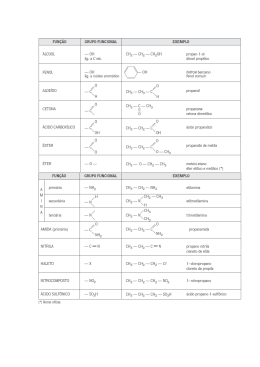
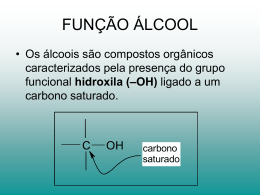
A. Isenmann
Princípios da Síntese Orgânica
5
Reações no grupo carbonila
5.1
Ácidos carboxílicos e seus derivados
5.1.1
Síntese de ácidos carboxílicos
O carbono do grupo carboxílico é o mais oxidado dentre todos os compostos orgânicos.
Assim, o preparo dos ácidos carboxílicos se dá, geralmente, por oxidação rigorosa (ver item
9.5.2) de outros compostos orgânicos.
5.1.2
Acidez - o equilíbrio prótico
Um resumo da acidez de diversas classes de compostos orânicos, entre outros, do ácido
carboxílico e de sua espécie protonada, encontra-se no anexo 2 deste livro (p. 898).
O entendimento da dependência da acidez (isto é, a facilidade de se ionizar por
desprotonação) é crucial para a maioria dos processos catalíticos, observados em ambiente
polar 216.
O
R C
+ H2O
Ka
O-
O
R C
O-
OH
Ácido carboxílico
+
R C
H3O+
O
Carboxilato
A mesomeria do carboxilato traz estabilidade ao ânion e explica a acidez elevada dos
ácidos carboxílicos, quando comparados com os alcoóis. Ocorre na verdade uma
deslocalização da carga negativa – um fato que será discutido criticamente a seguir.
Influência de entalpia e entropia no equilíbrio prótico dos ácidos carboxílicos
A constante deste equilíbrio é a constante de acidez, Ka , definida por 217:
Ka =
aRCOO− ⋅ aH O +
3
aRCOOH
RCOO − H 3O +
≈
[ RCOOH ]
;
log
!
1
= − log K a = pK a
Ka
(Lembre-se: quem descreve a força de um ácido em solução é a constante Ka e não o pH; o
pH é uma propriedade principalmente da água, ou seja, do solvente!)
As energias envolvidas nas desprotonações são bastante baixas, ficando na ordem de
grandeza das energias de Van der Waals ou da ativação de rotações em volta da ligação σ
de carbono-carbono (cerca de 20 kJ⋅mol-1). Para o ácido acético em água, à temperatura
216
V.Ferreira, P. Esteves, M.Vasconcellos, Ácidos e bases em química orgânica, Bookman 2007
A concentração da água, como está presente em grande excesso nestas soluções, pode ser considerada
constante e, portanto, entra no valor da constante Ka.
217
345
A. Isenmann
Princípios da Síntese Orgânica
ambiente, tem-se ∆G 0 = − R ⋅ T ⋅ ln K a = +27,2 kJ ⋅ mol −1 . Sendo assim, trata-se de uma
reação ligeiramente endergônica. No entanto, todas as protólises têm em comum o fato de
serem praticamente atérmicas, isto é, ∆H0 ≈ 0. Evidentemente se compensam as energias
que por si são de módulo alto: a energia que se gasta para liberar o próton e as energias
liberadas na criação de novas ligações, H+-solvente e carboxilato-solvente. São os solventes
mais polares que se organizam favoravelmente em volta dos íons. Resulta disso, um
complexo geralmente referido como "camada de solvente" ou "gaiola de solvente". No caso
do próton, são em média 4 moléculas de água; então seria mais correto notar "H3O+",
melhor ainda "H9O4+", em vez de "H+", como é o nosso costume. Independente da natureza
destas interações (ligação de hidrogênio, atração de Coulomb ou mesmo um caráter
covalente) pode-se afirmar que as reações de íons com a água são fortemente exotérmicas.
Como se explicam então as endergonias dos equilíbrios próticos?
Deve-se procurar a explicação nos efeitos entrópicos, envolvidos nos processos de
dissociação-associação. O grau de aumento em entropia causado pela desprotonação é igual
para todos os ácidos, já que uma molécula neutra produz um próton e um ânion carboxilato.
As diferenças se dão assim no grau de organização na camada do solvente em volta do
ânion. Um forte retirador de elétrons em posição α no ácido carboxílico (por exemplo, CF3) deslocaliza a carga negativa do ânion carboxilato. Desta forma o carboxilato fica mais
estável e não precisa de um grau de organização tão alto das moléculas de solvente ao seu
redor. Quer dizer, quanto mais distribuída a carga negativa no carboxilato, menor sua
camada de solvente.
Em geral vale a seguinte regra: um íon pequeno com alta carga pontual ("duro") é
estabilizado por uma espessa camada de solvente polar.
A organização de uma camada espessa de solvente tem por consequência uma redução na
entropia, que por sua vez é responsável pelo deslocamento do equilíbrio anotado acima,
para a esquerda.
O caminho desta argumentação termodinâmica é longo e complexo. Portanto, vamos
resumir todos esses efeitos, segundo sua argumentação lógica, fixados numa comparação
de dois derivados do ácido acético.
346
A. Isenmann
Princípios da Síntese Orgânica
Tabela 19.
Resumo dos efeitos termodinâmicos que explicam a diferença de
acidez entre ácido acético e ácido trifluoracético, a base dos diferentes comportamentos de
dissolução.
Critério
H3C-COOH
F3C-COOH
Efeitos térmicos, ∆H
≈0
≈0
Deslocalização de elétrons dentre os ânions
baixa
alta
carboxilatos
Localização pontual da carga no ânion
alta
baixa
Facilidade de estabelecer a camada de solvente
alta
baixa
Grau de organização
aumentou muito
aumentou pouco
diminui muito
diminui pouco
Entropia ∆S
mais endergônico menos endergônico
Força propulsora da dissolução, ∆G
Deslocamento do equilíbrio de dissociação
Para esquerda
Para direita
baixa
alta
⇒ Força do ácido
Ka (pKa)
baixo (alto)
alto (baixo)
As diferenças nos valores de pKa dos ácidos carboxílicos, conforme esta argumentação, têm
pouco a ver com as propriedades das moléculas em si (apesar disso, a argumentação com
efeitos indutivos e mesoméricos pelos grupos funcionais dos ácidos carboxílicos, é bastante
divulgada na literatura; exemplos sejam referidos abaixo). É mais satisfatório entender o
grau de dissociação e a acidez do sistema como propriedade do conjunto ácido/solvente,
sendo o resultado basicamente da entropia do sistema.
Quase todos os valores de pKa que se encontram na literatura subentendem uma solução
aquosa. A água tem uma constante dielétrica extremamente alta (εr ≈ 80), e assim
demonstra maior facilidade de organizar-se em volta de íons do que qualquer outro solvente
comum. Consequentemente os ácidos mostram o maior grau de dissociação em água 218.
Acidez e grau de dissociação dos ácidos carboxílicos
Os seguintes ácidos estão colocados em ordem de força decrescente.
Tabela 20.
Noção da força de ácidos carboxílicos
Ka
pKa
HCOOH (ácido fórmico)
1,8 · 10-4
3,74
H3CCOOH (ácido acético)
1,8 · 10-5
4,74
C6H5COOH (ácido benzóico)
6,3 · 10-5
4,20
Ácido
218
Um ácido bem conhecido mostra a forte dependência do grau de dissociação, do solvente: o ácido
clorídrico pode ser considerado completamente dissociado em H+ e Cl-, desde que estiver em solução
aquosa. Quando dissolvido em tolueno ou benzeno, em outro extremo, não conduz a corrente elétrica, pois
não é dissociado.
347
A. Isenmann
Princípios da Síntese Orgânica
Todos esses ácidos apresentam então pouca dissociação em água. O grau de dissociação α
é definido como parte dissociada em sua forma aniônica, em relação à quantidade total do
ácido dissolvido.
Para o ácido fórmico (1M), sendo o mais forte entre os ácidos carboxílicos simples, se
calcula
[HCOO ] ⋅100 =
−
α [%] ≈
[HCOOH ]
Ka
⋅ 100 = 1,34% .
[HCOOH ]
Efeito de substituintes sobre o pKa em ácidos benzóicos e ácidos alifáticos
Substituintes retiradores de elétrons (por exemplo, halogênios) abaixam a densidade
eletrônica e aumentam desta forma a acidez do ácido benzóico. O substituinte deveria ter
um efeito mais pronunciado em posição para 219 ao grupo -COOH do que em posição meta,
devido aos seus efeitos -I e -M (ver p. 316). Porém, esta regra não vale em todos os casos,
como se vê nos valores Ka de alguns ácidos benzóicos substituídos.
Tabela 21.
Força de alguns ácidos benzóicos substituídos.
COOH
R
Ka/posição
meta
para
-OCH3
-CH3
H
-Cl
-NO2
8,2·10-5
3,3·10-5
5,4·10-5
4,2·10-5
6,3·10-5
1,51·10-4
1,03·10-4
3,2·10-4
3,6·10-4
Mais complexa ainda se dá a influência dos grupos alquilas (ramificados) sobre a acidez.
Nestes deve-se considerar, além de todos os efeitos eletrônicos, um efeito de blindagem que
o grupo apolar exerce frente a água; a presença de grupos alifáticos apolares repele as
moléculas de solvente. E baixa solubilidade provoca baixo grau de dissociação do ácido
carboxílico.
Um pouco mais evidente é o caso em ácidos alifáticos, onde o carbono em posição α ao
grupo –COOH tem hibridação diferente. Comparando as constantes de acidez (em H2O, 25
°C) dos ácidos propiônico, acrílico e propargílico:
219
Os efeitos dos substituintes nas posições meta e para do ácido benzóico são especialmente bem estudados,
pois representam o ponto de partida da teoria de Hammett (ver notas de rodapé na p. 371 ou o livro didático
indicado na nota de rodapé 321) - um dos poucos tratados que reclama fornecer uma relação entre a cinética
e a termodinâmica, das reações de ácidos benzóicos e seus derivados. A posição orto não está referida neste
contexto porque entra em interação direta com o próton do grupo carboxílico.
348
A. Isenmann
Princípios da Síntese Orgânica
O
H3C
CH2
C
O
H2C
CH C
OH
O
HC
C
OH
C
pKa
:
4,88
4,25
OH
1,84
Caráter s do
carbono α
:
25%
33%
50%
O grau de dissociação nesta sequência aumenta devido à eletronegatividade do carbono α
que aumenta junto com o caráter s do orbital híbrido do carbono em posição α. Isto quer
dizer que um átomo mais eletronegativo em posição α promove a desprotonação, então
aumenta a acidez 220.
Um outro exemplo do efeito relativo que retiradores de elétrons exercem sobre grupo
carboxílico é visto na seguinte sequência (em água, 25 °C):
pKa
e também:
pKa
H3C-COOH
Cl-CH2-COOH
CHCl2-COOH
CCl3-COOH
4,76
2,86
1,29
0,65
H3C-COOH
F-CH2-COOH
4,76
2,66
Cl-CH2-COOH Br-CH2-COOH ICH2-COOH
2,86
2,90
3,16
Concluimos destes valores que ácido fluoracético é mais de 100 vezes mais forte do que o
próprio ácido acético!
5.1.3
Apresentação geral dos derivados de ácido carboxílico
Antes de entrar na discussão sobre a obtenção dos diversos derivados do ácido carboxílico,
sejam apresentadas e denominadas as variedades. Caso haja dúvidas sobre a pertença de
certo composto a esta classe é recomendado fazer o teste do número de oxidação no
carbono substituído (sobre o significado do NOX, recorra à p. 582): todos os derivados do
ácido carboxílico têm um carbono de NOX +3 (única exceção: o ácido fórmico, onde o
NOX do carbono é +2).
Por exemplo, uma nitrila, R-C≡N, é um derivado do ácido, enquanto a uréia, (H2N)2C=O,
não é (em vez disso, se deriva do ácido carbônico onde o carbono tem NOX +4).
A partir da estrutura comum,
220
Em analogia desta se explica também a acidez C-H diferenciada dos hidrocarbonetos, etano, eteno, etino,
ver p. 683).
349
A. Isenmann
Princípios da Síntese Orgânica
O
R C
O H,
têm-se os seguintes derivados possíveis:
1) Reposição do próton.
a) Por um metal: isto é feito por reação ácido-base, conforme discutida na p. 345,
fornecendo o sal do carboxilato 221:
O-
O
R C
M+
R C
O-
O
b) A reposição por -OH leva aos perácidos, por sua vez reagentes versáteis em oxidações e
epoxidações (exemplos, ver pp. 155, 224, 293, 427, 629, 662, 665)
2) Reposição do grupo hidroxila:
O
O
R C
R C
OH
X
a) por grupos contendo oxigênio ou enxofre:
X = OR´
Éster do ácido carboxílico
O
R C
OR´
O
C
O
Éster cíclico = “lactona”
X = O-Acila
= O-CO-R´
O
Anidrido do ácido
carboxílico
R
X = SR´
X = SH
Tioéster do ácido
carboxílico
Tioácido carboxílico
C
O
O
O
C
R´
R C
SR´
O tautomeria
OH
R C
R C
SH
S
b) por halogênio:
221
Uma curiosidade: todos os acetatos – até os dos metais mais pesados – são bem solúveis em água.
350
A. Isenmann
Princípios da Síntese Orgânica
X = F, Cl, Br, I
Haleto do ácido carboxílico R C
O
X
c) por funções de nitrogênio:
X = NH2
Amida do ácido carboxílico
O
R C
NH2
X = NHR´, NR´2
O
O
Amidas N-substituídas
R C
R C
NHR´ ;
O
C
NH (R´)
Cíclico: “lactama”
NR2´
O
X = NH-NH2
R C
Hidrazida
NH NH2
X = NHOH
X = N3
Ácido hidroxâmico
O
tautomeria
R C
NH OH
OH
R C
N OH
Azida do ácido carboxílico
O
O
R C
R C
N N N
N N N
3) Reposição do oxigênio carbonílico, (C=O)
a) por funções contendo oxigênio ou enxofre:
Ortoéster do ácido carboxílico
OR´
R C OR´
OR´
S
Éster do ácido tiocarboxílico
R C
OR´
351
A. Isenmann
Princípios da Síntese Orgânica
S
Éster do ácido ditiocarboxílico
R C
SH (R´)
b) por funções contendo nitrogênio:
Imidoéster
(= iminoéter)
NH
R C
R C
OR´
p. ex.:
NH
H3C
*
HCl
Acetamidina
NH2
R C
R C
NH NH2
ácido
C
NH2
NH
Amidrazona
X
OR´
e seus sais
Amidina do ácido
carboxílico
NH (R´)
(somente estável na
forma de aduto com R C
NH2 (HR´, R2´)
HCl = cloridrato)
Nitrila do
carboxílico
NH2
(H ou R´)
N NH2
R C N
5.2
Reações no carbono carboxílico
5.2.1
Formação de derivados de ácido carboxílico
O Dentro das substituições do grupo -OH em ácidos carboxílicos serão discutidas as
R C
reações onde há troca pelos grupos X = -OCH3 , -NH2 , -Cl , -OC(O)R e X NHR.
Todos os compostos têm em comum o grupo acila (em negrito), portanto essas reações de
substituição são, do ponto de vista do nucleófilo, chamadas de "acilações":
O
O
R C
X
+
Nu-
+
R C
X-
Nu
352
A. Isenmann
Princípios da Síntese Orgânica
O carbono do grupo acila é de arranjo plano, de hibridização sp², portanto pode ser atacado
com facilidade pelo nucleófilo (Nu-). Basta ter um bom grupo abandonador, X-, para que
possa ser substituído pelo grupo Nu-.
Esta é a reatividade geral que leva aos ésteres, derivado mais importante do ácido
carboxílico, caso o nucleófilo seja um álcool ou um alcóxido 222.
5.2.2
Os possíveis mecanismos da acilação: A→
→E versus E→
→A
O mecanismo Adição → Eliminação ("A→E"), sem dúvida alguma, é o mecanismo mais
comumente encontrado na formação dos derivados a partir do ácido carboxílico. O aduto
intermediário que contém um carbono quaternário geralmente não é uma espécie isolável e
o abandono do grupo X- ocorre imediatamente, restituindo o carbono sp2. Este mecanismo
se espera especialmente quando não há grandes impedimentos estéricos ao redor do grupo
carboxílico; também favorável (e especialmente desfavorável para a concorrência, E→A) é
a presença de retiradores de elétrons no grupo R.
δ+ O
R C
+ NuX
plano
OR C Nu
X
Complexo tetraédrico
intermediário
O
+
R C
X-
Nu
plano
O próprio ácido carboxílico reage somente sob catálise ácida. Uma catálise básica falha,
porque a base iria desprotonar o grupo –COOH imediatamente. O grupo –COO-, como
seria ilustrado na próxima página, não possui mais carbono de caráter eletrofílico,
precondição para o mecanismo A→E.
O mecanismo Eliminação → Adição ("E→A"; sua discussão no exemplo de um éster está
apresentada na p. 383) é o mecanismo mais raro. O intermediário carbonil cátion pode
também ser formulado como cátion oxônio, onde cada átomo tem um total de 8 elétrons de
valência.
Esse mecanismo pode ser observado onde o grupo carboxílico está numa posição de acesso
difícil (quer dizer, blindado por outros elementos estruturais do substrato) e quando no
grupo R (= posição α em relação ao grupo carboxílico) se encontra um grupo fornecedor de
elétrons.
222
Além desta via de acessar os ésteres, existe outra reação de substituição, levando ao mesmo produto. O
carboxilato pode ter o papel de nucleófilo e substituir um haleto alifático, um sulfato ou um sulfonato de
alquila (ver p. 32). Esta rota, porém, tem pouca relevância prática já que o carboxilato é um nucleófilo fraco e
duro e portanto não será discutida aqui.
353
A. Isenmann
Princípios da Síntese Orgânica
O
R C
X
5.2.3
R C O
- X-
R C O
Carbonil cátion
+ Nu-
O
R C
Nu
Cátion oxônio
(geometria linear)
Reatividade dos derivados de ácido carboxílico em acilações
Cada um dos derivados, R-C(O)X, tem uma reatividade diferenciada frente a nucleófilos,
conforme os efeitos eletrônicos típicos do substituinte X.
O carbono carboxílico quanto mais pobre em elétrons, mais facilmente ocorre o ataque pelo
nucleófilo.
Portanto, o grupo X
Aumenta a reatividade do derivado quando é retirador de elétrons (efeito da
eletronegatividade do átomo diretamente ligado ao carbono +3; "efeito -I"):
O > Cl, N > Br, S
Atenua a reatividade do derivado quando é doador de elétrons (disponibilidade de
pares de elétrons não-ligantes; "efeito +M"): N > O >> Cl, S 223.
Conforme já elucidado na substituição de aromáticos, o efeito M geralmente supera o efeito
I (p. 318). O conjunto destes efeitos permite estabelecer a seguinte sequência de reatividade
dos derivados do ácido carboxílico ante o ataque nucleofílico.
Figura 31. Reatividade relativa dos derivados do ácido carboxílico, frente nucleófilos
O
O
R C
O > R C
>
Cl
O
R C
R C
O
Anidrido
Cloreto do
ácido carboxílico do ácido
>
OR´
Éster
O
O
>
R
C
N
>
R C
> R C > R C
R C
NR´
O2
NH2
OH
O
Ácido
carboxílico
O
Amidas
Nitrila
Ânion
carboxilato
O ânion carboxilato, que está no final desta sequência, não demonstra nenhuma reatividade
frente nucleófilos “comuns”. Apenas com os nucleófilos mais fortes, tais como hidreto, H-,
ou carbânions, se consegue a acilação. Um exemplo para o primeiro caso é a redução de
carboxilatos com LiAlH4; um exemplo do segundo caso é a adição redutiva de compostos
organo-lítio (ver p. 382).
A sequência dada acima contém apenas os derivados mais comuns do ácido carboxílico.
Mas têm-se outros também, muitas vezes com papel de intermediário que facilita a entrada
do substituinte nucleofílico final. Sob esta luz pode ser visto o ceteno, R2C=C=O, por sua
vez o "derivado" mais reativo do ácido carboxílico (ver p. 386).
223
Os orbitais não-ligantes de Cl e S interagem menos intensamente com o grupo acila, porque estes são
elementos do terceiro período cujos orbitais 3p e 3d são bem maiores do que os orbitais 2p em C e O.
354
A. Isenmann
Princípios da Síntese Orgânica
Outros intermediários reativos do ácido carboxílico serão apresentados a seguir, sendo
estratégias modernas e poderosas para formar a nova ligação entre o carbono carboxílico e
o nucleófilo.
Reatividade dos ácidos carboxílicos aromáticos
Geralmente os substratos com grupo R = Ar são menos reativos do que os seus equivalentes
alifáticos. Isto se deve à conjugação entre os elétrons do anel aromático e os do grupo acila,
o que significa estabilização extra. Esta vantagem energética se perde quando o carbono sp²
se transforma em sp³ - como é o caso no estado de transição da sequência A→E. Por isso, o
morro de ativação é maior no substrato Ar-COOX, ou seja, a reação da substituição é mais
lenta.
Figura 32. Representação dos orbitais do tipo p,
na unidade Ar-C=O que entram em conjugação
com o anel aromático.
C
O
R
Mas existe também uma explicação trivial para reatividade, rendimento e velocidade baixa
com compostos aromáticos: eles têm uma solubilidade menor em ambiente polar, em
comparação com os parentes alifáticos, de pequenos grupos saturados. Já o nucleófilo é
uma espécie polar e, muitas vezes, a SN é feita com um sal soluto em ambiente aquoso.
Então a área de contato entre o substrato aromático e o reagente "Nu-" é pequena, ou seja,
as concentrações efetivas dos participantes, necessárias para a reação (na termodinâmica
chamadas de "atividades"), são baixas.
5.2.4
Métodos de ativação do grupo acila
Em muitas acilações é uma má estratégia usar o ácido carboxílico livre, pois o nucleófilo
com que se pretende fazer a reação, geralmente também representa uma base. Assim, a
reação ácido-base, que ocorre mais rapidamente do que o ataque nucleofílico, transforma o
ácido carboxílico no carboxilato. Como pode ser visto na sequência acima, o carboxilato
tem a reatividade mais baixa de todos os derivados, ou seja, praticamente não tem mais
carbono eletrofílico.
Então é preciso transformar o ácido carboxílico em um dos seus derivados mais reativos,
seja em uma etapa prévia ou "in situ" (= ativação intermediária, sem possibilidade de isolar
o derivado reativo). Isso implica que este tipo de ativação pode ser feito na presença do
nucleófilo que se pretende acoplar ao grupo acila.
Preparo do derivado reativo em uma etapa prévia
355
A. Isenmann
Princípios da Síntese Orgânica
a) Métodos padrões
São, na maioria, reações fáceis e completas que permitem a preparação destes derivados
diretamente a partir do ácido livre. Somente a obtenção da amida geralmente não é a partir
do ácido carboxílico, devido à reação ácido-base mencionada acima.
O
SOCl 2 ou
PCl5
Ac2O
O
R C
Cl
O
R C
O
R C
R C
R´ OH / H+
OH
R´2NH
O
O
R C
OR´
O
R C
NR´2
O cloreto do ácido carboxílico, sendo o derivado mais reativo frente a nucleófilos, é usado
frequentemente no laboratório de síntese e pode ser produzido por vários métodos. Como
material de partida se oferecem em primeira linha o próprio ácido carboxílico ou seu sal
alcalino. O método mais comumente usado é a reação do ácido (ou seu sal de sódio), com
os cloretos dos ácidos do fósforo ou do enxofre. A partir do ácido carboxílico livre se
obtêm os cloretos com os seguintes reagentes:
• SOCl2
Reagente de cloração mais utilizado 224, por ser mais barato; menos reativo do que PCl3 e
PCl5:
O
O
R C OH
+ SOCl2
R C
Cl
+ HCl
+ SO2
• PCl3
Reatividade média; cada mol reage com até três mols de grupos carboxílicos, compare
também quadro de vista geral sobre as reações do PCl3, no anexo 2 do livro:
O
3 R C OH
O
+ PCl3
3 R C Cl
+ H3PO3
• PCl5
Reagente de cloração mais poderoso do que PCl3, porém reage somente com um grupo
carboxílico:
O
R C OH
224
O
+ PCl5
R C Cl
+ POCl3
+ HCl
V.K. Yadav, K.G.Babu, J.Org.Chem. 69 (2004) 577-80.
356
A. Isenmann
Princípios da Síntese Orgânica
A primeira reação, usando SOCl2, muitas vezes ganha preferência no laboratório porque os
subprodutos desta são exclusivamente gases que podem ser facilmente separados da
mistura reacional. A evolução de HCl indica que apenas um cloro do reagente SOCl2 é
transferido para o ácido carboxílico.
Mecanismo:
O
O
R C
R C
OH
O
Cl
Cl S
Cl
OH
S OCl
O
O
R C
R C
- HCl
O
+ HCl
Cl
Cl
O
+
S
S
O
O
Embora a estequiometria desta reação ser 1 : 1, na prática sempre se aplica o reagente de
cloração em excesso. O cloreto de tionila que não reagiu pode ser facilmente removido e
separado do produto clorado, por destilação (SOCl2 é um líquido volátil; Teb= 75 °C). Este
procedimento somente pode dar problemas, onde o cloreto carboxílico também tiver
elevada volatilidade. Geralmente, a velocidade desta reação é suficientement alta; em casos
difíceis ajuda o efeito catalítico de DMF.
A equação com o cloreto de fósforo (III), conforme anotada na lista acima, não explica a
evolução de HCl gasoso, que na verdade sempre se observa. Isto se deve à formação do
anidrido misto em menores partes:
O próprio PCl3 pode ser visto como anidrido misto bastante reativo, a partir do ácido
fosfônico (H3PO3; ácido de força média) e o ácido clorídrico (ácido forte). Este reagente,
quando entrar em contato com o ácido carboxílico livre (ácido fraco), pode trocar a posição
na seguinte maneira: o ácido carboxílico fica preso no anidrido com o ácido fosfônico e o
HCl sai como ácido livre:
O
O
R C OH + Cl2P
Cl
R C O
PCl2
+ HCl
.
O anidrido entre ácido carboxílico e ácido fosfônico é igualmente bastante reativo e pode
ser usado, em analogia ao cloreto, para subsequentes substituições.
Todavia, a reação principal continua sendo a formação do anidrido misto, entre ácido
carboxílico e ácido clorídrico, conforme descrito acima.
Igualmente bem funciona a cloração com PCl5 que, ao contrário do PCl3, somente reage
com apenas uma alíquota de ácido carboxílico. O subproduto, POCl3, não tem mais o poder
como reagente de cloração, sob as condições aplicadas.
Porém, a reação com PCl5 funciona de forma mais favorável, ao ser conduzida entre o
carboxilato e PCl5, na fase sólida:
O
O
-
3 R C O Na
+
+ PCl5
3 R C Cl
+ 2 NaCl
+ NaPO3
357
A. Isenmann
Princípios da Síntese Orgânica
Esta síntese tem um rendimento molecular maior, pois fornece até três cloretos carboxílicos
a cada PCl5. Ela tem a vantagem de fornecer co-produtos sólidos que podem permanecer na
mistura reacional, porque são insolúveis e não interferem nas etapas subsequentes.
Os anidridos do ácido carboxílico, geralmente abreviados com "Ac2O" 225, podem ser
obtidos de várias maneiras. Um método certamente é o tratamento de um ácido carboxílico
com excesso de um anidrido que sempre está disponível em quantidade suficiente, o
anidrido acético. Este caminho já foi indicado no esquema na p. 356. Certamente uma
reação equilibrada, com rendimentos variáveis e não adequada para alguns destinos.
Em laboratório é também comum usar pentóxido de fósforo sendo um agente desidratante
forte. O subproduto é o óxido de fósforo pouco hidratado - um material resinoso e
geralmente insolúvel, então de fácil remoção.
2 AcOH + P4O10 → Ac2O + "(HO)2P4O9".
Anidridos também podem ser obtidos a partir dos cloretos de acila, já que estes são mais
reativos ainda frente nucleófilos. Para este fim aplica-se o sal carboxilato como nucleófilo
226
. O acetato, como sabemos do cap. 1, não é um bom nucleófilo. Mesmo assim, o grupo
acila o recebe voluntariamente, então este é um dos métodos mais utilizados no laboratório
de se produzir o anidrido, a partir do carboxilato de sódio e o cloreto de acila. O subproduto
é simplesmente sal de cozinha:
AcO-Na+ + H3C-C(O)Cl → RCO2C(O)CH3 + NaCl.
O
O
CH3 C
+
Cl
CH3 C
O- Na+
Nucleófilo
O
CH3 C
O
CH3
+ NaCl
C
O
Anidridos mistos, onde uma parte é o grupo acetil, pode-se obter por reação do ácido
carboxílico livre com ceteno (rota desenvolvida pela empresa Wacker em 1922; ver
também p. 386 e 390). O ceteno é um derivado muito mais reativo frente nucleófilos do que
o cloreto de acila, apresentado acima. Portanto, ele adiciona o grupo acetato em rendimento
de 100%.
AcOH + H2C=C=O → RCO2C(O)CH3.
225
"Ac" nesta fórmula, significa "Acila", então o grupo genérico R-C=O; um caso específico é H3C-C=O:
este grupo é chamado de "Acetila".
226
Lewis I. Krimen. Acetic Formic Anhydride. Org. Synth.; Coll. Vol. 6 (1988) 8.
358
A. Isenmann
Princípios da Síntese Orgânica
O anidrido mais produzido em escala industrial é o anidrido acético. Sua síntese é via
carbonilação de acetato de metila 227; as condições desta síntese são comparáveis à
carbonilação de Reppe, descrita na p. 190.
CH3CO2CH3 + CO → (CH3CO)2O
O segundo mais produzido é o anidrido maléico, por sua vez muito usado como monômero
em copolímeros hidrofílicos. É feito em escala industrial via oxidação catalítica de
benzeno, mais recentemente também a partir de butano. Os catalisadores de contato são
V2O5 e MoO3 no primeiro caso, no segundo óxidos mistos de vanádio e fósforo. Como as
temperaturas desta síntese são bastante altas, o maior perigo sempre é que a mistura pega
fogo (= oxidação completo até o CO2).
O
+ 7 O2
2
V2O5 / MoO3
2
O
+ 8 H2O
O
Anidrido ftálico, finalmente, se obtém via oxidação catalítica, ou do o-xileno ou do
naftaleno (processo de Gibbs):
O
CH3
+ 3 O2
CH3
O + 3 H2O
O
O
+ 4,5 O2
O + 2 CO2 + 2 H2O
O
Este é bastante usado como plastificante em diversos materiais poliméricos.
Note: os anidridos ftálico e maléico são mais estáveis (isto é, menos reativos) do que os
demais anidridos, já que a estrutura anelar de 5 membros é estatistica e
termodinamicamente favorável.
227
J. R. Zoeller, V.H. Agreda, S.L. Cook, N.L. Lafferty, S.W. Polichnowski, D.M. Pond. Eastman Chemical
Company Acetic Anhydride Process. Catalysis Today 13 (1992), 73-91.
359
A. Isenmann
Princípios da Síntese Orgânica
b) Métodos brandos e mais específicos
O cloreto de fosforila, POCl3, como não reage com o ácido carboxílico livre, mas somente
com o sal do ácido carboxílico, pode ser considerado sendo reagente de cloração mais
seletivo:
O
O
-
2 R C O Na
+
+ POCl3
2 R C Cl
+ NaCl
+ NaPO3
Sob condições especialmente brandas se obtém cloretos de ácidos com a combinação de
trifenilfosfina em tetracloreto de carbono 228: Do ponto de vista da trifenilfosfina essa
reação é mais uma vez apresentada na p. 795; ela é conhecida como "ativação de Appel",
onde se aproveita da grande preferência do fósforo pelo oxigênio.
O
O
R C OH
+ (C6H5)3P + CCl4
+ (C6H5)3PO + CHCl3
R C Cl
Essa e a próxima sintese destacam-se por não liberar HCl - que às vezes pode causar
reações paralelas.
Igualmente serve o cloreto do ácido cianúrico com apoio da base não nucleofílica NEt3,
sendo um sistema específico e brando fornecedor de cloro 229. O cloreto cianúrico é um
reagente mais acessível do que o cloreto de oxalila apresentado a seguir, porque tem
aplicações industriais importantes (compare na p. 328).
Cl
Cl
O
R
OH
+
N
Cl
NEt 3
N
N
Cl
- NHEt3
N
R
Cl
O
Cl
N
O
NHEt3
N
Cl
- NEt 3
R
Cl
+
N
HO
N
O
A síntese industrial de cloreto de acetila é por aquecimento de acetato de sódio com cloreto
de sulfurila:
O
2 H3C
O
-
+
C O Na
+ SO2Cl2
2 H3C
C Cl
+
N
Na2SO4
228
J. Blee, J.Am.Chem.Soc. 88 (1966) 3440.
K. Venkataraman, D. R. Wagle. "Cyanuric chloride: a useful reagent for converting carboxylic acids into
chlorides, esters, amides and peptides". Tetrahedron Letters 20 (1979) 3037–3040.
229
360
Cl
A. Isenmann
Princípios da Síntese Orgânica
Nos 20 anos passados o uso do seguinte reagente de cloração avançou muito 230 : o cloreto
de oxalila C2O2Cl2. Ele é de eletrofilia bastante baixa 231. Traços de DMF catalisam a
cloração do ácido carboxílico 232 e tornam este sendo um dos preferidos reagentes de
cloração nos laboratórios de síntese:
O
O
R
Cl
OH
Cl
O
O
Cat. DMF
R
Cl
+ CO2 + CO + HCl
Evidentemente e em analogia à cloração com cloreto de tionila, todos os subprodutos são
gases, o que facilita bastante sua remoção da mistura reacional e o isolamento/purificação
do cloreto de acila. Em comparação ao cloreto de tionila, o cloreto de oxalila reage mais
especificamente, ou seja, é um agente de cloração menos potente. Mas como (COCl)2 é
mais caro do que o SOCl2, até hoje é aplicado somente em pequena escala e em casos onde
o cloreto de tionila mostra reações laterais.
Embora dispor de dois átomos de cloro, o cloreto de oxalila reage com ácidos carboxílicos
somente na proporção de 1 : 1. A presença de um excesso de CaCO3 sólido foi descrita
como vantagem em casos onde o ácido de partida pode sofrer isomerizações. A separação
do cloreto de oxalila não reagido acontece por evaporação repetitiva do solvente que arrasta
os restos do cloreto de oxalila. Também aqui temos a restrição que o produto clorado não
deve ter uma pressão de vapor muito próxima à do C2O2Cl2, para não sofrer perdas
significativas nesta etapa de purificação.
Todavia, o uso do cloreto de oxalila (Teb = 63 – 64 °C; pressão de vapor p(50 °C) = 890
hPa) também tem aspectos negativos. É considerado tóxico, corrosivo e se decompõe
facilmente em contato com água; o armazenamento é mais seguro a baixas temperaturas, no
estado sólido (Tfus = 15 °C). Além disso, os intermediários nesta rota têm potencial
carcinogêneo.
O papel do catalisador, o DMF, fica mais claro com o seguinte esquema. Pode-se
identificar o carbono do DMF sendo o meio de transmitir o cloro, do cloreto de oxalila para
o grupo acila a ser ativado. Sua reação prévia com o cloreto de oxalila leva ao cloreto de
imínio que já conhecemos da síntese de Vilsmeyer-Haack (p. 305). Só aqui este cátion não
serve como eletrófilo, mas como meio de entregar o cloro ao grupo acila.
230
A. Balsamo, J.Med.Chem. 24 (1981) 525-32.
Synthetica Merck I, 369; A. Wissner, C.V. Grudzinska, J.Org.Chem. 43 (1978) 3972.
232
A.W. Burgstahler et al., Synthesis 1976, 767; G.A. Olah et al., Synthesis 1979, 58.
231
361
A. Isenmann
Princípios da Síntese Orgânica
O
O
N
O
H
Cl
O
Cl
Cl
O
O
N
O
N
Cl-
DMF
= Dimetilformamida
Cl
O
Cl
- CO - Cl-
- CO2
H
N
O
Cl
Eletrófilo e
transmissor do cloro
Cloreto imínio
Este intermediário, cloreto de imidoíla, reage então com o ácido carboxílico, onde ocorre a
troca de oxigênio pelo cloro. Isso regenera o catalisador e libera o produto desejado.
H
N
O
O
H
R
Cl
HO
-H
+
N
O
H
R
Cl
N
O
O
H
R
N
O
O
+
R
Cl
Cl-
Infelizmente, nem todo o cloreto de oxalila reage por este mecanismo. Alguma parte pode
também hidrolisar, sob a influência do ácido carboxílico (e traços de água). Essas formas
hidrolisadas, o monocloreto de oxalila e o ácido oxálico livre, são de baixa volatilidade.
Portanto, sua remoção da mistura reacional fica incompleta. Este fato deve ser levado em
consideração ao acrescentar o nucleófilo final, ao cloreto carboxílico. Caso o nosso objetivo
seja a formação de uma amida, a amina a ser acilada, pode ser protonada (= inativada) por
estes subprodutos.
Este problema pode ser contornado, ao aplicar a amina em excesso; alternativamente
acrescentar uma amina protetora (por exemplo, trietilamina) que neutraliza esses
subprodutos ácidos presentes na mistura. Formam-se sais que se precipitam do solvente
orgânico; sua separação ocorre facilmente por decantação numa etapa posterior de lavagem,
onde esses sais vão para a fase aquosa, enquanto o produto principal (em nosso exemplo a
amida) permanece na fase orgânica.
Outras aplicações do cloreto de oxalila na síntese orgânica:
• Reagente de acilação segundo Friedel-Crafts (ver p. 297), após decarbonilação
levando ao ácido benzóico 233;
• Junto ao DMSO serve como reagente brando de oxidação (oxidação de Swern, ver
p. 649).
233
P. E. Sokol, Org. Synth. 44 (1964) 69; Coll. Vol. 5: 706.
362
A. Isenmann
Princípios da Síntese Orgânica
Geralmente o cloreto carboxílico não permite estocagem prolongada. Sendo assim, ele é
feito logo antes da reação com o nucleófilo final, para minimizar contaminação e perdas
por hidrólise. A exclusão de umidade é de suma importância, em todos os métodos de
ativação prévia descritos aqui.
c) Métodos da ativação in situ do grupo carboxílico.
A azida tem uma reatividade comparável com o anidrido e então promove a acilação. As
azidas de acila geralmente são feitas a partir do cloreto de acila, só que essa reação não
atende o propósito de aumentar a reatividade, já que o cloreto de acila é suficientemente
reativo para a maioria das aplicações.
O
R C
O
O
NaN3
R C
R C
N N N
N N N
Cl
Acilacao
Azida
0 °C
...
No entanto, as azidas se consegue também - sob condições bastante brandas e em
rendimentos excelentes - diretamente a partir do ácido carboxílico, na presença de
catalisadores de trifenilfosfina e tricloroacetonitrila 234. Sendo assim, tem-se um método in
situ de ativação.
(Atenção: as azidas sofrem facilmente degradação de Curtius, ver p. 391).
Bastante eficaz é a ativação do ácido carboxílico por DCC (di-cicloexilcarbodiimida).
Trata-se de um agente dessecador forte que forma um intermediário derivado da uréia com
reatividade elevada. Isso implica que a condição para esta reação deve ser rigorosamente
anidro.
δ+
O
R C
OH
N C N
"DCC"
O
R C
N Hex
O C
NH Hex
Acilação
20 °C
...
O
- HN C NH
Hex Hex
O fator responsável para a alta reatividade do DCC 235 é a presença de duplas ligações
acumuladas (ver p. 131). Como se vê a seguir, trata-se de uma transformação in situ do
ácido livre para o seu anidrido 236. O método é aplicado desde mais de 40 anos 237, com
234
Doo Jang, Joong-Gon Kim. Direct Synthesis of Acyl Azides from Carboxylic Acids by the Combination
of Trichloroacetonitrile, Triphenylphosphine and Sodium Azide. Synlett. (2008)
235
Atenção: o nome ~imida para o DCC pode ser enganoso, porque tem nada a ver com a unidade estrutural
da "imida", -CO-NH-CO-.
236
B.J.Balcom, N.O. Petersen, J.Org.Chem. 54 (1989) 1922.
363
A. Isenmann
Princípios da Síntese Orgânica
muito sucesso, especialmente na etapa de ativação do grupo acila, na síntese de peptídeos
(ver p. 599). Como o subproduto, N,N´-diciclohexiluréia, é insolúvel na maioria dos
solventes, a purificação do produto é especialmente confortável.
O
R COOH + C6H11
N C N C6H11
N C6H11
R C O
NH C6H11
"DCC"
O-acilisouréia
+ R COOH
O
O
O
O
R C N
R C
NH C6H11
+
O
O C(NHC6H11)2
R C
C6H11
N-aciluréia
(indesejada)
O
Anidrida
Diciclohexiluréia
A reação com DCC tem um produto paralelo, a N-aciluréia, sendo não reativa no sentido de
uma SN no grupo acila. Felizmente, este caminho pode ser reprimido pela adição de Nhidroxisuccinimida que funciona como capturador e estabilisador da O-acilisouréia.
Um terceiro caminho de ativação, mais recente, é a reação com 1-hidroxibenzotriazona
seguida por um rearranjo para um derivado igualmente reativo à azida 238.
O
R C
OH
HO
o
O
+
N
N
N
R C O N
N
N
Acilação
O
R C N
N O
-
N
Todas estas ativações in situ têm em comum o fato de serem executadas sob condições
bastante brandas e, ao mesmo tempo, terem rendimentos altos. Portanto, são usadas como
metodologias para a síntese de peptídeos, a partir de aminoácidos (ver também p. 599).
237
238
H.G.Khorana, Chem.Rev. 53 (1953) 145.
X. Zhang et al. J.Org.Chem. 67 (2002) 9471-4.
364
...
A. Isenmann
Princípios da Síntese Orgânica
5.3
Reações dos derivados do ácido carboxílico
5.3.1
Reações do cloreto e do anidrido de acila
As reatividades dos derivados R-COCl e R-COAc frente nucleófilos, são bastante
semelhantes - o que justifica seu tratamento junto em um capítulo.
a) Reação com água
Como descrito acima, o cloreto/anidrido é reativo o suficiente para reagir com nucleófilos
até mesmo fracos. Por exemplo, o cloreto / o grupo acetato, podem ser substituídos em
pouco tempo, à temperatura ambiente, por água. Esta “saponificação”, o inverso da síntese
que consta do esquema acima, não ocorre com tal facilidade em ésteres e amidas, que
saponificam apenas ao serem atacados pelo nucleófilo consideravelmente mais forte, OH(ver p. 370 e 377, respectivamente).
b) Reação com alcoóis - "esterificação".
Estas reações ocorrem tipicamente pelo mecanismo A→E (p. 353). Conforme apresentado
lá, todas elas são a princípio reversíveis. Para aumentar o rendimento deve-se então
suprimir a reação reversa. Para este fim um dos produtos da reação, uma vez formado, deve
ser retirado da mistura reacional. O método padrão é a fixação do HCl, subproduto da
reação com o cloreto de acila, usando uma amina terciária ou piridina.
Aumento do rendimento da esterificação, a partir do cloreto de acila e um álcool:
O-
O
R C
+
CH3OH
Cl
Cl
O
CH3
R C O
H
- Cl-
O
R C
O CH3
- H+
R C
O CH3
H
Py
NH+ Cl-
Fixação como cloreto de piridínio
Além desta promoção termodinâmica, a reação é bastante acelerada pela amina. O seu
efeito catalítico pode ser entendido pela formação do seguinte complexo intermediário:
O
O
R C
R C
+ Py
N
Cl-
Cl
A formação deste complexo aumenta consideravelmente a reatividade do carbono do grupo
acila frente nucleófilos, porque o átomo de nitrogênio está presente como sal quaternário de
365
A. Isenmann
Princípios da Síntese Orgânica
amônio, então não tem efeito +M, mas somente –I. Este tipo de ativação é conhecido como
"catálise nucleofílica".
Existem exemplos de bases de Lewis quirais que podem ser usadas em lugar da piridina.
Elas servem para esterificar preferencialmente um enanciômero, a partir de uma mistura
racêmica do álcool 239. Sendo assim, esta estratégia encaixa-se na família dos catalisadores
quirais (catálise e indução assimética, ver as pp. 232 e 229, respectivamente).
Agora vamos considerar esta reação do ponto de vista do nucleófilo atacante, neste caso do
álcool. Para fins analíticos ou por motivos de proteção do grupo hidroxila podem-se reagir
alcoóis primários e secundários com uma mistura de anidrido acético e piridina, 1 : 1. A
reação ocorre rapida e quantitativamente. Os alcoóis terciários também reagem, porém
seguem um caminho diferente: eles reagem pelo mecanismo SN1, através do seu
carbocátion (pp. 50, 377 e 384).
Mais adiante encontraremos mais uma vez com a mistura de cloreto de acila e piridina,
sendo matéria de partida para cetenos (ver p. 386).
c) Reação de Schotten-Baumann
A formação mais completa e rápida, de ésteres a partir de derivados reativos do ácido
carboxílico, se obtém sob as "condições de Schotten-Baumann".
Usa-se esta reação na análise qualitativa, para identificar álccois. À temperatura ambiente a
maioria dos alcoóis está no estado líquido. Isto impede sua identificação pelo ponto de
fusão e, o que hoje pesa mais ainda, também a sua purificação por recristalização.
Frequentemente aplicada é a conversão quantitativa, do álcool com o cloreto do ácido 2,4dinitrobenzóico, em solução fortemente alcalina, sob refrigeração, fornecendo seu
respectivo éster. O éster, por sua vez, é um sólido à temperatura ambiente, no qual é
possível estimar a sua pureza através do ponto de fusão.
d) Aminólise em geral
A reação do cloreto de acila com amônia, aminas primárias ou secundárias, fornece as
respectivas amidas. A aminólise funciona de maneira semelhante à hidrólise:
H2O / OHC6H5 COCl +
Cloreto de benzoíla
C6H5 C NH2 C6H5
C6H5 NH2
- H+
- Cl-
C6H5 CO NH2 C6H5
Cl
Anilina
C6H5 CO NH2 C6H5
O-
C6H5 CO NH C6H5
Benzanilida
Neste exemplo verificamos mais uma vez a necessidade de "puxar" a reação equilibrada
para o lado direito, através do consumo do coproduto, HCl. E ainda: se não
acrescentássemos um agente sequestrador do ácido (por exemplo, piridina, aminas
terciárias, ou NaOH), o HCl liberado consomeria o restante da anilina: pela protonação se
239
G.C. Fu, Acc.Chem.Res. 33 (2000) 412-420.
366
A. Isenmann
Princípios da Síntese Orgânica
perde a nucleofilia do grupo -NH2; além disso, a anilina está sendo solubilizada na fase
aquosa (em forma do cloreto de anilínio), quer dizer, fica longe do substrato orgânico, o
cloreto de benzoíla. E como foi discutido na p. 32, misturas bifásicas implicam uma reação
lenta. Neste exemplo temos mais um argumento para usar uma base auxiliar (não
nucleofílica!): quando aplicada em excesso o produto da reação, a benzanilida, se precipita
em forma do seu sal ácido (última fórmula da primeira linha, do esquema acima, junto ao
ânion da base). Isto significa um aumento do rendimento por controle termodinâmico.
Justamente este efeito, a formação do sal a partir da amida, é o princípio da separação de
Hinsberg, descrita a seguir.
e) Separação de Hinsberg
Em muitas sínteses (compare p. 48) se obtém uma mistura de aminas primárias, secundárias
e terciárias. Uma tarefa pesada de purificação, pois as aminas são muito parecidas nas suas
propriedades físicas e químicas: basicidade, solubilidade e polaridade são praticamente
idênticas, então sua separação requer grandes esforços cromatográficos. Tal separação em
quantidades na escala de gramas é simplesmente impossível.
