónica que pode levar à cegueira. O
contágio ocorre por transmissão directa, pessoa a pessoa;
- as Infecções do trato genital - incluem cervicite, doenças inflamatórias pélvicas,
uretrites, epididimite e proctite. Nos homens, cerca de 50% dos casos de uretrite
não-gonocócica são causadas por Chlamydia trachomatis. Nas mulheres, 30% dos
casos manifestam-se sob a forma de cervicite; porém, as infecções assintomáticas
ocorrem em mais da metade dos casos e, em consequência, muitas não são
detectadas;
as Infecções neonatais - 60% das crianças nascidas de mães infectadas poderão
contaminar-se no momento do parto, e 18 a 50% podem desenvolver a doença,
nos primeiros dias de vida, uma conjuntivite; posteriormente, num período médio
de 3 meses, em 3 a 15% poderá ocorrer uma pneumonia associada ou não à
conjuntivite, com alteração do estado geral da criança.
Cerca de 30% das pneumopatias em recém-nascidos são causadas por Chlamydia
trachomatis.
Nos homens, a C. Trachomatis é responsável por cerca de 50% dos casos de
uretrite não-gonocócica e ainda por maior percentagem de uretrites pósgonocócicas. É a principal causa de epididimite em homens sexualmente activos
acima dos 35 anos de idade. Nos casos assintomáticos, pode ser diagnosticada ao
encontrar-se piúria ou queixa de disúria sem causa aparente.
A infecção uretral é comum nas mulheres. Casos de piúrias sintomáticas ou
assintomáticas sem causa aparente ou de cistites com cultura negativa são
altamente sugestivos de uretrite por Chlamydia trachomatis. O endométrio é o
órgão mais frequentemente infectado, e a maioria dos casos é assintomática.
Além de causar uretrites, cervicites, endometrites e doença inflamatória pélvica nas
mulheres, a infecção pode evoluir com complicações. As mais frequentes são a
infertilidade (fibrose das trompas de Falópio), a gestação ectópica e, menos
frequentemente, o chamado síndrome de Fitz-Hugh-Curtis (peri-hepatite). Estudos
realizados entre estas pacientes com quadros de infertilidade por lesões tubáricas e
gravidez ectópica demonstraram positividade três vezes maior para os anticorpos
contra a C. trachomatis do que na população normal.
As infecções do trato genital por Chlamydia trachomatis são consideradas as
principais causas das doenças sexualmente transmissíveis na Europa e nos Estados
Unidos, onde cerca de 3 a 4 milhões de casos são relatados por ano. Os índices de
transmissão do agente patogénico, são superiores aos da gonorréia e da sífilis, e a
sua ampla distribuição geográfica são preocupantes, justificando o uso de métodos
diagnósticos sensíveis e rápidos para a sua identificação.
Vários métodos estão disponíveis para a detecção de Chlamydia trachomatis em
amostras de material biológico: coloração histológica, cultura de células,
imunofluorescência e métodos imunoenzimáticos. Todos apresentam, porém,
restrições de sensibilidade ou especificidade, ou exigem um tempo prolongado para
a conclusão do diagnóstico.
Até alguns anos, por apresentar alta especificidade, a cultura de células Maconkey
era considerada o teste padrão para o diagnóstico da infecção por Chlamydia
trachomatis. Apesar de sua alta especificidade, a cultura apresenta algumas
restrições, como o longo tempo para obtenção do resultado e o facto de detectar
apenas bactérias vivas. Portanto, o resultado pode ser prejudicado por situações
inadequadas de colheita, armazenamento e/ou transporte, o que diminui a sua
sensibilidade em cerca de 70 a 80%.
Outra técnica utilizada para a detecção de Chlamydia trachomatis era a
imunofluorescência directa por anticorpos monoclonais específicos. Estsa técnica
apresenta uma sensibilidade maior do que a cultura, tendo entretanto a
desvantagem de apresentar reacções cruzadas, o que diminui a sua especificidade.
Os métodos imunoenzimáticos ou de imunofluorescência indirecta para a pesquisa
de anticorpos séricos, respectivamente, das classes IgG e IgM são menos sensíveis
e menos específicos do que os métodos citados acima, além do facto de não
permitirem a identificação das diferentes espécies do género Chlamydia nem serem
capazes de detectar anticorpos anti-Chlamydia pneumoniae.
A reacção em cadeia da polimerase (PCR) é um método de especificidade e
sensibilidade altas, que utiliza a amplificação do ácido nucléico da Chlamydia (DNAalvo), permitindo a sua detecção mesmo em concentrações ínfimas, da ordem de 1
a 10 microrganismos por mililitro de material biológico.
Técnicas de PCR e de captura híbrida são os métodos de escolha para o diagnóstico
da infecção por C. trachomatis. Podem ser realizadas em amostras endocervicais e
uretrais colhidas com swabs, acondicionadas em tubos apropriados e também em
amostras de urina (primeiro jacto).
Devemos porém ressaltar que a metodologia de reacção em cadeia da polimerase
pode se apresentar inconclusiva, especialmente na pesquisa em materiais
endocervicais e uretrais. Isso ocorre em cerca de 5% dos casos analisados por PCR,
pela presença de factores inibidores que interferem na reacção, inibindo a
actividade enzimática. Ainda não se tem a certeza da origem desses inibidores.
Acredita-se que se devam à presença de contaminantes bacterianos ou fúngicos.
Citomegalovírus
O citomegalovírus (CMV) é membro do grupo dos beta-herpesvírus. Hoje, é
reconhecido como um patogénio que afecta todas as faixas etárias, tendo caráter
endémico em todo o mundo e levando a graves lesões congénitas. A infecção pelo
CMV causa um grande espectro de manifestações clínicas, variando de evoluções
assintomáticas e infecções sublínicas a quadros de maior gravidade, dependendo da
condição imunológica do paciente.
Quando sintomática em pacientes imunocompetentes, geralmente evolui com um
quadro semelhante ao de mononucleose, com febre, linfadenopatia, alterações
hematológicas (leucopenia e trombocitopenia) e, muitas vezes, com sinais e
sintomas hepáticos, pulmonares, gastrointestinais ou neurológicos.
A transmissão pode dar-se por via transplacentária, respiratória, oral, venérea, por
intermédio do aleitamento, de transplante de órgãos ou transfusão sanguínea. O
antigénio pode ser detectado a partir da 4ª semana que se segue à infecção
(período de incubação). Na fase aguda, o vírus pode ser identificado em diferentes
secreções corporais. Os anticorpos da classe IgM aparecem logo no início da fase
aguda, e os da classe IgG, uma semana mais tarde.
Assim como no herpes simples e nos demais vírus do grupo herpes, a infecção pode
permanecer latente por toda a vida ou evoluir com episódios de reactivação. Depois
da primoinfecção, o vírus mantém-se de forma latente, com a virémia persistente
porém em níveis baixos. Os anticorpos desenvolvidos mantêm-se positivos por toda
a vida (IgG). Em situações que levem à diminuição da condição imunológica, pode
ocorrer a replicação viral com reactivação do quadro.
A infecção pelo CMV apresenta maior significado clínico nas mulheres grávidas, em
recém-nascidos (infecção congénita), nos imunossuprimidos, como os casos de
pacientes transplantados, portadores de neoplasias, pós-operatório de cirurgia
cardíaca, em curso de grandes agressões infecciosas e nos indivíduos com SIDA.
Normalmente, em indivíduos saudáveis, há dificuldade de correlacionar a infecção
por CMV ao episódio clínico da doença, pois a presença de anticorpos (IgG)
específicos para CMV tem uma prevalência muito alta (>60% da população de
adulto), e como já citado, as manifestações clínicas da infecção são muito variadas.
Embora o isolamento do vírus seja ainda considerado o melhor, outras técnicas
estão a ser desenvolvidas para a avaliação da virémia, que por ser intermitente,
exige exame de amostras consecutivas para permitir um diagnóstico mais seguro.
A colheita deve ser comparada entre amostra da fase aguda e da fase de
convalescença. Isto quer dizer que devem ser colhidas com 10 a 14 dias de
intervalo. A presença de anticorpos heterófilos e de factor reumatóide pode levar a
resultados falso-positivos dos anticorpos IgM. Nos casos neonatais, a transferência
transplacentária pode também induzir a falsa positividade.
As pesquisas geralmente baseiam-se na presença de CMV (particularmente das
inclusões virais). Aumentos significativos dos anticorpos IgG CMV específicos por
IFI ou EIA sugerem, mas não comprovam, infecção aguda ou reactivação de uma
infecção por CMV.
Anticorpos de baixa avidez fazem a distinção entre a resposta imune primária e a
reactivação da infecção por CMV (caracterizada por alta avidez de IgG). O teste de
avidez de anticorpos é um procedimento laboratorial que permite estimar o período
aproximado em que ocorreu a infecção.Percentagens de avidez inferiores a 30%
sugerem que a infecção ocorreu há menos de 2 meses. Percentagens superiores a
40% sugerem que a infecção ocorreu há mais de 3 meses e que os anticorpos IgM,
caso presentes, são residuais e desprovidos de significado clínico.O achado de
percentagens entre 30% e 40% é considerado indeterminado, já que não permite a
definição do período provável da infecção. Quando a avaliação está a ser realizada
em grávidas, o tempo de gestação deve ser considerado.
Anticorpos IgM específicos para CMV podem ser detectados em adultos por Enzima
imunoensaio (EIA), em ambas as infecções de CMV nas infecções primárias, em
cerca de 93 a 100% dos casos, e nas infecções reactivadas, em aproximadamente
40% dos casos. Porém, a resposta de IgM pode estar reduzida ou ausente em
pacientes imunocomprometidos com infecção activa. A maioria dos pacientes com
SIDA (95%) já é seropositiva para CMV antes da infecção pelo HIV ser
diagnosticada. Manifestações do sistema nervoso central e periférico causadas pela
infecção por CMV são muito raras. Entretanto, nos casos de SIDA, comumente são
diagnosticadas encefalites por CMV. Os índices dos anticorpos CMV podem ser
usados para diferenciar síntese intratectal da infiltração da barreira
hematoencefálica pelo CMV.
Os métodos mais rápidos, sensíveis e específicos para diagnóstico de CMV são os
de biologia molecular, a reacção em cadeia da polimerase (PCR) e a captura
híbrida, especialmente em recém-nascidos infectados congenitamente, em
amostras de medula óssea, análise de órgão sólidos para transplante, pacientes
imunocomprometidos, indivíduos imunocompetentes com infecção activa e dadores
de sangue. A evolução da PCR quantitativa para CMV tem monstrado que CMV DNA
em líquor é mais elevado em pacientes com CMV relacionado à polirradiculopatia do
que nos que sofrem de encefalites, e que a quantificação do CMV pode ser útil na
monitorização da terapia antiviral.
Sendo assim, os métodos moleculares para a detecção do vírus são importantes
aliados para identificar os pacientes com alto risco de desenvolvimento da doença.
Neste sentido, a qualificação da carga viral para o CMV pela técnica de reações em
cadeia de polimerase ou pela captura híbrida permite a definição da virémia e a
monitorização da terapêutica.
- Microscopia eletrónica do Citomegalovírus
CK Total
A creatinoquinase (CK), também chamada de creatina-fosfoquinase (CPK), é uma
enzima com vasta distribuição tissular, que desempenha importante papel
regulador no metabolismo intracelular dos tecidos contrácteis. Está presente
principalmente no músculo estriado, no tecido cardíaco e no cérebro.
Na electroforese, podem ser identificadas três isoenzimas, pela sua origem, e
também de forma numérica, de acordo com a migração na eletroforese. A CK-BB CK-1 - é a forma encontrada no cérebro; a CK-MB - CK-2 - é encontrada no
miocárdio e a CK-MM - CK-3 - é encontrada no músculo estriado. A CK-MM está
habitualmente presente no soro.
AUMENTO
MÚSCULO ESQUELÉTICO
MIOCÁRIO
CÉREBRO
ESTÔMAGO, ÍLIO E CÓLON
CK - BB%
CK - MB%
CK - MM%
0
1
97-98
96
1
22
2a3
0
99
77
0
4
Também podem ser encontradas outras formas, ditas isoenzimas variantes. Não
estão habitualmente presentes em indivíduos hígidos e não possuem tecido de
origem determinada. As duas maiores variantes conhecidas são denominadas
macro-CK, tipos 1 e 2. A macro-CK do tipo 1 é formada pela CK-BB, ou raramente
pela CK-MB, que se liga a uma imunoglobulina G ou A (raramente) dando origem a
macrocomplexos que correm electroforeticamente entre CK1 e CK2. É comum a sua
presença em idosos, especialmente em mulheres. A do tipo 2 parece ser um
complexo da CK mitocondrial, presente em pacientes que apresentam um quadro
de metástases tumorais ou outras enfermidades de alta gravidade. Apesar de não
serem doseadas, pois não existem ainda evidências de sua importância clínica, a
sua presença poderá, em alguns casos, afectar a análise da CK-MB, interferindo no
resultado final.
A sua maior utilização está no diagnóstico das lesões e doenças do músculo
esquelético e no enfarte agudo do miocárdio.
Encontra-se marcadamente elevada na distrofia muscular de Duchenne, com
elevações que variam de 20 a 200 vezes o limite superior da normalidade, exercício
físico intenso, polimiosite, dermatomiosites, miosites, miocardites, traumas
musculares, injeções intramusculares recentes e após crises convulsivas. Valores
muito elevados são encontrados também nas rabdomiólises, inclusive nas que têm
como causa a intoxicação por uso de cocaína.
Também pode encontrar-se elevada noutras situações, como acidente vascular
cerebral, embolia, enfarte e edema pulmonar, após cardioversão com múltiplos
choques, tosse grave, trabalho de parto, quadros de mixedema - hipotiroidismo,
nas neoplasias da mama, próstata e trato gastrointestinal, e noutras neoplasias em
estado avançado, no período pós-operatório imediato e na ingestão de grandes
quantidades de bebidas alcóolicas.
Os seus níveis séricos podem estar diminuídos em situações nas quais ocorra perda
de massa muscular, nas hepatopatias alcoólicas, na gravidez ectópica, nas doenças
do tecido conjuntivo, na artrite reumatóide, em pacientes idosos e acamados e na
terapia com esteróides. O repouso nocturno diminui os níveis séricos de CK em 10
a 20%.
No enfarte agudo do miocárdio (EAM), a CK total aumenta nas primeiras 4 a 6
horas após o início do quadro, apresentando um pico entre 18 a 24 horas e
permanecendo alterada por 48 a 72 horas após o episódio. A CK total pode estar
normal no período precoce pós-enfarte, quando a CK-MB já começa a elevar-se. Os
valores da CK podem aumentar entre 3 a 20 vezes os valores normais, dependendo
da localização e da extensão da área afectada.
Os valores de referência para a CK total é bastante ampla, variando com a idade,
estatura, actividade física e volume de massa muscular. No momento da
interpretação do resultado, isto deve ser levado em conta, pois alguns pacientes
podem ter, habitualmente, valores muito baixos, fazendo com que, nalguns casos,
mesmo estando dentro dos limites superiores de referência, já represente uma
elevação da CK total, na fase inicial do EAM.
CK-MB
Uma das isoenzimas da CK total, a CK-MB é considerada a análise de referência
para comparação com outros marcadores de lesão miocárdica. Para o seu
doseamento, utiliza-se um método no qual um anticorpo específico inibe a
isoenzima MM e a fração restante corresponde basicamente à MB. O seu
doseamento deve ser sempre acompanhado do doseamento de CK total. Para
avaliar melhor os resultados obtidos, pode-se avaliar o índice obtido pela divisão de
CK-MB/CK total x 100, que indica a percentagem de aumento da CK-MB em relação
à CK total.
Normalmente, a CK-MB representa 5 a 6% da CK total. Percentagens acima desses
valores são indicativos de origem miocárdica e associados a doença. Índices
superiores a 25% são raros, exigindo que se avaliem as interferências no método,
tais como a presença de CK 1 e 2.
É muito importante realizar a colheita de uma amostra de sangue logo no início dos
sintomas, quando os valores ainda estarão normais. Dessa maneira, servirão como
valores basais para estabelecer o ponto de partida da curva de acompanhamento.
The National Heart, Lung and Blood Institute recomenda a realização do
doseamento da CK total e da CK-MB em períodos de 6 a 8 horas, durante as
primeiras 24 horas após o episódio. Para estabelecer uma curva adequada de
acompanhamento, outros autores recomendam a colheita das amostras numa
sequência de 0-3-6-12 horas, seguida de doseamentos seriados cada período de 6
a 8 horas.
A evolução clássica da CK-MB durante a curva de dosagens seriadas é fundamental
para o diagnóstico de enfarte do miocárdio. O aumento inicial ocorre entre 3 a 8
horas após o início dos sintomas, atingindo o seu pico entre 12 a 24 horas e
declinando até a normalidade em 48 a 72 horas. Entretanto, o diagnóstico de
doença miocárdica deve-se pautar por um conjunto de achados clínicos, alterações
no electrocardiograma e dos marcadores bioquímicos cardíacos como CK, CK-MB,
troponina, mioglobina, entre outros. Cerca de 10 a 15% dos pacientes - geralmente
indivíduos com diminuição da massa muscular - evoluem com a CK-MB elevada
mas com a CK total normal.
Os valores encontrados no doseamento da CK-MB correlacionam-se com o tamanho
da área enfartada. Entretanto, apesar de altamente sensível, o doseamento é
incapaz de detectar pequenas áreas de necrose. Esta é a importância da realização
de curvas evolutivas de mais de um marcador bioquímico para auxiliar o
diagnóstico e o acompanhamento de possíveis complicações como o reenfarte.
Algumas outras lesões da musculatura cardíaca que não o enfarte do miocárdio
levam ao aumento dos níveis séricos da CK-MB, como as miocardites,
cardiomiopatias, cirurgia cardíaca de revascularização, troca de válvulas e
intervenções de defeitos congénitos. Outras condições que podem levar ao
aumento da CK MB incluem dermatomiosite, distrofia muscular de Duchenne e a
rabdomiólise.
Nestes casos, os valores costumam elevar-se menos e não se comportam em curva
crescente como no enfarte do miocárdio.
Clearance da Creatinina
A função renal normal depende da integridade de quatro aspectos da fisiologia
renal: o fluxo sanguíneo, a filtração glomerular, a função tubular e a
permeabilidade das vias urinárias. O rim tem como função básica a conservação de
fluídos, ou, por outras palavras, a concentração da urina. Além dessa função
básica, possui três importantes papéis, que são a depuração de substâncias tóxicas,
a manutenção do equilíbrio hidroeletrolítico interno e a produção de hormonas.
A função geral do rim pode ser avaliada pela clearance (depuração) renal de uma
determinada substância. Por definição teórica, a clearance é o volume de plasma a
partir do qual uma determinada substância pode ser totalmente depurada
(eliminada) na urina numa determinada unidade de tempo. Esse processo depende
da concentração sérica, da taxa de filtração glomerular e do fluxo plasmático renal.
É calculado a partir dos valores sérico e urinário da substância e do volume urinário
em 24 horas, sendo corrigido em relação à superfície corporal.
A clearance de moléculas pequenas não-ligadas à proteína que são filtradas
livremente pelos glomérulos, e não são secretadas ou reabsorvidas pelo sistema
tubular, como a inulina, fornece uma avaliação fiel da taxa de filtração glomerular.
Na prática, a clearance de creatinina é a escolhida para a avaliação da função renal.
A sua excreção não é influenciada pela dieta e é filtrada livremente pelos
glomérulos. Entretanto, além da secreção glomerular, há também uma secreção
tubular activa que é contrabalançada por um mecanismo de reabsorção tubular.
Assim, é possível utilizar a clearance de creatinina para avaliar adequadamente a
taxa de filtração glomerular, sendo um índice precoce da avaliação da função renal
superior à avaliação sérica isolada dos níveis de uréia e de creatinina. Além de
avaliar a função renal, são úteis também para o acompanhamento da evolução da
lesão renal e da resposta a terapêuticas.
A taxa de filtração glomerular é maior nos homens do que nas mulheres, já que os
homens possuem uma massa renal maior. Durante a gravidez, a taxa de filtração
aumenta em torno de 50%, retornando ao normal após o parto. Encontra-se
diminuída nos recém-nascidos e nas crianças até 5 meses de idade, por uma
imaturidade anatomofisiológica dos glomérulos. Os níveis da taxa de filtração
glomerular começam a diminuir progressivamente a partir da meia-idade, em
consequência da arterionefroesclerose, que leva à diminuição progressiva do
número de glomérulos. A clearance estará diminuído quando aproximadamente
50% dos nefronios estiverem lesados, indicando comprometimento da filtração
glomerular.
A colheita de sangue deve ser realizada em jejum ao final da colheita da urina de
24 horas para a correlação dos níveis séricos e urinários.
Clearance = Creatinina na urina (mg/dL) x Vol. (mL/min) /
Creatinina no soro (mg/dL)
Clearance Corrigida = Clearance x Factor (corrigida pela superfície corporal,
calculada em função do peso e da altura).
Cloro
O cloro é o anião de maior concentração no meio extracelular e desempenha o
principal papel na manutenção da neutralidade electroquímica do líquido
extracelular, incluindo o plasma. A maior parte do cloro ingerido é absorvido, e o
excesso é eliminado pelos rins. Existe uma discreta diminuição dos níveis séricos no
período pós-prandial, por aumento da formação de ácido (HCl) pelas células
parietais gástricas. Na maioria das vezes, a sua concentração é afectada pelas
mesmas condições que afetam a concentração do sódio.
O cloro sérico encontra-se aumentado na desidratação, nas perdas excessivas de
bicarbonato por perdas gastrointestinais baixas, na acidose tubular renal, na
insuficiência renal aguda, na alcalose respiratória, na excessiva reposição do íão por
hidratação venosa ou alimentação parenteral, nas situações de hiperfunção
adrenocortical, nalguns casos de hiperparatiroidismo primário e na intoxicação por
salicilatos.
O cloro sérico apresenta-se diminuído na hidratação acentuada, nas perdas
excessivas de cloro por via gastrointestinal, na cetoacidose diabética, na acidose
metabólica, nas nefropatias comperdas de sódio, no desvio do meio extracelular
para o intracelular, como o que ocorre na acidose respiratória compensada e na
alcalose metabólica. A hipocloremia é observada também na síndrome de Batten
(defeito de reabsorção); na crise de addison e na secreção inapropriada da ADH hormona antidiurética.
A determinação da concentração do cloro no suor é indicada na investigação da
fibrose cística, onde se encontra aumentada.
A excreção urinária está diminuída nas situações de grandes perdas
gastrointestinais, diminuição da ingestão de sal na dieta, período de retenção prémenstrual, sudorese excessiva, hiperfunção adrenocortical, retenções hídricas, nos
diferentes tipos de edema e na diabetes insipidus.
A excreção urinária está aumentada nas situações de uso de diuréticos, aumento da
ingestão de sal na dieta, diurese pós-menstrual, diurese maciça de qualquer
etiologia, doença túbulo-intersticial, depleção de potássio e insuficiência
adrenocortical.
Colesterol Total
É um esterol encontrado em todos os tecidos animais. Desempenha importantes
funções fisiológicas, incluindo a síntese de ácidos biliares, vitamina D, hormonas
esteróides e constituintes da dupla camada das membranas celulares.
O colesterol está presente na parede intestinal, proveniente de três fontes: dieta,
secreção biliar e intestinal e células. Alimentos de origem animal, em especial
carne, gema de ovos, frutos do mar e lacticínios, aumentam a quantidade de
colesterol na dieta. Praticamente todo o colesterol presente no intestino encontrase na forma livre, não-esterificado. Todo o colesterol esterificado proveniente da
dieta é rapidamente hidrolisado pelas esterases secretadas pelo pâncreas no
intestino delgado. Cerca de 30 a 60% do colesterol da dieta e do intestino são
absorvidos.
O colesterol total apresenta-se aumentado na hipercolesterolemia primária e
secundariamente no síndrome nefrótico, no hipotiroidismo, na diabetes mellitus, na
cirrose biliar primária e na hipoalbuminemia. Níveis baixos podem ser encontrados
na desnutrição e no hipertiroidismo.
A doença arterial coronária relaciona-se, em proporção direta e duplicada, com
níveis de colesterol séricos. Diferentes estudos corroboram a hipótese de que cada
1% de redução dos níveis de colesterol está associado à queda de 2% de risco de
doença arterial coronária.
O doseamento isolado de colesterol necessita de jejum, visto que os valores de
referência foram obtidos com jejum de 12 horas.
Em pacientes que apresentem valores alterados em relação aos desejáveis para a
idade, recomenda-se a validação com a repetição do doseamento dentro de um
intervalo mínimo de 7 dias e máximo de 2 meses.
Têm sido observadas variações sazonais do colesterol. Por exemplo, níveis séricos
são mais elevados no outono e no inverno e mais baixos no verão e na primavera.
Alguns factores podem interferir, como a postura antes e durante a colheita, stres e
ciclo menstrual.
Consultar Perfil Lipídico.
Colinesterase
É uma enzima cujo papel fundamental é a regulação dos impulsos nervosos através
da degradação da acetilcolina na junção neuromuscular e na sinapse nervosa.
Existem duas categorias de colinesterases: a acetilcolinesterase (colinesterase
verdadeira), que é encontrada nos eritrócitos, no pulmão e no tecido nervoso; e a
colinesterase sérica, sintetizada no fígado, também chamada de
pseudocolinesterase.
A sua determinação é útil na avaliação e no acompanhamento de pacientes com
intoxicação por organofosforados (inseticidas) que inibem a colinesterase
eritrocitária e diminuem os níveis da colinesterase sérica.
A colinesterase sérica está diminuída nas doenças parenquimatosas hepáticas
(hepatites virícas, cirrose), na insuficiência cardíaca congestiva, nos abscessos e
neoplasias. Os níveis baixos persistentes nos cirróticos têm sido apontados como
marcador de mau prognóstico. Valores diminuídos também são encontrados em
estados de desnutrição, infecções agudas, anemias, enfarte do miocárdio e
dermatomiosite. Diversas drogas como estrogénios, testosterona e contraceptivos
orais também podem interferir nos níveis da colinesterase sérica.
A colinesterase pode estar aumentada em pacientes obesos, em diabéticos e no
síndrome nefrótico.
Contagem de Addis
A contagem de Addis é realizada em amostras de urina de 12 ou 24 horas e
permite caracterizar valores mais precisos dos elementos anormais presentes na
urina, como aumento de eritrócitos e leucócitos e aumento das taxas de excreção
de cilindros, que podem surgir em quantidades pequenas demais para serem
detectadas numa amostra de urina aleatória de um exame de rotina.
A contagem é realizada no sedimento por meio de leitura microscópica em câmara
de Neubauer. É útil quando há suspeita de glomerulonefrite subclínica. São
encontrados valores elevados principalmente nos quadros de hematúria e nas
glomerulonefrites, em que apresentam um papel prognóstico.
Coombs Directa
O teste de Coombs directa é um exame que permite a identificação da presença de
anticorpos fixados sobre as hemácias. Técnicamente, baseia-se no facto de que os
anticorpos que recobrem as hemácias podem ser identificados pela adição de
anticorpos antigamaglobulina humana. Quando positivo, ou seja, quando indica a
presença de anticorpos aderidos às hemácias, formam-se pontes entre elas,
levando ao fenómeno visível de aglutinação.
O teste de Coombs contribui directamente para o diagnóstico da anemia autoimune, pois a sua positividade confirma que o anticorpo foi fixado in vivo à hemácia
do paciente, auxiliando dessa forma o diagnóstico diferencial com outras anemias
hemolíticas, como as causadas por alterações da hemoglobina ou da estrutura da
hemácia. É importante também no diagnóstico das anemias hemolíticas do recémnascido e das anemias induzidas por drogas.
Embora o teste de Coombs seja extremamente sensível, um resultado negativo não
exclui a presença de anticorpos ligados às hemácias.
Coombs Indirecta
O teste de Coombs indirecta permite a identificação de anticorpos antieritrocitários
no soro. É importante para a avaliação de grávidas Rh (-) (avaliação de
sensibilização), em pacientes com Rh (-) para avaliação da variante Du e nas fases
pré-transfusionais, especialmente em pacientes já transfundidos, em que pode ter
ocorrido sensibilização para Rh e outros sistemas.
O teste indirecto identifica in vitro diferentes anticorpos, de acordo com a fase do
teste que apresentou positividade. O teste é realizado em quatro diferentes etapas,
conhecidas como: fase fria (à temperatura ambiente) - geralmente anticorpos da
classe IgM; fase em meio protéico - identifica os anticorpos IgM e também
anticorpos incompletos (da classe IgG); fase quente (à temperatura de 37°C) detecta anticorpos que só reagem a essa temperatura (geralmente IgG); e a última
etapa, que identifica aglutininas da classe IgG e anticorpos que fixam o
complemento.
A ocorrência de aglutinação e/ou de hemólise durante quaisquer das etapas indica a
possibilidade da presença de anticorpos irregulares.
Creatinina
A creatinina não é formada pelo metabolismo corporal, sendo apenas um resultado
do metabolismo da creatina e, portanto, relacionada com a massa muscular. A
conversão da creatina em creatinina é praticamente constante, sendo que cerca de
2% da creatina total é convertida em creatinina em cada 24 horas.
A concentração sérica em indivíduos normais é praticamente constante,
apresentando uma variação em relação ao sexo e ao volume de massa muscular,
sendo portanto maior nos homens e nos atletas do que nas mulheres, nas crianças
e nos idosos. Normalmente, a sua excreção não é afectada pela dieta e pela
velocidade do fluxo urinário.
Os níveis séricos aumentam à medida que ocorre a diminuição da taxa de filtração
glomerular. Por isso, é utilizada como marcador da função renal. Os aumentos
tornam-se significativos quando existe uma perda de mais de 50% dos nefrónios
funcionantes, com diminuição expressiva da filtração glomerular. Níveis séricos
diminuídos podem ser encontrados em crianças e em condições em que ocorra uma
redução significativa da massa muscular.
A creatinina é filtrada livremente pelos glomérulos. Além da filtração glomerular, há
também uma secreção tubular activa contrabalançada por um mecanismo de
reabsorção tubular.
Na doença renal, à medida que a taxa de filtração glomerular diminui, a secreção
tubular activa conduz a uma proporção maior no volume de creatinina excretada
pela urina, tornando a utilização da clearance para avaliação da taxa de filtração
glomerular uma ferramenta menos precisa.
Uma outra variável que deve ser levada em conta aquando da avaliação da
clearance da creatinina é o uso de fármacos que possam interferir com a secreção
tubular, como os salicilatos, a cimetidina e a espirinolactona, entre outros.
Crescimeto
A hormona de crescimento é sintetizado pela hipófise anterior. É estimulada pela
GHRH (growth hormone - releasing hormone) e é inibida pelo GHIH (growth
hormone - inhibiting hormone) secretadas pelo hipotálamo. Actua em inúmeros
tecidos, como tecido mole, cartilagem e osso. A principal acção é promover a
síntese protéica e, consequentemente, o crescimento desses tecidos. Também
mobiliza gordura do tecido adiposo, aumenta a absorção intestinal de cálcio,
estimula a glicogenólise hepática e antagoniza os efeitos da insulina. Estas duas
últimas acções resultam num aumento da glicemia.
A GH induz à síntese hepática de somatomedinas (ou IGFs - insulin - like growth
factors), que são os mediadores da acção da GH na cartilagem e no osso.
A hipersecreção de GH deve-se a adenomas hipofisários, e os seus efeitos
dependem da idade do paciente. Quando ocorre antes do fecho das epífises dos
ossos longos, acarreta o crescimento desproporcional (gigantismo); quando ocorre
depois, leva a um aumento de ossos da face e das extremidades (acromegalia),
como também a um aumento de cartilagem e de tecidos moles.
O diagnóstico laboratorial da hipersecreção de GH requer a utilização de provas
funcionais, como o teste de supressão de GH com glicose para comprovação da
não-supressibilidade normal dessa hormona pela glicose. O doseamento basal de
GH é pouco útil nesse diagnóstico, em virtude de, nessa patologia, a secreção de
GH ser episódica.
A análise de IGF-I é um exame alternativo que tem melhor correlação com a
actividade clínica da doença do que o doseamento de GH. O doseamento seriado
dos dois é bastante útil na avaliação da eficácia terapêutica.
A prolactina pode estar elevada em 30% dos casos, devido à hipersecreção do
adenoma hipofisário. Portanto, os seus níveis séricos também devem ser avaliados.
A deficiência de GH em crianças resulta na baixa estatura (nanismo hipofisário),
definida como altura abaixo do terceiro percentil. Cursa com diminuição da
velocidade de crescimento e atraso da idade óssea. Pode ser consequente a doença
hipofisária primária ou a disfunção hipotalámica. A falência hipofisária primária
resulta de doenças genéticas (hipoplasia hipofisária, aplasia hipofisária,
hipopituitarismo familiar e deficiência familiar isolada de GH), trauma, tumores,
doenças granulomatosas, radioterapia do sistema nervoso central. A maioria dos
casos de deficiência de GH é de origem hipotalámica (traumas do parto, tumores ou
infecções).
O diagnóstico laboratorial da deficiência de GH requer a realização de provas de
estimulação, uma vez que os níveis de GH são geralmente baixos em estado basal.
Antes da realização dos testes de estimulação, é necessário investigar se o paciente
é eutiroidéio, uma vez que o hipotiroidismo causa uma resposta abaixo do normal
do GH ao estímulo.
Os estímulos farmacológicos mais comuns são: clonidina, piridostigmina (mestinon)
e insulina. O teste com insulina precisa ser cuidadosamente acompanhado com
doseamentos de glicemia capilar, devido ao risco de hipoglicemia grave. Devido à
possibilidade de ocorrência de resultados falso-negativos, geralmente são
realizados dois testes, com estímulos diferentes. Crianças com deficiência discreta
podem responder normalmente ao estímulo farmacológico. A dosagem de IGF-I
deve apoiar o diagnóstico, uma vez que níveis normais reduzem a possibilidade de
deficiência de GH.
GH (Hormona do Crescimento)
Indicado no diagnóstico e no acompanhamento da acromegalia e do gigantismo,
embora valores basais moderadamente elevados não confirmem o diagnóstico, que
necessita de comprovação com provas funcionais. Na avaliação do déficite de
crescimento, os testes de estimulação são fundamentais, pois níveis basais baixos
ou indetectáveis não são úteis para o diagnóstico. A alimentação, o exercício e o
stres de qualquer origem podem interferir nos resultados.
IGFBP3
(Proteína Carregadora das IGFS)
Entre as várias IGFBPs (IGF - binding proteins) descritas, a IGFBP3 é a principal
proteína carregadora dos factores de crescimento insulina-like. Os níveis dependem
do GH, e a IGFBP3 deve ser avaliada junto com a IGF1.
Somatomedina C ou IGF1
(Insulin - Like Growth Factor I)
É útil no diagnóstico e no seguimento dos pacientes com hipersomatotropismo
(acromegalia e gigantismo), no acompanhamento de pacientes em tratamento com
GH exógeno e no diagnóstico da deficiência da hormona de crescimento. A sua
interpretação deve levar em consideração mais a idade óssea do que a idade
cronológica.
Crioglutininas
As crioaglutininas são anticorpos capazes de aglutinar hemácias humanas. Acreditase que sejam formadas após exposição a microrganismos que apresentam grupos
antigénicos semelhantes aos encontrados nas hemácias. Esses anticorpos
necessitam da presença de complemento para a sua reacção.
Aparecem na população normal em títulos até 1/32. Em condições normais, exigem
refrigeração para que ocorra a reacção com as hemácias. Estas crioaglutininas
apresentam-se elevadas em diferentes situações, como nas infecções por
Mycoplasma pneumoniae, influenza, mononucleose infecciosa, doenças do
colagénio, artrite reumatóide e linfomas. Quando em títulos altos, podem
raramente, aglutinar hemácias em temperaturas próximas às do corpo humano,
podendo levar à anemia hemolítica. A presença das crioaglutininas pode interferir
na avaliação do grupo sanguíneo, na prova cruzada para transfusões, em análises
hematológicas e em reacções imunológicas.
A prova da crioaglutinina é utilizada para o diagnóstico de infecções pelo
Mycoplasma pneumoniae. Não é específica, porém apresenta-se positiva em 34 a
68% dos pacientes com a infecção. Títulos iguais ou maiores do que 1/128, na
presença de quadro clínico compatível, podem confirmar o diagnóstico. Em casos
duvidosos, recomenda-se um segundo doseamento, após 10 a 15 dias.
Crioglobulinas
As crioglobulinas são imunoglobulinas que têm a característica de se precipitar em
temperaturas abaixo de 37º C (4º a 30º C). As crioglobulinas são classificadas em
três tipos principais, de acordo com a sua composição, em tipos I, II e III. O
mecanismo pelo qual são produzidas é ainda desconhecido.
Quando presentes, os tipos I e II não apresentam sintomas clínicos importantes. Já
o tipo III, composto pela associação entre crioglobulinas, cursa com manifestações
importantes como fenômeno de Raynaud, artralgias, necrose de pele, púrpura
vascular, petéquias e nefropatia.
Nos três tipos de crioglobulinemia, é descrita a presença de neuropatia periférica e
de concentração sérica de complemento diminuídas, especialmente de C3 e C4,
(com maior frequência no tipo III).
As doenças associadas incluem doenças virícas, bacterianas e parasitárias, lúpus
eritematoso sistémico, poliartrite nodosa, síndrome de Sjögren, outras
manifestações auto-imunes, desordens linfoproliferativas e hepatite C.
Cryptococcus Neoformans
A presença do antigénio capsular polissacarídico do Criptococcus neoformans é
pesquisada por intermédio da aglutinação em látex, em amostras de soro e/ou
líquor. O antigénio criptocóccico está presente em 87% dos soros e em 99% dos
liquores de pacientes com Criptococose. É de grande importância no diagnóstico da
criptococose sistémica.
O método apresenta algumas limitações, como a reacção cruzada com antigénio de
Trichosporum beigelii e a presença de interferência não-específica, resultante da
presença de imunoglobulinas geradas por outras patologias como no caso da artrite
reumatóide.
Cultura de Anaeróbios
Os materiais clínicos adequados para a realização da cultura de bactérias
anaeróbias devem provir das cavidades fechadas do nosso organismo (coleções
líquidas, abcessos, sangue e liquídos biológicos em geral) ou seja, locais estéreis,
sem a presença da flora normal.
As bactérias anaeróbias causam uma variedade de infecções humanas, incluindo
peritonite, empiema, endocardite e artrite. As infecções anaeróbias são,
geralmente, de fonte endógena, representada pela própria flora normal. Entretanto,
apesar da grande variedade de anaeróbios da flora normal, as infecções são
limitadas a uma pequena quantidade de microrganismos, na qual se destacam o
isolamento do género Bacteroides spp. Estreptococcus spp., Prevotella spp. e
Clostridium perfringens.
Os materiais devem ser colhidos e inoculados em frascos anaeróbios de
hemocultura, até ao volume máximo permitido, que é de 8 mL. Quando o material
for sangue e liquídos biológicos, pode ser armazenado por um período de até 24
horas, à temperatura ambiente, em frascos anaeróbios.
Não poderão ser utilizados para pesquisar microrganismos anaeróbios materiais
provenientes de locais que normalmente participem da flora normal ou
transportados inadequadamente.
Cultura de Fezes
A coprocultura auxilia o clínico no diagnóstico da etiologia de diarréias bacterianas,
por meio do isolamento de patogénios entéricos.
As gastroenterites podem ser causadas por bactérias, vírus ou parasitas. Quando é
solicitada uma rotina de coprocultura, são procurados os agentes etiológicos mais
frequentes, tais como Shigella spp., Salmonela spp. e Escherichia coli
enteropatogénica. A E. coli enteropatogénica é pesquisada nos casos de diarréias
em crianças de até 4 anos de idade. A E.coli invasora é pesquisada em todas as
faixas etárias. A sua toxicidade é dada pela toxina que produz, a qual penetra na
mucosa intestinal, provocando diarréia aguda.
Em casos mais específicos, como as pesquisas de Campylobacter spp. e Yersinia
spp., o pedido médico deverá ser direcionado. Outros patogénios, como Aeromonas
spp. e Plesiomonas spp., podem também ser isolados. Cabe lembrar que algumas
espécies de Campylobacter spp. não crescem nas condições padronizadas para
coprocultura.
O crescimento abundante de germes como Pseudomonas aeruginosa, Candida spp.,
Staphylococcus aureus, entre outros, pode indicar pacientes tratados com
antibióticos de amplo espectro. Apesar de seu papel ainda não estar claramente
definido, sua presença é comunicada ao médico, indicando o seu predomínio.
A cultura deve ser realizada de preferência a partir de fezes frescas. Caso não seja
possível, pode-se enviar swab anal em gel de transporte ou fezes colhidas em meio
de transporte (tampão glicerol). Para a pesquisa de Campylobacter spp., são
adequadas apenas fezes frescas ou colhidas em gel de transporte. Os swabs com
meio de transporte e as fezes conservadas em tampão glicerinado podem ser
armazenados à temperatura ambiente até 24 horas.
Cultura de Materiais do Aparelho Génito-Urinário
Importante no diagnóstico laboratorial das uretrites, vaginites, endocervicites,
doenças sexualmente transmitidas e agentes bacterianos associados às infecções
do aparelho genital.
Secreção uretral, vaginal, urina do 1º jacto, esperma, secreção endometrial, fundode-saco uterino e secreção prostática são consideradas amostras clínicas
apropriadas.
As amostras clínicas colhidas com meio de transporte podem ser armazenadas até
24 horas à temperatura ambiente. As urinas e swabs colhidos sem meio de
transporte devem ser processados até 2 horas.
São considerados como crescimento patológico: Neisseria gonorrheiae,
Estreptococcus agalactiae, Haemophilus spp., Corynebacterium spp. e
enterobactérias.
A presença de Corynebacterium spp. e de enterobactérias é valorizada,
principalmente em crianças, mas também em adultos, quando presentes em
grandes quantidades e a critério do médico.
Cultura de Materiais do Aparelho Respiratório Inferior
Embora as infecções do aparelho respiratório inferior estejam entre as causas de
maior morbilidade e mortalidade dos pacientes, o diagnóstico dessas infecções é
frequentemente complicado pela contaminação da espécie clínica com a flora
normal.
A expectoração é submetida inicialmente a culturas, a fim de determinar o agente
etiológico das pneumonias. O material recebido no laboratório pode ser
expectoração, expectorada e/ou induzida. Outros materiais clínicos incluindo
aspirado traqueal, aspirado transtraqueal, lavado bronquico e lavado broncoalveolar
protegido e não-protegido.
A expectoração deve ser observada ao microscópio, para se avaliar a qualidade da
amostra e se é representativa do aparelho respiratório inferior, ou se contém
apenas saliva. O lavado broncoalveolar é obtido por meio de procedimentos
invasivos, sendo recomendado na suspeita de pneumonias nasocomiais em
pacientes com ventilação mecânica.
Os resultados da cultura de expectoração são interpretados com base na avaliação
da coloração de Gram preparada a partir da porção mais purulenta da amostra: se
contém mais de 25 polimorfonucleares/campo ou até 10 células epiteliais/campo,
observado através de objetiva de 10 vezes, o material é considerado satisfatório. A
informação mais simples envolve a quantificação de um volume maior ou igual a 10
células epiteliais, com a objetiva de 40 vezes, o que seria inaceitável para a cultura.
Nas culturas quantitativas, o volume do lavado broncoalveolar protegido colhido
frequentemente é de 0,01 mL a 0,001mL de secreção. A contagem de 103 UFC/mL
de um microrganismo corresponde a infecção.
Para o lavado broncoalveolar (LBA), a contagem de 10.000 UFC/mL ou mais de um
microrganismo específico correlaciona-se com pneumonia.
No lavado bronquico e no aspirado traqueal, a contagem de 106 colónias, ou seja,
1.000.000 UFC/mL, sugere um processo infeccioso.
Os microrganismos mais frequentemente isolados correspondem aos grupos dos
bastonetes gram-negativos não-fermentadores ou entéricos, os estafilococos e
enterococos. São eles: Staphylococcus aureus, Streptococcus pneumoniae,
Streptococcus pyogenes, Enterococcus spp., Haemophilus influenza, Moraxella
catarrhalis, Pseudomonas aeruginosa, Klebsiella pneumoniae e Rhodococcus equi.
Cultura de Materiais do Aparelho Respiratório Superior
Inclui as culturas de secreções da orofaringe, nasofaringe, ocular e do ouvido.
Auxilia os clínicos no diagnóstico das faringites bacterianas. A causa mais comum
de faringite é representada pelo Streptococcus pyogenes.
Em 7% dos casos, as secreções da nasofaringe são úteis no diagnóstico de
sinusites infecciosas e na detecção de portadores nasais de germes como
Staphylococcus aureus MRSA (estafilococos aureus resistentes à meticilina) e
Neisseria meningitidis. No caso da epiglotite, que têm evolução rápida e
progressiva com celulite, apresentam um grande potencial de obstrução das vias
respiratórias, e os seus agentes etiológicos são Haemophilus influenzae,
Streptococcus pneumoniae e Staphylococcus aureus.
Nas secreções oculares e ouvido, por possuírem a mesma mucosa de revestimento
do aparelho respiratório superior, são isolados os mesmos agentes e diagnosticadas
conjuntivites purulentas e otites. Estas infecções são mais comuns na infância e na
terceira idade.
Para as culturas da orofaringe, os resultados reportados são a presença do
crescimento de estreptococos beta-hemolíticos, Streptococcus pyogenes, outros
não-pyogenes e a ausência de estreptococos beta-hemolítico.
Nas secreções nasais, procura-se evidenciar a presença de Staphylococcus aureus,
Streptococcus pneumoniae e Haemophilus influenzae.
Nas secreções conjuntivais, a presença do crescimento bacteriano é avaliada,
juntamente com os dados clínicos, a presença de cirurgias ou próteses. Os
estafilococos coagulase-negativo e os bastonetes gram-negativos são os mais
frequentemente isolados, juntamente com Staphylococcus aureus, Streptococcus
pneumoniae e Haemophilus spp.
No diagnóstico das otites médias e externas, são utilizados secreções ou fluidos do
ouvido. As bactérias mais comunns são Pseudomonas spp. e outros microrganismos
provenientes da microflora respiratória.
Cultura de Micobactérias
A realização da cultura de micobactérias utiliza como princípio biológico a detecção
radiométrica do CO2 produzido pela actividade metabólica das micobactérias a
partir de meios de cultura específicos marcados com C14.
É destinada ao diagnóstico da tuberculose e das micobacterioses.
Pode ser realizada em expectoração, lavado bronquico, lavado broncoalveolar,
líquido ascítico, líquido pleural, líquido peritoneal, líquido cefalorraquidiano,
aspirado de medula óssea, sangue, fezes, biópsias e urina.
O uso de antituberculostáticos, e a presença de micobactérias não-adaptadas ao
crescimento em meio líquido ou à temperatura de 35ºC podem induzir a um
resultado falso-negativo.
Cultura de Outros Líquidos Biológicos
A cultura dos líquidos biológicos é utilizada para determinar a presença de agentes
infecciosos nos líquidos pleural, sinovial, ascítico e pericárdio, auxiliando no
diagnóstico etiológico de infecções como as pneumonias com derrame pleural,
pericardites e sinovites.
Os fluidos biológicos normalmente são estéreis, e a presença de um microrganismo
resulta quase sempre num agravamento do quadro clínico desses pacientes. O uso
de próteses e a terapêutica com imunossupressores têm contribuído muito para o
aumento da prevalência de positividade dessas amostras. As principais bactérias
isoladas incluem Neisseria spp., Streptococcus pneumoniae, Streptococos betahemolíticos e Staphylococcus aureus, podendo também ser isolados bastonetes
gram-negativos.
A presença de crescimento bacteriano de qualquer microrganismo após a incubação
é considerada patogénica, uma vez que esses líquidos são estéreis. Um número
pequeno de microrganismos na amostra enviada pode levar a um resultado falsonegativo.
Os líquidos biológicos destinados a cultura devem ser transportados ao laboratório
para sementeira em 2 horas após a colheita. Se esse tempo não puder ser
respeitado, inocular até o volume de 8 mL num frasco de hemocultura aeróbio ou
anaeróbio e enviar ao laboratório.
Cultura de Secreções, Abcessos, Tecido Subcutâneo,
Fragmento de Tecidos e Biópsias
Microrganismos residentes na pele e nas mucosas humanas, assim como no
ambiente, podem causar infecções quando inoculados em tecidos normalmente
estéreis ou em mucosas íntegras. Nem sempre é necessário existirem mecanismos
de virulência para o microrganismo causar a doença.
O material de biópsia enviado ao laboratório com mais frequência são os de
linfonodos, pulmão, fígado, fragmentos de tecidos obtidos por laparoscopia,
fragmentos ósseos, secreções de ferida cirúrgica, furúnculos e punção de
abscessos.
A cultura desses espécimes permite diagnosticar as infecções dos tecidos cutâneos
e subcutâneos e de órgãos mais profundos, tais como abscessos intra-abdominais
(incluindo as diverticulites), abscessos peritonsilares, cutâneos e esplênicos,
impetigo, foliculite, furúnculos, celulite, fasciite, erisipela e osteomielite. Permite
também obter a sensibilidade do microrganismo isolado aos antimicrobianos.
O material pode ser proveniente de secreções de pele, bolhosa, de impetigo, de
abscessos em geral, ferida cirúrgica, punção de agulha fina de órgãos e fragmentos
de tecidos. Os tecidos obtidos durante procedimento cirúrgico são os melhores
espécimes, já que os microrganismos são os agentes etiológicos da infecção. Os
materiais obtidos dessa forma devem ser enviados em frasco estéril contendo água
estéril, soro fisiológico ou lactato de ringer. Normalmente, a flora normal não
interfere com esta colheita de material. Em secreções purulentas e abscessos a
colheita deve ser feita por aspiração com seringa.
AUMENTO DA FORMAÇÃO
IMPETIGO
............
FOLICULITES
............
FURÚNCULOS
............
ERISIPELA
............
CELULITES
DIMINUIÇÃO DA SECREÇÃO
Staphylococcus aureus, Staphylococcus pyogenes.
............................................................
Staphylococcus aureus.
............................................................
Staphylococcus aureus.
............................................................
Staphylococcus pyogenes.
............................................................
Staphylococcus pyogenes, Staphylococcus aureus, Esterobactérias, Aeromonas
spp, Clostridium spp, Mycobacterium spp.
........................................................................
FASCIITE
Staphylococcus pyogenes, ou infecções sinérgicas por bactérias facultativas e
anaeróbias.
........................................................................
ABCESSOS
Staphylococcus aureus, Propionebacterium spp.
CUTÂNEOS
........................................................................
ABCESSOS
Staphylococcus aureus, Staphylococcus spp, Propionebacterium spp.
PERINEAL,
VULVOVAGINAL,
ESCROTAL
........................................................................
MIOSITES
Staphylococcus aureus, Staphylococcus spp.
...
...
...
...
...
...
...
...
AUMENTO DA FORMAÇÃO
DIMINUIÇÃO DA SECREÇÃO
OSTEOMIELITES
Staphylococcus aureus, Staphylococcus pyogenes, Streptococcus agalactiae,
Candida spp., Pseudomonas aeruginosas, Pamonella spp
...........................................................................
INSUFICIÊNCIA
Enterobactérias, Bactérias anaeróbias.
VASCULAR
...........................................................................
TRAUMA
Staphylococcus spp, Propionebacterium spp, Pseudomonas ssp,
ASSOCIADO
Staphylococcus aureus e Enterobactérias.
Cultura de Urina
Auxilia o clínico no diagnóstico das infecções urinárias. As urinas submetidas a
cultura são provenientes de pacientes com sintomas ou algum factor de risco para
o desenvolvimento de infecção urinária. As infecções do aparelho urinário têm
frequentemente como agentes etiológicos, bactérias com características de
crescimento rápido, como Escherichia coli, Enterococcus faecalis, Klebsiella
pneumoniae, Enterobacter cloacae, Proteus mirabilis, Pseudomonas spp. e
Staphylococcus saprophyticus, representando a maioria dos isolamentos em
pacientes hospitalizados, assim como da comunidade.
As culturas negativas são identificadas como ausência de crescimento bacteriano.
As prováveis contaminações são definidas como difteróides, estreptococos alfahemolíticos, lactobacilos, Estafilococos Coagulase-Negativa, ou crescimento de três
microrganismos diferentes.
As culturas são consideradas positivas quando a contagem de colônias for igual ou
superior a 105 UFC, com isolamento de um único agente etiológico, ou na presença
de dois microrganismos, quando houver indicação clínica, ou ainda com contagens
inferiores a 105 com o isolamento de um único microrganismo associado a um dado
clínico e/ou laboratorial.
Deve-se colher o 2º jacto urinário após higiene da genitália externa. Em crianças,
que não controlam a micção, fazer a higiene e colocar o saco colector adesivo, que
deve ser trocado a cada 30 minutos quando não tiver sido possível colher a urina. A
urina do paciente em uso de sonda vesical deve ser colhida na válvula lateral do
equipamento, após a desinfecção do mesmo.
A urina do 2º jacto pode ser da primeira micção ou de qualquer amostra urinária,
desde que o paciente retenha a urina por um período mínimo de 4 horas. A urina é
um fluido normalmente estéril; por conseguinte, uma colheita inapropriada pode
torná-la contaminada com a flora do períneo, da uretra ou da vagina.
As amostras permanecem estáveis até 2 horas após a colheita ou em geladeira
(2ºC - 8ºC).
Cultura de Fungos
O isolamento e a identificação de fungo em cultura são prova definitiva no
diagnóstico de infecções fúngicas, permitindo a escolha do tratamento adequado.
As diferentes amostras clínicas são semeadas nos meios de cultura apropriados
para isolamento e identificação de fungos.
As amostras deverão ser colhidas de maneira asséptica em frasco estéril, conforme
a natureza da amostra. Os materiais biológicos requeridos são:
- escamas de pele, unhas, pêlos (micoses superficiais e cutâneas);
- aspirado de lesão, secreções, biópsias de pele (micoses subcutâneas);
- expectoração, lavado brônquico, aspirado brônquico, escovado brônquico
(micoses sistémicas);
- secreções: pulmonar, vaginal, traqueal, orotraqueal, de lesões cutâneas,
abdominal, oral, de conjunctivas ou de qualquer outra localização (micoses
sistémicas);
- sangue, biópsia de qualquer órgão ou tecido, urina, líquor, líquidos sinovial,
ascítico, amniótico ou outros líquidos orgânicos (micoses sistémicas).
Os resultados serão interpretados de acordo com o tipo de material clínico
semeado, o local da lesão e a indicação clínica. Alguns fungos são parte da flora
normal, mas podem, ocasionalmente, causar doenças. Do mesmo modo, fungos
oportunistas podem ser apenas contaminantes como os reais causadores da
patologia em investigação. Muitas vezes, o médico prescritor deve ser consultado
sobre a condição clínica do paciente, para que se possa concluir qual o real valor do
isolamento de determinados fungos.
A presença de microrganismos da flora normal ou de infecção bacteriana
concomitante pode inibir o crescimento de fungos patogénicos.
Curva Glicémica
Também conhecida como prova de tolerância oral à glicose (PTOG), a curva
glicémica consiste na administração de 75 g de glicose - em solução aquosa a 25%
- por via oral, seguida de colheitas seriadas de sangue, nos tempos 0, 60 e 120
minutos, para o doseamento de glicose. Em crianças, administra-se 1,75 g/kg de
peso até a dose máxima de 75 g.
AUMENTO DA FORMAÇÃO
NORMAL
Inferior a 140 mg/dl
INTOLERANTE (tolerância diminuída)
intolerante à sobrecarga entre 140 e 200 mg/dl
DIABETES MELLITUS
Superior a 200 mg/dl
Para garantir a fidelidade dos resultados de um teste de sobrecarga oral à glicose,
devem ser tomados os seguintes cuidados:
- nos 3 dias que antecedem a prova, o paciente deve ingerir, pelo menos, 150 g de
carbohidratos por dia;
- o paciente deve estar realizar as suas actividades físicas habituais, mantendo-se
em regime alimentar normal, excepto pela adição da quantidade de carbohidratos
indicada no item anterior;
- o paciente não deve realizar medicação que interfira no metabolismo de
carbohidratos;
- durante o teste, o paciente deve manter-se em repouso e, se possível sem fumar;
- a prova deve ser realizada pela manhã, com o paciente em jejum de 8 a 10
horas.
Mesmo com todas essas precauções, é importante salientar que os resultados desta
prova, em testes realizados em dias diferentes, podem mostrar-se com vários
problemas. Assim, a interpretação deve ser feita sempre com cautela, por um
médico que conheça a história clínica do paciente.
Triagem Gestacional
O doseamento de glicose, os tempos de colheita e os critérios diagnósticos são
ligeiramente diferentes para mulheres grávidas. Em grávidas entre a 24ª e a 28ª
semana de gravidez, pode ser realizado um teste de rastreio em duas fases.
Na primeira, faz-se a glicemia em jejum ou a glicemia de 1 hora após a ingestão
oral de 50 g de glicose (sem necessidade de jejum prévio). Resultados de glicemia
de jejum iguais ou acima de 85 mg/dL ou após sobrecarga maior ou iguais a 140
mg/dL são considerados positivos e com indicação de realização de PTOG.
A segunda fase aplica-se nos casos em que se enquadrem nos critérios anteriores,
e é realizado a PTOG com 75 g de glicose.
As amostras de sangue são colhidas nos tempos basal , 60 e de 120 minutos. Os
limites de 126 mg/dL para glicemia basal e de 140 mg/dL após 2 horas são
considerados normais. O diagnóstico de diabetes gestacional será confirmado se
pelo menos um dos limites estabelecidos como normais for ultrapassado.
Consultar Glicose.
CYFRA 21-1
O Cyfra 21-1 é um marcador tumoral cuja presença caracteriza um mau
prognóstico nos carcinomas de células escamosas do pulmão. É o fragmento da
citoqueratina 19 encontrado no soro. Dados da literatura revelam que o Cyfra 21-1
não é um bom marcador tumoral de carcinoma de pequenas células do pulmão; é
porém um marcador de sensibilidade e especificidade melhores para outros
carcinomas do pulmão, como carcinomas de células escamosas, adenocarcinomas e
carcinomas de células grandes.
Dengue
Conhecida na literatura há cerca de 200 anos, caracteriza-se como doença
infecciosa febril aguda, de etiologia viral e evolução benigna na maioria dos casos.
A primeira epidemia foi relatada em ocorrências simultâneas nos três continentes
em 1779/1780.
Nos últimos 40 anos, mais de 3 milhões de casos de dengue foram registados no
mundo, com cerca de 58 mil mortes nesse período. As regiões tropical e subtropical
são as mais atingidas por esta doença que é causada por um vírus da família
Flaviviridae e transmitida pelos mosquitos Aedes aegypti e A. albopictus. No Brasil
e nas Américas, o A. aegypti é o mosquito epidemiologicamente mais importante.
Proveniente de África e domesticado por acção antropofílica, o Aedes aegypti é,
também, o vector responsável pela transmissão da febre amarela urbana. O
mosquito, de cor escura com manchas brancas, adaptou-se muito bem à vida
urbana. Possui hábitos diurnos, picando os indivíduos na parte da manhã e à tarde.
As fêmeas depositam os ovos em água limpa, onde as larvas podem proliferar-se a
partir de depósitos artificiais de água, como pneus e tanques.
O vírus de dengue é dividido em quatro sorotipos distintos, com características
antigénicas diferentes e isolados inicialmente de fontes diferentes: DEN-1, DEN-2,
DEN-3, DEN-4.
Na infecção humana, o quadro clínico pode ser assintomático, evoluir de forma
clássica, com febre, cefaléia de grande intensidade, artralgia, mialgia, rash
cutâneo, insónia e anorexia ou determinar manifestações hemorrágicas fatais,
sendo essa forma clínica denominada dengue hemorrágica, ocorrendo em menos de
0,2% dos casos de infeccção primária e em 90% dos casos que apresentam história
prévia.
A transmissão ao homem ocorre por meio da picada de mosquitos hematófagos
infectados. Não ocorre transmissão de pessoa a pessoa.
Os mecanismos que determinam a apresentação clínica da doença sob a forma
clássica ou hemorrágica ainda são desconhecidos. Entre as hipóteses
fisiopatogénicas da forma hemorrágica da doença, as mais aceitas são em função
da presença de anticorpos anti-virais: duas infecções sequenciais, por diferentes
sorotipos, com intervalo mínimo de 3 anos, são necessárias para que a resposta
imunológica do indivíduo sensibilizado seja amplifica da pela segunda infecção.
Outra hipótese aceite é a que relaciona as formas graves a uma maior virulência de
determinadas cepas virais.
No Brasil, as primeiras referências ao Dengue datam de 1916, em São Paulo, e em
1923 em Niterói, Rio de Janeiro. Entretanto, a primeira epidemia só foi relatada em
1982, em Roraima, em que foram registrados 11.000 casos. Em 1986, uma grande
epidemia ocorreu no estado do Rio de Janeiro, causada pelo serotipo DEN-1, até
então o único serotipo encontrado no Brasil. Foram identificados então mais de
33.000 casos em 1986 e 60.000 casos em 1987, todos de dengue clássico.
Na década de 1990, ocorreram vários casos de recrudescimento de DEN-1 no Rio
de Janeiro e a introdução da DEN-2, com alta incidência no início da década. No ano
de 1991, foram registados os primeiros casos de dengue hemorrágico no estado,
com 1.316 notificações e 8 óbitos.
Os meses de maior incidência de dengue no Brasil nos últimos anos, segundo dados
do Ministério da Saúde, são os cinco primeiros meses do ano, com pico de
incidência nos meses de Março, Abril e Maio. Recentemente, foram relatados nos
mídia (O Globo, 24 de março de 2001) casos de dengue causados pelo serotipo
DEN-3 no Rio de Janeiro.
A imunidade é serotipo-específica, ou seja, a infecção causada por um dos
serotipos não confere imunidade aos outros. Além disso, como já citado, a
probabilidade de ocorrência de formas mais graves da doença é maior em
indivíduos que apresentam infecção prévia. Sugere-se que esses indivíduos
desenvolvam anticorpos contra um dos serotipos que facilitariam a interacção
vírus-célula numa nova infecção viral.
Existe a tendência de pacientes que habitam regiões endémicas, quando
diagnosticados, apresentarem não a infecção primária, mas sim a secundária.
Portanto, a presença dos anticorpos é de grande importância, visto que na primeira
reinfecção e nas infecções subsequentes aparece uma alta ocorrência de
complicações.
Actualmente, utilizam-se testes de ELISA, que determinam simultaneamente a
presença de anticorpos IgM e IgG. A classificação da infecção como primária ou
secundária é pela análise da presença ou ausência de IgM e pela avaliação dos
títulos de IgG. Nos casos positivos, a detecção dos anticorpos IgM caracteriza a
infecção aguda. Apresentam-se positivos por volta do terceiro ou quinto dia após o
aparecimento da sintomatologia clínica, mantendo-se positivos cerca de 30 a 90
dias e declinando logo após, até níveis indetectáveis. Nalguns casos, mantêm-se
detectáveis até 8 meses após o quadro. Os anticorpos IgG apresentam-se positivos
por volta da segunda semana após o início do quadro, mantendo-se detectáveis por
toda a vida.
Os casos secundários caracterizamse pela presença de altos títulos de IgG, podendo
ou não apresentar níveis elevados de IgM. Recomenda-se que, nos casos clínicos
sugestivos da doença que apresentam serologia negativa, esta seja repetida com
intervalo de 1 semana, para afastar a possibilidade de não-detecção por níveis de
anticorpos muito baixos, ou em consequência da fase da doença primária ou
secundária em que a colheita foi realizada.
Além dos exames laboratoriais específicos, devem também ser avaliados, no
diagnóstico de dengue, exames complementares definindo trombocitopenia,
aumento de hematócrito e hipoproteinémia.
De grande utilidade são as aplicações recentes de testes imunocromatográficos,
reação em cadeia da polimerase (PCR) e hibridização de ácidos nucleicos para o
diagnóstico rápido, sensível e específico. O RNA viral pode ser detectado em soro,
líquido cerebroespinhal e tecidos, e os subtipos virais determinados por hibridização
dos produtos de reação em cadeia de polimerase (PCR) com sondas de
oligonucleotídeos tipo-específicas. A variação de linhagem e subtipos virais pode ser
identificada, em estudos de pesquisa, pela análise por RFLP (restriction fragment
length polymorphism) ou pela sequência de nucleotídeos dos produtos de PCR.
Estes ensaios encontram-se restristos, ainda não disponíveis para a maioria dos
laboratórios clínicos.
Desidrogenase Láctica
É uma enzima intracelular responsável pela oxidação reversa do lactato em
piruvato. É amplamente distribuída em todas as células do organismo,
concentrando-se mais especialmente no miocárdio, rim, fígado, hemácias e
músculos. Possui cinco formas de isoenzimas.
Os seus valores encontram-se elevados em todas as situações em que ocorre
grande destruição celular.
Os níveis séricos elevados encontrados em diferentes condições como anemia
megaloblástica e hemolítica, leucemias, linfoma, hemoglobinopatias, enfarte agudo
do miocárdio, enfarte pulmonar, insuficiência cardíaca congestiva, insuficiência
coronária, choque e hipóxia importantes, doenças musculares, lesões hepáticas,
neoplasias primárias ou secundárias (metastáticas), hepatites, icterícias obstrutivas
e cirrose.
Desordens das Supra-renais
O par de glândulas supra-renais localiza-se na borda superior dos rins (suprarenais). Cada supra-renal consiste no córtex (porção mais externa) e na medula
(porção mais interna). A medula secreta catecolaminas, em resposta ao estímulo
de fibras nervosas simpáticas pré-ganglionares.
O córtex compõe-se de três zonas: glomerular, fascicular e reticular. Secreta
corticosteróides que, dependendo da função dos esteróides, são classificados como
mineralocorticóides, glicocorticóides ou esteróides sexuais.
REGIÕES DA SUPRA-RENAL E PRODUÇÃO HORMONAL
CÓRTEX
ZONA GLOMERULAR
--> Mineralocorticóide
ZONA FASCICULAR
--> Glicocorticóides
ZONA RETICULAR
--> Esteróides sexuais
MEDULA
CATECOLAMINAS
--> Epenefrina, Noraepinefrina, Dopamina
Os glicocorticóides regulam o metabolismo dos carboidratos, das proteínas e das
gorduras. O cortisol é o principal glicocorticóide.
A hormona adrenocorticotrófica (ACTH) é a principal reguladora da produção e
secreção de cortisol. A ACTH é secretada pela hipófise anterior em resposta à
libertação, pelo hipotálamo, da hormona libertadora de corticotrofina (CRH). A
hormona antidiurético (ADH), a ocitocina e as catecolaminas podem influenciar o
ritmo diurno da ACTH. Febre, hipoglicemia, stres, distúrbios psicológicos podem
disparar a libertação de ACTH.
O cortisol circula na corrente sanguínea ligado a uma alfaglobulina chamada
transcortina ou CBG (cortisol binding globulin). Apenas uma pequena fracção circula
livre e é biologicamente activa. Possui um ritmo circadiano, com níveis elevados
pela manhã, caindo ao final do dia. Actua como feedback negativo no hipotálamo e
na hipófise anterior, diminuindo a libertação de CRH e de ACTH. É metabolizado
principalmente no fígado e seus metabolitos são excretados na urina como 17hidroxicorticosteróides (17-OH esteróides).
As hormonas sexuais circulam ligados a proteínas plasmáticas, principalmente à
SHBG (steroid hormone binding globulin). Apenas a fracção livre é biologicamente
activa. As hormonas sexuais sofrem metabolização hepática. Os androgénios são
excretados na urina como 17-cetosteróides (17-KS).
A aldosterona é o principal mineralocorticóide. É regulada por vários mecanismos,
sendo o principal o sistema renina-angiotensina. Em caso de depleção de volume,
ocorre libertação de renina das células justa-glomerulares das arteríolas aferentes
renais. A renina actua sobre o angiotensinogénio produzindo angiotensina I, por
sua vez convertida em angiotensina II pela enzima conversora de angiotensina
(ECA). A angiotensina II é convertida em angiotensina III, sendo ambas capazes de
se ligar ao receptor na célula-alvo e de ativar o ciclo fosfatidil inositol que estimula
a libertação do cálcio intracelular e a secreção de aldosterona.
Outros estímulos para a secreção de aldosterona são aumento do potássio sérico,
elevação dos estrogénios, redução do volume plasmático e redução do sódio sérico.
A aldosterona circula ligada a proteínas plasmáticas. Após sofrer metabolização
hepática, é excretada na urina como conjugados do ácido glicurónico.
Hipercortisolismo
Caracterizado pelos elevados níveis de cortisol e pela perda do ritmo circadiano.