A solução oferece uma derivatização seletiva das aminas, conhecida como separação de
Hinsberg 240. Em plena analogia à estratégia descrita acima, efetua-se uma conversão
quantitativa para amidas - esta vez formando sulfonamidas. O reagente é um cloreto de
ácido sulfônico. Quase todas as sulfonamidas (vulgarmente chamadas “sulfas”) são pouco
solúveis em água.
Reação da amina primária:
R
SO2 Cl +
R NH2
R
- HCl
SO2 NH
R
Sulfonamida
pouco solúvel
R
SO2 NH
NaOH
R
excesso
O
O
R
S
O
N R
R
S
O
N R
O
R
S
N R Na+
O
Sal da sulfonamida
= solúvel
Esta reação funciona rapida e quantitativamente.
A sulfa se torna solúvel ao acrescentar numa segunda etapa soda cáustica em excesso,
formando o amideto de sódio. A facilidade e o alto rendimento com que o desprotonamento
ocorre na sulfa se deve à estabilização do ânion por ressonância. É o forte efeito retirador
de elétrons do grupo sulfônico sobre o nitrogênio que facilita a distribuição da carga
negativa sobre três átomos, causando estabilidade termodinâmica no sal.
240
Embora a separação de Hinsberg não envolva ácidos carboxílicos, ela é apresentada neste local, justificado
pelo parentesco com a formação de amidas por aminólise, discutida logo acima.
367
A. Isenmann
Princípios da Síntese Orgânica
Já a reação entre o cloreto do ácido sulfônico e a amina secundária não ocorre com tanta
facilidade. Além disso, a sulfa, uma vez formada, não pode ser solubilizada de maneira
análoga à sulfa da amina primária. Simplesmente não há hidrogênio no N que pode ser
abstraído, daí não há formação do amideto.
A amina terciária, finalmente, não reage com o cloreto do ácido sulfônico sob as condições
aplicadas, então não fornece uma sulfa de baixa solubilidade. Como o método de Hinsberg
funciona em solução levemente ácida (HCl diluído), a amina terciária solubiliza-se
rapidamente no meio reacional em forma do seu hidrocloreto (também chamado de
cloridrato).
O reagente mais usado na separação de Hinsberg é o cloreto do ácido benzossulfônico, mas
também o cloreto de tosila (TsCl; velho conhecido como ativador de alcoóis no sentido de
SN, ver p. 36) tem certa importância.
f) Reatividade especial em anidridos cíclicos
Caso um substrato tenha dois grupos carboxílicos em distância adequada, o anidrido cíclico
pode se formar por condensação intramolecular:
O
HOOC
COOH
Ácido maléico
Ac2O
O
O
Anidrido maléico
HOOC
COOH
Ácido fumárico
No princípio, os dois grupos acilas do anidrido intramolecular podem reagir com o
nucleófilo. Porém, eles têm reatividades “diferentes”, o que permite interromper a reação
seletivamente após a entrada do primeiro nucleófilo. Assim é possível, sob certo cuidado na
estequiometria, fazer outras transformações posteriormente, no segundo grupo carboxila.
De qualquer maneira, as condições desta síntese devem ser bastante suaves e
estequiométricas, para não se correr o risco da dupla-acilação.
O seguinte exemplo, a partir de um anidrido cíclico, deve ilustrar como é possível fazer a
reação com apenas um Nu- (sendo o reagente metanol, um nucleófilo fraco). Logo após a
sua reação com o anidrido succínico, um grupo carboxila é liberado que, por sua vez, é bem
menos reativo frente ao metanol do que o anidrido do início. Desta forma, este não continua
reagindo com o metanol, mas pode ser isolado como mono-éster. Em etapa subsequente o
mono-éster pode ser ativado no sentido da segunda substituição nucleofílica.
368
A. Isenmann
O
O
O
O
Princípios da Síntese Orgânica
OMe
MeOH
MeOH
OMe
OMe
OH
O
Anidrido
succínico
O
O
não se forma
SOCl 2
O
OMe
Cl
....
O
A condensação do anidrido com amônia ou uma amina primária tem por objetivo produzir
uma imida. Essas reações geralmente não têm bons rendimentos nem são muito rápidas,
isto é, podem equilibrar-se após várias horas de reação. Todavia, pode-se afirmar que a
condensação intramolecular do anidrido é preferida de longe, comparada com uma
condensação intermolecular.
No exemplo a seguir mostrou-se especialmente favorável fazer a reação em forno de
microondas 241. Em poucos minutos se obtém a ftalimida com bons rendimentos 242.
241
Há aproximadamente 20 anos, quando foram desenvolvidos os primeiros reatores de microondas
para o laboratório de síntese orgânica. Desde então notou-se uma avalanche de publicações sobre o assunto,
pois inúmeras reações são aceleradas por esta forma de energia. Ao mesmo tempo, as temperaturas não são
muito altas. Rendimentos altos, tempos de reação curtos e poucos produtos paralelos são as vantagens desta
técnica.
Ver artigos de revisão:
C. O. Kappe, "Controlled Microwave Heating in Modern Organic Synthesis", Angew. Chem. Int. Ed. 2004,
43, 6250.
A. Loupy et al., New solvent-free organic synthesis using focussed microwaves, Synthesis 1998, 1213-34.
P. Lidström et al., Microwave assisted organic synthesis – a review, Tetrahedron 57 (2001) 9225-83.
K. Bougrin et al., Microwave-assisted solvent-free heterocyclic synthesis, Journal of Photochemistry and
Photobiology C: Photochemistry Reviews 6 (2005), 139-67.
D.V. Kuznetsov et al., Microwave Activation in Organic Synthesis, Russian Journal of Organic Chemistry, 41
(2005), 1719-49
Monografia:
C. Oliver Kappe, Alexander Stadler Microwaves in Organic and Medicinal Chemistry, Wiley-VCH 2005
242
S. Chandrasekhar, M. Takhi, G. Uma, Tetrahedron Lett. 38 (1997) 8089-8092.
369
A. Isenmann
Princípios da Síntese Orgânica
O
O
+ H2N
R
N
microondas (450 W)
5 min
O
Anidrido ftálico
5.3.2
O
Catalisador:
TaCl5/SiO2
sem solvente
Ftalimida
R
O
Reações dos ésteres e amidas
a) Saponificações de ésteres sob catálise básica.
A hidrólise de ésteres formando o ácido (ou seu sal) e um álcool é conhecida como
saponificação. Isto vem da reação-mãe que a humanidade aplica desde a antiguidade, para
produzir sabão, a partir de gordura animal, ao cozinhá-la junto com cinzas de madeira. O
que realmente ocorre é a hidrólise básica do triglicerídeo de ácidos graxos, sob influência
de KOH presente em alta concentração nas cinzas. Formam-se glicerina e os sais de
potássio dos ácidos graxos. Os últimos, por terem uma “cabeça” polar e uma “cauda”
apolar, mostram propriedades tensoativas. Elas acumulam-se na interface água/óleo de
maneira bem direcionada e assim abaixam a tensão interfacial entre as fases repelidas. A
presença de carboxilatos graxos pode levar até levar a emulsões estáveis 243. Todos nós
conhecemos e apreciamos este efeito compatibilizante, nos diversos atos de limpeza e
higiene pessoal do dia-a-dia.
243
Acima da concentração que é necessária para cobrir toda a interface plana, água/óleo ou água/ar, as
moléculas do sabão procuram outras formas de organizar-se. A mais conhecida é a micela, isto é, esferas submicroscópicas. A concentração limite inferior é chamada de CMC (concentração de micelas crítica). Micelas
podem ou não, conter a fase repelida no seu interior e desta forma estabilizar a dispersão fina das duas fases
repelentes, em forma de uma emulsão.
O seguinte gráfico contém as mais importantes formas de organização destes ânions anfifílicos, em ambiente
aquoso (sem a presença de óleo, para simplificar):
370
A. Isenmann
Princípios da Síntese Orgânica
O
H2C O C
HC O C(O)
+ 3 KOH
H2C OH
∆
HC OH
H2C O C
O
H2C OH
O
Óleo; gordura
C O- K+
+ 3
Carboxilato de potássio
("sabão")
Glicerina
A saponificação alcalina de ésteres é uma das reações mais bem estudadas, tanto do
mecanismo quanto das variações do substrato. A influência de substituintes X no anel
aromático de ésteres do tipo X−Ar-CO-OR´, particularmente, levou a uma sofisticação da
reatividade em reações orgânicas, conhecida como "Equação de Hammett" 244 ou “linear
free energy relationship” 245. É uma tentativa de quantificar a influência dos efeitos
indutivos, mesoméricos e estéricos 246, sobre a reatividade carboxílica. Apesar de inúmeras
tentativas (desde os anos 30 do século passado), porém, não se conhece ainda uma
expressão universal que relaciona, de maneira quantitativa e satisfatória, todos esses efeitos
com a reatividade.
"Catalisador" mais usado nesta síntese é o hidróxido de sódio:
Saponificação do éster:
O-
O
+
R C
OH-
OCH3
O
+
R C
CH3O-
OH
lento
R C OH
O
+
R C
OCH3
R COO- Na+
CH3O-
OH
+
CH3OH
Carboxilato de sódio,
precipita da mistura reacional
A última etapa deste mecanismo faz com que a reação seja praticamente quantitativa visto
que o produto é removido do equilíbrio reacional. A velocidade da saponificação é
v = k ⋅ Éster ⋅ OH − , então depende das concentrações do éster e do hidróxido. A palavra
"catalisador" foi escrita em aspas, por que o hidróxido, além de promover a reação de
hidrólise, também é consumido - o que não está conforme à definição rigorosa de um
catalisador.
[
][
]
244
L.P. Hammett, Physical Organic Chemistry, Reaction Rates, Equilibria, and Mechanisms, McGraw-Hill,
New York 1970.
245
Em uma revisão histórica-filosófica o trabalho de Hammett foi descrito como início da sub-disciplina
“Fisico-química orgânica”: K.N. Houk, J.K.Lee, Pure&Appl.Chem. 69 (1997) 237-239 ou
http://www.iupac.org/publications/pac/1997/pdf/6902x0237.pdf
246
R.W. Taft Jr., J. Am. Chem. Soc. 74 (1952) 3120-3128; ibid 75 (1953) 4231-4238.
371
A. Isenmann
Princípios da Síntese Orgânica
Um exemplo com relevância prática no laboratório é a saponificação do éster ftalato de
dietila com sódio metálico, um método rápido para se obter etanol absoluto, em pequena
escala. O etanol proveniente da destilação (de 92 a 96%; o restante é água) é tratado com
sódio metálico, na presença do éster etílico. A água presente na mistura azeotrópica reage
mais rapidamente do que o etanol, devido sua elevada acidez:
2 H2O + 2 Na
2 NaOH
+
H2
O sódio deve ser acrescentado aos poucos (perigo de incêndio!) e excesso deve ser evitado.
Já o hidróxido de sódio produzido por esta reação "catalisa" a saponificação do éster,
liberando etanol:
COO-
COOC2H5
2 Na+
+ 2 NaOH
+
2 C2H5OH
COO-
COOC2H5
Ftalato de dietila
Desta forma, o ponto azeotrópico do álcool hidratado pode ser superado com facilidade e a
água removida quantitativamente.
b) Transesterificações sob catálise básica.
De maneira semelhante à saponificação funciona a transesterificação em álcool, sob adição
de alcóxido. As quantidades da base não precisam ser estequiométricas, pois em cada etapa
se reforma um íon de alcóxido. Afinal tem-se um equilíbrio ácido-base que reforma o
mesmo tipo de alcóxido que tinha no início.
Exemplo:
O
R C
OCH3
+
Ph
CH2 O-
Alcóxido benzílico
O
+ CH3O-
R C
Solvente:
OCH2 Ph
Ph-CH2OH
Ph
CH2O-
+ CH3OH
O equilíbrio desta reação pode ser deslocado para a direita, desde que um dos produtos é
removido sucessivamente da mistura reacional. No caso de ésteres de metila com ponto de
ebulição alto, se oferece a remoção contínua do co-produto, o metanol, ao trabalhar a
temperaturas elevadas (≈ 80°C). Mais usado, portanto, é o metiléster e o metóxido de sódio
(ou potássio).
372
A. Isenmann
Princípios da Síntese Orgânica
O metóxido é também o catalisador atualmente mais usado na produção de biodiesel
por vários motivos:
• é barato,
• é mais eficaz do que o hidróxido,
• não arrasta muita umidade,
• promove a compatibilidade entre a fase polar e a gordura,
• produz poucos ácidos graxos livres,
• produz poucos resíduos que causam corrosão nos motores a combustão.
247
,
O metóxido usado neste processo podemos realmente chamar de catalisador, por que o
reagente em si para a transesterificação é o metanol, enquanto o consumo do metóxido fica
apenas em torno de 5 a 10% do metanol.
H2C O CO
H C* O CO
H2C O CO
Óleo; gordura
+
3 CH3OH
Catalisador:
[CH3O-]
H2C OH
HC OH
H2C OH
Glicerina
O
+ 3
C OCH3
Metilésteres
(Biodiesel)
A matéria prima desta reação é gordura, que pode ser de origem animal ou vegetal. No caso
da procedência animal, os ácidos graxos principais são: ácido palmítico, ácido esteárico
(ambos têm cadeias carbônicas saturadas) e o ácido oléico (contém uma dupla-ligação
C=C) 248. No caso de óleos vegetais, o teor em ácidos graxos com múltiplas insaturações é
elevado. A maioria dos ácidos graxos tem 14, 16 ou 18 - de qualquer maneira um número
par de carbonos.
Em geral se observam os seguintes padrões: quanto mais saturadas as cadeias carbônicas,
mais alto o ponto de fusão da gordura (e também dos demais ésteres). Bastante alto, por
exemplo, é o ponto de fusão do sebo de boi, por ser altamente saturado. Um passeio no
açougue revela que sebo de boi, a temperatura ambiente, é sólido. Esse é o motivo principal
de a matéria graxa não achar aplicação direta como combustível em motores a diesel.
Os metilésteres dos ácidos graxos, por outro lado, têm qualidades fisico-químicas bastante
semelhantes ao diesel de origem mineral (petróleo), ou seja, têm pontos de fusão em torno
de -30 °C. Uma vez efetuada a transesterificação, o biodiesel proveniente de gordura
saturada é até melhor do que da insaturada, devido à menor tendência para formar resíduos
resinosos (polimerização radicalar de alquenos, ver p. 164) e também, à maior estabilidade
frente auto-oxidação (p. 97). As reações silenciosas com o oxigênio do ar - condições que
se têm nos armazéns do biodiesel - levam à quebra da cadeia carbônica, nos locais da
insaturação. Oxidação subsequente dos aldeídos finalmente leva aos ácidos carboxílicos
livres, por sua vez altamente indesejados por serem produtos resinosos e corrosivos que
247
G.Knothe, J.Krahl, J.V.Gerpen, L.P.Ramos, Manual de Biodiesel, Edgard Blücher, São Paulo 2008
Uma curiosidade: evidentemente o carbono 2 na parte da glicerina é um centro assimétrico, caso estiver
esterificado com três diferentes cadeias carbônicas (este geralmente é o caso). Mesmo assim, a gordura
natural não gira a luz polarizada no polarímetro. Isso se deve às longas distâncias entre o centro assimétrico e
as unidades estruturais que causam a diferença.
248
373
A. Isenmann
Princípios da Síntese Orgânica
podem danificar as partes metálicas e as mangueiras e vedações de borracha do motor.
Portanto, o biodiesel de qualidade tem que ser aditivado, por antioxidantes (fenóis), entre
outros 249.
c) Saponificação, esterificação e transesterificação, sob catálise ácida
Os catalisadores ácidos mais usados para a saponificação de ésteres são o ácido
trifluoracético (TFA) e o ácido fluorídrico. O mecanismo pode ser formulado em cinco
etapas, todas elas reversíveis e equilibradas.
O
+
C6H5 C
H
+
K
OH
C6H5 C
OCH3
+ H2O
OH
C6H5 C OH2
OCH3
OCH3
OH
C6H5 C OH
O
H
k
(lento)
OH
OH
- CH3OH
C6H5 C
C6H5 C
CH3
OH
OH
O
- H+
C6H5 C
OH
A primeira reação se equilibra rapidamente. Sua constante de equilíbrio é
ÉsterH +
K=
; ao empregar um excesso de água pode-se formular a equação cinética
Éster ⋅ H +
da reação global como:
v = k ⋅ ÉsterH + = k ⋅ K ⋅ Éster ⋅ H + .
Enquanto a reversibilidade da saponificação básica, descrita mais em cima, foi impedida
pela insolubilidade do carboxilato, a saponificação ácida é livremente reversível. Isto
significa que a reação se desloca à direção reversa, ao trabalhar:
com um excesso de álcool; ou
removendo a água produzida.
Estas, ao mesmo tempo, são as medidas a serem tomadas para a esterificação, sob catálise
ácida.
A transesterificação, já mencionado no item anterior, segue os mesmos princípios: o álcool
liberado deve ser sequestrado continuamente.
249
Uma descrição qualitativa dos aditivos no biodiesel se encontra na página da Degussa AG, lider mundial
no setor.
http://www.degussa-biodiesel.com/
374
A. Isenmann
Princípios da Síntese Orgânica
OH
[ H+]
+ C2H5OH
R C
OH
+ CH3OH
R C
OC2H5
OCH3
Este tipo de transesterificação é o método principal na produção de poliésteres (o mais
produzido é o PET), a partir de um diéster, dietílico ou dimetílico, e um diol em excesso.
Importante na produção de poliésteres é a separação de qualquer umidade - quer como
impureza dos reagentes, quer como sub-produto da esterificação. Somente sob a condição
de separação rigorosa da água se consegue massas molares elevadas, uma precondição para
um material de construção de alta qualidade (módulos, resistência ao impacto e resistência
química altos).
Por outro lado, o método é pouco aceito (ainda) na produção de biodiesel, principalmente
devido aos custos elevados de ácidos, que devem ser bastante fortes, ao mesmo tempo
compatíveis com a matéria graxa. Em geral se mantém a incompatibilidade das fases aquosa e orgânica - durante todo o processo da transesterificação ácida, enquanto a catálise
básica (ver último ítem) sempre produz, em pequenas quantidades, carboxilatos dos ácidos
graxos, comumente conhecidos como "sabões". Agora entendemos porque catalisadores
básicos promovem a compatibilidade entre as fases da mistura reacional e, em
consequência, levam a um aumento da velocidade da reação. Além destes, são os ácidos
que causam os maiores estragos nas partes metálicas devido à corrosão - tanto no reator da
fábrica do biodiesel quanto no motor de combustão.
d) Formação da hidrazida, a partir da hidrazina 250:
O
C6H5 C
OCH3
O+ H2N NH2
Hidrazina
C6H5 C NH2 NH2
OC6H5 C NH NH2
OCH3
O
H
CH3
O
- CH3OH
C6H5 C
NH NH2
Benzidrazida
O segundo grupo -NH2 da hidrazina também pode reagir com um éster. Isto acontece,
especialmente ao gotejar solução de hidrazina sobre o éster, em geral, ao empregar um
excesso do éster. Essa segunda reação da hidrazina é mais pronunciada ainda quando
substituimos o éster para o cloreto do ácido carboxílico.
250
Não se deve confundir com a hidrazona, que é o produto de condensação entre hidrazina e uma
cetona/aldeído. Por exemplo, a 2,4-dinitrofenilhidrazona, R-CH=N-NH-(C6H3)(NO2)2, usada com finalidade
de caracterizar aldeídos e cetonas pelo ponto de fusão (ver também p. 411)
375
A. Isenmann
Princípios da Síntese Orgânica
e) Ésteres como grupos protetores
A saponificação ácida e também a básica de ésteres “comuns” requer condições bem
rigorosas, tanto de pH quanto de temperatura. Este fato limita o uso de ésteres como grupos
protetores na síntese orgânica, onde uma das precondições é a fácil remoção do protetor
após a reação desejada em outra parte do substrato. Entretanto, existe um número limitado
de ésteres que podem ser quebrados em seus componentes, sob condições mais suaves,
seguindo mecanismos especiais. Um destes é o éster de t-butila que se mostra estável em
ambiente básico, mas é facilmente hidrolisado em ambiente ácido. O cátion t-butílico tem
estabilidade suficiente para ser abandonado pelo éster protonado.
O
[H+]
R C
OH
R C
OBu-t
R COOH
O
+
(CH3)3C+
Bu-t
H2O
Mecanismo aAL1
(CH3)3COH
Os seguintes ésteres podem ser quebrados sob condições mais brandas ainda. Nestes casos
é melhor evitar a expressão "saponificação" - uma vez que um dos produtos destas reações
não representa mais um álcool. Em ambos os casos aplicam-se condições redutoras
seletivas. Note-se que o último éster pode ser usado como protetor, até na presença de
duplas ligações C=C no substrato, porque o zinco não ataca olefinas:
O
O
H2
R C
O CH2 Ph
Pd (C)
+ CH3Ph
R C
OH
Éster benzílico
O
R C
Cl
Zn
R COO- + H2C CCl2
O CH2 C Cl
Cl
Éster do tricloroetanol
MeOH
R COOH
Assim, os ésteres servem como protetores, particularmente de ácidos carboxílicos, somente
em segunda linha para alcoóis.
376
A. Isenmann
Princípios da Síntese Orgânica
f) Saponificação de amidas
A saponificação de amidas pode ser conduzida, em analogia aos ésteres, em meio alcalino
ou ácido. Porém, deve-se dar preferência à catálise ácida porque ela se torna irreversível,
por causa da formação do sal na última etapa da reação.
O
+
R C
H+
R C
OH + H O
2
OH
R C O
NH2
NH2
H
NH2
H
OH
R C OH
NH3
O
OH
- NH3
R C
OH
- H+
R C
OH
+ H+ irreversível
NH4+
Sal de amônio
(precipita)
g) Nitrilas a partir de amidas
A formação de nitrila a partir de amidas, funciona na presença de agentes dessecantes
fortes, em analogia à síntese dos anidridos descrita na p. 360:
∆
O
[ P4O10 ]
R C
NH2
- H2O
R C N
Ela é de execução mais fácil do que a reação “reversa” desta, a hidrólise das nitrilas,
conforme descrita a seguir.
5.3.3
Reações das nitrilas
a) Reações das nitrilas com nucleófilos fracos
Hidrólise para amidas ou ácidos carboxílicos
A hidrólise ocorre em duas etapas, primeiro para a carboxamida, depois para o
carboxilato/ácido carboxílico. Pode ser catalisada, tanto por bases (caminho comum)
quanto por ácidos fortes. Embora este ser muitas vezes o acesso preferido para os ácidos
carboxílicos, a aplicabilidade desta reação tem certas restrições e desvantagens.
A restrição geral é um rendimento moderado a baixo, que se deve à etapa subsequente da
síntese: a neutralização dos catalisadores, tanto dos ácidos quanto dos básicos. Formam-se
sais pouco solúveis em larga escala, acompanhados por reações paralelas e efeitos de
poluição do produto que geralmente é também sólido.
Além disso, a reação não pára na etapa da amida. Uma comparação das hidrólises de
acetonitrila e acetamida, ambas com cinéticas de segunda ordem sob catálise de hidróxido,
mostram velocidades bem comparáveis. A decomposição da acetonitrila é até um pouco
mais lenta:
377
A. Isenmann
Princípios da Síntese Orgânica
k[OH − ] = 1, 6 ⋅10−6 M −1s −1 para a acetonitrila e k[OH − ] = 7, 4 ⋅10−5 M −1s −1 para a acetamida.
Significa que não é possível obter-se a acetamida sendo um produto puro. Além disso, os
valores absolutos destas constantes documentam uma reação lenta. Temperaturas altas e
prolongadas (2 dias, sob refluxo) são necessários, para obter-se um rendimento aceitável.
Enfim, a saponificação alcalina não ocorre com facilidade. Um exemplo da sua aplicação é
a terceira etapa da síntese de Kolbe, descrita na p. 382.
Caminho da saponificação alcalina:
OH
R CN
+
OH
-
R C
O
R C
N
NH
+ H2O
- OH-
O
R C
NH2
+ OH- NH3
R COO-
Reagentes de baixo caráter nucleofílico necessitam então da ativação do grupo nitrila.
Neste sentido funciona a saponificação ácida da nitrila. Também ela é afetada por inerentes
desvantagens, porque pode ocorrer uma reação paralela: como a hidrólise é de caráter
exotérmico, então necessita de um controle rigoroso da temperatura, se não formam-se
excessivamente polímeros 251.
Caminho da saponificação ácida:
H
R C N H
Cátion nitrílio
H2O
O H
R C
R C
NH
O
OH
NH2
- H+
R C
NH2
Na parte inicial deste caminho, verificamos que a protonação do nitrogênio tem um efeito
ativador frente ao nucleófilo fraco, a água, porque aumenta a polarização δ+, no carbono do
grupo nitrila. A variedade dos catalisadores para reações de nitrilas com nucleófilos fracos
é grande. Na maioria dos casos, usa-se um ácido mineral forte como catalisador.
A escolha entre os dois métodos, catálise básica ou ácida, depende dos demais substituintes
no substrato, que podem ser sensíveis ou mudar a reatividade da nitrila, sob a influência
destes catalisadores. Para a maioria dos outros grupos funcionais, um ambiente fortemente
ácido é mais prejudicial (por exemplo, acetais ou aminais, ver pp. 403 e 409).
251
Revisão: V.Yu. Kukushkin, A.J.L. Pombeiro, Inorg. Chim. Acta 358 (2005) 1-21.
378
A. Isenmann
Princípios da Síntese Orgânica
Reação de nitrilas com alcoóis
Bastante divulgadas são as reações de nitrilas com alcoóis, sob catálise de HCl, que se
conhecem com reações de Pinner. O imidoéster que resulta desta adição aproveita da
estabilização pelo HCl. Ele pode até ser isolado, mas apenas em forma do cloridrato
(também chamado de hidrocloreto).
Por exemplo, a reação da nitrila com metanol ocorre de preferência sob catálise de HCl-gás
a seco.
R C N
+ HCl
-
R C N H Cl
NH2
NH
+ CH3OH
R C
R C
O CH3
O CH3
Cátion nitrílio
H
Cl-
Hidrocloreto do imidoéster
H2O
- HCl
- NH3
O
R C
O CH3
Ao aplicar um excesso de metanol pode-se formar o ortoéster:
NH2
NH
R C
R C
O CH3
Cl
-
O CH3
H
+ 2 CH3OH
- NH4Cl
OCH3
R C OCH3
OCH3
Ortoéster
Esta é a estratégia mais usada para conseguir a classe dos ortoésteres. O ortoéster do ácido
fórmico é usado como reagente para produzir cetais e acetais (ver item 5.5.6), em casos
onde uma reação direta do álcool com a cetona/aldeído fica difícil. Quando exposto ao
reagente de Grignard, abre-se o caminho de acessar aldeídos (p. 436). Além disso, os
ortoésteres têm certa importância como grupos protetores para o grupo carboxílico, porque
são altamente estáveis em ambiente alcalino, mas hidrolisam facilmente ao ser expostos a
ácidos diluídos, liberando o ácido carboxílico e três moléculas de álcool.
b) Reações das nitrilas com nucleófilos fortes
Nitrilas (uma vista geral, tanto sobre as sínteses das nitrilas quanto suas reações, encontrase no anexo 2 deste livro) são bastante estáveis e, portanto, pouco reativas. As reações no
grupo –C≡N evidentemente não ocorrem com a mesma facilidade que nos demais
379
A. Isenmann
Princípios da Síntese Orgânica
derivados do ácido carboxílico. Isto se vê, por exemplo, no próprio ânion cianeto (=
pseudo-haleto 252) que funciona como nucleófilo, sem perder sua tripla ligação.
Porém, nucleófilos fortes, tais como o hidreto ou um carbânion, conseguem reagir
diretamente com o carbono do grupo nitrila.
Reação com hidreto (compare p. 613):
LiAlH4
R CN
Redução total
R CH2 NH2
Amina primária
Reação com carbânion (ver também p. 615):
R CN
N
+ R´ Li
(ou Grignard)
R
C
+
Li
R´
O
H2O / H+
hidrólise
R
Sal da cetimina
(estável)
C
R´
Cetona
O segundo exemplo é uma síntese bem útil para cetonas. A nitrila somente reage com um
carbânion; a entrada de um segundo carbânion (como, por exemplo, está o caso nos ésteres)
é impedida por causa da carga negativa do complexo intermediário. Por esta razão não há
necessidade de se manter quantidade estequiométrica exata do reagente organometálico.
Qualquer excesso será imediatamente destruído na última etapa, a hidrólise.
c) Reação das nitrilas com eletrófilos: a reação de Ritter
A reação de Ritter é um método bastante simples para se obter amidas a partir de nitrilas.
H
OH
ou
CH2
+ R C N
H2SO4
H2O
N
O
R
Porém, é necessário um meio fortemente ácido, o que certamente limita o espectro das
aplicações deste procedimento simplificado (en inglês muitas vezes referido como “one pot
reaction”), especialmente quando haver outros grupos funcionais na molécula do substrato
252
A expressão "pseudo-haleto" vem da inorgânica e se deve à certa semelhança química entre os ânions
complexos, tais como CN-, N3-, SCN-, NCO-, e os ânions simples de F-, Cl-, Br- e I-. Sendo assim, os sais de
Ag+, Hg22+, Pb2+, Cu+ e Tl+ são pouco solúveis. Além disso, todos esses ânions funcionam como ligantes em
complexos estáveis de metais de transição. Com exceções existem também dimeros moleculares (X2) destas
espécies.
380
A. Isenmann
Princípios da Síntese Orgânica
(isto é, restrições similares à reação de Nef, p. 533, à alquilação de Friedel-Crafts, p. 296 e
à redução de Clemmensen, p. 609). O segundo reagente nesta reação pode ser um alqueno
ou um álcool a partir dos quais se forma um cabocátion. Este por sua vez é um eletrófilo
bastante forte que ataca o nitrogênio do grupo nitrila 253:
OH
H2SO4
OH2
- H2O
CH2
R
H2SO4
CN
N C
R
N C
R
H
N C
R
N
H2O
OH2
Tautomeria
[H +]
R
N
OH
R
H
H
N
OH
- H+
R
N
O
R
Como este mecanismo exige a formação de um carbocátion, a reação de Ritter não é viável
com alcoóis primários cujo carbocátion não tem estabilidade suficiente. Implica também
que o grupo alquila no nitrogênio do produto amida, sempre é ramificado, isto é, Csec.-N ou
Cterc.-N.
Uma outra restrição: alcoóis aromáticos (por exemplo, o álcool benzílico) também não
funcionam no sentido da reação de Ritter, já que outras reações do tipo alquilação de
Friedel-Crafts ocorrem mais rapidamente, no anel aromático.
Uma variação bastante utilizada, por outro lado, é a reação com ácido cianídrico, que
fornece a formamida. Devido à alta toxicidade deste gás essa síntese somente deve ser
executada em autoclave!
Note que o acoplamento entre o carbocátion e o ácido cianídrico ocorre na parte dura do
agrupamento C≡N (o cianeto é nucleófilo ambidente, ver p. 44), isto é, no nitrogênio.
Todavia, não podemos excluir a reação paralela no carbono (mole) deste grupo que levará à
nitrila terciária.
253
K. L. Reddy, Tetrahedron Lett. 44 (2003) 1453.
381
A. Isenmann
5.3.4
Princípios da Síntese Orgânica
Reações dos carboxilatos.
Ao ser atacado por um nucleófilo forte, qualquer derivado do ácido carboxílico da lista na
p. 354 reage. Muitas vezes o derivado menos eletrofílico de todos, o carboxilato, ganha
preferência neste tipo de adição nucleofílica.
a) Reação com LiAlH4
O hidreto, H-, é um dos melhores nucleófilos que existem. Sua basicidade e
polarizabilidade são bastante altas, seu volume é pequeno e os solventes tipicamente usados
(éteres) não conseguem solvatá-lo (compare os critérios da nucleofilia, p. 40). A reação
com LiAlH4 funciona bem com todos os derivados do ácido carboxílico. Como o número
de oxidação do carbono carboxílico cai de + 3 para -1, então trata-se formalmente de uma
redução (ver item 8.9). Resulta um álcool ou uma amina – em dependência do derivado
carboxílico usado.
Esta redução de carboxilatos tem certo valor preparativo, porque representa a primeira
etapa na síntese de Kolbe, para extensão da cadeia carbônica por um carbono.
Alongamento da cadeia carbônica em carboxilatos, segundo Kolbe:
R COO- Na+
LiAlH4
R CH2 OH
SOCl 2
R CH2 Cl
NaCN
SN2
R CH2 CN
NaOH / H2O
R CH2 COO- Na+
b) Compostos organo-lítio
Nos últimos anos os reagentes clássicos de Grignard (R-MgX, ver p. 432) vêm sido cada
vez mais substituídos pelos compostos organo-lítio. As velocidades e os rendimentos
muitas vezes aumentam, enquanto o número de reações paralelas diminui (ver p. 440).
Os carbânions proporcionados pelos compostos organo-lítio representam, em analogia ao
hidreto, excelentes nucleófilos. Também eles levam a uma redução do número de oxidação,
de +3 para -1 (exceção somente as nitrilas: NOX +3 para +1; ver p. 379) no carbono
carboxílico. Portanto, a reação pode ser chamada de "adição redutiva". Observe que traços
de água catalisam esta reação.
Mostrado isso na adição de R´Li ao carboxilato:
R COO-
R´Li
Traços
de água
O
R C
R´
Cetona
R´Li
Traços
de água
O- Li+
R C
R´
R´
H2O
Hidrólise
OH
R C
R´
R´
Álcool terciário
382
A. Isenmann
5.3.5
Princípios da Síntese Orgânica
Desvios do mecanismo padrão da substituição em compostos
carboxílicos
O mecanismo E→A
Todas as reações do tipo SN discutidas por enquanto, seguiram mecanismos onde a primeira
etapa foi a adição do nucleófilo, seguida pela eliminação do grupo abandonador, ou seja,
A→E. Em alguns exemplos a segunda etapa, a eliminação, até ficou ausente (ver logo
acima). Sem dúvida alguma, este é o mecanismo padrão da substituição em ácidos
carboxílicos, em geral no grupo carbonila. Importante para esta sequência reacional é:
1. O acesso livre do nucleófilo ao carbono carboxílico,
2. Os quatro grupos, ligados no carbono sp³ do complexo intermediário, não tenham alto
impedimento estérico. Esses dois primeiros argumentos quase sempre são amarrados
um a outro.
3. O grupo R no substrato R-COX seja um grupo retirador de elétrons; isso aumenta a
polarização positiva do carbono carboxílico e facilita o recebimento do nucleófilo.
4. Alternativamente, pode-se usar um nucleófilo muito poderoso que consegue atacar, até
num carbono pouco positivado.
Existe um número limitado de compostos que se desviam do padrão, procurando caminhos
da “segunda linha” (quer dizer, energeticamente mais exigente do que A→E).
É a alta exigência espacial mencionada no ponto 2 que impede o mecanismo
A→E no caso do ácido carboxílico do mesitileno e seus derivados (ver esquema a seguir).
Os grupos o-metilas do anel impedem a entrada de mais um vizinho no carbono do grupo
carboxílico. A molécula está bem blindada contra um ataque nucleofílico (o que
corresponde ao ponto 1 acima). Além disso, o anel aromático é rico em elétrons, então é
fornecedor de elétrons (ponto 3 da lista). Desta forma promove a formação de um cátion
intermediário após a exclusão do metanol. Somente depois o nucleófilo fraco entra em
ação: a água (ponto 4 da lista).
Enfim, a hidrólise deste éster não ocorre facilmente, porém pode ser forçada com H2SO4
bastante concentrado. O mecanismo é E→A (compare p. 353).
CH3
CH3
H2SO4
O
C
OCH3
CH3
H3C
H3C
CH3
O
C
- CH3OH
O CH3
CH3
H
H3C
C O
CH3
CH3
C O
H3C
+ H2O
- H+
H3C
CH3
O
C
OH
CH3
CH3
Oxônio-cátion
383
A. Isenmann
Princípios da Síntese Orgânica
As mesmas considerações valem para a esterificação deste ácido livre, ou seja, a reação
reversa da hidrólise. Ela não pode ser estabelecida por um álcool nem por um alcóxido, mas
sucede favoravelmente com diazometano (sobre as reações do diazometano, ver p. 875; sua
produção a partir do Diazald, ver p. 869):
CH3
O
+ CH2N2
C
OH
CH3
H3C
[ éter ]
- N2
H3C
CH3
O
C
OCH3
CH3
Note que essa não é uma SN, já que o oxigênio do grupo -OCH3 nunca saiu do grupo
carboxílico. Ver outro exemplo desta reatividade, logo a seguir.
Quebra da ligação O-alquila em ésteres
Um outro exemplo de reatividade fora do padrão será demonstrado no mesmo éster. A
seguinte substituição não ocorre no carbono carboxílico, mas é uma substituição
nucleofílica no grupo alcoólico (em negrito). Tentativas de transesterificação, até mesmo
com bons nucleófilos (tal como o tiofenolato), falham.
H3C
Éster
CH3
O
C
O CH3 +
CH3
[ DMSO ]
-
S
C6H5
Tiofenolato
SN2
CH3
O
+ H3C S C6H5
C
H3C
O
CH3
Carboxilato
Tioéter
Experimentos com ésteres deste tipo, marcados com 18O, não deixam dúvida sobre este
caminho. Neste exemplo o ânion do tiofenol ganhou maior reatividade como nucleófilo,
pelo uso de DMSO como solvente. Meios polares e apróticos deixam os ânions “nus” e
aumentam assim sua nucleofilia (ver p. 29). A substituição no grupo metila ocorre de
maneira sincronizada, sendo então uma reação SN2. Ao contrário do esperado, não se
obteve o tioéster, mas sim, o tioéter de metil.
A alternativa a este mecanismo é a substituição do tipo SN1 no éster, que é possível quando
as seguintes condições estão satisfeitas:
1) o solvente usado é de alta polaridade;
2) o substrato é um éster formado a partir de um ácido carboxílico bastante forte (isto é, o
grupo abandonador é muito bom)
3) o carbocátion intermediário tem um alto padrão de estabilidade (carbocátion terciário,
alílico ou benzílico; ver também p. 376).
384
A. Isenmann
CH3
H3C
C O
Princípios da Síntese Orgânica
O
O
CH3
C
H2O / acetona
SN1
CH3
H3C
C
-
+
C
O
CH3
NO2
NO2
H2O
O
CH3
H3C
18
= marcado pelo isótopo O
5.4
C OH
C
HO
+
CH3
NO2
Reações no carbono em posição α ao grupo carboxila
Os grupos carbonila (C=O) e carboxila (C(=O)O) são considerados retiradores de elétrons
(ver p. 316). Ele exerce os efeitos –I e –M sobre seu carbono vizinho (as definições destes
efeitos, ver p. 316). A consequência é uma acidez elevada da ligação C-H, em posição α.
Isto se deve à estabilização do produto desta desprotonação, ou seja, da base
correspondente, que é formalmente um carbânion. Podemos ver o composto carbonilado
com grupo CH em posição α, sendo um “pseudo-ácido”(p. 532).
H O
C
C
ou
H O
C
O
C O
C
O
C
+ base
C
C
ou
+ baseH+
O
C C O
O
C
C O
Uma importante classe de reações é a condensação de ésteres (p. 458) e outros compostos
com grupo carbonila. Por causa da grande importância preparativa, um capítulo inteiro
(cap. 6) é dedicado à sua discussão. Ela se baseia na formação do enolato que só é possível
por causa da acidez, mencionada acima.
Portanto, neste capítulo serão apenas apresentadas
•
As eliminações que fornecem o ceteno;
•
As degradações da cadeia carbônica por uma unidade metilênica;
•
A adição iônica de bromo à posição α em ácidos carboxílicos (reação de HellVolhard-Zelinsky).
385
A. Isenmann
Princípios da Síntese Orgânica
As reações radicalares, inclusive a quebra no carbono α que decorre no espectrômetro de
massas, ver p. 107.
5.4.1
Formação de cetenos
Certamente, ninguem jamais tinha um ceteno em mãos, porque esta espécie é muito reativa.
Falamos de espécies com a unidade estrutural C=C=O, que é formalmente o anidrido
intramolecular do ácido carboxílico. Sua caracterização vem logo a seguir (p. 388).
a) Cetenos a partir de cloretos de ácidos carboxílicos
Esta síntese requer a adição de uma base, sendo piridina a mais usada. Mas também
funciona com aminas alifáticas terciárias. Após de tudo que foi falado no cap. 5.2, é
evidente que essa base não deve ter boas qualidades nucleofílicas.
H
O
O
H3C
+
C
H3C
N
C
N
N
-
Cl
∆
Cl
H2C
C O
Ceteno
Cl-
+
Cloreto de
Piridínio
Ao tiver uma molécula com dois grupos cloretos de carbonila, pode-se esperar ciclização
(síntese de Blomquist, ver p. 473).
Lembre-se: a mistura do cloreto de acila com piridina é um dos melhores reagentes para
esterificações (ver p. 365), que neste exemplo seria uma "acetilação" 254.
Apesar de ser este o método de obtenção mais comum, existem também outras estratégias
para se obter cetenos:
b) Cetenos segundo Staudinger
O substrato desta síntese, o brometo de α-bromoacetila, é de fácil obtenção, a partir da
reação de Hell-Volhard-Zelinsky (ver p. 394). Em comparação à síntese com o cloreto de
acila, a síntese de Staudinger funciona a temperaturas mais brandas, porém, certos grupos
funcionais no substrato podem sofrer redução sob estas condições (remoção de grupos
fenólicos, pp. 598 e 603; óxidos de enxofre, p. 620; síntese de Reformatzky, pp. 394 e 503).
O
R CH C
Br
Br
+ Zn
- ZnBr2
H2C
C O
c) Pirólise do ácido acético (Wacker) ou da acetona (Schmidlin)
Estas reações são eliminações cis que pertencem às reações eletrocíclicas (pp. 146 e 204).
254
"Acilação" é a expressão para a reação com o grupo R-(C=O)-, em geral.
"Acetilação", no entanto, denomina a reação com o grupo H3C-(C=O)-.
386
A. Isenmann
Princípios da Síntese Orgânica
O
H3C
C
H2C
700 °C
OH
C O
+ H2O
Síntese de Wacker
C O
+ CH4
Síntese de Schmidlin
∆ H = +209 kJ mol-1
De maneira análoga:
H3C
O
[Cr / Ni]
CH3
780 °C
C
H2C
A acetona, ao passar por uma espiral quente de cromo-níquel, se decompõe em ceteno e
metano, com bons rendimentos. O equipamento especial é conhecido como “lâmpada de
ceteno”. A mistura dos gases que sai desta é lavada com acetona fria a -60°C. Assim, o
metano pode ser eliminado da mistura; embora o CH4 seja uma molécula bastante inerte,
essa etapa não deve ser dispensada, devido ao caráter reversível desta síntese.
d) Pirólise de diazocetonas
A partir da benzila (síntese ver p. 465) se consegue com quantidades sub-estequiométricas
de hidrazina a mono-hidrazona. Segue uma etapa de oxidação seletiva da hidrazona a parir
da qual uma molécula de N2 pode ser abstraída, liberando um carbeno. Como carbenos são
espécies extremamente instáveis (apenas 6 elétrons na camada de valência do carbono), a
última etapa desta síntese é um rearranjo no carbono com sexteto eletrônico, chamado
rearranjo de Wolff (ver a discussão dos demais rearranjos na p. 386). Esta síntese, segundo
Schroeter, fornece o difenilceteno.
Ph
C C
O O
Benzila
Ph
N2H4
Ph
C C
Ph
O N NH2
Monohidrazona
de benzila
HgO
-2H
Ph
C C
Ph
O N N
"Rearranjo de ∆
Wolff"
o - N2
Ph
O C C
Síntese de Schroeter
Ph
Podem-se obter cetenos em geral (sem a restrição de ter dois grupos fenilas), com a
seguinte estratégia:
O material de partida é uma diazocetona que é acessível via síntese de Arndt-Eistert
(cloreto de acila mais diazometano, ver p. 876; outros compostos α-diazocarboxílicos, ver
pp. 872 e 874). A temperaturas elevadas, especialmente na presença de catalisadores
387
A. Isenmann
Princípios da Síntese Orgânica
metálicos, as diazocetonas podem perder nitrogênio, N2. O produto desta etapa é um
acilcarbeno que tem um sexteto de elétrons no carbono. Conforme discutido mais
extensamente na p. 391, esta situação provoca um rearranjo, no caso, o rearranjo de Wolff.
R C C N N
O H
- N2
R C C
O H
R
o
O C C
Rearranjo de
H
Wolff
Acilcarbeno
5.4.2
As propriedades dos cetenos
O manuseio dos cetenos requer certo cuidado. O experimentador deve estar ciente de que o
próprio ceteno, H2C=C=O, é um gás (Teb. = -56 °C) muito tóxico e cancerígeno. A
molécula é muito reativa - uma característica que também foi observada na comparação de
dienos acumulados com dienos isolados (ver p. 131).
Os cetenos podem ser vistos como os derivados mais reativos do ácido carboxílico. Eles
adicionam muitos grupos HX na sua dupla ligação C=C.
Por exemplo:
R
C
δ+
C O
+
R
δ-
O
R C C
H OH
R
H
OH
De forma análoga reagem:
H OR , H NH2 , H SH , H
S
R , H Cl
Sobre a possibilidade de adicionar-se à dupla-ligação C=C, por via de cicloadição [2+2],
ver p. 245.
Os cetenos sempre devem ser preparados na hora, ou seja in situ, por causa da sua alta
reatividade. Além disso, eles são sintetizados e aplicados geralmente a temperaturas
bastante baixas. Não havendo um reagente adequado ao seu redor, eles se estabilizam por
dimerização. Já o produto desta, o diceteno (líquido, Teb. = 127 °C), tem estabilidade bem
maior, um fato que se reflete na alta exotermia (∆H = -113 kJ·mol-1) desta dimerização.
Todavia, a reatividade do diceteno ainda é suficientemente alta para reagir rapida e
completamente com nucleófilos, por exemplo, com etanol:
388
A. Isenmann
H2C
H2C
Princípios da Síntese Orgânica
C O
C O
H2C
δ+
O
C
O
δ-
H3C
+ C2H5OH
O
CH2 C
C O
lento
C
H2C
∆H = -113 kJ mol
OC2H5
-1
Diceteno
Ceteno
Acetilacetato de etila
Evidentemente esta reação não obedece as regras de Woodward-Hoffmann (ver item 3.5),
segundo as quais uma ciclização térmica [2 + 2] não deveria ser possível. Porém, quanto
mais alta a energia dos reagentes, mais frequentes são os casos de desvio das regras de
simetria dos orbitais. No caso da dimerização dos cetenos é mais razoável formular a
ciclização em etapas, via estruturas dipolares ou até radicalares, em vez de representá-la
com movimentos eletrônicos sincronizados.
O diceteno tem tantas facetas reacionais, quantos nomes:
é um anel de 4 membros incluindo um oxigênio (na nomenclatura tradicional: ~olídeo). Seu
nome, segundo a IUPAC, é 3-buteno-3-olídeo, sua classificação raciofuncional é βmetilenopropriolactona, ou seja, um enoléster cíclico. A reatividade alta do diceteno é, em
partes, uma consequência da tensão que existe em pequenos anéis, conhecida como tensão
de Pitzer 255. Seu carbono carboxílico reage prontamente com nucleófilos. Do ponto de
vista do reagente nucleofílico, foi adicionado o sínton 256 acetilacetonato,
H3CC(=O)CH2C(=O)+.
O
Nu-
O
O
O-
Nu
Enolato
Nu
H+ / H2O
O
O
Derivado da acetilacetona
(β-cetoéster, β−cetoamida, etc.)
Atenção ao desvio reacional: alguns cetenos substituídos podem também dimerizar para
1,3-ciclobutadiona! Logicamente esta tem uma reatividade diferente à do éster cíclico – na
verdade é mais estável e menos reativa.