O Síndrome de Cushing engloba as diversas causas de hipercortisolismo:
a administração exógena de cortisol ou de ACTH;
a secreção excessiva de ACTH pela hipófise anterior com consequente hiperplasia
do córtex adrenal e aumento na secreção de cortisol (doença de Cushing);
a secreção ectópica de ACTH ou de CRH por tumor não-hipofisário;
a hipersecreção autónoma de cortisol por adenoma ou carcinoma de adrenal;
a displasia nodular adrenocortical primária (com produção autónoma de
glicocorticóide e ACTH em níveis baixos ou normais).
A causa mais frequente do síndrome de Cushing é a terapia prolongada com
glicocorticóide, denominado síndrome de Cushing iatrogénico. Dois terços dos
pacientes com síndrome de Cushing endógeno têm doença de Cushing (adenoma
hipofisário secretor de ACTH).
Um terço dos casos do síndrome de Cushing por produção ectópica de ACTH devido
a tumores malignos não-hipofisários é causado por carcinoma de pulmão tipo oat
cell. Outras causas são: tumores de células das ilhotas pancreáticas, tumores
renais, timomas e adenomas brônquicos.
Menos de um terço das causas de síndrome de Cushing é de neoplasias adrenais.
Carcinomas adrenais podem secretar também grandes quantidades de androgénios.
As manifestações clínicas mais comuns do síndrome de Cushing são: acentuada
fraqueza e perda muscular, intolerância à glicose, obesidade central, face em lua,
pele fina, estrias violáceas, fragilidade capilar, atrazo de crescimento em crianças,
osteoporose, cifose, hipertensão arterial e distúrbios psiquiátricos. Nas mulheres,
podem ocorrer oligomenorréia, amenorréia, anovulação ou hirsutismo. Nos
homens, podem ocorrer diminuição de libido e/ou ginecomastia.
Laboratorialmente, o diagnóstico inicial é feito com a dosemento do cortisol basal,
ritmo circadiano, teste de supressão nocturna e doseamento do cortisol livre
urinário. A presença de hipercortisolismo deve ser confirmada pelo teste de
supressão com dexametasona 2 mg, e o diagnóstico etiológico requer o teste de
supressão com dexametasona 8 mg, além do doseamento de ACTH. O teste do CRH
também é utilizado, apesar da dificuldade de obtenção desse medicamento.
HIPERCORTISOLISMO
DOENÇA DE ADENOMA CRACINOMA
ACTH
IATROGÉNICO
CUSHING
SUPRASUPRAECTÓPICO
(ADENOMA
RENAL
RENAL
HIPOFISÁRIO)
Cortisol
Basal
Rítmo
Circadiano
ACTH
Cortisol
Livre
Urinário
17-OH
Esteróides
17 KS
(N)
(N)
A
A
ou N
ou N
( ) elevado (
A
A
) muito elevado ( )diminuído (A)ausente
A
Outros exames complementares são os testes dinamicos:
a supressão nocturna pela dexametasona (1 mg);
o teste de supressão com dexametasona (2 mg) (Liddle I);
o teste de supressão com dexametasona (8 mg) (Liddle II);
o teste de estimulação pelo CRH.
Insuficiência Adrenal
Caracteriza-se pela diminuição na produção de cortisol. Pode ser primária, com
ACTH elevada, ou secundária, com ACTH diminuído. A insuficiência secundária
geralmente resulta do uso prolongado de glicocorticóide como cortisona e
prednisolona, mas também pode ser devido a um tumor hipofisário. Em caso de
hipopituitarismo, outras hormonas podem estar comprometidos, como GH, TSH e
LH.
A insuficiência primária pode manifestar-se de forma aguda ou crónica. A forma
aguda é rara e a causa mais frequente é a hemorragia adrenal em pacientes com
terapia anticoagulante. Outras causas são: septicéemia, cirurgia, trauma, enfarte
do miocárdio e gravidez. Clinicamente, é caracterizada por astenia intensa, dores
graves na região lombar, pernas e abdómen, choque e insuficiência renal.
A insuficiência adrenal crónica é denominada doença de Addison, e a causa mais
comum é um processo de auto-imunidade. Outras causas são as infecções
granulomatosas como tuberculose, histoplasmose e amiloidose, sarcoma de Kaposi
e infiltração por outros patógenios como citomegalovírus. Clinicamente, a
insuficiência adrenal prolongada caracteriza-se por hiperpigmentação da pele,
especialmente nas cicatrizes e mucosa bucal, astenia, perda de peso, febre, mialgia
e hipotensão. Os achados laboratoriais podem não ser muito expressivos, a menos
que ocorra o choque, com hiponatremia, hipercalemia, hipoglicemia,
hemoconcentração e uremia.
Na insuficiência secundária devido a hiposecreção de ACTH, não ocorrem nem a
hiperpigmentação, nem alteração tão grave de volume e eletrólitos, uma vez que a
aldosterona não está sob controlo primário do ACTH.
AVALIAÇÃO LABORATORIAL DO HIPOCORTISOLISMO
PRIMÁRIO (Lesão
SECUNDÁRIO (Lesão do
Adrenal)
Hipotálamo ou Hipófise)
Cortisol (8HS)
N ou
Ritmo
A
A
ACTH
( ) elevado ( )diminuído (A)ausente
Em muitos casos de Insuficiência supra-renal moderada, os valores basais podem
ser compatíveis com a normalidade, havendo necessidade da realização do teste de
estímulo do cortisol com cortrosina (ACTH). O teste de estimulação do cortisol com
insulina só deve ser realizado após a constatação de que a adrenal é capaz de
responder ao estímulo com ACTH.
A discriminação entre insuficiência adrenal hipofisária ou hipotalámica pode ser
realizada com o teste do CRH. A resposta positiva sugere etiologia hipotalámica, e a
falta de resposta sugere etiologia hipofisária.
Síndrome Adrenogenital
Este síndrome engloba um grupo de patologias caracterizadas pelo excesso de
corticosteróides adrenais:
a Hiperplasia adrenal congénita;
a Hiperplasia adrenal de início tardio;
o Adenoma adrenal;
o Carcinoma adrenal.
Na maioria dos casos, ocorre hipersecreção de androgénios, que resulta em
virilização.
A hiperplasia adrenal congénita engloba inúmeros defeitos dos sistemas
enzimáticos que envolvem a síntese do cortisol, cuja deficiência leva ao aumento de
ACTH, com hiperplasia da adrenal e aumento de inúmeras hormonas
intermediárias.
O defeito de síntese mais frequente é a deficiência de 21-hidroxilase. Se o defeito é
completo, o paciente sofre acentuada perda de sal pela deficiência de aldosterona
com hipotensão arterial e insuficiência adrenal pela deficiência de glicorticóides.
Pode haver urgência no diagnóstico devido aos graves quadros de desidratação e
hipotensão em recém-nascidos. Pode ocorrer genitália externa ambígua no
nascimento. Os sinais de virilização podem ocorrer mais tarde, dependendo do grau
da deficiência enzimática. O principal exame diagnóstico é o doseamento de 17-OH
progesterona. Casos atípicos ou heterozigotos necessitam da prova de estímulo
com ACTH.
Outros exames utilizados são sulfato de deidroepiandrosterona (S-DHEA),
deidroepiandrosterona (DHEA), androstenediona, testosterona, renina e
aldosterona.
Outros defeitos de sistema enzimático são deficiência de 17-hidroxilase, 11hidroxilase, 3-beta-hidroxiesteróide desidrogenase e colesterol desmolase.
Na deficiência de 11-hidroxilase, além da virilização, ocorre hipertensão arterial
devido à formação de 11-desoxicorticosterona, um potente retentor de sódio.
Nestes casos, é importante o doseamento de 11-desoxicortisol.
Tumores produtores de excesso de androgénios são raros e podem ocorrer em
qualquer idade.
Hiperaldosteronismo
A aldosterona é secretada em resposta ao ACTH, à depleção de volume
intravascular ou à hiponatremia. Actua nos rins, reabsorvendo sódio e água à custa
da excreção de potássio, magnésio e hidrogénio, resultando em hipernatremia e
hipervolemia.
O hiperaldosteronismo primário ocorre quando há hipersecreção de aldosterona
pela supra-renal. Pode ser causado por adenoma unilateral (síndrome de Conn),
hiperplasia bilateral ou carcinoma. A principal manifestação clínica é a hipertensão
arterial. Os níveis de potássio encontram-se baixos ou no limite inferior da
normalidade e estão associados a fraqueza muscular, fadiga e poliúria nocturna;
algumas vezes, também, a polidipsia e intolerância à glicose. A perda urinária de
iões de hidrogénio pode acarretar alcalose metabólica.
O hiperaldosteronismo secundário ocorre em condições como insuficiência cardíaca
ou estenose da artéria renal, com consequente aumento da renina plasmática e da
aldosterona. Alguns pacientes, nesse caso, apresentam hipertensão, embora a
maioria dos casos curse com edema e sem hipertensão, relacionados a patologias
como síndrome nefrótico, cirrose com ascite e insuficiência cardíaca congestiva.
Hipoaldosteronismo
São raros os defeitos isolados na síntese de aldosterona. Defeitos parciais da
síntese aparecem na hiperplasia adrenal congénita em virtude da deficiência de 21hidroxilase.
Existem duas formas de deficiência isolada de aldosterona: o hipoaldosteronismo
idiopático e o hipoaldosteronismo hiporreninémico, sendo esste último o mais
frequente e geralmente associado a doença renal crónica, com diminuição da
libertação de renina por deficiência das células justaglomerulares, levando à
diminuição da aldosterona, hipercaliémia e acidose metabólica. Os níveis de sódio
geralmente estão normais ou baixos, e a restrição de sódio agrava as
manifestações clínicas. A Diabetes é um achado comum nestes pacientes.
Esta condição também é associada à deficiência de prostaglandina indometacina
induzida e é agravada pelo uso de heparina, bloqueadores do canal de cálcio ou
bloqueadores beta-adrenérgicos.
O principal sintoma da deficiência de aldosterona é a hipotensão arterial. O
hipoaldosteronismo idiopático é raro, e o paciente pode apresentar um quadro de
bloqueio cardíaco secundário a hipercaliémia ou hipotensão postural secundária a
hipovolemia.
ACTH (Hormona Adrenocorticotrófica)
Podem ocorrer picos de secreção pós-stres ou pós-alimentação. É indicado para o
diagnóstico da doença de Addison, em que se encontra extremamente elevado, e
no síndrome de Cushing, no qual ajuda no diagnóstico diferencial entre patologia
adrenal primária (adenoma ou carcinoma) com valores muito baixos e com o
hipercortisolismo ACTH dependente com níveis elevados. Colher entre as 7:00 e as
9:00 horas.
Cortisol
O seu doseamento é útil no diagnóstico de hipercortisolismo, sendo importante no
teste de supressão com dexametasona e no diagnóstico de insuficiência adrenal
primária (Addison) ou secundária, no qual o teste de cortirosina (ACTH) geralmente
é necessário. Convém lembrar que valores normais não descartam nenhuma das
patologias citadas. Deve ser dada atenção especial à possibilidade de outras
situações que podem elevar os níveis séricos de cortisol, como stres, obesidade,
alcoolismo, gravidez, uso de estrogénios e depressão endógena. Variações nas
proteínas séricas (globulina ligada ao cortisol - CBG) podem elevar ou diminuir os
níveis de cortisol. Colher entre as 7:00 e as 9:00 horas.
Cortisol Urinário Livre
Principal teste de triagem para hipercortisolismo. Detecta a fração de cortisol livre.
Deve ser avaliado em paralelo com o doseamento de creatinina urinária, em urina
de 24 horas, para validar a colheita.
11-Desoxicortisol Sérico (Composto S)
Também chamado composto S. Eleva-se no bloqueio da enzima 11-hidroxilase por
ser o precursor imediato do cortisol. Alguns pacientes com hirsutismo e níveis
basais normais de 11 desoxicortisol podem evidenciar defeitos parciais de síntese
no teste de estimulação com ACTH.
Deidroepiandrosterona (DHEA)
A deidroepiandrosterona, precursora da síntese de testosterona, é um androgénio
de fraca atividade. A sua utilização está indicada na avaliação do
hiperandrogenismo, como ocorre no hirsutismo, acne, hiperplasia congénita da
supra-renal, carcinoma de supra-renal e puberdade precoce.
A sua semi-vida é mais curta do que a do seu principal metabolito, o sulfato de
deidroepiandrosterona (S-DHEA). Isto permite que seja utilizada na avaliação de
respostas da supra-renal aos testes dinâmicos de estimulação e supressão. A hiperresposta do DHEA à estimulação com ACTH é utilizada para o diagnóstico de defeito
de síntese da supra-renal.
Os níveis séricos são significativamente alterados em condições de stres, variando
com a idade, com um pico na fase puberal.
Sulfato de Deidroepiandrosterona (S-DHEA)
O sulfato de deidroepiandrosterona (SDHEA) é o metabolito da DHEA, e os seus
níveis não sofrem grandes alterações com estimulação fisiológica, ao contrário do
que ocorre com a DHEA.
É encontrado em grande quantidade na circulação, e o seu doseamento pode
substituir o doseamento sérico dos 17-cetosteróides urinários. A sua utilidade
clínica está na avaliação do hiperandrogenismo, como ocorre no hirsutismo, no
acne, no carcinoma de supra-renal e no diagnóstico e controle terapêutico da
hiperplasia congénita da supra-renal.
Androstenediona
É um androgénio produzido pelas supra-renais e gonadas. Útil na avaliação do
hiperandrogenismo (hirsutismo, acne), no acompanhamento da hiperplasia
congénita da supra-renal e nos tumores virilizantes da supra-renal e ovário.
17-Hidroxiprogesterona (17-OHP)
É produzido pelos ovários e supra-renais e sofre variações de acordo com a fase do
ciclo menstrual. É importante na avaliação do hirsutismo e para diagnóstico e
acompanhamento da hiperplasia congénita da supra-renal por defeito de síntese da
21-hidroxilase. Casos duvidosos podem ser confirmados pelo teste de estimulação
com ACTH.
17-Cetosteróides (17-KS)
Utilizado na avaliação do hiperandrogenismo, uma vez que representa vários
metabolitos urinários de diferentes precursores. Nos homens, dois terços dos 17-KS
são derivados do córtex da supra-renal, e um terço, dos testículos. Na mulher, são
derivados praticamente da supra-renal. Os 17-KS elevam-se nos tumores adrenais
e gonodais, no síndrome do ovário poliquístico e na hiperplasia supra-renal.
17-Hidroxicorticosteróides (17-OHS)
Utilizado no diagnóstico de hipercortisolismo, uma vez que representa um grupo de
esteróides secretados pela adrenal, incluindo cortisol, 11-desoxicortisol e outros.
Várias situações podem elevar os seus níveis, como stres, obesidade e alcoolismo.
Aldosterona
Deve ser colhida após 1 hora de repouso em decúbito dorsal ou após 2 horas em
posição ortostática. São interferências comuns o uso de hipotensores e de
diuréticos, o teor de sódio na dieta e a postura.
É útil na avaliação da hipertensão e/ou hipotensão arterial. Importante para
diagnóstico de hiperaldosteronismo primário e secundário, hipoaldosteronismo,
síndrome de Bartter e pseudo-hipoaldosteronismo. Para localização de tumores,
podem ser colhidas amostras por cateterismo venoso.
No hiperaldosteronismo primário, a aldosterona encontra-se elevada. Já os níveis
de renina mostram-se baixos ou indetectáveis, associados com hipocaliémia.
Renina elevada com aldosterona elevada sugere hiperaldosteronismo secundário
(encontrado na hipertensão renovascular).
No síndrome de Bartter, a aldosterona e a renina podem estar elevadas em
pacientes normotensos, com potássio baixo.
No hipoaldosteronismo, constatamos elevação de potássio com níveis baixos de
aldosterona.
Na síndrome de resistência à ação da aldosterona (pseudo-hipoaldosteronismo),
encontra-se hipercaliémia com aldosterona em níveis elevados.
Renina
Deve ser colhida após 1 hora de repouso em decúbito dorsal ou após 2 horas em
posição ortostática. Pode ser colhida por cateterismo das veias renais.
Interferências comuns são o uso de diuréticos, de anti-hipertensivos e de
estrogénios, assim como a postura e o teor de sódio na dieta.
O seu doseamento é útil na avaliação da hipertensão e/ou hipotensão arterial. Tem
um papel importante no diagnóstico de hiperaldosteronismo primário e secundário,
hipoaldosteronismo e síndrome de Bartter, e no seguimento de portadores de
defeito da 21-hidroxilase em tratamento. Encontra-se diminuída no
hiperaldosteronismo primário e no hipoaldosteronismo hipo-reninémico, e elevada
no hiperaldosteronismo secundário e no síndrome de Bartter. Deve ser avaliada em
associação com os níveis de potássio e de aldosterona.
Catecolaminas e Ácido Vanil Mandélico
Utilizados na avaliação da hipertensão arterial. Úteis no diagnóstico de
feocromocitoma, neuroblastoma, ganglioneuroma, ganglioneuroblastoma e
gangliomas. Também empregues na monitorização do tratamento, para avaliar se
houve remoção completa do tumor, assim como na avaliação da possibilidade de
recidivas.
Devido à secreção intermitente de catecolaminas por alguns tumores, várias
colheitas devem ser realizadas num curto intervalo de tempo. Em 20 a 30% dos
neuroblastomas, o doseamento de AVM pode ser normal. É necessário observar as
inúmeras interferências causadas por drogas e pela dieta.
Ácido Vanil Mandélico (AVM)
Realizado em amostra de urina de 24 h, colhida em frasco especial contendo ácido
e após dieta especial. O uso de bebida alcoólica, de reserpina, de inibidores da MAO
(monoamino oxidase) e de contrastes radiológicos pode interferir, diminuindo os
seus valores. A ingestão de chocolate, chá, cafeína, banana, frutas cítricas,
vegetais, baunilha e anfetaminas pode elevar os valores.
Catecolaminas Urinárias
Realizado em amostra de urina de 24 h, colhida em frasco especial contendo ácido
e após dieta especial. O uso de guanetidinas e o uso crónico de inibidores da ECA
(enzima conversora da angiotensina), reserpina e bromoergocriptina podem
interferir e originar valores reduzidos. A ingestão de bebidas alcoólicas, chá, café,
banana, nicotina, teofilina, anfetaminas, levodopa, aldomete e isoproterenol pode
elevar os valores.
Digoxina
Os digitálicos são dos fármacos mais frequentemente prescritos na prática clínica.
Devido à sua capacidade de aumentar a força de contracção cardíaca, são usados
no controle de taquicardias supraventriculares e no tratamento da insuficiência
cardíaca congestiva. A digoxina é um dos digitálicos mais usada. Tem uma semivida de 24 a 48 horas (com uma média de 35 horas). Como a digoxina é eliminada
principalmente por meio de excreção renal, os pacientes com função renal reduzida
apresentam uma semi-vida sérica mais longa.
A digoxina deve ser avaliada até que o efeito terapêutico seja alcançado, no
acompanhamento da estabilidade e no uso adequado do fármaco pelo paciente e ao
primeiro sinal de aparecimento de efeito tóxico (como anorexia ou arritmia).
O doseamento em crianças é aproximadamente 50% maior do que o usado em
adultos, quando calculada em miligramas por quilograma de peso corporal. Doses
mais altas são necessárias por causa da maior relação de massa cardíaca com a
massa corporal em crianças. Estas doses mais altas refletem-se em concentrações
séricas mais altas em crianças.
A intoxicação digitálica acontece com frequência, já que o intervalo entre os níveis
terapêuticos e tóxicos é muito pequeno. Os sintomas clínicos mais comuns de
subredosagem são anorexia, náuseas e vômitos, visão turva e desorientação. A
manifestação miocárdica mais importante e comum da intoxicação digitálica é a
arritmia cardíaca. Frequentemente, o diagnóstico clínico de intoxicação digitálica
torna-se difícil em pacientes com patologias cardíacas graves, pois a maioria das
manifestações pode ser causada pela doença ou por farmacos. Por isso, o
doseamento de digoxina sérica é especialmente útil na interpretação de arritmias.
As maiores causas de intoxicação digitálica são a depleção de potássio e a
diminuição da função renal com a idade. Alterações da função renal, hipercalcémia,
alcalose, mixedema, hipomagnesemia, enfarte do miocárdio recente, hipóxia e
hipocaliémia podem aumentar a sensibilidade aos efeitos tóxicos da digoxina. O uso
de drogas como quinidina, verapamil, amiodarona, ciclosporina, espirinolactona,
entre outras, pode aumentar os níveis séricos por diminuição da clearance da
digoxina.
Quando diante de níveis séricos abaixo do esperado, deve-se avaliar a presença de
doença tiroidiana, má absorção, diminuição do fluxo sanguíneo intestinal por
arteriosclerose mesentérica e uso de drogas como laxantes, antiácidos, fenitoína,
neomicina e metoclopramida, que podem diminuir a absorção. Outro dado a ser
avaliado é o nível de adesão do paciente ao tratamento, tanto na frequência quanto
nas doses prescritas.
O tempo de colheita da amostra é um factor importante na determinação dos níveis
séricos de digoxina. Os níveis séricos elevam-se nitidamente durante a primeira
hora depois de uma dose oral. Isto é seguido por uma diminuição quando a
digoxina é levada para o miocárdio e outros tecidos. Normalmente, para absorção e
armazenamento, quando administrada por via oral, são necessárias 4 a 6 horas, e
por via endovenosa, cerca de 30 minutos. Depois disso, os níveis de digoxina no
soro tendem a estabilizar, passando a diminuir muito lentamente durante as 24 a
48 horas seguintes.
Não é possível determinar com precisão os níveis de digoxina em pacientes em fase
de troca de terapia de digitoxina para terapia com digoxina, visto que ambas as
drogas podem estar presentes no soro. Os anticorpos anti-digoxina usados na
maioria dos testes comerciais para doseamento de digoxina apresentam reacção
cruzada significativa com digitoxina (2% a 6% de reação cruzada, com alguns
casos que chegam a 40%) e outros digitálicos. O problema da reação cruzada é
exacerbado pelo facto de que níveis terapêuticos de digitoxina são
aproximadamente 10 vezes mais altos do que os da digoxina. O anticorpo antidigitoxina também apresenta reacção cruzada com digoxina (2% a 4% de reação
cruzada), mas, por causa dos níveis terapêuticos mais altos, a interferência da
digoxina é menos importante no doseamento de digitoxina.
Obtém-se a maior precisão diagnóstica colhendo a amostra de sangue num
momento padronizado durante a fase estável, quando os níveis de digoxina no soro
reflectem as concentrações cardíacas usuais. Normalmente, o equilíbrio é alcançado
num prazo de cerca de 5 dias, e a colheita é realizada imediatamente antes da
próxima dose, ou a qualquer momento, aquando da suspeita de intoxicação.
Doença de Chagas
A tripanossomíase americana ou doença de Chagas é uma parasitose endémica
causada pelo Trypanosoma cruzi. Pode cursar com infecções agudas ou crónicas.
A forma aguda geralmente é uma doença febril discreta. Pode evoluir,
especialmente em crianças, com um quadro mais exuberante, que é caracterizada
por febre, anorexia, edema da face e dos membros inferiores, linfadenopatias e
hepatoesplenomegalia discreta. Mais raramente ocorre miocardite.
Após a fase aguda, o paciente, permanece infectado e passa para uma fase crónica
assintomática. A doença crónica sintomática irá manisfestar-se anos ou mesmo
décadas após a fase aguda. As clássicas manifestações crónicas relacionam-se com
distúrbios de ritmo e condução cardíaca, tromboembolias, miocardiopatia de
chagas, megaesófago e megacólon.
Nas situações de imunossupressão, pode ocorrer a reactivação do quadro, que
muitas vezes é fatal.
A transmissão ocorre por vectores hematófagos, mas pode dar-se por via
congénita, transfusional e outras formas menos frequentes, como a inoculação
involuntária em laboratório.
Os métodos serológicos para diagnóstico da doença de Chagas apresentam
sensibilidade e especificidade elevadas, sendo úteis para o diagnóstico nas fases
aguda e crónica da doença. Entretanto, é possível a reacção positiva por
reactividade cruzada com as leishmanioses, especialmente com a forma visceral e
as formas cutaneo-mucosas da leishmaniose tegumentar, e com outros antigénios
em comum.
Por isso, é recomendável a realização de reacções por diferentes métodos como
hemaglutinação, imunofluorescência e enzima imunoensaio. Devem ser realizados
no mínimo dois métodos, para que se controlem mutuamente, visto que, em cada
método, são utilizados diferentes antigénios e formas do parasita, o que permite
diminuir a possibilidade de resultados falso-positivos. Resultados positivos devem
ser encontrados nos dois métodos utilizados para confirmar o diagnóstico. Nos
casos de apenas um método apresentar positividade, torna-se necessária a análise
clínica da história epidemiológica, achados clínicos e outros exames diagnósticos
complementares.
Na fase aguda, anticorpos das classes IgM e IgG são detectáveis. Na fase crónica,
são encontrados anticorpos da classe IgG. Os níveis de reactividade diferem para
cada método e de acordo com a finalidade do diagnóstico. Os valores considerados
para triagem de dadores de sangue são sempre inferiores aos considerados para o
diagnóstico clínico.
Electroforese da Hemoglobina
A realização da electroforese da hemoglobina permite a identificação das diferentes
fracções normais e patológicas da hemoglobina.
A hemoglobina é separada, de acordo com a sua migração, num meio sólido
(acetato de celulose), quando submetida a um campo eléctrico. A visualização das
diferentes posições é evidenciada por coloração.
É um método importante para investigar a presença das hemoglobinas anormais
como a hemoglobina S e C e nas talassemias que podem cursar com aumento da
HbA2 e com a presença da HbH. Permite também a quantificação das fracções
normais da hemoglobina como A, A2 e Fetal. As fracções podem ser quantificadas
por densitometria após eluição.
A electroforese é realizada em pH alcalino e pode ser realizada, de forma
complementar, também em pH ácido para melhorar a identificação de
hemoglobinopatias, já que outras fracções anómalas podem migrar na mesma
posição das hemoglobinas S e C como as HbD e HbE
Electroforese das Proteínas
O plasma humano contém mais de 500 proteínas identificáveis. Entre essas, estão
presentes proteínas transportadoras, anticorpos, enzimas, inibidores enzimáticos,
factores da coagulação e proteínas com outras funções. A avaliação das
concentrações de proteínas séricas e as proporções das diferentes fracções de
proteína têm considerável valor no diagnóstico de patologias agudas e crónicas.
A electroforese das proteínas no soro é uma técnica simples para separar as
proteínas do soro. É o teste de triagem mais utilizado para investigação de
anomalias das proteínas séricas. Em condições normais, são separadas cinco
bandas do soro: albumina, alfa-1, alfa-2, beta e gamaglobulinas. Eventualmente,
pode ser observada a presença da pré-albumina. O reconhecimento de
paraproteínas, normalmente encontradas nas gamapatias benignas ou malignas é o
uso diagnóstico mais importante para a electroforese das proteínas. Quando
alteradas, as bandas apresentam-se com padrões conhecidos para importantes
patologias.
A banda de albumina é relativamente homogenea, porém as demais são compostas
por uma mistura de diferentes proteínas.
Informações adicionais podem ser encontradas nos títulos referentes às diferentes
proteínas que compõem as bandas identificadas pela electoforese como: albumina,
alfa-1-antitripsina, alfa-1-glicoproteína ácida, haptoglobina, ceruroplasmina, entre
outras.
Pré-Albumina
Sintetizada pelo fígado, tem como função conhecida o transporte da tiroxina e
desempenhar um papel significativo no metabolismo da vitamina A. Forma um
complexo com o retinol, que se liga posteriormente à vitamina A. Devido à sua
baixa concentração no soro, com frequência não se traduz na electroforese das
proteínas séricas. Contudo, consegue ultrapassar a barreira hemato-encefálica e
pode também ser sintetizada por células do plexo coróide, o que explica seu
aparecimento frequente na electroforese do líquor cefalorraquidiano.
Os níveis de pré-albumina estão significativamente diminuídos em diversas
patologias hepáticas e aumentados em pacientes que usam esteróides, como
também na falência renal e durante a gravidez.
Por ter uma semi-vida muito curta e ser bastante sensível às variações do aporte
alimentar e ao estado funcional hepático, é considerada um bom marcador do
estado nutricional.
Albumina
É a proteína mais abundante no plasma, responsável por cerca de 60% da
concentração total de proteínas. É sintetizada exclusivamente pelo fígado,
aparecendo primeiro no citoplasma dos hepatócitos como um precursor chamado
pré-albumina. Possui um papel muito importante em diversas funções do
organismo, como o transporte de diferentes substâncias e em especial na
manutenção da pressão oncótica.
Foram descritas mais de 20 variantes genéticas de albumina. O tipo mais comum é
chamado albumina A. Essas albuminas variantes podem resultar numa faixa de
albumina larga na electroforese das proteína séricas ou podem dar origem a duas
faixas distintas (bis-albuminémia). Nenhuma dessas variantes foi ainda associada a
manifestações patológicas. Na rara síndrome de ausência congénita de albumina,
os pacientes podem apresentar edema moderado, mas podem poupar as
consequências hemodinámicas com a utilização de mecanismos compensatórios,
como o aumento das globulinas do plasma, que assumem algumas das funções da
albumina. O problema bioquímico principal nesses pacientes é uma alteração no
metabolismo lipídico, como o aumento de colesterol, fosfolipídios e outras
lipoproteínas.
Alfa-1-Globulinas
A alfa-1-antitripsina corresponde a cerca de 90% das proteínas que correm na faixa
das alfa-1-globulinas. A deficiência da alfa-1-antitripsina está associada ao
enfisema pulmonar e à cirrose hepática. Só é detectável pela electroforese quando
homozigótica; os estados heterozigóticos só podem ser identificados por técnicas
imunoenzimáticas que também são utilizadas para confirmação das deficiências
homozigóticas.
É uma das proteínas de fase aguda e pode ser encontrada noutros fluidos
orgânicos, como lágrimas, sémen, bile e líquido amniótico. Nos 10% restantes,
estão a alfa-1-glicoproteína ácida, a alfafetoproteína e outras proteínas. Os níveis
elevam-se nas doenças inflamatórias agudas e crónicas, neoplasias, após traumas
ou cirurgias e durante a gravidez ou terapia com estrogénios. Nos carcinomas
hepatocelulares, a elevação pode acontecer devido ao aumento da alfafetoproteína.
Alfa-2-Globulinas
Incluem a haptoglobina, a alfa-2-macroglobulina e a ceruloplasmina. Raramente se
encontram alterações nessa banda electroforética, já que a diminuição de um
componente é compensada pelos demais mesmo dentro dos valores de referência.
Níveis elevados de alfa-2-macroglobulina associados à diminuição da albumina
acontecem no síndrome nefrótico.
Os níveis de haptoglobina e de ceruloplasmina podem apresentar-se elevados em
numerosas situações que levam à reacção de fase aguda. Os níveis de haptoglobina
apresentam-se diminuídos nas hepatopatias graves, na anemia megaloblástica, nas
situações de aumento da hemoglobina livre, como na hemólise de eritrócitos ou na
reabsorção de grandes hematomas e na terapia com estrogénios e corticóides. Os
níveis de ceruloplasmina aumentam na terapia comestrogénios e encontram-se
diminuídos na doença de Wilson, na desnutrição, no síndrome nefrótico e nas
enteropatias com perda de proteínas.
Betaglobulinas
Composta pelas beta-lipoproteínas (LDL), transferrina, C3 e outros componentes do
complemento, beta-2-microglobulina e antitrombina III. A redução dessa banda é
rara. A anemia por défice de ferro leva ao aumento da transferrina. O
hipotiroidismo, a cirrose biliar, as nefropatias e alguns casos de diabetes mellitus
podem evidenciar-se pelo aumento de colesterol e consequente aumento das betalipoproteínas (LDL). A beta-globulina está elevada nos casos de icterícia obstrutiva
e menos frequentemente nalguns casos de hepatite. Quase sempre, está elevada
nos casos de cirrose hepática. Nesses casos, pode aparecer junto com sobreposição
ou fusão das bandas beta e gama pelo aumento de IgA, que ocorre nas cirroses
hepáticas, infecções de pele ou respiratórias e na artrite reumatóide. Elevações
causadas normalmente pelo aumento dos componentes do complemento podem
ocorrer na hipertensão maligna, doença de Cushing, poliartrite nodosa e
carcinomas.
Gamaglobulinas
Composta pelas imunoglobulinas, predominantemente pela IgG. As imunoglobulinas
A,M,D,E e proteína C reactiva encontram-se na área de junção beta-gama. A
ausência ou a diminuição da banda gama indica imunodeficiências congénitas ou
adquiridas. O aumento dessa banda sugere o aumento policlonal das
gamaglobulinas associadas a doenças inflamatórias crónicas, reacções imunes,
doenças hepáticas ou neoplasias disseminadas.
Bandas oligoclonais podem eventualmente ser observadas em infecções víricas
crónicas, nalgumas infecções bacterianas como as pneumonias por pneumococos e
as hepatites crónicas activas.
Tuberculose, sarcoidose, linfogranuloma venéreo e sífilis terciária são doenças
crónicas que levam ao aumento dessa banda. Artrite reumatóide, lúpus eritematoso
sistémico e outras colagenoses podem apresentar níveis normais a acentuadamente
aumentados, dependendo da fase de actividade da doença. Níveis aumentados
também são encontrados em linfomas malignos, doença de Hodgkin e leucemia
linfocítica crónica. Tipicamente, a macroglobulinemia de Waldenström e o mieloma
múltiplo exibem um pico homogéneo, que pode ou não resultar do aumento total
da banda gama.
As hepatopatias cursam frequentemente com aumento da banda das
gamaglobulinas. Na hepatite, verifica-se um aumento das beta e gamaglobulinas
com redução da albumina. Nas cirroses, o padrão mais sugestivo consiste na
elevação da gamaglobulina de base ampla, juntamente com a fusão da beta e da
gamaglobulina, sem a individualização dos picos da chamada fusão beta-gama.
Apenas cerca de 20% dos cirróticos apresentam a fusão completa, e cerca de 3%
apresentam a fusão parcial.
Alguns Padrões Electroforéticos Típicos
A diminuição das bandas da albumina, alfa-1, beta e gamaglobulinas com a
elevação da faixa alfa-2 é um padrão de perda seletiva de proteínas, como a que
ocorre no síndrome nefrótico. Entretanto, convém lembrar que é necessário uma
diminuição de pelo menos um terço na concentração da albumina para que seja
possível a visualização na electroforese. A alteração característica das reacções de
fase aguda consiste no aumento da alfa-1 e alfa-2-globulinas, quase sempre
associado à diminuição da albumina.
O aumento isolado da banda alfa-1 pode ser observado nas hepatites crónicas, nas
reacções de fase aguda acompanhadas de hemólise, na gravidez e durante a
terapia de estrogénios. O aumento predominante da banda alfa-2 pode ser
observado em patologias que cursam com vasculites como a artrite reumatóide ou
nas doenças por imunocomplexos.
Podem aparecer faixas normalmente não-visualizadas, como um pico que pode
surgir entre a albumina e a região alfa-1, como resultado de um aumento da
alfafetoproteína. Um grande aumento de proteína C reactiva numa reacção de fase
aguda severa pode gerar uma leve faixa na região gama. O aumento de lisozimas
observadas na leucemia monocítica pode produzir uma faixa depois da região de
gama.
O pico monoclonal apresenta-se na região gama, menos frequentemente na faixa
beta e, em raros casos, na região alfa-2. Consiste num pico alto e relativamente
delgado, homogéneo e fusiforme, que normalmente está associado aos casos de
mieloma ou são consequência de patologias como macroglobulinemia de
Waldeström, gamapatias monoclonais secundárias e idiopáticas.
Em contraste com os aumentos policlonais, as gamapatias monoclonais produzem
um padrão muito específico.
As bandas monoclonais ocorrem pelo aumento de clones de imunoglobulinas
distintas, secretadas por uma proliferação monoclonal de células plasmáticas,
acompanhada de quantidades progressivamente menores das imunoglobulinas
policlonais, à medida que as células plasmáticas normais são substituídas pelos
clones malignos.
AVALIAÇÃO LABORATORIAL DA ELECTROFORESE DAS PROTEÍNAS
Electroforese da Urina
A electroforese de proteínas da urina, assim como do soro, é uma técnica simples
para separar as proteínas presentes na urina. A indicação mais importante da
realização da electroforese de proteínas na urina é o reconhecimento de
paraproteínas nas gamapatias benignas ou malignas, especialmente no mieloma
múltiplo. Na presença de proteína de Bence Jones (cadeias leves), métodos
específicos de identificação, como a análise de cadeias kappa e lambda, podem ser
realizados para um diagnóstico mais específico.
Electroforese do Líquor
A realização da electroforese do líquido cefalorraquidiano (LCR) pode auxiliar na
quantificação da proporção albumina/globulinas. A sua maior utilidade está na
identificação de bandas oligoclonais presentes em 70 a 90% dos pacientes com
esclerose múltipla. Entretanto, bandas oligoclonais de IgG também podem ser
encontradas em diferentes situações como pan-encefalite esclerosante subaguda,
encefalite por caxumba, em pacientes com infecção pelo HIV, meningite
criptocócica, linfoma de Burkitt, neurossífilis, síndrome de Guillain-Barré,
carcinomatose meníngia, toxoplasmose e meningoencefalites víricas e bacterianas.
Na presença de lesões traumáticas do SNC, podem cursar nas fases iniciais com um
aumento dos níveis de alfa-2-globulina.
Como as alterações das imunoglobulinas podem ser derivadas de alterações
séricas, é importante a realização concomitante da electroforese do soro e do
líquor. Os casos duvidosos devem ser confirmados por outras técnicas, como a
imunofixação.
Enolase Neurónio-Específica (NSE)
A enolase neurónio-específica (NSE) é uma enzima glicolítica que existe na forma
de diversas isoenzimas diméricas: alfa-alfa, alfa-beta, beta-beta e gama-gama.
Dessas subunidades, as isoenzimas alfa-gama e gama-gama são as enolases
neurónio-específica que são encontradas principalmente nos neurónios e nas
células neuroendócrinas.
É um marcador diagnóstico de relativa especificidade (85%) para carcinoma de
pequenas células de pulmão. Pode ser usado para monitorizar os efeitos da terapia
e indicar recidivas antes das evidências clínicas.
É encontrado também noutras condições malignas como neuroblastoma,
melanoma, carcinoma de células das ilhotas pancreáticas e hipernefroma. No
neuroblastoma, o NSE correlaciona-se com o prognóstico, mas não é útil para o
acompanhamento das recidivas.
Epstein-Barr Vírus (EBV)
O vírus Epstein-Barr (EBV) é um herpesvírus humano conhecido por causar a
mononucleose infecciosa e também pela sua provável participação na etiologia de
alguns carcinomas (nasofaríngeo), linfomas (linfoma de Burkitt) e doenças
linfoproliferativas em pacientes imunodeprimidos. Recentemente, foi descrita o
síndrome ativo crónica severo, no qual a linfoproliferação continua activa. Já o
papel etiológico do EBV noutras patologias como artrites reumáticas, doença de
Hodgkin e síndrome de fadiga crónica ainda não está bem definido.
Na fase aguda da mononucleose infecciosa, anticorpos IgM e IgG para antigénios
precoces do EBV (EA), antígénios da capsíde viral (VCA) e antigénios nucleares
(EBNA) aparecem em sequência. A pesquisa mais utilizada é a de anticorpos IgM
contra VCA por enzima imunoensaio. A positividade indica infecção aguda.
Entretanto, a presença de anticorpos IgM para VCA ou IgM /IgG para EA, com
ausência ou baixa concentração de anticorpos contra EBNA, indica infecção recente
ou em curso.
A presença de anticorpos contra VCA e a relação IgG/ IgM inferior a 1 podem
auxiliar no diagnóstico de casos dificeis. A análise da avidez de anticorpos IgG
contra VCA do EBV também é útil em casos de reactivação ou infecção recente. A
persistência de EA e/ou VCA IgG em títulos altos indica infecção crónica pelo EBV.
A pesquisa de anticorpos específicos para EBV deve ser realizada nos quadros de
mononucleose para confirmação do diagnóstico ou nos casos de suspeita clínica que
cursa com anticorpos heterófilos negativos, não sendo diagnosticada pelo métodos
tradicionais, e também para o diagnóstico diferencial das patologias que podem
mimetizar um quadro de mononucleose (mononucleose-like), como hepatites
víricas agudas, colagenoses, síndrome de seroconversão do HIV-1, citomegália e
toxoplasmose.
A comparação de métodos de cultura de células e a amplificação genómica por PCR
indica a maior sensibilidade da técnica molecular (PCR ) na detecção de EBV. A
infecção crónica por EBV pode ser detectada pela presença do DNA-EBV em sangue
periférico por PCR. Em pacientes em tratamento profilático com imunoglobulinas, o
diagnóstico serológico pode acarretar problemas relativos a reacções falsopositivas.
A técnica de PCR usando células mononucleares de sangue periférico pode ser
usada para o diagnóstico preciso e precoce da infecção por EBV em pacientes
altamente vulneráveis, com doenças linfoproliferativas, nos quais a PCR se
apresenta altamente sensível, mesmo em amostras de saliva.
A PCR é útil em detectar o EBV noutras patologias tais como pneumonia intertiscial,
pericardite e miocardites a partir da análise de tecidos ou de líquidos. Entretanto,
deve-se ter o cuidado de proceder à avaliação qualitativa e quantitativa em
amostras colhidas em diferentes datas, além da correlação clínica para a
confirmação da relação entre os sintomas e a etiologia vírica.
Consultar Mononucleose Infecciosa
Eritropoietina
A eritropoietina (EPO) é um factor regulador na formação dos glóbulos vermelhos
do sangue. O rim é responsável por 90% da síntese da EPO circulante. Em
situações anormais, pode ser produzida noutros órgãos, como ocorre nos casos de
neoplasias.
O seu doseamento está indicado na avaliação das anemias, policitemias, como
marcador nalguns tumores e na monitorização dos níveis terapêuticos de EPOrecombinante.
Os níveis séricos estão diminuídos na policitemia vera, na insuficiência renal crónica
e na doença de Hodgkin. Níveis elevados podem ser encontrados em condições que
cursem com hipóxia, como em grandes altitudes, presença de hemoglobinas
anormais, cardiopatias e pneumopatias.
Exame Directo para Fungos
O exame microscópico directo de amostras clínicas tem como finalidade detectar a
presença de fungos em material colhido de pacientes com suspeita de infecção
fúngica. A presença de fungos pode ser detectada por meio de diferentes
colorações, tais como: hidróxido de potássio 20%, isolado ou acrescido de tinta
Parker ou de DMSO (dimetilsulfóxido) e Giemsa. A escolha da coloração varia
conforme o tipo de material a ser analisado e a suspeita clínica.
A visualização de estruturas fúngicas em material clínico é um importante
instrumento diagnóstico. O exame directo pode fornecer identificação completa ou
parcial do fungo (quando apenas o género ou o grupo pode ser identificado). Em
ambos os casos, proporciona informação necessária para a escolha da conduta
terapêutica.
Em certos casos, a identificação pelo exame directo é o único exame necessário
para o diagnóstico. São exemplos dessa situação a identificação dos agentes
etiológicos de algumas micoses, como pitríase versicolor (Malassezia furfur),
lobomicose (Loboa loboi), rinoesporodiose, adiaspiromicose.
Noutras micoses, o agente pode ser identificado pelo exame directo, como por
exemplo o Paracoccidioides brasiliensis e o Cryptococcus neoformans. Entretanto, é
necessário proceder à realização da cultura para fungos. Trata-se de um exame
indispensável, visto que os fungos podem permanecer presentes no exame directo
por longo tempo, mesmo após o tratamento, sem no entanto serem viáveis.
Portanto, não representa um bom parâmetro para controlo de cura.
As amostras deverão ser colhidas em frasco estéril, de maneira asséptica. Os
materiais biológicos necessários são:
escamas de pele, unhas e pêlos (micoses superficiais e cutâneas); aspirado de
lesões, secreções, biópsias de pele (micoses subcutâneas); expectoração, lavado
brônquico, aspirado brônquico, escovado brônquico (micoses sistémicas);
secreções: pulmonar, vaginal, traqueal, orotraqueal, de lesões cutâneas,
abdominal, oral, de conjuntivas ou de qualquer outra localização (micoses
sistémicas); sangue, biópsia de qualquer órgão ou tecido, urina, líquor, líquido
sinovial, ascítico, amniótico ou outros líquidos orgânicos (micoses sistémicas).
As infecções fúngicas podem atingir qualquer órgão ou tecido do corpo humano.
Portanto, qualquer material biológico pode ser analisado para a pesquisa de fungos.
Os resultados devem ser interpretados de acordo com o tipo de material e o local
da lesão. Muitas vezes, o fungo pode ser parte da flora local, não tendo significado
clínico.
Exame Microscópico Directo para Fungos com Tinta-da-China
É utilizado para a pesquisa de leveduras do género Cryptococcus em qualquer
material clínico. As amostras são coradas com tinta-da-china, com a finalidade de
visualizar as leveduras encapsuladas. A tinta-da-china não é capaz de penetrar na
cápsula dessas leveduras, mas é capaz de corar todo o material em negro,
permitindo o fácil reconhecimento das leveduras encapsuladas, que permanecem
claras. A sua presença é indicativa de criptococose, e o seu achado é fundamental
para confirmação diagnóstica, embora não permita garantir que as leveduras sejam
viáveis. A sua viabilidade terá que ser avaliada pela confirmação por realização da
cultura para fungos, visto que podem permanecer presentes no exame directo por
um longo tempo, mesmo após o tratamento, sem no entanto serem viáveis.
Portanto, não representam um bom parâmetro para controlo de cura.
Exame Parasitológico de Feses
A investigação da presença de parasitas nas fezes é realizada pela pesquisa de ovos
ou larvas de helmintas, e nas infecções por protozoários, quando se encontram
cistos ou oocistos nas fezes.
O exame parasitológico de fezes frescas possui uma excelente especificidade.
Entretanto, a sua sensibilidade só será adequada (95%) se forem solicitados
exames de pelo menos três amostras de fezes em dias distintos. Portanto, um
resultado negativo numa única amostra não elimina a possibilidade de uma
parasitose.
A positividade vai depender de diferentes factores. O estadio da infecção, o ciclo do
parasita, da eliminação intermitente de formas de resistências, da intensidade do
parasitismo e o exame propriamente dito, que utiliza apenas uma pequena amostra
de todo o material colhido, são alguns dos factores que interferem na possibilidade
de o exame se revelar positivo. Além disso, algumas parasitoses necessitam de
exames especiais para serem identificadas, como por exemplo o Enterobius
vermicularis, sendo também o caso da investigação da presença de trofozoitos.
São necessárias fezes frescas colhidas no frasco de plástico. No caso de fezes
sólidas ou pastosas, a quantidade deverá corresponder a 5 colheres plásticas
fornecidas com o frasco de colheita. Se as fezes estiverem liquefeitas, pelo menos
10 mL deverão ser fornecidos ao laboratório para análise.
As fezes deverão ser colhidas originalmente num recipiente limpo e a seguir
transferidas para o frasco fornecido pelo laboratório. O paciente não deve estar a
fazer laxantes ou ter sido submetido a contrastes radiológicos nos 3 dias anteriores
à colheita.
Durante a colheita, é importante evitar a contaminação pela urina, pois a sua
presença acelera a fermentação bacteriana, prejudicando a conservação.
ACHADOS PATOGÉNICOS
MAIS FREQUENTES
Ascaris lumbricoides Hymenolepis diminuta
Cryptosporidium sp.
Hymenolepis nana
Dietamoeba fragilis
Isospora beli
Entamoeba
Strongyloides
histolytica
stercolaris
Enterobios
Trichuris trichiura
vermicularis
Giardia lamblia
Schistosoma mansoni
ACHADOS NÃO
PATOGÉNICOS
Chilomastix mesnillii
Endolimax nana
Entamoeba coli
Iodamoeba butschlii
Exame Parasitológico de Feses/ MIF
Como estão presentes amostras de fezes de 3 dias diferentes, a sensibilidade do
exame aumenta. É útil na pesquisa de estrutura de resistência de helmintas (ovos e
larvas) e protozoários (cistos e oocistos).
As amostras de fezes são colhidas durante 3 dias consecutivos ou não (emissões
diferentes), em líquido conservante MIF (Merthiolate-Iodo-Formol). No casos de
fezes sólidas ou pastosas, deverá ser considerada como medida uma colher
fornecida com o frasco de colheita de cada emissão. Se as fezes estiverem
liquefeitas, deverão ser colhidos aproximadamente 2 ml de cada emissão.
As fezes deverão ser colocadas num frasco com líquido conservante (MIF), tendo-se
o cuidado de colocar pequenas porções de fezes de 3 emissões diferentes,
misturando bem todas as fezes. Deve-se evitar encher demais o frasco, pois as
fezes devem ficar totalmente cobertas pelo líquido conservante. Manter em local
fresco. Contra-indicados laxantes e enemas. O paciente não deve ter sido
submetido a contrastes radiológicos nos 3 dias anteriores à colheita.
Factor Reumatóide
O termo factor reumatóide (FR) engloba um grupo de auto-anticorpos das classes
IgG, IgM e IgA que tem em comum a capacidade de reagir com diferentes epítopos
da porção Fc da molécula da imunoglobulina G humana.
Apesar da pequena evolução no conhecimento dos mecanismos de ligação do FR
aos auto-antigénios e do seu envolvimento no processo patológico típico da artrite
reumatóide (AR), o FR IgM pode servir como marcador precoce na AR, apoiando-se
em dados que demonstram que o risco de desenvolvimento de AR aumenta de
forma proporcional ao aumento da concentração de FR em indivíduos normais. Os
pacientes com artrite que cursam com FR positivo, principalmente quando se
encontram em concentrações elevadas, correm maior risco de apresentar
complicações clínicas e uma menor resposta à terapia.
O FR está presente em cerca de 50-90% dos casos de AR clássicos, alguns meses
após o início da doença, e dessa percentagem, 17% em média apresentam-se
negativos nas fases mais precoces da doença. Durante a fase activa, são
encontradas concentrações mais elevadas, que começam a decair à medida que o
paciente evolui para a remissão clínica, mantendo-se positivas em níveis baixos e
estáveis e tornando a elevar-se nos períodos de reactivação da doença.
O FR está aumentado também em 75 a 95% dos quadros de síndrome de Sjögren,
em 50 a 60% dos pacientes com doença mista do tecido conjuntivo (DMTC), em 15
a 35% dos casos de lúpus eritematoso sistémico (LES), em 20 a 30% dos casos de
esclerodermia, noutros casos de colagenoses e noutras patologias, como nefropatia
e crioglobulinemia.
Sabe-se hoje que o FR não é produzido apenas sob condições patológicas, e uma
pequena parcela da população normal, especialmente os idosos, pode apresentar
positividade para FR. Essas percentagens de incidência, tanto nas patologias como
nos pacientes normais, assim como a ocorrência de falsos positivos, variam de
acordo com a sensibilidade e a especificidade do método utilizado.
Os ensaios tradicionais para investigação do FR empregavam partículas de látex
revestidas por imunoglobulina G humana (prova do látex) ou, na hemaglutinação
indirecta, hemácias de carneiro, revestidas por imunoglobulina de coelho (reacção
de Waller-Rose). A prova do látex era considerada mais sensível, e a reacção de
Waller- Rose, mais específica. Realizadas em conjunto, forneciam dados
complementares.
Actualmente, o método de referência para a pesquisa do FR é a nefelometria, que
fornece um resultado numérico em UI/mL, em vez dos resultados em títulos,
resultantes de diluições fornecidas pelo método anterior (látex), o que permite um
melhor acompanhamento dos pacientes. Com a nefelometria, podem ser
identificadas três classes de auto-anticorpos, isto é o FR das classes IgG, IgM e IgA.
A identificação e a quantificação da classe do FR que se encontra elevada podem
ser realizadas pelo método de ensaio imunoenzimático. A utilidade clínica dessa
individualização e quantificação tem sido cada vez mais explorada. Por exemplo, a
presença de FR IgA nas manifestações extra-articulares da AR com ausência de FR
IgM ou IgG; a predominância na AR de FR IgM, sendo que o FR IgG e IgA estão
geralmente presentes, mas em baixa frequência e quantidade. É rara a detecção
concomitante do FR das três classes noutra patologia que não a artrite reumatóide.
Afoiçameto das Hemácias ou Faucização das Hemácias
O teste de afoiçamento reproduz in vitro as condições de baixa tensão de oxigénio
que levam as hemácias que contêm hemoglobina S a sofrerem falcização. Para isso,
são utilizadas substâncias redutoras, como o metabissulfito de sódio.
É importante lembrar que o teste é positivo para anemia falciforme, na presença de
traço falcêmico e também para algumas outras variantes anormais da hemoglobina,
como a Hb Bart e a HbC Harlem. O teste pode ser falsamente negativo em baixas
concentrações de HbS e em altas concentrações de Hb fetal.
Fenitoína
Trata-se de uma droga anticonvulsivante, utilizada no tratamento da epilepsia, de
outros estados convulsivos secundários e nalguns casos de intoxicação digitálica e
arritmias cardíacas. Actua modulando o canal sináptico de sódio e prolongando a
inactivação, o que reduz a habilidade do neurónio de responder à alta frequência. O
efeito fisiológico dessa acção é a redução da transmissão sináptica central,
ajudando ao controlo de excitabilidade dos neurónios anormal.
A absorção do fármaco é lenta e por vezes incompleta. Cerca de 90% do fármaco
está ligada a proteínas. Apenas a fracção livre (cerca de 10%) é biologicamente
activa. É metabolizada no fígado, sendo a maior parte excretada na bilis na forma
de metabolitos inactivos, que são reabsorvidos no intestino e excretados na urina.
Independentemente de estar nos níveis normais, a fenitoína pode interferir na ação
de outros fármacos como anticoagulantes orais, ciclosporina, teofilina e
contraceptivos orais. Pode diminuir a concentração sérica ou a eficácia de outras
drogas, como a carbamazepina, ácido valpróico, primidona, corticosteróides,
dopamina e cloranfenicol.
Níveis baixos são encontrados, geralmente, nos casos de não-adesão ao tratamento
ou de utilização inadequada pelo paciente. Os problemas de absorção são mais
frequentes e importantes em crianças, especialmente até os primeiros 3 meses de
vida.
Níveis elevados encontrados em pacientes com uso crónico e adequadamente
monitorizados, sem alteração da posologia, podem ocorrer por interacções
medicamentosas ou infecções. As drogas que podem interagir, elevando os níveis
séricos de fenitoína, são, entre outras, antidepressivos tricíclicos, cloranfenicol,
cimetidina, amiodarona, sulfas, fenilbutazona, etanol, halotano e metronidazol.
Os valores de pico normalmente aparecem entre 4 a 5 horas após a última dose.
Entretanto, a grande variação inter-individual de metabolização faz com que os
níveis do fármaco necessários para alcançar os níveis terapêuticos e os momentos
de pico sejam extremamente variáveis. A semi-vida é de 20 a 40 horas nos adultos
e de cerca de 10 horas nas crianças.
São considerados níveis terapêuticos valores entre 10 a 20 mg/mL. Entretanto,
como já citado, a grande variação individual tanto na absorção quanto na
velocidade de metabolização do fármaco faz com que esses valores possam variar
de paciente para paciente, sendo aceite como nível terapêutico ideal aquele que
não apresente efeitos tóxicos e no qual não ocorram convulsões.
Os efeitos tóxicos manifestam-se por sonolência, ataxia, nistagmo, diplopia ,
disartria e confusão mental. Apesar das variações individuais, raramente se
observam sinais clínicos abaixo dos valores definidos como terapêuticos. O
nistagmo geralmente apresenta-se com valores acima de 20 mg/mL. Já a ataxia é
observada mais frequentemente em níveis entre de 25 a 30 mg/mL, e a disartria e
a sonolência, em níveis de 40 mg/mL
Recomenda-se que a colheita seja realizada imediatamente antes da dose seguinte
e com níveis estáveis (cerca de 7 dias de tratamento), ou a qualquer hora, caso se
suspeite de intoxicação.
Fenobarbital
É um barbitúrico de acção prolongada, frequentemente utilizado no tratamento dos
quadros de convulsões tónico-clónicas generalizadas e convulsões parciais simples
com sintomas motores, formas de epilepsia focal, bem como na ansiedade e na
insónia.
A monitorização dos níveis séricos é fundamental para se obterem os melhores
níveis terapêuticos, evitando-se os efeitos colaterais. Ou seja, é importante para
individualizar a posologia de cada paciente.
Níveis baixos são encontrados frequentemente por não adesão ao tratamento.
Porém, algumas interações medicamentosas também podem levar a concentrações
séricas baixas. Níveis séricos elevados são encontrados com o uso concomitante de
ácido valpróico, que inibe o metabolismo do fenobarbital, e nos casos de doses
inadequadas.
O fenobarbital é metabolizado pelo fígado e tem uma semi-vida longa, de 50 a 140
horas, o que leva ao risco de doses cumulativas. Cerca de 40 a 50% está ligado à
proteína.
O fenobarbital pode afetar o metabolismo da fenitoína, acelerar a clearance de
eliminação do cloranfenicol, teofilina, anticoagulantes orais, ciclosporina e
contraceptivos orais. Pode diminuir a concentração sérica ou o efeito da
fenilbutazona, griseofulvin, beta-bloqueadores, teofilina, corticóides,
antidepressivos triciclícos, quinidina, haloperidol e propoxifeno.
A furosemida, o ácido valpróico e os salicilatos podem inibir o metabolismo do
fenobarbital, levando a um aumento de sua concentração sérica. A sua actividade é
aumentada quando associada ao uso de bebidas alcoólicas, anti-histamínicos,
neurolépticos e antidepressivos tricíclicos. O uso de fenobarbital acelera a
degradação de outros agentes antiepilépticos, anticoagulantes orais, neurolépticos
e contraceptivos orais.
O uso de fenobarbital pode levar ao aumento da fosfatase alcalina, da gama GT e à
diminuição da bilirrubina e do cálcio sérico. O fenobarbital aumenta o metabolismo
da bilirrubina por indução enzimática e pode ser utilizado nos casos de
hiperbilirrubinémia congénita (não- hemolítica, familiar, não-obstrutiva).
Os pontos de equilíbrio são alcançados nos adultos em 10 a 25 dias e, em crianças,
entre 7 a 17 dias. A média é de 14 a 21 dias. O pico é atingido entre 6 a 18 horas
após a administração. Os efeitos colaterais mais frequentes são sonolência,
nistagmo, ataxia e disartria. Em doses elevadas, pode provocar o aumento da
frequência das convulsões.
Recomenda-se que a colheita seja realizada imediatamente antes da dose seguinte
e com níveis estáveis (cerca de 7 dias de tratamento), ou a qualquer hora caso se
suspeite de intoxicação.
Ferritina
A ferritina é a mais importante proteína de reserva do ferro e é encontrada em
todas as células, especialmente naquelas envolvidas na síntese de compostos
férricos e no metabolismo e na reserva do ferro. O doseamento da ferritina é o
mais fiel indicador da quantidade de ferro armazenada no organismo. A sua grande
utilidade clínica está no diagnóstico diferencial entre as anemias hipocrómicas e
microcíticas por deficiência de ferro de anemias por outras etiologias. Nesses casos,
a ferritina diminui antes das alterações dos níveis de ferro sérico e das alterações
morfológicas da série vermelha.
Entretanto, por fazer parte do grupo de proteínas de fase aguda, a ferritina elevase em resposta a infecções, traumatismos e inflamações agudas. A elevação ocorre
nas 24 a 48 horas iniciais, com um pico no terceiro dia, e mantém-se por algumas
semanas, o que pode dificultar a sua interpretação.
Os seus níveis podem elevar-se no excesso de ferro, em pacientes transfundidos e
em neoplasias, especialmente nas leucemias e linfomas e nos carcinomas de
mama, fígado, pulmão, cólon e próstata. Elevam-se também nas anemias
hemolíticas e megaloblásticas e nas lesões hepáticas, especialmente as lesões
alcoólicas. Cerca de 25% dos pacientes com hepatite crónica têm aumento da
ferritina.
Ferro
O ferro é absorvido principalmente na parte superior do duodeno e no jejuno. Uma
vez absorvido, liga-se à transferrina plasmática. A maior parte do ferro circulante é
captada pelos precursores eritróides na medula óssea para formar a hemoglobina.
A hemoglobina utiliza cerca de 80% do ferro corporal. O restante é armazenado no
interior das células reticulares da medula óssea, baço e fígado, 60% sob a forma de
ferritina e cerca de 40% como hemossiderina.
Portanto, a distribuição do ferro corporal é feita entre compartimentos: o funcional
- hemoglobina, mioglobina, enzimas heme e não-heme, o de transporte transferrina - e o de reserva - ferritina e hemossiderina.
A maior parte do ferro corporal é proveniente da dieta. É ingerido no estado férrico
e, para a sua absorção, precisa estar na forma reduzida (ferroso). Por isso, a
absorção é influenciada por factores redutores gastrointestinais, pH gástrico e
composição da dieta, como a presença de ácido ascórbico, que mantém o ferro no
estado ferroso. Outra fonte do ferro é a proveniente do processo de degradação da
hemoglobina, quando o ferro é libertado da hemoglobina e retorna à transferrina
plasmática.
Ao contrário de outros elementos, a homeostase do ferro é regulada pelo controlo
da absorção, estando directamente relacionada às reservas de ferro e ao nível de
eritropoiese. Quando as reservas de ferro diminuem ou o nível de eritropoiese
aumenta, a taxa de absorção de ferro aumenta de forma compensatória.
Os sinais clínicos da deficiência de ferro resultam de um longo período de
desequilíbrio no balanço de ferro.
Inicialmente ocorre uma depleção das reservas, sem alterações nos níveis de ferro
sérico, o que pode ser evidenciado pela redução dos níveis de ferritina. Essa
diminuição das reservas leva ao aumento da absorção intestinal de ferro. Num
segundo momento, com as reservas já depletadas, mas com o nível de
hemoglobina ainda normal, algumas alterações podem já ser evidenciadas, como a
presença de hemácias microcíticas no exame do sangue periférico, mesmo com
volume corpuscular médio (VCM) normal, presença de anisocitose, diminuição da
saturação de transferrina e da ferritina e aumento do TIBC (capacidade total de
combinação do ferro). Finalmente, os níveis de hemoglobina começam a cair,
instalando-se um quadro clássico de anemia ferropriva, com diminuição do ferro
sérico, da hemoglobina, do VCM, da saturação da transferrina e da ferritina.
A deficiência de ferro pode ocorrer por diferentes mecanismos:
- Por aumento das necessidades, como ocorre na gravidez, na lactação e na fase de
rápido crescimento.
- Por diminuição da ingestão, numa dieta pobre em proteínas animais.
- Por diminuição da absorção, como nos casos de cirurgias ou de patologias que
levem a má absorção.
- Por perda excessiva, como sangramentos agudos ou crónicos.
A perda sanguínea constitui a causa mais importante de deficiência de ferro em
adultos. Nos homens e nas mulheres na pós-menopausa, quase sempre é
consequência de perda sanguínea pelo tubo gastrointestinal, causada por patologias
benignas e malignas. O uso crónico de medicamentos como antiinflamatórios,
corticoesteróides, salicilatos e também o uso de álcool podem causar ou agravar os
quadros gastrointestinais. Nas mulheres, a hemorragia vaginal é também um factor
importante.
Quando a perda é aguda, leva a uma anemia normocrómica e normocítica, e,
quando crónica, a uma anemia microcítica hipocrómica. A hemorragia crónica quase
sempre esgota as reservas corporais de ferro, pelo esforço contínuo da medula em
restaurar os níveis circulantes de hemoglobina. A pesquisa de sangue oculto é um
exame importante na investigação dos pacientes com anemia ferropriva e deve ser
realizada em amostras múltiplas para impedir a possibilidade de negatividade
quando as perdas sanguíneas são intermitentes.
A deficiência de ferro por ingestão inadequada quase sempre é subclínica. A
infecção por ancilostomídeos é uma causa frequente de hemorragia.
FERRO SÉRICO DIMINUÍDO
Anemia Ferropriva
Doenças Crónicas
Neoplasias
Hipermenoréia
Hemorragias
FERRO SÉRICO AUMENTADO
Anemias Hemolíticas e Megaloblásticas
Aplasias Medulares
Necrose Hepática Maciça
Hemocromatose - Hemossiderose
Estrogenioterapia - Ferroterapia
A única anemia microcítica e hipocrómica em que as reservas de ferro estão
ausentes é a anemia ferropriva. Na maior parte dos casos, a avaliação dos
indicadores como ferro baixo, TIBC aumentado, ferritina e saturação de transferrina
diminuídas faz o diagnóstico. Entretanto, estes indicadores podem estar alterados
na presença de neoplasias, infecções, processos inflamatórios, alcoolismo,
desnutrição ou doença hepática, prejudicando a avaliação.
A determinação concomitante do ferro sérico e da transferrina permite a avaliação
da percentagem de saturação da transferrina, que é o melhor índice de avaliação
do armazenamento do ferro. A avaliação da capacidade de fixação do ferro (TIBC)
aumenta em resposta à diminuição do ião.
FERRO
Deficiência de ferro
Infecções Crónicas
Neoplasias
Menstruação
Gravidez
Hepatites
Nefropatias
Talassemia
TIBC
SATURAÇÃO DA
TRANSFERRINA
N
,N
Adaptado da tabela: Clinical Diagnosis and Management,
by Laboratory Methods- 19º edição -1999.
Fibrinogénio
O fibrinogénio (Factor I) é uma glicoproteína sintetizada no fígado e está envolvida
na etapa final da coagulação, que consiste na sua conversão em fibrina, sob a
acção da trombina.
Além do seu papel na coagulação, é uma importante proteína na resposta de fase
aguda. Portanto, pode estar elevado em diferentes patologias, como processos
inflamatórios e infecciosos agudos, traumas, neoplasias, pós-operatório, uso de
anticoncepcionais orais e síndrome nefrótico. Encontra-se também elevado por
influências genéticas, na gravidez e no tabagismo. No entanto, pode estar reduzido
devido à diminuição da produção hepática, em doenças hepáticas graves, ou por
aumento de consumo, com conversão excessiva do fibrinogénio em fibrina, sem
tempo para reposição adequada, como nos quadros de coagulação intravascular
disseminada. Pode também apresentar-se diminuído nos casos de fibrinólise
primária e secundária e devido ao uso de agentes fibrinolíticos.
Já foram identificadas diversas variantes hereditárias do fibrinogénio
(disfibrinogenemia). Os quadros podem variar entre alterações hemorrágicas,
tendência a distúrbios trombóticos ou indivíduos assintomáticos. A
disfibrinogenemia adquirida está associada a doenças hepáticas ou renais.
A sua avaliação tem um papel importante no diagnóstico diferencial das
coagulopatias adquiridas, na coagulação intravascular disseminada, na fibrinólise
primária e secundária, na disfibrinogenemia e na afibrinogenemia.
Fosfatase Ácida
As maiores concentrações da fosfatase ácida ocorrem na próstata, no fígado, na
medula óssea, nos eritrócitos e nas plaquetas.
Os pacientes com carcinoma da próstata confinados dentro da cápsula,
normalmente apresentam níveis normais da fosfatase ácida sérica; já nos casos
com metástases, mais da metade dos pacientes apresenta níveis elevados. Níveis
alterados podem também ser observados em pacientes com hipertrofia benigna de
próstata, retenção urinária e após manipulação prostática.
A fracção não-prostática encontra-se fisiologicamente elevada nas crianças em fase
de crescimento e patologicamente aumentada em condições em que existe um
hipermetabolismo ósseo. Valores elevados são encontrados na doença de Paget e
noutras patologias ósseas, no hiperparatiroidismo, nas metástases ósseas como no
carcinoma da mama, do pulmão, da tiróide, nos mielomas e em situações de
grande destruição de eritrócitos e de plaquetas. Elevam-se também em doenças
metabólicas como doença de Gaucher, doença de Niemann-Pick e em leucemias e
outras patologias hematológicas.
Consultar Fosfatase Ácida Prostática.
Fosfatase Ácida Prostática
A fosfatase ácida de maior importância clínica é a derivada da próstata. Dada a
importância clínica do aumento do nível sérico da fosfatase ácida no diagnóstico e
na monitorizaação da resposta ao tratamento e no aparecimento de metástases no
carcinoma da próstata, é necessário diferenciar-se especificamente entre aumentos
nas concentrações das fosfatases de origem prostática das formas de origem nãoprostática.
Certos inibidores podem aumentar a discriminação entre as duas formas. A fracção
prostática é fortemente inibida pelo tartarato, e a fração não-prostática é resistente
ao tartarato. Dessa forma, podemos obter valores mais específicos para o
acompanhamento dos casos de carcinoma prostático. Devemos lembrar que a
fosfatase ácida libertada pelas plaquetas apresenta resposta à inibição semelhante
à de origem prostática.