A altíssima reatividade dos cetenos não é de vantagem preparativa. Na prática é muitas
vezes mais fácil trabalhar com o dicloroceteno que, em analogia ao diclorocarbeno, é de
manuseio mais seguro. As etapas de síntese são semelhantes aos métodos ilustrados acima
(p. 386 e p. 386). O cloreto de dicloroacetila é tratado com amina terciária (que é base, mas
não é nuceófilo), formando o dicloroceteno cuja reatividade fica atenuada em comparação
com a substância-mãe, o ceteno. Este reage, por exemplo, com uma olefina, formando a
ciclobutanona (ver p. 245). Mas a vantagem preparativa se vê quando reage com um cis255
Não apenas o desvio dos ângulos no anel do ideal (109° para sp3, 120° para sp2), mas também a
proximidade dos substituintes laterais aumenta a energia interna e a reatividade em ciclos pequenos.
256
"Sínton" é a expressão para uma partícula hipotética, geralmente com carga, com a qual a molécula alvo
da síntese foi ampliada. Maiores informações, recorra ao capítulo sobre estrategias de síntese, cap. 7.
389
A. Isenmann
Princípios da Síntese Orgânica
1,3-dieno: esta vez a cicloadição "proibida", [2+2], realmente é supressa, enquanto a
cicloadição conforme as regras da simetria dos orbitais, [4+2], ganha em peso. Os
halogênios neste produto primário podem ser removidos redutivamente por zinco em pó
(cap. 8.6).
O
Cl2CH C
O
Produto [2+2] supresso
Cl
NEt 3
Cl
- HCl
Cl
Cl2C C O
Dicloroceteno
(menos reativo)
O
O
Zn
Cl
Cl
Produto principal [4+2]
5.4.3
Reações dos cetenos
A partir do ceteno, sendo o mais reativo dos derivados do ácido carboxílico, existem várias
possibilidades de reagir com nucleófilos. Em geral, o nucleófilo ataca o carbono que é
rodeado por duas duplas ligações (= hibridação sp). Do ponto de vista do nucleófilo a
reação com o ceteno é uma acilação.
+ H2O
O
R
CH2 C
OH
+ AcOH
O
R
CH2 C
O
H
R
δ+
C C O
C
OO
+ R´OH
R CH2 C
R
OR´
+ NH3
+ HCl
O
R CH2 C
NH2
O
R CH2 C
Cl
390
A. Isenmann
Princípios da Síntese Orgânica
As sínteses de Arndt-Eistert (ver mecanismo na p. 876) abrem um caminho para se
transformar um cloreto de ácido carboxílico em seu homólogo de cadeia carbônica maior,
contendo mais uma unidade metilênica no seu esqueleto. O interessante é que, nesta
transformação o carbono de acila (no esboço a seguir, em negrito) não deixa da sua função.
O carbono proveniente do diazometano fica inserido em posição α no produto:
O
CH2N2
R C
O
H
R C C O
Cl
R CH2 C
Cl
O efeito oposto, quer dizer, a diminuição do esqueleto carbônico, é o objetivo das sínteses a
seguir.
5.4.4
Degradação do esqueleto carbônico em derivados de ácido carboxílico
Todas as reações descritas a seguir têm por objetivo a diminuição da cadeia carbônica do
substrato por uma unidade. Elas envolvem uma etapa de rearranjo 257 que sempre se pode
esperar quando um átomo não consegue completar a camada de valência com 8 elétrons.
Além do mais, os substratos perdem uma unidade de CO2. Formalmente, trata-se de uma
oxidação, porém a parte oxidada, o gás carbônico, não é de interesse preparativo.
É bastante comum explicar o motivo para a decorrência de rearranjos, com um nitrogênio,
oxigênio ou carbono com 6 elétrons de valência. No entanto, a mudança de um grupo
vizinho quase sempre ocorre de maneira sincronizada com a saída do grupo abandonador
neste átomo. Até hoje falta a comprovação experimental para a formação de um nitreno,
oxeno ou carbeno livre. Caso o grupo vizinho seja opticamente ativo, sua configuração
absoluta se mantém durante a mudança. Um outro argumento a favor de um movimento
sincronizado provém da cinética com que estes rearranjos decorrem. Em todos os casos
observou-se uma reação muito mais rápida, do que se calculava a partir de uma barreira de
ativação para a formação do teórico nitreno/oxeno/carbeno livre.
Certamente podemos afirmar que os rearranjos que se observam ao redor de átomos com
falta de elétrons (os mencionados, inclusive os carbocátions, rearranjos de WagnerMeerwein, ver p. 19) são muito mais frequentes, do que rearranjos em radicais (exemplo na
p. 394) ou até em carbânions (ver exemplo na p. 429).
N
As degradações a serem discutidas a seguir ocorrem via rearranjo no nitreno, “
”.
Degradação de Curtius
O cloreto carboxílico pode ser substituído pelo “pseudo-haleto” azida, N3-, como já descrito
na p. 363:
257
Os demais rearranjos importantes na síntese orgânica serão tratados em contexto dos aldeídos/cetonas, na
p. 426 em diante.
391
A. Isenmann
Princípios da Síntese Orgânica
O
O
+ NaN3
R C
∆
o
R C
Cl
R N C O
- N2
N N N
Isocianato
Azida do ácido
carboxílico
O preparo da azida do ácido carboxílico é feito em uma mistura de acetona/água. Pode-se
isolar e purificar a azida, porém deve-se lembrar do perigo em trabalhar com quantidades
maiores, pois as azidas podem explodir! O tratamento térmico da azida em benzeno seco
leva ao isocianato, via rearranjo no sexteto do nitrogênio. Na presença de água o isocianato
sofre decomposição: ele é hidrolisado primeiramente para o ácido carbâmico, isto é, a
amida do ácido carbônico. Finalmente é a grande estabilidade do CO2 que leva à
decomposição do ácido carbâmico, deixando atrás o substrato com função de amina, porém
reduzido por uma unidade carbônica.
O
R N C O + H2O
R NH
∆
OH
H2O / H+
R NH2
+ CO2
Ácido carbâmico
(instável)
Aliás, a decomposição hidrolítica do isocianato com liberação do gás carbônico, conforme
descrito acima, é um dos processos que infla os poliuretanos durante a produção de espuma
258
.
Degradação de Hofmann
Uma amida, ao ser tratada com água de bromo em ambiente alcalino, pode ser facilmente
degradada. Na primeira etapa forma-se o eletrófilo, “Br+”, que ataca o nitrogênio e substitui
um dos seus hidrogênios. A base, OH-, abstrai o segundo hidrogênio do N com bastante
facilidade, uma vez que o nitrogênio é rodeado por retiradores de elétrons. Do ânion que
258
A produção mundial de isocianatos é bastante alta, devido ao alto valor industrial e econômico dos
poliuretanos. Os poliuretanos, formalmente produtos de condensação entre diisocianatos e di ou polióis, são
considerados dos mais versáteis materiais poliméricos. Só em 2000 foram produzidos 4,4 milhões de
toneladas de diisocianatos. Destes, 61,3% foram o metileno difenildiisocianato (MDI) e 34,1% o
toluenodiisocianato (TDI).
CH3
OCN
CH2
NCO
NCO
MDI
TDI
NCO
Formação do uretano:
δ+
R N C O
H O R´
O
R N C O
H
R´
Isocianatos alifáticos podem formar trimeros, conhecidos como biuretos. Entre os isômeros estruturais,
isocianato (R-N=C=O), cianatos (R-O-C≡N) e fulminatos [O=N=C]-, os isocianatos são os mais estáveis.
Todavia, todos eles são bastante reativos (ou até explosivos), devido às duplas-ligações acumuladas.
392
A. Isenmann
Princípios da Síntese Orgânica
resulta desta etapa pode sair o brometo, produzindo um nitreno altamente reativo no qual
ocorre o rearranjo.
"Br+"
de
Br2 / OH-
O
R C
-
- Br
- H2O
NH2
Amida
O
R C
H
N
+ OH- H2O
O
R C
N
O
- Br-
R N C O
Isocianato
(não isolável)
Br
Br
Pelos motivos descritos acima, o isocianato não pode ser isolado; ele se decompõe em
ambiente aquoso rapidamente e libera uma amina com esqueleto carbônico reduzido.
Degradação de Lossen
Material de partida:
O
R C
NH OH
Ácido hidroxâmico
O derivado O-acilado do ácido hidroxâmico pode ser facilmente degradado. O tratamento
com base promove a desprotonação do nitrogênio e a saída do ânion acetato, acompanhado
pelo rearranjo que fornece o isocianato.
A reação pode também ser conduzida a partir do ácido hidroxâmico livre, porém com mais
dificuldades (rendimento mais baixo, produtos paralelos). Isto se deve à qualidade inferior
do grupo abandonador, que neste caso seria o ânion hidróxido, OH-.
O
R C
O
O
OH-
R C
NH O C CH3
O
N O C CH3
- AcO-
R N C O
....
As próximas duas degradações percorrem estados radicalares. Como já dito acima são mais
raros do que os rearranjos em sextetos eletrônicos e cátions.
Degradação de Hunsdiecker
Ao tratar carboxilatos de prata com bromo (ou iodo), dissolvido em CCl4, eles
decarboxilam com facilidade, formando o respectivo haleto:
R COOAg + Br2
R
Br + AgBr + CO2
Esta degradação ocorre via radicais, mas não é uma reação em cadeia (compare as reações
do item 1.6). O benefício principal desta degradação se evidencia quando o carbono em
393
A. Isenmann
Princípios da Síntese Orgânica
posição α ao grupo carboxílico (no esquema acima representado pelo grupo R) é de difícil
acesso e/ou faz parte de um esqueleto rígido de carbonos. Assim, por exemplo, consegue-se
a introdução de bromo na posição de cabeça de ponte em compostos bicíclicos (R =
norbornila; ver p. 22). Isto é impossível por uma simples SN1 porque o esqueleto de
carbonos não tem a flexibilidade necessária para percorrer o estado de transição, ou seja,
não asume geometria trigonal plana. Uma introdução direta dos halogênios via substituição
radicalar é igualmente difícil, já que qualquer outro carbono seria bromado antes do da
cabeça de ponte. Além do mais, seria via radical σ (ver p. 53), o que é energeticamente
desfavorável.
Eletrólise de Kolbe
Os ânions carboxilatos, R-COO-, foram considerados os menos reativos de todos os
derivados do ácido carboxílico. A eletrólise de Kolbe funciona justamente com estes sais; o
contra-íon é geralmente Na+. Como todos os processos eletroquímicos, a eletrólise de Kolbe
funciona por transferência de elétrons singulares (SET). Como o substrato já é um ânion,
então a retirada de um elétron (= oxidação, sempre decorrendo no ânodo) fornece um
radical neutro. Os radicais sem carga podem então aproximar-se, sem repulsão eletrostática,
perder CO2 e reagir sob dimerização.
Além deste, existem outros aspectos nessa síntese, diferentes das demais degradações
discutidas acima:
1. Não há reagente químico. Este papel é cumprido por um eletrodo inerte.
2. Não há rearranjo no esqueleto carbônico por que não há sextetos de elétrons (os átomos
oxigênios destes radicais têm na verdade um alcance de 7 elétrons).
3. A cadeia carbônica do sal é diminuída por um carbono, como também nos exemplos
acima. Porém, os radicais formados recombinam imediatamente, formando o dimero, R-R.
O
Ânodo:
2 R C
O-
Cátodo:
2 H2O
O
- 2 e-
+ 2 e-
2 R C
O
2 OH-
- 2 CO2
2 R
R
R
+ H2
O rendimento do produto dimerizado, R-R, é alto porque não há reação mais rápida do que
a recombinação de radicais (ver p. 62). A dimerização dos radicais ocorre diretamente na
superfície do ânodo. Sob condições que serão discutidas no contexto de oxidação (p. 663)
os radicais R·, em vez de estabilizar-se por dimerização, podem também reagir com heteroátomos, caso estes estiverem presentes na solução.
5.4.5
A reação de Hell-Volhard-Zelinsky
Esta reação é de grande utilidade na química preparativa. Seletivamente em posição α ao
grupo carboxila é introduzido um halogênio, bromo ou cloro.
394
A. Isenmann
Princípios da Síntese Orgânica
Esta α-halogenação funciona ao tratar ácidos carboxílicos com excesso de halogênio e
quantidades catalíticas de fósforo vermelho. Numa etapa prévia forma-se o haleto de
fósforo (III). Este reagente substitui o grupo hidroxila e fornece o brometo de acila (p. 355):
R CH2 COOH
+ PBr3
O
OH
R CH2 C
- PBr2(OH)
R CH C
Br Tautomeria
Br
+ Br2
- Br-
Br
R CH C O H
Br
R CH2 COOH
Br
O
R CH C
OH
Produto final
- H+
Br
O
R CH C
Br
Observação: aqui não se trata de um processo radicalar, mas sim, de uma adição iônica do
bromo “Br+” na dupla ligação da forma enólica (ver p. 400). A disponibilidade do "Br+" é
assegurada pela presença do ácido de Lewis, PBr3 (compare p. 294).
Os ácidos α-halogenados, por sua vez, são reagentes muito versáteis, como se vê no
próximo esquema reacional. A primeira etapa de cada caminho representa a troca do αhalogênio por um outro nucleófilo.
395
A. Isenmann
H2C CH COOH
Ácido carboxílico
α,β-insaturado
Princípios da Síntese Orgânica
+ H+
- H2O
H 3C
CH COOH
H3C
OH
Ácido α-hidroxi carboxílico
+ NaSH
+ NaOH
- NaBr
H3C CH COOH
Ácido α-ciano carboxílico
(malonitrila)
1) + OH- ; + H2O ; - NH3
H 3C
δ−
+ NH3
- HBr
COOH
CH COOH
Ácido 1,3-dicarboxílico
(= Ácido metil-malônico)
CH COOH
Br
Ácido α-bromocarboxílico
2) + H+
H 3C
δ+
COOH
H2N
CH
CH3
SH
Ácido α-mercapto
carboxílico
- NaBr
CN
+ NaCN
- NaBr
CH COOH
Zn
R CH O
H2O
celulose
[OH-]
CH3
R CH C COOH
Ácido carboxílico
α,β-insaturado
de esqueleto ampliado
Síntese de Reformatzky
celulose-O CHR COO(carboximetilcelulose = espessante,
emulgador, colóide protetor)
D,L-alanina
(α-aminoácido)
Um destes caminhos, porém, segue um outro princípio mecanístico. A reação conhecida
como síntese de Reformatzky, conforme a definição dada no capítulo 6, é uma condensação
carbonílica. A ativação do componente nucleofílico, porém, é diferente das descritas no
cap. 6, portanto será discutida aqui. A reação de Reformatzky funciona especialmente bem
com α-bromoésteres. É a alta afinidade do zinco pelo bromo que faz esta reação alcançar
altos rendimentos.
Mecanismo:
A reação de Reformatzky é um acoplamento redutivo, no qual o zinco metálico tem o papel
de inverter a polarização no substrato α-bromado in situ. Enquanto o carbono α na
molécula de partida é altamente positivado (ele tem dois vizinhos retiradores, o Br e o
grupo carboxila), o zinco produz um caráter aniônico neste mesmo carbono! Uma inversão
semelhante, também conhecida como "Umpolung" (do alemão), é observada no preparo do
reagente de Grignard, onde o carbono do substrato R-X é positivado e no produto R-MgX
é negativamente polarizado. As condensações apresentadas no capítulo 6, por outro lado,
funcionam via ataque do aldeído ao α-carbono negativado do composto carboxílico ou
carboxilato, devidamente enolizados.
Sobre a formação e a estrutura do complexo intermediário organo-zinco se sabe pouco;
provavelmente é uma SET do zinco para o substrato, percorrendo estados radicalares;
existem evidências de um complexo dimêrico organo-zinco.
O carbono eletrofílico de um aldeído pode condensar-se com o carbono α negativado do
complexo metálico, liberando ao mesmo tempo o zinco. Resulta disso o composto βhidroxi que raras vezes pode ser isolado (em analogia à condensação ácida de aldeídos que
não pára no produto do aldol, ver p. 455). Ele desidrata facilmente e fornece o composto
396
A. Isenmann
Princípios da Síntese Orgânica
carboxílico α,β-insaturado, no exemplo a seguir o éster α,β-insaturado. Pode-se afirmar
que a reação de Reformatzky funciona porque o reagente organo-zinco reage somente com
aldeídos e cetonas, mas não com ésteres nem com α-bromoésteres. Este fato é crucial para
o funcionamento da síntese porque, caso contrário, a formação do complexo organo-zinco
seria acompanhada pela autocondensação do α-bromoéster.
Embora pareça um método perfeito para produzir novas ligações carbono-carbono, a síntese
de Reformatzky não é livre de desvantagens: como muitas outras reações organometálicas,
esta também percorre etapas altamente exotérmicas, com período de indução. As manchas
pretas de Zn, nos tetos em certos laboratórios, são testemunhos do difícil manuseio e os
perigos latentes desta reação. O uso de co-catalisadores organometálicos promete
melhoramentos na síntese de Reformatzky 259.
CH3
δ+
Br
CH
δ+
"Umpolung"
COOR´
Zn
δ+
BrZn
CH3
δ−
CH
COOR
R CH O
H2 O
- ZnBrOH
H CH3
R C CH
COOR
OH
Eliminação β - H2O
CH3
R CH
5.5
C
COOR
Aldeídos e Cetonas
Os focos principais deste livro são a reatividade química e o mecanismo da reação,
enquanto a organização por substâncias e reagentes fica em segundo plano. Sendo assim, os
diferentes métodos para produzir aldeídos e cetonas são dados a seguir, de forma resumida,
indicando apenas o capítulo onde serão discutidos.
5.5.1
Síntese de aldeídos
a) Síntese por oxidação (ver p. 638)
•
Oxidação de alcoóis primários, usando meios oxidantes seletivos. Reagentes: PCC
(piridina + HCl + CrO3, ver p. 647) ou reagente de Collins (CrO3 · 2 piridina, ver p.
647).
•
Quebra oxidativa de glicóis. Reagente: Pb(OAc)4 em meio aquoso (ver p. 651).
•
Ozonólise de olefinas, seguida por degradação redutiva. Reagente: O3 e Zn/AcOH
(ver p. 248)
•
Oxidação de cadeias laterais em compostos aromáticos. Reagente: SeO2 (ver p. 639)
259
J. D. Parrish, D. R. Shelton, R. D. Little, Titanocene(III)-Promoted Reformatsky Additions Org. Lett. 5
(2003) 3615-3617.
397
A. Isenmann
•
Princípios da Síntese Orgânica
Hidrólise de di-haletos geminais, R-CHX2 (ver p. 638).
b) Síntese por reação com organometálicos:
•
com reagente de Grignard. Substrato: ortoéster do ácido fórmico (ver p. 436)
c) Síntese por redução
•
Redução de Rosenmund. Substrato: cloreto carboxílico; reagente: H2 e catalisador
de Pd, desativado com BaSO4 (ver p. 614)
•
Redução de Stephen. Substrato: nitrila; reagente: SnCl2 em ambiente ácido (ver p.
615)
•
Redução com LiAlH(OC(CH3)3)3. Substrato: cloreto carboxílico (ver p. 614)
d) Síntese por substituição eletrofílica aromática (substrato = aromático rico em elétrons)
•
Reação de Gattermann. Reagentes: cianeto metálico e um ácido de Lewis (ver p.
304)
•
Reação de Gattermann-Koch. Reagentes: ácido clorossulfônico e ácido fórmico
(ver p. 304)
•
Reação de Vilsmeyer. Reagentes: DMF e cloreto de fosforila (ver p. 304)
•
Síntese de Reimer-Tiemann. Reagente: KOH em clorofórmio (ver p. 312)
5.5.2
Síntese de cetonas
a) Síntese por oxidação
•
Oxidação de alcoóis secundários. Reagente: CrO3 ou KMnO4 (ver também sínteses
indicadas no item 9.7.2)
•
Ozonólise com degradação redutiva. Reagente: Zn em AcOH (ver p. 248)
•
Oxidação de Oppenauer. Substrato: álcool secundário; reagentes: alcóxido de
alumínio e uma cetona de sacrifício em excesso (ver p. 604)
b) Síntese via compostos organometálicos (ver p. 377)
•
Reação de Grignard com nitrilas (ver p. 615)
•
Tratamento do cloreto de acila com compostos organo-cádmio (ver p. 615)
c) Síntese por substituição eletrofílica aromática:
•
Acilação de Friedel-Crafts. Reagentes: cloreto de acila ou anidrido carboxílico, e o
ácido de Lewis AlCl3 (ver p. 297)
•
Síntese de Hoesch. Reagentes: cianeto orgânico e um ácido de Lewis (ver p. 304)
398
A. Isenmann
Princípios da Síntese Orgânica
d) Síntese por descarboxilação:
• Substrato: β-cetoéster; hidrólise e decomposição térmica (ver p. 462).
e) Síntese por adição radicalar em olefinas:
• Reagente: aldeído (ver p. 79).
5.5.3
Estrutura eletrônica e reatividade dos compostos com grupo carbonila
O carbono do grupo carbonila tem hibridação sp², da qual resulta uma geometria plana e
ângulos de aproximadamente 120° entre ele e dois dos seus vizinhos.
Devido à polarização do grupo carbonila, δ+ no carbono e δ- no oxigênio, se abrem
principalmente três caminhos para reagir:
1. Eletrófilos podem atacar o oxigênio do grupo carbonila
2. Nucleófilos podem atacar diretamente o carbono do grupo carbonila
3. O efeito retirador de elétrons do grupo carbonila, aumenta a acidez e facilita a
substituição de hidrogênios em posição α.
A primeira possibilidade é rápida e reversível. Portanto não tem importância preparativa,
mas representa uma forma importante de ativação (isto é, protonação, catálise ácida) para
um subsequente ataque nucleofílico no carbono do grupo C=O+-H.
Aqui já se vê: as reações dos aldeídos e cetonas podem ser catalisadas por ácidos e também
por bases. A catálise ácida ocorre no oxigênio e aumenta indiretamente a carga parcial
positiva no carbono, o que facilita o ataque posterior do nucleófilo. Em ambiente básico,
por outro lado, tem-se nucleófilos bastante fortes. Eles conseguem atacar diretamente o
carbono do grupo C=O, enquanto a carga negativa se localiza no oxigênio.
É mostrada abaixo a sequência de reatividade de diferentes compostos com grupo carbonila
frente nucleófilos. Ela se explica pelos efeitos, indutivo (I) e mesomérico (M), do grupo
vizinho:
O
R C
>
Cl
>
R C
O C
R
>
R C
H
>
R C
CH3
O
O
O
O
O
R C
R C
O
R
N
R
R
O
Efeitos eletrônicos:
-I > +M
-I > +M
Referencial
+ I; -M
+M > -I
+M >> -I
Um catalisador ácido consegue protonar o oxigênio da carbonila e provoca o deslocamento
do respectivo composto para a esquerda desta sequência.
A catálise por bases fortes, por outro lado, não influencia na reatividade do grupo
carbonílico. Ela leva apenas à desprotonação da posição α do grupo carbonila, então
catalisa a formação do enolato. Este caminho, que corresponde ao ponto 3 da lista acima, é
de suma importância para todas as reações de condensação, apresentadas no capítulo 6 (ver
p. 454).
399
A. Isenmann
5.5.4
Princípios da Síntese Orgânica
A tautomeria ceto-enólica
"Tautomeria" é a expressão para o deslocamento reversível de um próton e uma ligação π
ao mesmo tempo. Ela não é uma mesomeria, mas sim, um verdadeiro equilíbrio entre dois
compostos isoméricos. Tautomerias são bastante comuns na química orgânica. Além da
tautomeria entre ceto e enol se observa este tipo de isomerização em:
•
azometina (= imina = base de Schiff)
•
amida (ou lactama)
•
endiol
•
nitro
•
nitroso
•
ácido hidroxâmico
enamina (ver p. 411)
ácido imido carboxílico (ver p. 100)
α-hidroxicetona (açúcares; grupo "reduton" da vitamina C; p. 81)
aci (ver p. 665)
oxima (aldoxima ou cetoxima) (ver p. 100 e 426)
carbiminodiol (p. 349)
Formas da catálise da tautomeria ceto-enólica
O seguinte esquema deixa claro que tanto ácidos quanto bases conseguem catalisar a
tautomeria. Um resumo da acidez de diversas classes de compostos orânicos, entre outros,
da cetona e de sua espécie protonada, encontra-se no anexo 2 deste livro (p. 898).
O
O
R CH C
R CH C
δ−
H
δ+
O
OH
- H+
Ânion enolato
+ H+
R CH C
forma ceto
+ H+
- H+
R CH C
+ H+
- H+
OH
OH
- H+
R CH2
C
+ H+
forma enol
R CH2 C
Cátion carbênio (oxônio)
Não só as formas ceto e enol têm importância na reatividade desta classe, mas também os
intermediários. No caminho da catálise básica, por exemplo, observamos um carbânion em
posição α ao grupo C=O que é estabilizado por mesomeria com o enolato. Essa é a forma
que reage mais facilmente com eletrófilos, dentre os quais o carbono positivado de outro
grupo C=O é o representante mais famoso (conteúdo principal do capítulo 6). Além de ser
intermediário reativo, o íon enolato pode também ser ligante em complexos de metais nos
quais funciona como quelato bidentado.
De qualquer maneira, seja catálise ácida ou básica, a tautomeria passa por uma forma
intermediária com carga. Portanto pode-se afirmar que a polaridade do ambiente (solvente,
teor de água, presença de sais) acelera esta reação.
400
A. Isenmann
Princípios da Síntese Orgânica
Não havendo particularidades estruturais no substrato, a forma ceto é dominante neste
equilíbrio 260. Por exemplo, apenas 0,00025% da acetona está presente na forma enólica, à
temperatura ambiente e na ausência de metais a serem complexados. Apesar desta posição
desfavorável no equilíbrio, a forma enólica já foi comprovada por vários métodos, em todos
os compostos que têm um grupo carbonila e pelo menos um hidrogênio em posição α:
•
A espectroscopia r.m.n. revela uma posição especial dos sinais 1H (δ de até 17
ppm).
•
Deslocamento das bandas de absorção no espectro infravermelho.
•
O composto carbonílico troca H por D na posição α, ao expor a D2O ou C2H5OD.
•
Pode-se adicionar bromo na dupla ligação da forma enólica.
A velocidade com que ocorre esta troca de hidrogênio, entre oxigênio e carbono α é
surpreendentemente baixa. Assim, em muitos compostos com grupos carbonilas é possível
determinar o teor em forma enólica, por titulação com Br2. A reprodução do enol pelo
equilíbrio dinâmico dentro do tempo experimental é desprezível (ver as constantes de
velocidade, listadas na Tabela 26, na p. 505), desde que a titulação for feita no banho de
gelo - recomendado abaixo zero graus.
forma enólica:
O H
CH C
- Br-
OH
O H
CH C δ+
CH C
Br
Br
Br
O
- HBr
CH C
Br
Br Br
forma ceto:
O
CH2 C
+ Br2
não há reação
Ao contrário da adição de bromo em alquenos, essa reação não pára na etapa do aduto
dibromo, mas elimina espontaneamente HBr (= eliminação β). Desta forma permanece
apenas um átomo de bromo (aquele em posição α) no produto bromado do enol. Como essa
bromação inclui uma etapa de desprendimento de uma molécula de ácido bromídrico, a
quantidade em enol pode ser facilmente determinada por titulação ácido-base.
Todavia, uma titulação desta deve ser executada no mínimo tempo possível, para não se
correr o perigo de uma sobreestimação do enol, devido a uma enolização repetida do
produto monobromado, -CHBr-C=O
-CBr=C-OH, que iria adicionar uma sobretaxa
de bromo.
260
Isso não vale, explicitamente, para os fenóis nem para os compostos 1,3-di-carbonílicos, como será
mostrada no cap. 6.5.1
401
A. Isenmann
Princípios da Síntese Orgânica
Observação: no ácido carboxílico também pode-se discutir uma tautomeria. Porém, a
adição de bromo não funciona tão facilmente porque a densidade eletrônica neste “enol” é
muito baixa. Portanto, a α-bromação precisa da presença de um catalisador (ver reação
HVZ, p. 394).
5.5.5
Reações de substituição em aldeídos e cetonas
As reações de substituição do oxigênio da ligação C=O por um outro carbono C´, formando
C=C´, ocorrem via complexos com heteroátomos do terceiro período (S, Si, P, também Ti).
Portanto, essas reações, cujo protótipo e a famosa reação de Wittig, será discutida
separadamente no capítulo 10 (vista geral na p. 832; ver também na p. 474).
Uma outra substituição, C=O → C=N-, é realizada na formação da hidrazona (ver p. 410 e
nota de rodapé na p. 375). Esse derivado tem importância na química analítica, por ser de
cristalização fácil.
O produto da substituição por amônia leva a compostos instáveis que podem ter significado
como intermediários ("imina", ver p. 408; também na condensação de Mannich, pp. 303,
415 e 491). O significado das bases de Schiff, cuja estrutura geral é R2C=N-R´, é discutido
na p. 413.
Bem mais importantes do que as substituições são as adições no grupo carbonila, conforme
apresentadas a seguir.
5.5.6
Reações de adição no grupo C=O
A adição em aldeídos e cetonas leva a um composto com carbono sp³, que é mais estável do
que foi visto nos ácidos carboxílicos; lá, com exceção dos ortoésteres, os produtos de
adição no grupo carboxila sofrem decomposição imediata (ver p. 353) e a instabilidade do
carbono sp³ foi explicada com base nos dois grupos fornecedores de elétrons (fortes efeitos
+M) que aumentam a densidade eletrônica no carbono a um grau que facilita o abandono de
um deles. Além disso, não podemos esquecer uma estabilidade extra que se dá, no caso do
carboxilato, por mesomeria. Portanto, como apresentado alí, a sequência reacional foi,
inevitavelmente, adição → eliminação.
Importante é que o nucleófilo ataca o carbono positivado do grupo carbonila, num ângulo
específico, conhecido como ângulo de Bürgi-Dunitz. Ele é, no caso ideal, de 107°, mas
pode ser variado dentro dos limites de 95 a 105°, quando for preciso para atribuir
reatividade do nucleófilo atacante, a grupos carbonilas em sistemas mais complexos. É
claro que o ângulo exato depende também da natureza e do tamanho do nucleófilo, mas em
princípio é uma propriedade intrínseca do grupo carbonila que se deve à posição dos lobos
do orbital molecular π* (= LUMO).
Compare também o dito na abordagem da indução assimétrica, p. 229 261.
261
Este ângulo de ataque é um dos fundamentos da teoria de Felkin e Nguyen Trong Anh, que refinaram as
regras de Cram da "indução assimétrica". Ver "Indução assimétrica", na p. 229.
402
A. Isenmann
Princípios da Síntese Orgânica
Nu107°
R2
C
O
R1
Reação com H2O:
Há formação do hidrato, um diol geminal. A formação é reversível e rápida. A maioria dos
compostos carbonílicos forma, mesmo com excesso de água, apenas uma pequena
porcentagem do seu hidrato. Somente na presença de grupos fortemente retiradores de
elétrons no esqueleto do composto carbonílico, o hidrato pode-se tornar a forma mais
dominante.
Um bom exemplo é o cloral (produzido em escala industrial, por introdução de cloro em
álcool etílico; é usado como tranquilizante):
Cl
Cl
O
C C
H
Cl
Cl OH
+ H2O
Cl
K = 2,7.104,
a 20 °C
Cloral
C
C OH
Cl H
Cloralidrato
Reação com ROH:
Na reação 1 : 1 formam-se hemiacetais, catalisada tanto por ácidos quanto por bases. Em
geral os hemiacetais são bastante sensíveis à hidrólise – independente do pH e/ou outros
sais presentes na solução.
O aldeído e (mais difícilmente) a cetona também podem reagir com duas moléculas de
álcool, formando o acetal ou cetal, respectivamente. Na adição da segunda unidade
alcoólica, apenas ácidos têm atividade catalítica. Mais corretamente, trata-se de uma
catálise específica do H+: o grupo hidroxila é protonado e pode ser abandonado em forma
de água. Esta etapa é mais lenta do que a subsequente adição do segundo álcool no
carbocátion. Portanto, a síntese mostra as dependências típicas de SN1.
O
+ CH3OH
CH2 C
CH2 C
Catalisador:
H+ ou OH-
aldeído / cetona
OH
+ CH3OH
OCH3
hemiacetal / hemicetal
Catalisador:
somente H+
OCH3
CH2 C
OCH3
acetal / cetal
Observação: para evitar a formação de α-haloéteres, o ácido HX usado na síntese do acetal
deve ter um ânion X- não nucleofílico. Isso é o caso, por exemplo, em HSO4-, H2PO4-, TsO-
403
A. Isenmann
Princípios da Síntese Orgânica
, mas não em Cl- ou Br- cuja nucleofilia já é considerável. Um outro benefício destes
ácidos, H2SO4, H3PO4 e TsOH, é o seu papel como retirador de água: todos eles são
higroscópicos. Ao retirar a água do sistema eles ajudam deslocar o equilíbrio da
acetalização para o lado direito.
Em casos mais difíceis, onde a cetona de partida tem grupos volumosos ou fornecedores de
elétrons, a etapa da formação do cetal a partir de álcool e H+ pode ser insatisfatória ou
mesmo impedida. Nestes casos se aplica com mais sucesso o ortoéster do ácido fórmico
(com catalisador NH4Cl), a partir do qual a transalcoxilação é possível (compare p. 380).
A formação de acetais representa o mais importante método de proteger o grupo carbonila
(ver também o grupo protetor THP, p. 499). Para este fim o glicol, HO-CH2-CH2-OH, é um
álcool bastante apropriado. Com esse diol forma-se um cetal/acetal cíclico,
com rendimentos melhores e velocidades maiores do que com dois alcoóis
R O
simples. Isto se deve à entropia ∆S e à entropia de ativação ∆S≠,
C
respectivamente. Lembre-se que no caso de alcoóis simples se unem três
O
moléculas; no caso do glicol somente duas, para formar o acetal.
Pela nossa surpresa, a polarização no carbono do acetal permanece como já era no
composto carbonilado de partida: no acetal também temos Cδ+. Prova disso é a reatividade
dos acetais frente alilsilanos e vinilsilanos; ver p. 824.
Enquanto os acetais/cetais são bastante resistentes frente reagentes básicos, a reconstituição
do grupo carbonila é muito fácil: a hidrólise em ácidos minerais diluídos é branda e
quantitativa.
Comparamos mais uma vez os aldeídos/cetonas com os ácidos carboxílicos. A estrutura
mais comparável aos acetais é o ortoéster. Interessante é a grande semelhança destes
compostos, em termos de estabilidade e função estratégica na síntese (embora o acesso ao
ortoéster é bastante diferente; ver p. 379).
Aproveitamos mais um pouco do assunto "proteção via acetal". Quando observar a reação,
desta vez do ponto de vista do álcool, podemos afirmar que um diol com grupos -OH
vicinais é especialmente adequado para a formação do acetal. Basta acrescentar um aldeído
ou uma cetona, para que se feche o ciclo de 5 membros (estável!) do acetal, conforme a
fórmula acima. Na prática usa-se a cetona mais barata e acessível - a acetona. Os substratos
a serem protegidos são abundantes: lembre-se que todos os açúcares e carboidratos naturais
contêm estes grupos de -OH vicinais. Sendo assim, a seguinte estratégia de proteção é
muito bem vinda e bastante usada pelo químico de substâncias naturais (ver também a
síntese da vitamina C, p. 228):
O
OH +
OH
H3C
[H+]
CH3
O
O
C CH3
CH3
404
A. Isenmann
Princípios da Síntese Orgânica
Os grupos -OH são perfeitamente protegidos contra qualquer atacante eletrofílico, desde
que o ambiente seja de pH > 7. Quando mudar para ambiente ácido, a acetona protetora é
clivada com facilidade (HCl diluído, 50 °C, 20 h).
A formação e quebra de acetais é um processo fundamental na natureza. As plantas
armazenam grande parte da energia, proveniente da fotosíntese, em forma dos carboidratos
amilose, amilopectina (juntos formam o "amido") e da celulose - materiais poliméricos
cujos monômeros de glicose são interligados por ligações acetais (aqui chamadas de
"ligações glicosídicas"). A quebra destas ligações glicosídicas ocorre nos estômagos dos
animais (ambiente ácido!) 262 e também nos digestores da indústria de madeira, onde
hidrólise parcial leva à celulose crua. Tudo isso é nada outro do que a hidrólise das ligações
acetálicas, reformando alcoóis, aldeído e cetona.
Além de hemiacetal e acetal existe uma terceira forma de ligação entre álcool e
aldeído/cetona, que tem igualmente a função de proteger: é a combinação com a forma
enólica, formando um enoléter (mais conhecido como "viniléter"), conforme descrito na
reação abaixo. Os enoléteres podem ser isolados e purificados. Eles são estáveis porque a
tautomeria para a forma ceto/aldo está impedida. A fixação na forma enólica provoca então
um reforço de todos os efeitos que esta forma confere às posições α e β do composto
carbonílico. Especialmente o ataque por eletrófilos em posição α é facilitado (este efeito é
ainda mais pronunciado em enaminas, ver p. 413).
Os viniléteres em outros contextos:
Polimerização vinílica e formação de poliviniléteres e polivinilalcoóis (p. 77);
O Tetrahidropirano (THP) é um enoléter cíclico; ele tem importância como grupo
protetor para alcoóis (p. 499).
Método padrão de preparo de enoléteres:
OCH3
R CH2 CH
OCH3
Piridina, H3PO4
- CH3OH
α
β
R CH CH O CH3
Enoléter
(= viniléter)
A abstração de metanol a partir do acetal representa uma eliminação β. Para facilitar a
desprotonação decorrendo em posição α se aplica uma base, favoravelmente piridina.
Sobre a utilidade dos enoléteres como ativadores da posição α em cetonas, estabilizadores
de cianidrinas (p. 406) e o seu papel de protetor do grupo -OH, após a reação com mais
uma unidade de álcool, ver p. 499.
262
Embora de ter estereoquímica diferente, as ligações glicosídicas são idênticas, na celulose e no amido. O
fato de o estômago humano consegue digerir o amido, mas não a celulose, tem sua explicação na
incompatibilidade das nossas enzimas com a ligação "β-glicosídica", mas também na alta cristalinidade e
estabilidade da estrutura terciária da celulose.
405
A. Isenmann
Princípios da Síntese Orgânica
Reação com R-SH:
Mercaptanos reagem, melhor ainda do que os alcoóis, com aldeídos e cetonas formando
tioacetais bastante estáveis. Eles até são resistentes frente ácidos minerais diluídos (sob
estas condições os cetais hidrolisam-se com a maior facilidade). Portanto, os tioacetais são
excelentes grupos protetores para a função carbonila e atendem a condições reacionais mais
variadas do que os acetais. Quando utilizados com esta finalidade, o enxofre pode ser
retirado por Hg2+, sob condições especialmente suaves, reconstituindo o aldeído / a cetona,
com rendimento excelente.
Outras aplicações dos tiocetais/tioacetais:
• Ao tratar o tiocetal/tioacetal com níquel de Raney e H2 ocorre a redução total para o
hidrocarboneto (ver p. 611).
• A polarização positiva do carbono aldeídico pode ser convertida em um carbânion!
Daí se abrem inúmeros caminhos para aumentar o esqueleto carbônico, já que esse
carbânion reage com os mais diversos síntons de carbono positivado. Para conseguir
esta "Umpolung", o tioacetal deve ser tratado com bases muito fortes (ver estratégia
de Corey e Seebach, p. 566 em diante).
Contudo, os tioacetais são compostos intermediários muito versáteis.
Reação com NaHSO3:
A reação de compostos carbonílicos com hidrogenossulfito (= bissulfito) de sódio é
reversível. Representa um bom método para purificar aldeídos e cetonas líquidos, que como
tais são de purificação difícil, enquanto os adutos do bissulfito são sólidos que podem ser
facilmente recristalizados.
Restrição: somente aldeídos e metilcetonas formam adutos cristalinos do hidrogenossulfito,
que são suficientemente estáveis para este procedimento.
O
R C
+ NaHSO3
Aldeído ou
metilcetona
(líquido)
O
R C
OH
SO3H
R C
SO3 Na+
α-hidroxissulfonato
(cristalino)
Reação com HCN:
A formação de cianidrinas (ver também p. 499) ocorre mais facilmente em solução aquosa
de pH ligeiramente básico. Ao operar com pH muito alto (> 10), o rendimento cai
drasticamente. Ainda poir seria um pH muito baixo (< 3), devido ao desprendimento de gás
cianídrico (perigo!). A reação é reversível, enquanto a sua velocidade aumenta com a
concentração de OH-:
406
A. Isenmann
O
Princípios da Síntese Orgânica
C N
R C
O
R C C N
H
+ H2O
- OH-
OH
R C C N
H
H
Aldeído
Cianidrina
A adição de cianeto em benzaldeído, formando a nitrila do ácido proveniente da amêndoa,
pode ser catalisada pela enzima emulsina, que se encontra no extrato de amêndoas amargas.
Forma-se o enanciômero L em 100% ee (isto é um exemplo para catálise assimétrica,
compare p. 232):
H
C
O
CN- , H+
Emulsina
Benzaldeído
CN
HO C
CN
H
+
H C OH
C6H5
C6H5
L-(+)-nitrila do ácido de amêndoa
Uma outra aplicação interessante das cianidrinas é descrita a seguir, tendo em vista a
expansão do anel em cetonas cíclicas (a própria cetona cíclica pode ser obtida por um dos
métodos descritos no item 6.3.1; a segunda etapa é uma redução descrita na p. 584, a
terceira o rearranjo de Wolff descrito na p. 387):
OH
O
KCN
CN
OH
H2 / PtO2
NH2
pH = 4
NaNO2
AcOH / H2O
O
Reação com alquenos:
A adição de um aldeído ou uma cetona a uma dupla-ligação C=C sob catálise ácida é
conhecida com reação de Prins 263. A espécie eletrofílica é, evidentemente, o composto
carbonílico protonado. O alqueno não precisa ser especialmente rico em elétrons ou
polarizado. A única restrição para um bom funcionamento é que o alqueno não seja muito
ramificado - por motivo estéricos e devido ao perigo de se polimerizar o alqueno por
iniciação catiônica (ver p. 167). Note-se que, ao empregar um excesso de aldeído, podem
ser adicionadas duas unidades do aldeído:
263
D.R. Adams, S.P. Bhatnagar, The Prins-Reaction, Synthesis 1977, 661.
407
A. Isenmann
Princípios da Síntese Orgânica
H
H
C C
C
+
HO
HO
C
C
C
R
R
R
R
R
Aldeído/cetona
protonado
-
C
H+
- H+
R
H
C
R
R
C
R
(adição de segunda
unidade de aldeído/cetona)
C O
+
O
OH
R
Álcool alílico
R
O
R
1,3-dioxano
A reação de Prins naturalmente não é uma síntese muito limpa, mas tem importância
industrial, para se obter o isopreno, o monômero da borracha natural: a adição de duas
unidade de formaldeído ao isobutileno fornece o 4,4-dimetil-1,3-dioxano. Sua
decomposição térmica no catalisador de fosfato de cálcio rende o isopreno em 77%.
CH3
C CH2 + 2 H2C O
H2O, H+
CH3
Isobuteno
O
[Ca3(PO4)2]
O
- H2O; - H2C=O
CH3
CH2
C CH CH2
Isopreno
Atenção:
Um outro produto se obtém a partir destes mesmos reagentes, quando aplicados sob
condições radicalares (reação de Paterno-Büchi, ver p. 79).
5.5.7
Adição de nitrogênio básico
A adição de nitrogênio ao grupo C=O é a reação precursora da substituição total do
oxigênio pelo nitrogênio. A reatividade diferenciada destes compostos e aplicações destes
em sínteses industriais justificam então um tratamento separado desta reação de adição.
Além disso, os produtos da N-adição têm certa importância para a caracterização dos
aldeídos e cetonas. A maioria dos adutos mencionados abaixo é sólida, à temperatura
ambiente.
408
A. Isenmann
Princípios da Síntese Orgânica
Reação com NH3:
No caso do aduto simples forma-se aldeidamônia, -CH(OH)NH2, que é bastante instável.
Ao trabalhar sob exclusão de água, isto é, com gás NH3 seco em solução etérica, a
aldeidamônia pode desidratar para a aldimina, R-CH=NH. A aldimina, por sua vez, é
bastante reativa e se estabiliza por trimerização ou polimerização (massa molar
descontrolada).
No caso do acetaldeído se tem:
NH
3 H3C
H3C
C
H
H
N
HC
HN
C
H
Acetaldimina
CH
CH3
NH
CH3
ou
CH
NH
x
CH3
(Não confunda este produto cíclico com a melamina, por sua vez o produto de condensação
da uréia, ver p. 328)
No caso especial do formaldeído a reação prossegue até a hexametilenotetramina. Este
produto é conhecido como “urotropina”, e acha uso em reações onde uma liberação
controlada de formaldeído e/ou amônia é desejada 264.
N
O
6 H C
+ 4 NH3
H
N
N
+ 6 H2O
N
Urotropina
Reação com aminas primárias:
Aminas primárias, R-NH2, formam iminas que são apenas estáveis quando a parte
hidrocarbônica do composto carbonílico for aromática. Em geral, a estrutura R-CH=N-R´ é
denominada base de Schiff.
264
Lembra-se da importância da urotropina, na marcha sistemática da separação de cátions, onde um grupo
de metais (Ga, Fe, In, Al, Be, Cr, U, Zr, Ti e La) pode ser seletivamente precipitado em forma dos seus
hidróxidos, devido ao pH bem controlado (= liberação de NH3 aos poucos).
409
A. Isenmann
Princípios da Síntese Orgânica
Reação com aminas secundárias:
Aminas secundárias, R2NH, reagem com a forma enólica do composto carbonílico,
formando uma enamina.
NR2
R CH C
R
Enamina
Sobre a importância "catalítica" da enamina, ver pp. 413 e 496; sobre a possibilidade de
formar o cátion imônio, ver as pp. 415 e 491. Sobre a tautomeria enamina-imina, ver p.
412.
Reação com hidroxilamina:
Com hidroxilamina, H2N-OH, em ambiente neutro, formam-se oximas 265, R2C=N-OH, que
têm certa importância estratégica porque sofrem facilmente o rearranjo de Beckmann (ver
p. 429).
Reação com hidrazina:
Com hidrazina, H2N-NH2, ocorre a condensação para hidrazona; ao usar um excesso de
composto carbonílico, o grupo NH2 restante na hidrazona condensa com outro grupo
carbonila, formando finalmente uma azina.
C O
C O
N2H4
C N NH2
C N N C
Hidrazina
Hidrazona
Azina
As hidrazonas têm aplicação na química analítica, onde tornam compostos carbonílicos em
derivados menos voláteis. Estes podem ser detectados e caracterizados com maior
facilidade, por exemplo via espectroscopia UV-VIS.
Em sínteses, as hidrazonas têm relevância na reação de Bamford-Stevens, levando a
compostos vinílicos (com finalidade de serem polimerizados; ver p. 873), como
intermediârios na redução de Wolff-Kishner (p. 610) e no rearranjo de Beckmann (pp. 102 e
429).
265
Uma rota alternativa para oximas alifáticas, é a reação entre nitritos, orgânicos ou inorgânicos, com
compostos de elevada acidez C-H. O ambiente geralmente é aquoso e o catalisador um ácido mineral. Essa
reação é referida em diferentes lugares de Organic Synthesis: Coll.Vol. 2 (1943) 202; Coll.Vol. 2 (1943) 204;
Coll.Vol. 2 (1943) 363; Coll.Vol. 3 (1955) 191; Coll.Vol. 3 (1955) 513.
410
A. Isenmann
Princípios da Síntese Orgânica
Reação com fenilidrazina, H2N NH Ar :
A fenilidrazina, especialmente a 2,4-dinitrofenilidrazina, é um reagente comum na química
analítica dos compostos carbonilados. Os produtos da condensação com aldeídos/cetonas,
as fenilidrazonas, são compostos de fácil cristalização. Assim, os aldeídos e cetonas podem
ser caracterizados pelo ponto de fusão, através dos seus derivados.
O
H2N NH C
NH2
A semicarbazida (fórmula acima) forma semicarbazonas, com a mesma finalidade que as
fenilidrazonas.
5.5.8
Mecanismos da N-adição em aldeídos e cetonas
Particularidades da adição de aminas primárias em aldeídos/cetonas
Todos os reagentes mencionados acima – fora as aminas secundárias - podem ser
generalizados pela fórmula H2N-Y, já que reagem da mesma maneira com o grupo
carbonila:
O
R C
+ H2N Y
[H+]
OH
R C NH Y
H
H
N Y
Elim. β
- H2O
R C
H
Derivado da
base de Schiff
O pH não deve ser muito baixo, porque desta forma o nitrogênio do reagente é protonado e
perde sua nucleofilia. Também não deve ser muito alto, onde a concorrência do nucleófilo
forte OH- se torna dominante. Para a maioria dessas reações existe portanto um valor ótimo
do pH que fica entre 5 e 9. A última etapa é bastante facilitada por agentes dessecantes.
Esta reação satisfaz todas as precondições para que ocorra em processos bioquímicos:
•
é viável em ambiente neutro,
•
tem uma exotermia moderada,
•
ocorre à temperatura ambiente.
Deve-se ressaltar que carbonilas aminadas, ao serem tratadas cuidadosamente com ácidos,
formam o íon imônio que apresenta uma série de reações subsequentes interessantes (ver
como exemplo a síntese de Strecker, na p. 412). O nome “base de Schiff” já implica esta
propriedade.
411
A. Isenmann
Princípios da Síntese Orgânica
Para a base de Schiff (= imina) existe uma forma tautomérica, a enamina. O mecanismo é
idêntico ao da tautomeria ceto-enólica (ver p. 400):
Tautomeria
C C N
C C N
R
R
H
H
Imina
(= base de Schiff)
Enamina
O equilíbrio é atingido mais rapidamente do que o ceto-enólico; em casos gerais, a imina
representa a estrutura dominante. As duas estruturas se destacam por terem reatividades
diferenciadas, frente reagentes organometálicos que geralmente são bases e nucleófilos ao
mesmo tempo. O carbono do grupo C=N da imina está sendo atacado por nucleófilos, tais
como reagentes de Grignard ou organo-lítio. Por outro lado, na forma da enamina
observamos reatividade do carbono em posição α. Os mesmos reagentes organometálicos
agem agora como bases, em vez de nucleófilos.
Comparando a imina/enamina com com a cetona/enol, podemos afirmar que o carbono α
em enaminas é aproximadamente 10 ordens de magnitude mais ácido. Sendo assim, pode
ser transformado com muita facilidade em uma espécie desprotonada e nucleofílica,
fazendo reações de condensação, entre outras 266. A desprotonação quantitativa por LDA
leva ao aza-enolato que pode ser alquilado por haletos de alquila numa segunda etapa, uma
reação elaborada por Corey e Enders 267 (compare também a estratégia de condensação
mista, por desprotonação quantitativa, p. 502).
Síntese de D,L-aminoácidos, segundo Strecker
É uma estratégia antiga de se aumentar a cadeia carbônica por uma unidade, via adição do
nucleófilo cianeto. A síntese de Strecker aproveita da situação deficitária em elétrons no
cátion imônio que assim consegue segurar firmemente o nucleófilo, CN-. A primeira etapa
então é irreversível. Sob estas condições se consegue a hidrólise posterior do grupo nitrila,
feita em solução ácida, com bons rendimentos. Os reagentes típicos da síntese de Strecker
são (NH4)2CO3/HCN ou NH4Cl/KCN na primeira, e HCl concentrado na segunda etapa.
H
H
R CH NH2 + CN-
R C NH2
CN
Cátion im ônio
α- aminonitrila
[ H2O ; H+ ]
H
R C NH2
R C NH3
COOH
COO
α - aminoácido
(racemato)
266
Ver estratégia de estabilização da forma enólica, via enamina, pp. 496 e 497. A não ser confundida com a
condensação de Mannich, p. 491, que ocorre em ambiente neutro a ácido.
267
E.J.Corey, D.Enders, Chemische Berichte 111 (1978) 1337.
412
A. Isenmann
Princípios da Síntese Orgânica
Comparando-se os adutos do cianeto, uma vez no cátion imônio, outra vez em aldeídos e
cetonas, pode-se afirmar que a α-aminonitrila é bem mais estável do que a cianidrina, RCH(OH)CN (p. 406). A partir de cetonas, porém, é mais difícil produzir aminoácidos
usando esta estratégia.
O produto desta síntese, aliás, é muitas vezes ilustrado com os grupos –NH2 e –COOH. Isto
não reflete a situação real, pois os aminoácidos existem em uma proporção inferior a 1%
nesta forma. Mais realística é a formulação com os grupos carregados, –NH3+ e –COO-. No
caso especial dos aminoácidos se estabeleceu a expressão “betaína” para esta forma (o
nome foi adotado da substância natural, (CH3)3N+-CH2-COO-, encontrado no suco da
beterraba, beta vulgaris, portanto, “betaína”; compare nota de rodapé 436 na p. 779).
Outras sínteses que ocorrem via compostos contendo a unidade –C=N•
•
•
As reduções (nestes casos: transferências de hidreto) de Leuckart-Wallach e de
Wolff-Kishner, ambas incluem adutos de N-bases em compostos carbonílicos. Elas
serão discutidas na p. 605.
A metilação de aminas primárias (e secundárias) segundo Eschweiler-Clarke (p.
606);
O cátion imônio pode funcionar como eletrófilo frente aromáticos ativados
(aminometilação de Mannich, pp. 303, 415 e 491).
Particularidades da reação de aldeídos/cetonas com aminas secundárias
Aminas primárias, como vimos acima, podem substituir completamente o oxigênio do
grupo C=O, formando uma dupla ligação, C=N (iminas). O mecanismo é uma adição do
nucleófilo, seguida pela abstração de água numa eliminação β.
Já na adição de aminas secundárias o nitrogênio da amina somente estabelece uma ligação
simples, por falta do segundo hidrogênio no N. Mesmo assim, é possível abstrair água ou,
como ilustrado no esquema abaixo, uma amina. Esta vez a eliminação β acontece entre os
carbonos e fornece, de maneira inequívoca, a enamina.
A precondição para esta reação é que o composto carbonílico seja “enolizável”, isto é,
disponha de pelo menos um hidrogênio no carbono em posição α. As fórmulas
mesoméricas apresentadas abaixo deixam claro por que a forma da enamina está
dominando a forma da imina: aqui a "imina" somente pode ser representada usando cargas o que é termodinamicamente desfavorável. Uma tautomeria entre as duas formas, como foi
visto na p. 411, aqui não é possível, porque a esfera coordenativa do nitrogênio já está
completa.
413
A. Isenmann
Princípios da Síntese Orgânica
CH3
O
R CH2 C
+ 2 HN
H
refluxo
- HN(CH3)2
CH3
H
Benzeno
- H2O
R CH CH N(CH3)2
N(CH3)2
R CH C
H
Aminal
N(CH3)2
R CH CH N(CH3)2
Enamina
As enaminas estabilizam então a forma enólica e deixam a posição α (em relação ao grupo
carbonila; no esquema em negrito) rica em elétrons. A segunda estrutura mesomérica
acima mostra claramente a alta densidade eletrônica neste carbono. Portanto, essas
enaminas podem ser consideradas bons nucleófilos, enquanto suas qualidades como bases
são apenas regulares.
Neste fato se baseiam importantes estratégias para condensações, onde esse carbono
nucleofílico ataca um outro carbono δ+, tipicamente providenciado por um outro grupo
carbonila intacto:
•
Condensações mistas (ver p. 464), onde o carbono positivado, de outra molécula
com função carbonila, representa o eletrófilo (fraco).
•
Condensação de Mannich (p. 491).
•
Condensação com o carbono β positivado em sistemas Michael (p. 527).
As enaminas se decompõem ao serem expostas à umidade. Além disso, são muito sensíveis
frente ácidos. Duas consequências práticas resultam desta instabilidade:
1)
O caminho de preparo descrito acima requer um separador de água (inglês: dean
stark trap), comumente usado em policondensações e secagens azeotrópicas.
Geralmente aproveita-se da propriedade de certos solventes orgânicos (benzeno,
tolueno, xilenos, clorofórmio, CCl4) de formar a quente uma mistura azeotrópica com a
água, enquanto são imiscíveis a frio. O processo físico é também conhecido como
"destilação azeotrópica" ou "arraste azeotrópico".
2)
As enaminas trazem todos os pré-requisitos para serem bons grupos protetores para
o grupo carbonila. A amina cíclica piperidina mostrou-se especialmente adequada neste
sentido (ver exemplo na p. 527).
Na forma da sua enamina, o composto carbonílico pode ser submetido a uma grande
variedade de reações desde que sejam conduzidas em ambiente básico - que é o caso na
maioria das reações das condensação descritas no capítulo 6 (especialmente pela estratégia
descrita na p. 496), sem afetar o carbono do grupo carbonila. A hidrólise posterior da
enamina, restaurando a carbonila, é fácil e branda (tampão de acetato = pH 4-5, temperatura
ambiente).
414
A. Isenmann
Princípios da Síntese Orgânica
"Dean Stark Trap" para separar água da mistura reacional via destilação azeotrópica. Os
solventes de arraste devem ter densidade é inferior à da água: benzeno, tolueno e os
xilenos são muito usados.
Formação do cátion imônio a partir de aminas secundárias
Aldeídos alifáticos que não possuem hidrogênio em posição α, obviamente não têm a
habilidade de formar o enol. O exemplo padrão é o formaldeído, que é discutido aqui
porque tem grande importância preparativa, especialmente na condensação de Mannich
(ver p. 491 e 304). Em uma primeira etapa ele adiciona uma amina secundária, formando
um semiaminal. Este, por sua vez, pode ser facilmente desidratado. O produto desta
eliminação β de água é o cátion imônio (também usada a expressão "cátion imínio", ver p.
826):
O
CH2
HNR2
pH 6
Formaldeído
H NR2
HO
CH2
R
Elim. β
- H2O
N
R
CH2
Imônio
Na forma do cátion imônio, a polarização positiva do carbono do formaldeído aumentou e
com isso também sua reatividade como "componente carbonílico” (expressão definida na p.
482), o que pode ser verificado na condensação de Mannich (ver p. 491 e questão 7 na p.
539).
Atenção: o efeito contrário se obtém ao se converter o formaldeído com uma amina
primária em uma base de Schiff, H2C=N-R. Neste caso, o nitrogênio dispõe de um par de
elétrons livre que age com efeito +M sobre os seus vizinhos. Assim, o carbono torna-se
menos positivado do que no aldeído livre, sendo, portanto, desativado na sua função de
415
A. Isenmann
Princípios da Síntese Orgânica
componente carbonílico. Neste efeito se baseia a condensação aldólica direcionada,
estratégia estabelecida por Wittig (ver p. 495).
5.5.9
O papel de compostos carbonílicos em reações multi-componente
Um campo cada vez mais amplo ocupam as reações multi-componente 268. Todas têm em
comum de funcionar com três ou mais reagentes que podem ser misturados ("one-potreaction"). Mesmo assim, eles reagem de maneira inequívoca, ou seja, a sequência
reacional entre os componentes é bem definida. Sendo assim, podemos chamá-las de
sínteses convergentes feitas em um único balão.
Os compostos carbonílicos têm um papel importante em quase todas essas reações multicomponente, portanto uma discussão desta classe de reações se justifica nesta seção.
Certamente uma das mais antigas reações multi-componentes é a condensação de Mannich
(pp. 303, 415 e 491) que, devido às reatividades vantajosas de todos os participantes, pode
ser realizada até sob condições fisiológicas.
Outros exemplos para reações multicomponente são a síntese de aminoácidos segundo
Strecker (p. 412) e a reação de Kindler (ver também p. 660):
O
O
N
130 °C, 3 h
+ S8
+
HN
O
TsOH
S
Além das duas sínteses descritas a seguir, vale mencionar a reação de Biginelli 269,
Bucherer-Bergs 270, Gewald 271, e também a historicamente importante síntese de piridina
segundo Hantzsch 272. Todas elas representam estratégias importantes na síntese de
compostos heterocíclicos - alguns são heteroaromáticos.
268
A. Dömling, Org. Chem. Highlights 5 (2004) April; A. Dömling, I. Ugi, Angew. Chem. Intl. Ed. 39 (2000)
3168.
Monografia: Jieping Zhu, Hugues Bienaymé, Multicomponent Reactions, Wiley-VCH 2005
269
H. Hazarkhani, B. Karimi, N-Bromosuccinimide as an Almost Neutral Catalyst for Efficient Synthesis of
Dihydropyrimidinones Under Microwave Irradiation, Synthesis, 2004, 1239-1242
I. Cepanec, M. Litvić, A. Bartolinčić, M. Lovrić, Tetrahedron 61 (2005) 4275-4280.
S. V. Ryabukhin, A. S. Plaskon, E. N. Ostapchuk, D. M. Volochnyuk, A. A. Tolmachev, Synthesis, 2007, 417427.
270
A ciclização de Bucherer-Bergs é muito semelhante à síntese de Strecker - sob adição de CO2.
R. Sarges, S. W. Goldstein, W. M. Welch, J. Med. Chem. 33 (1990) 1859-1865
C. Montagne, M. Shipman, Synlett, 2006, 2203-2206.
271
A etapa central do mecanismo é uma condensação de Knoevenagel.
H. Zhang, G. Yang, J. Chen, Z. Chen, Synlett, 2004, 3055-3059.
272
Também esta síntese contém uma etapa de condensação do tipo Knoevenagel.
Mecanismo: D. Pettersen, M. Marcolini, L. Bernardi, F. Fini, R. P. Herrera, V. Sgarzani, A. Ricci, J. Org.
Chem. 71 (2006) 6269-6272.
Artigo de revisão: R. A. Cherkasov and V. I. Galkin, Russ. Chem. Rev. 67 (1998) 857-882.
G. V. M. Sharma, K. L. Reddy, P. S. Lakshmi, P. R. Krishna, Synthesis, 2006, 55-58.
416
A. Isenmann
Princípios da Síntese Orgânica
Uma variação da síntese de Wittig, adicionando uma amina primária, leva a um produto
organofosforado estável; a reação é conhecida como síntese de Kabachnik-Fields 273 (ver p.
786).
Reação de Ugi
A reação de Ugi é especial, sob vários pontos de vista 274. O mais evidente é o fato de que
se usam quatro reagentes diferentes, um aldeído (1), um ácido (4), uma amina (2) e uma
isonitrila (3). Cada um destes compostos tem seu papel definido na cascata reacional onde a
maioria das etapas podem ser denominadas de condensações. Assim, o protótipo da reação
de Ugi, conforme mostrado a seguir, é referido na literatura como “U-4CC” (sendo
abreviação para Ugi 4 Component Condensation). O produto final é uma αaminoacilamida.
Esquema geral:
R2
O
R1
1
N
R1
+
NH2
H
R1
2
R2
C
+
H
N
N
R3
3
R2
H
O
O
+
OH
R4
4
R4
R1
H
N
N
R2
R3
O
α-aminoacilamida
Mecanismo:
Pode-se esperar um mecanismo iônico nesta reação, pelo fato de que um solvente polar
promove a reação. Esta é considerada a diferença mais fundamental em relação à reação de
Passerini, que será aprestentada a seguir. Alcoóis de baixa massa molar se mostram bons
solventes (a não ser que a reação seja executada "em fase sólida", ver abaixo). Ácidos de
Lewis podem ajudar como catalisadores, caso a reação seja impedida pela presença de
grupos R volumosos em um dos componentes. Os reagentes são aplicados em altas
concentrações e sob refrigeração. O andamento da reação pode ser controlado com bastante
facilidade; em poucos minutos, à temperatura ambiente, a reação de Ugi prossegue até altos
rendimentos.
Em primeira instância forma-se, além da espécie eletrofílica de imônio, um carboxilato que
por sua vez representa um nucleófilo (fraco). É uma reação ácido-base e, portanto, um
equilíbrio dinâmico. Em seguida a isonitrila entra em ação, atacando o carbono do íon
imônio:
273
S. Bhagat, A. K. Chakraborti, J. Org. Chem. 72 (2007) 1263-1270.
L. Weber, K. Illgen, M. Almstetter, Synlett, 1999, 366-374.
Uma aplicação interessante de microondas neste tipo de reação: H. Tye, M. Whittaker, Org. Biomol. Chem. 2
(2004) 813-815.
274
417
A. Isenmann
N
R1
H
N
R2
H
O
+
R4
H
R2
OH
+
NC +
R3
R4
N
R1
OH
R4
R2
O
+
H
O
O
H
R1
Princípios da Síntese Orgânica
R1
O
H
N
N
R2
R4
R3
O
Para maior esclarecimento, esta última etapa será referida mais uma vez, indicando o
movimento dos elétrons. A isonitrila 275 entra em ação com seu carbono nucleofílico. Essa
reatividade pode ser vista mais claramente na segunda fórmula mesomérica indicada a
seguir. A aparência de um zwitteríon é promovida pelo fato de que cada átomo ter um
octeto de elétrons 276. Isoeletrônico ao monóxido de carbono, o agrupamento RNC é de
geometria linear.
R3
N C
R3
N C
Fórmulas mesoméricas da isonitrila
Assim, a etapa central se explica como ataque nucleofílico da isonitrila, ainda apoiado
pelos elétrons do carboxilato. (O carbono 1 da isonitrila, durante esta etapa, é formalmente
oxidado!) O produto desta é instável e sofre logo e irreversivelmente um rearranjo que pode
ser descrito como migração do grupo acila (oriundo da componente 4), para o nitrogênio do
componente 2:
275
Isonitrilas, também chamdas de isocianetos, são umas das substâncias mais fedidas no laboratório
orgânico. Este fato certamente diminui o grau de popularidade da reação de Ugi. Isonitrilas são resistentes
frente bases fortes, mas reagem com facilidade ao ser expostas a soluções ácidas, formando formamidas, ou
até polímeros de estrutura má estudada. Sendo assim, a hidrólise ácida é um dos melhores métodos de limpar
um laboratório deste contaminante de olfato.
276
O grupo isonitrila age também (e em analogia ao grupo nitrila), como retirador de elétrons sobre o grupo
R3. Porém, esta reatividade não se efetua na reação de Ugi, ou seja, a ligação N-C3 não se quebra.
418
A. Isenmann
H
N
R2
O
O
C
H
R1
Princípios da Síntese Orgânica
-
O
R4
R4
R3
H
R1
O
R3
N
R2 O
°
N R2
H
N
N
H
N
R1
R4
O
R3
O
R4
R1
H
N
N
R2
R3
O
A etapa do rearranjo pode ser formulada por um estado de transição cíclico de 5 membros,
no qual o nitrogênio nucleofílico consegue atacar o carbono do grupo carboxílico, num
ângulo ideal (Bürgi-Dunitz, ver p. 402):
R4
R1
107°
N
H
N
O
H
O
R2
R3
Na literatura ainda espalhada é um rearranjo ("Rearranjo de Mumm"), que vai através de
um estado de transição igualmente cíclico e de 5 membros. Este, porém, não envolve o
nitrogênio proveniente do intermediário imônio, mas apenas as partes provenientes da
isonitrila e do carboxilato:
O
O
C
O
R4
C
N
R3
C
O
R4
C N
R3
Em toda consequência, não deve-se dar preferência a este mecanismo, já que o produto
deste não representa a ordem dos grupos R no produto corretamente e, além disso, leva à
estrutura de uma imida - o que não é o caso.
Uma outra medida que eleva o rendimento desta reação acima do normal, se baseia numa
técnica nova que se aplica aqui com sucesso: é possível fazê-la em “fase sólida”, o que
significa que um dos reagentes é fixado num polímero polifuncional, a ser solto somente
419
A. Isenmann
Princípios da Síntese Orgânica
após todas as etapas mecanísticas 277. Isto facilita bastante manuseio, limpeza e estabilidade
do produto e abre caminho para automatizar a reação. Em analogia à síntese de Merryfield
de oligopeptídeos (p. 599), esta reação é predestinada para a "química combinatória". Essa
nova expressão implica a possibilidade de ter acesso a uma grande variedade de derivados,
a base do mesmo tipo de reação. Isto pode ser verificado num cálculo simples: ao variar os
grupos R1 a R4, tendo apenas 3 alternativas em cada componente, já resultam 34 = 81
derivados!
Finalmente, vale destacar que essa reação é bastante acelerada quando está sendo
conduzida no forno de microondas 278. Em poucos minutos se consegue um rendimento
bom, permitindo desta forma a síntese de um grande número de derivados, em pouco
tempo.
Assim é possível fazer uma varredura (inglês: high throughput screening) de novos
aminoácidos e oligopeptídeos não-naturais, na busca de bioatividade medicinal. Entretanto,
a reação de Ugi estereosseletiva ainda é objeto da pesquisa atual.
Uma variação desta reação providencia um caminho elegante, levando a um
heteroaromático bicíclico que tem certa relevância como antiinflamatório 279. A reação
pode ser chamada de “U-3CC”, onde falta o componente 2, a amina primária, que fez parte
na condensação de Ugi clássica. O nitrogênio aromático da 2-aminopiridina assume o papel
desta amina.
O
R1
R1
R1
H
NH2
H
N
H
H+
CN
R3
R3
N C
N
- H2O
N
H
N
N
Imônio
R1
R3
N
N
R1
H
N
R3
N
N
H
N
Imidazo (1,2-a) piridina
Os intermediários desta reação não são isoláveis, porque a ciclização ocorre com bastante
facilidade. Isso é uma característica de todas as ciclizações, uma vez que a entropia
molecular não diminui tanto como numa reação intermolecular (ver cap. 6.3, para
277
P.Seneci, Solid-Phase Synthesis an Combinatorial Technologies, Wiley-VCH 2000.
A.W.Czarnik (editor), Solid Phase in Organic Synthesis, Wiley-VCH 2001
278
C.S.Graebin, Quim. Nova 28 (2005) 73.
279
K.Groebke, Synlett (1998) 661.
420
A. Isenmann
Princípios da Síntese Orgânica
explicação detalhada). A última etapa da cascata é uma tautomeria que está deslocada para
o lado direito, porque neste o sistema aromático se estende sobre os dois anéis.
Reação de Passerini
Muitas semelhanças com a reação de Ugi discutida acima, mostra a condensação de
Passerini 280. As condições típicas desta reação, porém, sugerem um mecanismo
sincronizado, mais doque um mecanismo iônico. A reação de Passerini ocorre melhor em
solventes apolares, com os reagentes em altas concentrações e à temperatura ambiente. É
uma reação de três componentes: um ácido carboxílico, um aldeído ou cetona e uma
isonitrila; o produto é uma α-hidroxicarboxamida.
C
O
+
R1
+
N
R2
O R
1
O
R4
R3
R4
OH
R2 R3
N
O
H
O
Mecanismo proposto:
Um papel fundamental nesta reação tem a criação de ligações de hidrogênio, evitando
assim a formação de intermediários com cargas:
O
O
+
R1
R2
O
H
O
R4
R2
O
O
R1
OH
R2
R4
O
O
C
O
R4
H
O
R1
N
+
R1
H
R3
C O
R2
R4
N
R3
H O
O
R1
R2
O
N
O R
1
R3
R4
O
R2 R3
N
H
O
R3
A estereosseletividade da reação ainda não está bem entendida 281; a sua flexibilidade
estrutural é grande, com as exceções de compostos com grupos R volumosos e cetonas α,β 280
281
C.K.Z.Andrade, S.C.S.Takada, P.A.Z.Suarez, M.B.Alves, Synlett (2006) 1539-41.
P.R.Andreana, C.C.Liu, S.L.Schreiber, Org.Lett. 6 (2004) 4231-3.
421
A. Isenmann
Princípios da Síntese Orgânica
insaturadas. Como a reação de Ugi, a síntese de Passerini também é adequada para um
planejamento combinatório de sínteses, à busca de produtos bioativos.
5.5.10 Reações redox dos aldeídos e cetonas
Todas as reações de oxidação e redução em compostos carbonilados serão discutidas nas
pp. 638 e 603, respectivamente.
Excepcionalmente, é discutida aqui a reação de Cannizzaro (1853) porque se trata de uma
redução e oxidação no mesmo composto. Este tipo de reação redox se chama
desproporcionamento. A precondição para o funcionamento desta reação é a ausência de
hidrogênios em posição α ao grupo carbonila. Também podemos formular assim: os
aldeídos que funcionam neste sentido não devem ser enolizáveis. Caso contrário, a
condensação entre duas moléculas (que é assunto da seção 6.1.1) ocorrerá
preferencialmente. Esta restrição rigorosa se explica pela dificuldade da transferência do
hidreto, esboçado na segunda etapa da reação a seguir. A reação de Cannizzaro é bastante
lenta, com a maioria dos aldeídos.
C
O + OHH
O-
δ−
O
C H
OH
δ+
C
H
lento
COOH
+
-
O CH2
Benzaldeído
COO- + HO CH2
Ácido benzóico
Álcool benzílico
O mecanismo desta reação é bastante complexo. Experimentos em solvente deuterado
comprovaram a ausência completa do solvente, na etapa da transferência do hidreto. Nestes
termos pode-se entender o funcionamento da reação de Cannizzaro, até em ambiente
aquoso.
Um outro fator surpreendente foi a influência, não apenas da concentração de OH-, mas
também do cátion presente em solução. Ca2+, Ba2+, e especialmente Tl+ mostram um
melhor funcionamento do que Na+ ou K+ 282. Portanto se crê que a transferência do hidreto
acontece através de um complexo formado entre o metal (ácido de Lewis) e duas moléculas
de aldeído. Desta forma, os ligantes aldeídicos tornam-se mais pobres em elétrons, um fato
que facilita a transferência do H-:
282
Isso chama nossa atenção, ao usar uma solução de formalina velha. Ela pode conter, além de trimeros de
paraformaldeído e polioximetileno, elevados graus de ácido fórmico (o metanol é presente todavia, porque foi
adicionado intencionalmente pela fábrica, devido ao efeito estabilizador). Portanto, é sempre recomendado
usá-la recém destilada.
422
A. Isenmann
Princípios da Síntese Orgânica
R
R
δ+
C
H
δ− O
C δ+
H
O
δ−
M
OH
Complexo ativado
A reação de Cannizzaro é aplicada - além de formaldeído e isobutiraldeído - quase
exclusivamente a aldeídos aromáticos.
É chamada reação de “Cannizzaro cruzada” quando for aplicada a dois aldeídos diferentes
(ambos sem α-H!). Quando um deles é o formaldeído, quase a totalidade do mesmo é
oxidado para o formiato, enquanto o outro aldeído é reduzido (= método suave para se
sintetizar um álcool primário).
Reação de Tichtchenko
Também essa reação representa um desproporcionamento. O reagente que desempenha o
papel de reagente e catalisador é o etóxido de alumínio, Al(OC2H5)3. Como o etóxido é
uma base mais forte do que OH-, o sistema deve ser anidro – caso contrário o reagente se
desfaz imediatamente.
O-
O
H3C
+ -OC2H5
C
H 3C
H
C OC2H5
H
Acetaldeído
O
δ−
O
+
H3C Cδ+
H
Transferência de
Hidreto
H3C
C
O C2H5
Etilacetato
+
H3C CH2 ORestituição
do catalisador
A vantagem desta reação é a possibilidade de executá-la mesmo na presença de hidrogênios
α, o que é impedido na reação de Cannizzaro. Ela também funciona, portanto, com
aldeídos alifáticos. Por outro lado, a reação de Tichtchenko não funciona com compostos
carbonilados duplamente ativados (ver p. 503). Uma aplicação industrial é a síntese de
acetato de etila, um importante solvente em tintas, a partir de acetaldeído (acessível via
processo de Wacker, a partir de etileno e oxigênio) e 3 a 5% de Al(OEt)3.
423
A. Isenmann
Princípios da Síntese Orgânica
Das reações de Cannizzaro e Tichtchenko podemos concluir que a acidez dos hidrogênios
em posição α ao grupo carbonila não é muito alta. Realmente, os valores pKa da acetona e
do acetaldeído são em torno de 20, dos demais compostos com grupo carbonila >20, devido
ao efeito +I dos grupos alquilas/arilas (ver também a tabela de acidez de compostos
orgânicos, no anexo 2 deste livro). Com o etóxido temos um reagente cuja basicidade não é
suficientemente alta para desprotonar cetonas e aldeídos em altas porcentagens (pKa de
etanol = 18). Como o etanol é ligeiramente mais ácido do que os substratos carbonilados
desta reação, então o equilíbrio
O
O
H3C
-
+ OC2H5
C
H
H2C
C
+ HOC2H5
H
está deslocado, na ordem de 100 vezes, para o lado esquerdo.
5.5.11 Reações em posição α
A maioria destas reações será discutida no capítulo 6, onde o foco preparativo é a criação
de uma nova ligação C-C. Aqui sejam mostrados apenas as halogenações e o nitrosamento.
A acidez do α-H se explica com o efeito retirador de elétrons, -I, do grupo carbonila.
Reações em posição α com eletrófilos são possíveis, tanto em ambiente ácido como em
ambiente básico; o substrato de partida no primeiro caso é o enol, e no segundo caso, o
enolato.
Reação do halofórmio
A reação do halofórmio (ver também p. 659) é importante, especialmente sob aspectos
analíticos. O reagente mais usado é iodo em NaOH.
424
A. Isenmann
O
1a)
R C
1b)
1c)
2)
Princípios da Síntese Orgânica
+ OH-
CH3
+ H2O
lento
O
R C CH2 I
O
R C CHI2
O
R C
+ OH+ H2O
+ OH+ H2O
+ OH-
CI3
OR C
CH2
+ I2
- Irápido
O
R C
CH2 I
OR C CH
I
OR C
O
+ I2
- I-
R C CHI2
O
+ I2
- I-
CHI2
R C
CI3
OR C
R COOH
CI3
+ CI3-
OH
3)
R COOH
+
CI3-
R COO- +
HCI3
Iodofórmio
(sólido)
A reação de iodofórmio ocorre preferencialmente via enolato, cuja formação é a etapa mais
lenta nesta sequência. Observa-se uma velocidade mais baixa na adição do primeiro iodo
(etapa 1a). Já na adição do segundo iodo (etapa 1b) os α-H´s percebem um efeito retirador
de elétrons, do grupo carbonila mais do iodo. Assim, eles podem ser abstraídos com maior
facilidade. Desta forma as etapas 1b e 1c ocorrem cada vez mais rapidamente, ou seja, a
reação tem caráter auto-catalítico.
O grupo -CI3, onde cada iodo age com efeito –I, pode ser abstraído na forma do carbânion
(etapa 2). A última etapa é uma reação de ácido-base onde se forma iodofórmio que se
precipita em forma de bonitos cristais amarelos. A formação destes cristais serve então
como teste qualitativo para metilcetonas. A utilidade preparativa desta reação (p. 659) é
relativamente pequena.
A bromação, em analogia à iodação, é uma reação auto-acelerada, onde produtos
intermediários não podem ser isolados. Ela ocorre geralmente sob catálise básica. A
metilcetona está diretamente bromada para a 1,1,1-tribromocetona 283.
Diferente na cloração: a reação pode ser catalisada por ácidos ou bases e pode ser parada no
produto mono, di ou triclorado. Na maioria das vezes ela ocorre com tanta facilidade que
simplesmente basta introduzir Cl2 gás sob refrigeração na cetona líquida. Sendo assim, a 1monocloroacetona é formada numa reação fácil e completa:
O
H3C
C
O
CH3
+ Cl2
H3 C
C
CH2Cl
+ HCl
283
Recomenda-se fazer esse tipo de reação, numa capela de boa ventilação: lembre-se que α-bromocetonas
têm efeito lacrimante (gás de defesa, usado pela polícia nos anos 90)!
425
A. Isenmann
Princípios da Síntese Orgânica
O produto desta reação, aliás, provoca lágrimas; portanto, tem sido usado pela polícia em
formulações de gases lacrimogêneos.
Nitrosamento
O nitrosamento (ver também p. 657) só funciona em ambiente ácido, pelo fato de o
eletrófilo, no caso o cátion nitrosil, apresentar-se na forma inativa do ácido nitroso, HNO2,
em pH mais alto ( NO+ + OH- → HNO2).
O
OH
R C CH2
+
N O
- H+
O
R C CH2 N O
Enol
R C CH N OH
Tautomeria
Nitroso-oxima
O O
+ H2O
R C C
Hidrólise
H
+
H2NOH
Hidroxilamina
O composto nitroso que se forma inicialmente está em equilíbrio tautomérico com a
aldoxima. A hidrólise desta fornece compostos (1,2)-dicarbonílicos que podem, por
exemplo, ser usados como complexantes de metais, com destino em catalisadores
homogêneos. Um produto 1,2-dicarbonilado é em geral de difícil acesso (explicação: ver p.
566), portanto é também um equivalente sintético valioso para muitas sínteses.
Esta reação é formalmente uma oxidação do substrato carbonílico em posição α,
acompanhada pela redução do reagente nitrosante para hidroxilamina. Devido ao pH ácido
do ambiente as qualidades nucleofílicas da hidroxilamina são bastante reduzidas, portanto
não se corre o perigo de produtos paralelos devido ao rearranjo de Beckmann (p. 429) que
se inicia com uma condensação entre o composto carbonilado e a hidroxilamina.
5.5.12 Rearranjos em compostos carbonílicos
Em outros contextos já foram mencionados os seguintes rearranjos:
•
No sexteto eletrônico do N: Curtius, Hofmann, Lossen, Hunsdiecker (p. 391);
material de partida: derivados do ácido carboxílico.
•
No sexteto eletrônico do C: Wagner-Meerwein (p. 19); material de partida: alquila
primária com bom grupo abandonador.
•
Igualmente no sexteto eletrônico do C: Wolff (p. 387); material de partida:
diazocetona.
426
A. Isenmann
•
Princípios da Síntese Orgânica
No sexteto eletrônico do O: processo “cumeno” (p. 94), também a rancificação de
gordura (p. 97); material de partida: hidroperóxidos aromáticos e alifáticos,
respectivamente.
Neste lugar cabem então mais quatro rearranjos, dos quais o primeiro decorre do sexteto do
oxigênio, os seguintes no sexteto do nitrogênio. Todos eles são importantes métodos
degradativos-oxidativos feitos em cetonas (“degradativo”, visto que o esqueleto carbônico
do substrato é quebrado em posição α ao grupo carbonila) 284. O último rearranjo
apresentado, o rearranjo de Favorskii, é um dos raros exemplos nos quais o esqueleto
carbônico se transforma ao redor de um carbânion.
Oxidação de Baeyer-Villiger
Esta reação tem grande valor preparativo (ver também p. 662). Um resumo da oxidação de
Baeyer-Villiger, junto com as demais possibilidades de oxidar cetonas, se encontra no
anexo 2 deste livro.
Cetona + H2O2 → Éster
Deve-se levar em consideração que H2O2 em alta concentração é perigoso e pode explodir.
É mais eficiente e menos perigoso usar perácidos, em vez da água oxigenada. Isto se deve
menos à sua reatividade do que à propriedade de misturar-se melhor com o substrato do que
a H2O2: a maioria das cetonas não se mistura bem com água (onde se dissolve a maior parte
do H2O2); portanto, o contato entre os reagentes fica restrito a uma área pequena e a reação
fica muito lenta. O perácido acético e o perácido benzóico são os oxidantes mais usados
neste sentido.
284
Já foram apresentados em outros lugares: degradação de metilcetonas por reação de halofórmio (pp. 424 e
659), e a contração de anéis (p. 658) em cetonas cíclicas.
427
A. Isenmann
Princípios da Síntese Orgânica
H
O
R
+
O C
C
O H
R´
C
R´
O
OH
+ H+
C
R´
R C O O
R C O O
HO O
R
O
R
R
Perácido
Cetona
H
caminho a:
OH
O
OH
C
R´
R C O
R C O O
HO
C R´
R
R
O
- H+
R C O
R
Éster
"íon oxênio"
O
H
caminho b:
OH
O
OH
O
C
R´
R C O R
R C O O
HO
-
R
C R´
- H+
R C O R
Éster
O
Embora o caminho a, com a formação do íon "oxênio", seja historicamente mais
importante, a existência de um oxigênio com sexteto de elétrons nunca foi comprovada, e
os argumentos para sua existência são cada vez mais fracos, pois a reação de BaeyerVilliger ocorre com bastante facilidade (visto que a formação de um íon oxênio livre deve
ser um processo altamente endotérmico). Hoje se acredita em um rearranjo sob movimento
sincronizado - saída do ácido carboxílico e mudança do grupo R - o que corresponde mais
ao caminho b, no esquema acima.
Caso haja dois grupos alternativos, geralmente o melhor doador de elétrons (quer dizer, o
grupo com maior efeito +I) muda de posição porque é ele que compensa melhor a falta de
elétrons no oxigênio. Além deste critério eletrônico, a facilidade de o grupo R se mudar
depende também do seu tamanho (efeito estérico). Assim, se conhecem casos de desvios,
onde o grupo R menos fornecedor de elétrons muda de posição.
A oxidação de Baeyer-Villiger também funciona com cetonas cíclicas, a partir das quais se
formam ésteres cíclicos (= lactonas), com um anel ampliado por mais um átomo, o
oxigênio:
O
[O]
BaeyerVilliger
O
O
428
A. Isenmann
Princípios da Síntese Orgânica
Rearranjo de Schmidt
Também a degradação segundo Schmidt e o rearranjo de Beckmann (ver abaixo) se aplicam
em cetonas. O esqueleto carbônico é rompido no local do grupo carbonila. Assim, estes
métodos representam vias alternativas e bem vindas à oxidação Baeyer-Villiger ou às
reações radicalares do tipo Norrish I (= cisão α, ver p. 107).
R´
C O
+
N3-
N3
R C OH
R
R´
+
+ H
- H2O
N N
C N
R
R´
+ H2O
o
- N2
- H+
O
OH
R C N
R´
R C NH
Amida
Sob aspectos mecanísticos, estas reações encaixam-se igualmente bem no item 5.4.4, onde
foram apresentados rearranjos no grupo carboxila.
Aviso: certas azidas, especialmente as dos metais pesados, são altamente explosivos!
Rearranjo de Beckmann
Uma cetona reage com hidroxilamina, em ambiente levemente alcalino, fornecendo uma
oxima (compare p. 411). Esta, por sua vez, pode ser protonada numa etapa posterior por um
ácido mineral concentrado (H2SO4 ou H3PO4), para depois desprender água. A questão de
qual dos grupos vai migrar para o nitrogênio com sexteto de elétrons, curiosamente não
depende das suas propriedades eletrônicas, mas apenas da posição relativa à água que saiu
do complexo. Sempre muda-se o grupo posicionado anti (ou E). Essa observação é um forte
indicativo para que as etapas 2 e 3 ocorram de maneira sincronizada.
R´
C O
R
1
+ H2NOH
- H2O
3
+ H2O
- H+
R´
C N
R
OH2
2
R C N
R´
- H+
R C N
R´
O
R
R C NH
C N
HO
R´
+ H+
C N
R
OH
Oxima
R´
R´
Amida
Caso se pretenda o produto isomérico deste, onde se mudar o grupo R em vez de R´, uma
isomerização in situ da oxima pode ser tentada. Para este fim, uma complexação da oxima
num metal de transição é o método mais promissor.
A última etapa representa uma tautomeria, formando uma amida estável.
Uma síntese industrial que decorre por este rearranjo, já foi apresentada na p. 100.
Rearranjo de Favorskii
A partir de uma cetona halogenada em posição α pode-se provocar um rearranjo que leva à
formação de um ácido carboxílico ou um éster, sob alongamento da cadeia carbônica.
429
R´
A. Isenmann
Princípios da Síntese Orgânica
O mecanismo deste rearranjo é bastante complexo. Inicialmente, uma base leva o substrato
ao enolato. Dos dois lados possíveis, o lado halogenado dispõe do próton mais ácido.
Porém, somente o enolato menos favorecido (caminho 1) leva à reação de Favorskii.
R Cl
O
H
1´
1
Base
Base
R Cl
O
R
Cl
O
H
O rearranjo funciona sob catálise básica e percorre um estado cíclico de pouca estabilidade
– uma ciclopropanona (etapa 3). A adição nucleofílica do etóxido, etapa 4, fornece um
intermediário com excesso de elétrons. Junto à tensão de pequenos anéis, o composto
cíclico torna-se instável e uma ligação C-C se rompe (etapa 5). Naquele momento pode-se
afirmar um intermediário de caráter carbaniônico, no exemplo a seguir no carbono em
posição bis-benzílica. Nesta etapa será definido o produto, visto que será quebrada aquela
ligação C-C que levará ao carbânion mais estável. O carbânion mais estável é geralmente o
carbono menos substituído ou então aquele carbono que recebe estabilização dos vizinhos
por mesomeria (= posição bis-benzílica), como mostrado no exemplo a seguir:
Ph Cl
O
2
-
EtO
Ph
Ph Cl
O
OEt
Ph
-
O
OEt
Ph
Ph
Ph
O
5
C OEt
Ph
Ph
EtO-
Ph
Ph
-
4
O
3
O
Ph
EtOH
Ph
COOEt
Ph
"carbânion"
Uma aplicação interessante deste rearranjo é o estreitamento de cetonas cíclicas (sendo uma
alternativa à síntese descrita na p. 658). A bromação ou cloração da posição α e o
tratamento com uma base prótica leva a uma cetona intermediária bicíclica. Caso a
molécula da partida for um ciclo de n carbonos, o biciclo seria [1,0,(n-1)], por sua vez
bastante instável e sujeito ao rearranjo de Favorskii. No exemplo a seguir, como também na
maioria das cetonas que são submetidas a esta reação, o biciclo é altamente simétrico, assim
não importa qual das ligações C-C quebra. O produto fica bem definido, no caso um ácido
(ou um éster) com um ciclo de (n-1) carbonos.
430
A. Isenmann
Princípios da Síntese Orgânica
O-
O
Cl
OH-
O
Cl
OH-
COOH
- Cl-
Este é um raro exemplo de um rearranjo decorrendo num carbânion 285. Por que é tão raro?
Deve-se procurar a explicação no par de elétrons não-ligante no carbânion, que precisa de
bastante espaço 286. A situação estérica no complexo ativado, portanto, é mais apertada do
que no carbocátion onde não há elétrons neste orbital, quer dizer, espaço desocupado. Além
disso, a migração 1,2 de um vizinho para um carbocátion envolve 2 elétrons, enquanto o
mesmo deslocamento num carbânion abrange 4 elétrons em movimento. Dois destes
somente cabem em um orbital antiligante - o que aumenta a energia do complexo ativado.
R
R
C C
1,2-migração
em carbocátions
C
C
1,2-migração
em carbânions
(menos favorável)
Aliás, a formação do próprio substrato desta reação, a cetona α-halogenada (p. 424), ocorre
via enolato, que também pode ser visto como estado carbaniônico (p. 400).
5.6
Reações dos compostos carbonilados com organometálicos
As reações de substratos com grupo carbonila (aldeídos, cetonas, derivados do ácido
carboxílico) com organometálicos têm um alto valor preparativo - a não dizer que elas
representam a classe mais versátil das reações dos compostos carbonílicos. Informe-se no
capítulo 10.3, sobre o preparo e as características principais (reatividade, estabilidade) dos
diversos compostos organometálicos. A parte negativa destes compostos "carbanóides" 287
reage com o carbono positivado do grupo carbonila, enquanto a parte metálica geralmente
forma um subproduto sem valor. As reações geralmente são bem controláveis e
direcionadas. Neste capítulo, então, conheceremos a mais importante estratégia para criar
uma nova ligação carbono-carbono.
285
Outros exemplos são referidos no contexo da ciclização de Ramberg-Bäcklund (p. 155) e na síntese de
Corey-Fuchs (p. 803).
286
A medição em ângulos de aminas revelou que um par de elétrons requer mais espaço do que um átomo de
hidrogênio!
287
A expressão ~óide é usada para exprimir semelhança. Carbanóide abrange então todos os compostos
organometálicos, pois todos têm um carbono rico em elétrons, não obstante quantos porcentos de caráter
iônico ou covalente. Detalhes no cap. 10.1.2.
431
A. Isenmann
5.6.1
Princípios da Síntese Orgânica
A reação de Grignard
A reação de Grignard (V. Grignard, Nancy; prêmio Nobel em 1912) é a mais importante e
mais versátil reação dos organometálicos. Em muitos livros é referida como "condensação",
mais no sentido de se criar uma nova ligação carbono-carbono do que no de uma reação
onde se libera água. Neste texto usaremos a expressão "condensação", em ambos os
sentidos, para reações das quais resulta uma nova ligação C-C (aqui e especialmente no
capítulo 6) e também em situações onde se forma uma molécula maior, sob exclusão de
água ou um a outra pequena molécula. Na sua definição mais ampla, a síntese de Grignard
pode ser descrita como reação entre um componente com carbono positivado e um
composto organo-magnésio.
Protótipo da reação de Grignard:
R
δ+ δ-
C O
R
δ-
δ+
+ R´ MgX
R
R C O MgX
R´
H2O / H+
- MgXOH
R
R C OH
R´
No início do século passado usavam-se compostos organo-zinco, que foram depois
substituídos pelos compostos organo-magnésio, por fornecerem rendimentos melhores.
Atualmente, em muitas reações de Grignard o Mg está sendo substituído pelo Li.
Obtém-se o reagente de Grignard, R´MgX, através da reação do brometo ou cloreto
orgânico e grânulos de magnésio metálico. A reação é fortemente exotérmica e deve ser
realizada sob refrigeração intensa (isto é, solvente volátil e condensador de Dimroth ou
Graham).
Os solventes mais comuns são (ver também Tabela 42, p. 715):
• Éter dietílico (Teb.= 35 °C) e
• THF (Teb = 65 °C).
Mais recentemente usam-se também:
• DME (= dimetoxietano; H3C-O-CH2CH2-O-CH3; Teb = 85 °C) e
• A amina TMEDA (= tetrametiletilenodiamina; (H3C)2N-CH2CH2-N(CH3)2; Teb =
120 °C ), com éxito.
Mas também outros solventes não nucleofílicos podem ser usados - desde que estejam
apróticos; exemplos são anisol ou dibutiléter. Os solventes próticos, dentre outros a água e
alcoóis, devem ser rigorosamente excluídos, porque levam ao hidróxido (alcóxido) de
magnésio, sob desprendimento de H2 - logo, produtos sem valor preparativo, por terem
apenas um O negativo, em vez de um C negativado.
A síntese do reagente de Grignard às vezes não quer começar, ou seja, percorre um período
de indução. Para ultrapassá-lo, a mistura reacional pode ser exposta a ultrassom; também
pode-se acrescentar traços de ácido de Lewis, preferencialmente uma gota de Br2, um grão
de MgBr2 anidro, CCl4 ou um grão de I2. Com o iodo, por exemplo, se consegue destruir a
camada oxídica protetora na superfície dos grânulos ou da fita do Mg metal e, assim, ativálo. Isto pode também ser feito por aquecimento prévio das raspas de magnésio junto com
um grão de I2 sobre a chama aberta. Além do mais, o MgI2 formado em traços ajuda a
remover os últimos restos de umidade da mistura. Deve-se lembrar que as sínteses
432
A. Isenmann
Princípios da Síntese Orgânica
orgânicas com os metais mais eletropositivos (álcali, álcalino-terrosos, Al) precisam de
condições rigorosamente anidro e livre de álcool ou qualquer outro solvente com atividade
prótica (ver pp. 165 e 690); igualmente recomendado é o trabalho sob atmosfera inerte, pois
o oxigênio do ar pode levar a produtos paralelos ou, no pior caso, ao incêndio.
Não é possível obter o reagente de Grignard a partir de 1,2-dihaletos. Nestes casos se
observa uma reação diferente, fornecendo o alqueno com altos rendimentos. O mecanismo
desta é em todo análogo à desalogenação com zinco (ver p. 152).