Os pacientes com carcinoma de próstata confinados dentro da cápsula
normalmente apresentam níveis normais da fosfatase ácida sérica; já nos casos
com metástases, mais da metade dos pacientes apresenta níveis elevados. Níveis
alterados podem ser observados em pacientes com hipertrofia benigna de próstata,
retenção urinária de monta e após manipulação prostática.
Fosfatase Alacalina
É uma enzima presente em praticamente todos os tecidos do organismo,
especialmente nas membranas das células dos túbulos renais, ossos (osteoblastos),
placenta, trato intestinal e fígado. Portanto, a fosfatase alcalina encontrada no soro
é resultado da presença de diferentes isoenzimas originadas em diferentes órgãos,
com predomínio das fracções ósseas e hepáticas.
Embora até hoje a sua função ainda não esteja bem definida, a fosfatase alcalina
parece estar envolvida no transporte de lípidos no intestino e nos processos de
calcificação óssea. A fosfatase alcalina óssea e a hepática partilham proteínas
estruturais, codificadas por um mesmo gene. A fosfatase intestinal só se expressa
em indivíduos dos grupos sanguíneos Lewis O e B. Além das isoenzimas conhecidas
- óssea, hepática, intestinal e placentária -, podem ser encontradas isoformas
patológicas, como a carcioplacentária ou Regan, que ocorre por uma depressão do
gene da fosfatase placentária em neoplasias (do ovário, do pulmão, trofoblásticas,
gastrointestinais, seminomas e doença de Hodgkin). Outras isoformas incomuns
têm sido descritas em várias neoplasias.
Na prática clínica, a grande utilidade está na investigação de doenças hepatobiliares
e nas doenças ósseas que cursam com aumento da actividade osteoblástica.
Como é totalmente excretada pela bilis, durante muito tempo pensou-se que sua
elevação nas patologias hepatobiliares resultava da falência de excreção da enzima.
Hoje sabe-se que a resposta hepática a qualquer tipo de agressão da árvore biliar é
sintetizar a fosfatase alcalina principalmente nos canalículos biliares. Isto explica a
sua marcada elevação nas patologias biliares.
A elevação tende a ser maior nas obstruções extra-hepáticas (litíase e carcinoma
da cabeça de pâncreas) do que nas intra-hepáticas (processos invasivos). Isto
acontece devido a uma combinação do aumento de produção associado a uma
diminuição da excreção. Por isso, é considerada um marcador importante para
processos obstrutivos hepáticos. Níveis elevados podem ser também encontrados
noutras lesões hepáticas activas e nas infiltrativas com níveis mais moderados de
elevação. Está aumentada nos carcinomas hepáticos primários e secundários.
Nas doenças ósseas, o maior aumento dos níveis séricos da fosfatase alcalina
aparece na doença de Paget (valores de 10 a 25 vezes o normal). Níveis
moderadamente elevados podem ser encontrados na osteomalácia, nalguns
tumores ósseos e no hiperparatiroidismo primário e secundário. As fracturas ósseas
levam a um aumento transitório, e na osteoporose os valores são normais.
Nas neoplasias, os níveis da fosfatase alcalina são úteis para avaliar a presença de
metástases envolvendo fígado e osso. Valores muito elevados são observados em
pacientes com lesões osteoblásticas como as encontradas no carcinoma da próstata
com metástases ósseas. Elevações menores são observadas quando as lesões são
osteolíticas, como as encontradas no carcinoma metastático da mama. Outras
condições malignas com infiltração hepática como leucemias, linfomas e sarcoma
podem cursar também com elevação da fosfatase alcalina.
Recém-nascidos e crianças, mas especialmente adolescentes, apresentam valores
significativamente mais elevados do que os adultos, devido ao crescimento ósseo.
Durante a fase de crescimento rápido da adolescência (puberdade), são
encontrados níveis extremamente elevados. Normalmente, os valores são
discretamente mais elevados nos homens do que nas mulheres, e essa diferença
desaparece durante e após a menopausa. Na população idosa, existe uma
diminuição dos níveis séricos habitualmente encontrados, como consequência do
aumento da incidência de osteoporose nessa faixa etária. Níveis elevados, duas a
três vezes os valores de referência, podem ser encontrados durante a gravidez,
especialmente no terceiro trimestre, devido à produção placentária.
Níveis diminuídos podem ser encontrados no hipotiroidismo, na anemia perniciosa,
nas hipofosfatémias e no uso de fármacos como contraceptivos orais. Aumento dos
níveis séricos podem ser encontrados após uma refeição com alimentos ricos em
gordura, especialmente em pacientes do grupo sanguíneo O ou B, devido à
elevação da fracção intestinal. Recomenda-se portanto que seja avaliada sempre
em jejum.
ALGUMAS CAUSAS DE FOFATASE ALCALINA ELEVADA
PUBERDADE
GRAVIDEZ
CRIANÇAS
DOENÇA DE PAGET
ACROMEGALIA
SARCOMA
HIPERPARATIROIDISMO
OSTEOMALÁCIA
MIELOFIBROSE
HIPERNEFROMAS
LEUCEMIAS
MIELOMA
ALCOOLISMO
CIRROSE
CIRROSE BILIAR PRIMÁRIA
COLANGITES
COLANGITES AUTO-IMUNES COLEDOCOLITÍASE
CA DO PÂNCREAS
COLECISTITES
COLANGITES
CA HEPÁTICO
METÁSTASES HEPÁTICAS
METÁSTASES ÓSSEAS
SARCOIDOSE
AMILOIDOSE
ABCESSOS HEPÁTICOS
HEPATITES
DROGAS COLESTÁTICAS
PANCREATITE
Fosfatase Alacalina Óssea
Os ossos encontram-se em constante processo metabólico de remodelação. Este,
inclui a degradação e reabsorção ósseas, que são mediadas pelos osteoclastos, e os
processos de estruturação e formação óssea mediada pela acção dos osteoblastos .
A remodelação óssea é necessária para a manutenção da saúde global e da firmeza
da trama óssea. Para isso, é importante um equilíbrio entre os processos de
reabsorção e formação ósseas.
Em estados de alteração do metabolismo ósseo, estses processos dissociam-se, ou
seja, perdem o equilíbrio. Quando a reabsorção excede a formação, leva a uma
perda de osso que pode conduzir a osteoporose ou a doenças do tecido ósseo,
como as lesões da doença de Paget. A utilização de marcadores bioquímicos
específicos para esses eventos de remodelação fornece dados analíticos em relação
à taxa de metabolismo, ou seja, do turn-over ósseo.
A fosfatase alcalina óssea é uma glicoproteína específica encontrada na superfície
dos osteoblastos. A sua função ainda não é de todo conhecida, porém o seu papel
na mineralização do esqueleto está confirmada.
A avaliação da actividade sérica da fosfatase alcalina óssea é um marcador de turnover ósseo que fornece informações úteis da remodelação óssea na osteoporose, na
doença de Paget e na monitorização de terapias preventivas e de reposição
hormonal ou de outras terapias.
A osteoporose é uma doença do metabolismo ósseo caracterizada pela remodelação
óssea anormal. Esta remodelação conduz a uma baixa da massa óssea e a uma
deterioração da microarquitetura do tecido ósseo, com aumento consequente da
suscetibilidade para fracturas. O tipo mais comum de osteoporose acontece nas
mulheres na menopausa, como resultado da deficiência de estrogénio produzida
pela cessação da função ovariana. A restauração dos níveis de estrogénio àqueles
compatíveis com a fase pré-menopausa, através da terapia de reposição hormonal,
previne a perda óssea e consequentemente a osteoporose.
A osteoporose pode também resultar de um pico de ganho de massa óssea
inadequado durante a fase de crescimento - um desequilíbrio ósseo relacionado à
idade, que remodela com excesso de reabsorção - e várias condições clínicas e
terapias que induzem a perda óssea ou a remodelamento ósseo desequilibrado,
incluindo doenças endócrinas como hipogonadismo, hipertiroidismo,
hiperparatiroidismo, hipercortisolismo, falência renal, metástases ósseas, doenças
gastrointestinais relacionadas com a nutrição e metabolismo mineral, doenças do
tecido conjuntivo, mieloma múltiplo, imobilização crónica, alcoolismo, tabagismo e
terapia crónica com heparina e corticóides.
A doença de Paget tem etiologia ainda desconhecida, porém hipóteses de etiologias
envolvendo factores genéticos e víricos têm sido apontadas. É uma desordem óssea
focal que produz dor e deformidade, com elevadas taxas de actividade de
remodelação óssea. As lesões ocorrem principalmente no cránio, coluna vertebral,
pelve e ossos longos, e podem resultar em fracturas e comprometimento
neurológico.
Fósforo
O fósforo é um dos constituintes mais abundantes do organismo, presente em
diferentes tecidos. Num adulto normal, a maior parte encontra-se no osso, e o
restante, nos tecidos moles e ligados a proteínas, lìpidos e carbohidratos. Participa
em diferentes processos metabólicos e está presente como fosfolípido em todas as
membranas celulares. A sua homeostase depende basicamente do controle da
absorção (intestino delgado), filtração e reabsorção renal e da reserva que é
armazenada no osso.
Cerca de 80 a 90% do fósforo ingerido é absorvido de forma activa no intestino. A
absorção é aumentada na diminuição da ingestão de cálcio, na acidez do conteúdo
intestinal e também pela acção da vitamina D e da hormona de crescimento.
A maior parte do fósforo absorvido é excretada na urina. É filtrado pelos glomérulos
e reabsorvido em grande parte pelo túbulo proximal, de modo que apenas cerca de
10 a 15% do fósforo filtrado alcança o túbulo distal. Nas situações em que há
diminuição da filtração de fósforo, ocorre aumento da reabsorção tubular, e, ao
contrário, quando aumenta a carga filtrada, diminui a reabsorção e aumenta a
depuração. Nos casos de lesão renal, à medida que a taxa de filtração glomerular
diminui, o fósforo é retido, reduzindo discretamente o nível de cálcio sérico, o que
estimula as paratiróides a secretar a paratormona (PTH). A ação da PTH no sistema
tubular reduz a reabsorção, na tentativa de restaurar os níveis normais de cálcio e
de fósforo. Com a evolução da lesão renal, a fracção de fósforo reabsorvido pelo
sistema tubular diminui, mantendo a depuração de fósforo constante, e por
conseguinte os níveis séricos normais. Quando a taxa de filtração chega a níveis
muito baixos, cerca de 20 mL/min, a excreção de fósforo não se mantém mais,
devido a esse mecanismo de compensação, levando a uma elevação do fósforo
sérico.
Os mecanismos de regulação do cálcio afetam também os níveis de fósforo, sendo
os seus valores directamente relacionados: a elevação de um significa a diminuição
do outro. Os níveis séricos de fósforo oscilam ao longo do dia: valores mínimos são
observados entre as 9 e as 12 horas, aumentando gradualmente até atingir um
platô à tarde, e apresentando um discreto pico em torno da meia-noite.
Os níveis de fósforo são mais altos nas crianças, e tendem a elevar-se nas
mulheres após a menopausa. Aumentam também com o exercício e na
desidratação. Os valores séricos diminuem com a ingestão de carbohidratos e
aumentam com a ingestão de fósforo, sendo portanto fundamental que a colheita
seja realizada em jejum.
O aumento do fósforo sérico ocorre pela diminuição da filtração glomerular,
aumento da reabsorção tubular renal e aporte exógeno ou endógeno. A diminuição
ocorre por doenças tubulares e aumento das perdas. Alterações nos níveis da
hormona paratiroidéia afectam a reabsorção renal do fósforo.
São causas de aumento dos níveis de fósforo no soro: desidratação, hipovolemia,
acromegalia, hipoparatiroidismo, pseudo-hipoparatiroidismo, hipervitaminose D,
metástases ósseas, sarcoidose, cirrose, embolia pulmonar, falência renal, após
manobras de ressuscitação e cetoacidose diabética.
Valores séricos diminuídos são encontrados no uso de diuréticos, antiácidos,
hiperparatiroidismo primário, septicémia, deficiência de vitamina D, acidose tubular
renal, síndrome de Fanconi, hemodialisados crónicos, após episódios de vómitos,
osteomalácia e por outras causas que levem à elevação de cálcio no soro.
Níveis urinários aumentados de fósforo podem ser encontrados no
hiperparatiroidismo, na acidose tubular renal e no uso de diuréticos. Níveis
diminuídos são encontrados no hipoparatiroidismo e no pseudo-hipoparatiroidismo.
Fragilidade Osmótica
Quando em meio hipotónico, os eritrócitos normais são capazes de resistir à
hemólise, aumentando o seu volume. A variação da forma leva a variação da
espessura das membranas dos eritrócitos, alterando a sua capacidade de resistir à
lise celular por variações da pressão osmótica do meio onde se encontram.
Os esferócitos têm fragilidade osmótica aumentada, pois apresentam uma
membrana mais escassa do que a membrana de um eritrócito normal, o que os
impede de acumular água no seu interior. Já os reticulócitos têm mais membrana,
o que os torna capazes de resistir melhor à hemólise, mostrando assim menor grau
de fragilidade osmótica.
Na curva de fragilidade osmótica, os eritrócitos são submetidas a concentrações
crescentes de cloreto de sódio. A percentagem de hemólise é avaliada pela
quantidade de hemoglobina livre na solução.
Frutosamina
Além da HbA, a glicose pode ligar-se a outras proteínas e globulinas por meio de
uma glicosilação não-enzimática. A proteína total, que após a ligação com a glicose,
se transforma numa cetamina estável, é denominada genericamente frutosamina.
Devido à sua semi-vida curta, de cerca de 30 dias, os resultados obtidos indicam a
média das glicemias nas 2 últimas semanas (1 a 3).
Os valores podem ser alterados em situações de perda ou diminuição da semi-vida
das proteínas. Não deve ser utilizada como diagnóstico e sim como monitorização
do controle da diabetes mellitus.
Os resultados dos doseamentos de frutosamina devem ser analisados juntamente
com os resultados de glicose e da hemoglobina glicosilada. A frutosamina mostrase elevada em todos os casos de diabetes fora de controle metabólico,
independentemente dos valores da glicemia de jejum. Os seus valores retornam
aos níveis de referência 20 dias após a estabilização da glicemia em níveis
adequados.
É útil no acompanhamento de casos de pacientes portadores de hemoglobinopatias
que interferem no doseamento da hemoglobina glicosilada.
Gama Glutamil Tanspeptidase (GGT)
A gama glutamil transpeptidase ou transferase (GGT) é uma enzima presente nas
membranas celulares e nas fracções microssómicas envolvidas no transporte de
aminoácidos através da membrana celular. Está presente em ordem decrescente de
abundância no túbulo proximal renal, fígado, pâncreas e intestino. Os níveis séricos
da GGT são principalmente de origem hepática. A sua semi-vida é de 7 a 10 dias,
aumentando para 28 dias nas lesões hepáticas ligadas ao álcool.
Os valores são aproximadamente 50% mais elevados nos homens do que nas
mulheres, e são directamente proporcionais à massa corporal, ao consumo de
álcool, ao fumo e ao nível de actividade física.
Por mecanismos ainda não muito bem esclarecidos, pacientes com diabetes
mellitus, hipertiroidismo, artrite reumatóide e doença pulmonar obstrutiva crónica
frequentemente apresentam valores aumentados de GGT.
Valores muito elevados são encontrados nos quadros de colestase crónica, como na
cirrose biliar primária ou na colangite esclerosante e noutras patologias hepáticas e
biliares.
Apresenta-se elevada em alcoólatras, mesmo sem hepatopatia, na obesidade e no
uso de fármacos como analgésicos, anticonvulsivantes, quimioterápios, estrogénio
e contraceptivos orais.
Nos períodos após enfarte agudo do miocárdio, a GGT pode permanecer alterada
por semanas.
Em estudos do gene humano, a GGT teve a sua sequência de nucleotídeos
identificada. Deste modo, puderam ser identificadas três principais formas de GGT
circulantes, que aparentemente, não são isoenzimas verdadeiras. Uma, de alto
peso molecular, aparece nos soros normais, na obstrução biliar e, com mais
frequência nos casos de neoplasia hepática. A segunda forma tem um peso
molecular intermediário e apresenta duas fracções: uma detectável em
hepatopatias, e a outra em obstruções das vias biliares. A terceira tem um baixo
peso molecular e ainda não teve a sua função definida.
Infelizmente, esses testes não estão disponíveis para uso clínico, pois o método
ainda não apresenta sensibilidade e especificidade adequadas.
Gasimetria Arterial
Para funcionarem normalmente, as actividades metabólicas necessitam de um pH
mantido em condições ideais. Este factor depende da manutenção do equilíbrio
ácido-base do organismo, obtido pela interacção dos mecanismos renais (controle
de concentração de bicarbonato) e pulmonares (controle de concentração de CO2).
Consequentemente, o estado do equilíbrio metabólico depende de mecanismos
respiratórios com uma troca gasosa adequada e boa oxigenação tecidual e um
tamponamento fornecido pelo rins.
Os distúrbios ácido-base que podem ocorrer são: acidose metabólica, acidose
respiratória, alcalose metabólica e alcalose respiratória. Inicialmente, pode parecer
que o conhecimento isolado da alteração da concentração de bicarbonato ou da
concentração de pCO2 conduza, respectivamente, a um diagnóstico de alteração
metabólica e respiratória. Entretanto, isto não é verdadeiro, visto que um distúrbio
primário pode levar a uma alteração secundária compensatória, que no final leva à
manutenção do pH para níveis adequados. Portanto, a análise do equilíbrio ácidobase depende da avaliação de um conjunto de diferentes parâmetros, que são
representados pelo exame gasimetria arterial. Esses parâmetros são:
- pH
- pCO2
- bicarbonato
- pO2
- saturação do O2
- bases em excesso
O primeiro parâmetro observado na análise da gasimetria arterial deve ser o pH.
Independentemente dos demais parâmetros, o pH determina se o paciente se
encontra em acidose ou alcalose. O valor limite do pH para essa definição é de 7,4.
Valores abaixo deste são considerados acidose, e acima alcalose. Apenas após a
avaliação desse parâmetro, outros, como bicarbonato e pCO2 poderão ser utilizados
para diagnosticar se a origem dos distúrbio é metabólica ou respiratória.
A concentração do bicarbonato, da pCO2 e do pH é interdependente. Portanto, a
alteração de um dos parâmetros leva ao movimento compensatório dos demais, na
busca do equilíbrio. Os níveis de bicarbonato associados aos mecanismos
respiratórios de retenção e eliminação do CO2 mantêm o equilíbrio ácido-base.
SITUAÇÃO
pH
BIRCABONATO
PCO2
Acidose
metabólica
<7.4
Baixo
Baixa
Acidose
respiratória
<7.4
Alto
Alta
Acidose
metabólica
>7.4
Alto
Alta
Acidose
respiratória
>7.4
Baixo
Baixa
CAUSAS FREQUENTES
Cetoacidose diabética, acidose
láctica (sépsis por gramnegativos), insuficiência renal,
paragem cardio-respiratória e
uso abusivos da aspirina.
DPCO, insuficiência
respiratória, paralisia dos
músculos respitetórios,
pneumonia e lesões do SNC.
Vómitos, outras causas de
perda
de ácido clorídrico e uso
abusivo de diuréticos.
Dor aguda e intensa,
ansiedade, hiperventilação,
estimulação
não pulmonar do centro
repirtório e uso de fármacos
Na acidose metabólica, a produção excessiva de ácido é tamponada pelo
bicarbonato, que é consumido nesse processo, levando a um aumento da
frequência respiratória para diminuir a pCO2 e compensar a perda de bicarbonato.
Na alcalose metabólica, existe uma perda de ácido, com aumento da concentração
do bicarbonato e de pCO2.
Na acidose respiratória, o aumento da concentração de CO2 leva a uma diminuição
do pH, o que induz a uma retenção renal de bicarbonato para compensar a equação
de equilíbrio.
A avaliação da pO2 é importante para avaliação do estado de perfusão tecidual.
Importante também é a determinação da percentagem de saturação da
hemoglobina pelo O2. Valores baixos de um ou de ambos os parâmetros acima
referidos, são indicação de patologia subjacente.
A queda da pO2 leva a acidose metabólica. As principais causas dessa condição são:
embolia pulmonar, enfarte agudo do miocárdio, anóxia tecidual secundária a
estados de hipoperfusão como situações cirúrgicas, septicémia, choque ou
insuficiência cardíaca grave, entre outras. Outra causa de hipoxia, são as situações
que levam à retenção do CO2, como as lesões pulmonares e a DPCO.
O excesso de bases representa o cálculo realizado a partir dos valores de pH, de
pCO2 e da concentração da hemoglobina, que significa o excesso ou o déficite de
bases, permitindo avaliar a gravidade do distúrbio metabólico. O excesso de bases
é encontrado nas alcaloses, e o déficite nas acidoses.
Gastrina
É uma hormona produzida pelas células G localizadas principalmente no antro e
duodeno proximal. Existem várias formas moleculares de gastrina. A principal
forma de gastrina na mucosa antral é um heptadecapeptídeo com 17 resíduos de
aminoácidos (G-17) que representa 90% da gastrina produzida nessa região. Já na
circulação, dois terços da gastrina sérica são representados por uma molécula
maior, com 34 aminoácidos (G-34).
Embora a G-17 tenha uma semi-vida mais curta que a G-34, ambas têm a mesma
intensidade de estimulação da secreção gástrica.
A sua avaliação é útil no diagnóstico do síndrome de Zollinger-Ellison, caracterizado
por ulceração péptica severa do trato gastrointestinal com hipersecreção ácida
gástrica, consequente à excessiva secreção de gastrina. Valores superiores a 1.000
pg/mL associados a hipercloridria são praticamente diagnóstico da presença desse
síndrome.
Além do síndrome de Zollinger-Ellison, níveis elevados podem ser detectados
também em situações que cursam com acloridria ou hipocloridria, como nas
anemias perniciosas, na gastrite atrófica e no carcinoma gástrico.
Nestes casos, os níveis não se encontram tão elevados quanto no Síndrome de
Zollinger-Ellison. O teste dinámico com infusão de cálcio ou secretina tem um papel
importante no diagnóstico diferencial entre as possíveis etiologias.
A ingestão de aminoácidos, drogas como carboneto, cloreto de cálcio e insulina
podem elevar os níveis de gastrina. Já o uso de atropina diminui os seus níveis
séricos.
Genotipagem da Hepatite C
Os vírus da hepatite C são um grupo de isolados ou linhagens com alto grau de
variabilidade. Esta heterogeneidade também pode ser observada no próprio
hospedeiro, do qual um simples isolado pode abrigar variantes víricas de múltiplas
sequências, as quais mostram apenas 92% de similaridade nas regiões que
codificam proteínas do envólucro. Isolados de determinados subtipos exibem
similaridades nucleotídicas em torno de 80% para o genoma completo ou regiões
não-estruturais, enquanto um ou mais subtipos podem ser classificados em alguns
tipos principais com similaridades em torno de 68%. Os seguintes genótipos são
caracterizados como principais tipos de HCV: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 e11. Estes
são subdivididos em diferentes subtipos possíveis de serem analisados pela
genotipagem para HCV:
-
1a, 1b, 1c
2a, 2b, 2c
3a, 3b
4a, 4b, 4c, 4d, 4e
5a
6a
7a, 7b
8a, 8b
9a
10a
11a
Evidências substanciais têm demonstrado a grande importância da tipagem e
subtipagem para a clínica. Estudos indicam que a infecção pelo HCV genótipo 1b
não apresenta resposta eficiente ao tratamento com interferon alfa (10% dos
pacientes tratados com resposta favorável), enquanto para os genótipos 1a, 2a, 2b
e 3a respondem de forma favorável (50 a 80% de resposta).
Tem sido demonstrado que infecções por HCV genótipo 1b evoluem rapidamente
para formas crónicas de hepatites, cirrose e carcinoma hepatocelular. A rápida
evolução da doença hepática crónica e cirrose em órgão transplantado é observada
para o subtipo 1b.
A genotipagem para HCV é obtida com a utilização de métodos de biologia
molecular e de sequenciamento genómico, os quais são úteis para definir os
genótipos e subtipos para estudos de epidemiologia molecular, estudos clínicos e
monitorização da hepatite C.
Genotipagem para HIV
Recentes avanços nos estudos sobre o HIV-1 orientados, principalmente, para a
quantificação da carga viral em plasma e células mononucleares do sangue
periférico de pacientes nos diferentes estádios da síndrome de imunodeficiência
adquirida (SIDA) têm evidenciado que a infecção pelo HIV-1 é um processo
dinámico, em que ocorre um equilíbrio entre a produção e a eliminação de
partículas virais. Este processo é observado, inclusive, na fase de lactência clínica,
caracterizada por ausência de sintomas, altos níveis de anticorpos circulantes e
níveis baixos ou não-detectados de virémia. A associação à elevada taxa de
mutação genómica do HIV-1 resulta no desenvolvimento de linhagens virais
mutantes, que são mais eficazmente selecionadas sob condições de pressão
selectiva exercida pelo sistema imunológico ou pela intervenção farmacológica
prolongada.
Essas variantes HIV-1 não são reconhecidas pelos mecanismos imunes efectores
(anticorpos neutralizantes e/ou linfócitos T citotóxicos) e podem apresentar
mutações nos genes codificantes para as enzimas protease ou transcriptase
reversa. Estas resultam em mutações de aminoácidos nas enzimas, selecionando,
desta forma, variantes resistentes aos anti-retrovirais.
Mutações no genoma do HIV que conferem resistência aos agentes anti-retrovirais
têm sido relatadas para todos os compostos usados no tratamento de pacientes
infectados. O início da infecção está associado a uma população homogenea de HIV
que é, usualmente, sensível aos anti-retrovirais. No entanto, as altas taxas de
mutação podem resultar em 106 variantes genéticas, entre as quais podem estar
presentes, em baixos níveis, as resistentes aos anti-retrovirais, antes do início da
terapia.
O significado clínico da resistência aos anti-retrovirais não está ainda totalmente
elucidado, porém atribui-se a ocorrência ao decréscimo na eficiência da
quimioterapia anti-retroviral.
A enzima transcriptase reversa é estudada mais extensamente para a intervenção
farmacológica. Ela é essencial para a replicação do HIV-1, uma vez que a expressão
das proteínas virais e a progénie viral dependem da conversão da cadeia simples de
RNA em cadeias duplas de DNA e a subsequente integração do cDNA viral ao
genoma da célula hospedeira.
Análogos aos inibidores nucleosídicos da transcriptase reversa 3'-azido-3'desoxitimidina (AZT; zidovudina); 2',3'-didesoxinosina (ddI; didanosina); 2',3'didesoxicitidina (ddC; zalcitabina); (-)-b-L- 2',3'-didesoxi-3'-tiacitidina (3TC;
lamivudina) e 2',3'-didesidro-3'-desoxitimidina (d4T; estavudina) são os fármacos
actualmente aprovadas para o tratamento da SIDA. Estes compostos actuam de
modo semelhante, após activação metabólica por fosforilação efectuada por
quinases intracelulares. As formas trifosfato resultantes, podem inibir a enzima
transcriptase reversa por competição pelo substrato ou podem actuar como cadeias
finais da reacção de transcrição reversa, inibindo a síntese do cDNA viral.
As mutações de resistência são divididas em primárias e secundárias. As primárias
devem ser analisadas em separado, já que são directamente responsáveis pela
diminuição acentuada na sensibilidade ao fármaco. Para utilização adequada da
genotipagem para HIV-1 é necessária uma avaliação criteriosa dos resultados.
As alterações nos códons abaixo podem definir resistência total ou parcial às
medicações anti-retrovirais.
Inibidores de transcriptase reversa análogos de nucleosídeos
- T215YF, M41L, K70R, D67N, L210W, K219Q, M184VI, V1181, E44D, K65R, L74V,
Y115F.
Inibidores de transcriptase reversa não-análogos de nucleosídeos
- K103N, V106A, V1081, Y181CI, Y188CLH, G190AS, P236L, L1001.
Inibidores de protease
- M461L, V82AFTS, 184V, L10FIRV, K20MR, L24I, V32I, M36I, 154V, A71VT,
G73AS, V77I, L90M, N88D, 147V.
Uma única mutação pode resultar em diferentes graus de sensibilidade viral ao
quimioterápio. A mutação na posição 184, com substituição de metionina por
valina, resulta num decréscimo de cerca de mil vezes na sensibilidade ao 3TC (alto
nível de resistência) e de quatro a oito vezes ao ddI e ao ddC (baixo nível de
resistência).
Trabalhos desenvolvidos por alguns investigadores têm evidenciado que o
desenvolvimento de mutações associadas à resistência viral à zidovudina segue um
padrão transitório: a mutação pode ocorrer primeiro em qualquer códon, enquanto
mutações adicionais acumulam-se lenta e progressivamente durante a terapia.
Outros investigadores têm observado, entretanto, que a resistência do HIV-1 à
zidovudina desenvolve-se de forma ordenada: em indivíduos assintomáticos em
tratamento com zidovudina, a mutação no códon 70 ocorre primeiro, mas é
substituída pela mutação, mais estável, no códon 215. Com o tratamento
prolongado, outras mutações nos códons 41, 67 e novamente 70 desenvolvem-se,
conferindo ao HIV maior resistência à zidovudina. Isto ocorre apenas com a
progressão da doença, uma vez que vírus altamente resistentes não têm sido
isolados de amostras biológicas de indivíduos assintomáticos.
Vários métodos laboratoriais têm sido utilizados para a detecção de mutações
associados à resistência à zidovudina, incluindo técnicas de clivagem por RNAse e
sistemas de hibridização RNA/RNA. Porém, a maioria dos estudos usa métodos
fundamentados na reacção em cadeia da polimerase (PCR), na qual o segmento do
genoma do HIV-1, codificado para transcriptase reversa e protease, é amplificado e
utilizado numa segunda reacção, empregando-se pares de primers especificamente
definidos para a amplificação de sequências de mutantes ou selvagens em códons
de interesse.
Os segmentos amplificados por PCR são sequenciados directamente utilizando-se
sequenciadores fluorescentes automatizados. Outro método para a análise
genotípica consiste no ensaio de hibridização com sondas (LIPA), que se baseia na
detecção de um sinal colorimétrico não-radioactivo, emitido pela hibridização do
produto de PCR do HIV-1 com sondas oligonucleotídicas, imobilizadas em fitas de
nitrocelulose. Entretanto, uma restrição ao seu emprego é polimorfismos muito
próximos, que dificultam a hibridização, impedindo a obtenção de resultados.
Glicose
A glicose é essencial para a função do cérebro e dos eritrócitos. O excesso de
glicose é armazenado na forma de glicogénio no fígado e nas células musculares.
A determinação de glicose no sangue é um dos exames mais solicitados aos
laboratórios clínicos e tem como finalidade diagnosticar e acompanhar o tratamento
de portadores de algum distúrbio no metabolismo de carbohidratos que levem a
situações de hipo, ou hiperglicemia.
Um dos problemas mais frequentes é a diabetes mellitus, que pode ser descrito
como um grupo de doenças metabólicas de diversas etiologias, caracterizado por
hiperglicemia, glicosúria e outras manifestações clínicas decorrentes do
comprometimento, principalmente, do sistema vascular e do sistema nervoso,
levando a lesões em múltiplos órgãos, em especial olhos, rins e coração.
A prevalência da diabetes mellitus tem vindo a aumentar acentuadamente nos
últimos anos. As causas apontadas para esse aumento são as mudanças de hábitos
de vida ocasionados pela acelerada urbanização, levando a um sedentarismo cada
vez maior, alimentação desequilibrada, obesidade e stres contínuo, que facilitam a
manifestação da doença em indivíduos geneticamente predispostos. Outro dado
importante é o aumento da esperança média de vida na população, que contribui
também para o aumento da prevalência da doença.
Estudos multicêntricos confirmam o aumento da prevalência e sugerem que os
indivíduos obesos apresentam o dobro de risco de desenvolver a doença; já aqueles
com parentes directos diabéticos apresentam risco triplicado. Outro dado
importante oriundo desses estudos é que metade dos pacientes que tiveram o
diagnóstico de diabetes confirmado desconhecia o facto e que 20% entre os que já
conheciam o diagnóstico da doença não faziam nenhum tipo de tratamento. O dado
de que metade dos pacientes diabéticos no nosso país vive com hiperglicemia levanos a um quadro desolador, no qual o risco de morbilidade e de mortalidade
aumenta significativamente por complicações vasculares, renais, neurológicas,
oftalmológicas e infecciosas.
Uma nova classificação e novos critérios diagnósticos de diabetes mellitus foram
propostos em maio de 2000 pela American Diabetes Association, e endossados pela
Organização Mundial de Saúde.
A classificação etiológica identifica quatro grupos distintos de diabetes
DIABETES TIPO I
Auto imunidade e idiopática
..........................................................................
DIABETES TIPO II
..........................................................................
OUTROS TIPOS ESPECÍFICOS
Defeitos genéticos na acção da insulina, defeitos genéticos da células beta, doenças do
pâncreas exócrino, endocrinopatias, induzido por fármacos, infecções, síndromes
genéticos associados e formas imunomediadas incomuns.
..........................................................................
DIABETES GESTACIONAL
..........................................................................
A diabetes tipo I apresenta duas formas clínicas. Uma, imunomediada, representa
90% dos casos e cursa com marcadores imunológicos de destruição das células
beta pancreáticas, como os anticorpos anti-ilhota, anti-insulina e anti-GAD, entre
outros. A outra corresponde a 10% dos casos que, por não terem etiologia
conhecida, são classificados como idiopáticos. Embora seja a principal
endocrinopatia diagnosticada na infância e na juventude, o termo diabetes infantojuvenil não deve ser utilizado, uma vez que de forma menos frequente, pode
também manifestar-se na idade adulta. O termo insulino-dependente também foi
abandonado, já que qualquer tipo de diabetes pode, levar à dependência em
relação à insulina, além do paciente apresentar grande tendência a cetoacidose e
coma.
A diabetes tipo II caracteriza-se pela resistência periférica à acção da insulina, forte
predisposição genética e familiar, e deficiência relativa de insulina, que aumenta
com a evolução da doença. A maior parte dos pacientes é obeso clássico ou
apresenta a chamada obesidade abdominal, que está associada ao aumento da
produção de ácidos gordos livres, levando a um maior afluxo hepático e provocando
hiperinsulinemia por diminuição da ligação e extração de insulina pelo fígado.
Ambos os quadros cursam com a resistência periférica da insulina. A glicemia
eleva-se de modo gradual e, durante os estágios iniciais, não induz a sintomas
clínicos significativos. Com isso, o paciente permanece sem diagnóstico por muito
tempo.
A diabetes gestacional é definidao como uma intolerância à glicose diagnosticada
durante a gravidez. A partir da sexta semana após o parto, nova avaliação deve ser
realizada para reclassificação do estado da paciente.
Os novos parâmetros diagnósticos para diabetes:
- quadro clínico de diabetes associado a uma glicemia casual de 200 mg/dL;
- glicemia de jejum superior a 126 mg/dL;
- glicemia 2 horas após sobrecarga oral (PTGO) de 200 mg/dL.
A avaliação do doseamento de glicose deve ser confirmada, pelo menos, em duas
ocasiões diferentes. O grupo de intolerantes inclui os indivíduos que se afastam da
normalidade (faixa de 110 a 125 mg/dL) mas não apresentam alterações
suficientes para serem considerados diabéticos.
Informações adicionais, consultar Curva glicémica.
NOVOS CRITÉRIOS DIAGNÓSTICOS DE DIABETES MELLITUS
PROVA DE
GLICEMIA
TOLERÂNCIA ORAL À
GLICEMIS DE JEJUM
CASUAL
GLICOSE
(AOS 120 MINUTOS)
NORMAL
até 109 mg/dl
até 139 mg/dl
Intolerante à
INTOLERANTE
intolerante em jejum
sobrecarga entre
(Tolerancia Diminuída) entre 110 e 125 mg/dl
140 e 200 mg/dl
DIABETES MELLITUS Acima de 126 mg/dl
> ou = 200 mg/dl
Acima de 200 mg/dl.
Mesmo a população aparentemente saudável deve ser submetida a exames, a fim
de obter o diagnóstico precoce da diabetes, o que favorece o tratamento. Casos
que devem ser investigados e/ou acompanhados:
- pacientes obesos,
- obesidade abdominal,
- sedentários,
- parentes de 1º grau de diabéticos,
- história de diabetes gestacional,
- história de macrossomia fetal e abortos de repetição,
- hipertensão arterial sistémica,
- resultados que indicam tolerancia diminuída à glicose,
- níveis aumentados de triglicerídeos e diminuídos de HDL colesterol.
Glicose-6-Fosfato Desidrogenase, Pesquisa
Este erro inato é causado por mutações no gene codificante para a enzima glicose6-fosfato desidrogenase (G6PD) que se encontra localizado no cromossomo X,
sendo portanto uma doença hereditária ligada ao sexo. Como a deficiência, está
ligada ao cromossomo feminino (X), para que ocorra a expressão total da doença o
gene não deve ser antagonizado por um cromossoma X normal. Portanto, a
manifestação é mais grave nos homens (XY) e num número reduzido de mulheres
que apresentam ambos os X alterados.
Mais de 400 mutações distintas já foram determinadas como responsáveis pela
deficiência de G6PD. Afecta cerca de 400.000 indivíduos no mundo.
A deficiência da glicose-6-fosfato desidrogenase (G6PD) altera directamente a
estabilidade das hemácias, tornando-as vulneráveis à desnaturação oxidativa da
hemoglobina, o que por sua vez leva a episódios hemolíticos intermitentes e à
presença de corpos de Heinz.
A susceptibilidade à hemólise dos portadores da deficiência pode ser aumentada
pela exposição a fármacos com propriedades oxidantes, em situações de agressões
víricas ou bacterianas e na presença de distúrbios metabólicos.
A apresentação clínica mais grave é a hemólise intravascular aguda, com
hemoglobinúria e icterícia após quadros infecciosos ou exposição a fármacos com
efeitos oxidantes (sulfonamidas, sulfonas, analgésicos, antipiréticos, antimaláricos
etc.). As manifestações clínicas podem aparecer ao nascimento ou os pacientes
podem permanecer assintomáticos por vários anos, conhecendo a doença após
infecções ou exposição aos medicamentos supracitados.
Quando o aparecimento dos sintomas é precoce, icterícia neonatal é comum e
desenvolve-se em 1 a 4 dias após o nascimento. Pode apresentar-se de forma
grave, evoluindo para comprometimento renal, sequelas cerebrais ou mesmo óbito.
O rastreamento neonatal é pertinente por se considerar a alta frequência do gene
defectivo na população e pela possibilidade de profilaxia de crises hemolíticas
causadas por fármacos e pela opção de medidas terapêuticas consensuais, levando
a um bom prognóstico.
Gonodotrofina Coriónica Humana
Os ensaios laboratoriais para o diagnóstico da gravidez doseiam a gonadotrofina
coriónica humana (HCG), que é uma hormona glicoprotéica produzida pelas células
do sinciciotrofoblasto da placenta. A molécula de HCG é composta por duas
subunidades: alfa e beta. A subunidade beta é a que confere a especificidade, uma
vez que a subunidade alfa é comum a outras hormonas como a LH, a FSH e o TSH.
Além das subunidades livres, outras moléculas HCG-relacionadas podem estar
presentes, tanto no soro como na urina de mulheres grávidas. Essas moléculas
podem apresentar grandes variações estruturais e são reconhecidas de modo
diferente por diversos imunoensaios para HCG.
Alguns ensaios detectam apenas a molécula inteira, que é a hormona
biologicamente activa, uma vez que, isoladamente, nenhuma das subunidades
exerce efeito hormonal. Outros detectam tanto a molécula inteira quanto a fração
beta livre. Outros, ainda, detectam, além das frações livres, todas as moléculas
HCG-relacionadas.
Esta é uma das razões para as variações passíveis de serem encontradas entre os
diversos métodos para doseamento de HCG. A variação é menos significativa na
gravidez normal e pode ser muito expressiva em gestações complicadas como
aborto espontâneo, pré-eclâmpsia, doença trofoblástica, síndrome de Down e
neoplasias do testículo, bexiga ou ovário. Nesses casos, a proporção de moléculas
de HCG fragmentadas ou de fração beta livre pode ser muito maior.
O doseamento de beta HCG também é útil como marcador tumoral, especialmente
no diagnóstico e no acompanhamento dos tumores trofoblásticos e testiculares. Os
ensaios capazes de dosear a molécula de beta HCG total e as suas fracções livres
são os mais indicados na detecção e na monitorização dessas patologias, já que
alguns tumores secretam, além da molécula total da HCG, uma grande quantidade
de fracções livres.
O carcinoma testicular apresenta um pico de incidência em homens entre os 30 e
40 anos, sendo 10 vezes mais frequente em pacientes com história de
criptorquidismo. Pode ser observada apenas a presença de nódulo indolor, ou ainda
de dor, disúria, perda de peso, feminilização, ginecomastia e diminuição da líbido.
Mais de 95% dos tumores testiculares originam-se nas células germinativas dos
túbulos seminíferos. Geralmente são malignos. Os tumores de células germinativas
podem ser divididos em seminomas e não-seminomas. Os não-seminomas mais
comuns são os tumores de células embrionárias, o teratoma e o coriocarcinoma. A
presença de alfafetoproteína (AFP) elevada indica a presença de um nãoseminoma, já que os seminomas não a secretam.
Detectar HCG elevada sugere a presença de um coriocarcinoma ou tumor de células
embrionárias, já que raramente a HCG é secretada por seminomas puros. Apenas 7
a 16% dos seminomas secretam HCG total, e cerca de 20 a 50% secreta também
as suas fracções livres. Por isso, como já citado, a utilização de métodos capazes
de detectar o HCG total e as suas frações aumenta a possibilidade diagnóstica. A
realização do exame histológico do material de punção testicular é fundamental
para diferenciar seminomas de não-seminomas.
Na gravidez normal, a secreção de HCG é detectável entre 7 a 10 dias após a
fertilização e eleva-se, alcançando níveis máximos nas 8 a 10 semanas de
gestação. Após a 12ª semana, os seus níveis começam a diminuir, até alcançarem
um platô.
Níveis inferiores a 5 mUI/mL podem ser encontrados em mulheres não-grávidas,
enquanto níveis superiores a 25 mUI/mL são interpretados como positivos. Os
valores intermediários entre 5 e 25 mUI/mL são considerados inconclusivos e
devem ser confirmados após 2 a 3 dias, pois no caso de existir gravidez, os valores
duplicam a cada 24 horas. Gestações múltiplas cursam com níveis mais elevados do
que a gestação normal.
Os níveis de HCG alcançados numa gestação normal são significativos, e a
realização de doseamentos seriados é importante para elucidar casos de gravidez
ectópica, que cursam com níveis bem menores.
Grupo Sanguíneo e Factor RH
A denominação grupos sanguíneos não se restringe apenas ao sistema de
antigénios encontrados nas hemácias, podendo também ser expressa por outros
constituintes sanguíneos como leucócitos, plaquetas e o próprio plasma. Esses
antigénios são definidos geneticamente, e de acordo com as combinações das suas
expressões na superfície das células sanguíneas, formam os sistemas que
identificam os diferentes grupos sanguíneos. Os principais antigénios eritrocitários e
os seus anticorpos correspondentes mais utilizados nas avaliações de imunohematologia de rotina são o sistema ABO e o Rhesus (Rh).
O sistema ABO tem uma característica peculiar. A maioria dos indivíduos normais
apresenta anticorpos (hemaglutininas potentes) contra os antigénios que não
possuem, e é nessa singularidade que os mecanismos de identificação do grupo
ABO se baseiam. Portanto, para classificar os diferentes grupos, podemos realizar a
chamada prova directa, em que são utilizados soros padrões que permitem
identificar a presença de um determinado antigénio na superfície das hemácias.
Uma outra forma é chamada prova reversa (confirmatória), na qual se utilizam
células com antigénios conhecidos, permitindo a pesquisa de anticorpos livres no
plasma ou no soro. É importante a realização das duas provas, para maior
segurança da classificação do grupo sanguíneo. Os grupos A e B apresentam
subgrupos de pouca importância clínica em relação às transfusões.
Nalgumas situações, é possível encontrarmos discordância entre as duas técnicas
e/ou dificuldades na classificação. Esses casos podem ser encontrados em
subgrupos mais fracos, com fenótipos raros. Entre as diferentes causas de
discordância na classificação ABO podemos citar os idosos e recém-nascidos, por
baixa actividade do antigénio (aglutinina), presença de auto-anticorpos frios,
imunossupressão, anticorpos ABO adquiridos passivamente ou, ainda mais
raramente, alterações dos antigénios em patologias graves como a depressão
antigénica observada em leucemias e noutras patologias, especialmente neoplasias.
Encontra-se também o antigénio B adquirido, associado aos carcinomas do cólon e
gástrico.
Grupo ABO
O
A
B
AB
Antigénios
presentes
H
A
B
AB
Anticorpos
naturais
anti-A e anti-B
anti-B
anti-A
-
% População
branca
45
40
11
4
% População
negra
49
27
20
4
A explicação da expressão do factor Rh é complexa e envolve a manifestação dos
diferentes antigénios em grupos de três. O sistema Rh foi assim denominado por
Wiener logo no início de sua descoberta de diferentes antigénios eritrocitários D-ce-C-E, e é baseado na sua teoria de hereditariedade dos antigénios Rh.
Posteriormente, outros autores propuseram a denominação sistema DCE, que
terminou por não ser utilizada na rotina.
Para simplificar a compreensão, consideremos que, do ponto de vista prático,
apenas se utiliza o soro anti-D para classificação dos grupos Rh positivos, negativos
e fracamente positivos (variante Du). Portanto, didáticamente, são indivíduos
Rh(+) (85% da população) os que apresentam o antígeno D, e Rh(-) (15% da
população) os que não apresentam o antígeno D. A variante Du é avaliada em
todos os casos negativos. Quando positivo, o indivíduo será tratado como Rh(+).
Haptoglobina
É uma glicoproteína produzida pelo fígado, que migra na região alfa-2-globulina, na
electroforese das proteínas, podendo ser identificados, pelo menos, três fenótipos
diferentes (HAP1, HAP2-1, HAP1-1).
A sua função é ligar-se à hemoglobina livre, libertada pela destruição intravascular
das células vermelhas, conservando o ferro e prevenindo a possível nefrotoxidade
causada pela hemoglobina durante a filtração glomerular.
O complexo formado pela haptoglobina e pela hemoglobina livre é removido pelo
sistema reticuloendotelial, no qual o grupo heme da hemoglobina é degradado em
ferro e em bilirrubina.
Em estados de acelerada hemólise intravascular, a haptoglobina começa a diminuir,
devido ao facto de estar a ser utilizada em maior velocidade do que a da reposição
hepática, o que leva ao aparecimento de dímeros de hemoglobina livre que serão
filtrados pelos rins. Uma vez filtrada, a hemoglobina livre é degradada pelas células
epiteliais tubulares, levando à formação de hemossiderina. O rim possui capacidade
limitada de filtração, em torno de 5 g de hemoglobina por dia. Quando ultrapassa
esse nível, a hemoglobina livre começa a aparecer na urina.
A hemoglobina livre pode ligar-se, também a uma outra proteína, a hemopexina,
que por sua vez se liga ao heme, transportando-o para o fígado, onde o ferro é
incorporado à ferritina e o heme segue a via metabólica das bilirrubinas. Assim
como a haptoglobina, pode ser consumida em estados de hemólise acelerada.
Os níveis de haptoglobina podem estar elevados em numerosas situações que
levam à reacção de fase aguda como no síndrome nefrótico, em que, dependendo
do fenótipo, podem aumentar de forma a compensar a perda de outras proteínas. A
mesma elevação pode ocorrer no uso de corticóides.
Apresentam-se diminuídos nas hepatopatias graves, nas situações de aumento da
hemoglobina livre pela hemólise aumentada de eritrócitos, como em
hemoglobinopatias, reacções transfusionais, anemia megaloblástica, eritropoiese
ineficaz associada a destruição medular de células vermelhas, queimaduras
extensas, coagulação intravascular disseminada, reabsorção de grandes
hematomas, síndrome nefrótico em que, dependendo do fenótipo, podem diminuir
os níveis séricos por perda urinária (geralmente fenótipo HAP1-1), e terapia com
estrogénios e corticóides.
Nas situações de hemólise severa aguda, a haptoglobina pode ser totalmente
consumida, necessitando de cerca de 1 semana para retornar aos níveis normais.
Já nos casos de hemólise crónica, como os que podem ser vistos em
hemoglobinopatias e na hemólise causada por válvulas cardíacas mecânicas, os
níveis séricos de haptoglobina decrescem mais lentamente, e o seu doseamento
seriado pode ser um melhor indicador para o acompanhamento do nível de
hemólise do que a avaliação isolada da hemoglobina.
Por ser uma proteína de fase aguda, a presença conjunta de processos
inflamatórios e de hemólise pode dificultar o diagnóstico em análises isoladas. Para
facilitar o diagnóstico diferencial, recomenda-se a avaliação de outras proteínas de
fase aguda, além do acompanhamento seriado.
HDL-Colesterol
O HDL-colesterol (colesterol contido nas HDL) é inversamente proporcional ao risco
de desenvolvimento de doenças coronárias. Níveis superiores a 60 mg/dL são
associados a um efeito protector, enquanto níveis séricos abaixo de 40 mg/dL
relacionam-se a risco mais elevado de desenvolvimento de doença coronária.
Esta capacidade protectora parece estar relacionada com o mecanismo de
transporte reverso do colesterol, no qual o HDL está envolvido, captando colesterol
não-esterificado dos tecidos periféricos pela acção enzimática da LCAT (lecitinacolesterol-acil-transferase) e formando as HDL maduras, que irão levar o colesterol
até ao fígado de forma directa ou transferindo-o para outras lipoproteínas, as VLDL.
No fígado, o colesterol é reutilizado em processos metabólicos ou excretado pela
bilis.
Os valores séricos variam de acordo com a idade e o sexo. Algumas situações
podem contribuir para a diminuição dos níveis séricos de HDL-colesterol, como
sedentarismo, tabagismo, diabetes, factores genéticos, obesidade e diversos
fármacos. O exercício e o uso moderado de flavonóides (contidos no vinho tinto)
têm sido apontados como factores que ajudariam a elevar os níveis séricos do HDLcolesterol.
Para cada redução de 5 mg/dL do HDL-colesterol abaixo da média, o risco de
doença coronária aumenta em 25%. Nas hepatopatias, os níveis séricos do HDLcolesterol podem encontrar-se diminuídos.
Consultar Perfil Lipídico
Helicobacter pylori
Em 1994, uma conferência de consenso declarou que a úlcera é uma doença
infecciosa e recomendou que o tratamento dos sintomas com antiácidos deveria ser
alterado para a erradiação antibiótica da infecção. A úlcera péptica é uma das
doenças mais comuns, afectando, aproximadamente, 50% da população do mundo.
O Helicobacter pylori está presente em quase 100% dos pacientes adultos com
úlcera duodenal e em aproximadamente 80% dos pacientes com úlcera gástrica. A
associação da úlcera gástrica com o carcinoma gástrico, suspeitada há muito
tempo, está agora a ser confirmada.
Em países em desenvolvimento, onde a maioria das crianças é infectada aos 10
anos de idade, as taxas de carcinoma gástrico são muito altas. Nos EUA e noutros
países desenvolvidos, padrões de higiene e a melhoria socioeconómica crescente da
população reduziram a incidência de infecção. Em paralelo, as taxas de úlcera
péptica e de carcinoma gástrico baixaram. Os antagonistas H2 receptores de
histamina diminuem a secreção ácida a curto prazo, mas as taxas de recaída
atingem 95% dos casos em 2 anos, determinando a retoma do tratamento.
Recentes achados em gastrites crónicas e infecção por Helicobacter pylori em
países desenvolvidos sugerem que:
- Gastrites por Helicobacter pylori são adquiridas na infância e na adolescência
(idade inferior a 20 anos) em mais de 50% dos casos;
- Risco e taxa de aquisição são mais altos na infância; posteriormente, ocorre um
declínio exponencial na taxa de aquisição;
- Novas infecções acontecem na maior idade, mas são bastante raras (incidência
anual de 0,4% em média);
- A taxa e o risco de infecção pelo Helicobacter pylori são altos em nascidos no
princípio do século XX, mas muito mais baixas entre os nascidos depois;
- Este declínio deve-se à diminuição na taxa e no risco de aquisição de gastrites por
Helicobacter pylori em particular na infância.
Todas as espécies de Helicobacter causam algum grau de inflamação persistente no
estômago dos mamíferos. São constatadas gastrites em praticamente todos os
humanos infectados, embora a maioria não tenha nenhum sintoma; 1 em 10
desenvolve a úlcera. O desenvolvimento de adenocarcinoma gástrico é três a doze
vezes mais provável em indivíduos infectados pelo Helicobacter pylori.
A identificação do Helicobacter pylori pela coloração de Gram e a detecção de
anticorpos anti-H. pylori por hemaglutinação ou fixação de complemento têm
sensibilidade relativamente alta e especificidade como a do teste de biópsia/urease.
Existe uma excelente correlação entre uma apresentação clínica clássica de
gastrites, a presença de Helicobacter pylori no estômago e a serologia alterada. A
sensibilidade e a especificidade são de 90%, e o valor preditivo de um resultado
negativo para anticorpos anti-Helicobacter pylori por enzima imunoensaio (EIA) é
elevado na infecção recente, excluindo-se uma população mais velha.
Combinações de antigénios diferentes melhoram a sensibilidade sem perda de
especificidade. Resultados positivos justificam uma tentativa empírica de curta
terapia com antimicrobianos em gastrites de origem desconhecida.
Pode ser esperada uma queda significativa das concentrações de anticorpos IgG
anti-H. pylori depois de terapia antibacteriana adequada. Pacientes assintomáticos
e sem tratamento continuam com seropositividade para IgG, mostrando a presença
de H. pylori mesmo após a resolução histológica. A erradiação de H. pylori está
associada à redução significativa do retorno de úlcera duodenal.
A sua avaliação é útil na diferenciação entre gastrites por Helicobacter pylori e
linfoma gástrico.
Recentemente, surgiram testes imunoenzimáticos para a detecção de antigénios
específicos de Helicobacter pylori. A investigação do antigénio Helicobacter pylori é
um método não-invasivo para a identificação das bactérias em amostras. O teste
tem altas sensibilidade (93,9%) e especificidade (96,3%), com uma precisão
diagnóstica de 95%. Este ensaio pode simplificar o diagnóstico preciso da infecção
por Helicobacter pylori.
A PCR é uma técnica altamente sensível e rápida de identificação de Helicobacter
pylori em espécimes clínicos.
Hemocultura
Quando uma bactéria vence as barreiras normais do hospedeiro e das células do
sistema reticuloendotelial, ela invade a corrente circulatória ou os vasos linfáticos,
podendo rapidamente disseminar-se e causar bacteriémia (presença de bactérias
no sangue). A bacteriémia pode ocorrer de forma transitória, intermitente ou
contínua.
Além disso, os seus produtos metabólicos interagem com os mecanismos de
resposta inflamatória, podendo levar à septicémia e ao choque, que é uma das
mais sérias complicações das doenças infecciosas.
A(s) bactéria(as) responsável(is) pode(m) ser identificada(s) pela realização da
cultura do sangue (hemocultura) e é (são) útil (eis) no diagnóstico etiológico e na
escolha da terapia.
Para o diagnóstico, é importante a colheita de mais de uma amostra (mínimo de 2,
ideal de 3), antes da administração de antimicrobianos. O número de amostras e o
intervalo entre as colheitas dependem do quadro clínico investigado. Nas
bacteriémias agudas e/ou contínuas, recomenda-se a colheita de 3 amostras com
intervalo de 1 a 2 horas. Já nas intermitentes, recomenda-se a colheita em
intervalos menores e antes ou imediatamente após o início do pico febril.
Para a colheita, deve-se fazer anti-sépsia da pele, com álcool a 70%, 2 vezes, e
esperar a acção do anti-séptico durante 2 minutos. Esta operação também pode ser
realizada utilizando-se uma primeira anti-sépsia com álcool a 70%; posteriormente,
utilizar álcool iodado. Puncionar a veia e colher o número de amostras no intervalo
de tempo indicado. Deve-se evitar a colheita de sangue na região inguinal.
O material biológico utilizado pode ser sangue arterial ou venoso, aspirado de
medula óssea ou de qualquer outro líquido biológico. Podem ser colhidos também
líquidos de cavidades fechadas para cultura de anaeróbios. Quando o material for
sangue, o volume colhido é um dos mais importantes parâmetros na detecção de
bactérias na corrente sanguínea. Colher os seguintes volumes nas diferentes faixas
etárias:
- crianças até 1 ano: 0,5 mL a 1,5 ml em cada frasco de cultura;
- crianças de 1 a 6 anos: 1,0 mL por cada ano de idade;
- adultos: 20 mL de sangue para cada amostra de hemocultura.
Quando for utilizada para cultura de líquidos biológicos, qualquer volume do
espécime pode ser utilizado.
O sangue colhido é acondicionado em frascos especiais com meio líquido e
diferentes de acordo com o tipo de bactéria a ser investigada (aeróbia ou
anaeróbia).
A rapidez na identificação etiológica do agente bacteriano é essencial para a
decisão precoce e adequada do tratamento específico. Os métodos modernos
automatizados permitem a disponibilização rápida dos resultados. Nesses métodos,
a presença de bactérias é detectada pelo CO2 produzido durante o crescimento
bacteriano, que irá modificar o sensor existente no fundo do frasco de cultura,
proporcionando a emissão de fluorescência, por sua vez detectada pelo aparelho
utilizado para leitura.
A presença de crescimento bacteriano no sangue do paciente indica bacteriémia
e/ou septicémia. Alguns agentes patogénios devem ser questionados quanto ao seu
poder patogénico, como por exemplo o estafilococos coagulase-negativos numa
única amostra, Propionebacterium acne e grupo Corynebacterium.
Nas infecções hospitalares, os patogénios mais encontrados são Staphylococcus
aureus, Escherichia coli, Klebsiella pneumoniae, Enterobacter spp., Pseudomonas
aeruginosa, Enterococcus spp. e estafilococos coagulase-negativos.
Nas endocardites, os agentes mais frequentemente isolados correspondem aos
Estreptococcus spp. alfa-hemolíticos ou mesmo beta-hemoliticos, assim como aos
Staphylococcus aureus e aos Stafilococcus coagulase-negativos.
O anticoagulante utilizado no meio de cultura (SPS) inactiva o sistema do
complemento, porém inibe o crescimento das Neisseria spp. e Gardnerella
vaginalis. As bactérias com exigencias especiais, tais como Brucella spp., Leptospira
spp., Bartonella spp., Legionella spp., Mycobacteria spp., não crescem nos meios
tradicionalmente usados para hemoculturas.
Hemocultura para Fungos
O isolamento e a identificação de fungos em cultura são prova definitiva no
diagnóstico de fungemia. O sangue venoso do paciente é inoculado num frasco
especial, que contém os nutrientes necessários ao crescimento adequado dos
fungos. Cada frasco contém um sensor capaz de detectar o decréscimo da
concentração de oxigénio, resultante do metabolismo e do crescimento do fungo
presente na amostra. Esse sensor é monitorizado por equipamento automatizado.
No fundo de cada frasco existe um fluorocromo que produz uma fluorescência que é
proporcional ao decréscimo do oxigénio na amostra. Na presença de fungos, ocorre
consumo de oxigénio e, consequentemente, aumento da fluorescência, que
provocará indicação de amostra positiva.
Os resultados devem ser interpretados correlacionando-se com o estado clínico do
paciente. Alguns fungos podem provocar fungemia transitória, sendo isolados em
amostra de hemocultura e com significado clínico duvidoso. Em caso de isolamento
de fungo patogénico, o resultado é liberado para o médico o mais rápido possível.
A presença de infecção bacteriana concomitante pode inibir o crescimento de
fungos, assim como o uso de medicação sistémica. Os frascos utilizados não são
selectivos para fungos e permitem também o crescimento de bactérias. Se
presentes, as bactérias de crescimento rápido podem mascarar a detecção dos
fungos. Alguns fungos, tais como Rodothorula rubra e Blastomyces dermatitidis,
podem não ser detectados por esse método, necessitando, para a sua identificação,
de outros métodos de cultura adicionais.
A identificação de presença de leveduras pelo método automatizado exige uma
espera de 7 dias, e os demais fungos, de 30 dias.
Para colher a amostra, fazer anti-sépsia da pele com álcool a 70%, 2 vezes, e
esperar a acção do anti-séptico durante 2 minutos. Puncionar a veia e colher o
sangue. O número de amostras e o intervalo de tempo de colheita entre elas são
determinados pelo próprio médico assistente. O volume recomendado é de 3 a 5
mL.
Hemoglobina A2, Pesquisa
A HbA2 é composta por dois pares de cadeias alfa e delta e é responsável por cerca
de 3% da hemoglobina do adulto normal. O nível de HbA2 aumenta gradualmente
durante o primeiro ano de vida, quando atinge os níveis de um adulto normal.
Os seus níveis podem estar aumentados nalgumas beta-talassemias, doença da
hemoglobina H e anemias megaloblásticas. A anemia por deficiência de ferro e as
sideroblásticas podem levar à diminuição da sua síntese.
Nos casos de beta-talassemias minor, a HbA2 está aumentada (>3,5%), com
presença de microcitose e hipocromia eritrocitária e níveis séricos de ferritina
normais.
Hemoglobina Fetal, Pesquisa
Cerca de 80% da hemoglobina do recém-nascido é constituída pela hemoglobina
fetal . Ela é composta por 2 cadeias alfa e 2 cadeias gama. Essa hemoglobina
possui uma maior afinidade para o oxigénio do que a do adulto.
Por volta dos 6 meses após o nascimento, quase toda a hemoglobina fetal é
substituída pela do adulto, a hemoglobina A (HbA), restando apenas cerca de 1 a
2% da fetal.
Níveis aumentados no adulto podem ser encontrados na persistência hereditária da
hemoglobina fetal, nas síndromes beta-talassémicas, na interação talassemia/outra
hemoglobinopatia, na anemia falciforme e nas síndromes mieloproliferativas.
Alterações discretas podem ser encontradas nas anemias aplásticas, na esferocitose
hereditária e na anemia megaloblástica.
Hemoglobina Glicosilada
O processo pelo qual a hemoglobina e outras proteínas se ligam à glicose é
denominado glicosilação e corresponde à adição de forma não-enzimática de
resíduos de açúcar a grupos de aminoácidos da proteína.
A glicosilação da hemoglobina ocorre durante os 120 dias do período de sobrevida
das hemácias. Entretanto, a glicose presente no sangue depende de um intervalo
de tempo para glicosilar a hemoglobina. A glicemia dos últimos 30 dias antes do
doseamento contribui com 50% da hemoglobina glicosilada doseada, e as glicemias
dos últimos meses (2 a 4), com 25%. O doseamento final, portanto, corresponde à
média ponderada dos níveis das glicemias das 6 a 8 últimas semanas antes do
doseamento.
No indivíduo adulto normal, a hemoglobina circulante é composta por 97% de HbA,
2,5% de HbA2 e 0,5% de HbF.
A hemoglobina predominante, a hemoglobina A, é formada por 2 subunidades alfa
e 2 subunidades beta. Por isso, as moléculas de açúcar podem ligar-se em
diferentes sítios da hemoglobina, produzindo distintas formas de hemoglobinas
glicadas ou glicosiladas.
A hemoglobina glicosilada HbA1 é uma hemoglobina de migração rápida na
electroforese, que tem um açúcar ligado ao local de maior reactividade para
glicação, o aminoácido N-terminal da cadeia beta. É encontrada em níveis
aumentados nos pacientes com hiperglicemia mantida e dentro dos limites de
referência em indivíduos normais.
De acordo com a metodologia empregue, podemos isolar apenas a HbA1 ou isolar
as suas diferentes fracções, que são designadas como HbA1a, HbA1b HbA1c.Como
a HbA1c representa cerca de 80% da HbA1, a sua avaliação reflete com fidelidade o
grau de glicosilação da hemoglobina. Além disso, a HbA1c é a única
irreversivelmente glicada e, portanto, suficientemente estável para ser utilizada
para monitorização dos níveis glicémicos.
A membrana da hemácia é totalmente permeável à glicose, expondo a hemoglobina
a concentrações de glicose similares às plasmáticas. É com essa exposição que
acontece a ligação da glicose com a valina N-terminal da cadeia beta da
hemoglobina A.
Embora existam diversos métodos para a quantificação da hemoglobina glicosilada,
tais métodos podem ser divididos em dois grandes grupos, de acordo com o
principio utilizado: a separa ção por diferenças estruturais (cromatografia
HPLC/coluna e electroforese) e a separação por diferença de carga (cromatografia
de troca iónica HPLC/coluna e método imunoenzimático). Os dois métodos mais
utilizados são o cromatográfico por troca iónica e a cromatografia de afinidade.
A cromatografia por troca iónica sofre a interferência de hemoglobinas variantes,
como a HbS e HbC, e também por drogas ou substâncias que alterem a carga da
hemoglobina. Já a cromatografia de afinidade não sofre a influência das
hemoglobinas variantes, nem de medicamentos. No entanto, não consegue
quantificar a fracção e sim o total de hemoglobinas glicosiladas. A literatura tem
apontado a quantificação da HbA1c por HPLC como método de referência pelo DCCT
(Diabetes Control and Complications Trial).
Recomenda-se a monitorização a cada 3 meses em todos os pacientes diabéticos.
Nalguns casos, como na diabetes gestacional ou com mudanças importantes do
esquema terapêutico, a monitorização poderá ser mais frequente (a cada 4
semanas).
As principais interferências são as patologias que alteram a semi-vida da hemácias,
hemoglobinopatias e nefropatias crónicas.
Hemoglobina H, Pesquisa
A hemoglobina H (HbH) pode ser encontrada na alfa-talassemia. É uma
hemoglobina composta por 4 cadeias beta. A doença manifesta-se por anemia
hemolítica crónica de grau moderado.
A sua presença pode ser detectada pela electroforese da hemoglobina ou pela
observação de corpos de inclusão que podem ser observados no sangue periférico
por coloração especial. As inclusões de hemoglobina H ocorrem pela precipitação de
cadeias em excesso.
Está também descrita uma forma adquirida da doença da HbH observada nalguns
pacientes com síndromes mieloproliferativos e mielodisplásicos.
Hemoglobina S, Pesquisa
A hemoglobina S pode apresentar-se de formas homozigótica (HbSS),
heterozigótica, (HbAS), ou ainda associada a outras variantes de hemoglobinas,
como a hemoglobina fetal e alfa ou beta-talassemia.
É possível a detecção da hemoglobina S pela electroforese da hemoglobina.
Entretanto, a presença necessita de ser confirmada por metodologias específicas,
visto que é possível observar outras fracções de hemoglobina na mesma posição,
como a HbD e a Hb Lepore, entre outras.
Estão disponíveis diferentes métodos para confirmação da HbS, como a
electroforese em pH ácido, a solubilidade e o teste de céluals falciformes.
A electroforese em pH alcalino com avaliação da percentagem de HbS é útil na
identificação e avaliação da concentração de HbS.
Hemoglobina, Solubilidade
Neste teste, investiga-se a presença de HbS, por meio da lise das hemácias e
redução da possível hemoglobina S presente, pela acção do ditionito de sódio.
Como a HbS é insolúvel em tampões inorgânicos concentrados, formam-se
polímeros que produzem opacidade.
O teste é útil como triagem para anemia falciforme, sendo positivo na presença de
HbS e de outras hemoglobinas também insolúveis como HbC, HbC Harlem e Hb
Bart.
Reações falso-positivas podem ocorrer devido à turvação ocasionada pela presença
de grande quantidade de corpos de Heinz e da presença de precipitação de
proteínas plasmáticas. Podem ocorrer falso-negativos na presença de pequenas
quantidades de HbS, alta concentração de hemoglobina fetal e anemia grave.
Hemoglobinas Instáveis
As hemoglobinas chamadas instáveis constituem um grupo composto por cerca de
80 diferentes tipos de hemoglobinas anormais, que cursam, nos seus portadores,
com anemias hemolíticas agudas ou crónicas. Podem ser identificadas pela
instabilidade das suas cadeias polipeptídicas, que se precipitam quando submetidas
ao calor, levando a uma reacção de floculação.
Esta instabilidade deve-se a uma ligação anormal entre as cadeias alfa e beta ou na
região do heme. A hemólise pode ser induzida por diferentes medicamentos e por
processos infecciosos bacterianos ou víricos.
Nestes pacientes, é frequente, após esplenectomia, a presença de corpos de Heinz
nas hemácias, por colorações especiais. É também um achado laboratorial comum
a presença de metaemoglobina proveniente da constante oxidação do heme.