Mecanismo da reação de Grignard
Apesar do seu alto valor preparativo e dos 100 anos de experiência com essa reação, o
mecanismo ainda não está perfeitamente entendido em todos os pormenores. As dúvidas já
começam no próprio reagente de Grignard, que pode se apresentar em estruturas diferentes,
dependendo das condições (solvente, temperatura) e da estrutura do grupo orgânico. Por
r.m.n. verificou-se que o brometo de metilmagnésio em éter dietílico se apresenta na forma
[(CH3)2Mg + MgBr2]; já o brometo de fenilmagnésio em éter dietílico existe em um
complexo tetraédrico em volta do metal, [PhMgBr ⋅ 2 Et2O], comprovado por difração de
raios-X.
Para a própria reação de Grignard existem muitas indicações para a existência de um
estado de transição (cíclico), no qual participam uma molécula do substrato carbonílico e
duas moléculas do reagente de Grignard. O argumento mais forte é fornecido pela cinética
da reação:
2
v = k ⋅ [Cetona ] ⋅ [Grignard ] .
Porém, a transferência do grupo R´ (no esquema abaixo, em negrito), de um Mg para um
outro Mg não foi comprovada. O último mecanismo proposto 288 inclui a transferência de
elétrons desemparelhados (SET), onde o magnésio é oxidado e o composto orgânico é
reduzido aos poucos. O ponto central deste mecanismo é que os radicais, uma vez
formados, não se movimentam livremente, mas ficam juntos, presos numa gaiola de
solvente. A última etapa é a recombinação dos radicais formando o produto, o alcóxido,
com rendimentos extraordinariamente bons (ver detalhes sobre o reagente de Grignard, no
cap. 10.3.2):
288
E.C.Ashby, The mechanisms of Grignard reagent addition to ketones, Accounts Chem.Res. 7 (1974) 272.
433
A. Isenmann
Princípios da Síntese Orgânica
Mecanismo antigo:
X
X
Mg
Mg
R´
R2C
R´
Mg
O
R´
R´
R2C
X
O
Mg X
Mecanismo novo:
R
R´MgX
+
C O
SET
R
[ R´MgX ]
+
C
R
R
O
Radicalânion
R
R´ C O- MgX+
R
[ MgX + R´ ]
Gaiola de solvente;
etapa rápida
5.6.2
Reações paralelas da reação de Grignard
Uma série de reações paralelas acompanha, infelizmente, a reação de Grignard. Um estado
de transição bem organizado é bastante desfavorável, do ponto de vista entrópico. O alto
grau de organização torna a reação principal demorada, o complexo fica vulnerável e os
caminhos para reações paralelas e indesejadas estão abertos. Quando, além disso, os
substituintes R, tanto no composto organometálico quanto no substrato carbonílico, são
volumosos ou ramificados, os rendimentos da reação principal podem cair a 20%!
A falha mais comum é a decomposição do reagente de Grignard, causada por traços de
umidade. Em geral, qualquer ácido de Brønsted (ácido carboxílico, água, fenóis, alcoóis,
tióis,...) consome o reagente, que se perde na forma de um sal inútil de magnésio e um
hidrocarboneto, R-H / Ar-H (R/Ar: proveniente do reagente de Grignard). Outra causa para
rendimentos baixos pode ser a presença de CO2 atmosférico, que reage rapidamente quando
estiver em contato com o composto organo-magnésio (p. 440); o oxigênio atmosférico
também reage, formando hidroperóxidos perigosos (p. 671). A maioria das sínteses é feita
em éter (dietiléter, THF ou dioxano), sob refluxo. Estes solventes produzem uma almofada
de vapores que cobre o meio reativo, por ser mais denso do que o ar. Em sínteses mais
difíceis e demoradas, porém, esta camada protetora não é suficiente e a reação de Grignard
deve ser feita sob atmosfera de argônio.
Além destas reações paralelas devido à contaminação por substâncias do ambiente, existem
mais dois caminhos paralelos, que nem mesmo com atmosfera inerte podem ser impedidos:
434
A. Isenmann
Princípios da Síntese Orgânica
Migração de prótons
Não apenas os ácidos clássicos de Brønsted, mas também o próprio substrato com grupo
carbonila pode decompor o reagente de Grignard por meio de uma reação prótica. Isto se
deve à elevada acidez do(s) hidrogênio(s) em posição α (pKa logo acima de 20).
X
Mg
R´
H
O
O- MgX+
R´ H
O
H2C
C
R
Enolato
C
CH2
+
R
H2O
- MgX(OH)
H3C
C
R
O substrato carbonilado não sofre grande mudança por esta reação, como se vê na etapa da
hidrólise deste esquema, mas o reagente de Grignard se perde.
Transferência de hidreto
Como a parte orgânica do reagente R-MgX já está negativada, então é facilitada a saída de
um hidreto, H-. O grupo R abandona neste caso um hidreto em posição β, para depois
perder o metal da posição α, também. O resultado desta “eliminação β ” é o alqueno.
O alvo do hidreto não é o magnésio, o que poderíamos esperar pela sua proximidade na
molécula, mas sim, é o substrato carbonilado. Essa reatividade será melhor entendida
quando estudarmos o efeito redutor dos hidretos metálicos (p. 621). A transferência do
hidreto representa uma redução do composto carbonílico, sendo o produto um álcool:
X
Mg
H3C
O
CR2
CH
CH2
H
H3C
CH CH2
Alqueno
(Oxidação)
+
R2C O- MgX+
H
H2O
R2CH OH
Álcool
(Produto de redução)
Nota-se que esta reação redox, como toda transferência de hidreto, é bastante lenta
(compare, por exemplo, a reação de Cannizzaro p. 422). Este fato abre caminhos para se
impedir ou, pelo menos, reprimir, essa reação paralela: por adição controlada de ácidos de
Lewis (jamais um ácido de Brønsted!). Geralmente, adiciona-se MgBr2 anidro, que
complexa no oxigênio do composto carbonílico. Por esta medida aumenta-se a eletrofilia do
carbono carbonílico - o que favorece em primeiro lugar o caminho principal da reação de
Grignard. O redimento pode ser até mesmo duplicado com esta medida.
435
A. Isenmann
Princípios da Síntese Orgânica
Esta reação paralela é um dos motivos para se subtituir hoje em dia o reagente clássico RMgX, por compostos organo-lítio 289. Os compostos RLi têm um caráter mais carbanóide
ainda do que RMgX; mesmo assim, eles geralmente não pedem a adição de ácidos de
Lewis. Assim, representam os nucleófilos mais reativos e mais “limpos”. Os demais
motivos de se usar reagentes R-Li são:
•
Melhores rendimentos na reação com cetonas com grupos R de alto impedimento
estérico;
•
Na reação com substratos carbonilados α,β-insaturados eles rendem mais aduto 1,2
e menos o aduto 1,4 (= orientação Michael, p. 520), em comparação ao reagente RMgX;
•
Seu manuseio é mais seguro, por serem menos pirofóricos (reação com água e
oxigênio é mais devagar do que em outros organometálicos).
Certamente uma desvantagem dos compostos R-Li é a sua elevada reatividade frente éteres
que - em quase todos os métodos clássicos - são usados como solventes. A Tabela 42 na p.
715 refere os tempos de meia-vida, para alguns sistemas típicos de RLi-éter.
Resumo das reações de compostos carbonilados com o reagente de Grignard
(Uma lista mais completa se encontra no anexo 2 deste livro.)
A seguinte tabela contém as mais importantes reações do reagente de Grignard com
compostos carbonílicos e carboxílicos. Em todos os casos, o carbono ligado ao oxigênio
sofre redução, pois perde a dupla-ligação com o oxigênio (eletronegatividade, EN = 3,5) e
ganha um novo vizinho carbono. Por esta razão, a reação de Grignard e as outras reações
de organometálicos com carbonos positivados se chamam também de “acoplamento
redutivo”. A variedade dos possíveis produtos e a flexibilidade da reação de Grignard é
impressionante.
289
Artigos de revisão: V.H. Gessner, C. Daschlein, C. Strohmann, Structure formation principles and
reactivity of organolithium compounds. Chem.Eur.J. 15 (2009), 3320-3334.
G. Wu, M. Huang, Organolithium reagentes in pharmaceutical asymmetric processes. Chem.Rev. 106 (2006),
2596-2616.
436
A. Isenmann
Princípios da Síntese Orgânica
Tabela 22.
Espectro dos produtos acessíveis pela reação de Grignard
Produto
Substrato
+ R-MgX
H2C O
Formaldeído
O
H2C CH2
Óxido de etileno
O
R´
+ R-MgX
Álcool primário
(+ -CH2-)
R CH2 CH2OH
Álcool primário
(+ -CH2-CH2-)
R
+ R-MgX
C
R CH2 OH
R´
H
Aldeído
O
+ 2 R-MgX
H C
O R´´
Éster do ácido fórmico
+ R-MgX
R´2C O
Cetona
O
R´
O
Éster
R´´
H C(OR´)3
Ortoéster do
ácido fórmico
R´ CN
Nitrila
CO2
Gelo seco
O2
+ R-MgX
+ R-MgX
+ R-MgX
+ R-MgX
" H + " qualquer ácido
de Brönsted
Álcool secundário
R
R CH OH
R CR´2
+ 2 R-MgX
C
CH OH
R´
CR2
R COO-
R O O- MgX+
+ R-MgX
Álcool terciário
OH
OH
(R´O)2CH R
Acetal
R
R´ C N
Álcool secundário simétrico
Álcool terciário
H+/H 2O
H+/H 2O
+
H /H 2O
+ R-MgX
R H
R CHO
Aldeído
R
R´
C O
Cetona
R COOH
Ácido carboxílico
R OH Álcool
Alcano
No início desta lista são referidos dois métodos de obtenção de alcoóis primários. Ambos
representam importantes estratégias para alongar a cadeia carbônica em R, por uma e duas
unidades, respectivamente. Com o alongamento das cadeias carbônicas em destaque, se
437
A. Isenmann
Princípios da Síntese Orgânica
entendem as duas linhas de síntese a seguir, com alcoóis primários, tanto de material de
partida como de produtos:
H2C O
1. SOCl 2
2. Mg
R OH
H2O
R CH2 O MgCl
R CH2 OH
R MgCl
O
R CH2 CH2 O MgCl
H2O
R CH2 CH2 OH
O epóxido (= oxirano) pode ser visto como um composto pseudo-carbonílico, também
referido como composto carbonílico "vinílogo". Portanto, tem a mesma polarização de um
grupo C=O, isto é, carbonos positivados. Caso seja usado um epóxido assimétrico,
O
R , o ataque nucleofílico do carbanóide ocorre com alta seletividade no carbono
menos substituído – principalmente por motivos estéricos.
A reatividade dos compostos carbonílicos frente ao reagente carbanóide depende da
polarização δ+ no carbono: quando mais positivado, mais fácil o ataque nucleofílico do
carbanóide. Assim, obtemos a seguinte sequência de reatividade na síntese de Grignard.
Figura 33. Reatividades relativas dos compostos carbonilados, frente o reagente
organometálico
O
R C
O
R C
Cl
>
O
O
R C
> R C
H
O
> R C
O
R > R C
OR´
O
> R C
> R COO
NH2
O
Os argumentos para essa reatividade diferenciada são as mesmas que foram estabelecidas
para a sequência de reatividade na acilação (Figura 31, na p. 354).
A reatividade relativamente baixa do éster se deve ao efeito +M do grupo alcoxila, que na
verdade supercompensa o efeito indutivo –I do oxigênio. Já no cloreto de acila, não se tem
mais a preponderância do efeito mesomérico. Lembre-se que os efeitos I e M são de
magnitudes comparáveis para os substituintes halogênios (ver p. 318). Os tamanhos dos
orbitais, π do carbono e n do cloro, são bastante desiguais, portanto o efeito +M do cloro
fica bastante reduzido. Ao mesmo tempo continua a diferença em eletronegatividade, entre
C e Cl, que causa um forte efeito -I no carbono. Por isso, o cloro é um forte retirador de
elétrons e então exerce efeito ativante na reação de Grignard.
O próprio ácido carboxílico não é apropriado para a reação de Grignard, devido seu
hidrogênio ácido que destrói o reagente R-MgX instantaneamente. Também o carboxilato
não fornece bons resultados, devido à carga negativa que repele o reagente carbanóide
438
A. Isenmann
Princípios da Síntese Orgânica
RMgX. Os derivados mais indicados são, portanto, o éster, o cloreto de acila (ambos
reagem duas vezes e fornecem alcoóis terciários, ver também esquema abaixo) e a nitrila
(que reage somente uma vez, fornecendo a cetona após hidrólise, ver também p. 615).
As sínteses de Grignard mais viáveis:
O
R C
Cl
+ R´ MgX
- MgXCl
O
R C
+ R´ MgX
R´
O
R C
R´
OH
H2O
R C
R´
O
R C
OR´´
R´
R´
+ R´ MgX
- R´´OMgX
R´ MgX
R C N
N+
MgBr
R´ C
H2O / H+
R
O
+ NH3
C
R
R´
Cetona
(não simétrica)
A obtenção de aldeídos via reação de Grignard parece impossível, já que o grupo carbonila
do próprio aldeído mostra alta reatividade frente ao reagente organometálico. Porém, existe
um substrato especial que permite parar a reação no estado certo de redução: é o ortoéster
do ácido fórmico 290. O acetal que resulta da reação com um equivalente do reagente de
Grignard é bastante estável e não reage com um segundo equivalente do reagente de
Grignard. Após a extinção da reação (inglês: quenching) com álcool e o tratamento com
água o acetal ainda continua estável (p. 403). Somente ao acidificar o ambiente ele se
degrada e fornece o aldeído de maneira segura e completa.
OCH3
R MgBr
+
H C(OCH3)3
Ortoéster do
ácido fórmico
R C
H
OCH3
Acetal
H+ / H2O
O
+
R C
2 CH3OH
H
Interessante também é o acesso aos ortoésteres (por sua vez um reagente bastante versátil,
ver pp. 379 e 403), via reação de Grignard. Funciona a partir do ortoéster do ácido
carbônico, C(OR)4; o mecanismo é semelhante ao da reação acima.
290
Após tudo que foi dito sobre o efeito indutivo, mesomérico e espacial, dos grupos -OCH3, é difícil
acreditar que o carbono em ortoésteres ainda está positivado (Cδ+). Sendo assim, a reatividade do ortoéster
frente o reagente carbanóide (reagente de Grignard; nucleófilo forte), nós surpreende, como também foi o
caso nos acetais, compare p. 403.
439
A. Isenmann
Princípios da Síntese Orgânica
OCH3
R MgBr
+
C(OCH3)4
R C OCH3
Ortoéster do
ácido carbônico
OCH3
Ortoéster
estável
Reações subseqüentes
O reagente R-MgX reage com bastante facilidade com a umidade, O2 e CO2 da atmosfera.
Portanto, as reações de Grignard mais demoradas e difíceis devem ser executadas sob
atmosfera inerte. É melhor usar argônio porque até o N2, geralmente muito inerte, é
vulnerável frente bases fortes e metais reativos, então pode reagir com o reagente de
Grignard, levando a nitretos indesejados.
Por outro lado, é possível promover justamente estas reações com H2O, O2 e CO2, e assim,
torná-las as reações principais. O valor preparativo da reação com gás carbônico é a
introdução do grupo carboxilato; isso implica um alongamento da cadeia carbônica do
substrato por uma unidade.
A reação com dióxido de carbono, chamada de carboxilação, que ocorre não só com os
reagentes Grignard, mas com todos os compostos organometálicos de caráter iônico ≥ 18%
(ver Tabela 36 na p. 682):
R M +
O C O
R COO-
+
M+
.
O clássico reagente de Grignard, R-MgX, reage com o CO2 da atmosfera (além do O2; o
valor da reação com O2 é comentado na p. 671). Na maioria das vezes trata-se de uma
reação paralela indesejada que deve ser reprimida por meio do trabalho sob gás inerte.
Geralmente é suficiente manter uma camada de vapor do próprio solvente, dietiléter, que é
mais pesado do que o ar e então protege a mistura reacional de Grignard, por uma
"almofada" de vapores. Mas em alguns casos essa reatividade pode ser usada de propósito,
para aumentar a cadeia carbônica do substrato R-M por uma unidade carbônica,
introduzindo o grupo -COO-.
O último exemplo da lista acima parece ser inútil, do ponto de vista preparativo. Mas tem
certa relevância analítica: segundo Zerewitinoff, podem ser determinados quantitativamente
hidrogênios ácidos em compostos orgânicos. O "Grignard de metila" desprende metano
que pode ser facilmente quantificado por volumetria. Segue um exemplo no qual a
substância ácida a ser analisada é um álcool:
R OH
+
CH3 MgX
R O- MgX+
+
CH4
Volume medido
Reação de compostos carbonílicos com outros reagentes organometálicos
Um método mais novo para se obter cetonas assimétricas, como alternativa bem vinda à
reação de Grignard com nitrilas (p. 615), é a partir do carboxilato. No entanto, o método
somente funciona com o composto organometálico mais reativo, R´Li (ver também p. 382).
440
A. Isenmann
Princípios da Síntese Orgânica
Neste caso, o acoplamento redutivo pára realmente no equivalente da cetona 291, porque o
intermediário é um diânion (isto é, duas cargas negativas quase vizinhas), que impede a
aproximação de mais um reagente carbaniônico, R´Li.
R COOH
Base
R COO
-
R´Li
R
O-Li+
R´
O-Li+
H2O / H+
R´Li
R
O-Li+
R´
R´
Não acontece
R
O
Cetona assimétrica
R´
A estratégia oposta é usada na síntese descrita a seguir, fornecendo igualmente uma cetona
assimétrica.
Ao aplicar organometálicos menos reativos que o reagente clássico RMgX, apenas os
derivados carbonílicos mais ativados reagem. Um bom exemplo são os reagentes organocádmio (ver outra aplicação na p. 615; sobre o reagente ver p. 758) que reagem unicamente
com cloretos de acila. Ao contrário do reagente RMgX, não há duplo ataque, mas a reação
pára na etapa da cetona; o reagente R´2Cd simplesmente é muito fraco para atacar o produto
primário mais uma vez.
O
R C
R´2Cd
O
R C
R´
Cl
O fato de que o substrato não reage com dois carbanóides torna esta estratégia até superior
à reação clássica de Grignard com nitrilas e fornece cetonas com rendimentos excelentes.
O reagente R´Li fornece então os nucleófilos mais fortes (e menos seletivos), enquanto o
R´2Cd é bastante fraco, mas muito seletivo. Existem também organometálicos de
reatividade intermediária (ver as reações típicas, no cap. 758). O método padrão para sua
obtenção é a metatese (p. 700). O reagente organo-titânio, por exemplo, é acessível a partir
de reagente de Grignard e cloreto de titânio. Este destaca-se por reagir seletivamente com
aldeídos, enquanto a reação com cetonas geralmente fica ausente 292 (lembre-se que isto não
foi possível com o reagente clássico organo-magnésio, que reage tanto com aldeídos quanto
com cetonas).
291
292
M.J.Jorgenson, Org.React. 18 (1970) 1.
M.T. Reetz, Organotitanium Reagents in Organic Synthesis, Springer Berlin 1986
441
A. Isenmann
5.7
Princípios da Síntese Orgânica
Exercícios. Assunto: reações no grupo carbonila
1) Em Tabela 19 (p. 345) se encontram resumidos os efeitos termodinâmicos que explicam
a diferença em acidez. Reproduza com suas próprias palavras, e em frases completas,
porque o ácido trifluoracético é mais forte do que o ácido acético.
2) Os seguintes compostos têm certa relevância na síntese e/ou na analítica orgânica.
Procure seus nomes triviais.
O
R C
RO C OR
NH NH2
OR´
R C OR´
OR´
R CH N NH2
R CH2 C
R´
OCH3
C
R C
OR
OCH3
O
O
OR
NH OH R
C N
O
O
NH (R´)
O
C
O
H2N
C
NH2
R
C
O
O
C
R´
3) A reação mais aplicada nos derivados do ácido carboxílico é a substituição nucleofílico
no carbono(+3).
a) Indique três derivados do ácido carboxílico que são mais reativos que a substância mãe.
b) Indique três derivados do ácido carboxílico que são menos reativos que a substância
mãe.
c) Rascunhe o mecanismo padrão desta substituição.
4) Quais métodos você conhece para ativar o ácido carboxílico livre, transformando-o em
seu cloreto?
5) O que se entende por "catálise nucleofílica"?
6) Explique o funcionamento da separação de uma mistura de aminas primárias,
secundárias e terciárias, conhecida como separação de Hinsberg.
7) a) Anote a reação global, da produção de biodiesel.
b) Quais são os possíveis catalisadores desta reação?
c) Procure argumentos que expliquam por que o metóxido de sódio atualmente ganha
preferência como catalisador, neste processo industrial.
8) Em um substrato de estrutura molecular mais complexa, pode surgir a necessidade de se
proteger um grupo ácido carboxílico.
a) Quais poderiam ser os motivos de se proteger o grupo ácido carboxílico?
b) Quais são os critérios a serem respeitados, ao introduzir um grupo protetor?
c) Quais são os métodos preferidos para se introduzir um protetor para o ácido carboxílico?
442
A. Isenmann
Princípios da Síntese Orgânica
9) Compare as reações de saponificação de ésteres, amidas e nitrilas, sob os seguintes
aspectos: possíveis catalisadores; temperatura; tempo de reação; rendimento.
10) A reação de Ritter é um método de derivatizar nitrilas. Descreva o funcionamento da
reação, indicando reagentes, princípio do mecanismo e produto.
11) Quais são os métodos mais comuns para produzir cetenos (in situ, logicamente)?
12) Na página 390 foi referida a reação entre o dicloroceteno e o ciclopentadieno.
Conforme alegado no capítulo 3.5.6, um dieno deste pode também reagir no sentido de uma
cicloadição [2+4]. O ceteno é bastante reativo e entrará neste esquema como dienófilo.
Certamente, as condições reacionais decidirão sobre a porcentagem com que este produto
se formará.
a) Esboce a reação [2+4], descrevendo a estrutura molecular do produto.
b) Na verdade, são dois produtos que resultam desta cicloadição (dois enanciômeros).
Desenhe esses produtos quirais e indique o carbono assimétrico.
13) "Degradação" é uma expressão vulgarmente usada quando algum objeto se transforma
e perde valor. Na química orgânica, no entanto, ela expressa uma reação que envolve a
fragmentação (= quebra) do esqueleto carbônico. Que o esqueleto carbônico em ácidos
carboxílicos e derivados não é um "santo gral", pode ser visto por meio de uma série de
degradações. Mencione esses métodos, seus reagentes e seus valores preparativos.
14) Compare a reatividade nas posições α, em cetonas e em ácidos carboxílicos.
15) O α-haleto do ácido carboxílico é um reagente versátil. Uma das suas aplicações mais
conhecidas é a reação de Reformatzky.
a) Descreva os reagentes e o produto desta síntese.
b) No mecanismo desta reação, qual o fato mais marcante?
c) Quais são as desvantagens e/ou perigos desta síntese?
16) Indique as reatividades em todas as partes das estruturas que são percorridas durante a
tautomeria ceto-enólica.
17) O método mais comum de proteger um grupo carbonila em uma síntese mais complexa
é via acetal.
a) Descreva a reação da formação do acetal, a partir de uma cetona.
b) Essa mesma reação tem relevância, também na química dos açúcares. Descreva a reação
e sua utilidade.
18) a) Como fazer um enoléter?
b) Para que servem os enoléteres (também chamados de viniléteres)?
c) Existe um derivado com nitrogênio que preenche o papel do enoléter?
19) a) Em qual/quais categoria(s) encaixa a reação clássica de Cannizzaro: ácido-base,
oxidação, redução, polimerização, complexação?
443
A. Isenmann
Princípios da Síntese Orgânica
b) Essa reação ocorre com muito facilidade? Justifique.
c) Quais saõ as desvantagens/restrições preparativas desta reação?
20) a) O que todos os compostos organometálicos têm em comum?
b) De que propriedade atômica depende a reatividade do carbono, em compostos
organometálicos?
c) Qual o caráter iônico das ligações nos seguintes compostos, em %? Oriente-se nas
eletronegatividades, indicadas na sua tabela periódica Tabela 34 na p. 682 (maiores
detalhes estão descritos no cap. 10.1.2).
NaCl, MgF2, B2O3, NH3 ?
Compare com o caráter iônico das ligações relevantes na química organometálica:
C-Li, C-Mg e C-Cu.
d) Que fator influência na reatividade de compostos organometálicos, além da diferença em
eletronegatividade?
21) Complete a seguinte tabela:
Substrato
C N
Reagente de Grignard
Produto
H5C2 MgBr
O
H5C2 MgBr
OC2H5
OCH3
H3CO C OCH3
H5C2 MgBr
OCH3
H2O
H5C2 MgBr
O
H7C3
C
H
CO2
CH2 COOH
22) Na p. 436 foram mencionados três derivados do ácido carboxílico que reagem de
maneira especialmente satisfatória com o reagente de Grignard: o cloreto de acila, o éster e
a nitrila.
a) Quais são os produtos destas reações?
b) Procure um argumento por que a nitrila somente reage uma vez com o reagente de
Grignard.
444
A. Isenmann
5.8
Princípios da Síntese Orgânica
Respostas dos exercícios de reações no grupo carbonila
1) A acidez destes compostos pode ser explicada com base das estabilidades dos seus
respectivos ânions, em solução aquosa. Um efeito entálpico (isto é, a diferença em
estabilidade entre o ácido e sua forma desprotonada, ∆H) é irrelevante, pois as reações são
praticamente atérmicas. Mas podemos esperar um efeito entrópico. Em geral um ácido fica
mais forte quando seu ânion (= base) fica mais estável. Realmente, o trifluoracetato se
destaca por ter uma carga negativa melhor distribuído do que o acetato, e isso aumenta
(ligeiramente) sua estabilidade. Também significa que não há necessidade de se criar uma
camada de solvente o que, por outro lado, é o caso do acetato, no qual a carga negativa fica
localizada no oxigênio e o grau de organização da água nas suas imediações é alto. Como
um alto grau de organização (∆S negativo) é desvantajoso para o andamento de uma
reação, então podemos esperar maior probabilidade para a formação do ânion
trifluoracetato do que do acetato dissolvido. Mais trifluoracetato implica mais prótons na
solução. O ácido mais forte é, portanto, o trifluoracético.
2) Indicado em parênteses: substância mãe do derivado. É recomendado identificar a
substância mãe através do número de oxidação (NOX) do carbono funcional:
NOX 1 = aldeído; NOX 2 = cetona; NOX 3 = ácido carboxílico e NOX 4 = ácido
carbônico.
OR
O
RO C OR
O
OR
R C
R CH2 C
OR´
Ortoéster
(ác. carboxílico)
C
R´
OCH3
Acetal
(cetona)
NH (R´)
Lactama
(ác. carboxílico)
O
OCH3
R C OR´
O
R C
Carbonato de
R C N
NH OH
tetraalquila
(Ortoéster do Ácido hidroxâmico Nitrila
(ác. carboxílico)
ác. carbônico) (ác. carboxílico)
NH NH2 R CH N NH2
Hidrazida
Hidrazona
(ác. carboxílico)
(aldeído)
OR´
C
O
O
H2N
Lactona
(ác. carboxílico)
C
O
NH2
R
Uréia
(ác. carbônico)
C
O
O
C
R´
Anidrido
(ác. carboxílico)
3) a) Ceteno, cloreto carboxílico, anidrido, azida, éster.
b) Amida, nitrila, carboxilatos de metais.
c)
O-
O
R C
+
X
plano
Nu
-
R C Nu
X
Complexo tetraédrico
intermediário
O
+
R C
X-
Nu
plano
445
A. Isenmann
Princípios da Síntese Orgânica
4) Os métodos padrões para a sua síntese, a partir do ácido carboxílico ou do carboxilato de
sódio, são:
• SOCl2 : reagente de cloração mais utilizado, por ser mais barato; menos reativo do
que PCl3 e PCl5.
• PCl3 : reatividade média; cada mol reage com até três mols de grupos carboxílicos.
• PCl5 : reagente de cloração mais poderoso do que PCl3, porém reage somente com
um equivalente de ácido carboxílico. Lembre-se que o sub-produto desta cloração, o
POCl3, não reage mais com o ácido carboxílico, mas reage mais duas vezes com o
carboxilato!
• Cloreto de oxalila, C2O2Cl2 um reagente cada vez mais procurado e também mais
em conta.
5) Em substituições nucleofílicas no grupo acila pode haver ataque de um nucleófilo que
não fornece um produto de substituição estável. Geralmente usa-se uma amina terciária
alifática ou piridina. Em ambos os casos, a nucleofilia é suficientemente alta para atacar o
carbono em cloretos de acila e anidridos; porém, o "produto" tem um nitrogênio
quaternário, isto é, uma carga positiva. Como isso é termodinamicamente desfavorável,
então a amina pode ser trocada com facilidade para um outro reagente nucleofílico que leva
a um produto sem carga e, portanto, mais estável.
6) O princípio da separação de Hinsberg é a alta reatividade das aminas frente o cloreto do
ácido sulfônico (que, por sua vez, é ainda mais reativo frente a nucleófilos do que o cloreto
do ácido carboxílico). Sendo assim, as aminas primária e secundária reagem
voluntariamente, enquanto a reação da amina terciária, devido a impedimentos estéricos, é
muito lenta, então pode ser separada da mistura na sua forma originária (ou em forma do
seu sal, NR3H+ Ar-SO3-. A mistura das sulfonamidas das aminas primária e secundária é
separada por filtração, já que ambas são substâncias sólidas e pouco solúveis. Finalmente,
um tratamento da mistura sólida com solução fortemente alcalina leva à desprotonação da
sulfonamida primária, que aumenta sua solubilidade e a leva para a fase aquosa. No
entanto, a sulfonamida secundária permanece insolúvel na água e pode então ser separada
por filtração.
7) a)
H2C
O CO
H C* O CO
H2C
+ 3 RO
-
O CO
Óleo; gordura
R = CH3 ou C2H5
H2C
OH
HC
OH
H2C
OH
Glicerina
O
+ 3
C OR
Ésteres dos ácidos graxos
(Biodiesel)
b) Essa reação é uma transesterificação, portanto pode ser catalisada, tanto por ácidos como
por bases. O ácido, no caso, ativa a parte proveniente do ácido graxo, protonando o
oxigênio duplamente ligado. Em consequência, a eletrofilia do carbono carboxílico
aumenta e pode ser atacado pelo nucleófilo (fraco) R-OH. O catalisador básico, por outro
lado, aumenta a reatividade do nucleófilo: mesmo sendo em pequena proporção, a base
446
A. Isenmann
Princípios da Síntese Orgânica
consegue desprotonar o álcool e o transforma em um alcóxido - um núcleófilo muito
melhor.
c) Embora a catálise ácida seja um método viável, a indústria ainda não a aplica em grande
escala, por vários motivos:
1) O ácido não promove a compatibilidade entre a gordura e a fase polar (onde se encontra
o reagente álcool, o produto glicerina, a umidade da matéria prima e os sais).
Consequência inevitável de uma mistura bifásica e incompatível é uma reação bastante
lenta.
2) É mais caro usar um ácido com certo caráter hidrofóbico (tal como ácido
toluenossulfônico), do que hidróxido ou alcóxido.
3) É difícil separar o catalisador ácido, do produto de biodiesel, para atender as exigências
das normas estabelecidas para um combustível de qualidade.
4) Restos do catalisador ácido (e também os ácidos graxos livres que sepre se formam em
menor grau) causam corrosão em elevada escala, tanto no equipamento da fábrica do
biodiesel, quanto nos motores de combustão dos consumidores.
Um catalisador básico, por outro lado, é
1) barato.
2) mais eficaz por promover a compatibilidade entre as fases do processo. Lembre-se que
os sais dos ácidos graxos formados em menor grau, são os nossos sabões clássicos, que
evidentemente compatibilizam gordura e a fase polar. Por outro lado, a presença de
sabões dificulta a purificação do biodiesel, já que a separação das fases após a reação é
dificultada, pela formação de uma emulsão (e espuma). Estes "poréns" mostram que o
processo alcalino não é livre de problemas operacionais, a serem melhorados
constantemente pelos engenheiros.
3) Devido à sua afinidade com a água, retira a umidade prejudicial da mistura reacional.
4) Os alcóxidos são catalisadores muito eficazes, acelerando o processo bastante e levando
a rendimentos muito altos que facilitam a purificação do produto final, o biodiesel.
8) a) Em algumas reações o efeito prótico do grupo -COOH pode representar um obstáculo:
quando o efeito tampão na mistura reacional diminui a reatividade do reagente.
Outro argumento: a solubilidade em ambiente apolar fica mais alta, ao transformar o ácido
carboxílico em um dos seus derivados (éster, ortoéster ou amida). Assim, podem esperar-se
velocidades mais altas com reagentes lipofílicos e rendimentos maiores, ao aplicar os
derivados hidrofobizados.
b) Um grupo protetor ideal
• deve ser de fácil introdução,
• resiste às condições da etapa reacional principal,
• direciona a reação principal e reprime os desvios reacionais e finalmente,
• deve ser de fácil remoção do substrato protegido, de maneira que nenhum outro
grupo funcional leve prejuízo.
c) Esterificação com t-butanol, tricloroetanol ou com álcool benzílico; ou usar o ortoéster.
447
A. Isenmann
Princípios da Síntese Orgânica
9)
Éster
ácidos (de Brønsted)
e bases
altas, em ambos os
casos
demorado em ambos
os casos
alto, especialmente
no caso da catálise
básica
possíveis
catalisadores
temperatura
tempo de reação
rendimento
Amida
ácidos (de Brønsted)
e bases
altas, em ambos os
casos
muito demorado
alto, especialmente
no caso da catálise
ácida
Nitrila
somente ácidos
fortes
altas
demorado
moderado
10) A reação de Ritter transforma nitrilas em amidas, usando um álcool terciário ou um
alqueno e um catalisador ácido de Lewis (reagentes muito semelhantes à alquilação de
Friedel-Crafts, ver p. 296). Em uma etapa prévia forma-se um carbocátion terciário,
relativamente estável, que ataca em seguida o nitrogênio da nitrila. Esse é um dos raros
exemplos onde há reação no heteroátomo - e não no C - de um derivado do ácido
carboxílico, formando um aduto estável. Neste caso forma-se, após desprotonação e
tautomeria, uma amida primária.
11) A substância mãe, o ceteno H2C=C=O, é produzido de maneira mais eficaz, na
"lâmpada de ceteno"; o material de partida é acetona que se desproporciona ao entrar em
contato com o fio metálico quente.
Um método aplicado a nível industrial é a pirólise de ácido acético.
Sínteses especiais e somente aplicadas em pequena escala são:
• Redução do brometo de α-bromo ácido carboxílico, R-CH2Br-COBr, usando zinco
em pó;
• Reação de benzila, Ph-CO-CO-Ph, com hidrazina, em seguida a redução, leva a um
composto diazo intermediário. Sua decomposição provoca um rearranjo (de Wolff),
fornecendo o ceteno de difenila.
12) a) A cicloadição [2+4] não precisa de reagentes com alta polaridade - pelo contrário,
alta polaridade em um dos participantes atrapalha o movimento sincronizado dos elétrons π
nessa reação eletrocíclica. Consequentemente, o ceteno entra nessa cicloadição só com seus
carbonos e deixa o oxigênio fora.
Cl2C C O
O
∆
Cl
Cl
b) Têm-se duas formas de cicloadição do dicloroceteno no ciclopentadieno que levam a
produtos cíclicos em proporções (probabilidades) iguais. Quando não há nenhuma
448
A. Isenmann
Princípios da Síntese Orgânica
estereosseletividade (isto é, ausência de matrizes, suportes ou complexantes quirais) a
síntese leva inevitavelmente a uma mistura racêmica de dois produtos. São estes:
σ
O
Cl
Cl
Cl
O
=
Cl
Cl
Cl
O
(Note que somente os carbonos em posição de cabeça de ponte são assimétricos.)
13) Podemos referir a degradação de Curtius, Hofmann, Lossen, Hunsdiecker e Kolbe, onde
as primeiras três percorrem estados de transição catiônicos (caso comum) e as últimas duas
são radicalares (muito raro, por outro lado, são rearranjos aniônicos, ver um único exemplo
na p. 429)
Curtius: Cloreto do ácido carboxílico + azida → isocianato → amina (com esqueleto de
uma unidade CH2 mais curto)
Hofmann: Amida + bromo (solução alcalina) → isocianato → amina (com esqueleto de
uma unidade CH2 mais curto)
Lossen: Ácido hidroxâmico + cloreto de acetila (H3C-COCl) → N-acetilamida →
isocianato → amina (com esqueleto de uma unidade CH2 mais curto)
Hunsdiecker: Carboxilato de prata + bromo → bromocarboxilato + AgBr → Brometo
orgânico (com esqueleto mais curto de um carbono) + CO2
Kolbe: Carboxilato (oxidação anódica) → descarboxilação → dimerização radicalar.
Resulta um hidrocarboneto com (2x-2) carbonos, sendo x = número de carbonos no
material de partida.
14) A posição α em cetonas é mais acessível para reações químicas, do que a posição α em
ácidos carboxílicos e derivados. Responsável é a tautomeria ceto-enólica que, no caso do
ácido carboxílico, é pouco desenvolvida, devido ao efeito +M do segundo oxigênio do
grupo carboxílico.
Um exemplo: A bromação em posição α é de fácil realização no caso da acetona (é só
aplicar uma solução aquosa alcalina de bromo, ver p. 424), enquanto a bromação do ácido
carboxílico (ou seu brometo) requer um catalisador eficiente, em forma de PBr3 (ver p.
394).
15) a) Éster do ácido α-bromocarboxílico + zinco em pó; na segunda etapa: acrescentar um
aldeído. Produto: Éster α,β -insaturado, com esqueleto carbônico ampliado pelo número de
carbonos do aldeído.
449
A. Isenmann
Princípios da Síntese Orgânica
b) Na primeira etapa, o acréscimo de zinco provoca uma inversão da polarização no
carbono α do derivado do éster.
c) Como toda reação organometálica (inclusive a famosa reação de Grignard), essa síntese
pode percorrer um período de indução. Neste momento o sistema está fora do seu equilíbrio
termodinâmico, ou seja, se encontra num estado supercrítico. Daí o experimentador pode
contar a qualquer momento com um começo súbito e veemente.
16)
E+
Base
Nu-
δ+
+ H+
- H+
O
R CH C
Nu-
+ H+
OH
- H+
R CH2
Base
OH
- H+
Ânion enolato
+ H+
R CH C
forma ceto
R CH C
R CH C
δ−
H
O
O
E+
- H+
OH
forma enol
+ H+
R CH2 C
C
Cátion oxônio-carbênio
17) a) A proteção do grupo C=O pode ser realizada por reação com álcool, de preferência
catalisada por ácidos. Especialmente a reação com o etilenoglicol provou-se útil, por ser
uma reação fácil e completa, para proteger aldeídos e cetonas. Forma-se um acetal cíclico,
por sua vez muito estável em ambiente básico. Igualmente inerte são os acetais frente
nucleófilos. A remoção do grupo protetor ocorre facilmente, ao tratar o acetal com ácidos
minerais diluídos (= o reverso da sua síntese; ver abaixo):
O
C
HO OH
+ H2C
Aldeído / cetona
CH2
Glicol
OH
[ H+ ]
O
C
OCH2 CH2OH
Hemiacetal / hemicetal
O
Acetal / cetal
b) O elemento estrutural mais marcante dos açúcares são os grupos hidroxilas situados em
carbonos vizinhos ("OHs vicinais"). Sendo essa a mesma estrutura do glicol, então deveria
ser possível a sua reação com um composto carbonílico. Na prática usa-se acetona para
proteger as hidroxilas vicinais. O composto fica estável durante as etapas feitas em
ambiente básico e pode ser abstraído em ambiente ácido.
450
A. Isenmann
Princípios da Síntese Orgânica
O
HC
OH
HC
OH
+
Unidade estrutural
típica em açúcares
H3C
[H+]
CH3
O
CH3
O
CH3
Açúcar protegido
Acetona
Uma etapa desta proteção foi apresentada, na síntese de vitamina C a partir da glicose (p.
228).
18) a) O método padrão é a partir de acetais/cetais:
OCH3
R CH2 CH
OCH3
Piridina, H3PO4
- CH3OH
α
β
R CH CH O CH3
Enoléter
(= viniléter)
b) Enoléteres mostram uma reatividade em posição α ao grupo carbonila que é superior à
da molécula de partida, por que têm mais semelhança com o enol do que com o
aldeído/cetona. Isto é vantajoso em todas as reações que visam uma reação em posição α e
onde o próprio grupo carbonila deve ficar fora do acontecimento.
c) As enaminas (forma tautomérica: base de Schiff), da unidade estrutural geral >C=C-N< ,
induzem o mesmo efeito ao composto carbonílico que os enoléteres. Elas deixam o carbono
α ainda mais rico em elétrons do que os enoléteres.
Aplicações: ver pp. 413; 496.
19) a) A reação de Cannizzaro é uma oxidação/redução. Isso ocorre a partir da mesma
substância (não da mesma molécula!), portanto pode ser classificada como
desproporcionamento, às vezes chamado de "reação auto-redox".
b) A reação não ocorre com facilidade: é uma reação difícil e lenta; enquanto houver
caminhos reacionais alternativos, o desproporcionamento de Cannizzaro fica em segunda
linha. A explicação é a transferência de hidreto a partir de uma molécula orgânica (quer
dizer, de origem C-H), o que sempre representa uma etapa difícil.
c) Além das desvantagens mencionadas no item b, é a necessidade de base forte como
catalisador: isso pode prejudicar outros grupos funcionais no substrato ou catalisar reações
paralelas. Algumas das restrições / insuficiências observadas na reação de Cannizzaro,
foram eliminadas com a técnica de Tichtchenko.
20) a) Todos os compostos organometálicos têm um carbono negativado (= carbaniônico,
ou melhor falado "carbanóide").
451
A. Isenmann
Princípios da Síntese Orgânica
b) A reatividade do composto organometálico geralmente aumenta junto à polarização da
ligação C-M. A medida para a polarização é a diferença em eletronegatividade (∆EN), dos
átomos vizinhos.
c)
NaCl
MgF2
B2O3
NH3
C-Li
C-Mg
C-Cu
2,8
2,7
1,5
0,9
1,5
1,3
0,6
∆EN
82
78
36
15
36
29
8
%
caráter
iônico
d) Segundo fator mais importante para a reatividade dos organometálicos é a
polarizabilidade dos participantes da ligação; provou-se mais reativa a ligação duro-mole
(quer dizer, de um átomo pequeno e pouco polarizável e outro átomo grande e altamente
polarizável), do que os contatos duro-duro ou mole-mole, que geralmente se destacom por
ter alta estabilidade.
Conclusão para compostos organometálicos: quando maior a ∆EN entre carbono e metal,
maior a camada eletrônica, então mais mole (polarizável) o carbono. Metais desta
característica são os mais eletropositivos, então Li, Na, K, Mg e Al. Ao mesmo tempo
podemos afirmar que, quanto menor o raio atômico do metal, menor também a
compatibilidade da ligação C-M, devido às diferenças em polarizabilidade. Assim, esperase reatividade especialmente alta de C-Li e C-Mg (ambas são combinações mole-duro).
Os dois fatos não apontam necessariamente na mesma direção: a ligação C-Li (∆EN alta; C
mole - Li duro) é mais reativa do que C-Ti (∆EN média; duro-duro) ou até C-Cu (∆EN
pequena; mole-mole). A reatividade aumenta novamente ao se ter uma ligação C-Hg ou CTl (∆EN pequena; C duro- metal mole).
21) Reações com reagente de Grignard:
Substrato
Reagente de Grignard
Produto
O
C N
C
H5C2 MgBr
C2H5
C2H5
O
H5C2 MgBr
C OH
OC2H5
C2H5
OCH3
H5C2 C OCH3
OCH3
H3CO C OCH3
H5C2 MgBr
OCH3
H2O
OCH3
Hidrólise
O
H5C2 C
OCH3
H5C2 MgBr
H3C
CH3
H7C3 MgBr
H7C3
C
OCH3
H C OCH3
OCH3
O
H
452
A. Isenmann
Princípios da Síntese Orgânica
CO2
CH2 MgBr
CH2 COOH
22)
a) A partir do cloreto do ácido carboxílico e do éster se obtém o mesmo produto - um
álcool terciário. São consumidos dois equivalentes do reagente organometálico (na prática
se usa um ligeiro excesso da quantidade calculada).
O
+ R´ MgX
R C
- MgXCl
Cl
O
R C
OR´´
+ R´ MgX
- R´´OMgX
O
R C
R´
+ R´ MgX
O
R C
R´
H2O
R´
O
R C
R´
+ R´ MgX
R C
R´
R´
O
R C
OH
R´
H2O
R´
OH
R C
R´
R´
A partir da nitrila se obtém uma cetona, enquanto somente um equivalente R-MgX foi
consumido.
b) A nitrila somente reage uma vez com o reagente de Grignard porque o primeiro produto
já possui uma carga negativa - o que repele o reagente carbaniônico. Assim, um segundo
ataque (que iria levar ao diânion) fica ausente. Na etapa da hidrólise final o nitrogênio é
substituído, conforme o esperado das iminas ou bases de Schiff (ver p. 412; 415).
R C N
O
R C
OR´´
+ R´ MgX
- MgXCl
+ R´ MgX
- R´´OMgX
O
R C
R´
+ R´ MgX
O
R C
R´
H2O
R´
O
R C
R´
+ R´ MgX
R´
R C
R´
R´
O
R C
OH
R´
H2O
OH
R C
R´
R´
453
A. Isenmann
6
Princípios da Síntese Orgânica
Reações de condensação de compostos com grupo
carbonila
A expressão “condensação” neste capítulo é usada exclusivamente para indicar a unificação
de dois compostos orgânicos, com formação de uma nova ligação carbono-carbono. Isto
pode ser efetuado pela aproximação de um carbono positivado a um carbono carbanóide de
outra molécula. O carbono positivado, como já deixa supor o título deste capítulo, é
disponibilizado pelo grupo carbonila.
6.1
A condensação de compostos carbonílicos com eles mesmos
("Autocondensação")
São três propriedades dos compostos carbonílicos que lhes conferem facilidade de fazer
autocondensação:
1) O carbono eletrofílico do grupo C=O;
2) O efeito retirador de elétrons que o grupo C=O exerce sobre o carbono em posição
α, elevando a acidez C-H nesta posição;
3) O equilíbrio ceto-enólico (ver p. 400) que igualmente afrouxa a ligação C-H na
posição α.
O primeiro argumento refere-se ao carbono aceitador de elétrons, ou seja, o receptor de um
carbono nucleofílico. O segundo e terceiro argumento falam do carbono nucleofílico que
disponibiliza os elétrons para a nova ligação C-C, a ser criada durante a autocondensação.
6.1.1
A autocondensação de aldeídos , “Condensação aldólica”
Esta reação funciona somente em ambientes de pH moderado; isto se reflete até na
natureza, sendo a condensação aldólica uma das estratégias principais para ampliar o
esqueleto carbônico em moléculas de seres vivos. Para ambos os caminhos, catálise básica
(mais comum) e catálise ácida, esta reação providencia exemplos importantes.
Condensação aldólica sob catálise básica
Sob catálise básica, conforme esperado da tautomeria apresentada no cap. 5.5.4, forma-se o
enolato, numa etapa que influencia a cinética da reação global. Como a primeira estrutura
mesomérica no esquema abaixo deixa esperar, o carbono β tem caráter carbanóide e então
pode atacar por um carbono positivado do grupo carbonila do segundo participante da
reação.
454
A. Isenmann
Princípios da Síntese Orgânica
O
O
H3C
C
H
H2C
+ H2O
H3C
CH CH2 C
-
H
O
C
H2C
H
C
H
O
H3C C
O-
O
+ OH-
H
O
+ H2O
H3C
- OH-
CH CH2 C
H
OH
"Aldol"
A dupla ligação do enolato é bastante rica em elétrons, portanto reage facilmente com o
carbono positivado do grupo carbonila, presente na outra molécula de aldeído. O produto é
um β-hidroxialdeído, ou simplesmente um “aldol”. Esta síntese é bastante fácil, mas a
reação é reversível.
Condensação aldólica sob catálise ácida
Sob catálise ácida a reação ocorre via enol. Desta vez o catalisador aumenta a eletrofilia do
grupo carbonila. Isto ocorre por protonação do oxigênio que faz com que o carbono fique
mais positivado (= eletrofílico). Assim, consegue-se atacar a dupla ligação do enol que por
sua vez representa um centro nucleofílico de qualidade média a fraca.
O
H3C
C
H
+ H+
OH
H3C
C
H
OH
- H+
H2C
C
H
OH
H3C
CH CH2 C
OH
Aldol protonado
H
O
- H2O
H3C
CH CH C
H
- H+
Crotonaldeído
A reação esta vez não pára no aldol, mas elimina prontamente água. Este comportamento é
mais pronunciado do que a desidratação de alcoóis secundários comuns, até mais fácil do
que em alcoóis terciários (que foi discutida no capítulo da síntese de alquenos, p. 144)!
Explicação: o produto desta desidratação tem alta estabilidade (= critério termodinâmico),
por ter duplas ligações em conjugação. A vantagem energética desta conjugação foi
455
A. Isenmann
Princípios da Síntese Orgânica
estimada em 17 kJ mol-1 para dienos (p. 131), então deve ser na mesma ordem de grandeza
para o aldeído α,β-insaturado formado aqui.
Uma técnica recente de polimerização que aproveita da condensação aldólica é descrita na
p. 842.
6.1.2
Autocondensação das cetonas
Comparando o carbono do grupo C=O em aldeídos e cetonas, pode-se perceber uma
eletrofilia inferior do carbono no caso da cetona. Isto se deve ao efeito +I do grupo alquila,
em comparação ao hidrogênio do aldeído. Além disso, um grupo alquila ocupa mais espaço
que H, isto é, funciona como blindagem do Cδ+ do grupo carbonila. Já estes fatores deixam
esperar menor facilidade para "condensações", no caso das cetonas.
CH3
C O
CH3
Efeito +I
Condensação sob catálise básica
A condensação básica de cetonas ocorre também através do carbanóide enolato, porém com
menos facilidade do que em aldeídos. O produto da condensação deve ser retirado
permanentemente da zona reativa. Somente assim, o equilíbrio pode ser deslocado para a
direita. Esta estratégia de deslocar reações equilibradas no sentido desejado foi chamada de
controle termodinâmico (ver p. 458) que em geral é possível quando todas as etapas da
cascata reacional são reversíveis. Outros exemplos neste capítulo, onde este controle é
aplicado com sucesso, são as descarboxilações (= remoção irreversível de CO2, p. 462) e as
reações onde se forma um β -dicarbonilânion (acetilacetonato, "acac", por exemplo, ver p.
510).
Realização experimental: a autocondensação da acetona ocorre especialmente bem sob
catálise com Ba(OH)2, um hidróxido de menor solubilidade:
O-
H3C
H3C
C CH2 +
O
C O
H3C
H3C
C CH2 C CH3
O
CH3
+ H2O
- OH-
OH
H3C
C CH2 C CH3
O
CH3
"Diacetonálcool"
A condensação é realizada de maneira mais adequada em um extrator Soxhlet:
456
A. Isenmann
Princípios da Síntese Orgânica
O produto, o diacetonálcool, tem um ponto de ebulição muito mais elevado (164 °C) do que
o material de partida, a acetona (Teb. = 56 °C). Consequentemente, só a acetona está sendo
evaporada e, ao completar o tubo do extrator, refluxada para o balão do fundo. Do
condensador a acetona goteja em cima do cartucho extrator que contém o catalisador
básico. A reação ocorre dentro do extrator onde se forma aproximadamente 1% do produto,
a cada ciclo. Logo que o líquido enche o extrator, o entorno puxa toda a mistura líquida
para baixo, no balão, onde agora o produto está se acumulando porque está fora do contato
com o catalisador. Cada um destes ciclos no Soxhlet leva 2 a 3 minutos e são necessários
cerca de 80 ciclos para obter um rendimento satisfatório.
Condensação sob catálise ácida
Sob catálise ácida, por outro lado, se obtém sem dificuldade o produto, já desidratado:
O
2 H3C
C CH3
[ H+ ]
- H2O
H3C
CH3
C
O
CH C
CH3
"Óxido de mesitila"
O mecanismo corresponde em todos os aspectos ao da condensação aldólica ácida.
Regioseletividade em cetonas assimétricas
Um problema inerente em cetonas assimétricas que não se dá com aldeídos, nem com os
ésteres, é o seguinte: a enolização do componente metilênico, R´-CO-R, pode ocorrer em
ambos os lados (distinguíveis) do grupo carbonila. Dentro de certos limites a enolização
pode ser direcionada pela escolha das condições reacionais. Controle cinético pode ser
457
A. Isenmann
Princípios da Síntese Orgânica
realizado quando a condensação é feita sob catálise básica. Neste caso o próton mais ácido
é abstraído mais rapidamente. Como se espera da estabilidade de carbânions, isto acontece
no carbono menos substituído. Por outro lado, em ambiente ácido a condensação da cetona
torna-se reversível e percorre estados de equilibração rápida entre ceto e enol. Neste caso
forma-se predominantemente o produto de condensação no lado mais ramificado (controle
termodinâmico). A autocondensação da etilmetilcetona (= butanona) sendo a cetona
assimétrica mais simples, deve ilustrar os diferentes produtos obtidos por este modo de
controle:
O
O
(base)
reação mais rápida
= controle cinético
O
(ácido)
OH O
OH
O
O
- H2O
O
O
- H2O
OH
Produto mais estável
= controle termodinâmico
O químico preparativo é bem aconselhado a planejar sua síntese de maneira que não seja
preciso considerar este tipo de controle regiosseletivo. É melhor bloqueiar seletivamente
um lado da cetona ou aplicar um dos métodos para direcionar condensações cruzadas (ver
p. 482).
6.1.3
Autocondensação de ésteres, Condensação de Claisen
Já a condensação de éteres não funciona sob catálise ácida. Além disso, é feita em ambiente
alcoólico (melhor ainda: sem solvente adicional), enquanto aldeídos e cetonas condensam
também na presença de água.
Deve-se usar um alcóxido como catalisador básico que seja do mesmo álcool que faz parte
do éster. Desta maneira o ataque nucleofílico no carbono do grupo acila (caminho 1) que
ocorre com maior frequência do que o ataque básico em posição α (caminho 2; reação
principal), não modifica o substrato - até mesmo se a transesterificação aconteça.
458
A. Isenmann
Princípios da Síntese Orgânica
O
H
CH2 C
2
H2C
OCH3
1
O-
O
C
H2 C
C
OCH3
+
CH3OH
OCH3
-
OCH3
H3C
+ H2C
C
H3C
C
OCH3
OCH3
O
H3 C
- CH3O-
O-
O
O
O
C CH2 C
OCH3
+ CH3O- H+
H3C
C
O
CH2 C
OCH3
OCH3
O
O
C
CH C
OCH3
OH3 C
O
C CH C
OCH3
Em comparação à condensação aldólica, a condensação de Claisen mostra algumas
particularidades:
1) O primeiro aduto dispõe de um abandonador apenas regular, sob as condições aplicadas:
CH3O-. Note-se que o meio reacional não é água, mas sim o álcool ou o próprio substrato, o
éster.
2) Forma-se um β -cetoéster, neste caso o acetoacetato de metila ("éster acetoacético"), que
por sua vez possui um grupo metileno duplamente ativado (ver também p. 503). A base
desprotona então quantitativamente o éster acetoacético e forma um sal que se precipita.
Esse sal é bastante estável, explicado pela formação de um complexo cíclico com o cátion
(de preferência duro), com a seguinte fórmula geral:
R
R CH2
OR´
O
Na
O
Nesta forma o β-cetoéster representa um ligante bidentado que confere estabilidade ao
complexo quelato 293. De fato, é uma remoção do produto do equilíbrio reacional que faz
com que a reação esteja deslocada para o lado do produto (= “controle termodinâmico”).
Ampliação da família das condensações de Claisen
Também é chamada “condensação de Claisen mista” onde um éster está condensado com
uma cetona, sob influência de uma base. No exemplo a seguir o alcóxido é uma base
293
Um parente do acetoacetato de metila é o ligante bastante usado para complexar íons de metais de
transição: "acac", ver logo abaixo.
459
A. Isenmann
Princípios da Síntese Orgânica
suficientemente forte para produzir uma pequena alíquota do enolato a partir da cetona
(lembre-se que o éster é menos α-ácido do que a cetona).
O
O
Ph
OEt
CH3
O
Ph
O
-
O
Ph
CH2
O
- H+
CH
Ph
+ H3C C
O
Ph
OEt
CH2
O
CH
H
pKa < 10
1,3-diona
= derivado da
acetilacetona, " acac"
Observação: todas as etapas são reversíveis. A formação do enolato estável no final pode
ser usado para "puxar" a reação para o lado direito, levando a um rendimento geralmente
satisfatório.
A base usada na condensação com ésteres não precisa ser necessariamente forte.
Realmente, a condensação pode até ocorrer “in vivo”: reações deste tipo se acham em
organismos animais, por exemplo, no ciclo do ácido cítrico e no ciclo dos ácidos graxos.
Muito eficazes se provaram aquelas bases que tenham poder nucleofílico especialmente
baixo. Muito citado neste sentido é DABCO (diazabiciclooctano), base que seria
apresentada no mecanismo da reação de Baylis-Hillman (ver p. 528).
Com a fosfina a condensação dos ésteres achou uma base poderosa, portanto pouco
seletiva. A condensação de Rauhut-Currier (p. 528) poderia ser apresentada, portanto, aqui
no capítulo das autocondensações, também.
Condensação de Dieckmann
Esta é uma forma especial da condensação de Claisen, uma condensação intramolecular
aplicada em ésteres de diácidos carboxílicos. Assim representa um método importante de
ciclização:
460
A. Isenmann
Princípios da Síntese Orgânica
O
C
COOC2H5
-
+
[ C2H5O Na ]
(CH2)n
- C2H5OH
COOC2H5
(CH2)n-1
O
CH C
OC2H5
Forma-se um composto cíclico 1,3-dicarbonilado. Este é somente o primeiro de mais 5
procedimentos descritos a partir da p. 471, para se obter anéis com número n de carbonos
diferentes. A condensação de Dieckmann fornece os melhores rendimentos com n = 4 – 6;
para valores de n > 6 deve-se aplicar o princípio da diluição de Ruggli-Ziegler (ver p. 470).
Tanto o éster quanto a nitrila são derivados do ácido carboxílico. Sob este aspecto deve-se
mencionar a autocondensação das nitrilas alifáticas, neste lugar. O catalisador é mais uma
vez uma base forte; a reação é conhecida como reação de Thorpe 294 :
R
C
N
R
C
NH
N
Base
R
C
N
R
NH2
R
C
N
R
enamina
imina
ou então a versão intramolecular (também conhecida como ciclização de Ziegler, ver p.
472):
C
C
N
Base
NH2
NH
N
C N
C N
A enamina que está em tautomeria com a imina, pode ser facilmente hidrolisada, formando
o grupo cetona.
A seguir sejam apresentadas duas reações consecutivas dos produtos da condensação de
Claisen que às vezes são planejadas, outras vezes ocorrem de maneira indesejada. Em
qualquer caso é importante que o químico preparativo saiba destas reações.
294
Revisão: J.P. Schaefer, J.J. Bloomfield, Organic Reactions 15 (1967) 1.
461
A. Isenmann
Princípios da Síntese Orgânica
“Quebra de ácido” do β-cetoéster
Como todas as etapas da condensação de Claisen são reversíveis, um β -cetoéster ou uma β dicetona também podem ser quebradas pelo caminho reverso - é só preciso deslocar o
equilíbrio da condensação para o lado esquerdo.
Atenção: a expressão "quebra de ácido" que se dá nesta reação, não se refere às condições
sob as quais a reação está sendo conduzida! Ela descreve o produto da quebra - que é o
ácido carboxílico.
Em meio fortemente alcalino se observa uma reação diferente daquela descrita acima:
O
H3C
C CH CH2 C6H5
H3C
COOH
+
COOEt
-
OH
CH CH2 C6H5
COOEt
+ OHHidrólise
- EtOH
H3C
COO-
+
CH2 CH2 C6H5
COOH
Essa reação representa então o reverso da condensação: ela libera o enolato e o composto
carbonílico – neste caso o acetato.
“Quebra de cetona” do β-cetoéster
Essa reação, típica para β-cetoésteres (β -cetoácidos) e 1,3-diésteres (1,3-diácidos), é
bastante aplicada no laboratório, porque faz parte da estratégia de "ativação", explicada
mais detalhadamente na p. 509.
O
H3C
C
O
CH
CH2
C
O
OC2H5
H 3C
+
1) H / H2O
C
CH2
CH2
2) ∆ : - CO2
A saponificação do β-cetoéster e sua descarboxilação térmica são feitos em ambiente
fracamente ácido ou básico. Resulta uma cetona. Essa reação é típica para compostos
duplamente ativados (ver p. 505), onde um dos grupos retiradores de elétrons é um ácido
carboxílico ou então pode ser convertido em tal, por hidrólise. Em condições levemente
ácidas pode-se formular um estado de transição cíclico, de 6 membros, o que explica a
facilidade do desprendimento do CO2.
462
A. Isenmann
Princípios da Síntese Orgânica
Lembre-se da regra: quando o estado de transição está estabilizado, a reação ocorre mais
facil e rápidamente! (literatura indicada: nota de rodapé No 71 na p. 147)
H
O
C
O
∆
C
- CO2
O
C C
C C
H
Enol
O
C
OH
Através de ligação de hidrogênio a transferência do H para o grupo carbonila em posição β
ocorre intramolecular.
Em condições levemente básicas o β-cetoácido também descarboxila, porém requer
temperaturas mais altas. Esta vez está percorrido um estado de transição de caráter
carbaniônico.
O R
R´´
C C
R´
O
C
O-
∆
- CO2
O
R´´
O
R
C C
R´
R´´
C C
R´
Enolato
O
R
H+
neutralizar
R
R´´
R´
Cetona
A "quebra de cetona" (= descarboxilação) sempre é concorrente da "quebra de ácido", ao se
trabalhar em ambiente aquoso alcalino. Se for o objetivo preparativo de promover a
"quebra de ácido" e reprimir a descarboxilação, o uso de alcóxido de sódio em álcool
anidro é aconselhável. Desta forma consegue-se o reverso da condensação de Claisen com
rendimento mais completo porque ocorre somente transesterificação, em vez de hidrólise
para o ácido carboxílico livre. Mesmo assim, a quebra de cetona não pode ser excluída
totalmente. Portanto, o valor preparativo da quebra de ácido do β-cetoéster é limitado;
muitas vezes se consegue o ácido com rendimento melhor via éster malônico 295 substituído
(ver p. 513).
Muito relacionada a esta quebra, é a reação de Japp-Klingemann que, por motivo didático,
está apresentada somente na p. 860.
Seguem dois exemplos onde a quebra de ácido representa a etapa central do mecanismo
(para a quebra de cetona se acham vários exemplos neste capítulo, ver p. 513 e as
referências dadas lá).
Síntese de Hüning
Esta sequência reacional, a partir do produto da acilação (ver p. 502) da cicloexanona, é
muito parecida com a síntese de Stetter apresentada logo a seguir. A primeira etapa é uma
"quebra de ácido", executada em ambiente fortemente básico.
295
Ambos os nomes estão divulgados na literatura portuguesa, malônico e malóico, representando (derivados
do ácido HOOC-CH2-COOH.
463
A. Isenmann
OH-
O
Princípios da Síntese Orgânica
O
C
R
1) OH2) H2O
O
R C
(CH2)5
COOH
Red.
R
COOH
A redução apresentada na última etapa pode ser por Clemmensen ou Wolff-Kishner (p. 609
e 610, respectivamente). Do ponto de vista do substrato acilante, R-C(=O)Cl, o resultado é
um aumento da cadeia carbônica por 6 carbonos, parecido à síntese de Stetter que segue
agora.
Síntese de Stetter
O substrato 1,3-cicloexadiona, pode ser facilmente obtido por redução da resorcina (= 1,3dihidroxibenzeno). Este composto contém um grupo metileno duplamente ativado (p. 505).
Nele pode ser efetuada uma alquilação (p. 510), seguida por uma "quebra de ácido" e uma
redução. O resultado final é um alargamento da cadeia carbônica por 6 unidades
metilênicas.
O
O
O
H
H
H
+ EtO Na
O
quantitativo
- EtOH
C2H5Br
H
C2H5
O
O
O
[ OH- ]
CH2
Clivagem
de ácido
CH2
COOH
CH3
H3C
Redução de
Clemmensen
CH2
COOH
Ácido caprílico
A terceira etapa, a "quebra de ácido" do anel, é a etapa mais difícil. Do seu bom
funcionamento depende, portanto, a viabilidade desta estratégia.
6.2
Autocondensações especiais no grupo carbonila
Em todos os exemplos, por enquanto, houve condensação entre o carbono do grupo
carbonila (componente carbonílico) e um carbono em posição α ao grupo carbonila
(componente metilênico). A seguir serão apresentadas estratégias para condensar
diretamente dois carbonos do grupo carbonila. Como a flexibilidade destas condensações é
bastante limitada, quer dizer, depende muito das demais propriedades estruturais, então
justifica chamá-las de "especiais".
464
A. Isenmann
6.2.1
Princípios da Síntese Orgânica
Condensação de benzoína e rearranjo do ácido benzílico
Com os aldeídos aromáticos se observa, na presença de íons cianeto, uma reação particular.
Trabalha-se em ambiente aquoso-alcoólico, ligeiramente básico (pH 8 – 9). Neste caso se
tem uma reação combinada que consiste da formação da cianidrina (p. 406) e logo em
seguida uma condensação:
CHO
+ CN-
O-
OH
C C N
C C N
H
H
CHO
O O-
O OH
C C
C C
CN H
H
Benzoína
Como o cianeto está abstraído do complexo na terceira etapa, então representa, segundo
adefinição clássica, um catalisador. Realmente, esta síntese requer somente 10 a 20% de
KCN, em relação ao aldeído. O produto se chama benzoína, e pertence à família das
aciloínas:
O OH
R´
C C
R
H
Aciloína
Este caminho é viável porque na segunda etapa se forma um carbânion que não só recebe
estabilização pelo grupo nitrila (retirador de elétrons), mas também pelo anel aromático
(estabilidade benzílica). Note-se que, através da adição do grupo ciano e desprotonação, a
polarização do carbono do benzaldeído se inverteu! Esta estratégia (p. 566) é de alto valor
preparativo e justifica o uso deste reagente, embora seja bastante tóxico. As condições
extraordinariamente brandas são mais uma vantagem desta síntese.
A benzoína/aciloína pode também ser obtida, a partir de ésteres. Só daí percorre um
mecanismo radicalar redutivo (ver p. 612).
Com a benzoina/aciloína temos um produto 1,2-bifuncional que em geral é de acesso difícil
(explicação: ver p. 566).
O seguinte derivado, por outro lado, não se consegue pela condensação de benzoína, já que
o aldeído, necessário para este fim, enoliza e finalmente fornece um outro produto (ver
questão No. 4, na p. 539).
465
A. Isenmann
Ph
CH2
CHO
Princípios da Síntese Orgânica
O OH
CN(H2O / EtOH / pH 8)
Ph
CH2 C CH CH2 Ph
Não funciona!
Existem três restrições gerais nesta condensação:
1) desproporcionamento de Cannizzaro é uma reação paralela inevitável,
2) A condensação da benzoína é reversível, portanto os rendimentos não são muito
altos (geralmente 50-60%)
3) O carbânion intermediário não pode ser apanhado por outros eletrófilos, portanto a
reação não serve para condensações mistas e direcionadas. A única exceção é a
reação viníloga com compostos carbonílicos α,β-insaturados (adição de Michael,
ver p. 519).
O desproporcionamento de Cannizzaro (p. 422) é uma reação bastante lenta, portanto
fornece pouco produto paralelo a esta síntese. Certamente mais grave, do ponto de vista
preparativo, é a terceira restrição. Mas existem duas variações que trazem melhoras,
exatamente neste ponto:
1) adição de cianeto de trimetilsilila, em vez de cianeto de potássio.
2) reação da cianidrina com etilviniléter (p. 499).
Por estas estratégias o intermediário fica bastante estabilizado; ele pode ser isolada,
purificado e desprotonado por uma base forte (pKa(C-H) ≈ 28), graças à alta robusteza do
grupo protetor contra bases e nucleófilos. Enfim, o carbânion produzido por estas vias pode
ser utilizado, não só em autocondensação, mas também em condensações cruzadas de
ampla gama (ver p. 499 e 501, respectivamente).
Rearranjo de benzoína
A partir da benzoína existem reações seguidas especiais que merecem ser discutidas:
A benzoína pode ser tratada com ácido nítrico, sendo oxidada a benzila. A benzila, por sua
vez, sofre um rearranjo para o ácido benzílico, sob catálise básica.
Benzoína
HNO3
Oxidação
O O
C6H5 C C C6H5
Benzila
Oo
O
C6H5 C C
C6H5 OH
O O-
OH-
δ+
C6H5 C C
C6H5
OH
OH
O
C6H5 C C
C6 H 5 O
Sal do ácido benzílico
466
A. Isenmann
6.2.2
Princípios da Síntese Orgânica
Síntese de pinacol e rearranjo de pinacol-pinacolona
O mecanismo desta condensação está relacionado à condensação de aciloína (ver p. 612).
Esta reação e similares, são induzidas pelo ataque “nucleofílico” de elétrons ao grupo
carbonila. Isto pode ocorrer de maneira direta, no cátodo de uma célula de eletrólise 296 ou,
mais comumente, fornecido por um metal não-nobre. Nos sistemas sódio metálico em NH3
líquido e Na/tolueno os “elétrons solvatados” se apresentam em bonita cor azul. A
diferença entre esta reação e as já discutidas é a dimerização de dois ânions-radicais, ao
invés de ataque de um carbânion ao carbono do grupo carbonila.
A condensação também funciona com outros metais (Al, Zn); um co-catalisador ácido e a
aplicação de ultra-som aceleram bastante essa síntese demorada 297. Finalmente seja
mencionada a possibilidade de se obter o pinacol por redução catódica (ambiente aquoso
ácido), em um processo semelhante à eletrólise de Kolbe (p. 663).
H3C
2
C O
+ Mg
(CH3)2C O
(CH3)2C O
H3C
Mg2+
2
Radical-ânion cetila
CH3
CH3
Dimerização
H3C
C O-
H3C
C O-
+
Mg
H
2+
H3C
C OH
H3C
C OH
CH3
Pinacol
CH3
Sob influência de ácidos o 1,2-diol se rearranja em uma cetona 298. Como mostrado no
exemplo a seguir é possível preparar cetonas de t-butila, por sua vez difícil de sintetizar nos
caminhos padrões (que seja, oxidação de alcoóis secundários, ver p. 644). A explicação é o
acesso difícil ao carbono em questão (o grupo t-butila é muito volumoso e pode então
bloquear o caminho para o oxidante); também consta-se uma inerente repelência
hidrofóbica, entre o substrato orgânico e o meio oxidante.
296
P.V.Gul´tyai, L.M.Korotaeva, Russian Chemical Bulletin 31 (1982) 156-160.
Z.Hongjun, L.Jitai, B.Yanjiang, L.Tongshuang, Pinacolization of aromatica aldeydes using
Zn/montmorillonite K10-ZnCl2 in aquous THF under ultrasound, Chemical Journal on Internet 5 (2003) 8.
298
Por causa da sua semelhança mecanística seja mencionado neste lugar um rearranjo que também se
observe em epóxidos. Sob influência de ácidos de Brønsted ou ácidos de Lewis o oxigênio é protonado e o
anel de três membros se abre (para as demais reações dos epóxidos, ver pp. 224 e 436). Intermediariamente
forma-se o carbocátion mais estável. Após a migração do hidreto resulta um aldeído ramificado em posição α:
297
O
H+
OH
OH
H
H
~H
O
CH C
H
467
A. Isenmann
Princípios da Síntese Orgânica
CH3
H3C
C OH
H3C
C OH
CH3
CH3
H+
H3C
C OH2
H3C
C OH
CH3
Pinacol
- H2O
H3C
C
H3C
C OH
CH3
CH3
CH3
CH3
H3C
C CH3
H3C
C CH3
H3C
C OH
H3C
C OH
Íon oxônio
(estável)
O
- H+
(CH3)3C
C
CH3
Pinacolona
Neste rearranjo forma-se intermediariamente um carbocátion. A sua estabilização
mesomérica faz com que esta reação ocorra com alta velocidade. Note-se que a segunda
estrutura mesomérica, um cátion oxônio, se destaca por ter octeto de elétrons em todos os
átomos, o que é especialmente favorável.
A facilidade de migração depende da qualidade do grupo como doador de elétrons (efeito
+I):
p-anisila > p-toluila > fenila > alquila terc. > alquila sec. > alquila prim. > H
Além disso, em dióis assimétricos 299, surge a questão de qual será o carbocátion formado
preferencialmente. Claro que o carbocátion mais estável ganha a preferência, ou seja, a
estabilidade decresce nas posições:
Benzílica, alílica > alquila terc. > alquila sec. > alquila prim. (ver p. 51).
Estes dois fatores, a qualidade do grupo em mudança e a estabilidade do carbocátion, estão
em concorrência.
Reação de McMurry
A rota mais conhecida para olefinas, a partir de cetonas é, sem dúvida, a reação de Wittig
(ver p. 769; outras rotas ver Figura 49 na p. 831). Todavia, a reação de McMurry representa
uma alternativa bem vinda quando se pretende produzir olefinas simétricas a partir de
cetonas 300. Nesta reação não há necessidade de compostos organofosforados que são todos
de alta toxicidade. É uma reação heterogênea usando um catalisador de contato de titânio
299
Relativamente novas são as técnicas que permitem condensações mistas do tipo pinacol. Um exemplo:
N.Kise, Y.Shiozawa, N.Ueda, Electroreductive cross pinacol coupling of aromatic ketones with aliphatic
ketones and aldeydes, Tetrahedron 63 (2007) 5415-26.
300
D. Lenoir, Synthesis 1989, 883.
468
A. Isenmann
Princípios da Síntese Orgânica
com baixo número de oxidação. O preparo do catalisador envolve uma redução de tricloreto
de titânio com LiAlH4 ou com potássio metálico. O sistema reacional tem propriedades
redutoras, portanto a reação de McMurry pode ser denominada de "olefinação redutiva".
O titânio, ainda mais que o fósforo, é um elemento com grande afinidade com o oxigênio.
Assim, a condensação de dois grupos carbonilas formando uma olefina ocorre diretamente,
sem ficar interrompida no estado de pinacol (ver p. 604). Até mesmo na presença de grupos
com alto impedimento espacial se obtêm alquenos simétricos com bons rendimentos. O que
exatamente acontece na superfície do catalisador não se sabe, mas trata-se de um processo
redutivo envolvendo a transferência indireta de elétrons desemparelhados (“SET”), do
metal (ou hidreto de metal) via titânio para o substrato orgânico.
OH OH
2
C O
TiCl3 / K
C C
THF
C C
Pinacol
Conhecemos o titânio como poderoso catalisador em sínteses orgânicas, desde os trabalhos
de K. Ziegler na década de 1950 (p. 169) que investigou a polimerização de alquenos. Mas
somente após as últimas duas décadas esse metal é reconhecido como altamente versátil,
também em outras áreas: além da reação apresentada aqui (que é um acoplamento e uma
redução), o Ti no ambiente do alisador de McMurry é valioso para transformar compostos
com grupo nitro secundário em cetonas (p. 535); também surgiu como estabilizador de
carbenos (p. 220) e na aplicação destes complexos em metilenações (p. 800), abrindo então
caminhos alternativos ao reagente de Wittig.
6.3
Excurso: Reações de ciclização
Este excurso se estende até a p. 482.
Sob o critério mecanístico uma separação deste item das demais condensações não se
justifica. Porém, as propriedades e utilidades dos compostos (macro-)cíclicos,
principalmente de sistemas não-conjugados contendo grupos carbonilas, e seu difícil acesso
por outras rotas, justifica a discussão destas sínteses e seus princípios fisico-químicos neste
lugar. As ciclizações de alquenos, cicloadição e alargamento de anéis insaturados já foram
referidos em outros lugares (p. 160; 210; 213; 223; 879;), além de outros métodos variados
(p. 102).
A estratégia geral é
X
Y
- XY
.
469
A. Isenmann
Princípios da Síntese Orgânica
Esta reação intramolecular (quer dizer, dentro da mesma molécula) sempre está em
concorrência com a reação intermolecular (= entre duas moléculas diferentes) que leva à
formação de dímeros, oligômeros e finalmente a polímeros de cadeia não-ramificada.
Na química preparativa, muitas destas ciclizações finalizam na classe das cetonas. Cetonas
cíclicas tem grande importância na indústria cosmética, onde são usadas como perfumes 301.
O princípio de diluição de Ruggli-Ziegler
Uma alta diluição é muitas vezes a única ferramenta para suprimir uma reação
intermolecular e então favorecer a reação intramolecular.
Discussão termodinâmica:
Na síntese de anéis de 3 e 4 membros o fator entrópico é favorável porque as duas partes
reativas da molécula estão “fixadas” em posições próximas, ou seja, o número de graus de
liberdade é pequeno. Especialmente no caso da anelação de 3 membros as colisões reativas
são frequentes – mais frequentes do que com 4 membros onde os centros reativos
naturalmente ficam mais distantes.
Isto implica, especialmente com 3 membros, uma reação rápida. O fator entálpico, por
outro lado, é extremamente desfavorável nestes anéis pequenos por causa da tensão, gerada
pelo desvio do ângulo preferido do carbono hibridizado sp³, de 109,5°. Bons rendimentos,
portanto, somente são esperados quando se consegue remover o produto da zona reacional
para que não ocorra a reação reversa. Como a maioria das condensações é de natureza
reversível, então a formação de ciclos com 3 e 4 membros por esta reação fica difícil ou até
impossível (as reações não-reversíveis que levam a ciclos pequenos com altos rendimentos,
ver nos caps. 3.5.1 e 3.5.3).
Em anéis maiores não há mais essa tensão interna que faz com que as reações
intermolecular e intramolecular mostrem os mesmos valores entálpicos. Porém, o termo
entrópico torna-se cada vez mais desfavorável, quer dizer, o número de graus de liberdade
aumenta e a probabilidade de encontrar dois grupos funcionais próximos fica muito
pequena.
Por isso, as ciclizações de anéis de tamanho médio, de 5 e 6 membros, são mais fáceis.
Nestes dois casos não há mais tensão e o aumento do grau de organização é moderado.
Discussão cinética:
As equações da velocidade são
v = k ⋅ [Substrato] , no caso da reação intramolecular e
301
A história das cetonas cíclicas, de anéis médios e grandes, começou com o isolamento de muscona e
zibetona, a partir de almíscar, um feromônio animal (Ruzicka 1926).
(CH2)7
CH2
H3 C
CH
O
HC
HC
O
(CH2)12
(CH2)7
Muscona
Zibetona
470
A. Isenmann
Princípios da Síntese Orgânica
2
v = k ⋅ [Substrato] , no caso da reação intermolecular.
Isto mostra claramente que uma alta diluição desfavorece a reação intermolecular (isto é, os
dois grupos funcionais provêm de diferentes moléculas) porque nesta condição um contato
reativo é menos provável. Por outro lado, os grupos funcionais, quando vêm da mesma
molécula, sempre têm a mesma probabilidade (embora da influência da viscosidade do
meio) de reagir - independente da concentração do reagente bifuncional. Isso é evidente
porque esses grupos não têm a liberdade de afastar-se livremente, um do outro. Em
conclusão, a reação intramolecular ganha sobre sua concorrente quando operar em alta
diluição. Especialmente os anéis maiores devem ser sintetizados, por este princípio, em alta
diluição.
As considerações termodinâmicas e cinéticas se encontram resumidas na tabela a seguir,
onde (+) significa favorável e (-) desfavorável. Fica evidente que a formação de ciclos de 4
membros é especialmente difícil: a maioria das reações iônicas - inclusive as condensações
apresentadas neste capítulo - falha e o único jeito de ciclizar e por intermediários reativos
(isto é, radicais) ou via reação eletrocíclica ([2+2], ver item 3.5.3). Muito pelo contrário, a
formação de ciclos com 5 e 6 membros é tão fácil que o rendimento quase sempre fica
acima do da reação bimolecular.
Tabela 23.
Considerações físico-químicas sobre a facilidade de formação de
anéis.
Tamanho do anel Critério cinético Critério termodinâmico
3
++
-4
5
++
+
6
+
++
7
+
+
6.3.1
Sínteses de cetonas cíclicas
Síntese de Ruzicka
Consiste na pirólise de diácidos carboxílicos, a partir dos seus sais de Ca ou Th (devido ao
preço elevado do tório, foram desenvolvidos rotas catalíticas com esse metal 302).
CH2 COOCH2 COO-
2+
Me
∆
CH2
C O
+ MeCO3
CH2
302
Tório, embora que não pareça, é um metal frequentemente usado na síntese. Além da síntese de anéis, é
aplicado com sucesso nos catalisadores do craqueamento do petróleo. Também faz parte do contato na síntese
dos commodities inorgânicos, tais como o ácido nítrico a partir da amônia e na síntese de ácido sulfúrico.
P. Patnaik. Handbook of Inorganic Chemical Compoundos. McGraw-Hill 2003, p. 931.
471
A. Isenmann
Princípios da Síntese Orgânica
Os ciclos de 5 e 6 membros são obtidos com bons rendimentos. O rendimento cai
constantemente, até o anel de 12 membros, acima do qual sobe novamente. Nesta última
subida para grandes anéis, porém, atinge-se apenas 5% de ciclização, no máximo.
Em alguns casos favoráveis pode-se forçar uma ciclização descarboxilante também a partir
do diácido carboxílico livre, sob catálise de CaO ou ThO2, ao aquecer com esses óxidos de
metais:
HOOC
∆
(CH2)4 COOH
O
Catalisador:
[CaO]
Ácido adípico
+ CO2 + H2O
Condensação de Dieckmann
Este método é aplicável para anéis de 5 a 8 membros (até ciclos de 4 membros são
possíveis, porém com rendimentos baixos). Ver também o esquema reacional na p. 460.
COOCH3
CH2 COOCH3
(CH3)3CO- K+
CH2 COOCH3
Xileno, ∆
CH
CH2
H+ / H2O
C O
∆ : - CO2
CH2
C O
CH2
Uma base forte e não nucleofílica, neste exemplo o butóxido terciário, induz a condensação
que claramente pertence à família de Claisen (p. 458). A descarboxilação foi apresentada
na p. 462 como "quebra de cetona".
Ciclização de α,ω-dinitrilas segundo Ziegler
Os compostos com anéis de 4 a 8 membros e também de 14 a 33 membros são obtidos com
rendimentos satisfatórios. No caso dos grandes anéis deve-se aplicar o princípio da
diluição. No entanto, para os anéis de tamanho médio, de 9 a 13 membros, os rendimentos
são muito baixos.
CH3
CH2 CN
CH2 CN
C6H5 N Li+
Éter
CH CN
CH2 C N
CH CN
CH2 C
H3O+
∆
N
- CO2
- NH3
CH2
C O
CH2
A condensação de Ziegler pode ser vista como versão intramolecular, da autocondensação
de nitrilas, conforme discutida na p. 459 (Reação de Thorpe).
472
A. Isenmann
Princípios da Síntese Orgânica
Ciclização de dicetenos segundo Blomquist
O
CH2 C
CH2 C
Cl
O
O
CH C O
+ 2 Et3N
- 2 HCl
CH C
C
CH C O
Cl
H+ / H2O
CH COOH
CH
C
O
CH
CH COOH
OH
CH2
C
O
CH2
∆
- CO2
CH2
C
O
Esta reação também não serve para ciclizar anéis de tamanho médio. Os rendimentos
máximos ficam com apenas 30%, mas a reação é de fácil execução.
Condensação de aciloína, segundo Prelog e Stoll
A condensação de aciloína é considerada sendo a melhor síntese de macrociclos 303. O
mecanismo desta reação foi discutido na p. 603.
O método clássico é o tratamento redutor de diésteres por metais de álcali, a temperaturas
elevadas: via diânion intermediário se consegue a ciclização. Nos últimos anos estabeleceuse o emprego de reagentes sililantes, por exemplo Me3SiCl, no sentido de grupos
ativadores, com os quais a variação da condensação de aciloína ganhou em universalidade.
Via compostos organosilanos é possível formar anéis de qualquer tamanho - até os de
tamanho médio. O papel do silício é:
• Apanhar o diânion, isto é, melhorando assim a eletrostática desvantajosa entre estes
grupes intermediários;
• A remoção do subproduto EtO-, cuja permanência poderia levar a produtos paralelos.
O procedimento serve então para sintetizar anéis de qualquer tamanho – inclusive os
médios. Na prática o diéster é gotejado sobre uma suspensão quente de sódio em xileno,
sob atmosfera inerte:
COOC2H5
COOC2H5
Na / Xileno
∆
C O
Zn
C O
CHOH
AcOH
CH2
Trata-se de uma reação na superfície do sódio. Acredita-se que os dois grupos de éster são
fixados frouxamente na superfície do mesmo grão de Na e desta forma têm uma chance
maior de reagir. Assim, um trabalho em alta diluição não é necessário. Pode-se obter
rendimentos ótimos (de até 90%), especialmente para anéis maiores.
303
J.J. Bloomfield, The acyloin condensation, Org.Reactions 23 (1976) 259.
473
A. Isenmann
Princípios da Síntese Orgânica
Reação de McMurry
A reação de McMurry (apresentada na p. 468) é um outro método de obtenção de ciclos
olefínicos a partir de dicetonas e dialdeídos. Ela ocorre na superfície de titânio finamente
dividido que foi produzido in situ a partir de TiCl3 e um redutor. O mecanismo percorre
ânions-radicais, em toda analogia à síntese de pinacol (p. 467) e à reação de Prelog-Stoll
(logo acima). Trata-se de um acoplamento redutivo para o 1,2-diol.
Uma variação é a reação com um ω-cetoéster 304 a partir do qual se forma o enoléter cíclico
que, após a quebra do éter, tautomeriza prontamente para a cetona:
R
C
O
O
OR´
C
R
TiCl3
Zn / Cu ou LiAlH4
OR´
C
R
C
O
C
C
Clivagem
+ 2 e-
Enoléter
Acilação de Friedel-Crafts intramolecular
Neste processo deve-se aplicar o princípio de diluição, sendo CS2 o solvente mais utilizado.
A reação-mãe e seu mecanismo foram apresentados na p. 297.
(CH2)n
n>7
C
O
(CH2)n
O
C
Cl
AlCl3
n<8
(CH2)n
O
C
Outras ciclizações
As ciclizações que se baseiam em reações eletrocíclicas são tratadas no cap. 3. A anelação
de Robinson e a condensação de Dieckmann são tratadas em outros lugares, neste mesmo
capítulo (ver pp. 526 e 460, respectivamente).
304
A letra grega ω indica que os dois grupos funcionais tenham distância máxima dentro da molécula do
substrato.
474
A. Isenmann
Princípios da Síntese Orgânica
Com 1,3-dibrometos obtêm-se ciclos de 3 membros, cuja síntese por outros métodos não é
fácil. As condições reacionais são, além disso, bastante brandas. Analisando o número de
oxidação dos carbonos (p. 582) em substrato e produto, essa reação se evidencia como
redução.
Ph
CH CH2 CH2
Br
Br
Zn / Cu
DMF
7-9°C
CH2
Ph
+ ZnBr2
CH
CH2
6.3.2 As regras de Baldwin - previsão de ciclizações por mecanismos
polares.
Como visto acima, existem muitas estratégias - a maioria destas bem mais antigas do que o
desenvolvimento das reações eletrocíclicas, apresentadas no cap. 3 - onde a ciclização é a
consequência de um ataque nucleofílico ou eletrofílico, intramolecular. Neste lugar cabe
então uma abordagem teórica sobre as ciclizações polares, uma coleção de regras
empíricas, estabelecidas por J. Baldwin, nos anos 70 do século passado. Em geral, essas
regras permitem estimativas sobre a facilidade cinética de reações que percorrem um estado
de transição cíclico. Note que na maioria das vezes essas reações são feitas de maneira
intramolecular, isto é, o produto é cíclico.
Vamos deixar claro que as regras de Baldwin não se aplicam a reações pericíclicas. São
pontos de partida e mecanismos totalmente diferentes - até que podemos afirmar que a
presença de elétrons π conjugados dificulta uma classificação segundo Baldwin, como
veremos abaixo (ver parte sobre ciclizações envolvendo o grupo enol).
Classificação e nomenclatura de Baldwin
A grande maioria das ciclizações ocorre via ataque nucleofílico. Devido à diversidade
destas reações, Baldwin procurou caracterizá-las por três parâmetros gerais:
1) O tamanho do menor anel em formação; indicado por um número arábico.
2) O posicionamento da ligação quebrada, em relação ao novo anel. Usam-se as
expressões exo, indicando que a ligação sacrificada mostrou para fora do novo anel;
endo, indicando que todos os átomos do grupo atacado fazem parte do novo anel.
3) A geometria daquele átomo que foi atacado pelo nucleófilo, é indicada por dig, trig
ou tet, dependendo se a sua coordenação antes da ciclização era digonal, trigonal ou
tetragonal, respectivamente. Em caso de carbono sendo o átomo atacado, então
trata-se de um centro sp (alquino, nitrila), sp² (alqueno, carbonila, imina) ou sp³
(carbono com quatro ligações simples), respectivamente.
Exemplos:
475
A. Isenmann
Princípios da Síntese Orgânica
Y
X-
X-
Y
5-exo-tet
CH2
6-endo-trig
X-
Y
6-exo-dig
Outros mecanismos do que o nucleofílico são raros:
Processo radicalar
(quebra homolítica)
3-exo-trig
4-endo-trig
Processo cationico
6-endo-trig
5-endo-trig
Note que muitos processos catiônicos não seguem rigorosamente as regras de Baldwin.
Mecanismos favoráveis e desfavoráveis
As preferências, ou seja, a rapidez e a facilidade com que as ciclizações ocorrem, são
resumidas na seguinte tabela, para os ciclos até 7 membros onde as regras de Baldwin
valem mais rigorosamente.
"+" significa favorável;
"-" desfavorável, ou seja, outras reações ganham preferência.
Esses critérios podem ser aplicados, especialmente quando fazer previsão do produto onde
há duas ou mais ciclizações concorrentes.
Tabela 24.
Membros no
novo ciclo
3
4
Probabilidade de ciclizações, organizada por tamanho do ciclo.
Posição do
Coordenação do átomo atacado
grupo atacado
exo/endo
tet
trig
dig
exo
+
+
endo
+
exo
+
+
endo
+
476
A. Isenmann
Princípios da Síntese Orgânica
5
+
+
+
exo
endo
exo
endo
exo
endo
6
7
+
- (!)
+
+
+
+
+
+
+
+
+
+
As fórmulas correspondentes a estes processos, a seguir organizadas por geometria do
centro atacado (Y):
Sistemas tetraédricos:
Y
-
X
X-
X-
Y
4-exo-tet
3-exo-tet
X-
Y
5-exo-tet
Y
6-exo-tet
todas as ciclizações exo são favoráveis.
Y
XY
7-exo-tet
Y
X5-endo-tet
X6-endo-tet
Essas duas são desfavoráveis.
Sistemas trigonais:
-
Y
X
3-exo-trig
X-
XY
4-exo-trig
X-
Y
5-exo-trig
X-
Y
6-exo-trig
Y
7-exo-trig
todas as ciclizações exo são favoráveis.
-
X
Y
Y
-
3-endo-trig
X
Y
4-endo-trig
X5-endo-trig
Y
X
-
6-endo-trig
Y
X7-endo-trig
Essas três são desfavoráveis.
477
A. Isenmann
Princípios da Síntese Orgânica
Sistemas digonais:
-
X
-
X
Y
X-
Y
4-exo-dig
3-exo-dig
Y
5-exo-dig
X-
X-
Y
6-exo-dig
Y
7-exo-dig
Essas são desfavoráveis.
-
X
Y
Y
-
X
3-endo-dig
Y
4-endo-dig
Y
X-
Y
-
X
5-endo-dig
X7-endo-dig
6-endo-dig
Todas as ciclizações endo são favoráveis.
Como já indicado em cima, as regras de Baldwin não devem ser usadas para fazer
afirmações absolutas, ou seja, falar em ciclizações permitidas e proibidas. Somente falam
se uma ciclização é provável ou não. As regras somente tem validade desde que o
nucleófilo pode atacar num ângulo, especificado pela geometria do centro atacado.
Em particular, isto são (afirmações feitos sobre o estado de transição):
• o ângulo ótimo de ataque de 180° 305, no caso do centro tet,
• 107° no centro trig (ver ângulo de Bürgi-Dunitz, p. 402)
• no centro dig é de 60°.
Estado de transição:
X-
Y
X-
Produto final:
Inversão de Walden
Ângulo X-C-Y = 180°
Y-
X
Bürgi-Dunitz
Y
X
109°
Ângulo X-C-Y = 107°
Y
XX
C Y
Ângulo X-C-Y = 60°
120°
C Y
Caso a geometria intramolecular inibir um posicionamento ótimo do nucleófilo, a
ciclização iria ficar ausente - mesmo se for provável pela reatividade química. Afinal,
305
Em analogia à inversão de Walden durante a SN2, ver p. 23.
478
A. Isenmann
Princípios da Síntese Orgânica
foram exatamente estes os critérios que levaram às conclusões generalizadas de Baldwin:
distorções de ângulos típicos ou alongamentos de ligações dentro do novo anel tornam certa
ciclização improvável. As posições e os tamanhos dos lobos dos MOs envolvidos no estado
de transição 306 devem garantir uma fácil e completa sobreposição.
Especialmente desfavoráveis são:
• 3-exo-dig, 4-exo-dig
• 3-endo-trig, 4-endo-trig e 5-endo-trig.
Atenção: as reações 5-endo-trig parecem bonitas no papel, mas não ocorrem (ver
exclamação na Tabela 24)!
Exemplo:
3
2
1
O-
4
5´
O
CH2
5
CH3
5-exo-trig
O
5-endo-trig
O
O
O-
O
CH3
O
O-
CH3
CH2
CH2
O
O
CH2
CH3
O
O
O
único produto
Um outro exemplo desta seletividade é o ataque intramolecular de um nitrogênio
nucleofílico num sistema Michael (ver cap. 6.6). Olhando nos MOs, podemos verificar que
cada uma das possibilidades tornou-se desfavorável à ciclização:
5-endo-trig
O
H
OMe
5-exo-trig
H
H
N
N
H
Lobo n mal alinhado com π*
O
OMe
Lobo n muito longe do π*
Reações endo-tet igualmente não levam à ciclização.
306
No nucleófilo: orbital n, com dois lobos parecidos ao orbital atómico p; no grupo atacado: um orbital
antiligante, seja σ* no caso do tet ou π* nos casos de trig e dig.
479
A. Isenmann
Princípios da Síntese Orgânica
Em alguns casos pode-se estimar até o tipo de catálise a ser aplicada para sucedir a
ciclização:
Molécula de partida:
O
Produto:
O
Base
O-
O
5-endo-trig
H+
O
H
H
HO
O
H
OH
OH
5-exo-trig
O
Ciclizações com o grupo enol
Os grupos enol ou enolato merecem uma consideração especial porque podem levar a
diferentes produtos cíclicos, quando aparecem como nucleófilo em uma molécula a ser
ciclizada. Lembre-se que este grupo é bidentado, quer dizer, pode reagir tanto no oxigênio
como no carbono negativado em posição α ao grupo carbonila. O primeiro caso, atacante
nucleofílico sendo o oxigênio, vamos denominar a seguir de "enolexo", o segundo caso,
ciclização com o carbono α, será chamado de "enolendo".