A presença de altas concentrações de hemoglobina fetal é uma limitação para essa
investigação.
Hemoglobinopatias
A hemoglobina é uma proteína composta por dois pares de cadeias polipeptídicas e
pelo heme (estrutura porfirínica que possui no interior um átomo de ferro mantido
em estado ferroso). As cadeias polipeptídicas são compostas por combinações entre
os diversos tipos de cadeias, resultando em hemoglobinas diferentes na sequência
de seus aminoácidos. As cadeias são identificadas como a (alfa), b (beta),g (gama),
d (delta) e as embrionárias e (épsílon) e z (zeta).
Cerca de 80% da hemoglobina do recém-nascido é constituída pela hemoglobina
fetal (duas cadeias a e duas cadeias g ). Essa hemoglobina possui maior afinidade
com o oxigénio do que a do adulto. Por volta dos 6 meses após o nascimento,
quase toda a hemoglobina fetal é substituída pela do adulto, a hemoglobina A
(HbA), restando apenas cerca de 1 a 2% da fetal.
A HbA é composta por dois pares de cadeias alfa e beta. Representa quase a
totalidade da hemoglobina do adulto, que pode apresentar mínimas fracções da
hemoglobina fetal e da chamada HbA2 (dois pares de cadeias alfa e delta).
Existe um grupo de hemoglobinas chamadas de anormais porque diferem do ponto
de vista estrutural e bioquímico da hemoglobina normal (HbA). As mutações são
genéticas, podendo ter o caráter homozigótico ou heterozigótico, e são
consequência de alterações dos genes estruturais (alterações qualitativas) ou dos
genes reguladores (alterações quantitativas) que comandam a síntese das cadeias
polipeptídicas.
Portanto, as alterações ditas qualitativas são as que afectam estruturalmente a
formação das cadeias, resultando em hemoglobinas bioquimicamente diferentes,
denominadas anormais ou variantes. São causadas na sua maioria pela substituição
de aminoácidos, sendo as mais comuns, no Ocidente, a hemoglobina falciforme
(HbS), que decorre da substituição do ácido glutámico por valina na posição 6 da
cadeia beta, e a hemoglobina C (HbC), decorrente da substituição do ácido
glutámico por lisina, também na cadeia beta.
Existem mais de 300 tipos diferentes de hemoglobinas anormais descritos, mas
apenas uma pequena parte delas está associada a manifestações clínicas e
hematológicas. Elas podem ser identificadas pela mobilidade electroforética e são
designadas por letras. A primeira a ser identificada foi a HbS, assim denominada
pela aparência característica em foice da hemácia falciforme que, em inglês, é
sickle (foice), o que explica a denominação HbS. Nos anos que se seguiram, foram
descritas muitas outras variantes. A partir de 1962, durante o Congresso da
Sociedade Internacional de Hematologia, estabeleceu-se a regra para a descrição
das variantes reconhecidas a partir daquela data. Ficou estabelecido que passariam
a receber designações seguindo o alfabeto (C, D, E, G, H) e que, quando com a
mesma mobilidade electroforética porém com estruturas diferentes, seriam
identificadas pela cidade onde foram descobertas.
As mutações dos genes reguladores promovem alterações quantitativas que levam
a anormalidades da síntese das hemoglobinas, com uma deficiência na produção de
uma ou mais cadeias polipeptídicas da hemoglobina. Esta ocorrência dá origem às
talassemias, classificadas de acordo com a cadeia que é produzida em taxa
reduzida como a-tal e b-tal, as mais frequentes. São descritas também a b-d-tal, dtal e a g d b -tal. Os quadros clínicos variam de assintomáticos a moderados, leves
e muito graves.
Mais informações de exames relacionados:
Curva de fragilidade osmótica
Electroforese de hemoglobina
Hemoglobina A2, S, H, fetal
Hemoglobinas instáveis
Hemograma
Hemoglobina, Solubilidade
Hemograma
O hemograma contempla diversas provas efectuadas, com a finalidade de avaliar
quantitativa e qualitativamente os componentes celulares do sangue. Os itens
avaliados incluem: eritrócitos, hemoglobina, hematócrito, índices hematimétricos,
leucócitos totais, contagem diferencial de leucócitos, plaquetas e exame
microscópico de esfregaço de sangue após coloração.
A análise quantitativa dos eritrócitos, leucócitos totais, plaquetas e a avaliação dos
índices hematimétricos são hoje realizados por meio de equipamentos
automatizados que combinam diferentes métodos de avaliação de alta tecnologia e
precisão à capacidade de análise de milhões de células, permitindo resultados mais
precisos.
A utilização desses equipamentos permite também a avaliação de índices
hematológicos e a visualização em histogramas que demonstram a distribuição dos
diferentes elementos analisados.
Essa característica possibilita a identificação de alguns parâmetros antes
impossíveis de serem avaliados ou que eram analisados subjetivamente, com a
visualização do esfregaço em lámina. Entre esses parâmetros, temos o índice de
anisocitose (RDW), a identificação de populações mistas de células, a anisocitose
plaquetária e alertas para possíveis alterações presentes na amostra examinada.
Esses alertas são específicos para alterações das séries vermelha, branca e das
plaquetas, como presença de blastos, granulócitos imaturos, desvio à esquerda,
linfocitos atípicos, agregados plaquetários, microcitose, hipocromia, entre outros.
Realizam ainda, por uma combinação de métodos de análise celular e coloração, a
contagem diferencial de leucócitos, que serve de orientação para o hematologista,
chamando a atenção para situações nas quais a avaliação deve ser mais cuidadosa.
A análise qualitativa é realizada pela avaliação da lámina corada, associada aos
resultados obtidos pela avaliação electrónica. A coloração das células diferencia em
detalhes as estruturas nucleares e citoplasmáticas, permitindo a avaliação do
tamanho das células, a relação núcleo/citoplasma, a forma do núcleo, a presença
de nucléolos, o padrão da cromatina e a coloração do citoplasma, a presença de
granulação, vacúolos e outras alterações morfológicas.
Os resultados auxiliam a identificação de doenças de origem primária ou secundária
de características agudas ou crónicas. São utilizados também para acompanhar a
evolução de uma variedade de doenças e para monitorizar os efeitos colaterais
decorrentes do uso de medicamentos.
A avaliação eritrocitária pode identificar processos anémicos, policitémicos,
alterações de forma e tamanho dos eritrócitos. A avaliação leucocitária pode
identificar processos inflamatórios, infecciosos, alérgicos, parasitários e leucémicos.
Pode também indicar a presença de elementos anormais e de linfocitos atípicos. A
avaliação plaquetária identifica processos de trombocitopenias adquiridas ou
hereditárias e trombocitoses.
SÉRIE VERMELHA
A contagem de eritrócitos é a primeira informação fornecida e é expressa em
milhões por mm3. Os níveis de eritrócitos iguais ou maiores que 6,20 milhões/mm3
em homens e iguais ou maiores que 5,70 milhões/mm3 em mulheres são
considerados poliglobulia.
A concentração de hemoglobina é expressa em g/dL, e a sua avaliação é de grande
importância pelo papel no transporte de oxigénio e por estar directamente
relacionada à anemia, sendo a melhor forma de avaliação laboratorial.
O volume relativo dos eritrócitos dentro do volume de sangue é fornecido pela
análise do hematócrito, que é expresso em percentagem.
A relação entre esses diferentes parâmetros pode ser obtida pela análise dos
índices hematimétricos que irão fornecer informações adicionais sobre as variações
de volume e concentração da hemoglobina. Os achados morfológicos do esfregaço
após coloração fornecem mais informações sobre conteúdo da hemoglobina, forma,
tamanho e inclusões eritrocitárias.
Índices Hematimétricos
Volume Corpuscular Médio - VCM
Avalia a média do tamanho (volume) dos eritrócitos, que estão no tamanho normal,
e são denominados normocíticos, diminuídos (microcíticos) ou aumentados
(macrocíticos). O achado de microcitose é comum em anemias por deficiência de
ferro, nas doenças crónicas e nas talassemias. O aparecimento de macrocitose
pode estar associado à presença de um grande número de reticulócitos, ao
tabagismo e à deficiência de vitamina B12 e de ácido fólico.
ALTERAÇÃO
DE TAMANHO
NORMOCITOSE
MICROCITOSE
MICROCITOSE DISCRETA
MICROCITOSE ACENTUADA
MACROCITOSE
MACROCITOSE ACENTUADA
ANISOCITOSE
ADULTOS
CRIANÇAS
ATÉ 3 ANOS
CRIANÇAS DE 4
A 14 ANOS
81,0
73,0
73,0
<ou = 81,0
<ou= 71,0
<ou= 73,0
80 a 80,9
71 a 72,9
73 a 74,9
< 60
< 60
< 60
> 90
> 99
> 99
>120
> 120
> 120
Presença de eritrócitos com volumes diferentes
Hemoglobina Corpuscular Média (HCM)
Índice hematimétrico que corresponde à média de hemoglobina por eritrócito. Pode
estar elevado na presença de macrocitose e diminuído na presença de eritrócitos
microcíticos.
Concentração da Hemoglobina Corpuscular Média (CHCM)
É a avaliação da hemoglobina encontrada em 100 mL de eritrócitos. Este índice
permite a avaliação do grau de saturação de hemoglobina no eritrócito. A saturação
da hemoglobina normal indica a presença de eritrócitos ditos normocrómicos.
Quando diminuída, teremos eritrócitos denominados hipocrómicos e, quando
aumentados, eritrócitos hipercrómicos.
RDW (Red Cell Distribution Width)
A variação do tamanho dos eritrócitos é analisada electronicamente pela variação
de impulsos obtidos durante a leitura. A análise dessa variação permite a obtenção
deste novo índice, que representa a amplitude de distribuição dos glóbulos
vermelhos, servindo como um índice de anisocitose, que se altera precocemente na
deficiência de ferro, mesmo antes da alteração de outros parâmetros, como a
alteração do VCM e a diminuição da hemoglobina.
Alterações da Cor (Características Tintoriais)
A coloração dos eritrócitos reflecte a concentração da hemoglobina e pode ser
ocasionada:
- pela diminuição da concentração de hemoglobina e consequente redução da cor,
que leva à chamada hipocromia;
- pela presença de células com diferentes concentrações de hemoglobina chamada
anisocromia;
- pela presença de um grande número de reticulócitos que caracteristicamente têm
uma cor azulada, que junto com a cor normal, produz a chamada policromasia.
POIQUILOCITOSE
Variação das formas dos eritrócitos que normalmente se apresentam numa
forma circular e com uma halo central claro.
...........................................................................
ELIPTÓCITOS/
Eritrócitos elípticos e ovalados, que ocorrem na ovolacitose hereditária
OVALÓCITOSE
(eliptocitóse) e podem também ser encontradas em anemias carenciadas
e mais raramente nas talassemias e outras anemias.
...........................................................................
ESFERÓCITOS
Eritrócitos pequenos, de forma esférica e hipercorada que aparece na esfericitose
hereditárias e nas animias hemolíticas auto-imunes.
...........................................................................
DACRIÓCITOS
Eritrócitos em forma de lágrima. Ocorrem provavelmente por atrazo da saída
da medula óssea. Presente na metaplasia mielóide, na anemia megaloblástica,
nas tatassemias e na esplenomegalia.
..........................................................................
CODÓCITOS
Células em forma de alvo. Ocorre um excesso de membrana, o que faz com
que a heoglobina se distribua num anél periférico, com uma zona densa
central. Encontradas nas talassemias, na hemoglobina C, icterícia obstrutiva
e na doença hepática severa.
..........................................................................
DREPANÓCITOS
Eritrócitos em forma de foice, caracteristica da anemia falsiforme.
..........................................................................
ACANTÓCITOS
Eritrócitos pequenos com projeções irregulares. Célula peculiar da betalipoproteinemia hereditária, presente também noutras dislipidemias, na cirrose
hepática, na hepatite do recám-nascido, na anemia hemolítica, após
esplenectomia e após administração de heparina.
..........................................................................
ESQUIZÓCIOTS
Fragmentos de eritrócitos de tamanhos diferentes e com fromas bizarras.
Observados em muitos casos de próteses valvulares e vasculares, microangiopatias, síndrome hemolítico-urêmico, nos casos de queimaduras graves e
na coagulação intravascular disseminada.
.
.
.
.
Inclusões e Outras Variações das Hemácias
As inclusões que podem ser observadas nos eritrócitos estão relacionadas com
diferentes patologias e são consequência do aumento ou de defeitos da
eritropoiese. Dependem, também, da capacidade do baço de retirar da circulação
as hemácias mal formadas. Outras alterações também podem ser observadas,
como a presença de eritroblastos e a formação de rouleaux.
ANEL DECABOT
Figura em forma de anel observada na anemias megaloblásticas, podendo,
nalguns casos, torcer-se, assuimindo um aspécto de oito (8). Pode também
ser observada noutras situações de eritropoiese anormal. É um filamento
fino, de cor vermelho-violeta, concêntrico em relaçãoa à membrana celular, que
resulta de restos mitóticos de mitoses anomalas.
...........................................................................
CORPÚSCULOS
DE HOWEL JOLLY
Corpúsculo de inclusão pequena, basófilo, restos nucleares de mitoses anomalas. São observados em pacientes esplenectomizados, nas anemias hemolíticas e megaloblásticas.
...........................................................................
PONTILHADO
BASÓFILO
Granulações variáveis em número e tamanho, de cor azulada, agregados de
ribissomas remanescentes. podem ser encontradas na intoxicação por metais
especialmente o chumbo, nas talassemias, e outras alterações da hemoglobina, nas mielodisplasias e noutras formas de anemia severa.
...........................................................................
CORPOS
DE HEINZ
Precipitados de hemoglobina desnaturada que podem ser encontrados aderidos
à membrana dos eritrócitos, em pacientes com anemia hemolítica por alguns
fármacos, na deficiência da glicose-6-fosfato desidrogenase e nos síndromes das
hemoglobinas instáveis. Para a sua visualização, é necessário coloração especial
com azul de cresil brilhante.
...........................................................................
CORPÚSCULO DE
PAPPENHEIMER
Associados a ribossomas remanescentes, apresentam-se como pontos
enegrecidos, agrupados ou localizados nas anemias hemolíticas e
sideroblásticas.
...........................................................................
ROULEAUX
Aglutinação dos eritrócitos que formam verdadeiras pilhas, podendo ser
observadas em lámina corada, são decorrentes da concentração elevada de
fibrinogénio ou de globulinas, especialmente nas gamapatias monoclonais.
Levam ao aumento da velocidade de hemossedimentação.
...........................................................................
ERITOBLASTOS
São eritrócitos nucleados que podem aparecer no sangue periférico devido
a grandes regenerações eritrocitárias, ou como consequências de infiltração
medular. Aparecem na anemia hemolítica e nas reações leuco-eritroblásticas
(fibrose e metástase medular).
SÉRIE BRANCA
A contagem global e diferencial de leucócitos e suas alterações quantitativas e
qualitativas são as principais informações fornecidas na análise da série branca. Os
leucócitos totais são expressos em mil/mm3. A contagem diferencial é de grande
importância, podendo definir perfis patológicos, e é fornecida pela análise conjunta
dos equipamentos automáticos e pela leitura do esfregaço corado, que avalia as
diferentes formas leucocitárias e as expressa de forma percentual (relativa) e em
mm3 (absoluta). A análise das alterações morfológicas dos leucócitos também é
realizada por observação microscópica do esfregaço corado. Os leucócitos podem
ser divididos em granulócitos (mielócito, metamielócito, com forma de bastonete,
neutrófilos, eosinófilos e basófilos), monócitos e linfócitos.
Alterações Morfológicas dos Leucócitos
Pelger-Hüet
É uma alteração hereditária autossómica dominante rara, que se caracteriza pelao
presença de neutrófilos hipossegmentados, sem que no entanto ocorra alteração da
função da célula. Os neutrófilos aparecem na periferia com discreta segmentação
(bilobulados) ou mesmo sem segmentação (como bastonetes). Como não conduz a
alterações funcionais, não apresenta repercussões clínicas. O diagnóstico assume
importância afim de evitar a sua interpretação como um desvio à esquerda. Pode
também ser encontrada nos eosinófilos. Existe, ainda, um quadro chamado de
pseudo-Pelger-Hüet, no qual esta alteração pode ser adquirida, sendo causada por
reacções a fármacos e em alguns casos de mielodisplasias e leucemias.
Alder-Reilly
É uma alteração hereditária autossómica recessiva, que se caracteriza por grânulos
grosseiros de cor púrpura, os quais podem ser encontrados em granulócitos,
monócitos e linfócitos.
May-Hegglin
Alteração hereditária autossómica dominante rara, na qual os neutrófilos
apresentam inclusão citoplasmática azulada de RNA, semelhante aos corpos de
Döhle, associada a trombocitopenia leve e à presença de plaquetas gigantes.
Chediak-Higashi
Doença autossómica recessiva rara, em que os granulócitos, monócitos e linfócitos
se apresentam com gránulos gigantes, associada a infecções piogénicas frequentes
e anormalidade funcional dos leucócitos, geralmente com sobrevida curta.
Alterações Adquiridas
Entre as alterações adquiridas, pode ser destacada a presença de corpúsculos de
Döhle, inclusões ovais azuladas encontradas na periferia do citoplasma.
Geralmente, encontram-se isolados e são consequência de ribossomas que
persistiram e costumam ser encontrados em infecções e grandes traumas. O
achado de granulações grosseiras (tóxicas) no citoplasma dos neutrófilos pode
acontecer em longos processos infecciosos. As vacuolizações citoplasmáticas
acontecem em resultado da depleção dos gránulos azurófilos no processo de
fagocitose.
Plaquetas
A avaliação das plaquetas pode ser feita de forma quantitativa, expressa em mm3,
e de modo qualitativo, pela avaliação das características analisadas no esfregaço
corado, o que permite a identificação de alterações morfológicas das plaquetas.
A utilização de equipamentos automáticos, além de fornecer contagens mais
precisas, permite que se obtenham informações em relação à presença de
anisocitose e agregados plaquetários, e também de índices plaquetários, que na sua
maioria ainda não são liberados para uso clínico. Entre eles, os que começam a ser
utilizados são o MPV (Mean Platelet Volume), considerado um índice de anisocitose
plaquetária, e o PDW (Platelet Distribution Width). Entretanto, ainda faltam maiores
dados de correlação clínica.
As alterações quantitativas podem ser tanto o aumento da quantidade de
plaquetas, chamada hiperplaquetémia, quanto a diminuição, denominada
plaquetopenia.
Consultar Plaquetas.
Hepatites
A hepatite vírica consiste numa doença inflamatória, com quadro clínico infeccioso.
São conhecidos vários tipos de vírus hepatotrópicos, com características físicoquímicas e epidemiológicas próprias para cada tipo e subtipos.
As infecções desenvolvem-se de forma transitória ou persistente, favorecendo a
evolução para a infecção aguda autolimitada, estado de portador crónico
assintomático e doença crónica.
Observa-se, com frequência, o desenvolvimento clínico associado às manifestações
sistémicas, semelhantes àquelas observadas em diferentes infecções, não
permitindo, dessa forma, o diagnóstico etiológico preciso.
Os principais agentes víricos hepatotrópicos causadores de hepatite são designados
como: vírus da hepatite A (HAV); vírus da hepatite B (HBV); vírus da hepatite C
(HCV); vírus da hepatite Delta (HDV); vírus da hepatite E (HEV) e vírus GBV-C
(HGV). Cada um apresenta características estruturais e genómicas que permitem
diferenciá-los, detectadas por diferentes metodologias laboratoriais com
sensibilidade e especificidade definidas.
Infecções persistentes causadas por HBV, HCV e HDV estão sempre associadas a
doença hepática crónica, podendo evoluir para cirrose e hepatocarcinoma.
Hepatite A
O vírus da hepatite A (HAV) classifica-se na família Picornaviridae e é transmitido
por via fecal-oral, determinando formas esporádicas e epidémicas de hepatite
aguda. Água e alimentos contaminados constituem as vias mais comuns em surtos
epidémicos. O período de incubação da doença é de 4 semanas, com evolução
clínica de forma branda a moderada e ausência de formas crónicas. A hepatite
fulminante é descrita em 0,2 a 0,7% dos indivíduos infectados. Em crianças na
faixa etária de 1 a 10 anos, a hepatite A é geralmente anictérica e assintomática.
A presença da imunoglobulina M específica (IgM anti-HAV) no sangue é quase
sempre concomitante ao período sintomático da hepatite aguda. Títulos elevados,
obtidos por metodologias de diluições seriadas ou por comparação aos valores
limites (cut off), são detectados na fase aguda e no início da convalescença. Títulos
menores são detectados 3 a 4 meses após o acometimento da doença e podem
persistir por mais de 6 meses, em 20 a 30% dos indivíduos, e até por 1 ano em 5 a
10%. Nos casos em que os sintomas precedem por poucos dias, a detecção de IgM
anti-HAV, recomenda-se a serologia pareada. A segunda colheita, feita após 15 a
20 dias, é útil para a confirmação do resultado reactivo, definindo o laudo de
positividade.
Os métodos actuais para a pesquisa em soro e plasma permitem a detecção
qualitativa e, eventualmente, um cálculo estimado, semiquantitativo, pela análise
comparativa da reactividade do teste com o valor limite (cut-off).
Anticorpos da classe IgG anti-HAV permitem a definição de infecção passada.
Resultados de seroconversão, traduzidos por anticorpos IgG não-reactivos numa
primeira análise e reactivos na segunda colheita, com intervalos de 20 a 30 dias,
estão associados a infecção recente.
A resposta imune aos diferentes antigénios estruturais, proteínas do capsídeo viral,
VP0, VP1 e VP3, ocorre a partir da quarta semana após a infecção, com títulos
máximos de anticorpos após a sétima semana.
A pesquisa do RNA-HAV em soro, plasma e suspensão fecal é um método pouco
utilizado, embora permita a detecção do genoma viral a partir do segundo dia após
a infecção, podendo prolongar-se até o 25º dia. A metodologia de PCR pode
detectar períodos mais longos de virémia (várias semanas), em concentrações
elevadas, de 104 a 106 partículas virais/mL de sangue.
A heterogeneidade de sequências genómicas de isolados víricos obtidos em
diferentes partes do mundo definiu, até ao momento, 7 genótipos e 1 sorotipo de
HAV.
Hepatite
A
Fase de
Incubação
Anticorpos
totais antiHAV
(serologia)
Anticorpos
IgM antiHAV
(serologia)
RNA-HAV
(pesquisa
em
suspenção
fecal
P: Positivo.
N: Negativa.
Fase aguda
Fase Crónica Resposta
sintomática
Canvalecência Sintomática ou eficaz à
ou
Assintomática vacinação
assintomática
N
N
P
P
P
N
P
P
N
Não há
comfirmação
de
evolução para
cronicidade.
São descritos
casos
esporádicos
de hepatite
fulminante e
de
reagudização.
P
N
N
Considerações
gerais
O HAV acomete
população
pediátrica com
evolução
assintomática e
população adulta
com
sintomatologia
evidente.
Resultado reactivo em primeira avaliação e comfirmado em segunda avaliação.
Resultado não-reactivo em primeira análise e confirmado em segunda avaliação.
Hepatite B
O vírus da hepatite B (HBV) pertence à família Hepadnaviridae e apresenta três
formas distintas: partículas defectivas esféricas, tubulares e o vírião, constituído
por um envólucro (antígeno de superfície do vírus da hepatite B - HBsAg),
nucleocapsídeo (antígeno do core - HBcAg e antigénio e - HBeAg), DNA de cadeia
dupla parcial e a enzima polimerase.
A transmissão ocorre por via parenteral, com a presença obrigatória de sangue ou
derivados. A transmissão sexual é eficaz, da mesma forma que a materno-fetal,
sendo a eficácia definida pelo grau de replicação viral ou pela quantidade de vírus,
avaliada pela concentração de DNA-HBV (carga viral) no sangue.
O HBV é detectado em sangue, saliva, leite materno, secreções vaginais, sémen e
líquido ascítico. A transmissão homossexual tem diminuído em consequência da
consciencialização e das acções efectuadas para controlar a epidemia de SIDA. A
transmissão heterossexual responde por mais de um terço dos casos novos nos
EUA. A taxa de portadores do HBV varia enormemente no mundo. A taxa global nos
EUA é 0,3%; em zonas da África, Filipinas, e Ásia, alcança 20%. O risco de adquirir
HBV após acidente com agulhas contaminadas varia de 20%, nos casos em que o
paciente é positivo para HBsAg, a 66%, quando o paciente é positivo para HBsAg e
HBeAg.
Estima-se a existência mundial de 300 milhões de portadores crónicos do vírus da
hepatite B (HBV). A história natural de infecção aguda por HBV varia de acordo com
a idade do paciente e o tempo de infecção. Em adultos, 95% dos casos evoluem
com vários graus de gravidade da doença aguda, havendo resolução espontânea na
maioria. Cerca de 5% de adultos e 90% de recém-nascidos infectados desenvolvem
hepatites crónicas.
O período de incubação para HBV varia de 45 a 180 dias. As características clínicas
da doença variam consideravelmente. A icterícia acontece em menos de 10% dos
casos em crianças abaixo de 5 anos de idade. Porém, a icterícia manifesta-se em
50% de crianças mais velhas e em adultos. Os sintomas incluem anorexia, náusea,
vómitos, queixas gripais e fadiga. Achados físicos variam de anormalidades
inespecíficas mínimas a icterícia e hepatomegalia e, ocasionalmente, características
extra-hepáticas que refletem fenômenos imunológicos, como vasculites, nefrites
por imunocomplexos, artrites e poliarterite nodosa.
A maioria dos adultos com infecção por HBV aguda apresenta recuperação total, e
cerca de 5%, especialmente os homens, desenvolve infecção crónica, que é
frequentemente assintomática. Dez a 20% desses pacientes podem progredir para
cirrose ou carcinoma hepático.
Três fases de replicação viral acontecem durante o curso da infecção por HBV,
especialmente em pacientes com hepatites crónicas.
Fase de Alta Replicação
Está associada à presença de HBsAg, HBeAg e HBV DNA. Ocorrem aumentos nas
aminotransferases, e, histologicamente, comprova-se a actividade inflamatória
moderada. O risco de evolução para cirrose é alto.
Fase de Baixa Replicação
Associada a perda do HBeAg, a diminuição ou a não-detecção de concentrações de
DNA-HBV. A soroconversão, com aparecimento de anti-HBe, pode indicar
diminuição da actividade inflamatória, uma vez que indica diminuição da replicação
vírica.
Fase Não-Replicante
Associada a ausência, a concentrações indetectáveis ou só detectáveis por meio de
técnicas ultra-sensíveis de marcadores de replicação viral, a inflamação apresentase diminuída, e os achados histológicos não são significativos.
O aumento das aminotransferases, especialmente da ALT (TGP), durante a hepatite
aguda B, varia de um aumento médio a moderado a um aumento notável, superior
a 100 vezes o valor da normalidade. As concentrações de ALT são normalmente
mais altas que as de AST. A concentração de bilirrubina sobe na maioria dos
pacientes com infecção aguda. A icterícia clínica manifesta-se em 50% de adultos
com concentrações de bilirrubina de 3,0 mg/dL. Concentrações maiores podem
acontecer. Uma elevação leve da fosfatase alcalina também é evidente.
Em pacientes que desenvolvem insuficiência hepática fulminante, uma queda rápida
em ALT e AST (TGO) pode levar à conclusão errónea de que a infecção hepática
está a melhorar, quando na realidade, há perda de hepatócitos. Aumentos nas
concentrações de aminotransferases durante mais de 6 meses são indicativos de
evolução para hepatite crónica.
O HBV pode determinar uma variedade de doenças hepáticas, incluindo infecção
aguda autolimitada, hepatite fulminante, hepatite crónica com progressão para
cirrose e falência hepática, hepatocarcinoma e estado de portador crónico
assintomático.
Sistemas de antigénios e anticorpos específicos são definidos como marcadores
biológicos e sorológicos do HBV e podem ser detectados por testes laboratoriais
sorológicos que apresentam alta sensibilidade e especificidade. O HBsAg e a IgM
anti-HBc são os principais marcadores em processos de infecção aguda, embora a
IgM anti-HBc possa permanecer reactiva por alguns meses, em determinados
indivíduos, após essa fase. O HBsAg pode ser detectado em soro e plasma, em
períodos de 3 a 13 semanas após o início da infecção, dependendo da carga viral à
qual o indivíduo foi exposto.
Marcadores da
Hepatite B
Fase de
Incubação
HBsAg
(antigénio de
envólucro
P
Anticorpo
Anti-HBs
N
Quando positivo,
é indicativo de
alta replicação
viral e
simultâneo ao
HBsAg, com
prognóstico
HBeAg
desfavorável.
(antigénio de
Negativo e
nucleosimultâneo ao
capsídeo viral)
HBsAg com
significado de
baixa replicação,
conferindo
melhor
prognóstico.
Fase aguda
sintomática ou
assintomática
Fase crónica
sintomática ou
assintomática
Resposta
eficaz à
Vacinação
P em alta
concentração
N
> 10 mU/ml
evoluindo para
superior a
100 mU/ml
N
10 mU/ml
após
a 1ª dose
100 mU/ml
após
a 2ª dose
1000 mU/ml
após
a 3ª dose
N
P em altos
títulos ou N: em
casos
de mutações da
região pré-core.
N
Convalecência
Baixa
P:em altas
concentração ou
concentrações
N
N
P ou N
Anticorpo
Anti-HBe
HBcAg (útil
em pesquisa)
IgM anti-HBc
Ac totais antiHBc
DNA-HBV
quantitativo
N ou P:baixa
concetração
N ou P
N ou P
N
N
P
P
N
P
N
N
P
N
N
N
N
N
P
P
N
P: define
replicação viral
activa com alta
produção de
vírus.
P
N
P
N
>500.000
N ou
cópias/ml (sem
N
<10.000cópias/ml
tratamento)
P: Positivo. Resultado reactivo na primeira avaliação e comfirmado na segunda avaliação.
N: Negativa. Resultado não-reactivo em primeira análise e confirmado em segunda avaliação.
DNA-HBV
Carga viral
>100.000
cópias/ml
>100.000
cópias/ml
A concentração máxima anterior ao desenvolvimento dos sintomas pode ser
definida por alguns métodos quantitativos ou estimada por métodos
semiquantitativos, quando comparada ao valor limite da reacção (cut-off). Estimase que 5 a 10% dos indivíduos infectados apresentem a IgM anti-HBc como único
marcador de infecção aguda. Outro antigénio estrutural, HBeAg, deve ser
pesquisado sempre após a confirmação do marcador HBsAg. A detecção do
antigénio, que pode ocorrer antes do desenvolvimento dos sintomas, define o grau
de infectividade e de replicação viral. A soroconversão para anti-HBe ocorre durante
as 5 primeiras semanas da doença, definindo a diminuição da replicação viral e,
consequentemente, do grau de infectividade.
A persistência do HBsAg, em concentrações elevadas, da ordem de dez a cem
vezes o valor limite, e a presença do HBeAg em análises seriadas, por períodos de
até 4 meses, associam-se à evolução para a infecção crónica. Anticorpos contra o
HBcAg, da classe IgG, são detectados de 12 a 20 dias após a fase aguda da
doença, persistindo por muitos anos. Esses anticorpos podem indicar infecção
passada, na ausência de HBsAg, ou infecção crónica, quando associados ao
antigénio de superfície.
Alguns estudos demonstram a presença de IgG anti-HBc como único marcador em
até 10% dos indivíduos. Este resultado apresenta significados diferentes que devem
ser analisados para determinadas situações:
- Baixas concentrações de IgG anti-HBc, geralmente inferiores a 10 vezes o valor
limite, sugerem infecção passada sem replicação viral;
- Altas concentrações de IgG anti-HBc, superiores a 10 vezes o valor limite,
sugerem infecção passada com replicação viral indetectável, sendo necessário,
nesses casos, o acompanhamento por meio da pesquisa do DNA HBV por PCR
qualitativo;
- Valores de IgG anti-HBc próximos ao valor limite sugerem reactividade
inespecífica.
Em todos os casos mencionados, deve-se avaliar a necessidade de análises em
novas colheitas, utilizando-se metodologias de diferentes procedências. A
complementação dos resultados, por meio da análise do HBeAg e anti-HBe, deve
ser realizada. Porém, a ausência do antigénio nem sempre está associada a
diminuição da replicação viral. A não-detecção do HBeAg em portadores com
elevada replicação viral é possível e associa-se às mutações do DNA HBV na região
do pré-core/core.
A detecção do DNA HBV em soro, plasma e tecido hepático auxilia na definição da
replicação viral efectiva (indivíduos HBsAg-negativo) e no diagnóstico precoce
(crianças nascidas de mães HBsAg-positivo). Após a detecção qualitativa,
recomenda-se a análise quantitativa da carga viral (DNA HBV). Essa avaliação
apresenta sensibilidade superior a 70% quando comparada à detecção do HBeAg,
sendo indicada para a monitorização do tratamento.
A pesquisa de anticorpos específicos para o HBsAg é indicada para definir a fase de
convalescença e redução drástica da replicação viral, sugerindo imunidade. A
recomendação para o diagnóstico laboratorial é a análise de pelo menos 2 amostras
de material biológico, soro ou plasma, em períodos diferentes, seguindo
metodologias tradicionais.
Hepatite C
O vírus da hepatite C (HCV) é classificado na família Flaviviridae. Apresenta RNA de
cadeia simples, polaridade positiva, com cerca de 9.400 nucleotídeos. Até a sua
identificação e caracterização em 1989, por Choo e colaboradores, os casos clínicos
não-diagnosticados sorologicamente para os vírus das hepatites A e B ou outros
vírus hepatotrópicos conhecidos (Epstein-Barr, febre amarela, citomegalovírus,
vírus da hepatite delta) eram rotulados como hepatite por vírus não-A e não-B.
Sabe-se, actualmente, que o HCV é o principal agente etiológico, responsável por
90 a 95% dos casos de hepatite pós-transfusional não-A e não-B. A transmissão
ocorre por via parenteral, por intermédio do sangue e derivados, pela utilização de
agulhas e seringas contaminadas e transplantes de órgãos e tecidos. A transmissão
sexual tem sido relatada, embora seja pouco frequente. Ocorrências esporádicas,
sem história prévia de transfusão ou outra causa aparente, representam cerca de
40% dos casos de hepatite C.
A infecção pelo HCV assemelha-se à causada pelo vírus B, e os sintomas iniciais da
doença são inespecíficos e/ou gastrointestinais, seguindo-se a icterícia. Os níveis de
alanina aminotransferase apresentam flutuações, com valores inferiores aos
observados nas hepatites A e B. O curso clínico da hepatite C é menos severo que o
da B, porém a evolução para a forma crónica da doença ocorre em 50% dos
pacientes infectados pelo vírus C, em comparação aos 5 a 10% dos casos de
indivíduos infectados pelo vírus B.
A detecção de anticorpos contra o HCV, em amostras de soro de pacientes
infectados, pode ser feita por ensaios imunológicos específicos. Porém, esses
anticorpos refletem a exposição prévia ao agente infeccioso e não podem ser
considerados marcadores da infecção actual. Não estão, ainda disponíveis métodos
imunológicos directos para a detecção de antigénios virais e métodos de cultura
celular para o isolamento viral. A quantificação dos níveis de alanina
aminotransferase, associada à sorologia, é utilizada como indicador auxiliar na
avaliação da infecção pelo HCV, porém não constitui medida directa da virémia.
A reacção em cadeia da polimerase (PCR), pela amplificação exponencial do ácido
nucleico viral, permite a detecção e a quantificação em amostras de soro ou
plasma, constituindo-se a medida directa para avaliação da virémia.
A capacidade de detectar e de quantificar a carga viral é altamente desejável e será
especificamente requerida nas seguintes condições: estabelecer o agente etiológico
em casos de infecção aguda, quando ensaios imunodiagnósticos são negativos;
identificação de indivíduos assintomáticos; monitorização da virémia em casos
crónicos, com fins prognósticos ou quando o imunodiagnóstico resulta em dados
inconsistentes; identificação de pacientes crónicos com elevada carga viral, que
possuem, portanto, alto risco de desenvolvimento do carcinoma hepatocelular;
monitorização da terapia antiviral; detecção do HCV em indivíduos anti-HCV
positivos que tenham desenvolvido auto-anticorpos; detecção do HCV em dadores e
receptores de transplantes hepáticos; avaliação da transmissão vertical do HCV.
As técnicas de rT-PCR e PCR são utilizadas para a detecção e a quantificação do
RNA viral ou do DNA pró-viral integrado ao genoma da célula hospedeira, em
amostras de soro ou plasma, tecido hepático e células do sangue periférico.
Rotineiramente, o método da rT-PCR é utilizado com boa sensibilidade e
especificidade como teste qualitativo e quantitativo em análises de soro ou plasma.
A metodologia utilizada permite a amplificação em quantidade do genoma viral
(RNA-HCV).
Marcadores da
Hepatite C
Fase da
incubação
Fase Aguda
Sintomática ou
Assintomática
P
Convalecência
Fase Crónica
Sintomática ou
Assintomática
P
Anti-HCV
N
P
RNA-HCV
P
P
N
P
qualitativo
RNA-HCV
> 50.0000
> 50.0000
> 100.000
N
carga viral
cópias/ml
cópias/ml
cópias/ml
P: Positivo. Resultado reactivo na primeira avaliação e comfirmado na segunda avaliação.
N: Negativa. Resultado não-reactivo na primeira análise e confirmado na segunda avaliação.
Comparação de alterações enzimáticas e marcadores em
diferentes estadios da hepatite C
Estadios da
Anticorpos
Anticorpos
ALT (TGP)
doença
ELISA
RIBA
Hepatite Crónica
Elevada
Positivo
Positivo
Portador da
Normal
Positivo
Positivo
Hepatite C
Hepatite C em
Normal
Positivo
Positivo
recuperação
Falso-positivo
Normal
Positivo
Negativo
para anti-HCV
HCV-RNA
(rT-PCR)
Positivo
Positivo
Negativo
Negativo
Os últimos estudos têm demonstrado que reacções contendo 10 ou mais cópias de
RNA-HCV são positivas. Este valor é equivalente a 2.000 cópias de RNA-HCV por
mililitro de soro ou plasma.
A genotipagem para HCV é obtida com a utilização de métodos em biologia
molecular e de sequenciamento genómico, os quais são úteis para definir os
genótipos e subtipos para estudos de epidemiologia molecular, estudo clínico e
monitorização da hepatite C.
Consultar Genotipagem para Hepatite C.
Hepatite D
O vírus da hepatite Delta (HDV), as co-infecções pelos vírus da hepatite B (HBV) e
HDV e as superinfecções por HDV em portadores do HBV podem ocorrer em
indivíduos procedentes de áreas endémicas.
A presença (infecção aguda) ou a ausência (infecção crónica) de IgM anti-HDV
deverá estar sempre associada a IgM anti-HBc ou a IgG anti-HBc, respectivamente.
Baixas concentrações de HDAg podem ser detectadas na fase aguda, e anticorpos
IgG anti-HDV, na fase crónica. A co-infecção HBV-HDV é diagnosticada pela
detecção transitória, e em baixas concentrações, de anticorpos específicos para o
HDV, principalmente da classe IgM, que poderão apresentar-se como os únicos
marcadores da infecção no período de declínio de HDAg e desenvolvimento de
anticorpos IgG. A presença constante de HBsAg e de IgG anti-HDV, em altos
títulos, indica evolução para a infecção crónica, com detecção e quantificação do
RNA HDV em soro ou plasma e detecção do HDAg em tecido hepático. Testes
específicos são desenvolvidos para a detecção do HDAg e RNA HDV no soro,
complementados pela maior sensibilidade do teste para IgM anti-HDV. A
monitorização do tratamento deve ser feita por testes quantitativos, que permitem
a detecção mínima de 10 cópias do genoma viral. O HDV apresenta diferentes
genótipos: I, II e III, sem haver, entretanto, correlação definida com a evolução
clínica.
Hepatite E
O vírus da hepatite E (HEV) é um dos agentes etiológicos de hepatites agudas por
veiculação hídrica. São descritos casos de evolução aguda e fulminante. Partículas
de HEV podem ser detectadas em suspensões fecais, na fase aguda da doença, por
métodos aplicados à pesquisa. Anticorpos IgM e IgA são detectados em soro e
plasma, por metodologias tradicionais, demonstrando percentagens variadas de
grupos populacionais com infecção aguda. Anticorpos IgG específicos para HEV
apresentam-se em altos títulos na fase aguda da doença, posteriormente ao
declínio de IgM. O RNA HEV pode ser detectado por PCR em soro, plasma ou
suspensão fecal até 40 a 50 dias após o início da doença, durante a fase aguda. A
detecção no material fecal é, entretanto, menos frequente (até 70%) do que nos
outros materiais biológicos (93%).
Hepatite G
A hepatite vírica é causada por diversos agentes, com seu próprio modo de
transmissão e replicação, e as doenças causadas por esses vírus diferem
significativamente em relação à severidade do dano hepático. Entretanto, várias
evidências laboratoriais e epidemiológicas têm sugerido a existência de agentes
adicionais, que podem ser transmitidos por via parenteral. Cerca de 10 a 20% de
casos de doença hepática é de etiologia desconhecida. Um agente em potencial
está associado ao soro de um cirurgião (com as iniciais "GB"), que tinha
desenvolvido hepatite aguda sem história epidemiológica conhecida. Estudos
experimentais de Deinhardt e cols. (1967) de inoculação em primatas (Saguinus
sp.) com o soro desse indivíduo mostraram que o material induziu hepatite nos
animais, e o agente envolvido é mencionado como agente GB.
Experiências adicionais de culturas de células levaram à caracterização de GB como
agente viral. Entretanto, a variação de hospedeiros primatas e experiências crosschallenge sugeriram que o agente GB era distinto dos vírus das hepatites
actualmente conhecidas, em humanos.
Além disso, anticorpos específicos aos vírus das hepatites A,B,C,E não eram
induzidos pela inoculação de GB em macacos, como não eram detectados em
imunoensaios.
Foi descrita a clonagem molecular de dois genomas, com características
semelhantes a flavivirus de macacos experimentalmente infectados. Esses genomas
representam dois vírus independentes: GB-vírus A (GBV-A) e GB-vírus B (GBV-B).
Têm sido descritos estudos de PCR para a detecção de GBV-A e GBV-B RNA e o uso
de imunoensaios para anticorpos específicos aos antigénios codificados pelos
genomas dos agentes GB.
Os genomas de GBV-A e GBV-B apresentam semelhança limitada na sequência de
nucleotídeos entre si (27%) e com o vírus da hepatite C (28%), nas regiões NS3
(helicase) e NS5B (RNA-polimerase dependente de RNA). Os seus genomas
apresentam respectivamente 9.493 e 9.143 nucleotídeos. Os genomas desses
agentes estão organizados de forma bastante semelhante a pestivírus e flavivírus,
com genes que codificam proteínas estruturais e não-estruturais em terminações 5'
e 3', respectivamente.
O grau de divergência na sequência entre GBV-A e GBV-B e outros membros da
família Flaviviridae demonstra que os agentes GB representam dois novos géneros
nessa família.
Um terceiro agente viral, recentemente notificado, foi identificado no soro de vários
pacientes com hepatite criptogénica (não A-E). Devido ao alto grau de identidade
com GBV-A (59% em nível de nucleotídeos e 64% em nível de aminoácidos), esse
vírus foi chamado GB-vírus C (GBV-C). Análises filogenéticas demonstraram que o
GBV-C é um membro adicional dos Flaviviridae, distinto do grupo HCV e mais
intimamente relacionado ao GBV-A..
A transmissão do HGV por meio de transfusões de sangue e por outras vias de
exposição parenteral, tais como em usuários de drogas injectáveis, tem sido
claramente estabelecida. A partir daí, conclui-se que muitos pacientes HGV-positivo
tenham sido co-infectados com HBV ou HCV, provavelmente devido aos factores de
risco compartilhados da infecção. O rastreio das doações de sangue para esses
vírus também elimina as unidades de sangue infectadas por HGV, reduzindo dessa
forma a incidência de hepatite pós-transfusional relacionada ao HGV.
Estudos recente, demonstram que a infecção por HGV está associada a hepatite na
maioria dos pacientes investigados. Há também pacientes com níveis normais de
transaminases que requerem estudos adicionais de sequenciamento para
determinar se são portadores ou pacientes em estado quiescente da doença.
A associação do vírus com a doença hepática crónica e a sua presença em
pacientes com dupla infecção por HBV ou HCV é irrefutável. Entretanto, a sua
associação e potencial envolvimento na hepatite fulminante e carcinoma
hepatocelular ainda estão a ser investigados.
Estudos retrospectivos evidenciaram os seguintes pontos:
- a doença relacionada ao HGV é geralmente branda, com níveis pouco elevados de
ALT (TGP);
- a infecção por HGV pode ser persistente e acompanhada de hepatite crónica;
- as infecções por HCV/HGV e HBV/HGV podem ocorrer simultaneamente e
resultam em co-infecções persistentes;
- a prevalência de HGV em dadores de sangue é maior do que HCV e não se
relaciona aos níveis de ALT presentes nos doadores;
- nas infecções duplas, os níveis de ALT são maiores e mais frequentemente
aumentados;
- indivíduos com infecções duplas crónicas podem apresentar severa
necroinflamação hepática;
- 6% e 10% de indivíduos com infecções crónicas respectivamente por HBV e HCV,
apresentam positividade para HGV.
A detecção do ácido nucléico viral a partir do soro de indivíduos infectados por HGV
é, actualmente, o método disponível para a identificação dos casos.
Comercialmente, existem kits que contêm primers dirigidos para o sequenciamento
das regiões NS5 e NCR (non-coding region) do vírus da hepatite G.
Intensos estudos que empregam peptídeos indicam que o uso de epítopos lineares
não resulta em reconhecimento fiável da infecção pelo vírus da hepatite G por
imunoensaios.
Hepatite por TTV
Partículas semelhantes a vírus são observadas com frequência em amostras
biológicas humanas sem, necessariamente, apresentarem correlação com alguma
patologia. Recentemente, uma dessas observações, originada num trabalho de
pesquisa no Japão, por Nishizawa e colaboradores, originou o chamado Transmited
Transfusion Vírus (TTV), caracterizado como uma partícula semelhante a vírus,
extraída de um paciente com doença hepática crónica, adquirida, provavelmente,
após transfusão sanguínea.
O TTV, vírus associado à hepatite pós-transfusional não A-não G, apresenta
estrutura genómica formada por DNA de cadeia simples e duas Open Reading
Frame (ORF), sugerindo relações filogenéticas com outros vírus da família
Circiniviridae e originando dois grupos genéticos com 16 genótipos distintos.
A replicação do TTV parece ocorrer em hepatócitos, onde foram detectados DNA
por métodos moleculares.
Novas metodologias foram desenvolvidas, com diferentes sensibilidades, permitindo
determinar altas prevalências de DNA-TTV em sangue de dadores sem doença
hepática aparente e em pacientes com hepatites associadas a outras etiologias
víricas - HCV e HBV, não havendo evidências de interferências na evolução da
doença hepática por essas co-infecções.
A via de transmissão parenteral parece ser a mais eficiente. Por outro lado, dados
epidemiológicos de alta prevalência em países como Escócia (1,9%) e Japão (12%)
sugerem a possibilidade de vias alternativas de transmissão.
A detecção de DNA-TTV em amostras de fezes colhidas em pacientes com hepatite
aguda e crónica demonstra a possibilidade de transmissão fecal-oral.
No Brasil, alguns dados demonstram a prevalência de DNA-TTV em 62% dos
indivíduos de uma casuística de 72 dadores de sangue e em 10% da população
geral.
No Japão, 12% dos dadores de sangue avaliados apresentaram DNA-TTV
detectável, 47% hepatites fulminantes não-A-G, 46% hepatopatias crónicas de
etiologia desconhecida, 39% carcinoma hepatocelular e 48% cirroses.
Embora a detecção de DNA-TTV tenha sido feita em tecidos hepáticos, não há
confirmação de sua patogenicidade para o fígado. Outro estudos têm sido
desenvolvidos para a definição de um novo agente hepatotrópico.
Herpes Simplex
A infecção pelos vírus Herpes simplex (HSV) está entre as infecções víricas com
maior prevalência na população mundial. Existem dois sorotipos diferenciados:
HSV1 e HSV2. São vírus DNA, da família Herpetoviridae, e reagem de forma
cruzada, pois os sorotipos possuem cerca de 50% de sequência homóloga. O
desenvolvimento, nas últimas décadas, de técnicas sorológicas que identificam e
diferenciam os sorotipos tem aumentado não só a possibilidade de diagnosticar e
tratar as infecções como também de compreender melhor a sua patogenia e meios
de transmissão, em especial do herpes perinatal.
A transmissão pode dar-se pelo contacto com superfícies mucosas infectadas por
soluções de continuidade da pele e mucosas, relações sexuais e durante o parto. A
disseminação do vírus ocorre pela migração centrífuga dos vírus através dos nervos
sensoriais periféricos. Na porta de entrada, na derme e na epiderme, ocorre o
processo de replicação, e as partículas virais são transportadas pela terminação
nervosa retrogradamente ao núcleo dos neurónios sensoriais. Conhece-se menos a
sucessão de eventos a partir deste ponto. Nalguns casos, ocorre a infecção com a
replicação viral e morte celular ao nível mucocutâneo. Noutros, o vírus fica em
estado de lactência. O detalhe dos mecanismos da persistência em lactência do
HSV e sua reactivação periódica permanecem obscuros.
O primeiro episódio de doença herpética, a primoinfecção, é normalmente
acompanhado de sinais e sintomas envolvendo lesões mucosas e extramucosas.
Apresenta longa duração dos sintomas e da permanência dos vírus na lesão e uma
taxa maior de complicações do que os episódios de reagudização ou recorrentes. A
gengivoestomatite aguda é a manifestação mais comum das primoinfecções. É mais
frequente entre 1 e os 4 anos de idade. O herpes labial e as úlceras da córnea são
as infecções sintomáticas recorrentes mais frequentes.
As manifestações clínicas e a evolução da infecção dependem da idade, da
localização, do estado imunológico do paciente e do tipo antigénico do vírus. A
exposição ao sol (luz ultravioleta), imunossupressão e traumas cutâneos ou do
gânglio podem levar à reactivação. Ocasionalmente, múltiplas linhagens do mesmo
subtipo viral são detectadas no mesmo paciente, principalmente os
imunodeprimidos. Este facto sugere a possibilidade de infecção exógena por
diferentes linhagens de um mesmo subtipo.
A infecção pelo tipo 1 émuitas vezes, adquirida mais cedo do que a do tipo 2. Cerca
de 90% dos adultos apresentam anticorpos contra HSV1 por volta dos 50 anos de
idade. Nas populações socioeconómicas desfavorecidas, a faixa etária decresce para
30 anos.
Os anticorpos contra o tipo 2 não são normalmente detectados até à puberdade. As
taxas de prevalência desses anticorpos correlacionam-se com a fase de vida sexual
activa, o que distingue um grupo do outro. A percentagem aumentou 30 pontos nos
últimos 12 anos nos Estados Unidos. Numa avaliação obstétrica, 25% da população
investigada tinham anticorpo positivo; entretanto, apenas 10% relatavam história
de lesão genital. Cerca de 50% dos adultos heterossexuais, com vida sexual activa,
apresenta anticorpos positivos, sendo a taxa 5% maior entre as mulheres.
O HSV tipo 1 está associado a uma variedade de infecções envolvendo lesões
mucocutâneas orolabiais, oftálmicas, meningoencefálicas, podendo eventualmente
causar lesões viscerais e genitais, enquanto o HSV tipo 2 (HSV2) normalmente
causa as infecções genitais sexualmente adquiridas. Ambos os tipos podem causar
lesões nas diferentes localizações, e a infecção clínica é indistinguível.
Tanto o HSV1 como o HSV2 podem ser responsáveis por lesões mucocutâneas
primárias em qualquer localização. A duração e a intensidade da infecção são
independentes do sorotipo envolvido. O tipo de vírus e a localização da
primoinfecção afectam a frequência e a probabilidade de recidiva. Estudos recentes
demonstram que tanto a frequência como a probabilidade são maiores quando a
infecção é causada pelo HSV2. A infecção genital por HSV2 ocorre com frequência
oito a dez vezes maior que a infecção genital por HSV1. Por outro lado, a infecção
orolabial por HSV1 ocorre mais frequentemente do que a infecção orolabial por
HSV2. A probabilidade de reactivação da infecção causada pelo HSV2 é duas vezes
maior.
Em indivíduos imunocompetentes, a infecção limita-se às localizações
mucocutanêas e ao gânglio sensorial. Em indivíduos imunodeprimidos, as lesões
causadas tanto pela primoinfecção como pelas reactivações tendem a ser mais
extensas e persistir por muito mais tempo do que nos indivíduos
imunocompetentes. Nesses pacientes, o quadro é grave, geralmente com
comprometimento esofágico, pneumonite intersticial e doença disseminada com
comprometimento visceral. A infecção pelo HSV2 é do tipo infecção oportunista
importante em indivíduos infectados pelo HIV. Calcula-se que até 90% desses são
co-infectados com HSV2.
Num pequeno número de casos, a infecção pelo HSV leva a encefalite vírica e a um
dano neurológico grave. O HSV, principalmente o tipo 1, pode causar encefalite em
adultos pela reactivação de infecção latente. As infecções mais agressivas, com
risco de vida, são a perinatal e as que ocorrem em indivíduos
imunocomprometidos, incluindo pacientes com SIDA.
Existem dados que demonstram que os pacientes que apresentam episódio
primário intenso e não tratado têm índices mais elevados de recorrência a longo
prazo.
As respostas imune, humoral e celular manifestam-se nas primeiras semanas e
persistem por toda a vida. Embora não possuam carácter imunizante, induzem a
manifestações mais leves e apresentam reacção cruzada entre os dois subtipos. A
sorologia permite a identificação de anticorpos IgM e IgG de forma qualitativa. A
avaliação deve ser realizada em sorologia pareada para melhor interpretação dos
resultados.
O isolamento viral em cultura de tecidos era o método de escolha para o
diagnóstico e tipagem do HSV. O HSV pode ser detectado em cultura depois de 2 a
8 dias, mas em vários casos, como nos de baixos títulos virais, cura das lesões ou
lesões atípicas, o vírus não pode ser isolado. A sensibilidade do isolamento do HSV
em cultura de tecido é de aproximadamente 105 vírus por mL.
A reacção em cadeia da polimerase (PCR) é o método de escolha para o diagnóstico
da infecção por HSV. É altamente sensível (até 5 vírus por ensaio), específica (98-
100%), e pode identificar o genótipo e a quantificação viral. A PCR quantitativa
pode ser útil para monitorizar a resposta à terapia antivírica. Os ensaios mais
usados para distinguir o tipo 1 do tipo 2 são os que utilizam a avaliação da
presença de anticorpos contra glicoproteínas do HSV1 (gG1) e do HSV2 (gG2).
Além disso, o ensaio realizado por PCR permite o diagnóstico utilizando-se
diferentes materiais como sangue, líquor, líquido amniótico, vilosidades coriónicas e
sangue fetal.
O líquido amniótico pode ser colhido a partir da 12ª semana até o final da gestação.
Porém, o período ideal para a colheita situa-se entre a 14ª e a 16ª semana. O
período ideal para a colheita de vilosidades trofoblásticos situa-se entre a 10ª e a
12ª semana de gestação. No entanto, esse prazo poderá estender-se até à 14ª
semana. O período ideal para a colheita de sangue fetal situa-se entre a 18ª e a
22ª semana de gestação.
Hidroxiprolina
Encontrada principalmente no colagénio da matriz óssea, a hidroxiprolina é o
aminoácido utilizado como marcador bioquímico do metabolismo ósseo, mais
especificamente da reabsorção óssea.
A hidroxiprolina libertada do osso na degradação do colagénio é metabolizada pelo
fígado (90%), e apenas 10% da hidroxiprolina são eliminados na urina.
A sua excreção urinária reflecte o grau de catabolismo ósseo, aumentando nos
momentos em que ocorre reabsorção óssea rápida. Níveis aumentados podem ser
encontrados na doença de Paget, na osteomalacia, em fracturas extensas em fase
de consolidação, nas neoplasias com metástase óssea, no hipertiroidismo, no
hiperparatiroidismo primário e secundário, na osteomielite aguda, na acromegalia e
na artrite reumatóide em actividade. Níveis normais ou ligeiramente alterados
podem ser encontrados na gravidez, na osteoporose, na doença renal e em doenças
inflamatórias cutâneas.
A dieta influencia directamente a taxa de libertação urinária de hidroxiprolina. Para
facilitar a colheita e evitar a realização de dieta preparatória, tem sido proposta a
avaliação em amostra de urina de 2 horas colhida após um jejum nocturno de 12
horas.
HIV 1
O síndrome da imunodeficiência adquirida (SIDA) é causado pelo vírus da
imunodeficiência humana tipo 1 (HIV-1), um retrovírus da família Retroviridae,
subfamília Lentivirinae, transmitido por contacto sexual, exposição a sangue
contaminado e derivados e da mãe infectada para o feto. Na infecção inicial há,
geralmente, um aumento significativo da virémia, posteriormente inibida pela
activação da resposta imune. As subsequentes concentrações no plasma reflectem
o equilíbrio alcançado entre vírus e hospedeiro, que pode durar alguns anos. Este
nível de interacção varia de pessoa para pessoa e pode ser preditivo de um curso
clínico de longa duração. Nessa fase clinicamente estável, indivíduos infectados
apresentam geralmente níveis de virémia baixos e persistentes e uma depleção
gradual de linfócitos T CD4(+) que pode levar a uma severa imunodeficiência, ao
aparecimento de múltiplas infecções oportunistas, a neoplasias e à morte.
O diagnóstico laboratorial pode ser feito pela pesquisa de anticorpos contra o HIV-1
por técnicas imunoenzimáticas e Western-blot. Essas técnicas, embora precisas,
apresentam limitações em determinados casos, tais como: crianças em idade até
15 meses, nas quais a permanência de anticorpos maternos, adquiridos na fase
gestacional através da placenta, no momento do parto ou na fase pós-parto,
através do colostro, pode determinar resultados falso-positivos; casos de infecção
recente, em períodos inferiores a 2 ou 3 meses, nos quais não houve, ainda, a
soroconversão, e casos que apresentam resultados indeterminados ou duvidosos.
Sorologia
Os testes sorológicos mais comuns são os imunoenzimáticos como ELISA (Enzyme
Linked Immunosorbent Assay) e ELFA (Enzyme Linked Fluorescent Assay); esses
testes que pesquisam anticorpos circulantes (anti-HIV) utilizam antigénios,
adsorvidos em fases sólidas, que podem ser de origem sintética, peptídeos
sintéticos (geralmente gp41 e p24 para o HIV-1 e gp36 para o HIV-2) ou o próprio
vírus inactivado. O método ELFA apresenta uma versão para a detecção simultânea
de anti-HIV e antígeno p24 (HIV-DUO), favorecendo o diagnóstico precoce de
infecção por HIV, antecipando-o em até 7 dias, quando comparado a métodos que
detectam apenas anti-HIV.
Os métodos enzimáticos são, preferencialmente, utilizados para triagens de
diferentes populações, em bancos de sangue e amostras sorológicas de indivíduos
com sintomatologia sugestiva ou mesmo assintomáticos e com história de situação
de risco.
Os testes imunoenzimáticos, licenciados e comercializados actualmente,
apresentam sensibilidade e especificidade que ultrapassam 98% a 99%.
Segundo recomendações do OMS, a entrega de um resultado de um teste
sorológico anti-HIV positivo só pode ser concretizada se pelo menos dois testes
forem realizados em paralelo, com a mesma amostra, por metodologias diferentes
(outro fabricante, outro tipo de antigénio ou diferente princípio metodológico) Se os
resultos forem positivos ou discordantes (+/ -), é necessário a realização de um
teste confirmatório, sendo o Western-blot o utilizado na rotina da maioria dos
laboratórios privados. Caso após estas duas etapas o resultado final seja positivo,
uma segunda amostra clínica deve ser solicitada para a repetição do procedimento
realizado na primeira amostra. Havendo discordância entre os resultados da 1ª
amostra com os obtidos na 2ª amostra, solicita-se uma nova colheita.
O teste confirmatório para a presença de anticorpos anti-HIV indicado em crianças
até dois anos de idade é a reacção em cadeia da polimerase (PCR) para a pesquisa
do cDNA-HIV.
Em indivíduos com alto risco de exposição ao HIV, uma reactividade intensa pelo
teste imunoenzimático apresenta um valor preditivo de 99%. Podem ocorrer
reactividades falso-positivas em testes imunoenzimáticos, principalmente em
pacientes com hepatite alcoólica, outras patologias em que ocorram anormalidades
imunológicas, neoplasias, mulheres multíparas e indivíduos politransfundidos, os
quais desenvolveram anticorpos contra antigénios HLA classe II, presentes em
linhagens de células onde há a replicação do HIV.
Os testes imunoenzimáticos para HIV-1 detectam 40% a 90% de infecções
causadas por HIV-2, e testes de ELISA licenciados para HIV-2 têm uma
sensibilidade de 99%. A opção mais utilizada actualmente são os testes
imunoenzimáticos que detectam, simultaneamente, anticorpos contra HIV1 e HIV2.
A detecção de antigénio p24 do HIV-1 circulante por teste imunoenzimático é
particularmente útil em determinadas situações. Nas primeiras semanas após a
infecção, o antigénio p24 está presente no soro, antes da detecção dos anticorpos.
Com o aparecimento destes, o antigénio p24 torna-se indetectável, permanecendo
assim por meses ou anos. O reaparecimento da antigenémia durante o curso da
infecção geralmente está associado a um prognóstico desfavorável.
Métodos para a detecção de antigénio p24, principalmente aqueles que utilizam
dissociação ácida, também têm sido utilizados para acompanhar pacientes em
tratamento antiviral, conforme demonstrado durante ensaios clínicos. A sua eficácia
porém é comprometida devido à baixa sensibilidade dos métodos actualmente
disponíveis, sendo substituídos pela detecção da carga viral por metodologias de
biologia molecular.
Western-Blot
O método de Western-Blot é amplamente utilizado como teste confirmatório dos
resultados obtidos em testes imunoenzimáticos. Outros testes também são
indicados, como imunofluorescência em células fixadas e o teste RIPA (Radio
Immuno Precipitation Assay). O Western-Blot utiliza antigénios do HIV, obtidos em
cultura de linhagem celular, separados eletroforecticamente em bandas distintas,
posteriormente transferidas para membrana de nitrocelulose. A reacção ocorre
entre os antigénios em contacto com os anticorpos, presentes no soro ou no
plasma de indivíduos infectados.
Padrões específicos de reacções podem ser identificados, e embora a definição de
testes positivos seja controversaa, a ausência de reacções específicas para os
diferentes antigénios do HIV confirma a reacção negativa.
Padrões de positividade (presença de anticorpos) podem ser definidos no teste
confirmatório de Western-Blot, pela visualização de bandas correspondentes às três
principais proteínas virais: a proteína do núcleo, p24 ou p31 e duas proteínas do
envólucro, gp41 e gp120/gp160 . Alternativamente, o Centers for Diseases Control
and Prevention-CDC recomenda que pelo menos uma das duas, p24 ou
gp120/gp160, seja identificada. Outras bandas de antigénios do HIV são p17, p31,
p51, p55, p66.
Padrões definindo casos indeterminados, quando apenas uma das três principais
bandas é visualizada no teste de Western-Blot, podem ocorrer, causando dúvidas
quanto à real interpretação. Indivíduos infectados, em fase avançada de SIDA,
podem não apresentar reactividade para p24, sugerindo resultado indeterminado.
Alguns podem apresentar esse padrão por longo período de tempo, sem evidências
de infecção por HIV. As interpretações mais correctas e apropriadas para os
resultados de testes de ELISA reactivos e Western-blot indeterminados envolvem
avaliações clínicas e acompanhamento laboratorial.
Testes de Western-Blot são desenvolvidos por diferentes laboratórios de produção
de kits e reagentes para diagnóstico. Isto origina a possibilidade de diferentes
critérios de interpretação que devem estar sempre sujeitos a avaliações
comparativas com aqueles recomendados por instituições de referência, tais como
o CDC.
A interpretação de resultados poderá acompanhar também os critérios
complementares como se seguem:
A presença ou a ausência de anticorpos contra HIV-1 em amostras de soro e/ou
plasma e a identificação de anticorpos presentes são determinadas pela
comparação de cada reacção com os controlos de reactividade baixa, alta e
ausente. Deverão ser seguidas três fases de interpretação: primeira - cada banda
de reacção que aparece na fita de nitrocelulose da amostra deverá ser identificada
comparativamente ao controlo padrão de alta reactividade; segunda - cada banda
deverá ser avaliada com base na intensidade de reacção; terceira - a interpretação
deverá ser finalizada pela associação dos padrões de reactividade.
Proposta de interpretação alternativa seguindo-se os critérios de interpretação do
Centers for Diseases Control and Prevention e da Association of State and
Territorial Public Health Laboratory Directors (ASTPHLD):
PADRÃO
Ausência da bandas
Presença de alguma banda sem atender ao critério de POSITIVO
Duas ou mais bandas das seguintes alternativas: p24, gp41 e
gp120/160.
Cada banda com reactividade maior ou igual ao padrão positivo.
Normalmente a banda para gp41 ou gp160 é difusa. Outras bandas
poderão ou não estar presentes.
INTERPRETAÇÃO
NEGATIVO
INDETERMINADO
POSITIVO
Nalgumas publicações, sugere-se que não há necessidade da presença de
reactividade para p31.
Outros estudos indicam como inadequada a interpretação de positividade para
reacções em que estejam ausentes quaisquer duas das bandas para p24, gp41 ou
gp 120/160 com padrão de reactividade igual ou maior do que controlo positivo. É
reconhecido que indivíduos com soroconversão recente podem apresentar padrões
distintos e incompletos de reacção mas que evoluirão para aumento de
reactividade, em número e intensidade, quando acompanhados por períodos de 4 a
6 meses. Os resultados com interpretação de POSITIVO apresentarão as bandas
p17, p31, p55, p66 e gp120.
Reactividades inespecíficas, que persistem mas não evoluem para padrões
completos, podem ocorrer, em particular nas regiões p17, p24, p55 e p66. As
reactividades inespecíficas podem ser atribuídas a auto-anticorpos ou a
reactividades com outros retrovirus humanos. Tem sido evidenciado que pacientes
com SIDA perdem a reactividade para p24 e p31, e que em crianças pode haver
falha de soroconversão e persistência de anticorpos maternos por vários meses.
Pacientes com doenças tumorais e em tratamento com imunossupressores podem
falhar na resposta com padrão de positividade em Western-Blot. Bandas
inespecíficas que não estejam acompanhadas das principais bandas de proteínas
virais, p24 e gp41/120/160, podem estar relacionadas com constituintes celulares
com pesos moleculares em torno de 70K, 51-55K (possivelmente originadas de HLA
DR) e 43 K (de origem HLA-ABC).
Carga Viral
A quantificação de partículas virais no sangue periférico, que define a carga viral,
pode ser estimada por níveis plasmáticos de antígeno p24, a principal proteína do
nucleocapsídeo viral; por métodos quantitativos de cultura celular para HIV-1 ou
pela quantificação directa do RNA viral no plasma, através de tecnologias de
amplificação do ácido nucléico ou de amplificação de sondas marcadas.
A utilização de testes quantitativos para antigénio p24 como parâmetro na
avaliação da carga viral é de valor limitado, uma vez que apenas 20% de indivíduos
assintomáticos e 40 a 50% de pacientes sintomáticos apresentam níveis
antigénicos detectáveis, mesmo utilizando-se procedimentos de dissociação de
imunocomplexos.
As metodologias de biologia molecular (RT-PCR, b-DNA, Nuclisens-NASBA) são
metodologias quantitativas hoje recomendadas para a avaliação da carga viral. Elas
permitem a quantificação directa do RNA viral em amostras de plasma.
A carga viral, definida em número de cópias do genoma viral por mililitro de
plasma, está correlacionada com o estadio da infecção, ao risco de evolução para
SIDA e à eficácia do tratamento anti-retroviral. Vários estudos demonstram que
níveis mais elevados de carga viral estão associados a maior risco de evolução da
doença, enquanto níveis mais baixos correlacionam-se a risco diminuído de
progressão clínica.
A avaliação da carga viral, associada a dados clínicos e a outros parâmetros
laboratoriais, permite ao clínico estabelecer condutas adequadas na monitorização
do tratamento anti-retroviral. As metodologias de biologia molecular podem
apresentar diferentes sensibilidades. A RT-PCR apresenta sensibilidade da ordem de
200 cópias de RNA viral, e a RT-PCR ultra-sensível, da ordem de 50 cópias.
É recomendada a realização de duas avaliações, com intervalos de 15 dias, para o
estabelecimento dos níveis de carga viral antes do tratamento. A eficácia do
tratamento anti-retroviral é acompanhada por avaliação 4 semanas após o início da
terapia. É esperado um declínio inicial rápido dos níveis da carga viral, que se
segue a uma diminuição em ritmo mais lento e moderado no decurso dos meses
subsequentes. A carga viral deve ser repetida rotineiramente a cada 3 ou 4 meses.
Nos momentos de mudança terapêutica ou início de novos esquemas, recomendase uma avaliação em 2 a 4 semanas após a alteração e a cada 10 a 12 semanas.
É indicado que a carga viral seja avaliada com o paciente em situação clínica
estável. Em casos de imunização recente, recomenda-se um intervalo mínimo de 4
semanas para a colheita da amostr. Sugere-se também que sejam utilizadas as
mesmas metodologias e o mesmo laboratório.
Homocisteína
A aterosclerose é uma doença da actualidade, e a sua consequência mais drástica é
a coronariopatia, uma das mais importantes causas de mortalidade nos países
desenvolvidos. Vários factores já se encontram descritos como factores de risco
para o aparecimento da doença, com inúmeras confirmações epidemiológicas.
Dessa forma, o tabagismo, a hipercolesterolemia, o sedentarismo, a obesidade, a
hipertensão arterial e a diabetes mellitus colaboram efectivamente para a
instalação de doença aterosclerótica. De forma revolucionária, agentes infecciosos
como Chlamydia pneumoniae, Helicobacter pylori e alguns vírus (herpes,
citomegalovírus e vírus sincicial respiratório) passaram também a contribuir na
causa de disfunções endoteliais e aterogénese.
Recentemente, um novo factor tem sido aventado no desenvolvimento dessa
doença: o aumento sérico de homocisteína (hiper-homocisteinémia). A hiperhomocisteinémia é, actualmente, considerada um factor de risco independente para
doença arterial coronária, doença vascular periférica, doença cerebrovascular e
tromboses. A evidência epidemiológica de que talvez esse aminoácido pudesse
estar envolvido no desenvolvimento da aterosclerose veio da observação de que
crianças com homocistinúria, um erro inato do metabolismo devido a um defeito
monogénico autossómico recessivo que cursa com aumento de homocisteína
plasmática e urinária, possuíam alto potencial aterogénico.
A homocisteína é um pequeno aminoácido sulfidrílico que ocupa posição reguladora
central no metabolismo da metionina. O plasma humano contém formas reduzidas
(1%) e oxidadas de homocisteína (98 a 99%), sendo que de 80 a 90% da forma
oxidada se encontram ligados a resíduos protéicos. A metileno tetraidrofolato
redutase (MTHFR) é uma importante enzima no metabolismo da homocisteína. A
sua actividade, quando diminuída, tem sido associada à hiper-homocisteinémia.
Uma das principais causas de diminuição da actividade é uma mutação (Ala 677>Val), que resulta na forma termolábil da enzima, que possui então 50% da
actividade normal. Deficiências de vitaminas do complexo B, seja por insuficiência
na dieta ou má absorção, estão associadas ao aumento de homocisteína sérica.
O doseamento de homocisteína na população geral de adultos saudáveis revela
concentrações que variam entre 5 a 15 mmol/L. Estudos recentes, demonstram que
o nível desejável deve estar abaixo de 10 mmol/L. Concentrações situadas acima
de 15 mmol/L são consideradas patológicas. Indivíduos com doença vascular
periférica, cerebral ou coronária possuem valores entre 15 a 25 mmol/L. Indivíduos
com insuficiência renal apresentam níveis séricos aumentados do aminoácido.
A grande vantagem em determinar o nível de homocisteína reside no facto de
podermos intervir em vias enzimáticas com suplementação dietética de vitaminas
B12 e B6 e ácido fólico, para atingirmos baixas concentrações séricas do
aminoácido. Os métodos habituais de doseamento de homocisteína nos laboratórios
clínicos são a cromatografia líquida (HPLC) ou o teste imunoenzimático com
anticorpos monoclonais e a análise de fluorescência polarizada.
Factores que aumentam os níveis da homocisteína:
Demográficos e Genéticos:
- Idade, sexo e étnia
- Níveis enzimáticos de metionina sintetase, MTFHR e cistatina sintetase
Adquiridos:
- Deficiência de vitamina B (B6, B12 e ácido fólico)
- Insuficiência renal
- Transplantes
- Hipotireoidismo
Outros:
Tabagismo, alcoolismo, sedentarismo e ingestão abusiva de café.
HPV
Os papilomavírus pertencem a um grupo de vírus que induzem à formação de
verrugas ou papilomas. Podem ocorrer em muitos vertebrados e, principalmente,
no homem. A natureza viral das verrugas no homem foi diagnosticada há mais de
80 anos.
Cada tipo viral exibe especificidade segundo os hospedeiros e o tecido epitelial que
infectam. Infecções benignas manifestam-se com formações de verrugas, e
algumas lesões associadas a tipos virais específicos podem progredir para
displasias, carcinomas e cancro invasivo.
A presença de tipos específicos de papiloma vírus humano (HPV) encontrados no
trato genital feminino e masculino está associada a doenças neoplásicas e a
carcinomas intra-epitelial da vulva, vagina, cérvixl e peniano.
O HPV apresenta como estrutura viral definida e completa um vírus de 52 a 55 nm
de diâmetro, contendo uma molécula de DNA de 8.000 pares de bases, organizados
em cadeia dupla, interna ao capsídeo protéico.
Pouco se conhece sobre o mecanismo de infecção pelo papiloma vírus. Receptores
estão presentes numa variedade de células, em diferentes espécies animais.
Entretanto, o tropismo específico a determinados tecidos é derivado da
permissividade à transcrição do genoma viral. Os níveis de expressão do oncogene
viral não sofrem alterações significativas durante a evolução do carcinoma in situ e
do cancro invasivo, sugerindo que essa progressão seja atribuída à acumulação de
mutações cromossómicas da célula hospedeira e a factores imunológicos que
podem influenciar aparecimento da infecção.
A análise das sequências de nucleotídeos do DNA permite a classificação em 70
genótipos diferentes. Dados epidemiológicos e experimentais demonstram a
associação de alto risco dos tipos HPV 16 e HPV 18 com a etiologia do cancro
anogenital, em especial o carcinoma cervical. Os tipos HPV 6 e HPV 11 associam-se
ao baixo risco, enquanto os tipos HPV 31, 33 e 35 relacionam-se ao risco
intermediário. Os sete genótipos identificados são descritos em 70% das neoplasias
cervicais. Outros tipos, 42, 43, 45, 51, 52, 56, 58, 59 e 68 podem ser identificados
nas outras lesões.
O DNA do HPV pode ser detectado em aproximadamente 10% das mulheres com
epitélio cervical normal. As lesões causadas pelo papilomavírus podem regredir,
persistir ou progredir com o tempo, ou de acordo com alterações imunológicas do
paciente. Estudos prospectivos demonstram que 15-28% dessa população podem
desenvolver neoplasia intra-epitelial, em 2 anos de acompanhamento.
Dados epidemiológicos sugerem que múltiplos parceiros sexuais e infecções por
HPV pertencentes ao grupo de alto risco são os factores mais importantes para o
desenvolvimento de carcinomas cervicais. Outros carcinogénios, tais como
metabolitos do tabaco e radiações ultravioleta, podem participar e favorecer a
evolução para carcinomas oral e cervical associados ao HPV.
Os métodos de biologia molecular são os mais sensíveis e específicos para a
complementação do diagnóstico da infecção por HPV. A utilização da técnica de
captura híbrida permite a detecção do DNA viral e a genotipagem por grupos de
risco. A pesquisa é feita em tecido e/ou células, permitindo a detecção antes do
surgimento de manifestações clínicas.
HTLV I/II
O vírus linfotrópico de células T humanas tipo I (HTLV I) foi o primeiro retrovírus
humano a ser descoberto. Descrito inicialmente nos Estados Unidos por Poiesz e
cols. no ano de 1980, está actualmente classificado na família Retroviridae. O
primeiro relato de infecção pelo HTLV I no Brasil foi feito por Kitagawa et al., após
estudo da soroprevalência de anticorpos anti-HTLV I em imigrantes japoneses que
viviam no país à 50 anos. Actualmente, são descritos casos em todo o país, tanto
em populações urbanas como em comunidades isoladas. Esse tipo de virus está
directamente associado a leucemia/linfoma de células T do adulto e a paraparesia
espástica tropical/mielopatia associada.
As proteínas do HTLV encontram-se codificadas nos genes gag (grupo antigénio),
pol (polimerase) e env (envelope), flanqueados por sequências terminais longas
repetidas (LTR), que contêm sinais importantes para o controle da expressão dos
genes virais. A região gag é inicialmente traduzida por uma proteína precursora
(p53) que é clivada em: proteína da matriz (Ma) ou p19, proteína do capsídeo (Ca)
ou p24 e proteína do nucleocapsídeo. A protease é codificada por uma sequência de
leitura aberta (open reading frame - ORF), situada entre a parte 3' da região gag e
a parte 5' da região pol. A integrase e a transcriptase reversa são codificadas na
região pol. De forma distinta dos outros retrovírus, este possui uma região
particular com cerca de 2 Kb, situada imediatamente acima da LTR 3', denominada
inicialmente pX, em razão da sua natureza desconhecida. Esta região contém pelo
menos 4 ORF que codificam as proteínas p40 tax, p27 Rex, p21 Rex, p12 I, p13 II,
p30 II.
O diagnóstico laboratorial da infecção por HTLV pode ser realizado por técnicas
sorológicas, tais como testes imunoenzimáticos (ELISA), testes de imunoblotting
(Western-Blot) e imunofluorescência indirecta (IFI), que detectam anticorpos para
as proteínas estruturais do vírus. Contudo, a técnica de reacção em cadeia da
polimerase (PCR) permite rápido acesso às sequências de DNA e RNA, celular e
viral, possibilitando o diagnóstico dos indivíduos que, embora portadores do vírus,
ainda não produzem anticorpos em níveis detectáveis. Esta é também a técnica
mais sensível para a detecção de sequências de provírus de retrovírus humanos,
mesmo quando o número de cópias é baixo. Além dessas técnicas, existem
métodos moleculares que são utilizados na quantificação do vírus circulante no
organismo do hospedeiro.
O risco de transmissão via amamentação e o grau de infecciosidade in vitro podem
ser avaliados através da detecção dos marcadores de infecção e replicação viral no
leite de mães portadoras do HTLV, por técnicas de PCR e cultura de células
mononucleares do leite materno (BMMC).
Imunofenotipagem de Leucemias
Desde o primeiro relato dessa doença como uma entidade clínica, em 1845, até os
dias de hoje, é notável a evolução ocorrida no entendimento da biologia das
leucemias. Durante esse período, as doenças hematológicas serviram como modelo
para o desenvolvimento dos conceitos actuais da imunologia, principalmente nas
áreas da diferenciação linfocitária e da genética molecular. De maneira
complementar, o desenvolvimento dessas áreas, juntamente com os avanços
técnicos no campo da imunologia, permitiram um maior entendimento das
leucemias.
As leucemias representam um grupo heterogéneo de doenças clonais de
precursores hematopoiéticos, caracterizadas por anomalias quantitativas e
qualitativas. Devido à extrema heterogeneidade das entidades englobadas sob a
mesma denominação, tanto no aspecto clínico como no comportamento biológico, é
fundamental a utilização de critérios diagnósticos precisos para a sua classificação.
Recentemente, métodos diagnósticos mais sofisticados (imunológicos e
moleculares) redefiniram a classificação das leucemias de acordo com a origem
celular, linfóide (B ou T e maturidade celular) e mielóide, além do tipo de anomalia
genética envolvida.
A classificação das leucemias segundo esse critério tem grande importância na
terapêutica e, portanto, na resposta obtida.
A determinação da origem celular irá influenciar directamente a conduta clínica, já
que a partir dela se saberá se a leucemia é linfóide ou mielóide, levando a dois
caminhos clínicos totalmente diferenciados. Além disso, o reconhecimento da
origem celular nas leucemias linfóides (B ou T) também dá uma informação
importante para a estratificação inicial da conduta clínica a ser adoptada.
Convém salientar que o reconhecimento do estadio de maturação diferencia entre
as formas agudas ou crónicas e é um factor relevante na avaliação do prognóstico
do paciente. Isso é bastante claro quando se trata de uma leucemia linfoblástica
aguda (LLA) de origem B. Nesses casos, a própria definição do estadio de
maturação está relacionada às translocações cromossómicas ocorridas. Além disso,
a informação obtida a partir do diagnóstico é ainda de grande utilidade para
orientar a terapêutica durante o processo de tratamento e principalmente após o
término, na procura de doença residual mínima (DRM).
A metodologia padrão para a classificação das leucemias inclui morfologia,
citoquímica, estudo de marcadores celulares (citometria de fluxo), citogenética e
análise molecular. Já a sua avaliação preliminar inclui o hemograma, que fornece
dados essenciais como a contagem global, diferencial, o comprometimento da
hematopoiese e a morfologia dos leucócitos.
Uma vez sugerido o diagnóstico de leucemia, é necessária a avaliação morfológica
do aspirado de medula óssea para a sua confirmação.
A classificação morfológica das leucemias agudas mais utilizada foi desenvolvida em
1976 por um grupo de hematologistas franceses, americanos e britânicos
(classificação FAB) e classifica a LLA em três subtipos, designados L1, L2 e L3. As
leucemias mielóides agudas (LMA) são caracterizadas por sete subtipos M0 a M7,
de acordo com o grau de maturação dos precursores mielóides envolvidos.
Citometria de Fluxo
Os marcadores de superfície celular são proteínas de membrana detectadas por
meio de anticorpos monoclonais marcados com substâncias fluorescentes. Essas
diferentes proteínas são expressas em diversas fases de maturação, o que permite
que sejam utilizadas como marcadores de tipo e estadio.
Além da caracterização dos antigénios expressos pelas células envolvidas, a
citometria de fluxo fornece informação quanto ao tamanho e à granulosidade
celular. O gráfico obtido permite uma análise preliminar da população investigada,
na qual se delineiam as áreas correspondentes a linfócitos, monócitos, granulócitos,
células plasmáticas e regiões de blastos linfóides ou mielóides, quando presentes.
Nas leucemias agudas, o exame está indicado para determinação da linhagem
celular, análise de clonalidade e do estado de maturação das células leucémicas,
expressão de padrões de antigénios aberrantes típicos de determinados grupos de
leucemias e acompanhamento do tratamento e detecção de doença residual mínima
(DRM).
Torna-se necessária a utilização de um painel mínimo de anticorpos monoclonais
contra antigénios específicos para determinação dos parâmetros descritos
anteriormente. Já um painel secundá rio, realizado a partir do painel inicial, pode
ser usado para a caracterizacão mais detalhada da patologia em questão e do
estadio de maturação da célula envolvida.
Geralmente, utiliza-se um painel primário mínimo de anticorpos monoclonais
selecionados, com o objetivo de se conseguir uma definição da linhagem celular,
que inclui a determinação dos antigénios intracitoplasmáticos mieloperoxidase
(MPO) para a determinação de linhagem mielóide e dos antígenos CD22 ou CD79 e
CD3 para a determinação inicial de linhagem linfocitária B ou T, respectivamente. A
enzima TdT (desoxinucleotidil terminal transferase) é um marcador
intracitoplasmático que está presente na maioria dos casos de leucemia linfóide
aguda (LLA) e que contribui para a confirmação da origem linfóide e blástica de
uma leucemia.
Com base nos mercadores celulares presentes nas células, é possível definir a
origem linfóide ou mielóide das leucemias e classificá-las de acordo com a tabela
abaixo:
LLA
LLA
LLA
LLA
LLAs de origem B
Pró-B
Comum (CALLA positivo)
Pré-B com a presença de imunogloglobulina intracitoplasmática
B madura (com imunoglobinas de superfície)
LLAs de origem T
LLA Pró-T
LLA Pré-T
LLA T madura (CD4+ ou CD8+)
LMA
M0, M1, M2, M3, M4, M5, M6 ou M7.
As leucemias crónicas fazem parte de um grupo de condições denominadas doenças
linfoproliferativas, que têm origem em precursores linfocíticos mais maduros. São
as leucemias mais comuns do adulto e na maioria dos casos, ocorrem por
proliferação dos linfocítos B. Entretanto, várias doenças crónicas que envolvem as
células T e as células NK (Natural Killer) também são encontradas. A distinção entre
leucemias crónicas primárias e outras doenças linfoproliferativas, como a fase
leucémica dos linfomas não-Hodgkin, leucemia linfoblástica aguda, leucemia
prolinfócitica e leucemia de células pilosas, entre outras, é necessária com vista ao
prognóstico e ao tratamento.
Os painéis utilizados para identificação das doenças linfoproliferativas crónicas
dividem-se entre os relacionados às leucemias e linfomas e permitem diferenciar
entre: leucemia linfóide crónica (LLC), leucemia prolinfocítica (LPL), leucemia de
células cabeludas (HCL), linfoma esplênico de zona marginal e associados, linfoma
de células do manto, linfoma folicular e discrasias de células plasmáticas: Linhagem
T: síndrome de Sezary, leucernia T do adulto (ATI), large granular leukemia (LGLL)
& linfoma hepatoesplênico (gama-delta), e finalmente, a avaliação de linfomas
CD30+.
O painel para leucemias agudas, leucemias crónicas e/ou linfoma e avaliação de
células CD34+ para transplante autólogo está disponível para análise em sangue,
medula óssea, líquor e líquidos corporais obtidos por punção. Para a realização em
medula óssea, o material é colhido pelo médico assistente ou no próprio laboratório
na quantidade mínima de 1 mL, colhido com heparina ou EDTA. Esse material deve
ser conservado à temperatura ambiente e enviado ao laboratório (no caso dda
colheita ser realizada pelo médico assistente), juntamente com o esfregaço de 2 a
5 láminas não-coradas.
Imunofenotipagem de Leucócitos
A imunofenotipagem linfocitária é realizada com a utilização de anticorpos
monoclonais que são capazes de reconhecer antigénios na membrana da célula.
Devido à capacidade de identificar e quantificar com segurança as populações
linfocitárias, é utilizada, na avaliação da imunodeficiência celular e na monitorização
de pacientes com SIDA.
No teste, é utilizado um grupo de anticorpos monoclonais conjugados. Os
anticorpos são selecionados para reconhecer diferentes populações e
subpopulações linfocitárias, permitindo estudar as populações de linfócito T (CD3) e
linfócito B (CD19) e as subpopulações de T, linfócito T helper (CD4) e linfócito T
supressor (CD8).
Em relação à SIDA, a contagem de linfócitos T CD4+ realizada em citómetros de
fluxo é, hoje, o teste recomendado para avaliação do estdiio da doença, orientando
decisões sobre o ínicio do tratamento, modificações da terapia antiviral e
tratamento profilático contra patogénios oportunistas.
Representa também um marcador prognóstico utilizado em conjunto com a análise
da carga viral, indicando a evolução clínica da doença e a expectativa de sobrevida.
No início da infecção vírica, há uma súbita diminuição da contagem de células CD4
e altos níveis de virémia plasmática (RNA-HIV). Ao longo de vários anos, há uma
diminuição gradual dos níveis de CD4, que se acentua por um período médio de 1,5
a 2 anos antes do diagnóstico da AIDS.
A doença avançada caracteriza-se pela contagem de CD4 abaixo de 200
células/mm3 e pelo desenvolvimento de infecções oportunistas, neoplasias,
síndrome de consumo e complicações neurológicas.
Alguns autores recomendam que os pacientes sintomáticos devem ser sempre
tratados, independentemente dos valores de CD4. Quando assintomáticos, a
decisão do tratamento está associada aos valores da carga viral e de CD4. Para
valores acima de 350 células/mm3, não há indicação de tratamento, e valores
abaixo desse limite indicam a necessidade de início do tratamento.
Os linfócitos T CD8+ também são afectados pela doença, observando-se níveis
aumentados na fase inicial e que retornam ao normal em meses. Com a evolução
da doença, tendem a diminuir em proporções menores que os dos linfócitos T
CD4+.
Vários factores podem influenciar a contagem de células CD4. Entre eles, variações
de métodos (que permitem valores normais entre 500 a 1.400 células por mililitro),
variações sazonais e diurnas, doenças intercorrentes e uso de corticóides. Existem
descrições que associan a co-infecção pelo HTLV-1 e a esplenectomia à contagem
enganosamente alta de CD4.
Imunoglobulina E Específica
O doseamento da imunoglobulina E (IgE total) é um bom teste laboratorial para a
triagem de processos alérgicos. Os níveis de IgE total encontram-se elevados na rinite
alérgica, na dermatite atópica e em muitos casos de asma. Porém, não é um teste
específico, pois existem outras situações clínicas que cursam com o aumento de IgE
sérica total, como parasitoses (helmintíases), mielomas, aspergilose, filariase
pulmonar e o síndrome de Wiskott-Aldrich. Por isso, torna-se necessário o diagnóstico
diferencial para alergia, por meio da pesquisa da IgE com actividade específica.
Além de diferenciar das outras possibilidades clínicas que apresentam aumento de IgE
total, a pesquisa da IgE específica é realizada para diferentes alergénios isolados,
permitindo identificar o alergénio específico, causador da sintomatologia clínica, com
uma eficiência acima de 90%, sem interferência de fármacos ou de parasitas.
O doseamento de IgE específica pode ser realizado para uma enorme variedade de
alergénios, que são agrupados em painéis de acordo com a sua natureza, para facilitar
a investigação. Os alergénios podem ser solicitados individualmente ou em painéis.
FRUTAS
Laranja
Limão
Ananás
Kiwi
Morango
Cacau
Tomate
f33
f208
f210
f84
f44
f93
f25
PEIXES E FRUTOS DO MAR
Peixe (bacalhau)
Camarão
f3
f24
Atum
Salmão
Mexilhão
f40
f41
f207
LACTICÍNEOS
Leite
Caseína
Alfa-lactoalbumina
Beta-lactoalbumina
f2
f78
f76
f77
PROTEÍNAS ANIMAIS
Carne de Porco
Carne de Vaca
Gema do Ovo
Clara do Ovo
f26
f27
f75
f1
GRÃOS
Glúten
Trigo
Milho
Amendoim
Grão de Soja
f79
f4
f8
f13
f14
INALANTES-EPITÉLIO
Pêlo de Cão
Pêlo de Gato
e2
e1
INALANTES-INSECTOS
Blatella germenica (barata)
i6
INALANTES-POEIRA
Hollister-stier-labs (poeira domociliar)
D. pteronysinnus
D. farinae
Bomia tropicalis
h2
d1
d2
rd 201
INALANTES-GRAMÍNEAS
Grama
Azevém
Rabo-de-gato
Erva-de-febre
Zaburro de alepo
g2
g5
g6
g8
g10
DROGAS
Penicilina G
Penucilina V
c1
c2
FUNGOS
Penicillium notatum
Cladosporium herbarum
Aspergillus fumigatus
Candida albicans
Alternaria alternata
m1
m2
m3
m5
m6
OCUPACIONAIS
Latéx (Hevea brasiliensis)
k 82
Imunoglobulinas
As imunoglobulinas ou anticorpos são um grupo de glicoproteínas presentes no soro
e nos líquidos orgánicos. São produzidas pelos linfócitos B, precursores que, depois
de sensibilizados, isto é, depois de terem entrado em contato com o antigénio,
originam os plasmócitos de diferentes linhagens e clones celulares, que irão
produzir as cinco frações de imunoglobulinas, denominadas imunoglobulinas A, G,
M, D e E. Apesar de apresentarem muitas semelhanças, diferem entre si no
tamanho, na composição de aminoácidos, no conteúdo de carbohidratos e na carga
eléctrica.
A estrutura básica de uma molécula de imunoglobulina (monómero) consiste em
duas cadeias polipeptídicas de cadeias leves e duas cadeias pesadas, sempre em
pares idênticos. As quatro cadeias mantêm-se unidas por ligações de pontes
dissulfeto, sendo que as duas leves (L) são menores e comuns a todas as classes
de imunoglobulinas. As cadeias pesadas (H) possuem um alto peso molecular,
contêm cerca de 440 aminoácidos e são maiores, com estruturas distintas em cada
classe ou subclasse.
As chamadas cadeias leves possuem um baixo peso molecular e contêm cerca de
220 aminoácidos. Apresentam-se em dois tipos distintos: kappa (K) e lambda (l).
Em cada imunoglobulina existem sempre duas idênticas de um único tipo, nunca as
duas simultaneamente.
As imunoglobulinas possuem dois locais idênticos de ligação com o antigénio. Um é
realizado pela cadeia H, e outro pela cadeia L. São bifuncionais, pois cada molécula
de imunoglobulina apresenta uma região que age na ligação com o antigénio,
enquanto a outra promove a ligação das imunoglobulinas às células do sistema
imune e ao sistema do complemento.
Imunoglobulina G
A IgG é a principal imunoglobulina do sangue, correspondendo a cerca de 70 a 75%
do total de imunoglobulinas. É monomérica e é o principal anticorpo nas respostas
imunes secundárias, quando são produzidas em grande quantidade. É a única que
atravessa a barreira placentária e confere um grande grau de imunidade passiva ao
recém-nascido.
Subclasses de Imunoglobulina G
Tanto a classe quanto as subclasses da imunoglobulina são determinadas pelo tipo
de cadeia pesada. A IgG apresenta quatro subclasses distintas: IgG1, IgG2, IgG3 e
IgG4, que possuem quatro cadeias pesadas, similares mas não-idênticas, já que
apresentam diferenças nas suas propriedades, tais como número de pontes
dissulfídicas e sequências de aminoácidos diferentes.
Apesar de apresentarem níveis séricos de imunoglobulinas normais, os pacientes
com baixa resistência a infecções de repetição, especialmente respiratórias, podem
apresentar uma deficiência específica de uma ou mais das subclasses de
imunoglobulinas.
As subclasses ocorrem na proporção de 66% de IgG1, 23% de IgG2, 7% de IgG3 e
4% de IgG4. A subclasse IgG1 é a principal subclasse nos adultos normais. A IgG2
é a única que não atravessa a barreira transplacentária, e sua deficiência está
muitas vezes associada à deficiência de IgA e especialmente a infecções víricas e
bacterianas, na maioria dos casos, do aparelho respiratório superior e inferior. A
IgG3 é a que se liga de modo mais efectivo ao complemento. A IgG1 e a IgG2,
respectivamente, ligam-se em ordem decrescente de ecfetividade, e a IgG4, em
muitos casos, falha inteiramente a ligação ao complemento pela via clássica. A
deficiência de IgG3 está associada a infecções respiratórias de repetição,
especialmente pelo Streptococcus pneumoniae e Haemophilus influenzae, e a
infecções das vias urinárias. A IgG4 apresenta níveis séricos elevados, em
condições atópicas. A sua deficiência também está associada a infecções do
aparelho respiratório superior e inferior, especialmente a doenças
broncopulmonares. Níveis séricos diminuídos de todas as subclasses podem ser
observados nas doenças auto-imunes e nas infecções pelo vírus HIV.
Imunoglobulina M
Representa 5 a 10% das imunoglobulinas totais Apresenta-se na forma de um
pentâmero e não possui subclasses. É reconhecida como o anticorpo precoce, visto
ser a primeira imunoglobulina encontrada na resposta a um estímulo antigénico.
Por isso, é útil no diagnóstico diferencial das infecções víricas ou parasitárias
agudas. Ao contrário da IgG, não atravessa a barreira placentária e é a única que o
recém-nascido sintetiza. Níveis séricos aumentados em recém-nascidos indicam
que a imunoglobulina M foi produzida pelo feto, sugerindo uma infecção pré-natal.
Como a IgM é pentamérica, ou seja, possui 10 sítios de ligação com o antigénio, é
mais eficiente em ligar-se aos antigénios e ao sistema do complemento. Essas
características, aliadas ao aparecimento precoce durante a evolução da infecção,
fazem dela um potente agente de combate às infecções.
Imunoglobulina A
A IgA representa cerca de 10 a 15% das imunoglobulinas totais. É a imunoglobulina
predominante nas secreções: saliva, lágrimas, secreções nasais, colostro, leite,
secreções traqueobrônquicas e gastrointestinais. A sua estrutura pode variar,
dando origem a duas subclasses: IgA1 e IgA2. A maior parte das IgAs é da
subclasse IgA1, a mais encontrada no soro, e a IgA2, mesmo em menor
quantidade, tem um papel importante, por se tratar da subclasse dominante nas
secreções, nas quais tem a função de proteger a mucosa de agressões. Não
atravessa a barreira transplacentária, mas devido à sua grande concentração no
colostro, tudo indica que contribua para a proteção dos recém-nascidos contra
infecções, principalmente do tubo gastrointestinal. Existe uma outra forma de
apresentação da IgA, chamada de IgA secretória (sIgA), na qual a imunoglobulina
A se apresenta associada a uma outra proteína, que é o componente secretório,
sintetizado pelas células epiteliais, servindo de receptor para a ligação da IgA.
Devido à sua presença nas membranas externas, os constituintes secretores da IgA
formam uma primeira linha de defesa contra agressões do ambiente externo.
Aparentemente, não conseguem activar o complemento pela via clássica, fazendo-o
pela via alternativa (sistema properdina).
Imunoglobulina D
Representam menos de 1% das imunoglobulinas plasmáticas. Aparecem em grande
quantidade na membrana dos linfócitos B circulantes. A função biológica ainda não
é bem conhecida, mas parece desempenhar um papel importante na diferenciação
dos linfócitos antigenicamente estimulados, sendo um dos principais receptores
para antigénios na superfície das células B.
Imunoglobulina E
Entre todas as classes de imunoglobulinas, a IgE é a que se encontra em menor
quantidade e está presente na superfície de basófilos e mastócitos, o que resulta na
libertação de mediadores da resposta de hipersensibilidade imediata. Tem um papel
importante na imunidade activa contra parasitas, especialmente os helmintas, e
nas doenças alérgicas. A IgE não atravessa a barreira placentária e não se liga ao
complemento pela via clássica, e sim pela via alternativa.
Alterações da Concentração das Imunoglobulinas
O aumento policlonal das imunoglobulinas séricas representa uma resposta normal
a uma série de patologias, como infecções, doenças hepáticas, doenças pulmonares
e alterações auto-imunes, entre outras. O aumento de IgG tende a predominar nas
respostas crónicas e nas doenças hepáticas; já os aumentos de IgA tendem a
predominar nas infecções de pele, aparelhos respiratório, intestinal e renal. A IgM
está relacionada às fases agudas das infecções primárias e às infecções intrauterinas. Na cirrose biliar primária, a IgM está marcadamente elevada, e algumas
vezes de forma conjunta com a IgG. A IgE normalmente encontra-se elevada na
asma e noutros quadros alérgicos, especialmente nas crianças. As infecções
bacterianas crónicas cursam com o aumento de todas as imunoglobulinas.
O aumento monoclonal das imunoglobulinas é causado por um único clone de
células plasmáticas, produzindo imunoglobulinas de estrutura idêntica. Estas
imunoglobulinas monoclonais são chamadas de paraproteínas e podem ser
polímeros, monómeros ou fragmentos da molécula de imunoglobulina. Esses
fragmentos costumam ser cadeias leves (proteínas de Bence Jones) e, mais
raramente, cadeias pesadas. A expressão gamapatia monoclonal é utilizada para
identificar o grupo de patologias em que são encontradas paraproteínas. Cerca de
60% das paraproteínas são produzidas por células oriundas do mieloma múltiplo ou
de um plasmocitoma solitário. Cerca de 15% são causadas pela superprodução,
pelos linfócitos B, principalmente em linfonodos, linfomas, leucemias linfocíticas
crónicas, macroglobulinemia de Waldenström ou mais raramente na doença de
cadeias pesadas. Cerca de 25% das paraproteinémias são benignas, e decorrem em
diferentes patologias ou muitas vezes de causas desconhecidas.
IMUNOGLOBULINAS MONOCLONAIS NO MIELOMA MÚLTIPLO
PROTEINÚRIA DE BENCE
PARAPROTEÍNA SÉRICA
INCIDÊNCIA
JONES
IgG
50%
60%
IgA
25%
70%
IgD
2%
100%
IgM
1%
100%
IgE
0,1%
a maior parte
Apenas cadeias leves
20%
100%
Biclonal
1%
Não-detectável
<1%
0%
Adaptada Tietz Textbook of Clinical Chemistry 3ª Edição - 1999.
Inibidor da C1-Esterase
O inibidor da C1-esterase actua na resposta inflamatória pela inactivação das
proteases do sistema complemento C1r e C1s, dos factores da coagulação XIa e
XIIa e da plasmina do sistema fibrinolitico.
A deficiência hereditária ou adquirida de C1-esterase leva a uma activação
desordenada de C1, com a produção de substâncias vasoactivas que causam
episódios recorrentes, com angioedema de pele, mucosas, faces, aparelhos
respiratório e gastrointestinal superior. Com a idade, o angioedema hereditário
pode precipitar situações de coagulação intravascular disseminada ou de falência de
múltiplos órgãos.
Existem duas variantes da doença: tipos I e II , e ambas são doenças autossómicas
dominantes. O tipo I corresponde a cerca de 85% dos casos e é caracterizado por
baixas concentrações plasmáticas do inibidor da C1 esterase. O tipo II corresponde
aos 15% restantes e caracteriza-se pela presença de concentrações normais ou até
elevadas do inibidor da C1 esterase, porém com comprometimento da actividade
funcional.
A variedade mais comum de deficiência adquirida ocorre por consumo aumentado,
em vez da síntese diminuída, e está associada a desordens linfoproliferativas
benignas ou malignas de células B. Uma forma menos comum é a associada a autoanticorpos contra o inibidor da C1-esterase.
Jejum
Quando nos referimos à necessidade de jejum, ou seja à privação de alimentação
antes da realização de uma avaliação laboratorial, este pode variar de acordo com
o exame solicitado, indo de um período determinado de horas, com 8 a 12 horas de
abstinência, ou apenas a restrição de ingestão alimentar matinal que antecede a
colheita de material para análise.
A maioria de exames hoje, dispensa longos períodos de jejum, sendo passíveis de
ser realizados desde que não tenha ocorrido alimentação recente (4horas) rica em
lipídios e proteínas que possam interferir ns sua análise.
Por outras palavras, poucos são os exames que exigem regras rigorosas de jejum
com o mínimo de 8 e o máximo de 14 horas para glicose, prova de tolerância à
glicose e testes funcionais endócrinos, de 12 horas para avaliação de lípidos,
especialmente triglicirídeos.
Entretanto, é necessário deixar claro que a maioria dos valores de referência é
obtida utilizando-se uma população saudável, em jejum pela manhã. Apesar disso,
na prática, constata-se que o doseamento dos diferentes analitos não sofre grande
interferência com o jejum, apresentando variações em torno de 5% nos resultados
decorrentes da alimentação, variações estas consideradas aceitáveis. Excepção
feita às restrições já citadas, permitindo a realização com vantagem da
extemporaneidade da colheita sanguínea.
Outro dado importante é o efeito do jejum prolongado, que pode afectar alguns
resultados de laboratório. Assim, tempos superiores a 14 horas podem influenciar a
doseamentos de glicose, insulina e hormonas. Após um jejum de 48 horas, a
libertação hepática de billirrubina pode aumentar em aproximadamente 240% do
seu valor. Com jejum prolongado, há uma diminuição de proteínas específicas,
como componente de C3 de complemento, pré-albumina e albumina. Os níveis
normais são rapidamente recuperados com ingestão de alimentos . As
concentrações de glicose, uréia, colesterol, triglicerídeos e apolipoproteínas
diminuem, e a creatinina e ácido úrico aumenta. Os níveis de hormonas tiroidéias
T3 e T4 diminuem.
LDL- Colesterol
O LDL- colesterol (colesterol contido nas lipoproteínas LDL) tem sido apontado por
diversos autores como um indicador de risco de desenvolvimento de arteriosclerose
melhor do que o colesterol total, já que está directamente envolvido no mecanismo
de desenvolvimento da lesão arterosclerótica. O endotélio tem participação activa
no processo de aterogénese, e sofre a acção dos factores de risco, como aumento
do LDL-colesterol, tabagismo, suscetibilidade hereditária, entre outros.
A disfunção endotelial, que se caracteriza por diminuição da resposta dos
mecanismos de vasoconstrição e vasodilatação arteriais sob a acção da
acetilicolina, parece decorrer de diferentes factores, por sua vez consequentes à
acção das LDL oxidadas. Estas são removidas pelos macrófagos com receptores
específicos, levando a um acúmulo de ésteres de colesterol no interior dos
macrófagos e levando à formação das células espumosas, as principais
responsáveis pela quantidade de colesterol na placa de ateroma.
Consultar Perfil Lipídico
Lipase
A lipase é a enzima digestiva produzida principalmente pelas células acinares do
pâncreas exócrino. Tem o papel fisiológico de hidrolisar as longas cadeias de
triglicerídeos no intestino delgado (lipólise).
A sua avaliação é essencial no diagnóstico das patologias pancreáticas. Ela aumenta
nas primeiras 8 horas após o início da agressão pancreática, atingindo valores mais
altos em 24 horas e mantendo-se elevada em torno de 7 a 14 dias. Os seus níveis
geralmente não permanecem elevados por mais de 2 semanas. Quando isto
acontece, sugerem complicações como abcessos e pseudocistos. Normalmente os
seus níveis elevam-se quase que paralelamente aos da amilase, um pouco mais
tarde, mantêm-se elevados por um período mais longo. O seu aumento não se
correlaciona necessariamente com a gravidade da doença.
O uso combinado da avaliação sérica da lipase e de amilase permite um melhor
diagnóstico. Cerca de 20% dos casos de pancreatite aguda cursam com níveis de
amilase normais e com lipase isoladamente elevada. Nas parotidites agudas, em
que a amilase pode apresentar-se elevada, os níveis séricos de lipase não se
alteram, auxiliando no diagnóstico diferencial.
A lipase é portanto um marcador mais específico de doença pancreática aguda do
que a amilase. Os seus níveis estão aumentados em pacientes com pancreatite
aguda e recorrente, abcesso ou pseudocisto pancreático, trauma, carcinoma de
pâncreas, obstrução dos ductos pancreáticos e no uso de fármacos (opiáceos). Está
também aumentada na maior parte das condições inflamatórias da cavidade
abdominal, doenças do trato biliar, abcessos abdominais e insuficiência renal aguda
e crónica (com menor frequência do que a amilase).
A lipase é filtrada pelos glomérulos, devido ao seu baixo peso molecular. Em
condições usuais, é totalmente reabsorvida pelos túbulos proximais, estando
ausente da urina de pacientes normais. Nos distúrbios renais que cursam com
alteração da capacidade de reabsorção tubular, a lipase pode ser detectada na
urina, numa relação inversa com a clearance da creatinina.
Lipoproteína (a)
A lipoproteína (a), ou Lp(a) é uma classe distinta de liproteínas, estruturalmente
relacionada com as lipoproteínas de baixa densidade (LDL), já que ambas as
classes de liproteínas possuem uma molécula de Apo B-100 por partícula e uma
composição lipídica similar.No entanto, ao contrário da LDL, a Lp(a) contém uma
proteína, a Apo (a), que se liga à Apo B-100.
A Apo (a) é estruturalmente semelhante ao plasminogénio. Apesar de não possuir
as mesmas características funcionais, compete com o plasminogénio nos seus locais
de acção, o que explicaria a sua capacidade de conduzir a um estado prótrombótico, ao impedir a formação da plasmina, a enzima responsável pela lise da
fibrina.
Outro dado importante é que, pela ligação entre a Apo B-100 e a Apo (a), a sua
velocidade de remoção da circulação fica diminuída, aumentando a sua
permanência na circulação, o que facilita a sua migração para a região
subendotelial.
É considerada por muitos autores um factor de risco para o desenvolvimento de
doença coronária obstructiva, de caráter genético, que sofre pouca influência dos
demais factores de risco já conhecidos. Portanto, níveis elevados constituem um
risco, independentemente do desenvolvimento de doença aterosclerótica.
Lipoproteínas
As lipoproteínas são complexos macromoleculares sintetizados no fígado e no
intestino delgado, que transportam o colesterol e os triglicerídeos através da
corrente sanguínea. São classificadas segundo características físico-químicas.
Lipoproteínas de Alta Densidade – HDL
As HDL são pequenas partículas constituídas por cerca de 50% de proteína,
especialmente Apo A I e II, e pouca quantidade de Apo C e Apo E, 20% de
colesterol, 30% de triglicerídeos e resíduos de fosfolípidos. A HDL pode ser
separada em duas subclasses principais: HDL 2 e HDL 3, que diferem em tamanho,
densidade e composição, especialmente em relação ao tipo de apoproteínas.
Cumprem o importante papel de transportar o colesterol até ao fígado directamente
ou transferindo ésteres de colesterol para outras lipoproteínas, especialmente as
VLDL. É atribuído à fracção HDL 2 o papel de proteção do desenvolvimento da
arteriosclerose.
Lipoproteínas de Baixa Densidade – LDL
As LDL representam 50% da massa total de lipoproteínas circulantes. São
partículas bem menores, tão pequenas, que mesmo quando em grande quantidade
não são capazes de turvar o plasma. O colesterol representa metade da massa da
LDL. Cerca de 25% são proteínas, especialmente Apo B-100 e pequenas
quantidades de Apo C; o restante é constituído de fosfolípidos e triglicerídeos. É a
lipoproteína que mais colesterol transporta. Tem a função de transportá-lo para
locais onde ele exerce uma função fisiológica, como por exemplo a síntese de
esteróides. São, na sua maior parte, produzidas a partir das lipoproteínas VLDL. A
sua concentração sérica tem uma relação directa com o aumento do risco de
aterogénese.
Lipoproteínas de Muito Baixa Densidade – VLDL
São partículas grandes, porém menores do que as partículas dos quilomícrons
produzidas no fígado. São constituídas por 50% de triglicerídeos, 40% de colesterol
e fosfolípidos e 10% de proteínas, principalmente Apo B-100, Apo C e alguma Apo
E. Têm como função o transporte dos triglicerídeos endógenos e do colesterol para
os tecidos periféricos para serem armazenados ou utilizados como fonte de energia.
Assim como os quilomícrons, são capazes de turvar o soro.
Quilomícrons
São grandes partículas produzidas pelas células intestinais, compostas por cerca de
85 a 95% de triglicerídeos de origem da dieta (exógeno), pouca quantidade de
colesterol livre e fosfolípidos e 1 a 2% de proteínas. Devido à sua proporção
lípido/proteína, os quilomícrons flutuam, dando ao plasma um aspecto leitoso,
formando ainda, sobre ele, uma camada cremosa, quando deixado em repouso.
Consultar Perfil lipídico.
Líquido Cefalorraquidiano - Líquor
O líquido cefalorraquidiano (LCR) é formado principalmente pelos plexos coróides.
Nos adultos, é produzido a uma taxa de 20 mL/h, o que corresponde a
aproximadamente 500 mL/24 h. Como o volume do LCR é de cerca de 100 a 150
mL, isso significa que é renovado em média a cada 6 horas. Entre as suas
diferentes funções, a principal é proteger mecanicamente o tecido cerebral. Além
disso, actua como um lubrificante, evitando atrito com o crânio, realiza a recolha de
resíduos, faz circular os nutrientes e varia a sua produção de acordo com a pressão
intracraniana. A composição do líquor é controlada pelas barreiras hematoencefálica
e hematoliquórica, que também protegem contra a invasão de agentes externos.
EXAME MICROSCÓPICO
O líquor normal é límpido, cristalino, inodoro e com aspecto de água. De acordo
com as diferentes patologias, essas características alteram-se. Apresenta-se
opalescente ou turvo pelo aumento de bactérias, fungos, eritrócitos e leucócitos. A
cor é resultante da presença de bilirrubina, eritrócitos, hemoglobina, leucócitos ou
proteínas.
Na hemorragia subaracnóidea, o aspecto é hemorrágico vermelho turvo. Esta
coloração também poderá ocorrer nos acidentes de punção, sendo o diagnóstico
diferencial a prova dos 3 tubos, cujo aspecto na hemorragia é uniforme e no
acidente de punção tende a clarear a cada tubo. A presença de coágulo nos
acidentes de punção, o aspecto do sobrenadante após a centrifugação, que nas
hemorragias se apresenta xantocrómico, enquanto nos acidentes é límpido,
também auxiliam no diagnóstico diferencial.
Nas meningites bacterianas, o líquor apresenta-se turvo, amarelo e, por vezes,
xantocrómico, após centrifugação. Já nos casos de meningites víricas, a cor
geralmente varia de esbranquiçada a incolor após a centrifugação.
EXAME BIOQUÍMICO
Cloro
Qualquer condição que altere os níveis séricos de cloreto também irão afetar o nível
de cloreto no LCR. Os cloretos no LCR são normalmente 1 a 2 vezes maiores do que
os séricos. Níveis diminuídos são encontrados nas meningites tuberculosa e
bacteriana e na criptococose.
Glicose
Os níveis de glicose no LCR correspondem a cerca de 2/3 da glicose sanguínea de
jejum. A proporção normal de glicose LCR/plasma pode variar de 0,3 a 0,9. São
considerados valores anormais de glicose no LCR resultados inferiores a 40 mg/dL
e/ou relações inferiores a 0,3. A diminuição dos níveis da glicose no líquor é um
dado importante no diagnóstico das meningites bacterianas, tuberculosas e
fúngicas, nas quais encontramos geralmente valores baixos a muito baixos. Já nas
meningites víricas, os níveis variam de normais a discretamente baixos. Outras
patologias que cursam com níveis diminuídos são neoplasias com comprometimento
meníngeo, sarcoidose, hemorragia subaracnóide e hipoglicemia sistémica, entre
outras. Níveis elevados de glicose no LCR não possuem significado clínico,
reflectindo aumento dos níveis da glicemia sistémica. Acidentes de punção podem,
ocasionalmente, causar aumento da glicose no LCR.
Proteína
Das proteínas encontradas no líquor, mais de 80% são provenientes do plasma.
Normalmente, equivalem a valores inferiores a 1% do nível sanguíneo. O aumento
dos níveis de proteínas no líquor é um bom indicador, embora não-específico, da
presença de doença.
As proteínas no LCR podem estar elevadas em diferentes patologias, como
meningites, especialmente as bacterianas, doenças neurológicas, hemorragias e
tumores, entre outras. A elevação pode ser decorrente da alteração da
permeabilidade da barreira hematoencefálica, da diminuição dos mecanismos de
reabsorção, de uma obstrução mecânica do fluxo do LCR ou do aumento na síntese
de imunoglobulina intratecal.
Os níveis podem estar diminuídos em crianças entre os 6 meses e 2 anos de idade
e em condições associadas a um turnover aumentado, como acontece nas punções
com remoção de grandes volumes, traumas com perda de líiquor e aumento da
pressão intracraniana.
É importante lembrar a variação da concentração de proteína de acordo com o local
da punção, pois os valores encontrados são menores nos ventrículos e maiores na
região lombar, assim como também ocorrem drásticas variações nos recémnascidos.
Para avaliação da integridade da barreira hematoencefálica, pode-se utilizar um
índice obtido pela proporção entre os níveis de albumina no líquor (mg/dL) e no
soro (g/L). Normalmente, o valor encontrado é menor que 9. Valores maiores
indicam alterações da barreira, que podem variar de discretas a severas, de acordo
com os índices encontrados. São considerados discretos valores entre 9 e 14,
moderados entre 14 e 30, e acima de 30, um comprometimento severo. Índices
ligeiramente alterados são encontrados em crianças de até 6 meses, traduzindo
imaturidade da barreira hematoencefálica. Os acidentes de punção invalidam a
utilização destes índice.
NÍVEIS DE PROTEÍNA
PUNÇÃO VENTRICULAR
PUNÇÃO CISTERNAS
PUNÇÃO LOMBAR
RECÉM-NASCIDOS
PREMATUROS
EM ADULTOS
5 a 15 mg/dl
15 a 25 mg/dl
15 a 45 mg/dl
Até 150 mg/dl
Até 500 mg/dl
EXAME CITOLÓGICO
A análise citológica do LCR é composta de duas fases distintas: a citometria, em
que é feita a análise quantitativa das células, e a citologia, em que é feita a
contagem diferencial em lámina corada. Convém lembrar a importância do exame
citológico nas meningopatias leucémicas (mais frequente na leucemia linfoblástica
aguda), tanto no diagnóstico como no acompanhamento do tratamento.
As meningites bacterianas agudas apresentam grande celularidade (geralmente
acima de 500 leucócitos/mm3) e com predomínio de polimorfonucleares. Já as de
origem vírica, fúngica ou tuberculosa apresentam celularidade menor e um
predomínio de células mononucleres, podendo no entanto, nas primeiras 24/36
horas manter um predomínio de polimorfonucleres.
CITOMETRIA
Até 5 leocóciotos/mm3
Adultos
0 eritócitos/mm3
Até 30 leocócitos/mm3
Recém-nascidos
0 eritócitos /mm3
Até 10 leocócitos/mm3
1 mês a 1 ano
0 eritócitos /mm3
Até 8 leocócitos/mm3
1 a 4 anos
0 eritócitos /mm3
Até 5 leocócitos/mm3
Acima de 5 anos
0 eritócitos /mm3
CITOLOGIA
POLIMORFONUCLEARES
Adultos
2%
Crianças
10%
MONONUCLEARES
Adultos
98%
Crianças
90%
Outras Avaliações
Índice imunoglobina
líquor/soro
Doenças neorológicas, HIV, meningites fúngicas.
Esclerose múltipla, infecções do sistema nervoso central,
síndrome de Guilain-Barré, mielite transversa,
carcinomatose meníngea.
Diagnóstico diferencial etiológico de meningites e
Ácido Láctico
traumatismo craniano.
Tumores, acidentes vasculares, meningites, convulsões,
Creatinofofoquitase
traumatismo craniano.
Lesão cerebral por hipóxia, diferencial de acidente de
Desidrogenase Láctica
punção e hemorragias, meningites bacterianas.
Outros: VDRL/FTA-Abs, Vírus HIV, Toxoplasmose, Gram e culturas, e a pesquisa de
antigénios bacterianos rápidos.
Electroforese de
Proteínas
Líquido Pericárdico
Em condições normais, encontram-se cerca de 10 a 50 mL de líquido no espaço
pericárdico. É um filtrado plasmático produzido por um processo transudativo. O
derrame pericárdico (aumento da quantidade de líquido) pode ser encontrado em
processos inflamatórios, malignos ou hemorrágicos. Entre as causas mais comuns,
encontramos pericardite bacteriana, vírica, tuberculosa e fúngica, infecções por
micoplasma e relacionada com a SIDA, carcinomas metastáticos e linfomas, enfarte
do miocárdio, hemorragias por trauma, distúrbios da coagulação, doenças do foro
reumático, lúpus eritematoso sistémico e distúrbios metabólicos como uremia e
mixedema.
EXAME MICROSCÓPICO
O líquido pericárdico normal é amarelo claro e límpido. Apresenta-se hemorrágico
em processos infecciosos, malignos, traumas, distúrbios da coagulação, derrame de
aneurisma aórtico, colagenoses, pericardite hemorrágica idiopática e pós-enfarte.
Entretanto, pode significar também um acidente durante a punção. O diagnóstico
diferencial será realizado pelo hematócrito, que nos acidentes será similar ao do
sangue periférico, ao contrário do encontrado nas efusões hemorrágicas
verdadeiras. Além disso, o sangue da efusão não coagula, enquanto o do acidente
de punção sim. Já as efusões por processos metabólicos é clara e com uma cor
amarelo-pálida.
Grandes volumes (acima de 350 mL) sugerem processos malignos, uremia e
processos inflamatórios ligados à SIDA.
EXAME BIOQUÍMICO
Proteínas
A determinação de proteínas tem pouco valor para o diagnóstico diferencial das
efusões pericárdicas.
Glicose
Podem ser encontrados níveis de glicose inferiores a 40 mg/dL nos derrames
bacterianos, na tuberculose, na artrite reumatóide e nos processos malignos
metastáticos. A sua avaliação tem pouco valor para o diagnóstico diferencial.
EXAME CITOLÓGICO
A análise citológica é composta de duas fases distintas: a citometria, em que é feita
a análise quantitativa das células, e a citologia, em que é feita a contagem
diferencial em lámina corada. A presença de células de aspecto morfológico
suspeito determina a indicação de citopatologia para células neoplásicas. As
metástases dos carcinomas de pulmão e de mama são as mais observadas.
ADULTOS
CITOMETRIA
<500 leucócitos/mm3
0 eritócitos /mm3
EM ADULTOS
POLIMORFONUCLEARES
<25%
Outras Avaliações
A presença de anticorpos antinucleares, em títulos altos, tem sido descrita nos
derrames associados ao lúpus eritematoso sistémico. No entanto, não são
específicos para o diagnóstico dessa patologia.
A análise pela coloração de Gram apresenta uma sensibilidade de 50%, e a cultura,
de 80%, para o diagnóstico de pericardite bacteriana. Já a cultura e a coloração
para tuberculose (BAAR) apresentam uma sensibilidade de aproximadamente 50%,
que pode chegar a 90% quando analisado o tecido pericárdico em vez do líquido.
Líquido Peritonial
É um ultrafiltrado do plasma que ocupa a cavidade peritoneal, e a sua produção
depende da permeabilidade vascular e da pressão oncótica, sendo portanto um
transudado. Nesse espaço revestido por mesotélio, normalmente existem cerca de
50 mL de líquido presente. Aumentos acima desse nível já são considerados
alterações e podem ocorrer em patologias peritoneais, primárias ou não. Pacientes
com volumes aumentados são considerados portadores de ascite, e o líquido é
chamado de líquido ascítico.
EXAME BIOQUÍMICO
Amilase
Os valores de amilase são similares aos níveis séricos. Valores superior a três vezes
o valor sérico é uma boa evidência de origem pancreática, como pancreatite aguda,
pseudocisto pancreático ou traumatismo. A presença de úlcera péptica perfurada,
perfuração intestinal, trombose mesentérica e necrose de anças intestinais também
pode produzir níveis elevados.
Glicose
Os valores de glicose são similares aos séricos, apresentando-se diminuídos em
30% a 60% dos casos de peritonite tuberculosa e em cerca de 50% dos casos de
carcinomatose peritoneal. Os pacientes hiperglicémicos cursam com valores de
glicose aumentados no líquido ascítico.
Proteínas
A determinação dos níveis de proteínas é um dos dados utilizados para diferenciar
os exsudados dos transudados. No entanto, no líquido ascítico, esse parâmetro não
funciona de forma satisfatória. Nos transudados encontramos níveis de proteína
mais baixos (< 50% do valor sérico) e nos exsudados mais altos. Não é raro que as
amostras infectadas ou relacionadas com processos malignos apresentem
concentrações de proteínas na faixa do transudado. Muitos pacientes com ascite
por cirrose ou insuficiência cardíaca possuem taxas de proteínas na faixa do
exsudado. O Índice obtido pela relação da albumina no soro-albumina do líquido
ascítico é considerado um bom parâmetro na diferenciação da cirrose das outras
formas de efusão peritoneal.
TRANSUDADOS
Insificiência cardíaca congestiva, cirrose hepática, hipoproteínas (síndrome nefrótico)
...................................................................................
EXUDADOS
Peritonites bacterianas primárias e secundárias, tuberculose, hepatomas, carcinoma
metástico, linfomas e mesomielomas, traumas, pancreatite e peritonite biliar.
EXAME CITOLÓGICO
A análise citológica é composta de duas fases distintas: a citometria, em que é feita
a análise quantitativa das células, e a citologia em que é feita a contagem
diferencial em lámina corada. A presença de células de aspecto morfológico
suspeito, determina a indicação de citopatologia para células neoplásicas. Os
carcinomas metastáticos do pulmão e da mama são os mais observados.
ADULTOS
CITOMETRIA
<500 leucócitos/mm3
0 eritócitos /mm3
CITOLOGIA
POLIMORFONUCLEARES
<25%
Outras Avaliações
Segundo alguns autores, o colesterol tem um valor moderado na diferenciação
entre a ascite de origem maligna e a ascite por cirrose, utilizando-se como valor
limite 45 a 48 mg/dL.
Níveis elevados de desidrogenase láctica são encontrados nas ascites malignas. O
índice entre os valores de LDH líquido ascítico/LDH soro superior a 0,6 possui uma
sensibilidade de 80% no diagnóstico diferencial de neoplasias.
Valores de bilirrubina superiores a 6,0 mg/dL e uma proporção de bilirrubina
líquido/soro superior a 1,0 sugerem ruptura da vesícula biliar, causando
coliperitonite.
Líquido Pleural
A cavidade pleural é revestida pelos mesotélios das pleuras visceral e parietal.
Normalmente, contém uma pequena quantidade de líquido, dito pleural, o que
permite o movimento de uma membrana contra a outra. O líquido pleural é um
filtrado plasmático produzido continuamente pela pleura parietal. Quando ocorre
acumulação de líquido, é denominado derrame pleural, que é o resultado do
desequilíbrio entre a produção e a reabsorção do líquido.
EXAME MICROSCÓPICO
O líquido pleural é límpido, inodoro, amarelo-pálido e não coagula. Apresenta-se
hemorrágico nos processos traumáticos e malignos e no enfarte pulmonar. Pode
também acontecer num acidente durante a punção, sendo o diagnóstico diferencial
feito pela presença de pequenos coágulos e pela característica de ir clareando com
a drenagem continuada. Pode aparecer também noutras situações, como traumas,
distúrbios da coagulação e ruptura de aneurisma da aórta. Na realização do
hematócrito, este, apresentará um valor próximo a 50% do sangue periférico no
caso de hemotórax. Já nos processos metabólicos, o líquido pleural é claro e tem
cor amarelo-pálida.
Grandes volumes (acima de 350 mL) sugerem processos malignos, uremia e
processos inflamatórios ligados à SIDA.
EXAME BIOQUÍMICO
Amilase
Níveis de amilase acima dos níveis séricos indicam a presença de pancreatite
aguda, pseudoquisto do pâncreas, ruptura esofágica ou, nalguns casos, derrames
de origem maligna.
Glicose
A diminuição dos níveis de glicose para valores inferiores a 60 mg/dL ou um índice
glicose líquido pleural/glicose soro inferior a 0,5 são mais frequentes e acentuadas
nos derrames pleurais da artrite reumatóide e nos macroscopicamente purulentos
(empiemas). Em neoplasias, tuberculose, lúpus eritematoso sistémico e infecções
bacterianas não-purulentas, apresentam valores baixos num pequeno número de
casos.
LDH
É um dos parâmetros para diagnóstico diferencial entre exsudados e transudados.
Os níveis de LDH são sempre analisados em relação aos valores séricos (líquido
pleural/soro), obtendo-se um índice que é maior do que 0,6 nos exsudados e
menor do que 0,6 nos transudados. A presença de níveis diminuídos de LDH
durante a evolução dos processos inflamatórios indica uma boa evolução e um bom
prognóstico. Em contrapartida, níveis aumentados indicam uma evolução
inadequada e sugerem que se mude para uma conduta terapêutica mais agressiva.
Proteínas
A determinação dos níveis de proteínas no líquido pleural só tem significado clínico
como um dos dados utilizados para diferenciar os exsudados dos transudados.
O transudado acontece por factores mecânicos, como aumento da pressão
hidrostática ou diminuição da pressão oncótica, que influenciam o processo de
formação e reabsorção dos líquidos e apresentam níveis protéicos 50% menores do
que os do plasma.
O exsudado acontece por aumento da permeabilidade capilar e diminuição da
reabsorção linfática, e apresenta níveis protéicos maiores. Os transudados são
geralmente bilaterais e são encontrados na cirrose hepática, no síndrome nefrótico
e na insuficiência cardíaca congestiva. Os exsudados são mais frequentemente
unilaterais e aparecem nas infecções bacterianas, neoplasias e doenças do
colagénio.
EXAME CITOLÓGICO
A análise citológica é composta de duas fases distintas: a citometria, em que é feita
a análise quantitativa das células, e a citologia, em que é feita a contagem
diferencial em lámina corada. A presença de células de aspecto morfológico
suspeito determina a indicação de citopatologia para células neoplásicas.
Normalmente, encontram-se células mesoteliais nos processos inflamatórios. A
presença de eosinofilia (>10%) pode resultar de diferentes causas, como reacção a
fármacos, síndromes de hipersensibilidade, doenças reumatáticas, traumatismos,
enfarte pulmonar e doença de Hodgkin. Nestas situações, pode ser observada a
presença dos cristais de Charcot-Leyden, derivados dos eosinófilos.
TRANSUDADOS
EXSUDADOS
CITOMETRIA
<1.000 leucócitos/mm3
0 eritócitos /mm3
<1.000 leucócitos/mm3
0 eritócitos /mm3
CITOLOGIA
POLIMORFONUCLEARES 20%
MONONUCLEARES 80%
PREDOMÍNEO DE
POLIMORFONUCLEARES
Líquido Sinovial
É normalmente claro, transparente e viscoso. A sua quantidade média em
condições normais é de 1,0 mL. Na sua composição encontramos: um ultrafiltrado
plasmático, com ausência dos elementos da coagulação e o ácido hialurónico,
produzido pelas células sinoviais, responsável pela viscosidade característica do
líquido sinovial. A análise do líquido sinovial incluí as análises macroscópica,
citológica e bioquímica e outras investigações específicas.
EXAME BIOQUÍMICO
Mucina
O teste consiste na adição de ácido acético e na observação da formação ou não do
coágulo de mucina. A formação de um coágulo de mucina firme e compacto revela
um grau de viscosidade normal. Já a formação de um coágulo entre regular e fraco,
em fragmentos dispersos e numa solução turva, reflecte diluição e
despolimerização do ácido hialurónico, ou seja, uma alteração da viscosidade, que é
uma característica inespecífica de várias artrites inflamatórias.
Glicose
As concentrações no líquido sinovial são semelhantes às plasmáticas. Por isso, a
interpretação adequada dos valores de glicose do líquido sinovial requer uma
comparação com os níveis séricos da glicose em jejum. Em condições normais e na
maioria das condições não inflamatórias, a diferença é menor que 10 mg/dL.
Diferenças significativas são encontradas nos processos inflamatórios e infecciosos.
Os valores da glicose podem ser mascarados pela glicólise, aquando da presença de
um grande número de leucócitos.
Proteínas
A concentração normal de proteínas varia de 1,2 a 2,5 g/dL. Valores aumentados
são encontrados em processos inflamatórios e sépticos. Durante a fase
inflamatória, proteínas maiores, como o fibrinogénio, entram no líquido sinovial.
EXAME CITOLÓGICO
A análise citológica do líquido sinovial é composta de duas fases distintas: a
citometria, em que é feita a análise quantitativa das células, e a citologia, em que é
feita a contagem diferencial em lámina corada. Normalmente, é pancicelular, com
predomínio de mononucleares. Durante a análise microscópica por luz polarizada, é
avaliada a presença de cristais, livres ou no interior das células, que podem ajudar
no diagnóstico de artropatias induzidas por cristais. Permite também identificar os
cristais como os de urato monossódico (gota), pirofosfato de cálcio
(condrocalcinose ou pseudogota) e outros, como apatita e oxalato de cálcio.
LEUCÓCITOS
NORMAIS
Até 200/ml
INFLAMATÓRIOS
2000 a 75.000
NÃO-INFLAMATÓRIOS
200 a 2.0000
SÉPTICOS
>100.000
POLIMORFONUCLEARES
<25%
>50%
<25%
>85%
EXAME MICROSCÓPICO
É normalmente claro, transparente ou levemente amarelo. A turvação pode dar-se
pela presença de células (leucócitos e eritócitos) ou pela presença de cristais ou
fibrina.
A alteração de cor mais frequente é a avermelhada, que pode ser causada por uma
hemartrose verdadeira ou por um acidente durante a punção. A diferenciação dá-se
pelo facto de que, nos casos de acidente de punção, a coloração varia, clareando no
fim da colheita, e pela presença de coágulos. A coloração esverdeada pode ser
observada em artrites sépticas.
A viscosidade está directamente relacionada à quantidade de ácido hialurónico e
encontra-se diminuída nos processos inflamatórios. A observação faz-se durante a
colheita do material, pela velocidade de queda pela agulha de punção e analisada
laboratorialmente pelo teste da mucina.
A presença de coágulos é outro dado importante da avaliação macroscópica. Em
condições normais, o líquido sinovial não coagula. Portanto, a presença de coágulos
é indicativa de processos inflamatórios.
Outras Avaliações
Os níveis de ácido úrico no líquido sinovial geralmente são similares aos séricos e,
portanto, não possuem valor no diagnóstico. O dosemento da desidrogenase láctica
encontra-se elevada na artrite reumatóide, na gota e nas artrites infecciosas. É
provável que isso se deva à presença de um grande número de leucócitos, o que
prejudica sua aplicabilidade clínica.
Alguns autores mencionam a possibilidade de identificação do factor reumatóide e
de anticorpos antinuclerares no líquido sinovial, sem contudo terem uma definição
clínica clara da importância desses achados. Níveis de complemento diminuídos,
sem correlação com níveis séricos, têm sido descritos em artrites séptica e
reumatóide e no lúpus sistémico. A presença de imunocomplexos acontece em
diferentes patologias e parece ter relação com a actividade da doença.
Listeria
É causada pela Listeria monocytogenes, bacilo Gram-positivo que pode ser
encontrado no solo, na vegetação e em reservatórios animais. A infecção humana
ocorre geralmente, durante a gravidez, ou em condições de alteração da
imunidade, especialmente da imunidade celular. Trabalhos recentes, evidenciam
como principal fonte de contaminação a ingestão de alimentos contaminados. De
forma distinta de outros patogénios alimentares, a Listeria monocytogenes não
causa distúrbios gastrointestinais, mas desencadeia síndromes invasivos como
meningites, septicémias e partos de nados-mortos.
Os anticorpos são pesquisados por reacções de aglutinação. Títulos positivos devem
ser considerados com cuidado, devido à possibilidade de reacção cruzada com
outras bactérias.
Títulos de até 1/160 podem ser encontrados na população geral. Portanto, são
considerados significativos títulos iguais ou superiores a 1/160. Recomenda-se
sempre realizar a análise em mais de uma amostra, com intervalo de 10 a 15 dias.
Variações superiores a 4 vezes o título anterior são indicativas de infecção recente.
A identificação da bactéria por hemocultura e material vaginal após aborto e os
exames histopatológicos da placenta podem ser realizados para confirmação do
diagnóstico.
Litio
O mecanismo de acção de sais de lítio na estabilização do humor é desconhecido.
Não é conhecido também o seu papel na fisiologia normal. Em níveis terapêuticos,
não altera o humor do indivíduo normal. A relação com iões potássio e sódio tem
experimentalmente sugerido uma possível influência na polarização de membrana
na transmissão neurológica. O carbonato de lítio é administrado por via oral no
tratamento de alterações de humor, particularmente na psicose maniacodepressiva.
O doseamento de lítio sérico é utilizada na monitorização de pacientes para avaliar
a adesão do paciente à terapia, níveis terapêuticos e avaliar estados de intoxicação
pelo fármaco. O principal objetcivo é a manutenção do níveis terapêuticos. Os
sintomas de intoxicação por lítio incluem anorexia, náuseas, vómitos, diarréia,
letargia, ataxia, sonolência, tremores, fraqueza muscular, arritmias cardíacas e
coma.
Apresenta uma semi-vida de 24 horas em adultos saudáveis. É absorvido
rápidamente no trato gastrointestinal, apresentando pico dos níveis séricos cerca de
2 a 4 horas após a absorção. Cerca de metade da dose é excretada pela urina após
6 a 12 horas. Depois disso, a excreção continua lentamente durante 10 a 14 dias.
Os diuréticos, em especial os tiazídicos, podem aumentar os níveis séricos, pois a
depleção de sódio diminui a clearance renal do lítio.
A colheita deve ser sempre realizada imediatamente antes da dose ou entre 6 a 12
horas após a última dose.
Magnésio
O magnésio é um dos catiões inorgânicos mais abundantes no organismo. É
essencial para diversos processos físico-químicos, sendo um co-factor para diversas
enzimas intracelulares. A sua concentração é maior no meio intracelular do que no
extracelular. A absorção é feita principalmente pelo intestino delgado, sendo a
distribuição em 50% nos ossos, menos de 1% no sangue, e o restante em tecidos
moles. A homeostase é mantida por excreção renal e regulada pela reabsorção
tubular.
Convém lembrar que os níveis séricos podem manter-se inalterados até que ocorra
cerca de 20% de depleção do magnésio do organismo. O doseamento sérico do
magnésio não reflecte directamente a sua concentração intracelular. Têm sido
desenvolvidos testes para o doseamento de magnésio intracelular. Actualmente, na
investigação da hipomagnesemia, podemos dispor da avaliação sérica e da
excreção urinária de 24 horas.