A maioria das ciclizações são condensações aldólicas ou alquilações intramoleculares do
grupo enol; os princípios e as regras que resultam para essas duas reações são semelhantes:
480
A. Isenmann
Princípios da Síntese Orgânica
Condensação aldólica intramolecular:
Alquilação intramolecular de enolatos:
(enolendo)-exo-tet
-
Y
O
(enolendo)-exo-trig
+ Y-
Y
-
O
O
Y-
O
Ciclos de 6 e 7 membros: favorável
Ciclos de 3 a 5 membros: desfavorável
(enolexo)-exo-tet
-
Y
O
(enolexo)-exo-trig
+ Y-
O
Y
O
O
Y-
Ciclos de 3 e 7 membros: favorável
Remarcável é que as reações 5-(enolendo)-exo-... são desfavoráveis, enquanto as
ciclizações para o anel maior, 6-(enolendo)-exo-... são favoráveis. A explicação dá mais
uma vez a posição dos lobos dos MOs envolvidos, a ser mostrado para uma reação de
alquilação:
Br
t-BuO-K+
O
Br
-
O
?
- Br-
O
> 70%
5-(enolexo)-exo-tet
Y
-
O
lobo longe do centro tet
O
0%
5-(enolendo)-exo-tet
Y
O-
O
lobo perto ao centro tet
Na ciclização com 6 membros, por outro lado, tanto a 6-(enolendo)-exo-tet como 6(enolexo)-exo-tet tornam-se favoráveis, devido à melhor aproximação ao centro tet. Mesmo
assim, observa-se uma forte preponderância do primeiro caminho:
481
A. Isenmann
Princípios da Síntese Orgânica
t-BuO-K+
O
Br
-
O
?
Br
- Br-
O
> 95%
6-(enolexo)-exo-tet
O
0%
6-(enolendo)-exo-tet
Para o grupo enol (ou enolato) resultam regras que são ligeiramente diferentes das
estabelecidas para atacantes simples (Tabela 24):
Tabela 25.
do ciclo.
Membros no
ciclo
3
4
5
6
7
Probabilidade de ciclizações no grupo enol, organizada por tamanho
Grupo atacado:
exo-tet
(enolendo)
(enolexo)
+
+
+
+
+
+
+
Grupo atacado:
exo-trig
(enolendo)
(enolexo)
+
+
+
+ (!)
+
+
+
Restrições nas regras de Baldwin
A teoria de Baldwin tem várias restrições:
•
As regras de Baldwin se limitam à cinética, mas falam nada sobre a estabilidade
termodinâmica dos produtos cíclicos (nem sobre a posição de equilíbrios, por sua vez
pura termodinâmica). Em caso de uma reação cineticamente favorecida, a sua reversa é
igualmente favorecida. Reações alternativas podem seguir ou até ocorrer em vez da
ciclização, se forem favorecidas pela termodinâmica.
•
Cátions ou moléculas contendo elementos do 3° período muitas vezes não seguem as
regras de Baldwin.
•
Ciclizações que evidentemente desobedecem as regras de Baldwin, provavelmente
são produto de uma reação intermolecular.
•
Centros nucleofílicos com hibridação sp² merecem uma consideração aparte porque
a forma do orbital não-ligante é diferente. Isto vale especialmente para os enolatos.
•
As regras não valem para ciclizações em sistemas de elétrons π conjugados. Lá
deveríamos confiar exclusivamente nas considerações feitos a base da simetria dos
orbitais moleculares (regras de Woodward-Hoffman, ver p. 210).
Fim do excurso “Reações de ciclização”.
6.4
Condensações mistas (= condensações cruzadas)
A reação dada na p. 459, a condensação de Claisen entre uma cetona e um éster, já
encaixou neste assunto "condensação mista", significa que a molécula-alvo foi feita a partir
482
A. Isenmann
Princípios da Síntese Orgânica
de duas espécies diferentes. Aquela síntese funcionava devido à maior acidez na posição α
da cetona.
Um dos objetivos principais da síntese orgânica é evitar a formação de uma mistura de
vários produtos.
No caso geral das condensações a partir de dois substratos diferentes tem-se o problema
inerente de que ambos os reagentes podem exercer as duas funções, tanto:
nucleófilo (também chamado componente metilênico ou pseudo-ácido), quanto
eletrófilo (também chamado componente carbonílico ou aceitador).
O objetivo principal em condensações de dois componentes diferentes é então direcionar as
reatividades, isto é, definir o papel do eletrófilo e do nucleófilo, para que se forme somente
um dos quatro produtos possíveis.
Aliás, essa necessidade não se deu nos exemplos discutidos na primeira parte deste
capítulo, onde os dois componentes eram simétricos e/ou idênticos.
A dificuldade inerente se vê nas reatividades como eletrófilo (do carbono do grupo C=O) e
nucleófilo (do carbono em posição α ao grupo C=O) de cada composto, qualidades que
geralmente não se opõem, mas aumentam juntas:
Enolizabilidade
Eletrofilia
Amida
És ter
Cetona
Anidrido
Cloreto de acila
Aldeído
Portanto, foram desenvolvidas as seguintes estratégias para se obter um único produto de
condensação definido:
1. Utilização de um pseudo-ácido, com baixa tendência de condensação consigo
mesmo, e/ou um aceitador sem α-H. Aceitadores típicos são aldeídos, cetonas e
ésteres aromáticos, ésteres do ácido carbônico, ácido fórmico e ácido oxálico. As
condensações que aproveitam desta estratégia são descritas nas pp. 484 a 490.
2. Ativação ou desativação da função carbonílica por reação com aminas, apresentada
nas páginas 491 a 495.
3. Ativação do pseudo-ácido pela estabilização da forma enólica, seja na forma do
enoléter ou da enamina. Esta estratégia está apresentada a partir da p. 495.
4. Com bases muito fortes se consegue desprotonar o pseudo-ácido quantitativamente,
em uma etapa preliminar. Esta estratégia está apresentada nas pp. 502 adiante.
5. Emprego de um pseudo-ácido duplamente ativado que enoliza e desprotona muito
mais fácil do que o componente carbonílico. Essas reações, comumente chamdas de
"Condensações de Knoevenagel", são apresentadas nas pp. 505 a 518.
483
A. Isenmann
Princípios da Síntese Orgânica
Uma estratégia utilizando intermediários organometálicos do Zn já foi apresentada com a
síntese de Reformatzky (ver p. 394), uma outra seria discutida na p. 533, usando
nitroalcanos que só podem fazer o papel do pseudo-ácido.
6.4.1 Condensações de pseudo-ácidos com baixa tendência de
autocondensação e aceitadores sem α-H
Condensação de Claisen-Schmidt
É uma estratégia para se condensar uma cetona com um aldeído de maneira controlada.
Controle significa que cetonas sempre mostram uma reatividade carbonílica inferior à dos
aldeídos, portanto representam em condensações sempre o componente metilênico. A
elevada atividade carbonílica dos aldeídos se deve ao menor efeito +I do H, em
comparação ao grupo alquila da cetona. A diferença, porém, não é grande - o que implica
que uma condensação de Claisen-Schmidt pode levar a uma mistura de diferentes produtos.
O seguinte exemplo é uma condensação de cetona com aldeído aromático. A falta de
hidrogênio em posição α faz com que este aldeído não tenha reatividade como componente
metilênico, impedindo assim uma autocondensação. O produto, portanto, é bem definido.
O-
O
H3C
C CH2
Ar
H
O
- H2O
C
+
H3 C
H3 C
C CH2 CH Ar
O
H+
OH
H3C
C CH2 CH Ar
O
C CH CH Ar
O
Cetona α,β− insaturada
O catalisador é uma base que, neste caso, não precisa ser muito forte. Basta desprotonar
uma pequena alíquota da cetona (compare também texto junto à tabela dos valores de pKa,
na p. 905). Tão logo o enolato se forme e o aldeído ou outra molécula de cetona disputam à
posição α do enolato, geralmente o aldeído ganha - especialmente quando aplicado em
excesso.
A reação de Cannizzaro (ver p. 422) é muito lenta, por isso não representa uma
concorrência séria nesta reação.
Para provocar uma condensação de sentido invertido poderíamos pensar em um aldeído
mais uma cetona sem α-H, tal como a benzofenona. Mas esta estratégia geralmente falha: a
cetona não reage como componente carbonílico e o produto misto não se forma. Em vez
deste se forma o condensado de dois aldeídos (autocondensação, ver p. 454).
Existe, porém, um jeito de forçar o aldeído no papel do componente metilênico,
desativando sua reatividade carbonílica. O método mais comum é a desativação por aminas
primárias. Forma-se na primeira etapa a base de Schiff que pode ser desprotonada na
segunda etapa usando uma base forte, por exemplo, LDA (= diisopropilamideto de lítio 307):
307
LDA, quando usado em pequenas quantidades, é feito "in situ", a partir de diisopropilamina (dissolvida em
THF, -50°C) e n-butilítio. O comércio atende com LDA dissolvido em solventes poares apróticos (THF,
éteres) ou apolares. É uma base forte (pKa = 36) que serve para desprotonar quantitativamente alcoóis e
484
A. Isenmann
R CH2 CHO
Princípios da Síntese Orgânica
R´ NH2
LDA
R CH2 CH NR´
-H
Base de Schiff
R CH
+
CH NR´
Li+
A tendência deste ânion de sofrer autocondensação é baixa. Assim, a reação com o grupo
carbonila de outra origem leva ao N-análogo do aldol que, se for desejado, pode ser
facilmente transformado no aldeído α,β-insaturado. Por arraste a vapor d´água, na presença
de ácido oxálico, esta etapa é quantitativa.
R
HC
R´
N
R
H
C
HC
C
O
N
O
H
C
R´
+
Li
Li+
R
- R´NH2
HC
C
C
O
- H2O
Síntese do ácido cinâmico segundo Perkin
Anidrido + benzaldeído → ácido cinâmico.
A condensação clássica de Perkin funciona com anidrido acético como componente
metilênico e benzaldeído como aceitador. A mistura reacional deve ser anidro; acetato de
sódio tem o papel da base, geralmente funcionando melhor quando estiverem presentes
traços de aminas terciárias. No produto de condensação a posição do grupo carboxílico, em
relação ao grupo fenila, é seletivamente trans, mesmo quando estiver presente um grupo
volumoso em posição 2 308.
O
C
H3C
C O C
O
CH3
O
+ AcO- AcOH
H3C
O
H3C
C O C CH2
O
-
C O
O
CH CH
Ph
H
O
O
C
H
H3C
Ph
OPh
O
C
O
CH
C
CH2
∆
- AcOH
Eliminação β
COOCH CH
Ph
Sal do ácido cinâmico
Um catalisador alternativo ao acetato é a piridina.
compostos carbonílicos com hidrogênio em posição α. Sua vantagem preparativa: essa base é de baixa
nucleofilia.
308
G. Solladié, Y.Pasturel-Jacopé, J.Maignan, Tetrahedron 59 (2003) 3315-21.
485
O
A. Isenmann
Princípios da Síntese Orgânica
O único produto desta síntese é o ácido cinâmico da configuração E. Isto muda
drasticamente quando o aldeído aromático é o aldeído salicílico. Sem parar na etapa do
derivado cinâmico, ocorre a condesação intramolecular, formando cumarina. Isto é somente
possível a partir do derivado cinâmico da configuração Z.
CHO
+ Ac2O
OH
Perkin
CH
OH
CH
COOH
- H2O
"Z"
O
O
Cumarina
A seguir uma descrição - um pouco mais detalhada - sobre o mecanismo desta
condensação. Deve-se atentar que a pesquisa mecanística fornece valiosas informações que
representam os fundamentos para a otimização da síntese, novos catalisadores e até novos
derivados.
Conforme a explicação nos clássicos livros didáticos, tem-se na primeira etapa uma adição
do enolato do anidrido acético, ao carbono eletrofílico do benzaldeído. A explicação do
funcionamento desta síntese foi, por dezenas de anos, que o acetato quando aplicado na
ausência de solvente prótico age como base forte, enquanto em soluções aquosas os sais de
ácidos carboxílicos são bases fracas 309, devido à camada de solvente em volta dos ânions
(ver p. 345).
Uma reinvestigação mecanística recente 310, porém, evidenciou pontos fracos nesta
explicação:
1. sob qualquer condição o anidrido acético é muito menos ácido (pKa(C-H) ≈ 20) do que
o ácido acético (pKa(O-H) = 4,5)
2. uma desprotonação do anidrido acético levará a uma espécie altamente instável que
logo sofrerá decomposição em carbeno e acetato.
Com base em ensaios com a base t-butóxido e DMF como solvente, um novo mecanismo
foi sugerido no qual o diacetato do benzaldeído tem um papel central:
309
310
D.D. Perrin, pKa Prediction for organic acids and bases, Chapman & Hall London 1981
S. Chandrasekhar, P. Karri, Tetrahedron Lett. 47 (2006) 2249-51.
486
A. Isenmann
Princípios da Síntese Orgânica
O
1)
OAc
Ph
H
+
Ph
Ac2O
OAc
pKa > 20
-
O
O
2)
O
Ph
O
Ph
O
O
-
O
OAc
H
O
pKa = 11
-
O
3)
O
Ph
O
O
OAc O
4)
Ph
O
OO
+
Ph
H
Ph
O
Ph
O
O
OAc
Ph
OAc
OAc
Ph - PhCHO
OPh
O-
OPh
O
5)
Ph
OAc O
O
Ph
COO-
- AcO
Ph
COO-
A reação na etapa 3 envolve o benzaldeído como catalisador (!), já que na etapa 5 estará
desprendido. Isto está de acordo com a observação que a condensação de Perkin não
funciona bem ao se aplicar um excesso de anidrido acético que, de fato, diminui a
concentração deste catalisador (= etapa 1).
O segundo argumento crítico de Chandrasekhar tornou-se até o ponto central no estudo
cinético de Kasprzyk 311, que postulou um ataque nucleofílico de aminas terciárias, no
carbono do anidrido acético, seguido pela substituição do acetato e a eliminação para o
ceteno. Somente depois, o acetato pode agir como nucleófilo e atacar o ceteno, fornecendo
o ânion do anidrido. Por este caminho conseguem-se explicar, tanto a dependência da
nucleofilia da amina usada, como a ocorrência de produtos paralelos (que são basicamente
acetona e CO2).
311
H. Kasprzyk, S. Kinastowski, React. Kinet. Catal. Lett. 77 (2002) 3-12.
487
A. Isenmann
Princípios da Síntese Orgânica
O
H3C
1)
O-
lento
C
H3C
OCOCH3
O
H3C
rápido
+ CH3COO-
C
C OCOCH3
NR3
R3N
2)
O
H3C
NR3
H2C
+ CH3COO-
C
NR3
+ CH3COO- HNR3+
C O
Ceteno intermediário
-
3) CH3COO + H2C
C O
rápido
OH2C
O
H2C
C OCOCH3
C OCOCH3
De qualquer maneira, a condensação de Perkin requer condições forçadas, isto é, altas
temperaturas (110 a 180 °C) e acima de 10 horas de reação. Tipicamente funciona sob
refluxo do anidrido acético que é reagente e solvente, ao mesmo tempo.
Síntese de pentaeritritol
O formaldeído encaixa no grupo dos aceitadores sem α-H. Ao mesmo tempo, destaca-se
por ser mais reativo, quer dizer, mais eletrofílico, do que outros aldeídos. Outra
condensação que leva essa reatividade em conta, será apresentada na p. 491.
Acetaldeído + formaldeído + base → Pentaeritritol + ácido fórmico
O
H3C
C
[ Base ]
+ H2C O
HO
CH2
O
CH2 C
H
HO
CH2
O
CH C
- HB
H
H
Pseudo-ácido
H2C
+ B-
O
Aceitador
OH
H2C
Base
O
O
(HO CH2)3C
C
H
+ HCHO
- HCOOH
Redox
CH2
HO CH2
C
CH2 OH
CH2
OH
Pentaeritritol
O único papel possível do formaldeído pode ser o de “aceitador” porque não tem α-H. Ele
deve ser aplicado em excesso. O acetaldeído é o pseudo-ácido (= componente metilênico) e
entra três vezes em ação - até esgotarem seus hidrogênios em posição α. Após essas três
condensações, finalmente, pode-se efetivar a lenta reação de Cannizzaro (ver p. 422).
O que facilita esta reação é o fato de que o formaldeído é melhor eletrófilo do que o
acetaldeído, visto que um grupo metila é considerado melhor fornecedor de elétrons do que
488
A. Isenmann
Princípios da Síntese Orgânica
um simples hidrogênio (ver p. 46). Se fosse contrário, uma condensação do acetaldeído
entre si, isto é, sem participação de algum formaldeído, seria a consequência.
Pela condensação entre formaldeído e aldeídos com esqueleto carbônico ramificado em
posição α se obtém 1,3-dióis com bons rendimentos:
R
H
C
R
δ+
R
2 H2C O
C
[ Base ]
CHO
R
CH2OH
+
HCOOH
CH2OH
Condensação com o éster do ácido fórmico
Esta reação também é chamada de “α-formilação”, mas não pode ser confundida com a
formilação de Vilsmeyer que foi discutida no contexto dos compostos aromáticos (p. 304).
Mais uma vez, um dos dois participantes não possui α-hidrogênios, então somente pode ter
o papel do aceitador.
O
Ph C CH3
O
+
H C
[ C 2H5O- Na+ ]
O
Ph
C
OC2H5
Acetofenona
= pseudo-ácido
OCH2 CH
OC2H5
Aceitador
O
Ph C
CH2 CH O
+
HOC2H5
Um olhar no produto desta síntese revela um grupo metileno que pode ser considerado
duplo ativado (no esquema em negrito; ver também p. 503), logo um pseudo-ácido
especialmente bom. Porém, este grupo não reage notavelmente neste sentido, acoplando
com mais um ou até dois aceitadores de formiato. Isto é surpreendente, mas a explicação
deve-se procurar no efeito do anel aromático que funciona como doador de elétrons, desta
forma diminui a acidez do composto 1,3-dicarbonila (ver também as restrições discutidas
na p. 490).
Condensação com ésteres do ácido oxálico
Ésteres do ácido oxálico, além de não terem um grupo metileno em posição α, são
aceitadores mais fortes do que um éster simples. Isto se deve à presença do segundo grupo
acila (= retirador de elétrons) em posição α. Assim, o carbono do grupo carboxila fica mais
positivado, quer dizer, mais eletrofílico do que um éster comum. Resumindo, o éster do
ácido oxálico sempre tem o papel do componente carbonílico, então é bastante apropriado
para condensações mistas. O componente metilênico pode ser escolhido quase sem
restrição: um aldeído, uma cetona ou outros ésteres – o produto sempre fica definido.
489
A. Isenmann
Princípios da Síntese Orgânica
O O
O
H3 C
CH2 C
+
C2H5O C C OC2H5
OC2H5
O O
+ C 2H5O- Na+
H3 C
- C 2H5OH
CH C C OC2H5
COOC2H5
Aceitador
Pseudo-ácido
120 °C
O
- CO
O
H5C2O C CH C OC2H5
CH3
Éster 2-metilmalônico
Ao aquecer o produto primário acima de 100 °C acontece uma descarbonilação, isto é, o
desprendimento de monóxido de carbono. Resulta um derivado do ácido malônico 2substituído (a degradação é especialmente fácil em compostos monossubstituídos). Assim,
esta estratégia serve para produzir β -oxoésteres e ésteres malônicos monossubstituídos.
Note que esses últimos não podem ser obtidos, de maneira satisfatória, via alquilação do
próprio éster malônico.
Condensação com o éster do ácido carbônico
Ésteres do ácido carbônico podem reagir com cetonas para β -oxoésteres e com nitrilas para
ésteres do ácido cianoacético.
Os ésteres do ácido carbônico são cancerígenos!
O
O
O
C δ+
α + C2H5O
OC2H5
+ C 2H5O- Na+
- C 2H5OH
Dietilcarbonato
H
C
OC2H5
O
Outras condensações mistas com ésteres
As condensações com os ésteres dos ácido fórmico, oxálico, benzóico e carbônico, podem
ser consideradas sendo variações mistas da reação de Claisen. Nenhum destes ésteres
dispõe de hidrogênios em posição α ao grupo carboxila. Então, outros ésteres ou cetonas
podem ser usados como componente pseudo-ácido (note que estes últimos não devem ser
muito reativos, para não correr o risco de entrarem em auto-condensação).
Mais sensível às condições reacionais (solvente, temperatura, base), mas também viável é a
maioria das condensações mistas entre ésteres e cetonas e entre ésteres e nitrilas. Nestes
casos o éster tem predileção para ser o componente carbonílico:
490
A. Isenmann
Princípios da Síntese Orgânica
R3
O
O
1
+
R C
3
R CH2
R1
C
R
4
C
CH
O
OR2
R4
C
+
R2OH
+
R2OH
O
β -dicetona
R3
O
1
+
R C
R3 CH2 C N
OR2
R1
C
CH
C
O
N
β-oxocarbonitrila
Restrições:
Em todos esses casos podem ser atingidos rendimentos de no máximo 50%, então deve-se
perguntar o que limita as condensações do tipo Claisen.
Todas elas têm que lidar com o problema que o produto da condensação é um composto
duplamente ativado (ver p. 503) e então têm um elevado potencial como pseudo-ácido.
Realmente, a sua desprotonação ocorre facil e rapidamente, daí mais um componente
carbonílico pode condensar nele.
Para reprimir esta reação paralela deve-se usar:
1) pequena quantidade de base (no exemplo acima, o etóxido);
2) evitar um excesso do componente carbonílico na mistura.
Além desta "supercarbonilação" existem outros perigos que podem prejudicar o
rendimento. Todos os compostos 1,3-dicarbonilas podem sofrer “quebra de ácido” e
“quebra de éster” (ver p. 462), especialmente durante um tratamento prolongado a
temperaturas altas e sob influência de uma base (que é essencial nas condensações de
Claisen).
6.4.2
Condensações mistas via desativação do componente carbonílico
Condensação de Mannich
A condensação de Mannich (1917) 312, segundo os pontos dados na introdução do item 6.4,
encaixa nas categorias 1, 2: ela usa um aceitador sem α-H e a reatividade do grupo
carbonila é modulada por aminas.
componente pseudo-ácido + formaldeído + amina (sec.) → componente β-aminometilado
Emquanto a maioria dos aldeídos, cetonas e éteres mostra somente baixa a moderada
reatividade nas condensações, o problema experimental no formaldeído é contrário: temos
312
F.F. Blicke, The Mannich Reaction. Org. Reactions 1 (1942) 303.
491
A. Isenmann
Princípios da Síntese Orgânica
que moderar sua reatividade excessiva 313. Mantendo sua alta reatividade poderia causar
condensações repetitivas, em compostos metilênicos que tenham mais de um α-H. Além
disso, o formaldeído, ao ser tratado com base, facilmente sofre desproporcionamento de
Cannizzaro ou até polimeriza formando polioximetileno, -[CH2-O]-x (algumas destas
possibilidades foram apresentadas na síntese do pentaeritritol, ver p. 488). Neste sentido a
reação de Mannich, também chamada de "aminoalquilação", representa uma técnica para
desativar o formaldeído, no sentido destas possíveis reações paralelas, usando uma amina.
A preferência deve ser dada a uma amina secundária porque com aminas primárias ou até
mesmo amônia abrem-se caminhos para produtos paralelos: nestes casos podem reagir
todos os hidrogênios disponíveis no N, como será mostrado mais a frente (ver método de
Wittig, p. 495; também ver questão No. 7, na p. 539), com o formaldeído 314. Isso levará
inevitavelmente a uma mistura de produtos. A amina, por ser aplicada em forma do seu sal,
cloreto de amônio R2NH2+ Cl-, totalmente solúvel no ambiente aquoso, solvente
exclusivamente usado na condensação de Mannich. A amina geralmente é aplicada em
quantidade 1 a 1, em relação ao formaldeído (embora tenha as características de catalisador,
ver mais adiante).
Ao mesmo tempo, a reação do formaldeído com a amina representa uma ativação do
formaldeído, no sentido de eletrófilo que na seguida pode ser atacado por um componente
metilênico (ver também p. 415). Isto se vê claramente na sua segunda forma mesomérica,
no esquema a seguir, que mostra o produto intermediário, o cátion imínio 315.
Mesmo que todos os reagentes estiverem presentes desde o início, observe-se a adição da
amina ao formaldeído sendo a reação mais rápida de todas:
H3C
NH
H3C
+ H2C
+ H+
O
- H2O
H 3C
H3C
N CH2
H 3C
N CH2
H3C
Cátion imínio
A presença de um componente nucleofílico (= C-H pseudo-ácido = metilênico), sob as
condições ligeiramente ácidas, leva à condensação com o cátion imínio. A forma reativa do
componente metilênico, nesta situação, é certamente o enol (ver p. 400). O produto desta
etapa é a “Base de Mannich”. O tratamento térmico desta base, finalmente, leva à
eliminação da amina. Esta recebe assim o caráter de catalisador, porque entrou e saiu da
mesma forma, da cadeia reacional. Resulta então uma cetona α,β -insaturada. Do ponto de
vista do componente metilênico houve uma ampliação da cadeia carbônica, por uma
unidade metilênica, =CH2.
313
Em caso de outros aldeídos usados nesta condensação, ver H.E.Zaugg, Recent Synthetic Methods
Involving Intermolecular α-Amidoalkylation at Carbon, Synthesis 1970, 49.
314
Em vez da amina secundária pode se aplicar, também com sucesso, uma amida, R-CO-NHR´. Neste caso
se obtém, como espécie eletrofílica, um sal de acilimíniometileno, e a reação com o composto pseudo-ácido é
chamado de "α-amidometilação", em vez de α-aminometilação.
315
Os sais de imínio (também referidos como sais de imônio) representam síntons bastante versáteis. Ver H.
Böhme, H.G. Viehe, Iminium Salts Organic Chemistry, Adv. Org.Chem. 9 (1976) 253.
492
A. Isenmann
O
Princípios da Síntese Orgânica
OH
[H +]
R C CH3
δ−
R C CH2
O H
CH3
R C CH CH2 N
H3C
NH +
H3C
H2C
+ H+
O
- H2O
Ciclo catalítico
H3C
δ+
N CH2
H3C
"Imônio"
CH3
Base de Mannich
Eliminação β
- (CH3)2NH
∆
+ Base
O
R C CH CH2
Se for desejado, a base de Mannich também pode ser isolada; ela é relativamente estável e
pode ser armazenada, especialmente em forma do seu hidrocloreto. Uma redução adequada
transforma o grupo carbonila em um álcool; através desta rota tem-se acesso ao β aminoálcool (muitos derivados desta classe são substâncias bioativas, de importância
farmacêutica).
Como visto no esquema acima, a amina pode ser elimidada da base de Mannich - o que
leva ao produto com unidade O=C-C=CH2, e daí um composto carbonílico α,β -insaturado.
Em analogia aos demais representantes desta unidade estrutural (acroleína, acrilatos, ácido
acrílico e metacrílico,...) esse produto é bastante reativo e versátil. Pode ser usado em
adições de Michael (p. 523), ciclizações de Diels-Alder (p. 252) e polimerizações (p. 77 e p.
837), por exemplo. Claro que uma substância tão reativa é susceptível a uma série de
reações paralelas e indesejadas. Então podemos ver a base de Mannich como grupo protetor
para o agrupamento acrílico que somente deve ser liberado na hora da sua utilização.
A partir de cetonas cíclicas a reação de Mannich fornece uma cetona cíclica com grupo
metileno exocíclico em posição α ao grupo carbonila. Este composto dificilmente se
consegue por outros métodos (a reação de Wittig com Ph3P=CH2 , apresentada na p. 769,
não funciona bem na vizinhança de grupos carbonilas). Note-se que uma quaternização do
nitrogênio – iodeto de metila ou sulfato de dimetila são os reagentes mais usados para este
fim - facilita bastante a última etapa, isto é, a liberação da amina e da espécie insaturada (a
rigor, essa etapa representa uma variação da eliminação de Hofmann, ver p. 142):
493
A. Isenmann
Princípios da Síntese Orgânica
NR2
O
R´
CH2
H
O
H2C=O
HNR2
R´
Mannich
CH2
H
O
O MeNR2
NR2
MeI
R´
Quaternização
R´
CH2
H
CH2
- MeNR2
Grupo metileno
exocíclico
= muito reativo
Caso o componente metilênico disponha de mais de um grupo metilênico reativo (que é o
caso, por exemplo, na acetona ou na cicloexanona), este deve ser empregado em excesso de
4 para 1, em relação ao formaldeído, para reprimir a ocorrência de duplas bases de
Mannich. No exemplo dado acima este perigo foi impedido pela presença do grupo R´ que
representa um bloqueio do grupo metileno em posição α.
Todavia, caso o componente metilênico seja uma cetona, temos que lidar com o problema
da regiosseletividade. Numa cetona assimétrica, com grupo metila de um lado e um grupo
alquila mais comprido do outro lado do grupo carbonila, a condensação de Mannich sob
catálise ácida acontece de preferência no grupo alquila mais comprido, devido ao efeito
indutivo +I do grupo alquílico (p. 46). Portanto, se obtém geralmente a base de Mannich
mais ramificada (em analogia à condensação aldólica, p. 455).
O componente metilênico (= pseudo-ácido) nesta reação não se restringe a cetonas, mas
pode ser largamente variado. Entre os compostos com ligação C-H ácida, os mais usados
são aldeídos, compostos nitro alifáticos, HCN e acetileno. A única condição para se obter a
base de Mannich com rendimento satisfatório é a nucleofilia da amina que tem que ser mais
alta do que a do composto C-H-ácido (no exemplo acima, isto é o enol 316). Caso contrário,
o formaldeído poderia diretamente reagir com o pseudo-ácido, numa condensação aldólica
comum. Por este motivo não se consegue a base de Mannich a partir do éster malônico
pseudo-ácido forte, pKa ≈ 13,3, ver tabela na p. 505), formaldeído e dialquilamina.
Além dos pseudo-ácidos mencionados, podem também ser usados compostos aromáticos
que são ativados no sentido da substituição eletrofílica (p. 322), tais como fenóis ou certos
compostos heteroaromáticos. Um exemplo seja a síntese de gramina, um alcalóide natural,
com efeito tóxico já em baixas concentrações (ver também questão No. 16, na p. 543), por
aminoalquilação do indol:
CH2 N(CH3)2
+ HCHO
N
Indol
H
+ HN(CH3)2
- H2O
N
H
Gramina
316
Esta condição, ao mesmo tempo, explica porque a execução da condensação de Mannich em ambiente
básico não é favorável, pois o enolato (ao contrário do enol) é um concorrente potente à amina primária, em
termos de nucleofilia.
494
A. Isenmann
Princípios da Síntese Orgânica
O grande valor da reação de Mannich é o fato de que todas as etapas reacionais ocorrem
sob condições bastante brandas. É possível obter-se a "base de Mannich", até sob condições
in vivo, ou seja, em células vivas. De fato essa reação faz parte de ciclos biológicos mais
complexos cuja finalidade é a ampliação do esqueleto carbônico.
A síntese de Mannich geralmente se restringe ao formaldeído sendo componente
carbonílico. Raramente esta síntese é ampliada a outros aldeídos, um dos quais pode ser o
aldeído succínico. Com este aldeído se abre um caminho interessante que leva a uma
variedade de alcalóides (ver questão No. 8, na p. 539).
Desativação da função carbonílica por aminas primárias
Esta estratégia para condensações mistas foi desenvolvida por Wittig (1964):
Aldeído + amina primária + base forte + cetona → aldeído α,β-insaturado
A transformação do aldeído em uma base de Schiff (ver p. 411) reduz drasticamente a sua
capacidade como componente carbonílico porque o par de elétrons não-ligantes no N
exerce um forte efeito +M. Este efeito doador de elétrons percebe, em primeira linha, o seu
vizinho direto, o carbono carbonílico. Entretanto, a base de Schiff é bastante apropriada
para uma condensação cruzada. Em uma etapa preliminar a base de Schiff é desprotonada
pelo diisopropilamideto de lítio, LDA, uma base bastante forte (ver nota de rodapé 307, na
p. 484). Assim, ela torna-se ativada na sua função como componente metilênico. Esta
primeira etapa da reação exige a ausência de substâncias próticas (especialmente água e
alcoóis). Caso contrário se perderá a base LDA, pela desprotonação preferencial destes,
fornecendo hidróxido ou alcóxidos que, por sua vez, não têm basicidade suficiente para
ativar a base de Schiff.
O papel de componente carbonílico pode então ser preenchido por qualquer aldeído ou
cetona, mesmo sendo um composto de baixa eletrofilia (ver p. 456). No exemplo a seguir a
condensação do acetaldeído com benzofenona que, certamente, é um componente de baixa
reatividade no carbono carbonílico:
H3C
CHO
+
H2N
H3C
+ LDA
CH N
- LDAH+
Base de Schiff
1) (C6H5)2CO
2) H2O
- LiOH
6.4.3
C6H5
C6H5
C
OH
CH2 CH N
+ H+ / H2O
- C6H11NH2
H2C
CH N
Componente metilênico
(C6H5)2C
CH CHO
- H2O
Ativação do pseudo-ácido pela estabilização da forma enólica
495
A. Isenmann
Princípios da Síntese Orgânica
Não só as condensações carbonílicas mistas, mas também as alquilações e acilações, da
posição α de um composto com grupo carbonila enolizável, sofrem das desvantagens
alegadas na p. 482, podendo levar a uma mistura feia de produtos. Na maioria das vezes a
autocondensação do composto com grupo carbonila é dominante, enquanto
alquilação/acilação planejada ficam em segundo plano.
Conforme elucidado no tratado da condensação aldólica (p. 454), a etapa central das
condensações é o ataque de um carbono nucleofílico em posição α ao grupo carbonila, a
um carbono positivado qualquer. Para se tornar nucleofílico, este carbono α tem duas
possibilidades: ou em forma do enol ou em forma do enolato. A primeira possibilidade é o
assunto deste parágrafo, a segunda será apresentada na p. 502.
Vamos então falar sobre estratégias de reprimir reações paralelas por estabilização da forma
enólica, de maneira controlada (item 3 da lista na p. 482):
•
enol estabilizado na forma de enaminas,
•
enol estabilizado na forma de enoléteres,
•
enol estabilizado na forma de éter silílico (mecanismo na p. 500).
•
enol estabilizado na forma de viniléster do ácido fosfórico, uma variação da
ativação de cetonas α-halogenados segundo Perkov, ver p. 793.
•
enol estabilizado em forma de alil e vinilsilanos, conforme descrito no cap. 10.9.5,
na p. 820.
Em todos esses casos introduz-se um grupo funcional auxiliar que estabiliza o enol,
deixando assim o carbono C=C-X mais nucleofílico. Como o grupo X tem apenas caráter
temporário, podemos chamá-lo de ativador da posição α, ou então protetor do grupo
carbonila!
Alquilação e acilação de cetonas, via enamina
Por reação da cetona com uma amina secundária se obtém a enamina cuja dupla ligação é
bastante rica em elétrons. Pirolidina e piperidina são as aminas mais usadas para este fim.
A segunda etapa da reação abaixo é uma acilação: o carbono acílico do segundo reagente
pode facilmente atacar no carbono 2 da enamina (segunda etapa do esquema abaixo).
Forma-se intermediariamente o íon imônio que, em seguida, hidrolisa facilmente,
restaurando a função da cetona.
O
+ H+
- H2O
+
N
O
N
N
O
C
δ−
R
- Cl-
O
C
Cl
O
C
H2O
R
R
H
Pirolidina
Enamina
Ion imônio
β-Dicetona
496
A. Isenmann
Princípios da Síntese Orgânica
Como a primeira etapa é uma reação em equilíbrio, a água liberada durante a formação da
enamina deve ser retirada continuamente, por destilação azeotrópica.
O ponto fraco nesta estratégia é, mais uma vez, a insuficiência em regiosseletividade, no
caso de cetona assimétrica, onde duas enaminas diferentes podem ser formadas na primeira
etapa. Um melhoramento, mas não uma solução perfeita com respeito à regiosseletividade,
promete a estabilização em forma de enoléteres (p. 498) que podem ser produzidos mais
seletivamente.
Ao contrário da acilação de enaminas (= acoplamento com R-COCl), que é uma reação
limpa, a alquilação (= acoplamento com R-Cl) é mais complicada e afetada com
rendimentos baixos e produtos paralelos. Os motivos serão discutidos mais detalhadamente
na p. 527.
Condensação carbonílica cruzada, entre enamina e um aceitador com grupo carbonila
Enoléteres e enaminas (ver pp. 403 e 413, respectivamente) representam as formas clássicas
de se fixar cetonas e aldeídos na sua forma enólica. Lembre-se que a atividade metilênica
(= nucleofílica) depende essencialmente da disponibilidade da forma enólica, portanto estes
derivados são bastante ativados para condensações com aceitadores. O exemplo dado a
seguir usufrui da ativação pela morfolina, uma amina secundária, especialmente adequada
devido à alta solubilidade no ambiente 317.
Atenção: aminas primárias não servem para esta finalidade. Em vez da enamina, forma-se
uma base de Schiff cuja acidez em posição α fica até inferior à da cetona de partida (p.
495)!
A etapa da formação da enamina é catalisada por ácidos; a posição do equilíbrio pode ser
manipulada por destilação azeotrópica da água liberada nesta etapa.
O
O
O
O
N
H
Morfolina
N
N
CHO
+ H2O
[H+]
Hidrólis e
O
317
P.W.Hickmott, Enamines, Tetrahedron 38 (1982) 1975.
497
A. Isenmann
Princípios da Síntese Orgânica
Esta síntese, porém, não está livre de caminhos paralelos. A penúltima estrutura do
esquema acima ilustra o perigo que se corre neste tipo de ativação feita numa cetona: o
outro lado do grupo carbonila pode também reagir com um aceitador. Devido à conjugação
das duplas-ligações sua estabilidade é mais alta, então sua reatividade fica abaixo da
enamina da partida, mas ainda acima de uma cetona não-aminada. Portanto, deve-se
acrescentar o componente aceitador em quantidade equimolar à enamina.
Outras reações a partir das enaminas são apresentadas nas pp. 411, 413 e 496.
Alquilação e acilação de cetonas, via enoléteres
A ativação do composto carbonílico via enoléter é muitas vezes o caminho preferido, sendo
melhor do que o caminho via enamina, devido a problemas em regiosseletividade. O
método da vinilação pelo cloreto de trialquilsilila (p. 500), aliás, tem que lidar com essas
dificuldades, também.
Interessante é que os enoléteres podem ser obtidos por caminhos diferentes, quer dizer,
existem alternativas ao esquema padrão, reação da cetona com um hetroátomo nucleofílico,
O, N ou Si. Estes caminhos serão apresentados a seguir. Alternativa mais recente é o
enoléter estabilizado pelo fósforo (ver método de Perkow, p. 792).
a) Eliminação de álcool a partir de acetais (método clássico, ver também p. 405):
OR´
R CH2 CH
OR´
0,5% H3PO4
- R´OH
OR´
R CH CH
Enoléter
Acetal
b) Adição nucleofílica de um alcóxido em acetileno
A adição de alcóxidos em derivados do acetileno fornece o enolato. O álcool que serve
como solvente nesta reação fornece o próton necessário para formar o enoléter 318:
R
R C C
R + R´O
-
OR´
C
C
R
+ R´OH
- R´O-
OR´
R CH C
Enoléter
R
O enoléter (ver também p. 405), uma vez formado, é bastante estável em meio básico até
neutro. Ele representa um excelente componente metilênico em reações de condensação,
tanto com outros compostos carbonílicos, quanto com haletos de alquila e haletos de acila.
318
Do ponto de vista do álcool esta reação é uma vinilação no oxigênio.
498
A. Isenmann
Princípios da Síntese Orgânica
Para que servem enoléteres?
A utilidade sintética dos enoléteres, também chamados de viniléteres, não se restringe à
ativação do componente metilênico em condensações. Eles também servem como grupos
protetores para alcoóis, como pode ser mostrado com o reagente γ-pirano.
O 3,4-dihidro-2H-pirano (nome trivial: γ-pirano) pode ser obtido a partir de
tetraidrofurfurilálcool (barato), a 300 °C no contato catalítico de Al2O3. Ele é um enoléter
que pode ser alcoxilado com bons rendimentos:
δδ+
+
HO
R
[H+]
R
O
O
álcool protegido
O
γ-pirano
Esta reação ocorre com bastante facilidade; assim, o γ-pirano representa um bom grupo
protetor para o grupo hidroxila, seja em alcoóis primários, secundários ou fenólicos,
conforme discutido nos acetais (p. 402). O protetor chamado de "THP", de tetraidropiranil,
é estável em ambiente básico e pode ser removido por ácidos diluídos, sob condições
brandas.
Enoléter também servem como estabilizador da cianidrina (ver p. 406).
Na abordagem da condensação de aciloína (p. 465) foi mencionado um método que permite
ampliar esta síntese às condensações cruzadas: a conversão com etilviniléter, levando a um
intermediário que resiste aos ataques de nucleófilos e bases (Stork, 1971). Lembre-se que a
água é um nucleófilo, também; é responsável pela fácil degradação da cianidrina por
hidrólise, sob as condições da condensação de benzoína. Justamente isso pode ser evitado,
pelo uso de um enoléter.
OH
R CH
CN
Cianidrina
(qualquer)
OC2H5
CH CH2
OC2H5
O CH CH3
R CH
LDA
CN
Cianidrina protegida
(estabilidade do Acetal)
OC2H5
OC2H5
O CH CH3 R´X
R C
O CH CH3
CN
R C
R´
CN
H2O / H+
O
R C
R´
Pode-se afirmar que o grupo -OH foi protegido, de maneira eficaz, pela reação com o
enoléter. Assim é possível isolar e purificar o intermediário que demonstra a estabilidade
típica dos acetais (p. 403).
499
A. Isenmann
Princípios da Síntese Orgânica
Através da conversão com etilviniléter podem ser protegidas cianidrinas de uma grande
variedade de aldeídos (exceção: o formaldeído) 319. De maneira segura elas podem ser
desprotonadas por meio de base forte e submetidas ao ataque de carbono eletrofílico. Além
da alquilação mostrada no esquema, pode-se aplicar também uma acilação ou uma
condensação com composto carbonílico comum, pois o carbânion é um excelente
nucleófilo. Em última etapa o acetal e a cianidrina podem ser hidrolisados sob condições
brandas.
Caso se pretenda a condensação com o carbânion proveniente do formaldeído, deve-se dar
preferência a um outro derivado da cianidrina, à dietilaminoacetonitrila,
(C2H5)2N-CH2-C≡N (observe a diferença ao reagente de Mannich, p. 491!)
Condensação com sililenoléter
Um método mais recente para atribuir o papel metilênico a um composto carbonílico é a
formação do sililenoléter.
A reação do enol (ou enolato) com cloreto de trimetilsilila, reagente sililante mais utilizado,
acontece exclusivamente no oxigênio. A formação do sililenoléter é quantitativa. A
explicação está relacionada, em analogia ao dito na p. 512, às polarizabilidades dos
reagentes: o silício duro procura o oxigênio duro que, por sua vez, é menos polarizável do
que o carbono em posição α - quer no enol ou no enolato. Assim, a reação com agente
sililante é uma boa alternativa à enamina (ver pp. 413 e 491), para a estabilização da forma
enólica:
Si(CH3)3
O
O
OH
+ (CH3)3Si
Cl
C C
Enol
Enoléter de trimetilsilila
Os mais promissores são os trimetilsililenoléteres porque reagem com alta
regioseletividade. Além disso, o cloreto de trimetilsilila é o reagente sililante mais barato e
abundante (ver síntese industrial, p. 807). O grupo silila pode ser removido sob condições
brandas, na presença de ácidos de Lewis (neste exemplo: TiCl4).
O
δ+
Si(CH3)3
O
C6H5 CH2 C
+ δ−
H
+ TiCl4
- (CH3)3SiCl
O
C6H5 CH2 CH
TiCl3
O
H2O
OH
O
C6H5 CH2 CH
68%
319
S. Hünig, Chimia 36 (1982) 1.
500
A. Isenmann
Princípios da Síntese Orgânica
Estabilização de cinaidrinas por silano
Em analogia ao método de estabilizar uma cianidrina via enoléter (p. 499), o mesmo efeito
estabilizante se consegue por silanos, de maneira elegante. Assim, temos um método
poderoso para produzir um carbono nucleofílico (carbânion), numa posição que, quando
pensamos ao aldeído de partida, é contra sua naturesa: é o carbono do grupo carbonila!
Com esta proteção e ativação se abrem caminhos para se obter compostos 1,2-bifuncionais
por reação de condensação mista (as dificuldades inerentes em acessar este tipo de produto
é explicado em maiores detalhes na p. 566; a estratégia aplicada é "Umpolung", isto é,
transformar o δ+C do intermediário, em δ-C).
O reagente, Me3Si-CN, se obtém facilmente pela reação do cloreto de trimetilsilila e KCN
em solvente polar aprótico, o N-metilpirrolidona. Esse cianeto de trimetilsilila reage com
benzaldeído ou outro aldeído, fornecendo a α-trimetilsililoxinitrila, com bom rendimento.
A acidez C-H desta cianidrina protegida não é muito alta (pKa em torno de 28), mas pode
ser desprotonada quantitativamente com bases fortes (LDA, dietilamida de lítio, n-BuLi,
etc.). Resulta um carbânion que pode ser submetido à alquilação, em geral, a qualquer
condensação com carbono eletrofílico.
O
CN
(CH3)3SiCN
Ph
H
Ph
CH
OSi(CH3)3
α-trimetilsililoxinitrila
(cianidrina protegida)
Benzaldeído
CN
[LiN(C2H5)2]
Ph
- H+
C
OSi(CH3)3
Desprotonação quantitativa
O
CN R1
δ+
R1
R2
Ph
C
C
(CH3)3SiO
O
R2
+ H2O
- CN- (CH3)3SiOH
Ph
O
R1
C
C
R2
OH
Benzoína
ou
δ+
+ R
Br
- Br-
CN
Ph
C
(CH3)3SiO
R
1) Ph2N.2HF
2) OH-
O
Ph
R
Desoxibenzoína
(Cetona)
Após ter efetuada a etapa da condensação, o composto organo-silício é hidrolisado e o
produto carbonílico liberado. Como o sílício estabelece uma ligação extremamente estável
com flúor, então a presença de fluoretos facilita a abstração do grupo protetor de
trimetilsilila (ver também pp. 822 e 829).
Por esta estratégia se consegue:
• benzoínas, a partir dos reagentes benzaldeído e uma cetona,
• cetonas, quando o reagente eletrofílico for um haleto, R-X.
501
A. Isenmann
•
Princípios da Síntese Orgânica
1,2-dicetonas, Ar-CO-CO-R, derivados da benzila, quando o reagente eletrofílico
for um cloreto de ácido, R-COX.
6.4.4 Condensação cruzada após desprotonação quantitativa do pseudoácido.
A estratégia no. 4, da lista dada na introdução à condensação cruzada (p. 482), prevê a
desprotonação quantitativa e rápida, do componente metilênico que tem certa acidez C-H
em posição α ao grupo carbonila. Somente no enolato que resulta da reação quantitativa é
possível efetuar uma condensação de maneira segura. A alquilação e acilação de compostos
carbonílicos simples são exemplos perfeitos onde esta estratégia é aplicada com sucesso.
Essencial é o uso de uma base bastante forte, para desprotonar o enolato de maneira
quantitativa. Neste sentido, podem ser consideradas muito fortes as seguintes bases:
•
•
•
•
NaNH2 em xileno,
NaH em benzeno,
t-butóxido em t-butanol,
LDA em THF.
Todas essas bases são fortes o suficiente, para desprotonar uma cetona simples, cujo pKa
certamente não é abaixo de 20 (ver tabela na p. 505 e no anexo 2 deste livro).
Em uma condensação por esta estratégia podemos esperar rendimentos mais altos (de até
70%; pela estratégia de Wittig, p. 495, até mais de 70%), do que nas condensações feitas
pelos outros caminhos descritos na p. 464.