O quadro de depleção é mais frequente do que os de intoxicação. Os sinais clínicos
da depleção só se manifestam quando os níveis séricos se encontram muito
comprometidos, ou seja, em valores abaixo de 1mEq/L. As causas de depleção
podem ser: má absorção, desnutrição, diarréia severa, uso de sonda nasogástrica
de demora sem reposição líquida adequada, alcoolismo, pancreatite aguda,
hiperalimentação parenteral prolongada, diálise crónica, hiper- e
hipoparatiroidismo, hiperaldosteronismo, cetoacidose diabética, lactação
abundante, gestação (2º e 3º trimestres) e raros casos idiopáticos. Os sinais
clínicos de hipomagnesemia são: fraqueza, tremores, irritabilidade, delírio,
convulsões, tetania e alterações no electrocardiograma.
Geralmente, a depleção é acompanhada por hipocalcémia. Uma das indicações para
avaliação do nível sérico de magnésio é a presença de hipocalcémia ou de
hipocaliémiaa que não responde à reposição.
Níveis séricos do magnésio são considerados prognósticos na insuficiência cardíaca
congestiva e no período pós-enfarte do miocárdio, além de diversos relatos de uma
boa correlação entre níveis normais de magnésio e o sucesso nas manobras de
ressuscitação.
A associação de terapia com aminoglicosídeos e ciclosporina com hipomagnesémia
indica a sua monitorização aquando da necessidade de terapia com esses fármacos.
Como já citado, os quadros de hipermagnesémia são menos frequentes. Os
aumentos de níveis séricos de magnésio podem ocorrer nas desidratações severas,
na insuficiência renal, na insuficiência adrenocortical, na doença de Addison, em
grandes traumas teciduais, no hipotiroidismo, no lúpus eritematoso sistémico e no
mieloma múltiplo. O uso abundante de antiácidos e de enemas ricos em magnésio
também pode, mais raramente, levar ao aumento sérico. Os sinais clínicos são:
diminuição de reflexos, sonolência, arritmias e paragem cardíaca. O aumento do
nível sérico de magnésio potencializa os efeitos cardíacos da hipercaliémia.
Cerca de 40% do consumo diário de magnésio é absorvido e excretado pela urina.
O equilíbrio é mantido pela acção reguladora da reabsorção tubular.
A grande vantagem da avaliação dos níveis urinários está na avaliação da redução
dos níveis de magnésio e no acompanhamento da reposição terapêutica. O
magnésio urinário diminui antes do que o sérico. Níveis diminuídos são encontrados
nas dietas pobres em magnésio, síndromes disabsortivas e alterações da função
tubular renal. Níveis elevados geralmente estão ligados ao uso de fármacos,
especialmente diuréticos.
Marcadores Tumorais
O marcador tumoral perfeito seria aquele que fosse produzido somente por um
tecido e secretado em quantidades mensuráveis em fluidos corporais, só seria
positivo na presença de uma neoplasia maligna e deveria ser capaz de identificá-la
antes de sua expansão além do seu local de origem. Os seus níveis séricos
deveriam reflectir o tamanho do tumor, permitir caracterizar o seu tipo e
estadiamento e reflectir respostas ao tratamento e à progressão da doença. Esse
marcador tumoral perfeito ainda não existe. Se existisse, poderia ser usado como
triagem para a presença da neoplasia oculta em indivíduos assintomáticos,
permitindo o diagnóstico e o tratamento precoce.
Na prática, a maioria dos marcadores tumorais encontra-se em baixas
concentrações em indivíduos normais e em quantidades mais altas durante
processos inflamatórios e outras condições malignas e não-malignas. Por isso, o
seu papel mais importante não está no diagnóstico da neoplasia, e sim como um
co-factor, orientador e confirmatório, do diagnóstico, com um papel definido na
avaliação das recidivas, na resposta à terapia e na avaliação do prognóstico de
evolução do tumor.
Os marcadores tumorais são divididos em 5 categorias:
- Enzimas e proteínas
- Glicoproteínas
- Glicoproteínas mucinas
- Hormonas
- Moléculas do sistema imune.
Enzimas e Proteínas
A NSE (enolase neurônio-específica), na sua forma gama, está elevada nos
soros dos pacientes com neuroblastoma, carcinoma pulmonar de pequenas células,
melanoma, carcinoma de células da ilhota pancreática e hipernefroma. No
neuroblastoma, a NSE correlaciona-se com o prognóstico, mas não é útil para o
acompanhamento das recidivas. O uso primário de NSE está no carcinoma
pulmonar de pequenas células. Cerca de 70% desses pacientes apresentam níveis
altos de NSE. A NSE pode ser usada para monitorizar os efeitos da terapia e a
avaliação de recaídas antes das evidências clínicas.
Elevações da desidrogenase láctica são notáveis em quase todas as
malignidades. Os valores encontrados na neoplasia sobrepõem-se com valores em
doenças benignas. Não tem nenhum valor como um marcador tumoral de triagem,
entretanto, tem utilidade limitada na monitorização da terapia em malignidades
hematológicas. São encontrados níveis extremamente altos nos casos de leucemias
em crianças e nos casos de linfoma não-Hodgkin nos quais o tratamento fracassou.
Os níveis de ferritina podem elevar-se em neoplasias, especialmente na doença de
Hodgkin, nas leucemias agudas, nos carcinomas de mama, fígado, pulmão, cólon e
recto, em tumores da próstata e testículos e no mieloma múltiplo. São úteis na
monitorização da evolução da doença.
Os níveis de fosfatase alcalina são úteis em neoplasias para avaliar a presença de
metástases envolvendo fígado e osso. Valores muito elevados são observados em
pacientes com lesões osteoblásticas, como as encontradas no carcinoma de
próstata com metástase óssea. Aumentos menores são observados quando as
lesões são osteolíticas, como as encontradas no carcinoma metastático de mama.
Outras condições malignas com infiltração hepática como leucemias, linfomas e
sarcoma podem cursar também com elevação da fosfatase alcalina. A sua elevação
pode ocorrer também pela presença de isoformas patológicas.
Os níveis de fosfatase ácida podem estar alterados em pacientes com carcinoma
de próstata. Os que se encontram confinados dentro da cápsula normalmente
apresentam níveis normais; já nos casos em que há metástases, mais da metade
dos pacientes apresenta níveis elevados. Níveis alterados podem ser observados
em pacientes com hipertrofia benigna de próstata, retenção urinária e após
manipulação prostática. A fracção não-prostática encontra-se elevada em condições
em que existe um hipermetabolismo ósseo, como nas metástases ósseas no
carcinoma da mama, pulmão, tiróide, mielomas e em situações de grande
destruição de eritrócitos e de plaquetas em patologias hematológicas malignas.
Glicoproteínas
As glicoproteínas são marcadores tumorais derivados de tecido fetal ou placentário,
encontrados em pequenas quantidades no tecido do adulto normal. Portanto, esses
marcadores não são específicos para nenhum tumor. Exemplos de marcadores
tumorais dessa classe são antigénio carcinoembrionário (CEA), alfafetoproteína
(AFP), gonadotrofina coriónica humana, (HCG), antigénio polipeptídio tecidual
(TPA), antigénio do carcinoma de células escamosas (SCC-A) e antigénio específico
da próstata (PSA).
O CEA foi primeiro identificado em 1965 em extratos de carcinoma de cólon
humano e em células de cólon fetais. Existe em baixos níveis na mucosa do cólon
normal, pulmão e tecido da mama, e é encontrado no soro associado com várias
malignidades. É usado especialmente na monitorização de tumores
gastrointestinais, particularmente no carcinoma colorrectal. Cerca de 63% de
pacientes com carcinoma colorretal têm elevações de CEA. Quando presente, o CEA
correlaciona-se histologicamente com a fase do tumor. Níveis pré-operatórios muito
altos são prognósticos de altas taxas de retorno e baixas taxas de sobrevivência. Se
o tumor secreta CEA, este pode ser usado para monitorizar a eficácia da remoção
cirúrgica do tumor, bem como para monitorizar a recidiva da doença.
A sua avaliação não é recomendada para screening por causa da incidência de
elevação de CEA noutras doenças inflamatórias.
Consultar Antígeno Carcinoembrionário.
A AFP é a principal glicoproteína plasmática precoce do feto humano. Encontra-se
elevada no soro fetal, no soro materno e no soro de adultos com hepatomas e
teratoblastomas testiculares.
Nem todos os hepatomas ou teratoblastomas produzem AFP, mas se sintetizam,
fazem-no em grandes quantidades. Nem sempre as elevações de AFP estão
associadas a malignidade; os níveis podem estar elevados em doenças
inflamatórias do fígado e intestino. É inútil como screening por causa das
elevações significativas em condições benignas.
Consultar Alfa-1-Fetoproteína.
A HCG é secretada através do sinciciotrofoblasto placentário. A cadeia alfa dessa
molécula compartilha a sequência homóloga com a hormona luteinizinante (LH),
mas a cadeia beta é única. A beta-HCG, é normalmente encontrada no soro e na
urina durante a gravidez. Porém, pode também estar presente em 10% dos
pacientes com doença inflamatória intestinal benigna, úlcera duodenal e cirrose
hepática. Além disso, a beta-HCG é encontrada em quase 100% dos pacientes com
tumores trofoblásticos e em 10% a 40% de tumores de células não-germinativas,
como carcinoma do pulmão, mama, trato GI e ovário. Em pacientes com tumores
trofoblásticos (células germinativas) como seminomas, teratomas e
coriocarcinomas, a beta-HCG é muito útil quer para o diagnóstico, como para a
monitorização da terapia, prevendo o aparecimento de metástases e predizendo o
fracasso do tratamento ou recidivas da doença. Quando avaliado em combinação
com AFP, torna-se particularmente útil na detecção dos seminonas.
Consultar Gonadotrofina Coriônica.
O TPA aparece no soro de pacientes com carcinoma de células escamosas da
cabeça e pescoço, pulmão e bexiga, mas também é encontrado em condições
benignas, processos cicatriciais, gravidez e doenças inflamatórias. Além disso, o
TPA pode ser encontrado em 20% das doenças benignas da mama, sendo por isso
não-específico para o diagnóstico ou a monitorização de carcinoma.
O SCC-A, subfracção do antigénio tumoral TA-4, está aumentado nos carcinomas
de células escamosas do útero, endométrio e noutros carcinomas da área genital.
TA-4 e SCC-A também estão presentes em níveis altos em tumores de células
escamosas de cabeça e pescoço, pulmão e cérvix. O SCC-A é útil na monitorização
da terapia nesses tumores, mas não para o diagnóstico.
O PSA é uma glicoproteína com actividade enzimática proteolítica que dissolve gel
seminal depois da ejaculação. O PSA é encontrado no tecido prostático normal,
benigno e maligno e no plasma seminal, e é produzido no citoplasma das células
acinares prostáticas e no epitélio ductal. Os níveis de PSA são elevados no
carcinoma da próstata. Também aparecem níveis de PSA altos na hipertrofia
benigna da próstata e nas prostatites agudas ou crónicas. Os níveis de PSA
correlacionam-se directamente com o volume da próstata, com a fase do carcinoma
e com a resposta à terapia. O carcinoma de próstata é a única forma de carcinoma
em homens nos quais o PSA é detectável no soro. Por isso, o doseamento de PSA é
recomendado, em combinação com o exame rectal digital, para a investigação do
carcinoma da próstata.
Glicoproteínas Mucinas
Glicoproteínas mucinas são antigénios de superfície celular de alto peso molecular.
Elas são compostas por 60 a 80% de carbohidratos e têm uma semelhança
estrutural com os antigénios de grupo sanguíneo Lewis A e B. As glicoproteínas
mucinas expressas na superfície epitelial incluem CA 15-3, MCA, CA 19-9 e CA 125.
O CA 15-3 é expresso durante a diferenciação mamária e é encontrado em células
mamárias lactentes, epitélio pulmonar, carcinoma de mama, ovário, pâncreas,
estômago e fígado. Podem ser encontrados níveis baixos de CA 15-3 em condições
não-malignas como hepatites crónicas, cirrose, sarcoidose, tuberculose e lúpus
eritematoso sistémico. São detectados níveis elevados de CA 15-3 em carcinomas
de mama, ovário, pâncreas, estômago e fígado.
A sua utilização está indicada no acompanhamento do carcinoma da mama,
especialmente no rastreamento da presença de metástases ósseas. Os seus níveis
diminuem em resposta à quimioterapia. Doseamentos consecutivos do CA 15-3 têm
previsto recaídas de carcinoma da mama antes da sua demonstração pelo exame
clínico.
Consultar CA 15-3.
O MCA é encontrado na maioria das células de carcinoma da mama,
independentemente do grau histológico. Os níveis são mais altos em metástases do
carcinoma da mama, do que as alterações encontradas nos níveis do CA 15-3.
O CA 19-9 é uma muciglicoproteína idêntica em estrutura com antigénio Lewis A, e
a expressão do CA 19-9 depende da expressão do antigénio Lewis. O CA 19-9 é
encontrado nas pancreatites agudas e crónicas, na doença hepática benigna, no
carcinoma do pâncreas e outras patologias malignas. A sua maior indicação está no
acompanhamento do carcinoma do pâncreas. As diminuições dos valores séricos
depois de ressecção cirúrgica demonstram que estas foram eficazes, e a avaliação
periódica prevê a recorrência 3 a 9 meses antes dos sintomas clínicos aparecerem.
Consultar CA 19-9.
O CA 125 é uma muciglicoproteína grande com baixo teor de carbohidrato que se
expressa no epitélio do cólon embrionário e é encontrada em várias doenças
benignas e malignas. A monitorização dos níveis de CA 125 é muito útil durante
tratamento do carcinoma do ovário para mulheres de todas as idades.
Consultar CA 125.
Hormonas
A calcitonina é uma hormona produzida pelas células C da tiróide e desempenha
um papel na regulação do cálcio. A calcitonina está presente em altas
concentrações na gravidez e em várias doenças benignas, como hipertiróidismo,
doença de Paget e anemia perniciosa. Além disso, a calcitonina está elevada em
neoplasias malignas específicas como carcinoma da mama, hepatoma,
hipernefroma e carcinoma do pulmão, mas está notavelmente elevada no
carcinoma medular da tiróide. Como um marcador tumoral para o carcinoma
medular da tiróide, o nível de calcitonina correlaciona-se com a gravidade da
doença e é útil para monitorizar a terapia, além de poder ser usado como triagem
nas famílias com transmissão autossómica dominante do carcinoma medular da
tiróide.
A tireoglobulina é uma glicoproteína produzida pelas células foliculares da tiróide
e é necessária para proteólise e libertação da tiroxina (T4) e da triiodotironina (T3)
na circulação. Níveis altos de tireoglobulina estão presentes em quase todas as
doenças da tiróide, sendo portanto inúteis para screening de doença benigna ou
maligna. Porém, a tireoglobulina é um marcador tumoral útil, depois de
tiróidectomia total ou radioterapia, quando níveis de tireoglobulina podem antecipar
o aparecimento de metástases.
O ácido vanil mandélico (VMA) e o ácido homovanílico (HVA) são encontrados
na urina nos casos de feocromocitoma e neuroblastoma. Os níveis pré-tratamento
correlacionam-se com a fase da doença, e as determinações consecutivas são úteis
para a monitorização da terapia.
A PTH-RP (paratormona, proteína relacionada) é secretada principalmente por
tumores que cursam com hipercalcémias malignas, como carcinoma epidermóide
do pulmão, carcinoma da mama e do córtex renal e outros tumores epiteliais.
Moléculas do sistema imune
As imunoglobulinas monoclonais (proteínas M) foram os primeiros marcadores
tumorais conhecidos. Elas são reconhecidas pela electroforese de proteína do soro
ou da urina e caracterizadas por imunofixação no soro ou na urina como
imunoglobulinas (IgG , IgA, IgM, IgD, IgE, ou cadeias leves livres k (kappa) ou l
(lambda). As proteínas M estão presentes em quase 1% dos adultos, mas cerca de
25% dessas proteínas têm significado indeterminado. Cerca de 50% dessas
proteínas M identificadas indica o diagnóstico de mieloma múltiplo.
Aproximadamente 4% dos pacientes com imunoglobulinas monoclonais têm
macroglobulinemia de Waldenström, doença maligna de linfócitos B que secretam
grandes quantidades de IgM. Quase 15% dos pacientes com proteínas M têm
doença maligna linfoproliferativa de células B, como leucemia linfocítica crónica ou
linfoma.
A beta 2-microglobulina está situada na superfície da membrana de quase todas
as células nucleadas e é libertada na circulação durante o turnover da membrana. A
B2-M ajuda a prever os fracassos de tratamento em pacientes com linfoma e
mieloma múltiplo, tendo relação com o tamanho do tumor e um valor prognóstico.
Oncogenes e produtos de genes como marcadores tumorais
A próxima geração de marcadores tumorais descoberta deverá incluir a descoberta
de mutações em oncogenes, quantificações de proteínas codificadas através desses
oncogenes, ou talvez auto-anticorpos produzidos pelas oncoproteínas na
translocação cromossómica, algumas das quais podem ser descobertas através de
técnicas de citogenética e também através de estudos usando hibridização com
sondas radioactivas, inclusive bcr/abl na leucemia mielogénica crónica, bcl-2 em
linfomas foliculares e myc em linfomas e outras leucemias
Genes supressores do tumor (TSGs) regulam o crescimento das células, parando a
sua proliferação. Mutações em TSGs conhecidas envolvidas com neoplasias incluem
inactivação do gene de Rb encontrado no retinoblastoma familiar, gene de APC na
polipose familiar do cólon, WT-1 no tumor de Willms e p53 encontrado numa
grande variedade de tumores (epiteliais, leucemia, linfoma, sarcoma e
neurogénios). Um ensaio imunofluorimétrico para quantificação da proteína p53
tem demonstrado a sua presença no carcinoma do ovário e no carcinoma da mama.
Como já citado, não há nenhum marcador tumoral perfeito, e por isso, não devem
ser usados para screening da presença de neoplasias malignas.
O PSA é actualmente o único marcador aprovado pelo FDA, em combinação com o
toque rectal para triagem do carcinoma da próstata. A AFP é apropriadamente
usada como um teste de triagem em populações de risco (chineses, japoneses e
esquimós do Alasca). A calcitonina pode ser usada como um teste de screening
para carcinoma em famílias de pacientes com carcinoma medular da tiróide.
Vários testes são eficazes no diagnóstico diferencial de tumores específicos. A AFP e
a beta-HCG são úteis no diagnóstico diferencial de tumores de células germinativas
não-seminomas, quando utilizadas na clínica de forma apropriada. O CA 125 é
usado na avaliação de massas ovarianas, mas com reservas. Embora o CA125
tenha-se mostrado elevado antes da descoberta clínica do carcinoma do ovário,
menos de 50% dos pacientes com doença inicial apresentam elevações nos níveis
de CA 125. Por outro lado, em mulheres na pré-menopausa, várias condições
benignas são associadas a elevações moderadas de CA 125. Uma combinação de
ensaios que usam CA 125, CA 15-3 e TAG72 (anticorpo monoclonal específico para
fragmento de gonadotrofina urinária) demonstrou uma especificidade de 99,9%,
para a detecção do carcinoma do ovário em estadios precoces, mas os números de
pacientes foram considerados insuficientes para extrapolar o resultado para a
população em geral.
Proteínas M detectadas por electroforese de proteína no soro não são úteis para
screening para mieloma, porque só 50% dos pacientes que apresentam proteína
monoclonal têm mieloma múltiplo. O diagnóstico, o prognóstico e a monitorização
da terapia dependem não só da descoberta de uma proteína monoclonal mas
também da caracterização do tipo de imunoglobulina. Pacientes com mieloma IgA
apresentam taxa de sobrevida significativamente reduzida e complicações mais
severas da doença do que os pacientes com mieloma IgG ou doença de cadeias
leves.
Os marcadores tumorais citados têm aplicabilidade na monitorização da progressão
da doença ou da eficácia da terapia. A frequência da monitorização não é padrão,
mas uma frequência apropriada deveria testar mensalmente no período pósoperatório, durante os primeiros 6 meses, a cada 2 meses durante mais 6 meses,
trimestralmente durante o ano seguinte e 2 vezes ao ano nos anos subsequentes.
Metahemoglobina
A hemoglobina é continuamente oxidada do estado ferroso para o estado férrico
devido ao processo dinâmico de libertação de oxigénio para os tecidos.
Diariamente, cerca de 1% da hemoglobina total é convertida espontaneamente de
oxihemoglobina em metahemoglobina.
Em indíviduos normais, os níveis de metahemoglobina no sangue não ultrapassam
3% da hemoglobina total. Portanto, a condição clínica denominada
metahemoglobinemia é aquela em que o paciente apresenta níveis acima dos 3%
de hemoglobina oxidada para a forma férrica. Essa forma da hemoglobina é incapaz
de transportar o oxigénio. Esta característica dá ao sangue uma tonalidade
vermelha escura, devido à presença da hemoglobina desoxigenada, que em níveis
acima de 5% levam a cianose clínica.
As causas da metahemoglobinemia podem ser deficiência enzimática, tóxica ou por
anomalia molecular.
A deficiência enzimática hereditária da NAD-diaforese é descrita como
metahemoglobinemia congénita. Essa enzima participa numa etapa da via principal
da redução da hemoglobina e a sua ausência resulta no acúmulo do pigmento
pardo nas hemácias circulantes. Os heterozigóticos normalmente não apresentam
sinais clínicos da deficiência.
Entretanto, as manifestações podem ocorrer de forma intermitente, induzidas por
fármacos ou por exposição a poluição oxidante. O curso da doença é benigno,
devendo-se proteger esses pacientes de factores desencadeantes da
metaemoglobinémia, na verdade, os mesmos que afectam os indivíduos normais.
Na metahemoglobinémia tóxica, o quadro é provocado pelo uso de diferentes
fármacos, compostos químicos, derivados de benzeno e outras substâncias tóxicas.
A poluição é uma causa importante. Nestes pacientes, é possível observar os
produtos da agressão oxidativa no interior dos eritrócitos sob a forma de corpos de
Heinz.
Ambas as etiologias - metahemoglobinemia tóxica e metahemoglobinemia por
deficiência enzimática hereditária, podem ser tratadas por agentes redutores, como
a vitamina C, ou por via parenteral, pelo azul de metileno.
A metahemoglobinemia por anormalidade molecular resulta da presença de uma
hemoglobina anormal a hemoglobina M.
Geralmente, o diagnóstico das metahemoglobinemias é clínico. A sua confirmação é
realizada pela pesquisa laboratorial da presença de metahemoglobinemia.
Microalbuminúria
A albumina é excretada na urina em quantidade contínua, e encontra-se
aumentada em alguns pacientes com diabetes mellitus. Foi verificado que, nesses
pacientes, existe um período lactente entre o início da lesão renal e a doença renal
instalada. Durante essa fase, a lesão glomerular expressa-se por meio da excreção
aumentada da albumina. No entanto, essas quantidades são muito pequenas para
serem detectadas pelos testes laboratoriais padrões e mesmo nas investigações de
proteinúria nas 24 horas. Por isso, a imunoturbidimetria/nefelometria, método mais
sensível, é preferível para proceder à investigação, visto que os testes tradicionais
só são positivos em níveis de 150 a 200 mg/L.
A microalbuminúria pode ser definida como uma elevação persistente da albumina
urinária, acima de 20 µg por minuto. Aparece antes da manifestação clínica da
proteinúria, detectável pelos métodos tradicionais, e tem de ser revelado um
marcador preditivo do aumento de falência renal e mortalidade precoce dos
pacientes diabéticos, em muitos e diferentes estudos detalhados e rigorosos.
Alguns estudos relatam a presença de microalbuminúria 5 anos antes da
manifestação clínica clássica do quadro de diabetes mellitus. Outros sugerem a sua
utilização na monitorização do aparecimento da lesão renal em pacientes com
hipertensão, lúpus eritematoso sistémico e durante a gravidez nas suspeitas de
quadros de pré-eclâmpsia.
Albuminúria
<20ug por minuto
<30 mg/24h
Microalbuminúria
<20 a 200 ug por minuto
30-300 gm/24h
Macroalbuminúria
>200 ug por minuto
>300 mg/24h
A excreção de albumina é mais alta em fumadores do que em não-fumadores e
diminui 30-50% durante a noite, possivelmente devido à pressão sanguínea e a
taxas de filtração glomerular mais baixas à noite.
Devem ser doseadas, no mínimo, 3 amostras em datas variadas no período de 1
mês, e pelo menos 2 devem ser positivas para a confirmação do diagnóstico de
microalbuminúria. A colheita de múltiplas amostras torna-se necessária, visto que a
excreção pode variar de um dia para o outro. A variação biológica entre as
amostras, durante um período de 1 mês, segundo a literatura, pode ser de 20 a
40%.
São factores de interferência o exercício físico, infecção urinária, doença aguda e
qualquer outro factor que leve a resultados falso-positivos de proteína na urina.
Alguns autores sugerem que a colheita deve ser realizada à noite (urina nas 12
horas nocturnas) para que ocorra em repouso, evitando a interferência do exercício
nos valores da albumina urinária.
A presença de microalbuminúria identifica os pacientes com maior risco de
desenvolvimento de nefropatia diabética. A incidência de nefropatia diabética é de
aproximadamente 30% para pacientes com diabetes mellitus insulino-dependente
(DMID) e varia entre 25% a 60% nos pacientes com diabetes mellitus Tipo II nãoinsulino-dependente (DMNID).
Nos pacientes com DMID, a microalbuminúria predispõe ao desenvolvimento de
proteinúria persistente em 80% e grave lesão renal subsequente. Em contraste, a
maioria dos pacientes com excreção de albumina urinária menor do que 30 mg/dia
permanece saudável.
O risco de progressão para doença renal em pacientes com microalbuminúria é 20
vezes mais alto do que nos pacientes com excreção normal.
A hipertensão é o principal factor preditivo para o desenvolvimento de
microalbuminúria. Recentes estudos acompanharam pacientes que tinham
microalbuminúria, hipertensão, resistência periférica à insulina e hipertrofia renal e
cardíaca. O risco de macroalbuminúria (>300 mg/24 h) e de declínio da taxa de
filtração glomerular é de 80% em 10 anos, em comparação com os 5% de risco
desse tipo de evolução verificados nos pacientes que cursam com albuminúria
normal. Já nos pacientes diabéticos com microalbuminúria, normotensos ou em
tratamento precoce com antihipertensivos com inibidores da enzima angiotensina
convertase, observa-se um atrazo no início da macroalbuminúria e na diminuição
da taxa de filtração glomerular.
É recomendado que os pacientes diabéticos pesquisem a microalbuminúria pelo
menos uma vez por ano.
Microbiologia
O laboratório de microbiologia tem a finalidade primordial de auxiliar no diagnóstico
etiológico das doenças infecciosas. Para que isto ocorra de maneira satisfatória, a
qualidade da colheita, do armazenamento e do transporte do material clínico é
extremamente importante.
Todos os materiais devem ser obtidos antes do início da terapia antimicrobiana.
Devem ser colhidos no local onde se espera encontrar o microrganismo, e com a
menor contaminação externa possível. Deve-se colher a amostra em quantidade
suficiente, seguindo as instruções específicas para cada local. Antes de colher o
material, o local deve ser limpo com soro fisiológico, tendo o cuidado de não se
utilizarem substâncias anti-sépticas.
O material colhido, salvo instruções específicas, deve ser acondicionado em
recipiente estéril. Quanto mais precoce for a colheita, maiores são as
probabilidades de isolamento do agente etiológico. Os dado clínicos, assim como o
local e a hora da colheita, são informações importantes para o tratamento
adequado da amostra.
Para informações mais detalhadas, consultar nos títulos:
- CULTURA DE ANAERÓBICOS
- CULTURA DE FEZES
- CULTURA DE LÍQUOR
- CULTURA DE MICOBACTÉRIAS
- CULTURA DE OUTROS LÍQUIDOS BIOLÓGICOS
- CULTURA DE PONTA DE CATETER
- CULTURA DE SECREÇÕES, ABCESSOS, TECIDO SUBCUTÂNEO, FRAGMENTO
DE TECIDOS E BIÓPSIAS
- CULTURA DO APARELHO GENITOURINÁRIO
- CULTURA DO APARELHO RESPIRATÓRIO SUPERIOR
- CULTURA DO APARELHO RESPIRATÓRIO INFERIOR
- CULTURA DE URINA
- HEMOCULTURA
Mioglobina
A mioglobina é uma proteína de baixo peso molecular, encontrada nos músculos
esqueléticos e cardíacos. É uma proteína que se liga ao oxigénio, actuando como
reserva de oxigénio, o que facilita a sua movimentação dentro das células
musculares. Uma lesão celular do músculo esquelético ou cardíaco leva à libertação
de mioglobina para a circulação sanguínea.
Nos pacientes com enfarte agudo do miocárdio (EAM), podemos encontrar níveis de
mioglobina elevados já nas primeiras horas após enfarte, com pico entre as 6 e as
9 horas, retomando os níveis normais em 24 a 48 horas após o enfarte. A elevação
da mioglobina é extremamente precoce quando comparada com outras enzimas
cardíacas, devido ao seu baixo peso molecular, que permite um rápido
deslocamento para a circulação. Esta mesma característica é responsável pela sua
curta permanência em níveis alterados, já que é rapidamente filtrada e eliminada
pelos rins.
O doseamento de mioglobina é apontada como um marcador de rastreio no
diagnóstico precoce do EAM, devido à sua rápida elevação após a lesão miocárdica.
Como já mencionado, valores elevados de mioglobina podem ser encontrados
também nas lesões do músculo esquelético, não tendo portanto uma especificidade
para lesões cardíacas. Devido à sua característica de rápida eliminação renal,
resultados alterados obtidos em pacientes com patologias renais devem ser
interpretados com cuidado, pois podem ser causados pela diminuição da eliminação
renal.
Mononucleose Infecciosa
Doença infecciosa aguda causada pelo vírus Epstein-Barr (EBV), apresenta uma
maior incidência em crianças, adolescentes e adultos jovens. Durante o curso da
doença, aparecem anticorpos heterófilos, capazes de aglutinar eritrócitos de
carneiro e de cavalo, que são uma característica dessa infecção. São anticorpos da
classe IgM não-específicos para mononucleose, podendo estar presentes noutras
patologias.
O diagnóstico é feito pelo conjunto dos sinais clínicos associados a positividade para
anticorpos heterófilos. Diante de um quadro sugestivo que apresente a pesquisa
negativa para esses anticorpos, torna-se necessária a pesquisa de anticorpos
específicos para EBV, visto que mais de 70% das crianças e cerca de 10% dos
adultos não desenvolvem anticorpos heterófilos. Os achados clínicos mais
frequentes são febre, adenomegalia, linfocitose com alto grau de linfócitos atípicoas
(mais de 50%, geralmente com presença de células de Downey), esplenomegalia e
hepatomegalia.
O monoslideteste, também conhecido como reação de Paul e Bunnell, é uma
reacção de hemaglutinação que detecta a presença de anticorpos heterófilos
utilizando eritrócitos de cavalo, sendo útil como teste de triagem para
mononucleose. É sensível, e positiva logo nas primeiras semanas, mantendo-se
positivo por cerca de 12 semanas. O monoslideteste não é um exame específico
para EBV, não sendo útil na avaliação da doença crónica.
A reacção de Paul-Bunnell pesquisa a presença de anticorpos heterófilos contra
eritrócitos de carneiro. São considerados sugestivos de mononucleose títulos
superiores a 1/56. Esses anticorpos podem pertencer a outro grupo de anticorpos
heterófilos, como os encontrados na doença do soro e no soro normal (anticorpos
de Forssman). Como os anticorpos heterófilos que surgem na mononucleose
possuem a característica de serem absorvidos pelo eritrócito de boi e de não serem
absorvidos pelo rim de cobaia, a reacção de Paul-Bunnell-Davidsohn explora essa
característica, permitindo a exclusão de outros anticorpos heterófilos, servindo
dessa forma como teste confirmatório. Uma pequena percentagem de falsopositivos foi descrito em linfomas, na leucemia linfocítica aguda, na hepatite
infecciosa, no carcinoma do pâncreas, na infecção por citomegalovírus, na artrite
reumatóide e na rubéola.
A pesquisa de anticorpos para o vírus EBV torna-se necessária para a confirmação
do diagnóstico da mononucleose naqueles casos nos quais os pacientes apresentam
alterações clínicas sugestivas porém sem os achados hematológicos clássicos e os
títulos negativos para anticorpos heterófilos. São anticorpos específicos que
aparecem no final do período de incubação, atingindo títulos mais baixos durante a
fase de recuperação, que irão persistir por toda a vida como indicadores de
imunidade para essa doença.
Os anticorpos específicos para EBV devem ser pesquisados também para o
diagnóstico diferencial das patologias que podem mimetizar um quadro de
mononucleose, como pode acontecer em quadros de hepatites víricas agudas,
colagenoses, síndrome de soroconversão do HIV-1, infecção por citomegalovírus e
toxoplasmose.
Consultar Eptein-Barr, Vírus.
Mycobacterium tuberculosis
A infecção por Mycobacterium tuberculosis, o bacilo causador da tuberculose
identificado em 1882, é a causa de infecção e da doença que se desenvolvem em
pelo menos dois estadios:
Fase Activa - em que o paciente é altamente infeccioso;
Fase Lactente - fase não-contagiosa.
Para realizar o diagnóstico laboratorial, é necessário isolar o microrganismo
pertencente ao complexo Mycobacterium tuberculosis. O diagnóstico definitivo de
tuberculose no aparelho respiratório é realizado pela concentração, digestão e
descontaminação de amostras do trato respiratório (expectoração normal ou
induzida, lavado broncoalveolar ou lavado gástrico). Métodos de rotina, por meio da
utilização de culturas, requerem pelo menos 3 semanas e usualmente 4 a 6
semanas para o resultado final. A avaliação microscópica, ou bacterioscópica, é o
método mais rápido para a detecção de micobactérias, permitindo o resultado em
poucas horas. Entretanto, é pouco sensível e inespecífico, não permitindo a
identificação entre as diferentes espécies.
Estima-se que sejam necessários 5.000 a 10.000 microrganismos por mililitro de
expectoração para a positividade em bacterioscopia, e que apenas 50% dos casos
diagnosticados clinicamente apresentem cultura positiva. Técnicas imunológicas e
sorológicas são limitadas devido à baixa sensibilidade e especificidade.
Outras avaliações também são úteis, tais como a análise do ácido micótico por
cromatografia líquida de alta performance, como complementar à cultura e a testes
bioquímicos. Sondas de ácidos nucléicos espécie-específicas representam um
auxílio para confirmações rápidas de resultados de culturas para as diferentes
espécies de micobactérias.
O desenvolvimento de métodos de biologia molecular, específicos e sensíveis,
permite alternativas para o diagnóstico mais rápido e seguro da tuberculose por
meio da detecção do genoma bacteriano directamente nas amostras biológicas.
As reacções de amplificação isotérmica de rRNA e a reacção em cadeia da
polimerase (PCR) - Amplicor PCR têm sido amplamente utilizadas para a detecção
de ácidos nucléicos de M. tuberculosis em amostras biológicas do aparelho
respiratório.
O princípio do teste de PCR está na execução de quatro processos principais:
preparação da amostra, amplificação do DNA-alvo, hibridização do produto
amplificado à sonda específica e detecção do produto final.
As amostras de expectoraação normal ou induzida, lavado broncoalveolar e lavado
brônquico são recomendadas para a avaliação por PCR. Outros materiais podem ser
utilizados, podendo ocorrer perda na sensibilidade do método. Resultados falsonegativos podem ocorrer por interferência de constituintes existentes nas amostras
biológicas que alterem a actividade da enzima DNA polimerase.
Resultados fidedignos dependem da colheita e de procedimentos de transporte
adequados.
A sensibilidade analítica do teste de PCR para DNA purificado é de cinco ou mais
cópias de DNA de Mycobacterium tuberculosis purificado. A sensibilidade para
células de M.tuberculosis é igual a 40 células.
Estudos comparativos demonstram a sensibilidade da PCR de 90%,
significativamente superior à bacterioscopia, que é de 67%. Esses dados
demonstram que não deve ser excluída a possibilidade de diagnóstico de
tuberculose em resultados negativos individuais, bacterioscopia ou PCR.
A possibilidade de detecção de DNA em materiais negativos em cultura
pertencentes a pacientes anteriormente positivos e com história de terapia reforça
a idéia de detecção de microrganismos não-viáveis.
Os métodos em biologia molecular para M.tuberculosis mostram grande
sensibilidade e utilidade para o diagnóstico laboratorial de tuberculose pulmonar. A
alta especificidade do método é de grande valia para diferenciar M.tuberculosis das
outras micobactérias, que não a tuberculosis, uma vez que as terapias para essas
infecções são diferentes. A PCR tem, na prática clínica, um impacto significativo
para a conducta com os pacientes supostamente infectados com M. tuberculosis.
A PCR permite o diagnóstico rápido, em 24 a 72 horas, e a administração da terapia
correta, propiciando a melhoria do paciente e reduzindo o uso de fármacos, assim
como as necessidades de isolamento e de investigações adicionais.
NTX - N - Telopeptídeo Tipo 1
O osso encontra-se em constante processo metabólico de remodelação. Isso inclui
a degradação e reabsorção ósseas, que são mediadas pelos osteoclastos, e os
processos de estruturação e formação óssea, mediados pela acção dos
osteoblastos.
A avaliação dos produtos específicos da degradação da matriz óssea fornece dados
sobre o turnover ósseo, ou seja, o índice de metabolismo ósseo. Cerca de 90% da
matriz orgânica do tecido ósseo são formados por colagénio do tipo I, uma proteína
helicoidal com ligação cruzada nos terminais N e C da molécula e que forma a
estrutura básica e a resistência do tecido ósseo.
Os N-telopeptídeos de ligação cruzada com colágeno tipo I (NTx) são um indicador
sensível da reabsorção óssea. São o produto final da actividade osteoclástica,
encontrados na urina. Níveis elevados de NTx indicam elevada reabsorção óssea. O
seu doseamento é importante na osteoporose, em mulheres na pós-menopausa, na
doença de Paget e para a monitorização de terapêutica anti-reabsortiva. Convém
lembrar que, mesmo sendo um bom marcador do metabolismo ósseo, valores
normais não afastam a existência de osteoporose nem a necessidade de
tratamento.
Paratiróide
A maioria dos indivíduos possui dois pares de glândulas paratiróides, localizadas
posteriormente aos polos superior e inferior da glândula tiróide. Elas são
responsáveis pela secreção da hormona paratiroidéia (PTH), que posteriormente, é
degradado nos tecidos periféricos, especialmente fígado e rins, produzindo os
fragmentos aminoterminal e carboxiterminal. O fragmento aminoterminal tem uma
semi-vida mais curta do que o carboxiterminal. Contudo, apenas a molécula intacta
e o fragmento aminoterminal possuem actividade biológica.
A síntese e a libertação do PTH dependem dos níveis de cálcio ionizado: a
hipercalcémia bloqueia e a hipocalcémia aumenta a secrecção de PTH. A
hipocalcémia prolongada da insuficiência renal crónica acarreta um
hiperparatiroidismo secundário.
A PTH promove a conversão de 25-hidroxivitamina D (25-OHD) em 1,25dihidroxivitamina D (1,25-dihidroxicolecalciferol). Nos rins, o metabolito
dihidroxivitamina D estimula directamente a captação intestinal de cálcio.
A PTH também promove a libertação de cálcio do osso, além de aumentar a
reabsorção e diminuir a excreção renal de cálcio. Promove ainda a excreção de AMP
cíclico, hidroxiprolina, sódio, potássio e bicarbonato, além de inibir a reabsorção de
fosfato, aumentando a fosfatúria.
O aumento dos níveis séricos de PTH causa manifestações clínicas de
hipercalcémia. Casos leves necessitam de monitorização laboratorial periódica,
tanto sérica como urinária. Casos graves são classificados como emergência
endocrinológica, com risco de vida, necessitando de intervenção médica imediata. A
secreção inadequada de PTH causa hipocalcémia, cujo diagnóstico e tratamento
precoce também são vitais.
Hiperparatiroidismo
O hiperparatiroidismo (HPT) pode ser classificado como primário, secundário ou
terciário. O HPT primário caracteriza-se por aumento de PTH, hipercalcémia,
hipofosfatémia e hipercalciúria, e pode ser causado por hiperplasias, adenoma ou
carcinoma das paratiróides.
O HPT secundário decorre geralmente de insuficiência renal crónica ou deficiência
de vitamina D (por deficiência na ingestão ou resistência à sua acção), que, devido
à hipocalcémia prolongada, gera um aumento compensatório de PTH.
Laboratorialmente, os achados mais clássicos são hipocalcémia, hiperfosfatémia e
aumento de PTH.
O HPT terciário surge no decurso de um período prolongado de HPT secundário com
o desenvolvimento de autonomia na secreção de PTH e consequente hipercalcémia.
No pseudo-hipoparatiroidismo, os baixos níveis de cálcio sérico disparam a secreção
de PTH. Contudo, nem os rins nem os ossos respondem ao estímulo, devido a
defeito nos receptores hormonais, resultando em hipocalcémia persistente, apesar
do aumento de PTH.
A PTHrP (PTH-proteína relacionada) é uma molécula semelhante à fracção
aminoterminal da PTH. É secretada por tecidos neoplásicos, como o carcinoma de
células escamosas do pulmão, gerando um pseudo-hiperparatiroidismo, com
hipercalcémia grave e níveis baixos ou indetectáveis de PTH.
Níveis de PTH elevados também ocorrem em 80 a 90% dos pacientes com o
síndrome de neoplasia endócrina múltipla familiar tipo I (MENS tipo I), em
indivíduos que apresentam adenoma ou hiperplasia das paratiróides associados a
tumores hipofisários e pancreáticos.
As manifestações clínicas de hipercalcémia são variadas e afectam principalmente o
sistema nervoso central, os rins, o coração e o aparelho gastrointestinal. São
comuns manifestações como depressão, alucinações, coma, diminuição da filtração
glomerular, poliúria, nictúria, polidipsia, nefrolitíase, fadiga, anorexia, náuseas, dor
abdominal, constipação e úlcera péptica. O aumento de enzimas pancreáticas pode
levar à pancreatite. Podem ocorrer também dores ósseas, fracturas, osteíte fibrosa
cística e precipitação de cálcio em tecidos moles como pulmões, rins, vasos
sanguíneos, articulações e córnea.
Hipoparatiroidismo
Após cirurgias de adenoma ou hiperplasia de paratiróide, em pacientes com doença
óssea avançada, podem ocorrer hipocalcémia severa e tetania. É o chamado
fenómeno do osso faminto, decorrente da rápida mineralização óssea e
consequente depleção do cálcio ionizado circulante. Essa condição é geralmente
transitória.
Inúmeros fármacos usados em quimioterapia podem alterar a função das
paratiróides, acarretando hipocalcémia, hipomagnesémia e diminuição da PTH.
O hipoparatiroidismo primário definitivo pode seguir-se à remoção cirúrgica das
paratiróides ou à auto-imunidade.
O síndrome de Di George é uma patologia congénita rara, que cursa com
deficiência de PTH, devido à aplasia das paratiróides e do timo. Geralmente, a
deficiência neonatal de PTH provoca a morte por hipocalcémia grave e infecções
persistentes.
O hipoparatiroidismo funcional com hipocalcémia é observado com frequência em
crianças, especialmente pré-termo e de baixo peso ao nascimento. Isso é atribuído
a uma imaturidade temporária ou à não-reactividade das glândulas paratiróides aos
baixos níveis de cálcio por várias horas após o nascimento.
A secreção fetal de PTH pode ser inibida in útero em resposta à hipercalcémia
materna.
O termo pseudo-hipoparatiroidismo é utilizado para casos de resistência à acção da
PTH nos tecidos alvos, cursando com hipocalcémia, hiperfosfatémia e hiperplasia de
paratiróides. Clinicamente, caracteriza-se por baixa estatura, obesidade, face
redonda e 4º e 5º metacarpianos curtos. Este síndrome é conhecido como
osteodistrofia hereditária de Albright.
O hipoparatiroidismo idiopático abrange inúmeras anomalias. O início da doença
pode ocorrer na infância precoce ou mais tardiamente. Embora tenha sido relatada
a presença de anticorpos circulantes, a etiologia ainda não está claramente
definida.
Ocorrem ainda defeitos de libertação e de atividade da PTH, devido a vários outros
factores, como a diminuição de magnésio sérico, que impede a libertação da PTH e
diminui o seu efeito hipercalcémico. Pacientes desnutridos como os alcoólicos têm
tendência a hipomagnesémia.
As manifestações clínicas dependerão da intensidade e da duração da hipocalcémia.
A hipocalcémia crónica pode ser assintomática, desenvolver sintomas precocemente
ou apresentar doenças dermatológicas, como monilíase e psoríase. Os sintomas
encontrados mais frequentemente são irritabilidade neuromuscular, tetania,
fraqueza muscular, laringoespasmo, broncoespasmo, manifestações psiquiátricas
(ansiedade, irritabilidade e demência), arritmias, hipotensão, insuficiência cardíaca
congestiva, catarata, fadiga, dores ou espasmos musculares. São comuns
calcificações ectópicas.
Paratormona (PTH) - Molécula Intacta
É útil no diagnóstico diferencial das hipercalcémias: hiperparatiroidismo primário,
hiperparatiroidismo secundário (na insuficiência renal crónica) e hipercalcémias das
doenças malignas. O doseamento de PTH deve ser incluído na rotina de
investigação de litíase renal, devido à possibilidade de diagnosticar
hiperparatiroidismo.
No hipoparatiroidismo, os níveis de cálcio encontram-se baixos, com PTH em níveis
baixos ou indetectáveis. O hipoparatiroidismo transitório ou permanente pode
ocorrer no pós-operatório de cirurgias da tiróide.
Hiperparatiroidismo
primário
Hiperparatiroidismo
secundário
Hipercalcémias
malignas
CÁLCIO SÉRICO
FÓSFORO SÉRICO
PTH
PTH-RP (Paratormona - Proteína Relacionada)
É secretada principalmente por tumores malignos. A PTH-rp tem uma estrutura
semelhante à PTH e é útil no diagnóstico diferencial do hiperparatiroidismo primário
e das hipercalcémias malignas. Não se eleva na insuficiência renal crónica.
Hiperparatiroidismo
primário
CÁLCIO SÉRICO
FÓSFORO SÉRICO
PTH
PTH - RP
Hipercalcémias
malignas
---
Osteocalcina
A osteocalcina é uma proteína sintetizada pelos osteoblastos. É encontrada na
matriz óssea, nos dentes e no sangue. Como a sua função se relaciona à ligação do
cálcio à matriz óssea, é utilizada como um marcador bioquímico da formação óssea.
A sua concentração sérica reflecte a actividade dos osteoblastos maduros e
representa uma pequena percentagem da parcela sintetizada pelos osteoblastos,
uma vez que a maior parte dessa fracção está ligada à hidroxiapatita na matriz
óssea.
Os níveis séricos variam de acordo com o sexo, a idade e o horário de colheita da
amostra. Concentrações elevadas são encontradas na infância e na puberdade, com
pico durante a puberdade e o declínio da fase adulta. Nas mulheres após a
menopausa, os níveis séricos tendem aumentar.
O doseamento da osteocalcina está indicada na investigação das doenças ósseas,
como osteoporose pós-menopausa, hiperparatiroidismo e doença de Paget, que
cursam com níveis elevados. Também se encontra aumentada em pacientes com
fracturas que necessitem de redução aberta, com inserção de pinos e nos casos de
metástases ósseas não-tratadas. Níveis diminuídos são encontrados em pacientes
com uso crónico de glicocorticóides, na osteoporose senil e no hiperparatiroidismo.
A reconhecida variação dos níveis de osteocalcina, relacionada à instabilidade após
a colheita, determina cuidados especiais na colheita e no armazenamento das
amostras.
AMPc
A maior parte do AMPc encontrado na urina provém da acção da paratormona
(PTH) nos túbulos renais. O doseamento é importante para o diagnóstico do
hiperparatiroidismo primário. Atenção especial deve ser dada à possibilidade de
resultados falso-positivos em pacientes com hipercalcémia tumoral, pelo estímulo
realizado pelo peptídeo PTH-rp. Portanto, no diagnóstico diferencial de
hipercalcémia, o seu doseamento tem muito pouco interesse.
Pesquisa de Oxiúros
A fêmea do Enterobius vermicularis põe os seus ovos na região perianal durante a
noite, e não no lúmen intestinal. Por isso, o exame de fezes de rotina é quase
sempre ineficaz (negativo). Assim, deve ser utilizado o método de pesquisa na fita
adesiva (método de Graham, swab anal para a pesquisa de oxiúros).
O material deverá ser colhido algum tempo após o paciente se ter deitado, ou pela
manhã, ao acordar, antes de qualquer higiene. Abrir a prega anal do paciente e
aplicar a parte adesiva da fita transparente na região. Em seguida, distendê-la com
a face adesiva sobre uma lámina de vidro (fornecida pelo laboratório), de maneira
que fique bem distendida, lisa e sem bolhas de ar.
Pâncreas
Insulina
A insulina é uma hormona peptídica produzida pelas células beta das ilhotas de
Langerhans do pâncreas. A sua secreção é estimulada pela hiperglicemia. Estímulos
nervosos e hormonais também controlam a sua secreção.
A sua principal função é o aumento da permeabilidade das células à glicose,
resultando na diminuição dos níveis plasmáticos. A intensidade de resposta à
insulina depende do número de receptores e da afinidade destes pela insulina.
Algumas condições podem cursar com hiperinsulinismo sem hipoglicemia, como
acontece na obesidade e noutros síndromes de resistência insulínica.
Um doseamento de insulina inapropriadamente elevado (maior que 10 mU/mL)
com hipoglicemia (glicemia menor que 40 mg/dL) sugere o diagnóstico de
insulinoma.
É bastante útil a determinação da relação insulina/glicemia, que normalmente é
menor do que 0,3. Uma relação maior que 0,3 sugere a presença de insulinoma. É
importante lembrar que, na presença de anticorpos anti-insulina, o doseamento de
insulina fica prejudicado.
As hipoglicemias podem ser classificadas em três grupos:
Tipo I
Pacientes diabéticos em tratamento com uso de insulina ou de fármacos relacionados.
...................................................................................
Tipo II
Insulioma
Jehum
Fictícia (auto-administração de insulina)
Tumores endócrinos que levam à deficiência de hormonas contra-reguladores.
Doença hepática, Indução por fármacos.
...................................................................................
Tipo III
Idiopatica
Reactiva
Hipersecreção de insulina pós alimentação
Hiprsensibilidade dos tecidos periféricos à acção de insulina
Deficiência de mecanismos contra-reguladores
Alimentar
Em consequência da gastrectomia
A hipoglicemia de jejum é definida como a presença de níveis baixos de glicose,
ocorrendo em jejum, geralmente 6 horas ou mais após a alimentação. A
hipoglicemia reactiva acontece em resposta à alimentação, num estado pósabsortivo. Geralmente, ocorre 2 a 4 horas após a alimentação. O teste de tolerância
à glicose realizado até o tempo de 5 horas é uma possibilidade, embora não
definitiva, de diagnóstico nestes casos.
Peptídio C
Após a clivagem da pró-insulina, são libertadas a insulina e uma sequência de
aminoácidos denominada peptídio C. O peptídio C é secretado em concentrações
equimolares à insulina. Não tem acção fisiológica conhecida, e o seu interesse
clínico está na avaliação da capacidade secretora das células beta pancreáticas,
especialmente quando o doseamento de insulina está prejudicado pela presença de
anticorpos endógenos.
É particularmente útil no diagnóstico da hipoglicemia fictícia, na qual o peptídio C é
indetectável e os níveis de insulina se mostram elevados.
Parasitoses Intestinais
As chamadas doenças parasitárias ainda são responsáveis por um alto índice de
morbilidade em grande parte do mundo. Apesar do grande avanço tecnológico, do
alto padrão educacional, da boa nutrição e de boas condições sanitárias, mesmo
países desenvolvidos estão sujeitos a doenças parasitárias.
Nos últimos anos, a investigação e o tratamento dessas doenças têm tido um
interesse renovado. A globalização permite uma rápida deslocação de pessoas pelo
mundo, como viajantes e migrantes de áreas endémicas. Além disso, o facto de
terem sido achados patógenios emergentes e reemergentes em pacientes
imunocomprometidos por diferentes motivos, especialmente em pacientes com
SIDA, fez com que parasitas anteriormente sem importância clínica em humanos,
como os coccídeos intestinais Isospora belli, Cryptosporidium parvum e Sarcocystis
hominis, fossem observados.
Para um diagnóstico parasitológico preciso, é importante ter em consideração
factores que condicionam as parasitoses, como o mecanismo de transmissão, a
biologia, o clima e as condições sanitárias, além da patogenia. O exame clínico é o
primeiro passo para o diagnóstico, mas o laboratório é essencial nessa definição,
estabelecendo a espécie de parasita presente no paciente.
Amebas
Flagelos
Coccídeos
Neomatódeos
Trematódeos
Cestódeos
AUMENTO DA FORMAÇÃO
PROTOZOÁRIOS
Entamoeba histolytica, Entemoeba coli, Endolimax nana,
Dientamoeba fragilis e lendomoeba butschii
Giardia lamblia, Chilomaxtix mesnilli
Cryptosporidium parvum, Isopora belli
HELMINTAS
Ascaris lumbricoides, ancilostomídeos, Enterorobius vermucularis,
Strongyloides, stercolaris, Trichuris trichiura
Schistosoma mansoni
Taenia sp., Hymenolepis diminuta, Hymenolepis nana
É no aparelho digestivo que a grande maioria dos parasitas do homem encontra o
seu hábitat adequado. Cada parasitose no entanto tem a sua peculiaridade,
dependendo da biologia do helminta ou protozoário a ser pesquisado. Por esta
razão, não existe um método único, capaz de identificar com precisão todas as
formas de parasitas.
Assim, na falta desse método onivalente, dentro dos descritos na literatura, temos
de lançar mão de diferentes métodos diagnósticos isolados electivos para um
determinado parasita, ou combinados, para poder realizar um diagnóstico mais
preciso de acordo com o parasita investigado. Os melhores resultados são obtidos
com a utilização combinada dos métodos de Hoffmann e colaboradores e Kato-Katz.
O método de Hoffmann e cols., apesar de simples sedimentação, apresenta
excelentes resultados na detecção de ovos, cistos e larvas.
Na pesquisa específica do helminta Schistosoma mansoni, o método mais eficaz é o
Kato-Katz, que permite uma avaliação da carga parasitária, graças à contagem do
número de ovos por grama de fezes. Ainda mencionando a esquistossomose, há
autores que dizem que devem ser realizados pelo menos seis exames de fezes com
resultados negativos para que seja requisitada uma biópsia rectal. Neste caso, o
material é colhido pelo médico e remetido ao laboratório para análise.
Outro método recomendado, especialmente nos casos de pacientes com eosinofilia
muito alta, é o Baermann-Moraes, específico para a pesquisa de formas larvares de
nematódeos, principalmente o Strongyloides stercoralis, que é eliminado nas fezes
na sua forma larvar.
Para a pesquisa de helmintas como a Taenia sp., que elimina proglotes,
recomenda-se a tamização (simples peneiração das fezes) ou a identificação de
vermes, em que será feita a identificação dos proglotes de Taenia sp. e de vermes
adultos de outros helmintas.
Em crianças, a literatura relata um alto índice de contaminação com Enterobius
vermicularis, cuja fêmea realiza a oviposição durante a noite, na região perianal.
Nesses casos, o exame parasitológico costuma apresentar-se negativo, e a técnica
indicada é a fita gomada transparente, aderida a uma lámina, chamada de método
de Graham.
A investigação da presença de helmintas nas fezes é realizada pela pesquisa de
ovos ou larvas. Já as infecções por protozoários são diagnosticadas quando se
encontram trofozoítos, cistos ou oocistos nas fezes.
O exame parasitológico mais simples é o que permite a detecção de ovos e larvas
de helmintas e cistos de protozoários nas fezes frescas. A eliminação intermitente
de formas de resistência, a intensidade dos parasitas e o exame que utiliza apenas
uma pequena amostra do material oferecido são alguns dos factores que interferem
na positividade do exame. Isto deve-se ao facto de as técnicas existentes
possuírem em geral óptima especificidade, embora a sensibilidade só se mostre
adequada se forem solicitados exames em pelo menos três amostras de fezes de
dias distintos.
AUMENTO DA FORMAÇÃO
Parasitas
Intestinais
Ascaris
lunbricoides
Trichuris
trichiura
Ancilostomídeos
Schistosoma
Hoffmann
Identificação
Safranina
Kato- Biópsia BaermannTamização de vermes Graham MIF Metatoxilina azul de
Katz
retal
Moraes
adultos
metileno
X
X
Ñ
Ñ
X
Ñ
X
Ñ
Ñ
X
X
Ñ
Ñ
X
Ñ
X
Ñ
Ñ
Ñ
Ñ
X
Ñ
X
Ñ
Ñ
Ñ
X
Ñ
Ñ
Ñ
X
X
X
Ñ
mansoni
Enterobios
vermucularis
Strongyloides
stercopalis
Taenia sp.
Girardia
lablia
Entemoeba
histolytica
Entemoeba
coli
Endolimax
nana
Iodamoemba
butschii
Chilomastix
mesnilli
Cryptosporidium parvum
Isospora beli
Ñ
Ñ
Ñ
X
Ñ
Ñ
X
X
Ñ
Ñ
Ñ
X
X
Ñ
Ñ
X
X
Ñ
Ñ
Ñ
X
Ñ
Ñ
Ñ
Ñ
Ñ
Ñ
X
X
Ñ
X
Ñ
Ñ
Ñ
Ñ
Ñ
Ñ
X
X
Ñ
X
Ñ
Ñ
Ñ
Ñ
Ñ
Ñ
X
X
Ñ
X
Ñ
Ñ
Ñ
Ñ
Ñ
Ñ
X
X
Ñ
X
Ñ
Ñ
Ñ
Ñ
Ñ
Ñ
X
X
Ñ
X
Ñ
Ñ
Ñ
Ñ
Ñ
Ñ
X
X
Ñ
Ñ
Ñ
Ñ
Ñ
Ñ
Ñ
Ñ
Ñ
X
Ñ
Ñ
Ñ
Ñ
Ñ
X=Eletivo / =Possível / Ñ=não-indicado
Ñ
X
A técnica do MIF (Methiolate/Iodo/Formol) é uma metodologia que possibilita o
achado de estruturas de resistência de helmintas e protozoários. As amostras de
fezes devem ser colhidas em 3 dias distintos, num recipiente contendo o
conservante MIF. Esse conservante contém formol, razão pela qual as fezes não
necessitam de conservação em frigorífico.
É também muito útil em crianças, por apresentar um alto índice de positividade
para Giardia lamblia, protozoário que tem um ciclo biológico com período sem
eliminação de quistos que pode chegar a 15 dias.
Nos quadros clínicos compatíveis com amebíase, nos quais o exame parasitológico
se apresentou negativo, a literatura recomenda que seja solicitada uma pesquisa de
trofozoítos (formas vegetativas), por intermédio de coloração das fezes diarréicas
pela hematoxilina férrica. Nesses casos, o material (fezes diarréicas) deve ser
colhido em líquido conservante, para que as formas vegetativas sejam preservadas,
visto que os trofozoítos se deterioram quase que imediatamente após a emissão.
O achado de formas de resistência de protozoários considerados comensais não
constitui uma necessidade de tratamento, mas revela que os pacientes estiveram
susceptíveis à contaminação orofecal.
Parasitas oportunistas, como os coccídeos intestinais (Cryptosporidium parvum e
Isospora belli), podem ser reconhecidos por meio de técnicas de coloração como a
safranina-azul de metileno. Nesses casos, as fezes devem ser colhidas com o
conservante formol a 10%, para que suas formas sejam preservadas.
Paternidade
A investigação de paternidade por meio da análise de DNA (material genético
presente no núcleo de todas as células), desenvolvido no início da década de 1980,
tem sido, desde então, largamente utilizada como elemento fundamental em
disputas jurídicas (herança e noutros processos frequentemente encontrados em
tribunais de família).
O teste de investigação de paternidade baseia-se no facto de que todos nós
recebemos metade do material genético (alelos) de cada um dos progenitores
biológicos. São estas informações que determinam as nossas características
biológicas. O conhecimento desse material genético no genoma humano revelou
uma série de sequências de DNA que, devido à sua abundância e variabilidade, se
transformaram em marcadores genéticos individuais (impressão digital molecular).
Os marcadores genéticos utilizados na investigação de paternidade foram definidos
a partir de estudos moleculares e populacionais, nos quais os índices de
variabilidade e de frequência foram avaliados nas raças humanas.
Bancos de dados e a análise de frequência desses marcadores foram
cuidadosamente construídos e são, até hoje, a base da estatística empregue na
análise de DNA fingerprinting em casos de disputa de paternidade, em que
alcançam níveis de confiança superiores a 99,99%.
Actualmente, dois métodos são utilizados para a investigação de paternidade:
PCR/STR e westhern blot. Este segundo método é mais antigo, porém ainda é
utilizado na complementariedade de exames realizados por PCR.
O PCR/STR, o mais utilizado no momento, tem como base a amplificação de
repetições de DNA no genoma humano denominadas STR (short tandem repeats).
As STRs são repetições de três a sete bases de DNA que se constituem em
marcadores muito polimórficos, amplamente distribuídos em todo o genoma.
Os STRs são diferenciados pelo número de cópias dessas sequências repetitivas
contidas num fragmento de DNA amplificado por PCR. Cada indivíduo tem um alelo
herdado da mãe e outro do pai. Assim, serão identificados dois tipos de repetições
para cada STR analisado. Devido à grande eficácia deste método na detecção e
distinção das diferenças no genoma humano, o poder de exclusão de paternidade é
extremamente alto (>99,999 %), o que leva, nos casos de não-exclusão, a
probabilidades extremamente elevadas de determinar a paternidade biológica.
A estimativa quantitativa destes dados é obtida com a análise de no mínimo 12
STRs. O índice de paternidade (ip), que são probabilidades estatísticas de um
suposto pai ser o pai biológico da criança em questão, comparado com um
indivíduo aleatório, é calculado a partir do resultado obtido em cada um dos 12 STR
analisados.
O método de investigação de paternidade pela técnica de STR/PCR tem como uma
das principais vantagens a utilização de uma quantidade muito menor de DNA, o
que possibilita a utilização de DNA extraído de raspagem bucal, por exemplo, sem a
necessidade de colher sangue venoso.
Perfil Lipídico
Segundo as recomendações dos últimos Concensos Internaconais, o termo
tradicional - lipidograma – deveria ser abandonado, passando a adoptar-se a
denominação de perfil lipídico, que é composto pelos doseamentos de colesterol
total (CT), triglicerídeos (TG), HDL - colesterol (HDL-C) e LDL - colesterol (LDL-C).
O doseamento de lípidos totais deveria também ser abolida, por se dispor dos
vários doseamentos específicos de cada componente isoladamente. A realização de
rotina da electroforese de lipoproteínas para a investigação do risco de doença
arterial coronariana é apontada como desnecessária. A sua indicação fica restrita à
investigação de alguns casos específicos, como as hipolipidemias, e ao diagnóstico
diferencial de hiperlipidémias dos tipos II b e III de Fredrickson.
Os Consensos recomendam que os adultos com idades acima de 20 anos realizem o
perfil lipídico. Nos casos de resultados dentro das faixas desejáveis e sem outros
factores de risco, a avaliação deve ser repetida a cada 5 anos, desde que os hábitos
de vida e as condições clínicas não se alterem. Nos casos com valores alterados a
avaliação deverá ser repetida de acordo com os graus de risco e o médico
prescritor.
Em crianças e em adolescentes, a avaliação deve ser realizada entre os 2 e os 19
anos de idade, não como rastreio, mas sim, a critério do médico, quando existirem
na família situações como doenças arteriais coronárias, cerebrovasculares ou
periféricas precoces ou parentes próximos com níveis de colesterol total igual ou
superior a 300 mg/dL ou de triglicerídeos igual ou superior a 400 mg/ dL.
No adulto, deve ser investigado na presença de pancreatite aguda, xantomatose,
obesidade ou outros factores de risco de doença coronária. Os factores de risco
coronários incluem: sexo masculino; história familiar prematura de doença
cardiovascular; fumo; hipertensão; níveis baixos de HDL-colesterol; diabetes
mellitus; doença cerebrovascular ou vascular periférica oclusiva; obesidade e
sedentarismo.
A doença arterial coronariana relaciona-se em proporção directa e duplicada com
níveis de colesterol séricos. Diferentes estudos corroboram a hipótese de que cada
1% de redução dos níveis de colesterol está associado à queda de 2% de risco de
doença arterial crónica. Outros estudos baseados em angiografias demonstram que
a diminuição de 26% dos níveis de colesterol LDL relacionou-se com uma menor
progressão da doença arterial crónica em 49% dos casos, estabilização das lesões
em 33% dos casos, regressão em 18% dos casos e com diminuição de 47% de
doenças coronárias.
Estudos clínicos e epidemiológicos têm demonstrado que o aumento das
concentrações dos níveis de triglicerídeos pode ser considerado um factor de risco
independente para aterosclerose. A dificuldade dessa avaliação deve-se às diversas
partículas ricas em triglicerídeos.
Os doseamentos de lipídios são passíveis de variações tecnicamente conhecidas.
Essas variações podem ser consideradas analíticas, quando estão relacionadas à
metodologia e aos procedimentos laboratoriais, e pré-analíticas, quando
relacionadas a factores intrínsecos, estilo de vida, uso de medicações, doenças
associadas, procedimentos de colheita e manipulação da amostra. Os factores préanalíticos são reconhecidos como os principais responsáveis pela variabilidade dos
resultados.
Para minimizar os efeitos dos factores pré-analíticos sobre os resultados dos
doseamentos de lípidos, vários cuidados devem ser tomados. O paciente deve
realizar os doseamentos no seu estado metabólico normal e estável. Para isso, pelo
menos nas 2 semanas que antecedem a colheita, a alimentação, o peso e os
exercícios não devem fugir do habitual. Caso contrário, os resultados obtidos
poderão não representar os níveis habituais do paciente. A ingestão de álcool deve
ser evitada pelo menos nas últimas 72 horas, pois interfere directamente nos
valores dos lípidos, especialmente dos triglicerídeos.
O exame não deve ser realizado antes de 8 semanas posteriores à recuperação de
traumas, cirurgias, infecções bacterianas e víricas agudas ou doenças crónicas
debilitantes. Nos casos de EAM e AVC, a amostra deve ser obtida logo nas primeiras
24 horas ou somente depois de decorridas 8 semanas da fase aguda, pois no
intervalo do 2º dia à 7ª semana, os valores normalmente encontram-se reduzidos.
Nas grávidas, os valores encontram-se habitualmente elevados. Portanto, a
avaliação só deve ser realizada 3 meses após o parto.
É muito importante avaliar sempre o uso concomitante de medicamentos.
Para uma correcta avaliação e acompanhamento dos exames que fazem parte do
perfil lipídico, a qualidade da amostra é fundamental. A colheita deve ser realizada
após 12 a 14 horas de jejum; água e medicamentos que não possam ser suspensos
podem ser ingeridos.
Períodos de jejum inferiores a 9 horas e superiores a 14 horas não são
recomendados. Nos casos de jejum inferiores a 9 horas, ocorrem uma diminuição
de 2 a 4% do LDL-C, de 1 a 4% do HDL-C, e um aumento de 2 a 4% do TG. O
doseamento isolado de colesterol não necessita de jejum, que entretanto, deve ser
solicitado, visto que os valores de referência foram obtidos com jejum de 12 horas.
Em pacientes que apresentem valores alterados em relação aos desejáveis para a
idade, recomenda-se a validação com a repetição do doseamento dentro de um
intervalo mínimo de 7 dias e máximo de 2 meses.
AMOSTRA DO
PACIENTE
BIOLÓGICO
COMPORTAMENTAL
CLÍNICO
USO DE FÁRMACOS
FONTES DE VARIAÇÃO PRÉ-ANALÍTICA
Tempo de jejum, anticoagulante, condições de armazenamento e
transporte, punção venosa, identificação do material, instruções e
preparação antes da colheita.
Intra-individual, idade, sexo, raça.
Dieta, tabagismo, exercício, obesidade, stres, consumo de álcool e de
cafeína
Doença metabólica e endócrina, doenças hepática, renal aguda, etc.
Anti-hipertensivos, imunossupressores, esteróides sexuais.
Vários autores recomendam que, para um diagnóstico definitivo, o doseamento
deve ser repetido num intervalo de 8 a 15 dias, quando encontrado um valor
alterado. Se o resultado obtido na segunda avaliação exceder os limites de variação
aceitáveis (à volta de 3 a 5% para o colesterol total, 10% para o HDL- colesterol e
até 20% para os triglicerídeos), recomenda-se uma terceira avaliação antes de se
afirmar um diagnóstico definitivo. O valor a ser considerado, será a média dos
valores mais próximos.
Recomenda-se também que os doseamentos sejam realizados num mesmo
laboratório, possibilitando assim a comparação com a diminuição da variabilidade
analítica.
DOENÇAS QUE INTERFEREM NOS VALORES DOS LIPÍDIOS
Hipertireoidismo Insuficiêncis renal crónica
Hipertireoidismo
Atresia biliar congénita
Diabetes mellitus
Síndrome nefrótico
Doenças de
Armazenamento
Lúpus eritematoso
sistémico
FÁRMACOSS QUE INTERFEREM NO DOSEAMENTO E NOS VALORES DOS LÍPIDOS
Anti-hipertensores: tiazidas, clortalidona, espironolactona,
Ácido acetil salicílico
betabloqueadores.
Imunossupressores: ciclosporina, prednisolona,
Ácido ascórbico
prednisona
Esteróides: estrógenios, progestágenos,
Amiodarona
contraceptivos orais.
Anticonvulsivantes
Alopurinol
PERFIL LIPÍDICO - VALORES DE REFERÊNCIA EM ADULTOS (>ou = 20 ANOS)
COLESTEROL TOTAL
LDL-C
HDL-C
TRIGLICERÍDEOS
Valores em mg/dL
Desejáveis
< 200
< 130
> 40
< 150
Limítreofes
200 – 239
130 - 159
150 - 199
Aumentados
> ou = 240
> ou = 160
> ou = 200
Directrizes internacionais para o tratamento de dislipidémias e prevenção da aterosclerose
PERFIL LIPÍDICO - VALORES DE REFERÊNCIA ENTRE 2 E 19 ANOS
COLESTEROL TOTAL
LDL-C
Valores em mg/dL
HDL-C
TRIGLICERÍDEOS
10 anos >ou= 40
10 a 19 anos >ou= 35
-
10 anos <ou= 100
10 a 19 anos <ou= 130
Limítreofes
170 – 199
110 - 129
10 anos >ou= 100
Aumentados
> ou = 200
> ou = 130
10 a 19 anos >ou= 130
Directrizes internacionais para o tratamento de dislipidémias e prevenção da aterosclerose
Desejáveis
< 170
< 110
Pesquisa da Células LE
A pesquisa de células LE é um teste citomorfológico, uma forma indirecta de avaliar
a presença de anticorpos antinucleares. A sua formação ocorre em duas fases
distintas. Inicialmente, acontece a interacção do núcleo com o anticorpo
antinuclear, geralmente da classe IgG. O núcleo já sensibilizado é fagocitado por
leucócitos íntegros, especialmente neutrófilos e monócitos, na presença da fracção
C1 do complemento, dando origem à célula LE.
A positividade do teste dá-se pelo aparecimento de leucócitos com inclusões
homogéneas, violáceas, amorfas, de rosetas (diversos leucócitos envolvendo
material nuclear amorfo ou ainda de corpos nucleares amorfos livres).
É um fenómeno inespecífico, que ocorre em cerca de 60 a 80% dos casos de lúpus
eritematoso sistémico, mas que pode ser encontrado noutras colagenoses e em
reacções ao uso de diversos medicamentos.
Pesquisa de Coccídeos
Os coccídeos (Cryptosporidium parvum e Isospora belli) são geralmente parasitas
oportunistas, causadores de diarréia, que tiveram sua incidência aumentada com o
advento da SIDA e com a evolução de outras patologias que cursam com o
comprometimento imunológico.
Nos pacientes imunocompetentes, geralmente evoluem para a resolução
espontânea, sem uso de medicamentos.
As fezes devem ser colhidas em recipiente com conservante - formol a 10%.
Pesquisa de Elementos Anormais das Fezes
É pesquisada a presença de eritrócitos, leucócitos e muco e é avaliado o pH. Como
os leucócitos e os eritrócitos são rapidamente destruídos no lúmen intestinal,
quando presentes, sugerem a presença de lesões intestinais mais altas, associadas
ao aumento do trânsito intestinal, ou de lesões de partes mais baixas do cólon,
como colites ulcerativas, disenterias bacilares, diverticulite e tuberculose intestinal.
A presença de leucócitos isolados sugere infecções bacterianas, colites inflamatórias
e, quando em menor quantidade, pode estar associada a lesões parasitárias.
As fezes frescas devem ser colhidas em frasco de plástico. No caso de fezes sólidas
ou pastosas, a quantidade deverá corresponder a 5 colheres plásticas fornecidas
com o frasco de colheita. Se as fezes estiverem liquefeitas, pelo menos 10 mL
deverão ser fornecidos ao laboratório para análise.
As fezes deverão ser colhidas originalmente num recipiente limpo, e a seguir
transferidas para o frasco colector. O paciente não deve estar a usar laxantes nem
ter sido submetido ao uso de contrastes radiológicos nos 3 dias anteriores à
colheita.
Durante a colheita, é importante evitar a contaminação pela urina, pois a sua
presença acelera a fermentação bacteriana, prejudicando a conservação.
Pesquisa de Gordura Fecal
A presença de gordura fecal é indicativa de má absorção de gorduras pelo intestino.
É importante como diagnóstico de triagem da esteatorréia associada a patologias
do intestino delgado, fibrose quística do pâncreas, pancreatite crónica, doenças
inflamatórias intestinais, enteropatias víricas, bacterianas e parasitárias.
Fezes frescas devem ser colhidas em frasco de plástico. No caso de fezes sólidas ou
pastosas, a quantidade deverá corresponder a 5 colheres plásticas fornecidas com o
frasco de colheita. Se as fezes estiverem liquefeitas, deverão ser fornecidos ao
laboratório pelo menos 10 mL para análise.
As fezes deverão ser colhidas originalmente num recipiente limpo e a seguir
transferidas para o frasco colector. O paciente não deve estar a usar laxantes nem
ter sido submetido ao uso de contrastes radiológicos nos 3 dias anteriores à
colheita.
Pesquisa de Larvas
Alguns helmintas eliminam formas larvares em vez de ovos, como acontece nos
casos de Strongyloides stercoralis, helminta responsável por um alto índice de
eosinofilia.
Fezes frescas devem ser colhidas em frasco de plástico. No caso de fezes sólidas ou
pastosas, a quantidade deverá corresponder a 5 colheres plásticas fornecidas com o
frasco de colheita. Se as fezes estiverem liquefeitas, pelo menos 10 mL deverão ser
fornecidos ao laboratório para análise.
As fezes deverão ser colhidas originalmente num recipiente limpo, e a seguir
transferidas para o frasco colector. O paciente não deve estar a usar laxantes nem
ter sido submetido ao uso de contrastes radiológicos nos 3 dias anteriores à
colleita.
Durante a colheita, é importante evitar a contaminação pela urina, pois a sua
presença acelera a fermentação bacteriana, prejudicando a conservação.
Pesquisa de Rotavírus
Apesar de desconhecermos as percentagens exactas da incidência de
gastroenterites causadas pelo rotavírus, sabemos que se trata do maior
responsável pelos episódios de diarréia infantil no mundo e que, de acordo com a
bibliografia, raramente se manifesta em adultos. A transmissão é feita por via fecaloral, apresentando um pico de incidência nos meses de inverno.
As fezes frescas devem ser colhidas em frasco de plástico. No caso de fezes sólidas
ou pastosas, a quantidade deverá corresponder a 5 colheres plásticas, fornecidas
com o frasco de colheita. Se as fezes estiverem liquefeitas, pelo menos 10 mL
deverão ser fornecidos ao laboratório para análise.
As fezes deverão ser colhidas originalmente num recipiente limpo, e a seguir
transferidas para o frasco colector. O paciente não deve estar a usar laxantes nem
ter sido submetido ao uso de contrastes radiológicos nos 3 dias anteriores à
colheita.
Durante a colheita, é importante evitar a contaminação pela urina, pois a sua
presença acelera a fermentação bacteriana, prejudicando a conservação.
Como é feita em crianças e lactentes, a colheita pode ser realizada directamente
das fraldas, desde que não estejam contaminadas por urina.
Pesquisa de Sangue Oculto
A pesquisa de sangue oculto nas fezes é útil para a identificação de lesões do tubo
gastrointestinal que cursam sem sangramento clínicamente visível. As causas mais
comuns são sangramentos oriundos de úlceras gástricas e duodenais, gastrite,
ulcerações medicamentosas da mucosa gastrointestinal (antiinflamatórios,
aspirina), neoplasias gástricas ou do cólon, diverticulite, colites, algumas
parasitoses, hemorragias de boca ou trato respiratório superior deglutidas.
É utilizada também no diagnóstico de anemias ferroprivas, resistente ao
tratamento, especialmente em crianças.
Para um resultado fidedigno, o exame deverá ser realizado de forma seriada em
dias diferentes, tanto para certificar a negatividade, um falso-positivo ou o carácter
crónico do sangramento.
Casos de falsos-positivos podem ocorrer pela presença de mioglobina e/ou
hemoglobina de origem de carne animal, assim como também do uso de alimentos
ricos em peroxidases. Falsos-negativos podem acontecer no uso de vitamina C e
agentes antioxidantes.
Uma pequena quantidade de sangue é fisiologicamente perdida pelo tubo digestivo,
cerca de 2,0 a 2,5 mL/dia. Os métodos de investigação têm sensibilidade para
identificar apenas valores acima de 5 mL.
Para realizar adequadamente o exame, o paciente deverá manter a dieta por 3 dias
consecutivos. A dieta deve ser isenta de carne e derivados que possam conter
hemoglobina. Deve-se evitar o excesso de clorofila (vegetais verdes) e suspender o
uso de medicamentos que contenham ferro, bismuto ou cobre, assim como aspirina
e antiinflamatórios.
Pesquisa de Trofozóitos
Os trofozoítos são formas vegetativas dos protozoários. Pesquisá-los é importante
em fezes diarréicas, com o objetivo de identificar Giardia intestinalis, Entamoeba
histolytica, Dientamoeba fragilis e outros protozoários intestinais, os quais muitas
vezes não são evidenciados pelos exames parasitológicos de rotina, que realizam a
pesquisa de formas de resistência (quistos).
Devem ser colhidas fezes diarréicas, recém-emitidas, em recipiente com líquido
conservante.
Plaquetas
As plaquetas têm forma discóide, e têm origem na fragmentação do citoplasma dos
megacariócitos na medula óssea. Têm uma importante participação na fase inicial
da hemostase, por meio dos mecanismos de adesão plaquetária à superfície
estranha, agregação das plaquetas entre si, formando o tampão plaquetário,
activação da coagulação plasmática pela exposição do factor plaquetário 3,
libertação do factor plaquetário 4, e também pela absorção de factores de
coagulação na sua atmosfera. Também é libertado o factor mitogénico plaquetário
responsável pela multiplicação acelerada das células do endotélio vascular. Em todo
o processo, estão envolvidos mecanismos como adesão e agregação paquetárias.
A avaliação das plaquetas pode ser feita de forma quantitativa, expressa em mm3,
e de modo qualitativo, pela avaliação das características analisadas no esfregaço
corado, o que permite a identificação de alterações morfológicas das plaquetas.
As alterações quantitativas podem ser tanto o aumento da quantidade de
plaquetas, chamada trombocitemia, quanto a diminuição, denominada
trombocitopenia.
As alterações qualitativas podem ser observadas em processos de produção
acelerada de plaquetas. Plaquetas de diferentes tamanhos caracterizam uma
anisocitose plaquetária, comum nos síndromes mieloproliferativos, mielodisplásicos
e disfunções plaquetárias. Plaquetas gigantes (macroplaquetas) ou com morfologia
bizarra (plaquetas dismórficas) demonstram um turnover acelerado e são
encontradas quando ocorre grande destruição periférica, como nos casos de
púrpura trombocitopénica idiopática, tromboses importantes e síndrome de Bernard
Soulier.
Um fenómeno de aglutinação de plaquetas in vitro pode ocorrer devido à presença
de anticorpos dirigidos a antigénios na membrana de plaqueta. O EDTA leva à
quelação de cálcio e expõe esses antigénios, permite a ligação aos anticorpos,
levando à aglutinação das plaquetas e a um resultado falsamente diminuído.
Porém, a colheita e a realização imediata da avaliação das plaquetas ou a
confirmação em material colhido com citrato afastam a possibilidade dessa
interferência.
Consultar Hemograma.
Pneumocystis carinii
O Pneumocystis carinii é um parasita oportunista que, em imunossuprimidos, causa
um quadro de pneumonia grave e de grande morbilidade. As evidências indicam
que a maioria das crianças entra em contacto com o parasita, desenvolvendo
diferentes níveis de reacções - de assintomáticas e subclínicas a clinicamente
moderadas. O P. carinii pode então persistir num estado lactente até uma
reactivação, em situação de baixa imunidade.
Até há pouco tempo, eram considerados população de risco crianças prematuras,
pacientes com doenças hematopoiéticas malignas, com deficiência de
imunoglobulinas, e pacientes a usarem fármacos imunossupressores. Após o
advento da epidemia da SIDA, ocorreu um aumento da prevalência da pneumonia
pelo Pneumocystis carinii, já que cerca de 60 a 85% desses pacientes desenvolve o
quadro de pneumonia pelo P.carinii, sendo considerada a infecção oportunista mais
comum nesse grupo.
Ao microscópio, três formas evolutivas do P.carinii podem ser identificadas:
trofozoítos, esporos e quistos (forma de mais fácil reconhecimento).
O diagnóstico clínico da pneumonia pelo P.carinii normalmente baseava-se nas
evidências radiológicas e na observação microscópica de material respiratório
usando diferentes técnicas de coloração. Actualmente, é possível a identificação de
trofozoítos e quistos de P.carinii em material de origem respiratória como a
expectoração espontânea ou induzida e o lavado broncoalveolar por
imunofluorescência directa, utilizando-se anticorpos monoclonais, com alto grau de
especificidade.
Porfirina
As porfirias são um grupo de patologias resultantes de distúrbios enzimáticos na
síntese do heme. Os defeitos podem ser congénitos, como erros inatos do
metabolismo, ou adquiridos, por disfunções eritrocitárias e hepáticas causadas por
patologias metabólicas ou exposição a agentes tóxicos. Das diferentes espécies de
porfirinas, apenas três têm significado clínico: as uroporfirinas, as coproporfirinas e
as protoporfirinas. O ácido deltaaminolevulínico e o porfobilinogénio são
precursores metabólicos das porfirinas. Por isso, podem ser úteis no diagnóstico
dessa patologia. Como podem apresentar-se normais nos períodos de lactência, a
sua maior indicação é a avaliação, especialmente nas fases de ataques agudos. São
muito úteis no diagnóstico da forma intermitente aguda.
Como existem diferentes enzimas envolvidas neste processo, o defeito em cada
passo dessa síntese corresponde a uma manifestação distinta da doença. Segundo
a classificação de Moore, as porfirias são divididas em agudas e não-agudas, com
base nas manifestações clínicas e no padrão de produção e excreção de porfirinas e
de seus precursores.
Porfirias agudas ou hepáticas - Porfiria intermitente aguda, coproporfirina
hereditária e porfirina variegada, que têm em comum as manifestações abdominais
e alterações neurológicas e psiquiátricas.
Porfirias não-agudas ou eritropoiéticas - Porfiria hepatocutânea, protoporfirina
hematopoiética e porfirina congénita, que têm em comum as manifestações
cutâneas, com fotossensibilidade solar.
Os sintomas são dependentes do órgão atingido e podem incluir lesões de pele,
alterações hepáticas e distúrbios do sistema nervoso central. Alguns factores
podem desencadear a doença, como o uso de medicamentos, hormonas, sulfas,
benzodiazepinas e álcool, além de dietas rígidas para emagrecimento.
Não há tratamento específico para nenhuma das diferentes formas de porfirias. O
tratamento é sintomático, e em muitos casos a única solução é o transplante
hepático.
O aumento da excreção do porfobilinogénio na urina reflecte uma crise aguda de
porfiria.
A avaliação é realizada numa amostra de urina recém-emitida, que deve ser
mantida envolta em papel de alumínio para protecção contra a luz.
Potássio
O potássio é o catião de maior concentração nos líquidos intracelulares. Apenas
cerca de 2% do potássio corpóreo é extracelular. A relação entre as concentrações
do potássio no meio intracelular e extracelular é normalmente de 38:1. A
manutenção dessa relação é muito importante para o funcionamento
neuromuscular normal. Cerca de 90% do potássio ingerido é absorvido pelo trato
gastrointestinal, sendo 80% excretados pelos rins, e o restante, pelas fezes. A
quantidade excretada pela fezes aumenta como compensação, em situações
clínicas de insuficiência renal. Os níveis séricos variam em ritmo circadiano, com
maiores concentrações pela manhã entre as 8 e as 9 horas, e menores entre as 15
e as 16 horas.
Existe uma pequena variação entre os níveis séricos e plasmáticos, sendo os níveis
séricos cerca de 0,4 mEq/L maiores do que os níveis plasmáticos. Essa diferença é
atribuída em parte à libertação de potássio pelas plaquetas durante a coagulação.
Nos casos de pacientes com contagem de leucócitos e/ou plaquetas elevadas, essa
diferença pode ser maior. Os cuidados durante a colheita são fundamentais,
podendo levar a variações de até 20% dos valores, caso haja demora no uso do
garrote ou no exercício de abrir e fechar a mão prolongado, ou ainda a ocorrência
de hemólise.
A deficiência de potássio (hipocaliémia) prejudica a função neuromuscular, tendo
como sinais clínicos fadiga, mialgia e fraqueza muscular, especialmente nos
membros inferiores; labilidade emocional, fraqueza muscular progressiva,
hipoventilação, íleo paralítico, hiporreflexia, paralisia, alterações no
electrocardiograma, taquicardia com alterações na onda T (achatamento ou
inversão), depressão do segmento ST e, nos casos mais graves, prolongamento do
intervalo PR, arritmias ventriculares e paragem cardíaca podem ser evidenciados.
Os riscos são maiores nos pacientes com isquemia miocárdica ou hipertrofia
ventricular esquerda. A hipocaliémia predispõe à intoxicação digitálica.
O déficite de potássio sérico pode ser causado por uma redistribuição do potássio
entre os meios intra- e extracelular e por déficite verdadeiro de potássio. As causas
mais comuns de alteração de distribuição de potássio são resposta à
insulinoterapia, alcalose, grandes leucocitoses, excesso de beta-adrenérgico e
hipotermia. As causas de perda real de potássio podem ser subdivididas em causa
com perda renal e sem perda renal. Entre as hipocaliémias com perda renal estão
acidose tubular renal, necrose tubular aguda, fármacos, hipomagnesémia, uso de
mineralocorticóides, doença de Cushing, aldosteronismo primário e secundário, uso
de corticóides, diuréticos e outros fármacos, como anfotericina B e teofilina em
altas doses. As hipocaliémias sem perda renal incluem ingestão inadequada,
alcoolismo, má absorção, hidratação e alimentação parenterais inadequadas,
perdas gastrointestinais por vómitos, diarréia, laxantes, fístulas e sonda
nasogástrica prolongada, queimaduras e sudorese excessiva. Na cetoacidose
diabética, há perda de potássio por diurese osmótica causada pela hiperglicemia e
pelo desvio para o meio intracelular pela insulinoterapia. Porém, essas alterações
são mascaradas, no início do quadro, pela desidratação.
É importante lembrar que a hipercaliémia é muito mais importante clinicamente na
presença de pH alcalino do que ácido. Para cada aumento do pH de 0,1, o potássio
diminui 0,6 mmoL.
A hipercaliémia leva a uma despolarização da membrana celular, com sinais clínicos
como confusão mental, fraqueza, paralisia flácida, hipoventilação por fraqueza da
musculatura respiratória e irritabilidade muscular. No electrocardiograma, podemse observar bradicardia, alterações na onda T (apiculada), desaparecimento das
ondas P, aumento dos intervalos PR e QRS, fibrilação ventricular ou assistolia.
Esta condição é mais frequente em pacientes hospitalizados, especialmente os
idosos. Alguns estudos apontam a incidência de 15% de hipercaliémia numa
investigação de um grupo de pacientes internados acima de 70 anos de idade.
Reflecte geralmente excreção renal inadequada, mobilização do potássio dos
tecidos, excesso de aporte oral ou de administração parenteral. A insuficiência renal
é a causa mais comum de hipercalemia. Outras causas são grandes traumas,
neoplasias malignas, anemias hemolíticas graves, 50% dos casos de doença de
Addison e uso de antiinflamatórios, especialmente nos renais crónicos. A
desidratação pode causar uma pseudo- hipercaliémia .
Portanto, a avaliação dos níveis séricos de potássio é útil na avaliação do equilíbrio
electrolítico, especialmente em pacientes idosos, em hiperalimentação parenteral,
em uso de diuréticos, nas doenças renais e nos pacientes em hemodiálise. É
importante fazer a avaliação também nos quadros de arritmias cardíacas, fraquezas
musculares, encefalopatias de origem hepática, monitorização da cetoacidose na
diabetes mellitus e na reposição intravenosa de líquido.
Em condições normais, os rins excretam entre 80 a 90% do potássio oriundo da
dieta, variando com a alimentação. Os diuréticos são a causa mais frequente do
aumento da perda de potássio por via urinária. Outras causas de perdas renais são
as secundárias à acidose tubular renal, à alcalose metabólica e ao excesso de
mineralocorticóides. Em pacientes com hipocaliémia, perdas urinárias superiores a
20 mEq/L sugerem que os rins são a fonte de perda ou uma causa aguda para
hipocaliémia, já que podem demorar de 1 a 3 semanas para conseguir manter de
forma efetiva a concentração de potássio. Caso, nessa mesma situação, os níveis
urinários de potássio sejam inferiores a 20 mEq/L , o mais provável é que não
sejam os rins a maior fonte de perda; nessas condições, a deplecção já deve durar
semanas.
As condições associadas à diminuição dos níveis de potássio na urina incluem
perdas gastrointestinais, distribuição entre os meio extra e intracelular, ingestão
inadequada de potássio e doença renal com oligúria. Níveis elevados de potássio na
urina podem ser encontrados no aldosteronismo primário e secundário, no uso de
ACTH e corticóides, na doença tubular renal e na síndrome de Cushing.
Prolactina
A sua função é o início e a manutenção da lactação pós-parto. Ao contrário de
outras hormonas hipofisárias, a prolactina é regulada mais pela inibição do que pelo
estímulo. O factor de inibição da prolactina é a dopamina, um neurotransmissor
hipotalámico que controla a sua síntese por meio da inibição da secreção. A
prolactina é secretada episodicamente, com níveis mais elevados durante o sono.
Os seus níveis elevam-se durante a gravidez.
A lactação ocorre quando os níveis elevados de estrogénio da gestação começam a
diminuir. Durante a amamentação, os níveis retornam gradualmente aos níveis prégravídicos.
Hipoprolactinemia
Ocorre no pan-hipopituitarismo. Clinicamente, os níveis baixos de prolactina
manifestam-se pela impossibilidade de amamentar no pós-parto.
Hiperprolactinemia
O adenoma hipersecretor de prolactina (prolactinoma) é a disfunção mais frequente
da hipófise anterior. Os tumores podem ser microadenomas ou macroadenomas, de
crescimento lento ou rápido, secretores apenas de prolactina ou de prolactina e GH.
A hipersecreção de prolactina pode ser consequência da ausência de inibição pela
dopamina, como nos casos de craniofaringioma, compressão da haste hipofisária
por um adenoma ou formação granulomatosa. Nestas condições, o transporte da
dopamina secretada pelo hipotálamo por meio da haste hipofisária está
prejudicado, o que diminui sua concentração na hipófise anterior.
Alguns fármacos bloqueiam a ação da dopamina, como as fenotiazidas, enquanto
fármacos como a metildopa impedem a sua síntese. A hiperprolactinemia pode
ocorrer ainda como consequência do efeito estimulador do estrogénio e do TRH,
respectivamente em casos de terapia com estrogénios ou hipotiroidismo primário.
Stress, sela vazia, doença hepática grave, ovários poliquísticos, doença de Cushing,
insuficiência renal crónica, estímulo mamário, trauma da parede torácica, fármacos
antidepressivos, anti-hipertensivos e metoclopramida também acarretam elevação
da prolactina. Há ainda a hiperprolactinemia idiopática.
As manifestações clínicas da hiperprolactinemia são galactorréia não relacionada ao
pós-parto ou a amamentação, disfunção eréctil em homens e alterações menstruais
em mulheres.
O TAC do cránio ou a ressonância magnética são necessários na complementação
do diagnóstico.
Níveis basais de prolactina superiores a 200 ng/mL sugerem fortemente adenoma,
embora níveis pouco elevados possam ser encontrados.
Grandes tumores com níveis baixos de prolactina podem indicar compressão da
haste hipofisária, com bloqueio da inibição dopaminérgica, em vez de adenoma
hipersecretor de prolactina.
Doseamento de Prolactina
Deve ser colhida, de preferência, na forma de pool de 2 a 3 amostras e, quando
possível, com o paciente em repouso. Útil no diagnóstico de galactorréia, alterações
menstruais, infertilidade e hipogonadismo em mulheres, bem como na avaliação de
ginecomastia, impotência, infertilidade e hipogonadismo masculino. Importante na
avaliação da reserva hipofisária em ambos os sexos.
Macroprolactinémia
Várias isoformas de prolactina com diferentes actividades biológicas podem ser
identificadas no soro. A forma monomérica corresponde à maior parte da prolactina
doseada em indivíduos normais.
A pesquisa de macroprolactina realizada por cromatografia é importante para
definir, dentro da população hiperprolactinémica, os casos consequentes à presença
de agregados moleculares como a big-big prolactin. Estes compostos possuem
pouca actividade biológica, o que justifica a hiperprolactinemia oligossintomática ou
assintomática, que na grande maioria não necessita de tratamento.
O doseamento de prolactina por diferentes metodologias numa amostra de soro de
paciente com macroprolactina positiva pode fornecer resultados bastante
diferentes, uma vez que alguns métodos detectam todas as isoformas, enquanto
outros não.
Proteína C Reativa
As reacções inflamatórias são uma resposta do organismo aos diferentes tipos de
agressão tecidual, como as infecções víricas, bacterianas, parasitárias ou fúngicas,
e às agressões físicas, químicas e imunológicas.
Diante de uma reacção inflamatória, os monócitos secretam substâncias como IL-6,
IL-1 e TNF, que levam ao hepatócito a informação da necessidade de síntese das
denominadas proteínas de fase aguda. A determinação da concentração plasmática
dessas proteínas ajuda, clinicamente, a avaliar a presença, a extensão e a
actividade do processo inflamatório e a monitorizar a evolução e a resposta
terapêutica.
A proteína C reactiva (PCR) é uma das proteínas plasmáticas de fase aguda. Foi
identificada no soro de pacientes com pneumonia pneumocócica, em reacção de
precipitação do polissacarídeo C da parede do pneumococo (Gram-positivo), agente
etiológico da patologia. É usada rotineiramente para monitorizar a resposta de fase
aguda, sendo considerada uma das mais sensíveis, por apresentar algumas
características, como semi-vida curta (entre 8 a 12 horas) e valores normais muito
baixos (< 0,5 mg/dL), que em resposta a estímulos inflamatórios, podem atingir
valores até 100 vezes o normal em menos de 24 horas. Além de se elevar
rapidamente após o estímulo inflamatório (4 a 6 horas), na ausência de estímulo
crónico, normaliza-se em 3 a 4 dias.
A proteína C reactiva deve ser utilizada como método auxiliar no diagnóstico,
controle terapêutico e acompanhamento de diversas patologias, uma vez que é o
mais sensível e precoce indicador de processos inflamatórios resultantes de
infecções, carcinomas, necrose tecidual e cirurgias. Depois de 24 horas, a
velocidade de hemossedimentação (VS) é complementar à PCR.
Durante muitos anos, a PCR foi utilizada apenas no contexto de avaliação de
processos inflamatórios, mas actualmente, tem assumido outros papéis importantes
na clínica. Por exemplo, podemos citar que, como marcador de mortalidade nos
primeiros 24 meses após enfarte agudo do miocárdio (EAM), foi mais valiosa que as
enzimas cardíacas. Além disso, altos níveis séricos de PCR puderam ser
considerados factores preditivos de ruptura cardíaca subaguda pós-EAM. Nove
pacientes que apresentavam este tipo de complicação foram comparados ao grupo
controle de 28 pacientes enfartados sem complicações. No grupo com ruptura
cardíaca, níveis elevados de PCR, superiores a 20 mg/dL, foram evidenciados no
segundo dia pós-EAM. Um marcador tradicional de lesão muscular, a enzima CK,
não mostrou diferença significativa entre os dois grupos. A sensibilidade diagnóstica
de altos níveis séricos de PCR, predizem uma possível ruptura cardíaca pós-EAM, foi
de 89%, garantindo o seu uso na prática cardíaca.
Com a recente descoberta de componentes inflamatórios na arteriosclerose, a PCR
foi proposta como indicador de risco para doença coronária e acidentes vasculares
cerebrais. O risco revelado pelos altos nivéis séricos de PCR, é independente de
factores ligados à dislipidémia e pode ser reduzido pelo uso de aspirina como
tratamento profilático. Estas novas hipóteses de doenças arterioescleróticas
associadas à possibilidade de terapêutica preventiva abrem novas perspectivas, que
devem ser consideradas. Da mesma forma, níveis aumentados de PCR parecem
estar relacionados a eventos coronários em pacientes com angina estável ou
instável.
Noutras áreas da medicina, como a infecciologia e a cirurgia, a importância da PCR
também deve ser levada em conta. O uso do doseamento da PCR foi avaliado num
estudo envolvendo 193 casos de endocardite infecciosa. Níveis elevados de PCR
puderam ser evidenciados em casos que cursavam com complicações, levando à
conclusão de que a PCR é um bom marcador prognóstico e pode ser utilizada para
monitorizar a resposta à terapia antimicrobiana em endocardites infecciosas.
Na recuperação cirúrgica, a PCR aumenta nas primeiras 4 a 6 horas, revelando
picos séricos por volta das 48 a 72 horas pós-operatório, em concentrações de 2,5
a 3,5 mg/dL. Em cirurgias que cursam com evolução favorável, os níveis de PCR
normalizam-se por volta do sétimo dia após o procedimento cirúrgico. Se existirem
complicações, os valores da PCR permanecem elevados, e podem atingir níveis
superiores a 3,5 mg/dL.
Em estados inflamatórios crónicos, as concentrações de PCR podem persistir altas
indefinidamente. Em casos de LES e outras doenças do colagénio, colites
ulcerativas e leucemias, as concentrações são geralmente normais, porém
marcadamente mais altas nas infecções bacterianas do que nas víricas, auxiliando
no diagnóstico diferencial.
Na febre reumática, a PCR é um bom parâmetro de reagudização, pois persiste em
concentrações elevadas (>4 mg/dL) quando a doença está activa, embora decaindo
a níveis normais durante a remissão.
Encontram-se níveis elevados de PCR e de haptoglobina em pacientes com
espondilite anquilosante HLA-B27 clinicamente activa; mas nem a PCR nem a
haptoglobulina estão elevadas nos casos activos com HLA-B27, em que são
encontradas concentrações elevadas de IgA.
Aproximadamente 60% de recém-nascidos saudáveis podem apresentar,
normalmente, concentrações de PCR acima de 1 mg/dL durante os primeiros 20
dias de vida. Consequentemente, as faixas de referência de adultos não são
adequadas para crianças.
O doseamento pela técnica de nefelometria, ou imunoturbidimetria, utilizando
anticorpos monoclonais anti-PCR, ao contrário dos métodos tradicionais
qualitativos, origina resultados quantitativos (mg/dL) que facilitam a interpretação
clínica e o acompanhamento laboratorial de cada caso.
Proteína Bence Jones
A proteína de Bence Jones é formada por dímeros de cadeias leves (kappa e
lambda) de imunoglobulinas monoclonais encontradas na urina. A presença ou a
ausência da proteína de Bence Jones na urina depende da taxa e da quantidade de
síntese de cadeias leves e do estado renal do paciente.
É encontrada em 60 a 70% de pacientes com mieloma múltiplo, macroglobulinemia
de Waldenstrom (30%), em 20% de doenças linfoproliferativas e também em
aproximadamente 10% das gamapatias monoclonais benignas. Pode estar presente
também na amiloidose sistémica.
Aproximadamente 20% dos mielomas múltiplos secretam apenas cadeias leves
livres (proteína de Bence Jones), e cerca de 50% dos pacientes com mieloma tem
imunoglobulinas monoclonais no soro e cadeias leves livres na urina. A
monitorização da proteína de Bence Jones é útil na avaliação de recidivas da
doença, na transição para uma forma mais agressiva e na resposta à terapia. A
presença e a quantidade de proteína de Bence Jones influenciam o prognóstico.
Os critérios do National Cancer Institute para a avaliação de mielomas apontam
como resposta objectiva uma diminuição de 50% da concentração de
imunoglobulinas monoclonais no soro ou da excreção de cadeias leves na urina de
24 horas. The Southwest Oncology Group define como resposta objetiva a
diminuição de 75% de imunoglobulinas monoclonais no soro (para níveis menores
que 25 g/L) e diminuição de 90% de cadeias leves monoclonais na urina. As
respostas devem manter-se pelo menos por 4 semanas e devem ser acompanhadas
por doseamentos normais de cálcio sérico, valores de albumina sérica equivalentes
a 30 g/L e nenhuma progressão de doença óssea. Pacientes com diminuição de
50% a 74% da produção de imunoglobulinas monoclonais têm bom prognóstico.
A proteinúria de Bence Jones tem um efeito nefrotóxico. Assim, a sua presença
aumenta o risco de insuficiência renal. Pacientes com mieloma que secretam só
proteína de Bence Jones têm pior prognóstico. Mieloma de cadeias lambda têm
prognóstico pior do que os de cadeias kappa.
Proteína S
Os estados de deficiência de proteína S assemelham-se clinicamente aos da
deficiência de proteína C.
A deficiência, quando homozigótica, manifesta-se na fase neonatal como púrpura
fulminante e coagulação intravascular disseminada. Quando heterozigótica, têm
sido descritos episódios tromboembólicos venosos e de trombose vascular cerebral,
especialmente quando associados a outros factores de risco.
A proteína S é também dependente de vitamina K, sendo sintetizada pelo fígado e,
em menor quantidade, pelas células endoteliais e por megacariócitos.
Duas formas da proteína S estão presentes na circulação: a livre e a conjugada. A
porção livre corresponde a aproximadamente 40% da concentração plasmática da
proteína e representa a forma funcionalmente activa, enquanto a outra (60%)
circula conjugada à proteína ligadora do C4b, um componente do sistema
complemento.
Proteínas Totais e Fracções
O plasma humano contém diversas proteínas identificáveis, que representam um
papel importante na manutenção da pressão osmótica e em diferentes funções
como proteínas transportadoras, anticorpos, enzimas, inibidores enzimáticos,
factores da coagulação, entre outras. A avaliação dos seus níveis séricos é de
grande utilidade na avaliação do estado nutricional e da presença de doenças
sistémicas agudas ou crónicas.
O doseamento isolado de proteínas totais tem pouco valor, já que a alteração numa
das fracções pode ser compensada por alteração oposta de outra fracção, como
ocorre nas inflamações crónicas, em que há diminuição de albumina com aumento
de gamaglobulina. Geralmente, o valor isolado da proteína total tem utilidade
médica em grandes elevações como no mieloma múltiplo ou na diminuição
acentuada dos seus níveis, como os que são encontrados nos estados graves de
desnutrição, perdas como no síndrome nefrótico e enteropatias com perdas de
proteínas, ou na alteração da síntese protéica, que ocorre nas doenças hepáticas
graves.
O doseamento das fracções albumina e gamaglobulina e a avaliação da relação
albumina/globulina auxiliam na orientação diagnóstica em alterações sistémicas
com diminuição da albumina, como estados carenciais, perdas renais, distúrbios
intestinais e hepáticos e quadros de aumento das imunoglobulinas, como as
gamapatias e os processos infecciosos crónicos.
Consultar Albumina
Eletroforese de proteínas.
Proteinúria
Pequenas quantidades de proteína oriundas do plasma e do trato urinário podem
ser encontradas na urina normal.
Falsas proteinúrias podem acontecer pela contaminação da urina com sangue ou
secreções genitais. A proteinúria funcional pode surgir, geralmente com valores
pouco elevados, num quadro febril importante, exercício vigoroso, posição
ortostática, desidratação e uso de drogas, entre outras situações.
A proteinúria é um indicador importante da presença de patologia renal e pode
resultar de lesão glomerular, comprometimento tubular, degradação do tecido renal
ou concentrações excessivas de proteínas de baixo peso molecular (gamapatias
monoclonais).
Consultar Urina-EAS.
Eletroforese de Proteínas.
PSA
A incidência mundial do carcinoma da próstata, é ascendente. As estáticas
demomonstram que o carcinoma da próstata é hoje, a segunda maior causa de
morte por carcinoma nos homens. Esta tendência deve manter-se, devido ao
aumento da esperança de vida da população.
A epidemiologia do carcinoma da próstata é complexa, e poucos factores de risco
foram estabelecidos. Alguns estudos documentaram como factores de risco:
- Idade,
- Raça e etnia,
- História familiar,
- Dieta,
- Produção de androgénio e outros factores, como vasectomia e hiperplasia benigna
da próstata.
Idade
O carcinoma da próstata é particularmente comum em homens idosos. É
clinicamente insuspeito, frequentemente detectado em autópsias. A sua prevalência
é cerca de 40% em homens acima dos 70 anos de idade. Desconhece-se a
participação de algum mecanismo fisiológico concomitantemente ao
envelhecimento, necessário ao desenvolvimento e à progressão do carcinoma da
próstata, ou se esses processos são tão lentos que requerem muitos anos para se
tornarem clinicamente evidentes. A elevação da incidência de carcinoma da
próstata em relação à idade conduziu à especulação de que os processos de
envelhecimento podem estar relacionados biologicamente com o desenvolvimento
do carcinoma da próstata.
Raça e Etnia
Foram observadas grandes variações na incidência e na mortalidade de carcinoma
da próstata entre países e grupos raciais. Americanos e africanos têm as taxas de
mortalidade mais altas do mundo, seguidos por brancos de países escandinavos.
Homens asiáticos têm as taxas mais baixas. As grandes diferenças raciais na
incidência de carcinoma da próstata e mortalidade fornecem dados importantes na
etiologia da doença.
História familiar
Estudos epidemiológicos demonstraram que pacientes que relatam história de
carcinoma da próstata em pais, irmãos ou filhos têm duas a três vezes aumentado
o risco de desenvolver a doença, com estimativas de risco mais altas associadas a
múltiplos parentes afetados ou em idades jovens.
Algumas análises apoiam a possibilidade de factores genéticos herdados como
explicação desse fenómeno. Porém, não foram ainda obtidos dados decisivos da
possível herança de genes no cromossomo X. Vários estudos populacionais
apontam um risco maior de carcinoma da próstata em homens com irmãos
afetados quando comparados com pais afetados, aumentando a hipótese de uma
herança ligada ao cromossomo X.
Dieta
A modificação do aporte dietético é, potencialmente, um método promissor de
controle de carcinoma da próstata. A possibilidade de que a dieta tenha um papel
importante na etiologia da doença é apoiada por dados epidemiológicos, tais como:
- a incidência de carcinoma da próstata tem aumentado no Japão, onde mudanças
dietéticas significativas aconteceram nas últimas três décadas;
- a incidência da doença é mais elevada em japoneses e imigrantes chineses nos
Estados Unidos;
- a incidência de carcinoma da próstata está correlacionada com outros carcinomas
relacionados com a dieta, como carcinoma do cólon.
O componente dietético principal associado ao carcinoma da próstata é a gordura.
Dados epidemiológicos globais são consistentes, mostrando a associação com
gordura dietética, especialmente a gordura saturada, embora alguns estudos não
apoiem essa relação.
Foram apontados vários mecanismos para o efeito da gordura no desenvolvimento
do carcinoma da próstata, inclusive efeitos na composição de membrana celular,
síntese de prostaglandina e níveis de andrógenios no sangue ou tecido. Porém,
esses mecanismos são especulativos, e faltam provas críticas e dados
experimentais.
A relação de risco de carcinoma da próstata para vários micronutrientes é, ainda,
incerta. O papel da vitamina A ou dos seus precursores, particularmente o
betacaroteno, é obscuro. Alguns estudos mostram uma associação negativa, e
outros mostram não haver associação ou existir até mesmo uma associação
positiva. Recentes estudos sugerem efeitos protectores no uso de selénio e
vitaminas D e E. O selénio está envolvido na biossíntese da testosterona e pode
estimular a produção de hormonas pituitárias e adrenais. Está envolvido, também,
em vias metabólicas antioxidantes, e a sua deficiência pode expor as células
prostáticas a tensões oxidativas que podem ser carcinogénicas.
Androgénios
Os androgénios têm um papel no desenvolvimento do carcinoma da próstata. São
hormonas necessárias ao crescimento, à manutenção e à actividade funcional das
células prostáticas. A castração ou a terapia com estrogénio podem ter um efeito
paliativo no carcinoma da próstata. Porém, os resultados de estudos
epidemiológicos da avaliação de níveis séricos de androgénio foram incompatíveis.
Essa inconsistência pode reflectir a fraca correlação entre níveis circulantes de
androgénio e níveis intracelulares. Pode reflectir também, a fase imprópria para
doseamento do androgénio. A maioria dos doseamentos é realizada em pacientes
idosos, porém os níveis de androgénio são de grande valor na adolescência ou no
adulto jovem.
Outros Factores
Uma gama extensa de outras exposições exógenas e características do hospedeiro
foi apontada no papel etiológico do desenvolvimento do carcinoma da próstata,
inclusive peso ao nascer, obesidade, altura, hiperplasia benigna de próstata,
consumo de álcool, tabagismo, exposição a radiação, agentes infecciosos,
vasectomia e níveis de actividade física, embora as evidências que apoiem um
papel importante para esses factores sejam bastante ténues.
O exame rectal digital (toque rectal) era o único teste disponível para a detecção
precoce do carcinoma da próstata. Recentes estudos sugeriram que a sensibilidade
desse procedimento é de aproximadamente 30%, e sua especificidade de 40% para
a doença órgão-limitada.
A introdução do doseamentos sérico do antígeno específico da próstata (PSA) no
final na década de 1980 marcou um grande avanço no campo do diagnóstico e do
acompanhamento dos pacientes com carcinoma da próstata.
O PSA é uma proteína produzida pelo epitélio da próstata e das glândulas
periuretrais, agindo do ponto de vista funcional, como uma protease envolvida na
liquefação do coágulo seminal. Embora apareça em altas concentrações no líquido
seminal, é ainda desconhecido o mecanismo pelo qual o PSA passa para a
circulação. Acreditava-se inicialmente, que o PSA se originava unicamente na
próstata; porém, baixas concentrações têm sido detectadas em glândulas
periuretrais, mamárias, salivares e pancreáticas, leite, soro de mulheres, líquido
amniótico e outros fluidos corporais.
Num estudo recente, foi avaliado um grupo de mulheres com lesões benignas e
carcinoma da mama antes e após cirurgia. O PSA foi detectado, nessas pacientes,
usando-se técnicas ultra-sensíveis. As concentrações de PSA foram correlacionadas
com os achados histológicos. As pacientes com carcinoma da mama apresentaram
concentrações mais altas de PSA, avaliadas no período pré-operatório, com
diminuição após a cirurgia, que porém, não se mostrou significativa. Mulheres com
lesões benignas de mama, equivalentes a um terço dos casos estudados, também
apresentaram valores de PSA no soro. Portanto, a expressão de PSA no soro não
distingue doenças benignas e malignas de mama, mas talvez possa ser valiosa no
acompanhamento da doença.
O PSA apresenta-se, na sua maior parte, na forma de complexos formados com
enzimas inibidoras da protease como a alfa-1-antiquimiotripsina e a alfa-2macroglobulina. Uma menor porção circula na forma livre, representando
geralmente menos de 30% do PSA total. Tanto a forma livre como a ligada às
proteínas podem ser medidas por técnicas de imunoensaio. Ambas as formas são
enzimaticamente inactivas no soro.
Em associação com o toque rectal, o doseamento do PSA conduziu a um
diagnóstico de carcinoma da próstata mais precoce em pacientes mais jovens e em
maior número de pacientes diagnosticados. Além disso, o PSA também é útil para
monitorizar a resposta à terapia.
Porém, o PSA tem as suas próprias limitações em relação à sensibilidade e à
especificidade, sendo a sua principal desvantagem a ausência de especificidade
como marcador tumoral. Além disso, essa característica faz com que exista uma
grande faixa de valores comuns às doenças da próstata, tanto benignas como
malignas.
Nos últimos anos, têm sido desenvolvidos vários ensaios de PSA com sensibilidade
crescente, e a avaliação da fracção livre do PSA tem demonstrado oferecer uma
especificidade melhor do que o doseamento isolado do PSA total.
Preconiza-se hoje, que níveis de PSA no soro normal não deveriam exceder 4
ng/mL. Entretanto, alguns estudos têm demonstrado uma prevalência moderada de
carcinoma da próstata detectado por biópsias em pacientes com níveis de PSA total
entre 2,6 e 4,0 ng/mL. Níveis entre 4 e 10 ng/mL, geralmente, levam à
probabilidade de 20% de risco de carcinoma; níveis superiores a 10 ng/mL têm
acima de 50%, e níveis superiores a 40 ng/mL estão associados a doença
metastática óssea. Porém, convém lembrar a correlação com a idade, na qual
níveis até 6,5 ng/mL, em homens acima de 70 anos, podem ser normais.
A análise integrada de vários testes e diferentes índices é útil para aumentar a
sensibilidade e a especificidade da investigação. Para aumentar a especificidade do
PSA, especialmente na faixa cinzenta (entre 4,0 e 10,0 ng/dL), utiliza-se a soma de
diferentes recursos, como:
- PSA ajustado à idade;
- Densidade do PSA;
- A velocidade do PSA;
- Relação PSA livre/ PSA total.
PSA Ajustado à Idade
A idade é um factor importante em níveis de PSA crescentes. Por isso, alguns
médicos usam níveis ajustados à idade, definindo valores diferentes, normais para
cada grupo etário. Esse método sugere que homens abaixo de 50 anos de idade
deveriam ter PSA abaixo de 2,5 ng/ml, enquanto níveis de até 6,5 ng/ml seriam
considerados normais para homens acima de 70 anos.
Densidade Prostática
Segundo Stamey e cols., cada grama de tecido prostático na hipertrofia benigna da
prostata (HBP) liberta 0,31ng de PSA, enquanto cada grama no carcinoma da
próstata liberta 3,5 ng de PSA. Normalmente, é aceite como limite de uma próstata
normal o volume de 25 g. A densidade do PSA é calculada segundo a definição de
Benson e cols., em que:
dPSA =PSA/volume prostático (obtido por ultra-som transrectal).
Densidades acima de 0,12 g sugerem a presença de carcinoma da próstata e
obrigam à realização de biópsia confirmatória. Já as menores que 0,12 g indicam
HBP. Antigamente considerava-se como ponto de cut-off o valor de 0,15g; hoje já
consideramos o valor de 0,12 g, e existe uma tendência para se considerar 0,10g
como ponto de corte ideal.
Entre os factores que podem interferir na análise da densidade do PSA estão a
variabilidade dos dados de avaliação do volume prostático pelo ultra-som,
diferenças na proporção do conteúdo glandular e estromal (glândula com o mesmo
peso, mas com conteúdo glandular maior irá produzir mais PSA) e outros factores
locais, como manipulação prostática, infecções urinárias e prostatites.
A Velocidade do PSA
A velocidade do PSA é outro dado importante, e signific a avaliação dos valores do
PSA num período de tempo definido. É esperado um aumento de 0,75 ng/mL/ano.
Se o tempo de duplicação do PSA é superior a 10 meses, há uma recidiva local;
quando o tempo de duplicação é inferior a 6 meses, indica recidiva à distância.
É considerado um parâmetro útil para avaliação de doença residual em pacientes
após cirurgia.
Relação PSA Livre/ PSA Total
O doseamento de PSA livre e a relação PSA livre/PSA total são utilizadas para
aumentar a sensibilidade de detecção do carcinoma da próstata, quando o PSA total
é normal (entre 2,6 e 4,0 ng/mL), e melhoram a especificidade quando o PSA total
está aumentado, particularmente na faixa entre 4,0 e 10,0 ng/mL. Trinta a 50%
dos pacientes com PSA total entre 4,0 e 10,0 ng/mL já possuem doença extra
prostática na altura da cirurgia. Valores dessa relação acima de 0,15 sugerem
hipertrofia prostática benigna, e abaixo desse nível indicariam investigação mais
intensa, na procura de uma possível neoplasia prostática.
O prognóstico do carcinoma da próstata depende de muitos factores. Se for
diagnosticado precocemente, pode ser curado aproximadamente em 90% dos
casos. São considerados factores importantes de prognóstico a ausência de
metástases, idade, fase clínica, extensão do tumor e níveis séricos de PSA.
O acompanhamento do pós-operatório de prostatectomia radical deve ser realizado
pela avaliação do PSA, pois o aumento dos seus níveis ocorre nas recidivas, muito
antes de qualquer evidência clínica ou detecção por métodos de imagem. Nódulos
palpáveis ao toque rectal são considerados recidiva local. A recidiva à distância é
evidenciada por adenopatia pélvica na tomografia computadorizada, aumento de
volume e do número de gânglios e alterações na cintilografia óssea. Na recidiva
local, o PSA aumenta 2 anos após a cirurgia. Na sistémica, o PSA aumenta antes de
1 ano, após a cirurgia.
O acompanhamento das recidivas após a cirurgia é feito pelo doseamento do PSA
no pré-operatório, 3 semanas após a cirurgia, trimestral nos primeiros 2 anos,
semestral entre 2 a 5 anos e a partir daí, com avaliação anual.
O efeito das doenças genitourinárias e da manipulação urológica nos valores do PSA
tem sido investigado e discutido. Tem sido relatado aumento dos níveis de PSA nas
prostatites agudas e subclínicas, na retenção urinária, citoscopia, cateterização
uretral, após ejaculação, biópsia prostática e após massagem prostática vigorosa,
porém em graus variados. Acredita-se ser prudente a colheita de soro para
doseamento de PSA antes dos procedimentos descritos, para afastar a possibilidade
de interferência. Relata-se o período de 48 horas para os valores voltarem a níveis
basais após a ejaculação, 1 a 6 semanas após a massagem prostática, e 72 horas
após o toque rectal.
Alguns trabalhos demonstram a redução de 50% dos valores de PSA 48 horas após
a resolução dos quadros de retenção urinária. São necessários cerca de 6 a 8
semanas para a normalização dos níveis de PSA após a resolução do quadro de
prostatite aguda e 30 a 40 dias após biópsias. Outro relato importante é a
possibilidade de alteração do PSA em ciclistas, usuários de bicicleta ergométrica e
praticantes de hipismo. Recomenda-se a suspensão dessas actividades 1 a 2
semanas antes da colheita da amostra, especialmente em pacientes com hipertrofia
benigna da próstata.
O rastreio anual por doseamento de PSA, para carcinoma da próstata é
recomendado pela American Urological Association e pela American Cancer Society,
para homens a partir de 50 anos, ou nos casos com história familiar ou ascendência
africana, a partir dos 40 anos. O rastreio deve ser realizado pelo exame da próstata
por toque rectal e pelo dosemento de PSA. Essa associação aumenta a sua
sensibilidade e especificidade, visto que cerca de 15% das neoplasias da próstata
pode não produzir aumento dos níveis de PSA, assim como, noutros casos, o PSA
pode estar alterado antes da identificação do tumor pelo toque rectal.
Existe a necessidade urgente de se identificar novos marcadores que, utilizando
sistemas baseados em critérios moleculares, celulares e histológicos, conduziriam a
um diagnóstico mais preciso. A meta dessa combinação é identificar, com precisão,
pacientes em diferentes estágios do carcinoma da próstata, desde os
aparentemente localizados, que requerem terapia local, aos que requerem terapia
local e sistémica. Têm sido realizadas tentativas para identificar novos marcadores,
como por exemplo o antígenio de membrana próstata-específico (PSMA), proposto
recentemente como novo marcador sérico para o carcinoma da próstata (ainda não
disponível para uso de rotina), com valor prognóstico clínico, não observado para
outros marcadores. Outro passo será a identificação de algumas alterações
moleculares e celulares, na fase pré-maligna e nas células malignas, utilizando-se
métodos como imunocitoquímica e RT-PCR.
Rastreio Neonatal
O rastreio neonatal de doenças genéticas é importante para a identificação precoce
de indivíduos com doenças metabólicas, particularmente as patologias passíveis de
manuseamento dietético ou de tratamento medicamentoso.
O rastreio com amostras de sangue colhidas em papel de filtro a partir da punção
do calcanhar do recém-nascido - o teste do pézinho - foi implementado na década
de 1960, com os testes para fenilcetonúria (PKU), doença do xarope de bordo,
homocistenúria, galactosemia e hipotiroidismo congénito. À medida que novas
metodologias se desenvolveram, testes relativos a outras doenças foram sendo
incluídos paulatinamente no programa de rastreio neonatal.
O sucesso das práticas de triagem depende do cumprimento de várias etapas
como: esclarecimento da comunidade médica e familiar, controlo da qualidade das
amostras sanguíneas e das técnicas laboratoriais e um pronto acompanhamento
dos recém-nascidos que se mostrem positivos nos testes diagnósticos.
É fundamental que o médico prescritor esteja consciente dos procedimentos de
aplicação do teste e dos protocolos individuais do laboratório que o efectua.
Resultados falso-positivos podem ocorrer devido a uma série de causas:
prematuridade, hemotransfusão, aplicação precoce do teste, inadequabilidade préanalitica da amostra como superaquecimento, uso de antibióticos, etc. Nesses
casos, são sempre provocadas angústias desnecessárias.
Os pais do recém-nascido devem estar cientes dos benefícios da realização do
exame. É necessário que os profissionais médicos e os pais compreendam que um
teste positivo para uma determinada doença triada não é sinónimo de recémnascido portador da doença, mas sim que há a necessidade de avaliação profunda
imediata.
O raciocínio inverso também deve ser considerado, ou seja, apesar de os testes
serem, na sua maioria, sensíveis e específicos, qualquer recém-nascido ou lactente
que se apresente sintomático, mesmo com rastreio negativo, deve ser reavaliado.
Em termos pragmáticos, o tempo para a obtenção da amostra tem sido
padronizado para prover o potencial máximo de detecção do maior número de
patologias. Condições detectáveis a partir da aferição de marcadores protéicos que
estão presentes ao nascimento (por exemplo: hemoglobinopatias, IgM para
toxoplasmose, deficiência de galactose 1-fosfato-uridil transferase e deficiência de
biotinidase) são adequadamente rastreadas, independentemente da época de
realização do teste.
Por outro lado, doenças para as quais a detecção adequada depende da
acumulação de uma substância específica decorrente de um metabolismo alterado
estão completamente vinculadas à época de obtenção da amostra, facto que torna
crítica a confiabilidade do rastreio neonatal. Nesse grupo, estão incluídas os
doseamentos de tiroxina (T4) e de hormona tireotrófica (TSH) para hipotiroidismo
congénito, de fenilalanina para PKU, de galactose para galactosemia e de 17hidroxiprogesterona (17-OHP) para hiperplasia congénita da supra-renal.
O timing óptimo para o rastreio é entre 48-72 horas após o nascimento,
preferencialmente após a ingestão de dieta láctea.
Patologia
Incidência
Locus
cromossómico
Deficiência
enzimática
1:12.000
12q22-q24
Finilalanina
hihoxilase
1:4.000
Maioria dos casos
de causa nãocongénita
Não existente
PKU
Hipretiroidismo
congénito
Incidência
A instituição precoce de
dieta previne a ocorrência de atrazo mental
A instituição precoce de
hormonoterapia previne
a ocorrência de atrazo
mental e do crescimento
Hiperplasia
congénita da
supra-renal
1:12.000
6p21.3
21-hidroxilase
(90% dos casos)
1:375 (HbSS)
11p21.3
HbSS
1:250.000
3p25
Biotinodase
1:60.000
9p12.3
Galactose 1fosfato-urudil
transferase
1:2.500
-1:9.000
7q31
Não existente
1:1.000
-1:8.000
Causa infecciosa
Não existente
Hemoglobinopatias
Deficiência de
biotinidase
Galactosemia
Fibrose quística
(IRT)
Toxoplasmose
cingênita (IgM)
O tratamento precoce
previne o excesso de
virilização, a baixa estatura e a dificiência de
cortisol (resto da vida)
A instituição precoce de
medidas de suporte diminui a morbi-mortilidade
O uso de biotina oral
previne e diminui a
sintomatologia
Alto índice de sequelas
cognitivas/endocrinológicas
Avaliação rotineira do
aparelho respiratório
O tratamento específico
interrompe a doença
aguda.
Cuidados universais no manuseamento de sangue são observados. O local da
punção deve ser a região lateral da superfície plantar do calcanhar, e não a curva
posterior. Medidas como o aquecimento prévio e o manuseamento do pé a ser
puncionado abaixo do nível cardíaco aumentam o fluxo sanguíneo local. A assépsia
do local é feita com álcool, evitando-se o excesso, sob o risco de diluição da
amostra colhida e consequente alteração do resultado de alguns testes.
A tabela na página anterior mostra os testes laboratoriais disponíveis na rotina, a
incidência das doenças em questão, o locus cromossómico envolvido e sua
deficiência enzimática, assim como considerações terapêuticas.
Outros exames adicionais podem também ser solicitados na mesma amostra de
sangue do recém-nascido, colhida em papel de filtro, tais como: cromatografia de
aminoácidos, glicose-6-fosfato desidrogenase, deficiência de desidrogenase dos
ácidos gordos de cadeia média, sorologia para SIDA, sífilis, rubéola, citomegaloviros
e doença de Chagas.
Resistência à Proteína C Activada
A resistência à proteína C activada é causada por um defeito hereditário,
autossómico dominante, ligado ao gene do factor V, onde ocorre uma troca de
aminoácidos (arginina por glutamina) no local onde o factor Va é clivado pela
proteína C activada, tornando o factor Va mutante (factor V Leiden) e assim
resistente à inactivação pela proteína C.
A mutação resulta num aumento importante do risco de trombose. Estudos
demonstram que, provavelmente esse defeito corresponda a cerca de 20 a 60%
dos casos de trombose de etiologia não-esclarecida. O risco trombótico aumenta
significativamente se associado a outros factores de risco, como o uso de
contraceptivos orais, gravidez e em idosos.
Reticulócitos
Os eritrócitos são formados a partir de uma célula mãe na medula óssea.
Estimuladas pela eritropoetina, essas células diferenciam-se, dando origem a uma
sucessão de divisões mitóticas com um contínuo processo de diferenciação, até à
expulsão do núcleo do eritroblasto, dando agora origem ao reticulócito. Esse
processo ocorre num período de 72 horas. Nas 48 horas seguintes, o reticulócito
em maturação transforma-se num eritrócito.
Portanto, o reticulócito é uma célula jovem que representa uma fase intermédia
entre os eritroblastos da medula óssea e os eritrócitos maduros, anucleados e já
totalmente hemoglobinizados. Por ainda não estarem totalmente maduros, os
reticulócitos apresentam-se na periferia como células um pouco maiores que os
eritrócitos e com uma coloração azul-acinzentada, que se deve à existência de
material nuclear residual de cor azulada associada à cor avermelhada da
hemoglobina.
A sua avaliação é importante, pois serve como indicador da produção de eritrócitos
pela medula óssea. As causas mais comuns de reticulocitose são as hemorragias
agudas, as anemias hemolíticas agudas e crónicas e a resposta ao tratamento de
reposição de ferro, folato e vitamina B12. Uma contagem diminuída de reticulócitos
pode ocorrer nas anemias aplásticas, na invasão medular e nas anemias por
carencias antes do respectivo tratamento.
Retracção do Coágulo
A retracção é a fase final do processo de coagulação e está directamente ligada à
actividade funcional adequada das plaquetas. O coágulo pode estar alterado em
volume na presença de plaquetopenia e nas anemias.
A avaliação da retracção tem maior utilidade na avaliação de deficiências funcionais
das plaquetas. É quando a retracção se encontra diminuída, mesmo na presença de
um número normal de plaquetas. Pode ser influenciada pela quantidade de
trombina e da fibrinogénio e por valores alterados do hematócrito.
Rubéola
Causada por um vírus RNA do género Rubivirus, a rubéola continua a manifestar-se
na população, apesar das campanhas de vacinação, que conseguiram diminuir a
incidência da doença.
É uma doença normalmente moderada, com complicações pouco frequentes, que
pode ser assintomática em cerca de 50% dos casos ou cursar com manifestações
clínicas discretas. No entanto, quando ocorre em gestantes suscetíveis,
especialmente durante o primeiro trimestre e, com menor frequência, no segundo
trimestre de gravidez, pode levar ao aborto espontâneo ou ao síndrome de rubéola
congénita, com comprometimentos cardíacos, oculares, auditivos e do sistema
nervoso fetal. Os riscos abortivos e teratogénicos da infecção em mulheres grávidas
tornam de grande importância a investigação pré-natal de anticorpos contra o vírus
da rubéola.
O contágio ocorre por via respiratória, e o período de incubação, de 2 a 3 semanas,
é seguido por sintomas virais e rash cutâneo maculopapular, com linfadenopatia
suboccipital. Os anticorpos anti-rubéola são detectáveis logo após o
desaparecimento do rash cutâneo. Os primeiros a aparecer são da classe IgM,
detectáveis cerca de 4 a 5 semanas após a infecção (ou vacinação). Actualmente,
métodos ultra-sensíveis possibilitam a sua detecção por mais tempo (6 meses ou
mais). A seguir, aparecem os da classe IgG, que, quer por infecção natural ou por
vacinação, persistem pelo resto da vida.
A infecção quase sempre confere imunidade permanente. Entretanto, a reinfecção
pode ocorrer, especialmente nos indivíduos vacinados, apresentando aumento da
concentração de anticorpos da classe IgG. A resposta de anticorpos da classe IgM
está tipicamente ausente ou baixa, mas pode acontecer, embora raramente, o que
dificulta significativamente a sua interpretação.
Anticorpos IgM são detectados por EIA em 100% dos pacientes entre 11 e 25 dias
depois do exantema; em 60% a 80% dos indivíduos 15 a 25 dias após a vacinação,
e em 90% a 97% das crianças com rubéola congénita, entre 2 semanas e 3 meses
após o nascimento. O anticorpo materno IgG, adquirido passivamente, desaparece
após 6 a 7 meses. O feto não desenvolve IgM antes de 18 a 20 semanas de
gestação. A imunidade activa é raramente adquirida antes dos 2 anos de idade.
Nas investigações de possíveis infecções fetais e pós-natais, é necessário evitar
reacções falso-positivas para IgM pela presença de factor reumatóide,
mononucleose infecciosa, infecção por parvovírus e citomegalovírus. Em alguns
casos, as mulheres grávidas podem ser reactivas para anticorpos IgM para rubéola,
citomegalovírus, varicela-zoster e sarampo. Todos os resultados de IgM positivos
devem ser confirmados por mais de um método em soros duplos e comparados
com a história clínica detalhada.
O diagnóstico laboratorial é realizado por técnicas imunoenzimáticas que avaliam e
quantificam a presença de anticorpos IgM e IgG, com a finalidade de diferenciar
entre infecção aguda, passada, congénita ou vacinação. As novas técnicas
imunoenzimáticas eliminaram a possibilidade de resultados falso-positivos e falsonegativos.
Pesquisam anticorpos IgG e IgM com maior sensibilidade, permitindo uma detecção
mais precoce e efectiva por maior período de tempo. No entanto, a grande
sensibilidade desses testes, ao tornar possível a detecção de anticorpos IgM,
mesmo em níveis baixos, por um longo período de tempo após a fase aguda, fez
com que a presença de IgM não seja suficiente para o diagnóstico da doença em
fase aguda.
A presença de soroconversão é conclusiva de infecção aguda. A presença de
anticorpos IgM indica infecção aguda. Porém, pode ser atribuída a níveis residuais
de infecção passada ou reacção pós-vacinação. Actualmente, para definir a fase da
doença, dispomos da avaliação dos testes de avidez dos anticorpos IgG. Estes
testes baseiam-se na característica de baixa avidez que os anticorpos apresentam
pelo antigénio, durante o início da resposta imunológica. Portanto, na infecção
recente, estão presentes os anticorpos IgG de baixa avidez, e nas infecções mais
antigas, encontramos os de alta avidez. Consideram-se de baixa avidez índices
inferiores a 30%, que indicam que a infecção ocorreu nos últimos 3 meses. Índices
superiores a 60% são considerados de alta avidez, apontando para uma infecção
ocorrida há mais de 3 meses. Valores entre 30% e 60% não permitem a
caracterização da fase da doença.
Sífilis
Doença infecto-contagiosa, essencialmente transmitida pelo contágio sexual, que
tem como agente etiológico o Treponema pallidum. É um patogénio exclusivamente
humano, com carácter infectante apenas na fase aguda da doença. Após o
contágio, a infecção apresenta um período de incubação médio de 3 semanas, após
o qual se manifesta a lesão inicial, o cancro duro, com repercussão ganglionar
inguinal bilateral e indolor, que evolui para auto-resolução, mesmo se não tratada,
em cerca de 1 a 2 meses, sem deixar cicatrizes. Esta fase é denominada sífilis
primária.
Cerca de 2 a 3 meses após, aparecem as lesões generalizadas da sífilis secundária,
que se caracterizam por erupção cutânea generalizada com envolvimento palmoplantar. Caso não-tratada, assume carácter sistémico, evoluindo crónicamente, com
períodos de actividade e de lactência.
Em cerca de um terço dos pacientes não-tratados ocorre um estágio de
manifestações, dito terciário, que se caracteriza por lesões parenquimatosas,
musculoesqueléticas, mucocutâneas progressivas, lesões aórticas ou sintomáticas
do sistema nervoso central.
Cerca de 10% dos pacientes que apresentam a forma primária, caso não-tratados,
evoluirão para neurossífilis. A neurossífilis assintomática é a forma mais comum de
apresentação. Não há sinais ou sintomas clínicos. Acredita-se que os pacientes que
apresentam alterações no líquor, mesmo sem sintomatologia, durante as fases
iniciais da doença, tenham mais chances de evoluir para síndromes neurológicos
tardios. A progressão das alterações neurológicas pode dar-se com quadros de
meningite sifilítica, sífilis meningovascular, meningoencefalite sifilítica, tabes
dorsalis e sífilis medular.
Portanto, a sífilis pode manifestar-se no sistema nervoso tanto de forma aguda
como crónica, e mesmo vários anos após a forma primária. Deve-se dar atenção a
essa hipótese, já que a SIDA levou a um aumento da incidência e a formas clínicas
atípicas de evolução mais agudas e mais graves dessa doença. As características
laboratoriais consistem no achado de alterações do líquor como pleocitose,
aumento de proteínas, redução da glicose ou positividade para a reacção de VDRL.
O diagnóstico laboratorial da sífilis é feito pela pesquisa directa do treponema, ou
pela pesquisa de anticorpos formados durante a infecção, que podem ser de dois
tipos: anticorpos não-treponémicos e anticorpos treponémicos.
A pesquisa directa, realizada por microscopia de campo escuro, apesar de
altamente específica, tem indicação limitada, podendo ser realizada na fase
primária, directamente do cancro duro do órgão genital ou noutras localizações.
Durante a fase secundária, o material pode ser obtido das lesões cutâneas e no
líquido amniótico, placenta, muco nasal e lesões cutâneas de recém-nascidos para
a investigação de sífilis congênita.
A pesquisa de anticorpos não-treponémicos, inespecífica para o diagnóstico, é feita
pela reação de VDRL (Veneral Disease Research Laboratories test), que se mostra
positiva numa provável reacção cruzada contra a cardiolipina, componente presente
em vários tecidos, sendo indicada como exame de triagem. Estão presentes nas
primeiras semanas da doença e, quando em títulos iguais ou maiores de 1/16,
sugerem fortemente casos de sífilis; títulos inferiores, geralmente até 1/8, são
encontrados em diferentes patologias, especialmente no lúpus eritematoso
sistémico e como títulos residuais (cicatriz sorológica) de sífilis anteriormente
tratada. O tempo de negativação depende directamente da fase em que foi iniciado
o tratamento, podendo nalguns casos, permanecer indefinidamente com baixos
títulos flutuantes.
A pesquisa de anticorpos treponémicos, que são específicos contra o Treponema
pallidum, é indicada como testes confirmatórios e pode ser realizada pela
imunofluorescência indirecta (FTA-ABS) e pela hemaglutinação passiva (TPHA).
O FTA-ABS (fluorescent treponemal antibody absorption) é o mais sensível,
auxiliando no diagnóstico de diferentes estágios da doença. Permite a pesquisa de
anticorpos IgG e IgM, fundamental na investigação diagnóstica da sífilis congênita,
assim como na avaliação do estágio da doença. Quando positivos, permanecem por
toda a vida como cicatriz sorológica. Quando negativos, afastam o diagnóstico de
sífilis. Apesar de sua alta especificidade, existem casos de reacções falso-positivas
em 2% da população normal em pacientes com lúpus eritematoso sistémico,
durante a gravidez, na lepra, mononucleose, leptospirose, artrite reumatóide,
cirrose biliar primária e doenças associadas à produção de globulinas anormais.
A reacção de hemaglutinação é, também, considerada um teste confirmatório.
Resultados falso-positivos são encontrados em diferentes patologias. A sua
sensibilidade é similar à do FTA-ABS, com excepção da investigação da fase
primária, na qual é menos sensível. Assim como no FTA-ABS, se positivos, os
anticorpos permanecem por toda a vida como cicatriz sorológica.
O diagnóstico da sífilis congênita baseia-se na presença de anticorpos IgM, sendo o
método de escolha a pesquisa do FTA-ABS IgM. No entanto, um resultado negativo
não afasta a possibilidade de infecção, já que a positividade só acontece em cerca
de 80% dos casos. A persistência de reacções sorológicas positivas, treponémicas e
não-treponémicas, por mais de 6 meses após o nascimento é altamente indicativa
de sífilis congênita.
Sódio
O sódio é o maior catião e a principal partícula osmótica do meio extracelular. Por
isso, é considerado o íão mais importante do organismo, tanto do ponto de vista
quantitativo quanto pela sua importância na manutenção do equilíbrio osmótico e
da electro-neutralidade. As alterações da concentração do sódio extracelular
resultam em alterações da osmolaridade, que por sua vez, influenciam a
distribuição da água corporal.
Os distúrbios da homostase do sódio podem ocorrer por excessiva perda, ganho ou
retenção de sódio ou por excessiva perda, ganho ou retenção de água.
Entre as causas de hiponatremia, o uso de diuréticos e a reposição de líquidos
intravenosos hipotónicos são as mais frequentes. As perdas gastrointestinais por
vómito, diarréia e drenagem por sonda nasogástrica, sudorese e queimaduras
extensas também são importantes fontes de depleção de sódio. Síndrome nefrótica,
cirrose, hipoalbuminemia grave, insuficiência cardíaca congestiva e insuficiência
renal aguda com oligúria ou crónica com acidose também levam à hiponatremia.
Algumas outras patologias podem cursar com hiponatremia, como hipotiroidismo,
doença de Addison, distúrbios da secreção de vasopressina e da hormona (ADH)
antidiurético que ocorrem em pacientes com doenças crónicas, dor, stress físico ou
emocional, neoplasias e distúrbios metabólicos do sistema nervoso central. A
velocidade da diminuição do sódio é proporcional à gravidade dos sintomas clínicos
observados.
A concentração de sódio pode estar também diminuída em condições de
hiperglicemia, nas quais o líquido é atraído do meio intracelular para o extracelular,
levando a uma diluição. O aumento excessivo de triglicerídeos e de proteínas
também pode ser a causa de hiponatremias por diluição (falsas hiponatremias).
Reposição deficiente oral ou venosa de água, diurese excessiva (diurese osmótica e
diabetes insipidus central ou nefrogénico), perda pela pele, pulmão ou tubo
gastrointestinal sem reposição líquida, superdosagem parenteral por hidratação
endovenosa ou hiperalimentação parenteral, síndrome de Cushing e o
hiperaldosteronismo primário podem levar a hipernatremia.
O sódio é livremente filtrado pelos glomérulos, e cerca de 60% do sódio filtrado é
reabsorvido no túbulo proximal. Também ocorre reabsorção de sódio ao longo dos
outros segmentos dos nefrónios. Receptores localizados nas células justaglomerulares são sensíveis à diminuição da pressão arterial ou da concentração de
sódio no filtrado e estimulam a secreção de renina. A renina estimula as suprarenais a produzirem aldosterona, que regula a reabsorção de sódio e a secreção de
potássio. Deste modo, quando ocorre uma diminuição do nível de sódio ou quando
o rim é hipoperfundido, os túbulos distais sob a influência da aldosterona retêm
sódio.
Portanto, a avaliação da concentração do sódio urinário é importante como apoio ao
diagnóstico diferencial das hiponatremias em causas renais e extra-renais e dos
casos de oligúria.
Níveis urinários aumentados são encontrados na doença de Addison, nas nefrites
com perdas de sal, nas dietas com teor excessivo de sódio, na acidose tubular
renal, no uso de diuréticos e no sindrome de secreção inapropriada da hormona
antidiurética, entre outras condições.
Níveis diminuídos podem existir na desidratação, na insuficiência cardíaca
congestiva, nas hepatopatias, no síndrome nefrótico, em situações que levem à
diminuição da taxa de filtração glomerular e em dietas pobres em sódio.
SÓDIO URINÁRIO NA AVALIAÇÃO DE OLIGÚRIAS
Pré-renal
< 20 mEq/ml
Renal
> 30 mEq/ml
Pós-renal
> 30 mEq/ml
Tempo de Protrombina
Os fármacos anticoagulantes orais actuam sobre os factores da coagulação
pertencentes ao sistema extrínseco da coagulação. Por isso, o (TP) tempo de
protrombina é o exame de eleição para a monitorização da terapêutica com estes
fármacos.
Por avaliar a via extrínseca, o TP pode estar elevado na deficiência isolada do factor
VII, na presença de anticorpos inibidores circulantes e em patologias que afectem o
processo de absorção, síntese e metabolização da vitamina K, visto que a produção
desse factor é dependente dessa vitamina. Pode apresentar-se alterado também,
quando ocorre um comprometimento da fase final da via comum (X, V, II e I).
Como teste de referência para o acompanhamento da anticoagulação oral, o TP não
fornecia a uniformidade desejada. As tromboplastinas utilizadas (inicialmente tecido
humano e actualmente oriundas de tecido animal) geravam resultados que
variavam amplamente em comparações intra e interlaboratoriais. Estas variações
representavam um grande entrave ao acompanhamento adequado dos pacientes.
Por este motivo, depois de diferentes tentativas de padronização, em 1983, a
Organização Mundial da Saúde (OMS) estabeleceu, em conjunto com o Comité
Internacional de Trombose e Hemostasia e a Comissão Internacional de
Padronização em Hematologia, a recomendação para a utilização mundial do ISI
(International Sensibility Index) e a conversão dos resultados obtidos em INR
(International Normalized Ratio).
Os fabricantes mundiais de tromboplastina foram orientados para padronizar o seu
reagente, comparando as suas tromboplastinas com a tromboplastina de referência
mundial da OMS. Com esses dados, podem calcular o índice de sensibilidade
internacional (ISI) para cada lote de tromboplastina produzido. Esse valor de ISI,
fornecido pelo fabricante em cada lote enviado, é utilizado para o cálculo do INR
(razão normalizada internacional). Quanto maior o ISI, menor a sensibilidade do
reagente.
O INR é obtido por um cálculo que divide o valor do TP encontrado na amostra do
paciente pelo resultado do TP de um pool de plasmas normais, elevados ao ISI.
Portanto, na prática, ele passa a funcionar como um TP padronizado intra e inter
laboratorialmente.
A alteração do TP com o uso de anticoagulantes orais é obtida em média 3 a 5 dias
após o início da administração. Durante este período, a avaliação do TP deve ser
feita diariamente, até que o INR alcance o valor terapêutico preconizado como ideal
para a condição clínica de que se está a tratar. Além disso, a avaliação deverá
manter a frequência diária, até que se comprove a estabilidade dos valores obtidos.
O acompanhamento do paciente pelos valores do INR só deve ser feito em
pacientes com resultados já estabilizados.
O horário ideal para a colheita do sangue para avaliação do TP está directamente
relacionado com o horário da administração do medicamento. Os principais
protocolos apontam para que os anticoagulantes devam ser administrados à tarde
(18 h) e o sangue colhido na manhã seguinte (até as 10 h), de modo a garantir a
absorção adequada do medicamento. No entanto, na prática, a melhor indicação é
que o paciente tome o medicamento sempre no mesmo horário e faça a colheita no
mesmo prazo em que realizou as anteriores.
REFERENCIAIS DE ALVOS TERAPÊUTICOS
Maioria das situações com indicação de anticoagulação
2.0-3.0
Prevenção e tratamento de trombose venosa
Embolia pulmonar, sistêmica
Embolia arterial pós-operatória
Infarto agudo do miocardio
Doença de válvula cardíaca
Fibrilação atrial
..................................................................
Prótese cardíaca
2.5-3.5
..................................................................
Formas recidivantes de trombose venosa profunda
3.0-4.0
Formas recidivantes de embolia pulmonar
..................................................................
Adaptação das recomendações do American College of Physicians, National Heart Lung, and Blood
Institute e British Society for Haemalotogy
Diversos factores podem interferir na acção dos anticoagulantes orais. O primeiro
problema, e o mais comum, é a adesão adequada do paciente ao tratamento.
Outros factores importantes são o teor de vitamina K da dieta, a dose em relação à
massa corporal, interacções medicamentosas, integridade da função hepática e
comportamento metabólico individual quanto ao fármaco.
Em relação ao uso de outros fármacos, sabemos que existe a interferência com a
interacção entre drogas que podem potenciar, diminuir ou inibir a acção dos
anticoagulantes orais, por diferentes mecanismos, alterando a sua acção
terapêutica.
FÁRMACOS QUE ALTERAM A ACÇÃO DOS ANTICOAGULANTES ORAIS
POTENCIALIZAM
Alguns antibióticos, anti-inflamatórios, ácido acetilsalicílico, antidepressivos tricíclicos
anti-agragantes plaquetários, cimitidina e outros fármacoas com acção no tracto
gastrointestinal, hormonas teroidéias, anti-lipémicos, imunossupressores, inibidores
de MAO, entre outras.
........................................................................................
INIBEM
Alguns antibióticos, antiácidos, contraceptivos orais, barbitúricos, antifúngicos,
álcool, diuréticos, corticosteróides, anti-histamínicos, esteróides, entre outros.
........................................................................................
DIMINUEM
Laxantes, Vitamina C.
Tempo de Coagulação
O tempo de coagulação é um teste de baixa sensibilidade e de reprodutibilidade
muito variável, sendo afectado principalmente por alterações da via intrínseca, do
fibrinogénio e fibrina. Pode estar elevado no decurso de heparinoterapia.
Este teste é substituído pela realização do tempo de tromboplastina parcial activado
(TTPA), que fornece um resultado fidedigno das alterações de via intrínseca.
Tempo de Sangria
É um indicador de alterações numéricas (quantitativas) e funcionais (qualitativas)
das plaquetas. Geralmente, mantém-se normal, mesmo quando as plaquetas se
encontram diminuídas, mas acima do limite de 100.000/mm3.
Em pacientes com plaquetopenia, a variação do tempo de sangria mantém uma boa
correlação com os valores das plaquetas.
Valores alterados podem ser encontrados nos defeitos congénitos das plaquetas,
como a trombastenia de Glanzmann, e nos adquiridos, como nos quadros de
uremia e síndromes mieloproliferativos.
Tempo de Tromboplastina Parcial Activada
O tempo de tromboplastina parcial activada (TTPA) avalia defeitos da via intrínseca
da coagulação, podendo, portanto, constatar a deficiência dos factores VIII, IX, XI
e XII. É útil também no controle do uso terapêutico de heparina e na avaliação da
presença de anticoagulantes circulantes.
Pode apresentar-se alterado também quando ocorre comprometimento da fase final
da via comum (X, V, II e I).
O achado de TTPA prolongado na presença de TP normal indica a possível
deficiência dos factores XII, XI, IX, VIII. Ao contrário, TTPA normal na presença de
TP prolongado indica comprometimento do factor VII.
Quando ambos (TTPA e TP) estão alterados, indicam comprometimento da fase
final da via comum, ou seja, dos factores X, V, II e I. Já ambos normais indicam
pacientes sem alterações ou comprometimento do factor XIII.
Teofilina
A teofilina é um potente broncodilatador amplamente utilizado em doenças
pulmonares obstrutivas agudas e crónicas. Além do efeito broncodilatador, possui
efeitos no aumento da clearance mucociliar, efeitos vasodilatadores, diuréticos, sob
a contracção diafragmática, e um efeito miocárdio inotrópico positivo.
O nível sérico terapêutico está entre 10 a 20 mg/mL. Apresenta uma semi-vida de
cerca de 8 horas em não-tabagistas e de 5 horas em tabagistas. A metabolização é
extremamente individualizada, indicando a necessidade de monitorização adequada
das concentrações séricas. São observadas grandes discrepâncias entre a dose
administrada e as concentrações séricas em indivíduos submetidos a doses
idênticas. Cerca de 90% do fármaco é metabolizado no fígado, e cerca de 60%
encontra-se ligada a proteínas. O estado de equilíbrio é alcançado por volta das 48
a 72 horas. A teofilina atravessa a barreira transplacentária, podendo ser
teratogénica.
Os efeitos colaterais mais comuns são náuseas, vómitos, diarreia, taquicardia,
arritmias, convulsões e hemorragias gastrointestinais.
O uso de teofilina pode diminuir a acção da fenitoína, do lítio e de bloqueadores
neuromusculares. O uso concomitante de cimetidina, alopuridol, propranolol,
contraceptivos orais, amiodarona, lincomicina e infecções pelo vírus influenza ou
mesmo a vacinação para esse vírus podem aumentar a concentração da teofilina.
Rifampicina, fenitoína e fenobarbital podem diminuir a concentração da teofilina.
Os estados de insuficiência cardíaca, doenças hepáticas, febre prolongada e
obesidade aumentam a semi-vida da teofilina, e consequentemente os seus níveis
séricos. A semi-vida está diminuída em fumadores, existindo casos que indicam a
necessidade de uma quantidade 1,5 a 2 vezes maior do fármaco para se obter o
mesmo efeito que se consegue em pacientes não-fumadores. A semi-vida está
diminuída também em recém-nascidos.
A colheita deve ser realizada 2 horas após a administração oral ou 30 minutos após
a administração por via endovenosa.
Teste de HAM
O teste de Ham consiste numa prova de lise ácida realizada em soro acidificado a
37º C, na qual as hemácias comprometidas, ao contrário das normais, sofrem lise
na presença de complemento. É um teste pouco sensível mas com alta
especificidade.
É utilizado na investigação da hemoglobinúria paroxística nocturna (HPN).
A HPN é uma patologia adquirida, em que a medula óssea produz hemácias com
membranas celulares defeituosas, o que as torna sensíveis à lise pelo
complemento.
Clinicamente, estes pacientes apresentam hemólise intravascular crónica, e muitos
deles cursam com episódios de trombose venosa recidivante.
Teste Triplo (rastreio pré-natal)
Até alguns anos atrás, se uma mulher, durante a gravidez, quisesse saber se seu
bebé nasceria normal, a única opção seria submeter-se a exames de carácter
invasivo. Ainda hoje, esses procedimentos estão em uso e envolvem riscos de 0,5 a
1,5%. Ou seja, 1 a 3 mulheres em cada 200 submetidas a punção correm algum
risco, que pode ser desde uma infecção até danos de continuidade da gestação.
Apesar de pequena, essa incidência de risco já seria pelo menos três vezes maior
do que a probabilidade de nascimento de um bebé com problemas.
Com a divulgação, pelos meios de comunicação, de métodos diagnósticos prénatais, muitas mulheres resolveram submeter-se ao procedimento,
independentemente da idade, basicamente por conta de ansiedades em relação à
gravidez. A grande maioria dessas pessoas encontra-se no grupo chamado de baixo
risco, ou seja, abaixo dos 35 anos. Nesse caso, pela pequena probabilidade de
nascimento de um bebé com anomalia cromossómica, o procedimento não seria
indicado. Os exames invasivos só seriam então indicados quando o risco de
anormalidade fosse maior do que o risco do procedimento. É por isso que, em
geral, mulheres com 35 anos de idade ou mais, primíparas, são encaminhadas aos
exames de punção.
Novas alternativas de investigação por meio de exames não-invasivos avaliam
estatisticamente os riscos de gestação do feto com anomalia cromossómica.
O rastreio bioquímico, ou teste triplo (TT), é uma dessas alternativas. Nele, são
doseados a alfafetoproteína, a gonadotrofina coriónica e o estriol livre, numa
amostra de sangue da gestante, colhida entre 15 e 19 semanas de gestação. Por
intermédio desse exame, é possível detectar cerca de 60% das anomalias
cromossómicas. Além disso, quando colhido entre 16-18 semanas, também é
possível avaliar riscos para algumas malformações, como defeitos do tubo neural e
da parede abdominal.
O cálculo de riscos no exame tem como base o risco de nascimento de um portador
da síndrome de Down, pela idade materna (probabilidade inicial), sendo recalculado
pelos resultados dos doseamentos hormonais realizados.
Um exame tendo como resultado risco aumentado não significa que o feto
apresente alterações, apenas indica que estatisticamente estará justificada a
realização dos procedimentos tradicionais para complementação e definição
diagnóstica, como a biópsia de vilo corial ou a amniocentese.
Isto fica claro quando se observa um grupo de gestantes que fizeram o exame.
Cerca de 5% (do grupo de gestantes até 34 anos) e 30% (do grupo com idade de
35 anos ou mais no parto) foram selecionadas para punção de líquido amniótico. De
todas as gestantes que fizeram o exame invasivo, apenas 1 a 2 em cada 100
tiveram confirmação de alteração no feto.
Outros marcadores indirectos para as anomalias cromossómicas também têm sido
utilizados, em especial a medida ultra-sonográfica da translucência nucal (TN) com
11-13 semanas de gestação (rastreio biofísico), com detecção de quase 70% dos
fetos com anomalia cromossómica e 50% dos fetos com síndrome de Down.
Os exames de rastreio já foram aplicados em centenas de milhares de gestantes
em todo o mundo e têm mostrado excelentes resultados, especialmente hoje,
quando, ao contrário do que ocorria no passado, os casos de gestações tardias em
primíparas são cada vez mais frequentes.
É uma maneira de, prevenindo punções desnecessárias, detectar no imenso grupo
de gestantes de baixo risco, que não fariam nenhum exame específico da parte
genética, aquelas que estariam correndo risco de estar a gerar um bebé com
anomalia cromossómica. Deste modo, o casal poderá ter maiores informações
quanto ao bem-estar de seu bebé e eventuais opções quanto à gestação. Os
métodos são simples, seguros, e têm causado grande impacto em todo o mundo na
prevenção do atrazo mental e das malformações congénitas ligadas a anomalias
cromossómicas.
Convém salientar alguns aspectos dos exames de rastreio:
- Fazem parte da rotina pré-natal e são principalmente indicados para gestantes
jovens e sem histórico de anomalia cromossómica
- Os resultados refletem probabilidades (maior ou menor) de estar a gerar um feto
com anomalia cromossómica. Um exame alterado indica investigação adicional,
que, na maioria das vezes, apenas confirma a normalidade do bebé; exames
normais diminuem riscos, mas não garantem a normalidade do bebé em todos os
aspectos. Considerando a limitação dos exames em detectar todos os fetos com
anomalias cromossómicas, a realização de testes de rastreio em sequência (TN com
11-13 semanas, seguido por TT com 15-19 semanas e complementado por ultrasonografia morfológica com 20-22 semanas) tem sido de grande utilidade para
tranquilizar a maioria dos casais. Resultados normais em todos os exames
diminuem acentuadamente os riscos de anomalias. Por outro lado, cada novo
exame oferece mais uma possibilidade de detectar falsas normalidades de um
exame anterior.
Tiróide
A regulação da função tiroideia começa no hipotálamo, com a secreção da hormona
libertadora de tireotrofina (TRH), que por sua vez estimula a síntese e a libertação
da hormona estimuladora da tiróide (TSH).
A secreção de TSH é regulada não apenas pela TRH, mas também pelos níveis de
T4 e T3 livres circulantes. Inúmeros fármacos, bem como algumas hormonas,
podem interferir na função tiroideia.
Hipertiroidismo
O hipertiroidismo decorre do excesso de hormonas tiroideias circulantes.
Laboratorialmente, caracteriza-se por TSH suprimido e T3 e/ou T4 elevados. Uma
vez que as hormonas tiroidéias circulam no sangue ligados às proteínas, e que
apenas a fracção livre da hormona é metabolicamente activa, nos casos que
cursam com a diminuição da TBG (thyroid binding globulin), podemos encontrar
apenas as fracções livres de T3 e T4 elevadas, enquanto os níveis séricos do T3 e
T4 totais podem estar em níveis normais ou até baixos.
A causa mais comum é a doença de Basedow Graves, ou bócio difuso tóxico (85%
dos casos). É uma doença auto-imune causada por auto-anticorpos circulantes
dirigidos contra receptores na superfície da célula tiroidéia: anticorpos
estimuladores da tiróide (TSAB). Podem ocorrer anticorpos que não estimulam a
função tiroidéia, mas apenas se ligam ao receptor de TSH. A diversidade de acção
desses anticorpos gera variações na expressão clínica da doença, bem como uma
variedade de denominações e testes para detectá-los. O denominado anticorpo
anti-receptor de TSH (TRAB) identifica a presença de anticorpos que se ligam ao
receptor de TSH, independentemente da sua acção ser estimulatória ou não.
Outras etiologias do hipertiroidismo são bócio nodular tóxico, tumores secretores de
HCG, tumores hipofisários, carcinomas tiroidéios, ingestão excessiva de iodo,
hipertiroidismo fictício por uso de T3 e/ou T4 e tiróidite.
Os sintomas clínicos de tirotoxicose estão relacionados com efeitos catabólicos e
hipermetabólicos causados pelo aumento da actividade em vários tecidos e maior
sensibilidade às catecolaminas:
perda de peso e de massa muscular, dispnéia, fadiga, nervosismo, irritabilidade,
insónia, tremor, intolerância ao calor, sudorese excessiva, taquicardia, palpitações,
pele quente e húmida.
A maioria dos pacientes tem manifestações de oftalmia não-infiltrativa, com olhar
brilhante e lid lag. A oftalmia infiltrativa manifesta-se com protrusão do globo
ocular (exoftalmia). Pode ocorrer ainda edema pré-tibial.
Hipotiroidismo
O hipotiroidismo surge quando os níveis das hormonas tiroidéias são insuficientes
para preencher as necessidades metabólicas das células. Laboratorialmente,
caracteriza-se pela elevação dos níveis séricos do TSH e pela diminuição dos níveis
de T3 e T4.
O hipotiroidismo pode ser congénito ou adquirido. O hipotiroidismo primário
consiste na deficiência tiroidéia propriamente dita, e é responsável pela maioria dos
casos de hipotiroidismo congénito, que ocorre aproximadamente em 1 por cada
4.000 nascimentos.
O hipotiroidismo secundário é consequente à falência hipofisária na secreção de
TSH e, geralmente, deve-se a um tumor hipofisário. Já o hipotiroidismo terciário
resulta da falência hipotalâmica na secreção de TRH, geralmente causada por
tumor, insuficiência vascular, infecção, processo infiltrativo ou trauma.
O hipotiroidismo adquirido é a secreção inadequada das hormonas tiroidéias devido
ao dano da tiróide, como os causados por tiroidite crónica, cirurgia, tratamento com
iodo radioactivo para hipertiroidismo ou carcinoma, atrofia idiopática e carcinoma
metastático.
A tiroidite crónica, chamada tiroidite de Hashimoto, é a causa mais frequente de
hipotiroidismo primário adquirido. É uma doença auto-imune resultante da
infiltração da glândula tiróide por linfócitos, células plasmáticas e tecido conjuntivo.
Pode ser geneticamente determinada e leva à produção de linfócitos sensibilizados
e anticorpos contra a tiróide, podendo eventualmente, causar a destruição do
tecido tiroidéio. Acima dos 50 anos de idade, o número de novos casos
diagnosticados tem aumentado exponencialmente. A manifestação clínica pode
variar, e o paciente pode ser hipotiroideu, eutiroideu ou hipertiroideu.
Clinicamente, o hipotiroidismo congénito ou cretinismo está associado a pescoço
curto e largo, língua aumentada e protrusa, pernas curtas, abdómen distendido,
voz ou choro rouco, pele seca, letargia e atrazo mental. Estes sintomas
manifestam-se caso o tratamento do recém-nascido não seja iniciado
precocemente.
A deficiência adquirida das hormonas tiroidéias torna mais lentos os processos
metabólicos, provocando fadiga, atrazo mental, alterações de personalidade, déficit
de memória, intolerância ao frio, dispnéia de esforço, rouquidão, constipação e
parestesias.
O hipotiroidismo grave é denominado mixedema e carateriza-se pela infiltração da
pele por mucopolissacarídeos, com a face edemaciada, especialmente em torno dos
olhos. A língua apresenta-se aumentada, e o espessamento das cordas vocais leva
à rouquidão. Ocorrem bradicardia e aumento de peso, a contractilidade miocárdica
é reduzida, e a frequência cardíaca torna-se mais lenta. A anemia pode estar
presente e ser consequência de hipometabolismo, de redução das necessidades de
oxigénio e de diminuição da eritropoietina.
O coma mixedematoso é a apresentação mais severa do hipotiroidismo: coma,
hipotermia, hipoglicemia, hipotensão, hiponatremia e falência respiratória com alta
taxa de mortalidade.
Tiroidites
As tiroidites podem ser classificadas como aguda, subaguda, crónica e fibrótica (ou
tiroidite de Riedel). As manifestações clínicas variam de acordo com o tipo de
tiroidite.
A tiroidite aguda é rara, e caracteriza-se por abcesso e supuração da tiróide. O
início é abrupto, com febre, calafrios e mal-estar. Geralmente, é provocada por
uma infecção bacteriana, particularmente por estafilococos, estreptococos ou
pneumococos, por disseminação de focos sépticos ou secundária a lesão do
pescoço.
A tiroidite subaguda, conhecida como tiroidite de Quervain, é possivelmente
causada por um vírus, podendo ser acompanhada de infecção do tracto respiratório
superior, dor na região da tiróide e febre.
A tireidite crónica, também conhecida como linfocítica, ou doença de Hashimoto, é
uma doença auto-imune com intenso infiltrado inflamatório crónico da tiróide. A
presença de auto-anticorpos conduz, eventualmente a destruição do tecido tiroideu.
As manifestações da tiroidite de Hashimoto são extremamente variáveis, podendo
ser do tipo hipo, hiper- ou eutiroidismo. O sinal principal é a presença de um bócio
indolor. Em estadios finais, quando a fibrose é importante, o paciente pode não ter
bócio.
A tiroidite de Riedel é de etiologia desconhecida e caracteriza-se por fibrose
extensa.
Neoplasias Tiroidéias
Podem ocorrer isoladamente ou associadas a bócios nodulares benignos. Alguns
adenomas são normofuncionantes, e outros são hiperfuncionantes. Os adenomas
foliculares são tumores encapsulados, benignos, e os mais encontrados.
O carcinoma tiroideu é mais frequente em pacientes submetidos a irradiação do
pescoço na infância. Os tumores malignos são classificados como papiloma,
folicular, indiferenciado, medular e epidermóide. O carcinoma papiloma é o mais
frequente.
O carcinoma medular é responsável por 5% dos carcinomas e é derivado das
células parafoliculares da tiróide. Pode ocorrer esporadicamente ou ter um padrão
familiar, associado a outras neoplasias endócrinas.
Hormona Estimulante da Tiróide (TSH)
É muito útil no diagnóstico do hipotiroidismo primário, sendo a primeira a ser
alterada. Na fase inicial da doença, apenas o TSH se encontra elevado, enquanto os
níveis séricos do T3 e T4 permanecem normais. Com a introdução de ensaios ultrasensíveis, o doseamento de TSH tornou-se valioso para a detecção do
hipertiroidismo, substituindo em muitos casos a prova do TRH.
Tiroxina (T4)
Anticoncepcionais, gravidez e beta-bloqueadores elevam os níveis de T4, sem que
isso necessariamente signifique doença tiroidéia. Outros factores que podem
interferir são o uso de hormonas tiroidéias, alteração congénita dos níveis de TBG,
uso de salicilatos, diazepam e corticóides, desnutrição, patologias hepáticas ou
renais, outras drogas e a presença de anticorpos anti-T4.
É útil no diagnóstico do hiper- e do hipotiroidismo. Nos casos de hipotiroidismo
primário, é a segunda alteração laboratorial a surgir. Após a elevação do TSH,
ocorre a diminuição do T4, podendo o T3 ainda permanecer em níveis normais.
Apenas nos estados mais avançados da doença ocorrerá diminuição do T3.
Tiroxina Livre (T4L)
A fracção livre reflete o efeito metabólico da hormona, sendo indicada para
avaliação do hiper- e do hipotiroidismo, minimizando a influência das proteínas
séricas. Torna-se assim, mais valiosa do que o doseamento do T4 total,
especialmente em grávidas ou em mulheres a fazer anticoncepcionais. É apontado
como uma interferência o uso de hormona exógena, de drogas antitiroidéias e de
beta-bloqueadores.
Triiodotironina (T3)
Útil no diagnóstico de hipertiroidismo. No hipotiroidismo, é a última a ser alterada,
podendo ainda permanecer normal mesmo com TSH elevado e T4 diminuído. Pode
estar diminuída nas doenças graves em geral, no uso de beta-bloqueadores ou
corticóides.
Triiodotironina Livre (T3L)
A grande indicação do T3L é o diagnóstico e o acompanhamento do paciente
hipertiroideu.
Globulina Ligadora da Tiroxina (TBG)
É a principal proteína transportadora das hormonas da tiróide. O aumento ou a
diminuição conduz a uma resposta paralela da T4 e T3 totais.
São consideradas as causas mais comuns de aumento da TBG: gravidez, uso de
estrógenios, hepatite aguda, salicilatos, diazepam, fenilbutazona e factores
congénitos.
Síndrome nefrótico, andrógenios, corticóides, cirrose hepática, desnutrição e
factores congénitos são considerados as causas mais comuns de diminuição de
TBG.
Tireoglobulina (TG)
Varia com o estado funcional da glândula e encontra-se elevada no hipertiroidismo,
em tiroidites e em carcinomas da tiróide. O seu principal valor é no seguimento de
carcinomas pós-cirúgicos (especialmente papiloma, folicular e misto papilomafolicular).
Para isso, é fundamental o dosemento pré-operatório da tireoglobulina e do seu
anticorpo, permitindo um acompanhamento da queda dos níveis de TG no pós-
operatório. Se no pré-operatório os níveis de TG forem muito baixos, ou o anticorpo
for positivo, o teste não terá importância no acompanhamento do caso específico.
Calcitonina
O seu doseamento é útil para o diagnóstico dos casos de carcinoma medular da
tiróide, dos quais cerca de 10% é familiar e normalmente integra os componentes
da neoplasia endócrina múltipla tipo II.
Aproximadamente 30% dos carcinomas medulares da tiróide apresentam níveis
basais normais. Por isso, é necessário o teste de estímulo com pentagastrina para o
diagnóstico. A calcitonina pode estar elevada nos carcinomas pulmonar, do
pâncreas e da mama, na insuficiência renal, anemia perniciosa, cirrose, síndrome
de Zollinger-Ellison, gravidez a termo e recém-nascidos.
Anticorpos antitiroideus
A pesquisa de anticorpos antiroideus é também de grande importância nas doenças
auto-imunes da tiróide. Os mais pesquisados são os anticorpos antitiroglobulina
(anti-TG), antiperoxidase (anti-TPO) e anti-receptor do TSH (Trab).
Consultar Anticorpos Antireóidianos.
PATOLOGIAS
HIPERTIROIDISMO
HIPOTIROIDISMO
PRIMÁRIO
HIPOTIROIDISMO
SECUNDÁRIO
HIPOTIROIDISMO
TERCIÁRIO
TSH
T3
T3L
T4
T4L
TRH
Suprimido
Hiper-reactivo
Não-reactivo
Gradualmente
reactivo
Interferências Medicamentosas
FÁRMACOS
T4 TOTAL T3 TOTAL
Amiodarona
Ácido iopanóico
TBG
N
N/
N/
N
Danazol
N
N
Fenilbutazona
Fenobarbital
-
Furosemida
N/
N/
N
N
N/
N/
Lítio
N/
N
Propiltiouracil
N/
N/
Resorcina
N/
N
N
N
-
N
-
N
N/
-
N
-
Propanolol
N
-
Iodo
Salicilatos
N/
N/
Heparina
Metoclopramida
-
N/
ou
N
Fenitoína
Glicocorticóides
-
N
Estrogénio
N
-
N/
N/
Diazepam
Dopamina
N
Carbamazepina
TRH
N/
-
N/
Androgénios
Anfetamina
TSH
-
-
-
N ou
-
-
-
-
-
-
Sulfoniluréia
N/
-
-
-
Toxoplasmose
A toxoplasmose é uma zoonose causada pelo Toxoplasma gondii. É um parasita de
vida intracelular obrigatória. A transmissão aos humanos normalmente ocorre por
ingestão de oocistos em alimentos crus ou mal cozidos, água não-potável, contacto
com animais infectados e por via transplacentária.
A infecção é, geralmente, assintomática ou subclínica, com raros casos graves, que
se manifestam especialmente em indivíduos imunocomprometidos e na gravidez,
levando a riscos abortivos e teratogénicos. No feto, causa lesões cerebrais e
oculares, de gravidade variável, dependendo da fase da gestação em que ocorreu a
infecção. No adulto, na fase aguda, raros casos mais graves podem evoluir com
coriorretinite e comprometimento cerebral. Indivíduos infectados não-tratados na
fase aguda podem evoluir para a doença crónica, por persistência da forma quística
do parasita, podendo ocorrer a activação da doença em situações de
comprometimento do estado imunológico.
Aliado ao facto de que a maioria dos casos não apresenta dados clínicos
significativos, existe uma grande incidência de anticorpos IgG na população,
apresentando positividade em cerca de 70 a 90% dos adultos investigados. Isso
cria uma dificuldade para se estabelecer o diagnóstico clínico e sorológico, o que
torna a realização e a interpretação da sorologia para toxoplasmose de grande
importância, especialmente na possibilidade de infecção congénita.
A investigação sorológica para toxoplasmose deve ser sempre realizada no período
pré-natal. A detecção de anticorpos IgG, indicando infecção passada, afasta o risco
de possibilidade de toxoplasmose congénita. Mulheres com sorologia negativa
devem ser consideradas grupos de risco e acompanhadas durante toda a gestação.
O diagnóstico sorológico baseia-se na demonstração de anticorpos específicos
contra antigénios do Toxoplasma gondii no soro dos pacientes infectados.
Anticorpos IgG e IgM podem ser detectados por imunofluorescência indireta (IFI),
hemaglutinação e testes imunoenzimáticos.
Os anticorpos da classe IgG aparecem de 1 a 2 semanas após a infecção e
persistem por toda a vida. A detecção de anticorpos da classe IgM é utilizada para
diagnóstico de infecção aguda e geralmente surge cerca de 5 dias após o contacto
com o parasita, desaparecendo após algumas semanas ou meses. Porém, baixos
títulos de IgM podem permanecer por um tempo superior a 1 ano.
A grande sensibilidade desses testes torna possível a detecção de anticorpos IgM,
mesmo em níveis baixos, por longo período de tempo após a fase aguda, fazendo
com que a presença de IgM não seja suficiente para o diagnóstico da doença em
fase aguda.
Este facto pode levar a incertezas quando os anticorpos IgM e IgG são detectados
numa primeira amostra de sangue de uma gestante, podendo tratar-se de uma
infecção adquirida anteriormente à gravidez, que não traz riscos para o feto, ou de
uma infecção aguda, que requer intervenção e tratamento.
Portanto, actualmente, para definir a fase da doença, dispomos da avaliação dos
testes de avidez dos anticorpos IgG. Esses testes baseiam-se na característica de
baixa avidez que os anticorpos apresentam pelo antigénio, durante o início da
resposta imunológica. Portanto, na infecção recente, estão presentes os anticorpos
IgG de baixa avidez, e nas infecções mais antigas, encontramos os de alta avidez.
Consideram-se de baixa avidez índices inferiores a 30%, que indicam que a
infecção ocorreu nos últimos 4 meses; índices superiores a 60% são considerados
de alta avidez, apontando para uma infecção ocorrida há mais de 4 meses. Valores
entre 30% e 60% não permitem a caracterização da fase da doença.
O diagnóstico de infecção fetal é, actualmente, realizado pela pesquisa de
toxoplasma no líquido amniótico pela técnica de PCR. O doseamento de IgM em
amostras colhidas por punção do cordão umbilical, além de ser realizada somente a
partir da 22ª semana de gestação, é pouco sensível, não detectando baixos níveis
de IgM pelos métodos disponíveis.
O diagnóstico da infecção congénita em recém-nascidos é dificultado pela
positividade para IgG, indicando transferência transplacentária de anticorpos
maternos que permanecem positivos por cerca de 6 a 18 meses. A presença de
IgM, que não tem passagem transplacentária, pode indicar infecção fetal, porém
não é suficiente para se estabelecer o diagnóstico, visto que o feto pode produzir
anticorpos IgM tardiamente. Actualmente, o método de escolha para o diagnóstico
de toxoplasmose congénita é a pesquisa de toxoplasma no sangue periférico do
recém-nascido ou do cordão umbilical pela técnica de PCR.
Transaminase Oxaloacética
A aspartato aminotransferase - AST, antigamente denominada transaminase
oxaloacética, é encontrada em diversos órgãos e tecidos, incluindo coração, fígado,
músculo esquelético e eritrócitos.
Está presente no citoplasma e também nas mitocôndrias, e portanto a sua elevação
indica um comprometimento celular mais profundo. No caso do hepatócito, isso
revela-se por uma elevação por tempo mais prolongado no curso das hepatites
virícas agudas e uma elevação selectiva nos casos de hepatites alcoólicas,
metástases hepáticas e necroses medicamentosas e isquémicas.
Aumentos da AST no soro são muitas vezes encontrados no enfarte agudo do
miocárdio, elevando-se nas primeiras 12 horas e apresentando um pico sérico após
cerca de 24 horas, com retorno aos valores normais num período de 3 a 5 dias.
Valores discretamente elevados podem ser encontrados também no enfarte
pulmonar, no enfarte renal ou em casos de grandes tumores, na embolia pulmonar,
distrofias musculares, dermatomiosite, traumas da musculatura esquelética, no
pós-operatório, especialmente de cirurgias cardíacas, cirrose alcoólica, hepatite
induzida por fármacos, mononucleose infecciosa, citomegaloviroses, anemias
hemolíticas, pancreatite aguda e acidente vascular cerebral.
Transaminase Pirúvica
A alanina aminotransferase - ALT, antigamente denominada transaminase pirúvica,
é encontrada abundantemente no fígado, em quantidades moderadas no rim e em
pequenas quantidades no coração e na musculatura esquelética.
A sua origem é predominantemente citoplasmática, fazendo com que se eleve
rapidamente após a lesão hepática, tornando-a um marcador sensível da função do
fígado. Como marcador hepatocelular, apresenta valores alterados em patologias
que cursam com necrose do hepatócito, como hepatites virícas, mononucleose,
citomegalovirose e hepatites medicamentosas. Entretanto, é um marcador menos
sensível que a AST para hepatopatias alcoólicas, cirrose activa, obstruções extrahepáticas e lesões metastáticas do fígado.
Pode apresentar-se elevada, em situações de trauma da musculatura esquelética,
miosites e miocardites, e normal ou discretamente elevada nos casos de enfarte
agudo do miocárdio.
Em recém-nascidos, podem ser encontrados valores superiores aos de referência, o
que é atribuído à imaturidade dos hepatócitos nos recém-nascidos, que apresentam
as membranas celulares mais permeáveis. Os valores igualam os níveis do adulto
por volta dos 3 meses de idade.
Transferrina
A transferrina é uma proteína de transporte e leva o ferro no plasma e no líquido
extracelular para suprir as necessidades teciduais. Aparece como uma banda
distinta na electroforese de proteínas e é o maior componente da fracção
betaglobulina. A maior parte é sintetizada pelo fígado, e o restante, por diferentes
locais. A ligação com o ferro é estável em condições fisiológicas, mas a dissociação
pode ocorrer em meio ácido. É capaz de se ligar a outros elementos, como cobre,
zinco, cobalto e cálcio, mas com exceção da ligação ao cobre, não há significado
fisiológico.
É responsável pelo transporte do ferro do seu local de absorção a nível intestinal ou
nos locais de catabolismo da hemoglobina para os precursores de células vermelhas
na medula óssea ou para os locais de armazenamento de ferro no sistema
reticuloendotelial na medula óssea, no fígado e no baço. Após a libertação do ferro,
a transferrina retorna à circulação e é reciclada. A sua semi-vida é de 8 dias. Além
da função de transporte, a transferrina minimiza os níveis de ferro livre no plasma,
a perda urinária de ferro, e previne os potenciais efeitos tóxicos de níveis elevados
de ferro livre circulante.
O organismo contém cerca de 3 a 5 gramas de ferro, porém apenas 3 a 5
miligramas são encontrados no plasma. A maioria apresenta-se ligada à
transferrina. No entanto, uma pequena parcela pode ligar-se a outras proteínas,
como a albumina. O nível de ferro livre circulante é muito pequeno, portanto o ferro
sérico avaliado reflecte basicamente o que se encontra ligado à transferrina.
A transferrina apresenta grande polimorfismo genético, e têm sido identificadas
variantes pela electroforese. Essas variantes são denominadas de acordo com a sua
posição de migração na electroforese comparadas com o tipo mais comum,
conhecido como transferrina C. Apesar dessas variantes não estarem associadas a
patologias, em alguns casos podem migrar junto com outros componentes na
electroforese, mascarando os resultados. Raramente, quadros congénitos de
atransferrinémia resultam em níveis praticamente indetectáveis de transferrina,
associada a sobrecarga férrica e a anemia severa por dificuldade de mobilização
dos stoques do ferro orgânico.
A diminuição dos níveis de transferrina pode ser observada nas doenças hepáticas e
em situações clínicas com perdas protéicas, como certas enteropatias, síndrome
nefrótico e desnutrição, além de ser um bom marcador de desnutrição em
pacientes hospitalizados. Níveis baixos podem ser encontrados numa variedade de
estados inflamatórios agudos e crónicos e em casos de malignidade.
O doseamento de transferrina é importante na avaliação das anemias. Na anemia
ferropriva, o nível de transferrina está elevado, mas a sua percentagem de
saturação é baixa. Na anemia das doenças crónicas, a transferrina apresenta-se
normal, e a percentagem de saturação está aumentada. Níveis elevados também
podem ser encontrados nos estadios iniciais de hepatites agudas, na gravidez e no
uso de estrogénios. Nas hemocromatoses idiopáticas e hemossideroses por
sucessivas transfusões sanguíneas, são observados níveis elevados de saturação
(acima de 90%).
A transferrina apresenta-se aumentada na deficiência crónica de ferro nãocomplicada, alterando-se simultaneamente ou por vezes um pouco antes das
alterações dos níveis séricos do ferro. Entretanto, a sua correlação clínica não é
inteiramente satisfatória, visto que, em cerca de 30 a 40% dos pacientes com
anemia ferropriva crónica, podem ser encontrados valores dentro dos limites da
normalidade.
A transferrina não é uma das proteínas de fase aguda. Portanto, apresenta-se
diminuída mesmo nos casos de doenças agudas ou crónicas graves, que podem
cursar com deficiência de ferro sérico. Justamente por isso, trata-se de um bom
parâmetro para acompanhamento.
Triglicerídeos
Os triglicerídeos circulantes são provenientes da dieta (fonte exógena) e do fígado
(fonte endógena). Triglicerídeos, ésteres de ácidos gordos de glicerol, representam
a maior quantidade de gordura no organismo. A sua função primária é armazenar e
providenciar energia para as células. A concentração de triglicerídeos do plasma é
dada pelo balanço entre as taxas de entrada e de eliminação dessas moléculas no
organismo. As concentrações de triglicerídeos no plasma variam conforme a idade e
o sexo. Aumentos moderados ocorrem durante o crescimento e o desenvolvimento.
Doseamentos de triglicerídeos são usadas para avaliar hiperlipidemias. Altas
concentrações podem ocorrer com hipoparatiroidismo, síndrome nefrótico, doenças
de depósitos de glicogénio e diabetes mellitus.
Concentrações extremamente elevadas de triglicerídeos são muitas vezes
encontradas em casos de pancreatite aguda. Alguns fármacos, como
anticoncepcionais orais e estrogénio, podem levar a resultados falsamente
elevados.
O papel dos triglicerídeos no risco de desenvolvimento de doença arterial coronária
tem sido bastante discutido.
Até agora, os trabalhos apontavam os triglicerídeos não como factores de risco
independentes, mas sim como associados à presença de outros factores de risco,
variando inversamente com os valores de HDL-colesterol e directamente com os
níveis séricos do LDL-colesterol.
Estudos clínicos e epidemiológicos mais recentes demonstraram que o aumento das
concentrações dos níveis de triglicerídeos pode ser considerado um factor de risco
independente para aterosclerose. A dificuldade dessa avaliação deve-se às diversas
partículas ricas em triglicerídeos.
Cabe lembrar que níveis séricos aumentados de triglicerídeos aumentam a
adesividade plaquetária, favorecendo a trombogénese.
Troponinas
As troponinas são constituídas por três diferentes proteínas (C,T e I) que estão
presentes nos músculos esquelético e cardíaco, onde são elementos importantes no
processo contrátil. São codificadas por genes diferentes, sendo que a troponina C
se expressa de forma idêntica, e as troponinas T e I, de forma diferenciada nas
duas localizações.
Essa característica das troponinas T e I permitiu a identificação por anticorpos
monoclonais e a sua utilização no diagnóstico diferencial do enfarte agudo do
miocárdio (EAM).
Outra característica importante é que as troponinas não são detectadas em
pacientes hígidos, fazendo com que mesmo pequenos níveis detectados na fase
inicial da lesão sinalizem de forma precoce a presença de injúria miocárdica.
A troponina I é a mais específica para lesões do músculo cardíaco, eleva-se entre 4
a 6 horas e atinge o pico em torno de 12 horas após a lesão miocárdica,
permanecendo elevada no soro por um período de 3 a 10 dias. Esse marcador tem
sido apontado como o marcador de lesão miocárdica mais próximo do ideal,
demonstrando claramente o seu valor prognóstico tanto no enfarte como na angina
instável. Vários estudos demonstraram a sua positividade na presença de
microenfartes indetectáveis por outros marcadores.
A troponina T apresenta-se alterada nas lesões do músculo cardíaco, aparece no
soro, após o início dos sintomas, com curva semelhante à do aparecimento da CKMB, eleva-se entre 3 a 12 horas e atinge o pico em torno de 24 horas após a lesão
miocárdica, com a característica de se manter elevada por mais tempo, 12 a 14
dias.
Devido à sua alta concentração nos músculos cardíacos, pela sua alteração precoce
e por normalmente não estar detectável na circulação, o doseamento das
troponinas, especialmente a troponina I, tem sido utilizada com alta sensibilidade e
especificidade como um novo marcador de lesão miocárdica.
Uréia Sérica
A uréia é um produto do catabolismo de aminoácidos e proteínas. Gerada no fígado,
é a principal fonte de excreção do nitrogénio do organismo. É difundida através da
maioria das membranas celulares, e a sua maior parte é excretada pela urina,
sendo que pequenas quantidades podem ser excretadas pelo suor e degradadas por
bactérias intestinais.
É livremente filtrada pelos glomérulos e é dependente da velocidade do fluxo
urinário, ligado directamente ao grau de hidratação. Grande parte da uréia filtrada
é reabsorvida passivamente nos túbulos proximais.
No indivíduo saudável, a sua concentração varia de acordo com diferentes factores
tais como o conteúdo protéico da dieta e a hidratação.
Os níveis séricos da uréia são alterados por diferentes formas de acção sobre o seu
metabolismo. Os glicocorticóides e a hormona tireidéia aumentam, e os
androgénios e a hormona de crescimento diminuem os seus níveis séricos.
Apesar de ser um marcador da função renal, é considerada menos eficiente do que
a creatinina pelos diferentes factores não-renais que podem afectar a sua
concentração. No entanto, a sua elevação é mais precoce, e não sofre com a
variação da massa muscular. A avalição conjunta com a creatinina é útil no
diagnóstico diferencial das causas de lesão renal.
Os aumentos dos níveis séricos da uréia podem ser classificados, de acordo com a
sua origem, como pré-renais, renais e pós-renais. O quadro abaixo apresenta essa
classificação.
UREMIA PRÉ-RENAL Níveis aumentados de produção
(Função renal normal) de uréia ou diminuição
do fluxo sanguíneo
UREMIA RENAL
Doença renal intrínseca
UREMIA PÓS-RENAL Obstrução do fluxo renal
(Reabsorção da uréia )
Catabolismo protéico aumentado,
ingestão excessiva de proteínas,
choque traumático ou hemorrágico,
desidratação, descompensação cardíaca
aguda, absorção de grandes
hemorragias, infecções maciças ou
toxémia.
Doença renal glomerular ou tubular
aguda ou crónica ou lesão
parenquimatosa difusa.
Obstrução do tracto urinário por cálculo,
obstrução externa, tumores de bexiga,
tumores ou hipertrofia da próstata,
defeitos congénitos de bexiga ou uretra
Os níveis séricos diminuídos são mais raros e decorrem de importante restrição da
ingestão de proteínas, desidratração, reposição excessiva de líquidos, durante a
gestação e nas doenças hepáticas graves por diminuição da síntese da uréia.
Urina Tipo II e Sedimento urinário
O exame rotineiro de urina é um método simples, não-invasivo, capaz de fornecer
uma variedade de informações úteis em relação a patologias envolvendo os rins, o
trato urinário e, por dados indirectos, algumas patologias sistémicas.
Sumário de urina, exame de urina tipoII e sedimento urinário são alguns dos
sinónimos empregues na identificação desse exame.
Apesar de simples, diferentes técnicas encontram-se envolvidas na sua realização,
em quatro etapas distintas:
- avaliação da amostra,
- análise física,
- análise química,
- análise microscópica do sedimento.
Avaliação da Amostra
Como na maioria dos exames laboratoriais, a qualidade dos resultados depende da
colheita.
A urina deverá ter sido colhida recentemente, com um volume mínimo de 20 mL,
sem adição de preservativos, refrigerada e nunca congelada, para garantir a sua
melhor preservação. Deve estar claramente identificada e colhida em recipiente
adequado.
A colheita deverá ser realizada após assepsia da área genital, desprezando-se o
primeiro jacto e colhendo-se o jacto intermediário. O recomendável é a colheita da
primeira micção da manhã ou uma amostra com pelo menos quatro horas de
intervalo da última micção, em recipiente de plástico esterilizado. Se necessário, a
amostra poderá ser colhida a qualquer hora, lembrando-se da existência, durante o
dia, de variações em relação à dieta, exercício físico, concentração da urina e uso
de medicamentos.
O exame do primeiro jacto da urina é recomendado quando o objetivo é a
investigação do trato urinário inferior, mais especificamente da uretra. A urina de
primeiro jacto contém células e bactérias presentes na uretra, tornando-a uma boa
amostra indirecta para outras avaliações, como as uretrites com pouca secreção. A
diferença de celularidade encontrada entre o primeiro e segundo jactos auxilia a
localizar a origem do processo.
Análise Física
Aspecto
O aspecto normal é límpido. Entretanto, uma ligeira turvação não é
necessariamente patológica, podendo ser decorrente da precipitação de cristais e
de sais amorfos não-patológicos.
A turvação patológica pode ser consequência da presença de células epiteliais,
leucócitos, eritrócitos, cristais, bactérias e leveduras.
Pode ocorrer a presença de depósito por excesso de muco em função de processos
inflamatórios do trato urinário inferior ou do trato genital, ou pela presença de
grande quantidade de outros elementos anormais.
Cor
A cor habitual da urina é amarelo citrino, o que se deve, na sua maior parte, ao
pigmento urocromo. Essa coloração pode apresentar variações em situações como
a diluição por uma grande ingestão de líquidos, que torna a urina amarelo-pálida.
Uma cor mais escura pode ocorrer por privação de líquidos.
Portanto, a cor da urina pode servir como avaliação indirecta do grau de hidratação
e da capacidade de concentração urinária.
O uso de diversos medicamentos e a ingestão de corantes alimentares também
podem causar alteração da cor da urina.
Há numerosas possibilidades de variação de cor, sendo a mais frequente a cor
avermelhada (rosa, vermelha, vermelho-acastanhada). Em mulheres, deve-se
sempre afastar a possibilidade de contaminação vaginal. A cor avermelhada pode
acontecer na presença de medicamentos, eritrócitos, hemoglobina,
metaemoglobina e mioglobina. As porfirias também podem cursar com coloração
vermelha ou púrpura da urina.
Também é frequente a cor âmbar ou amarelo-acastanhada, pela presença de
bilirrubina, levando a urina a apresentar-se verde-escura em quadros mais graves.
Densidade
A densidade ajuda a avaliar a função de filtração e concentração renais, bem como
o estado de hidratação do corpo. Depende directamente da proporção de solutos
urinários presentes (cloreto, creatinina, glicose, fosfatos, proteínas, sódio, sulfatos,
uréia, ácido úrico) e o volume de água. Normalmente varia entre 1.015 a 1.030.
Densidades diminuídas podem ser encontradas na administração excessiva de
líquidos por via intravenosa, reabsorção de edemas e transudados, insuficiência
renal crónica, uso de drogas, quadros de hipotermia, aumento da pressão
intracraniana, diabetes insipidus e hipertensão maligna.
Densidades elevadas podem ser encontradas na desidratação, diarréia, vómitos,
febre, diabetes mellitus, glomerulonefrite, insuficiência cardíaca congestiva,
insuficiência supra-renal, proteinúria, síndrome de secreção inapropriada de
hormona antidiurético, toxémia gravídica, uropatias obstructivas e no uso de
algumas substâncias, como contrastes radiológicos e sacarose.
Análise Química
Corpos Cetónicos
Os corpos cetónicos são um subproduto do metabolismo da gordura e dos ácidos
gordos que proporciona fonte de energia para as células quando as reservas de
glicose estão diminuidas ou quando a glicose não pode penetrar nas células devido
à falta de insulina.
Os corpos cetónicos que passam para a corrente sanguínea são quase totalmente
metabolizados no fígado. Quando são formados em velocidade maior do que o
normal, são excretados na urina. O jejum ou a dieta podem determinar o
aparecimento de corpos cetónicos na urina. O uso de alguns fármacos pode levar a
falso-negativos, entre elas o captopril, a levodopa e o paraldeído.
Bilirrubina
Aumentadas nas situações em que ocorre o aumento da bilirrubina sérica
conjugada e sua consequente presença na urina. Portanto, valores elevados podem
ser encontrados em doenças hepáticas e biliares, lesões parenquimatosas,
obstruções intra e extra hepáticas, neoplasias hepáticas ou do trato biliar. Ao
contrário, estará sempre ausente nas ictéricias por hemólise. Alguns casos de
doença biliar obstrutiva crónica podem cursar com níveis alterados de bilirrubina
sérica e ausência de bilirrubina na urina. Falso-negativos podem ser induzidos pelo
uso de ácido ascórbico e exposição da urina à luz intensa por longo tempo.
Hemoglobina
A presença de hemoglobina na urina pode ser proveniente de diferentes estados de
hemólise intravascular, em que uma quantidade excessiva de hemoglobina satura a
capacidade de ligação com a haptoglobina. Nessas condições, fica livre no plasma,
sendo filtrada pelo glomérulo e em parte reabsorvida pelo sistema tubular. A
restante é excretada na urina.
A outra causa é a presença de eritrócitos libertados no trato urinário por pequenos
traumas, exercícios extenuantes ou patologias das vias urinárias, em que as
hemácias são lisadas, libertando hemoglobina. A verdadeira hemoglobinúria é rara,
sendo mais frequente a segunda situação, em que a hemoglobinúria é
acompanhada pela presença de hematúria.
Glicose
A glicose presente na urina reflecte os níveis séricos da glicose associados à
capacidade de filtração glomerular e de reabsorção tubular. Normalmente, a
glicosúria só se manifesta quando os níveis séricos se encontram acima de 160/180
mg/dL.
A glicosúria pode ser causada tanto pelo diabetes mellitus como por outras
patologias, como doenças renais que afectem a reabsorção tubular e nos quadros
de hiperglicemia de outras origens que não a diabética.
Nitritos
A presença de nitritos na urina indica infecção das vias urinárias, causadas por
microrganismos que reduzem os nitratos a nitritos. O achado de reacção positiva
indica a presença de infecção nas vias urinárias, principalmente por bactérias
entéricas.
pH
Avalia a capacidade de manutenção renal da concentração de iões de hidrogénio no
plasma e líquidos extracelulares. Participando do equilíbrio ácido-base, os rins,
quando em funcionamento normal, excretam o excesso de iões de hidrogénio na
urina. Portanto, o pH da urina reflecte o pH plasmático e é um indicador da função
tubular renal. Normalmente varia entre 5,0 a 8,0.
Valores elevados podem ser encontrados na alcalose respiratória, em dietas com
grande ingestão de vegetais e frutas cítricas, hiperémese ou o uso prolongado de
sonda nasogátrica, na presença de cálculos renais, infecção das vias urinárias,
especialmente por microrganismos que utilizam uréia (Proteus e Pseudomonas sp.),
síndrome de Cushing, hiperaldosteronismo, hipocalemia, insuficiência renal,
síndrome de Fanconi e subredosagem de alcalinos. Os fármacos também podem
alterar o pH urinário, como os diuréticos e a terapia alcalina (bicarbonatos).
Valores diminuídos podem ser encontrados em acidose metabólica e respiratória,
perda de potássio, dieta rica em proteínas, infecção das vias urinárias por
Escherichia coli, diarréias severas, diminuição de cloro, fenilcetonúria e tuberculose
renal. O uso de anestésicos e de ácido ascórbico, assim como de outros fármacos,
pode diminuir o pH urinário.
Proteínas
Em indivíduos normais, uma pequena quantidade de proteína é filtrada pelo
glomérulo (albumina, alfa-1 e alfa-2-globulinas), sendo a sua maior parte
reabsorvida por via tubular e eliminada em pequenas quantidades pela urina. São
considerados normais, valores de até 150 mg/ 24 h.
O aumento da quantidade de proteínas na urina é indicador inicial de patologia
renal. Entretanto, não são todas as patologias renais que cursam com proteinúria, a
qual não é uma condição exclusiva de doença renal, podendo aparecer em
patologias não-renais e em algumas condições fisiológicas.
As proteínas são excretadas em velocidades diferentes e em momentos variáveis
durante o período de 24 horas, sendo maior durante o dia e menores durante a
noite.
As proteinúrias podem ser classificadas, quanto à sua origem, como pré-renal,
renal e pós- renal.
Pré-renal
Algumas patologias não-renais, como hemorragia, estados febris, algumas
endocrinopatias, distúrbios convulsivos, neoplasias, queimaduras extensas,
mioglobinúria, hemoglobinúria, mielomas, superexposição a certas substâncias
(ácido sulfossalicílico, arsénico, chumbo, éter, fenol, mercúrio, opiáceos), lesão do
sistema nervoso central, leucemia (mielocítica crónica), obstrução intestinal,
reacção de hipersensibilidade, toxémia, toxinas bacterianas (difteria, escarlatina,
estreptocócica aguda, febre tifóide e pneumonia) podem levar a proteinúrias ditas
pré-renais.
A proteinúria transitória pode surgir em consequüência de estados não-patológicos,
como stress físico (exercícios intensos) ou emocional, desidratação, dieta (proteínas
em excesso), exposição ao frio e posição do corpo (proteinúria ortostática).
Renal
Pode ocorrer devido a um comprometimento glomerular, tubular ou intersticial,
glomerulonefrites, síndrome nefrótico, nefropatia diabética, hipertensão (maligna,
renovascular), amiloidose, doença poliquística, lúpus eritematoso sistémico,
nefropatia membranosa, pielonefrite (crónica), tumores, malformações congénitas,
síndrome de Goodpasture, trombose da veia renal, acidose tubular renal, necrose
tubular aguda, intoxicação por metais pesados e alguns fármacos.
Pós-renal
Contaminação por material da área genital (uretral e genital), infecções do trato
urinário superior e inferior (uretrites e cistites) e prostatites.
Urobilinogénio
O urobilinogénio é um produto de redução formado pela acção de bactérias sobre a
bilirrubina conjugada no trato gastrointestinal. A maior parte do urobilinogénio é
excretada nas fezes. Uma pequena parte é reabsorvida através da via enterohepática e reexcretrada na bile e na urina. Os níveis urinários de urobilinogénio
geralmente são maiores do início até meio da tarde, mantendo-se em níveis
inferiores a 1 mg/dL. O aumento do urobilinogénio na urina indica a presença de
processos hemolíticos, disfunção hepática ou porfirinúria.
Análise Microscópica do Sedimento
Células Epiteliais
É comum o achado de algumas células epiteliais. Podem ser de três tipos distintos:
células escamosas, transacionais e dos túbulos renais. A maioria não tem
significado clínico, representando uma descamação de células velhas do
revestimento epitelial do trato urinário. O achado de células com atípias nucleares
ou morfológicas pode indicar a presença de processos neoplásicos necessitando de
investigação específica. A presença de fragmentos epiteliais e de células de origem
tubular pode estar ligada a processos de necrose tubular aguda e a lesões
isquémicas renais, entre outras lesões do rim.
Eritrócitos
Podem estar presentes em pequena quantidade na urina normal (2 a 10 por
campo). A presença de hematúria indica lesões inflamatórias, infecciosas ou
traumáticas dos rins ou vias urinárias. Deve-se sempre excluir contaminação por
via genital. O exercício extenuante pode levar a hematúria discreta.
A forma da apresentação dos eritrócitos, segundo alguns autores, pode indicar a
sua origem, servindo como um diagnóstico diferencial de hematúrias de origens
glomerular e não-glomerular. Quando se apresentam na sua forma esférica
habitual, seriam de origem mais distal no trato urinário; quando crenadas
(irregulares), teriam origem glomerular.
Leucócitos
Podem estar presentes em pequena quantidade na urina normal (2 a 10 por
campo). Normalmente neutrófilos. Quantidades aumentadas indicam a presença de
lesões inflamatórias, infecciosas ou traumáticas em qualquer nível do trato urinário.
Deve-se sempre excluir contaminação por via genital.
Cilindros
São elementos exclusivamente renais compostos por proteínas e moldados
principalmente na luz dos túbulos contornados distais e túbulos colectores.
Indivíduos normais, principalmente após exercícios extenuantes, febre e uso de
diuréticos, podem apresentar pequena quantidade de cilindros, geralmente hialinos.
A sua formação é influenciada pelos elementos presentes no filtrado e pelo tempo
de permanência dentro do túbulo. Nas doenças renais, apresentam-se em grandes
quantidades e em diferentes formas, de acordo com o local da sua formação.
Os mais comuns são os cilindros hialinos. São compostos principalmente pelas
proteínas de Tamm-Horsfall, considerados normais em pequenas quantidades (0 a
2) e em maior quantidade em situações como febre, desidratação, stress e
exercício físico intenso.
Os cilíndros podem estar presentes em diferentes patologias como os hemáticos
(doença renal intrínseca), leucocitários (pielonefrites), de células epiteliais (lesões
túbulos renais), granulosos (doença renal glomerular ou tubular e algumas
situações fisiológicas) e céreos (insuficiência renal, rejeição a transplantes e
doenças renais agudas e estase do fluxo urinário).
Cristais
São um achado frequente na análise do sedimento urinário normal, raramente com
significado clínico e com ligação directa com a dieta.
Alguns cristais representam um sinal de distúrbios físico-químicos na urina ou têm
significado clínico específico, como os de cistina, leucina, tirosina e fosfato de
amoníaco magnesiano. Podem também ser observados cristais de origem
medicamentosa e de componentes de contrastes urológicos.
A cistina está ligada a um defeito metabólico, designado por cistinúria, e é
responsável por cerca de 1% dos cálculos urinários. Como a tirosina e a leucina são
resultado de catabolismo protéico, o seu aparecimento na urina sob a forma de
cristais pode indicar necrose ou degeneração tecidual importante. Os cristais de
fosfato amoníaco magnesiano estão relacionados a infecções por bactérias
produtoras de urease.
Apesar de não existir uma relação directa entre a presença de cristais e o
desenvolvimento de cálculos, alguns autores apontam a existência de diferenças
morfológicas entre os cristais dos pacientes que desenvolvem calculose com uma
apresentação de formas maiores, agregadas e bizarras.
Muco
Produzido pelo epitélio do túbulo renal e células epiteliais. A presença excessiva de
muco decorre de processos inflamatórios do trato urinário inferior ou do trato
genital.
Velocidade de Sedimentação
A velocidade de sedimentação (VS) reflecte o resultado entre as forças envolvidas
no movimento de sedimentação dos eritrócitos e os mecanismos oponentes
exercidos por substâncias plasmáticas, principalmente o fibrinogénio e as proteínas
de fase aguda.
A capacidade de agregação dos eritrócitos depende de factores ligados às mesmas,
como a força de coesão entre os eritrócitos e a sua carga eléctrica, que tem uma
força repulsiva que mantém os eritrócitos afastados em condições normais, e
factores plasmáticos que têm como função atenuar o efeito das forças repulsivas.
A presença de processos inflamatórios leva a uma agregação maior dos eritrócitos,
formando agregados conhecidos como rouleaux. Esse fenómeno favorece o
aumento da velocidade de sedimentação dos eritrócitos.
O aumento da concentração plasmática de imunoglobulinas e fibrinogénio leva a
uma diminuição da força repulsiva entre os eritrócitos, facilitando a agregação e
aumentando portanto a VS. A presença de proteínas anómalas, como no mieloma,
de eritrócitos alterados em número, forma ou tamanho e o uso de medicamentos
podem levar a uma alteração da VS, mesmo na ausência de resposta de fase
aguda. As principais alterações que podem levar a um aumento significativo da VS
(=100mm na 1ª hora) são processos infecciosos, doenças do tecido conjunctivo,
neoplasias e doenças renais.
A velocidade de sedimentação (VS) é um indicador não-específico de infecção e
lesão tecidual. É útil para monitorizar a inflamação crónica, inclusive a actividade
da doença como na artrite reumatóide. A VS é mais útil do que a proteína C
reactiva para o diagnóstico e a monitorização da polimialgia reumática e a artrite de
células gigantes, em que se encontra frequentemente elevada durante a recaída.
Homens entre 45-64 anos com VS no limite superior têm duas vezes mais risco de
morte de doença coronária do que os homens com VS na faixa inferior, depois de
ajustar outros factores de risco.
O método tem alta sensibilidade com baixa especificidade, o que leva a alterações
em inúmeras situações patológicas e em algumas situações fisiológicas como
período menstrual, gravidez, temperatura, sexo e idade.
ALTERAÇÕES DA VELOCIDADE DE SEDIMENTAÇÃO
ELEVADA
Infeceções bacterianas
Hepatite aguda, hepatopatia crónica
Pancreatites, colites e ilites, peritonites
Processos inflamatórios agudos e crónicos
Febre reumática
Lúpus eritematoso sistémico
Artrite reumatóide
Vasculites e dermatomiosites
Anemias graves
Leucemias e linfomas
DIMINUÍDA
Policitemia
Hemonoglobinopatia
Esferocitose
Alterações da forma dos eritrócitos
Microcitose
Hipofibrinogenemia
Insuficiência cardíaca
Cardiopatia congénita
Desnutrição grave
Lesões hepáticas graves
Metástases
Síndrome nefrótico, glomerulonefrite
aguda, pielonefrite
Tiroidites
Mieloma, crioglobulinémia e
macroglobulinémia
Necrose tecidual (cirurgias, queimaduras,
quimioterapia e radioterpia)
Uso de heparina
Uso de antiflamatórios
Vitamina B12
A vitamina B12 (cianocobalamina) é encontrada em praticamente todos os
alimentos de origem animal. É separada das proteínas animais pela acção da
secreção gástrica. Uma vez libertada, forma com o factor intrínseco (FI), um
complexo cobalamina-FI. Essa associação é indispensável para a sua absorção, pois
adere-se aos locais especifícos nas células epiteliais do íleo terminal, permitindo a
absorção da cobalamina num processo que demora várias horas para se completar.
Participa nos mecanismos de manutenção da hematopoiese, no metabolismo do
ácido fólico e na função neurológica. É transportada no plasma ligado a proteínas
chamadas de transcobalamina e armazenada principalmente no fígado.
A sua deficiência leva a anemia megaloblástica, a alterações dos níveis de ácido
fólico eritrocitário e a neuropatias.
Pode ocorrer nas deficiências da dieta, especialmente em dieta vegetariana estrita,
deficiência de factor intrínseco, síndromes de má absorção, alcoolismo, uso de
fármacos (contraceptivos orais, anticonvulsivantes e aspirina) e raros casos de
ausência congénita de transcobalamina.
Baixar