Exemplo:
Alquilação de uma cetona simétrica (se fosse assimétrica, certamente resultará uma mistura
de produtos, devido à formação de dois enolatos diferentes).
O
O
NH2-
O
H2C CH CH2 Cl
- Cl-
H
CH2 CH CH2
55%
Existe uma série de dificuldades preparativas que reprimem o rendimento desta síntese:
1. Certa quantidade de autocondensação da cetona é inevitável.
2. Além da monoalquilação também ocorre a dialquilação do enolato.
3. A baixa regioseletividade, inerente na cetona, impede o emprego de cetonas
assimétricas - apesar do que foi alegado na p. 457, sobre o controle termodinâmico
e/ou cinético da enolização em cetonas assimétricas.
502
A. Isenmann
Princípios da Síntese Orgânica
São conhecidas também as alquilações de aldeídos, ésteres e nitrilas, sendo afetadas com as
mesmas dificuldades e reações paralelas.
Condensação de Stobbe
Também a condensação de Stobbe funciona somente após a desprotonação quantitativa do
componente metilênico (pseudo-ácido). O pseudo-ácido é o éster succínico (= composto
1,4-dicarboxila). Ao contrário dos compostos homólogos, 1,3-dicarboxilas (p. 505), este
não tem um grupo metileno duplamente ativado, consequentemente sua acidez C-H
continua bastante baixa. Portanto, deve-se usar uma base muito forte (por exemplo, NaH,
Ph3C- Na+ ou LDA).
O procedimento requer o uso de quantidades estequiométricas de todos os participantes,
inclusive a base. O componente 1, que tem o papel do pseudo-ácido é tratado na primeira
etapa com a base, acrescentando a base aos poucos mas não muito devagar, para formar o
enolato. Sob controle de um indicador especial se consegue acertar o ponto de equivalência
desta reação ácido-base.
Na segunda etapa o composto 2 cujo papel é componente carbonílico é gotejado nesta
mistura até completar a reação.
Uma falta de base nesta condensação provocará a auto-condensação do composto 1. Isto é,
uma parte funciona como componente metilênico, outra parte como componente
carbonílico. Um excesso de base provocará uma condensação do composto 2 com outras
moléculas da mesma espécie, pela mesma razão. No caso ideal se aplica a base (no
exemplo a seguir o t-butóxido) em quantidades adequadas para retirar um próton em cada
éster succínico; uma segunda desprotonação do ânion não acontece por ser muito menos
ácido.
Essa síntese funciona porque a base carbanóide que resulta da primeira etapa, não tem força
e velocidade suficiente para desprotonar o composto 2.
COOEt
CH2
O
COOEt
(CH3)3CO- K+
CH2
R2C O
CH
CH2
COOEt
Éster succínico
(componente metilênico)
Base
EtO C
C
1)
+
CR2 H / H2O
CH2
COO-
O
CR2
CH2
2)
O
C
OEt
COOH
C
CR2
O-
H2C
1)
COOEt
H
CH CR2
COOEt
Enolato
quantitativo
COOEt
COOH
O
Lactona
R2C
CH CH2 COOH
Ácido β,γ-insaturado
∆
- CO2
2)
COOH
R2C
C
CH3
503
A. Isenmann
Princípios da Síntese Orgânica
Na última etapa deste exemplo surge a dúvida de qual dos grupos carboxilas seria
descarboxilado. Porém, observa-se apenas um produto – aquele onde se forma o ácido β,γinsaturado. Isto se deve ao fato de que no estado de transição da descarboxilação o grupo
abandonador está em conjugação com a dupla ligação. Assim, o CO2 pode sair com mais
facilidade, provavelmente em forma de radical, que neste caso é estabilizado pela posição
alílica.
Síntese de Reformatzky
Esta síntese (ver também p. 394) encaixa neste capítulo porque numa etapa preliminar o
éster α-bromado é transformado quantitativamente numa espécie carbanóide. Somente
depois é acrescentado o componente aceitador, geralmente um aldeído.
Éster α-bromado + composto carbonílico + zinco → β -hidroxiéster
Br
CH2 COOCH3
+
Zn
[ Br Zn
CH2 COOCH3 ]
ZnBr + CH2 COOCH3
quantitativo!
O-
O
R C
+ CH2 COOCH3
H
R C CH2 COOCH3
H
[ H3O+ ]
OH
R C CH2 COOH
H
- H2O
R CH CH COOH
O produto primário, um β-hidroxiéster, pode ser facilmente desidratado e hidrolisado para o
ácido α,β -insaturado.
O papel do zinco nesta reação foi a inversão da polarização no substrato α-bromado 320.
Este termo se refere à polarização do carbono α que tem δ+ no substrato e δ- após a reação
com o metal. A reação com o Zn é bastante rápida. Isso é decissivo para o sucesso desta
síntese porque uma metalação, se fosse lenta, levaria ao produto de autocondensação do
éster α-bromado.
Outras sínteses que aproveitam do efeito de "Umpolung" são quase todas as reações
organometálicas – especialmente aquelas com metais muito eletropositivos (preparo do
reagente de Grignard: R-X + Mg → R-MgX). Uma outra síntese onde ocorre a inversão
δ+
C → δ-C, onde o carbono do grupo carbonila torna-se um sínton carbaniônico é a
“condensação de aciloína”, apresentada na p. 465.
320
D. Seebach, Chimia 34 (1980) 185.
504
A. Isenmann
Princípios da Síntese Orgânica
6.5
Pseudo-ácidos com grupo metileno duplamente ativado
6.5.1
As particularidades dos compostos 1,3-dicarbonílicos
Uma reatividade especial se espera de compostos com grupo metileno que tem dois
vizinhos retiradores de elétrons. Nestes casos a acidez C-H fica elevada, pois a carga
negativa da base conjugada pode ser distribuída, através de mesomeria, entre os dois
vizinhos.
Especialmente as condensações, também conhecidas como condensações de Knoevenagel
(p. 513), aproveitam da elevada acidez destes compostos e de sua reatividade especial. Para
uma vista geral dos compostos orgânicos e sua acidez, recorra o anexo 2 deste livro, p. 905.
Os seguintes três efeitos contribuem para a sua reatividade:
1. O grupo metileno em posição α (= posição 2) torna-se muito mais ácido ainda
quando estiver presente um segundo retirador de elétrons em posição 3. O seu valor
pKa cai em aproximadamente 10 unidades, em comparação com o composto com
apenas um retirador de elétrons (geralmente um grupo carbonila).
2. A troca entre a forma ceto e enólica é acelerada.
3. A forma enólica está sendo favorecida, podendo até prevalecer no equilíbrio com a
forma ceto.
A seguinte tabela deve esclarecer os pontos 1 e 2 321. Os compostos simples ativados são
comparados com duplos ativados, em termos das suas constantes de acidez e de velocidades
de desprotonação.
EWG C
H
k1
k -1
EWG C
+ H+
Ka =
k1
k -1
EWG = Electron Withdrawing Group
= Retirador de eétrons
Tabela 26.
Acidez e velocidade prótica de compostos com um e dois grupos
retiradores de elétrons (EWG).
Composto
pKa
k1 (s-1)
H2C(NO2)2
Dinitrometano
4
8,3 ⋅ 10-1
H2C(COCH3)2
Acetilacetona
8,8 1,7 ⋅ 10-2
H3C-NO2
Nitrometano
10,2 4,3 ⋅ 10-8
H3CCO-CH2-COOC2H5 Acetoacetato de etila
10,7 1,2 ⋅ 10-3
H2C(CN)2
Dinitrila do ácido malônico
12
1,5 ⋅ 10-2
321
Os valores destas tabelas foram aceitos de Peter Sykes, A Guidebook to Mechanisms in Organic
Chemistry. 5th Ed., Longman, London 1982.
505
A. Isenmann
Princípios da Síntese Orgânica
H2C(COOC2H5)2
H3C-CO-CH3
Dietilmalonato
Acetona
2,5 ⋅ 10-5
4,7 ⋅ 10-10
13,3
20
Todos esses compostos (menos a acetona) podem ser desprotonados, quase
quantitativamente, por bases comumente existentes em ambiente aquoso (isto é, os
hidróxidos solúveis dos metais não-nobres). Lembre-se do pKa da água (= ácido do OH-) ≅
15. Sendo assim, no caso do acetoacetato de etila temos o equilíbrio:
O
H3C
O
CH2
OEt
+ OH-
O
K
H3C
O
CH
OEt
+ H2O
,
onde
K (acetoacetato) 10−10,7
K= a
= −15 ≈ 2 ⋅104 .
K a (água )
10
Sobre a seleção da base a ser usada para a desprotonação do composto 1,3-dicarbonila, ver
p. 515.
Pode-se ainda ver que não apenas o grupo carbonila pode ativar o grupo metileno, mas
também outros grupos retiradores, tais como ciano e nitro; menos pronunciado é o efeito
EWG dos halogênios (ver mecanismo da reação halofórmio, na p. 424); até mesmo
sistemas aromáticos que transferem o efeito dos seus elétrons π para a posição benzílica,
agem neste sentido.
Aparência da forma enólica em compostos duplamente ativados
O terceio efeito citado acima (p. 505) que trata da importância da forma enólica, acha sua
explicação no fato de que um estado cíclico do enol está sendo estabilizado por uma ligação
de hidrogênio intramolecular:
H
O
O
O
C
C
C
CH2
O
C
CH
Forma enólica do
composto 1,3-dicarbonila
O deslocamento do equilíbrio termodinâmico a favor do enol pode ser verificado nos
seguintes valores:
506
A. Isenmann
Tabela 27.
Composto
Princípios da Síntese Orgânica
Porcentagem da forma enólica, sem a presença de solventes
% enol (na fase líquida)
H3C-CO-CH3
Acetona
1,5 ⋅ 10-4
H2C(COOC2H5)2
Dietilmalonato
7,7 ⋅ 10-3
NC-CH2-COOC2H5
Cianoacetato de etila
2,5 ⋅ 10-1
C6H10O
Ciclohexanona
H3CCO-CH2-COOC2H5 Acetoacetato de etila
7,5
30,0
H3CCO-CH(C6H5)-COOC2H5
H2C(COCH3)2
1,2
Acetilacetona
C6H5CO-CH2-COCH3 Fenilacetilacetona
76,4
89,2
O teor da forma enólica depende da temperatura e principalmente do solvente. Observa-se a
estabilização da forma enólica por solventes de caráter apolar. No exemplo da acetilacetona
sejam ilustrados os efeitos do solvente (embora que, em muitas condensações, prefere-se
trabalhar sem solvente adicional; os próprios componentes da condensação são líquidos e
devem ser usados em concentrações mais altas possíveis, para maximizar o rendimento).
Tabela 28.
Solvente
Estabilização da forma enólica por solventes.
% enol
15
H2O
58
CH3CN
sem solvente, em fase líquida
80
92
C6H14
sem solvente, em fase gasosa
92
Compostos bis-ativados que têm relevância na síntese por condensação
N C
CH2 C N
Dinitrila do ácido malônico
507
A. Isenmann
Princípios da Síntese Orgânica
O
C2H5 O C
CH2 C N
O
C2H5 O C
O
CH2 C OC2H5
C6H5 CH2 C N
O
H3C
O
H3C
CH3
O
C CH2 C OC2H5
O
(RO)2P
Dietilmalonato (malonato de dietila)
Cianeto de benzila
O
C CH2 C
Nitriloacetato de etila
O
CH2 C OR
Acetilacetona
Acetoacetato de etila
Éster do ácido fosfônico
O último composto da lista é de elevado interesse: a condensação dos dois hidrogênios no
grupo metileno funciona especialmente bem, com uma grande variedade de compostos
carbonílicos, levando a um acoplamento sob formação de um dupla-ligação C=C. Esta
reação é conhecida como Horner-Emmons, uma variação da famosa reação de Wittig. Ela é
discutida na p. 783.
Esses compostos, ao serem empregados em condensações, sempre representam o
componente pseudo-ácido. O primeiro produto de condensação é um composto com grupo
hidroxila em posição 2. Mais característico ainda e na maioria dos casos o produto
desejado, é a dupla-condensação com o componente carbonílico, seguindo o esquema:
C O + H2C
retirador 1 [base]
retirador 2
retirador 1
HO C
CH
retirador 2
∆
- H2O
retirador 1
C
C
retirador 2
Este comportamente pode ser comparado com a já discutida condensação aldólica, onde
sob catálise ácida o aldol não pude ser isolado, mas logo perdeu água formando um
composto carbonilado α,β-insturado (ver p. 455). Porém, tem-se certas diferenças às
condensações discutidas aqui:
1) A condensação de Knoevenagel é uma condensação mista, portanto o espectro de
produtos acessíveis é muito mais amplo.
2) A escolha do catalisador é mais sutil na condensação de Knoevenagel: enquanto na
condensação aldólica se usa qualquer ácido de Brønsted, a reação de Knoevenagel
508
A. Isenmann
Princípios da Síntese Orgânica
anda sob condições especialmente brandas usando catalisadores básicas. tais como
acetato de amônio, β -alanina, piridina ou piperidina 322.
Por causa de reações consecutivas interessantes sejam apresentadas as três sínteses a seguir;
elas se encaixam perfeitamente nesta classe de ativação, enquanto o eletrófilo (essa
denominação é mais coerente do que "componente carbonílico", neste caso) é de natureza
diferenciada.
Éster - um grupo retirador de elétrons especial
Entre todos os grupos retiradores de elétrons (EWG) um significado especial pode ser
atribuído ao grupo –COOR. O reagente metilênico contendo o grupo éster pode ser, nos
casos mais simples, um β-cetoéster ou um diéster do ácido malônico. Após terminar a
condensação com outro reagente carbonílico o produto é um novo éster que em seguida
pode ser hidrolisado. Resulta então o grupo ácido carboxílico que sofre facilmente
degradação: basta aquecer a cerca de 120°C e o composto perde CO2. Esta descarboxilação
(apresentada mais detalhadamente na p. 513; não confundir com descarbonilação da p. 489)
pode ser vista como pirólise ou eliminação cis (p. 147). Do ponto de vista preparativo o
agrupamento éster, no esquema a seguir em negrito, somente tem o papel de ativador. Isto é
o oposto de um grupo protetor. Podemos observar que o grupo éster somente serve para
definir quem age como componente metilênico, mas não faz parte do produto final. Isto é
uma importante estratégia dentre as condensações mistas.
O
H3C
CH2
+ base
OEt
C
R´
O
O
O
H3C
CH
C
δ+
O
O
OEt
H3C
1 : 1
O
H3C
CH
O
R´
R
C
C
OH
CH
O
Hidrólise
H2O/H+
O
H3C
OEt
CH
O
C
O
R´
R
C
OH
- CO2
H3C
R´
R
R
C
C
OEt
R´
R
CH2
OH
C
OH
OH
Note-se que o ácido carboxílico livre geralmente não pode ser usado no sentido de ativador
(exceção ver p. 513), já que o catalisador básico aplicado na primeira etapa o desprotona
imediatamente. Desta forma o catalisador se perde e o grupo carboxilato formado não tem
mais o poder de retirador de elétrons.
322
G. Jones, Org.Reactions 15 (1967) 204.
509
A. Isenmann
Princípios da Síntese Orgânica
Reações típicas dos compostos com grupo metileno duplamente ativado
Especialmente a alquilação e a acilação da posição α em compostos carbonílicos
(componente metilênico) são estratégias de suma importância, na confecção do esqueleto
carbônico da molécula alvo. Por isso, estas reações sejam referidas em primeiro lugar.
δ+
O
R Cl
O
C C
δ+
R COCl
Enolato
C C
R
O
O
C C C
+
Cl-
R + Cl-
Ambas as reações, com haletos de alquila e haletos de acila, respectivamente, somente
funcionam quando o enolato for preparado quantitativamente. Por isso são usados
geralmente compostos com grupo metileno duplamente ativado. Se isso não for
estabelecido temos que contar com uma série de produtos paralelos, conforme ilustrado a
seguir.
1. Alquilação em posição α ao grupo carbonila
O carbono em posição α ao grupo carbonila tem uma polarização natural negativa. Isto se
deve à segunda fórmula de mesomeria do enolato, no esquema a seguir. Podemos então
pensar em uma reação nesta posição com um carbono positivado, por exemplo, um haleto
de alquila.
O
O
R
Base
R
R´
R´
R´´
X
O
R
R´
O
R
R´´
CH
R´
não funciona!
Pelos seguintes motivos esta sintese não funciona:
1) O enolato do substrato pode reagir com outra molécula igual, fornecendo o produto da
autocondensação, em vez do desejado produto alquilado.
2) O equilíbrio da forma enólica (ou enolato) em compostos carbonílicos simples fica ao
lado esquerdo (= ceto ou aldo) - além do mais a enolização é uma reação lenta. Portanto
deve-se adicionar um catalisador - de preferência uma base. A nucleofilia desta base,
porém, pode provocar a formação de produtos paralelos (por exemplo, éteres, ver
síntese de Williamson, p. 136)
510
A. Isenmann
Princípios da Síntese Orgânica
3) O produto desta reação pode se alquilado mais uma vez, sua reatividade é comparável à
do substrato.
4) Cetonas geralmente dispõem de hidrogênios ácidos em ambos os lados do grupo
carbonila. Em casos de cetonas assimétricas isto leva a uma mistura de produtos.
Uma solução razoável é a dupla ativação do substrato, na maioria dos casos práticos
efetuada pelo grupo de éster, em posição β. Assim, a posição α torna-se ácida o suficiente
(exclusão do motivo No. 1, da lista negativa) para ser desprotonada rapida e facilmente,
usando, por exemplo, alcóxidos. Assim, forma-se quantitativamente o enolato (exclusão do
motivo No. 2). De maneira mais vantajosa e limpa o enolato se produz numa etapa
preliminar; sobras de base podem ser removidas da mistura (exclusão do motivo No. 3),
antes de acrescentar o haleto de alquila.
Atenção: uma reação análoga com haleto aromático, Ar-X, isto é, uma arilação do grupo
metileno desprotonado, não funciona . Explicação: ver Questão 10, p. 543.
O
O
O
R´´
Br
EtO
EtO2C
EtO2C
EtO2C
R´
R´
R´
quant.
R
R´´
R
R
2a etapa
1a etapa
Hidrólise
O
EtO2C
R
R´
O
1) OH- / H2O
R´´
2) H+
H
H
O
O
R´
R
R´´
O
O
- CO2
R
R´
R
R´´
R´
R´´
Da mesma maneira funciona a condensação com o éster do ácido malônico:
COOEt
R X
+ CH2
[ C2H5O Na ]
COOEt
COOEt
R CH
COOEt
Hidrólise e
Descarboxilação
1) H+ / H2O
2) ∆ : - CO2
R CH2 COOH
Maloato de dietil
Em uma primeira etapa o malonato deve ser desprotonado quantitativamente. Isto é muito
fácil por causa da alta acidez do grupo metileno. Porém, excesso de base (neste caso o
etóxido) devem ser evitados, para não provocar a perda de reagente por reações paralelas
(síntese de éter segundo Williamson, ver p. 136).
A saponificação do éster pode ser feita em ambiente ácido ou básico. A descarboxilação do
ácido carboxílico livre, porém, é mais viável em ambiente ácido. Forma-se um ácido
carboxílico cuja redução com LiAlH4 e tratamento com SOCl2 fornece um haleto onde a
511
A. Isenmann
Princípios da Síntese Orgânica
cadeia carbônica foi aumentada por dois átomos de carbono, em comparação ao material de
partida.
Regiosseletividade do carbono nucleofílico
Essa consideração não tem restrição aos compostos metilênicos duplamente ativados, mas
também vale para compostos com somente um grupo carbonila.
Na alquilação descrita acima temos que lidar com o fato de que o enolato é um ânion
bidentado. Este caráter ambidente faz com que a condensação, entre o enolato e um
composto com carbono eletrofílico, nem sempre forneça uma nova ligação C-C. Pode
ocorrer uma reação no oxigênio, também. O conceito que explica as proporções com que se
formam os produtos é mais uma vez a polarizabilidade dos reagentes, como já foi elucidado
em outros compostos bidentados (C≡N, NO2, N≡O, DMSO; p. 44). Em solvente aprótico
observa-se uma dureza diferenciada dos seguintes reagentes eletrofílicos R-X (em
parênteses já indicada a porcentagem do produto O-alquilado a partir do propiofenolato, um
enolato bastante estabilizado; a alquilação foi feita em HMPT 323, solvente polar aprótico, e
se refere ao esquema a seguir):
R-I (15%) < R-Br (40%) < R-Cl (67%) < R-OTs (85%).
macio ...................................................................duro
Os haletos de alquila devem ter carbono primário ou no máximo secundário. A reação em si
é uma SN2 (ver p. 17). Resumindo pode-se afirmar que na maioria dos casos não se observa
a O-alquilação, mas sim, a condensação no carbono α.
OC
C
O
C
-
C
duro
macio
+ R
CH2 OTs
duro
O CH2 R
C C
+ R
CH2 I
macio
O
R CH2 C
C
323
Valores adaptados de Organikum, Organisch-chemisches Grundpraktikum, VEB Berlin 1988, p.182.
Esta obra é altamente recomendada porque acompanha o químico preparativo nas suas tarefas diárias, de
laboratório. Existe na segunda edição, traduzida para o Português:
H.G.O.Becker, W.Berger, G.Domschke, Organikum - Química orgânica experimental, Fundação Calouste
Gulbenkian, Lisboa 1997.
512
A. Isenmann
Princípios da Síntese Orgânica
O conceito macio-duro aplicado aqui corresponde plenamente à teoria dos orbitais de
fronteira (ver p. 207 com notas de rodapé; nota de rodapé na p. 45). Centros duros reagem
de preferência sob controle de carga, enquanto centros macios reagem sob controle dos
orbitais.
2. Condensação clássica de Knoevenagel
Esta condensação funciona com o éster do ácido malônico, o malonato de dietila. A reação
requer uma catálise por ácidos ou bases fracos, para estabelecer condições mais suaves
possível. Assim, estabelecem-se condições de controle cinético, isto é, a reação mais rápida
entre as possíveis reações paralelas é favorecida. Neste caso a reação mais rápida é a
enolização do malonato; reações tais como a autocondensação do componente carbonílico
fica em segunda linha. Esta última possibilidade, o perigo da autocondensação, não se dá
no exemplo a seguir, porque o aldeído aromático não tem H em posição α:
COOC2H5
C6H5 CHO + H2C
COOC2H5
COOC2H5
C6H5 CH C
COOC2H5
[ H+ ]
[Base suave]
- H2O
COOC2H5
C6H5 CH C
COOC2H5
COOH
C6H5 CH C
COOH
∆
- CO2
C6H5 CH CH COOH
Ácido cinâmico
Neste exemplo pode-se verificar que ácidos 1,3-dicarboxílicos (e também β -cetoácidos)
desprendem facilmente CO2. A etapa da descarboxilação, descrita na p. 462 como "quebra
de cetona", pode ser efetuada por temperaturas levemente elevadas. Seu significado para o
planejamento de síntese, ver p. 509; outras descarboxilações (do éster acil-malônico, quebra
cetônica de β -cetoéster,...) ver as pp. 503, 516, 471, 472, 472, 473, 523, 555, 462, 511, 514.
Para a condensação clássica de Knoevenagel, quando for planejada com descarboxilação
logo a seguir, existe uma maneira elegante de dispensar a etapa da hidrólise, por sua vez
essencial para desprender o CO2 a partir do éster. O fato de o catalisador da primeira etapa
ser bastante suave e específico permite o emprego do ácido malônico livre, em vez do seu
éster! Assim, esta síntese pode ser feita em uma só etapa (“one-pot-reaction”). Note-se que
esse atalho somente é possível, devido à seleção de uma "base" especial, sendo catalisador
da enolização do ácido malônico livre (lembre-se que, ao desprotonar o ácido carboxílico, o
grupo -COO- praticamente perde todas as suas qualidades como retirador de elétrons; então
o composto metilênico deixa de ser "duplamente ativado").
De maneira análoga funciona a condensação entre acetilacetato de etila ou cianacetato de
etila (em vez do éster malônico) e aldeídos. O cianacetato de etila é obtido através da
reação de clorocetato de etila com cianeto de sódio.
513
A. Isenmann
Princípios da Síntese Orgânica
COOC2H5
+ H2C
R CHO
R CH C
R CH C
- H2O
C N
COOC2H5
COOC2H5
[Base suave]
CN
COOH
[ H+ ]
∆
R CH C
- CO2
C N
CN
R CH CH C N
Nitrila α,β-insaturada
3. Acilação de compostos duplamente ativados
Exemplos para a alquilação de compostos com grupo metileno duplamente ativado já
encontramos na p. 510 e na síntese de Stetter (p. 464). Essencial para um bom rendimento
foi a desprotonação quantitativa do componente metilênico numa etapa prévia. Isto se
consegue, em caso de pseudo-ácidos duplamente ativados, com uma base de força média;
no exemplo da síntese de Stetter foi usado etóxido para este fim. Caso o pseudo-ácido não
for duplamente ativado, então a base deve ser consideravelmente mais forte (ver exemplo
na p. 502).
Aqui seja referida a possibilidade de acilar o ânion do composto metilênico - em toda
analogia à alquilação. O exemplo seja o diéster do ácido malônico, então claramente um
pseudo-ácido duplamente ativado. A primeira etapa é mais uma vez a desprotonação
completa do malonato que se consegue com um alcóxido. Em seguida acresecenta-se o
cloreto de acila que condensa rapida e completamente.
Interessante no produto desta reação são os dois grupos de éster, ambas em posição β ao
grupo de cetona. Isto significa, após a sua hidrólise para grupos ácidos carboxílicos, que
ambas podem ser abandonadas em forma de CO2. Ou seja, duas vezes pode ser efetuada a
"quebra de cetona" (p. 462).
COOR
CH2
RO- Na+
COOR
O
Maloato
1) H+ / H2O
2) ∆ : - CO2
COOR
COOR
CH
CH
C
OR
O
O COOR
C6H5 COCl
C6H5 C CH
- Cl-
C
COOR
OR
Enolato, quantitativo
O
C6H5 C CH2
COOH
∆
- CO2
O
C6H5 C CH3
Metilcetona
Do ponto de vista do cloreto de acila, a condensação do éster malônico levou a um
alargamento por apenas um carbono (em negrito).
514
A. Isenmann
Princípios da Síntese Orgânica
A tendência atual na síntese orgânica é a diminuição de etapas de baixo "rendimento
atômico", onde for possível. O que significa isso? Dos reagentes que se empregam numa
síntese devem permanecer, se for possível, todos os átomos no produto. Em termos de
rendimento atômico esta síntese certamente é um péssimo exemplo: não só as duas partes
alcoólicas R-OH se perdem, mas também e irreversivelmente, dois carbonos em forma de
gás carbônico. Considerando também a problemática do efeito estufa, promovido pelo CO2,
esta síntese é pouca favorável e certamente ficará restrita à pequena escala.
Todavia, o benefício estrutural desta síntese é evidente: acrescentou-se um grupo metila,
sob redução do carbono carboxílico a um grupo carbonila. Isto é o resultado inverso da
reação halofórmio, ver p. 424.
Como mostrado várias vezes, o carbânion do malonato é estabilizado por mesomeria. A
forte participação da segunda fórmula de ressonância desta mesomeria tem também como
consequência a acilação no oxigênio. Esta reação, muitas vezes indesejada, é especialmente
pronunciada na presença de piridina.
A acilação de ésteres do ácido malônico representa uma alternativa preparativa, para a
síntese de cetonas via reagente organo-cádmio (ver p. 440).
Por analogia, a acilação do acetoacetato de etila (e também da acetilacetona, conforme
discutido a seguir) fornece a β -dicetona.
Acilação de compostos 1,3-dicarbonilados
O
O
O
O
O
OEt
Acetilacetona ("acac")
Acetilacetato de etila
EtO
O
OEt
Dietiléster do ácido malônico
Esta síntese é de alto valor preparativo porque leva a novos compostos 1,3-dicarbonilas que
são assimétricos. Ocorre via condensação entre o éster malônico e um derivado ativado do
ácido carboxílico, seguido por descarboxilação. A diferença entre essa e as condensações
apresentadas acima é somente a natureza do aceitador (componente eletrofílico), onde o
grupo retirador de elétrons tem um NOX mais alto.
O catalisador preferido é o hidróxido de magnésio, Mg(OH)2, que confere a
regioseletividade correta a esta condensação. Isto requer uma explicação mais detalhada.
Ao desprotonar o éster malônico (ou a acetilacetona) o ânion não se afasta muito do cátion
proveniente da base. Caso este cátion tiver valências para estabilizar o ânion em forma de
um ligante quelato, um complexo entre o metal e dois oxigênios iria se formar, isto é, um
complexo entre um ácido duro e duas bases duras. O ânion, por sua vez, não só tem centros
básicos duros, mas também um centro mole: o carbono negativado do enolato. Este, por
ficar não-complexado, pode ser acessado pelo componente carbonílico nesta condensação.
O cloreto de ácido, componente carbonílico, é acrescentado aos poucos através do funil de
adição.
515
A. Isenmann
Princípios da Síntese Orgânica
Nu- duro
E+ duro
O-
O
R COCl
OEt
Componente eletrofílico
Nu- macio
Componente metilênico
Em caso de o catalisador ser NaOH ou KOH, mas também Ba(OH)2, o contra-íon é Na+,
K+ ou Ba2+ e a complexação do enolato fica ausente; daí o cloreto do ácido se liga
preferencialmente ao oxigênio negativado, com a consequência que a condensação
intencionada falhou. Por outro lado, o Mg2+ complexa e bloqueia os oxigênios que
representam as bases duras. O componente carbonílico somente pode se ligar ao carbono
negativado – bem no sentido das condensações discutidas neste capítulo.
A diferença em regiosseletividade seja ilustrado no exemplo do acetilacetato de etila:
O
O
Na+ O-
R
O
C
Cl
R
O
O
OEt
Transmetalação
OEt
+ Mg2+
- Na+
O
Mg
O
Produto O-acilado
O
O
R C
O
OEt
Cl
OEt
Complexo quelato
R
O
Produto O-acilado
1. Hidrólise
2. Descarboxil.
O
R
O
Caso o composto duplamente ativado seja acetilacetona, a etapa da descarboxilação é
naturalmente impedida (ver questão 13, na p. 539).
Síntese do éster glicídico, segundo Darzens
Esta síntese certamente é uma reação extraordinária, porém encaixa neste capítulo porque o
material de partida representa um composto com grupo metileno duplamente ativado que
pode ser desprotonado com facilidade.
Como já apresentado na p. 224, os epóxidos em geral são muito valiosos na síntese
orgânica, uma vez que são bastante reativos (reagem principalmente com nucleófilos;
516
A. Isenmann
Princípios da Síntese Orgânica
fazem polimerizações) e também devido a sua estrutura 1,2-bifuncional. Por exemplo, a
substância bioativa etanolamina, H2N-CH2-CH2-OH, cuja estrutura tem um álcool e uma
amina em posição 2, pode ser feita a partir de óxido de etileno. Portanto, os epóxidos são
considerados, junto com os ácidos α-halogenados, como mais importantes síntons 1,2bifuncionais (ver nota de rodapé na p. 389).
Pelos mesmos motivos, um epóxido que tem um grupo de éster em posição α, isto é, um
éster glicídico, é um composto intermediário importante:
R
H
C CH COOCH3
1
O2
Éster glicídico
Há duas possibilidades:
1. Quebrando as duas ligações C-O do epóxido revela o acesso pelo éster α,β-insaturado +
oxigênio. Como será discutido mais detalhadamente no cap. 6.6.1, essa dupla-ligação C=C
(= sistema Michael) reage de preferência com nucleófilos, portanto o oxigênio é
providenciado pela mistura H2O2 + base → HOO- (enquanto as epoxidações que foram
apresentadas na p. 224 funcionam com oxigênio eletrofílico).
2. Quebrando uma ligação C-O e uma C-C do epóxido revela um composto carbonílico
mais um carbeno em posição α do éster. Mesmo se a reação não percorre o estado de
carbeno livre, a reação representa uma inserção na ligação C=O (compare as pp. 158, 220 e
880), em um carbeno latente. Esta síntese é conhecida como reação de Darzens e funciona
com
Éster α-clorado + alcóxido (= base) + aldeído .
Segundo as regras de Baldwin (p. 475) essa ciclização é favorável (3-exo-tet):
517
A. Isenmann
Princípios da Síntese Orgânica
O
Desprotonação
quantitativa
Cl
CH2 COOCH3
+ CH3O- CH3OH
R C
Cl
CH COOCH3
H2O / OH-
C CH COOCH3
H
O
-
- Cl
Cl
R
H
Carbeno latente
R
H
C
CH COOCH3
O
-
R
H+
C CH COOH
O
Éster glicídico
R
H
C
∆
CH
O
C O
- CO2
H O
R
H
C
C
H
OH
O
R CH2 C
H
Enol
Uma das possíveis reações consecutivas do éster glicídico já foi incluído no esquema
acima: a saponificação seguida pela descarboxilação ("quebra de cetona", ver p. 462). O
resultado desta síntese é um enol que tautomeriza para o aldeído. Neste reconhecemos a
prolongação da cadeia carbônica, em comparação ao aldeído de partida, por um grupo
metileno.
Observação: as reatividades discutidas nesta síntese têm certa semelhança com as
discutidas na p. 220.
6.6
O sistema Michael
Denomina-se sistema Michael quando uma dupla-ligação C=C está em conjugação com
uma ligação múltipla C=X ou C≡X altamente polarizada (isto é, um grupo carbonila,
carboxila ou nitrila).
Reatividade:
Dentro dos compostos carbonílicos, os α,β -insaturados (unidade estrutural C=C-C=O) têm
um papel especial. Trata-se de um sistema de duplas ligações conjugadas, então devem ser
consideradas as paricularidades do efeito M (ver p. 318) que se propaga através dos
elétrons π deste sistema. A distribuição das polarizações neste sistema é δ+ δ- δ+ δ-..., é
um exemplo do princípio vinílogo, segundo o qual certa polarização (ou reatividade) se
propaga de maneira ondular ao longo de grupos vinilas (= duplas ligações conjugadas), ver
gráfico na p. 318.
Uma molécula com a estrutura C=C-C=O e as devidas polarizações são dadas no seguinte
esquema, já incluindo sua reatividade frente nucleófilos. Em princípio, o nucleófilo tem
duas possibilidades de atacar, onde
518
A. Isenmann
Princípios da Síntese Orgânica
o caminho 1 é referido como sentido "normal", "direto", ou "adição nucleofílica
1,2";
o caminho 2 representa o sentido "Michael", "conjugado", "vinílogo" ou "adição
nucleofílica 1,4".
•
•
δ−
δ+ δ− δ+
C
C
2
O
C
1
NuDeste quadro concluímos que existe mais um carbono positivado, além do carbono do
grupo carbonila: o carbono em posição β. No primeiro olhar essa polarização parece
estranha, já que o carbono β faz parte de uma dupla ligação da qual nós esperamos uma
densidade eletrônica elevada (ver item 2.1) 324. Mas sua reatividade frente nucleófilos é tão
pronunciada que nos temos que liberar-nos da impressão que qualquer dupla-ligação C=C
seja rica em elétrons. Realmente, o nucleófilo consegue adicionar-se neste carbono com
bastante facilidade.
A carga negativa que se forma intermediariamente em posição α pode ser deslocalizada por
mesomeria, conforme indicado a seguir:
Nu
+
C
C
C
O
O
O
-
Nu
C
C
C
Nu
C
C
C
H+
H
Nu
C
C
O
C
Os compostos carbonílicos α,β-insaturados que reagem neste sentido se chamam sistemas
Michael e a reação é a adição de Michael (1887) 325.
6.6.1
Adições nucleofílicas no sistema Michael
O caso especial da adição de alilsilanos e vinilsilanos será apresentado no capítulo dos
organossilanos (ver p. 820).
Adição de HBr
O próprio reagente, HBr, indica que esta reação ocorre sob catálise ácida. Pode-se afirmar
então que a adição Michael não só acontece em ambiente básico (que é o caso geral), mas
também é possível em meio ácido.
324
Por esta razão nos não podemos representar o orbital π desta molécula por duas "bananas", uma em cima e
outra em baixo do plano atômico, de acordo com a imagem clássica para olefinas com dupla-ligação C=C
isolada.
325
Arthur Michael era americano, portanto pronuncie “Maicl”.
519
A. Isenmann
O
C
Princípios da Síntese Orgânica
+ H+
C C
OH
OH
C C C
C C C
+ Br-
+ Br-
OH
C
H
OH
C C
C
Br
C C
O
C C C
Br
Br
A primeira etapa é a adição reversível de H+ no oxigênio do grupo carbonila. Resulta um
cátion que pode ser atacado pelo brometo nas duas posições indicadas (ver também questão
16 do cap. 5). Cada uma das adições de HBr é de caráter reversível. Porém, a entrada do
brometo em posição β é favorecida porque existe um equilíbrio subsequente que fica
deslocado para o lado direito. Assim, a adição do brometo em posição β é o produto
principal da reação. Em resumo, a função carbonila tem apenas o papel de orientador, mas
não participa da reação.
Reação com reagente de Grignard
1) R MgX
2) H2O
R´
O
OH
R´
C C C
R´
R´´
"normal"
R
C C C
R´
R´´
1) R2CuLi
2) H2O
R´ H
O
sentido de Michael
R C C C
R´
R´´
Esta reação mostra claramente duas características dos compostos carbonílicos α,β insaturados:
1. A adição “normal” e a adição de Michael são concorrentes, muitas vezes têm-se
reatividades comparáveis.
2. O carbono positivado em posição β representa um centro “macio” enquanto o
carbono do grupo carbonila é um centro eletrofílico mais “duro” (para a definição
de duro e macio segundo Pearson, recorra às pp. 40 e 685).
520
A. Isenmann
Princípios da Síntese Orgânica
Com a escolha certa da parte metálica do reagente de Grignard é possível direcionar o
produto de prevalência: a reação de Michael requer um reagente organometálico macio
porque o carbono β é um centro macio 326.
O reagente de Grignard com magnésio é um reagente duro, igual ao composto R-Li, então
ataca preferencialmente o carbono do grupo carbonila (duro). Existe uma técnica
incrivelmente simples de converter o caráter duro do organometálico em um nucleófilo
macio: basta a presença de sais de Cu(I), sem necessidade de produzir e isolar o composto
organometálico misto. O carbânion se desenvolve in situ para um centro básico macio.
Por isso se obtém bons resultados da adição de Michael com reagente organo-cuproso,
também chamado reagente de Corey-House (ver p. 750). Sua preparação é fácil e de alto
rendimento, pela transmetalação ou metatese, como descrita a seguir (ver também p. 700).
Atualmente, em muitas reações clássicas o reagente organo-magnésio é reposto por este
reagente organo-cuproso:
R X
Li
- LiX
Inversão da
polarização
R Li
(duro)
CuX
Transmetalação
R2 CuLi
Lítio-dialquil-cobre
Reagente de Corey-House
(mole)
Um outro exemplo reacional, onde a polarizabilidade de nucleófilo e eletrófilo determina a
regiosseletividade, é apresentado na p. 515.
A regiosseletividade depende então das naturezas de ambos os participantes, do substrato
com estrutura C=C-C=O e do atacante organometálico. Além disso, existem critérios para a
reatividade que somente são relacionados à estrutura do composto carbonílico α,β insaturado. O produto típico para compostos carbonílicos simples (caminho 1, no esquema
acima) se obtém preferencialmente quando o grupo R´ é muito volumoso e R´´ é um
hidrogênio (= aldeído). O produto 2 é o principal quando R´= H e R´´ = alquila, alcóxido
(isto é, cetona ou éster).
326
Em um conceito mais moderno dos orbitais de fronteira o HOMO foi substituído pelo FERMO (Frontier
Effective-for Reaction Molecular Orbital), com a vantagem de descrever a forma, direção e tamanho dos
HOMOs, em cada um dos centros nucleofílicos, em ânions bidentados. O enolato, sem dúvida, é um ânion
bidentado que tem dois sítios reativos - o oxigênio e o carbono em posição β. É claro que o conceito de
HOMO (inventado por Fukui, ver também p. 207) não é adequado para diferenciar entre as reatividades
diferenciadas dentro da mesma molécula, visto que somente existe um único HOMO a cada molécula. Ao
invés deste, podem-se criar tantos FERMOs que precisam, para explicar as diferentes reatividades dos sítios,
predizer ângulos de novas ligações (sejam covalentes ou coordenativas, quer dizer, complexos metálicos) e
até distâncias de novas ligações que resultam da reação, neste caso, entre o composto carbonílico e o reagente
organometálico.
Artigo original: R.R.Silva, J.M.Santos, T.C.Ramalho, J.D.Figueroa, Concerning the FERMO concept and
Pearson´s HSAB principle, J.Braz.Chem.Soc. 17 (2006) 223-6.
521
A. Isenmann
Princípios da Síntese Orgânica
Arilação e vinilação de alquenos usando catalisadores de paládio
O desenvolvimento recente das reações sob catálise de paládio de baixo número de
oxidação (NOX) levou a uma ampliação considerável da família das reações do tipo Heck
(ver cap. 4.3.7), por sua vez conhecida desde os anos 70 do século passado. Jeffery 327
descreve não só a arilação de olefinas, mas também a sua vinilação, usando catalisadores
complexos de paládio. O substrato, no caso um aromático ou composto vinila contendo
cloro, bromo ou iodo, é submetido a um tratamento prévio com Pd(OAc)2. Esse tem por
consequência uma inversão da reatividade no respectivo carbono insaturado que tornou-se
por esta medida nucleofílico. Na segunda etapa este nucleófilo ataca uma dupla-ligação
C=C polarizada de um sistema Michael. A reação foi testada com sucesso com acrilato de
metila, metilvinilcetona e acroleína. O mais interessante é o fato que a estereoquímica nas
duplas-ligações de ambos os substratos, vinílico e carbonílico, se mantêm no produto que
se forma com bons redimentos e isento de isomerização cis-trans. Do ponto de vista do
aromático/vinílico, o halogênio foi substituído pelo sistema Michael. Do ponto de vista do
composto carbonílico α,β-insaturado, a prolongação da cadeia carbônica por um sínton
insaturado é uma ampliação bem vinda à condensação de Michael descrita a seguir e
também à alquilação com o sistema Michael (p. 527).
Anote que o cloreto de tetrabutilamônio tem duas funções: é catalisador de transferência
das fases (ver p. 32), aqui entre uma fase sólida e uma líquida; também é complexante do
Pd(II) e ajuda manter o catalisador em solução e ativo. No capítulo sobre a catálise de
Ziegler (p. 172) um reagente deste foi chamado de co-catalisador.
R´
Cl
+
C
O
Solvente: DMF,
Temp.amb.
Rend: > 95%
R
Cl
R´
+
C
K2CO3;
(NBu4)+Cl Catalisador:
Pd(OAc) 2
R´
O
R
O
R
C
C
R´
O
R
R = H, CH3, OCH3
Quando falamos em "compostos vinílicos", automaticamente pensamos em polimerizações
(ver p. 164). E realmente, temos que contar com produtos paralelos oligoméricos,
especialmente quando operar com excesso em reagente vinílico. Note que a polarização do
reagente Michael, se propaga através do efeito mesomérico (= efeito M; representação na
Figura 29, p. 318) aos novos carbonos do produto, quase sem sofrer enfraquecimento.
Sendo assim, o próprio produto torna-se um concorrente, em receber uma nova unidade
vinílica.
327
T.Jeffrey, Tetrahedron Lett. 26 (1985) 2667.
522
A. Isenmann
Princípios da Síntese Orgânica
Adição de Michael com outros nucleofilos
Também outros nucleófilos, tais como NaCN ou HCN 328, anilina e mercaptanos, aminas
secundárias, reagentes organometálicos de caráter mole (por exemplo, RZnI 329) - todos se
adicionam no carbono em posição 4. A explicação é novamente o caráter macio destes
reagentes que preferem reagir com o centro macio do substrato carbonílico insaturado.
As quinonas podem ser vistas, de certa maneira, como sistemas Michael duplamente
ativados. Assim, as adições de nucleófilos devem ser especialmente fáceis.
O
Nu-
Nu- = CN-, H2N
Ph, R
SH
O
Com a alilação de Hosomi-Sakurai (p. 829) será apresentado o acoplamento com um
nucleófilo organossilício.
6.6.2
Condensação de Michael
Sob condensação de Michael se entende a adição de um componente metilênico
nucleofílico, na posição β de um composto carbonílico α,β -insaturado. Em outras palavras:
condensação de Michael é um acoplamento C-C onde o aceitador é um sistema Michael.
Note que os aceitadores de Michael geralmente são citotóxicos e carcinogênicos, já que eles
se adicionam prontamente no DNA. Uma alquilação deste tipo acarreta, como sabemos
hoje, mutações graves.
Em geral, é vantajoso estabilizar o nucleófilo em forma do enol ou enolato para que tenha o
suficiente tempo para reagir com o sistema conjugado. Neste sentido oferecem-se os enóis
estabilizados como enamina, enoléter ou sililenoléter (p. 495); igualmente adequados são
compostos 1,3-dicarbonilados, apresentados como duplamente ativados (p. 505).
A reação do enolato com o composto carbonílico α,β -insaturado do esquema a seguir
fornece um δ-cetoácido. Este exemplo inclui de uma vez, a descarboxilação do produto
primário (p. 462), sendo a sequência reacional mais comumente aplicada.
328
Hidrocianamento de compostos carbonilados insaturados, ver J.A. McRae, R.A.B. Bannard, Org.Synth.
Coll. Vol. IV (1963) 393. Na reação de Nagata usa-se cianeto de dietilalumínio como fonte do CN
nucleofílico: W. Nagata, M. Yoshioka, Org.React. 25 (1977) 255.
329
A.L. de Souza, I.S. Resck, J.Braz.Chem.Soc. 13 (2002) 233.
523
A. Isenmann
Princípios da Síntese Orgânica
O
O
H3C
O
+ C2H5O-
C CH2 C
OC2H5
H3C
- C2H5OH
O
H
C CH CH2
CH C
COOC2H5
O
OC2H5
1) H2C CH C
O
H3C
OC2H5
C CH
COOC2H5
Nucleófilo
Hidrólise /
Descarboxil.
H+ / H2O
- CO2
2) + C2H5OH ; - C2H5O-
O
H3C
O
C CH2 CH2
CH2 C
OH
δ-cetoácido
O rendimento desta síntese muitas vezes pode ser duplicado quando se adiciona NH3 ou
aminas primárias como catalisadores. Elas têm duas funções:
1. Sua propriedade como base (fraca) é suficiente para desprotonar em partes o
componente com grupo metileno, já que este pseudo-ácido é duplamente ativado
(ver valores pKa, na Tabela 26, p. 505).
2. Elas reagem em uma etapa preliminar com o composto carbonílico α,β -insaturado,
formando uma imina (ver p. 411). Desta forma, sua qualidade como componente
carbonílico fica drasticamente reduzida. (compare o dito sobre aminas
desativadoras, na p. 495)
Mais um exemplo de condensação do tipo Michael:
A síntese de dimedona
524
A. Isenmann
COOC2H5
CH2
COOC2H5
Princípios da Síntese Orgânica
COOC2H5
+ C2H5O-
COOC2H5
CH
- C2H5OH
CH COOC2H5
COOC2H5
Éster malóico
(CH3)2C
H3C
O
C
CH C
CH C
H3C
OCH3
CH3
Óxido de mesitila
O
C2H5O C
COOC2H5
CH COOC2H5
+ C2H5OH
- C2H5O-
(CH3)2C
CH C O
+ C2H5O-
O
OC2H5
(CH3)2C
- C2H5OH
CH2
CH2 C
CH2 C
O
CH3
O
C2H5O C
O
O
CH C
(CH3)2C
-
- C2H5O
O
1) Hidrólise
CH2
2) Descarboxilação
CH2 C
O
O
OH
Dimedona
O produto, dimedona 330, se precipita em forma de cristais amarelos. É um derivado da dihidro-resorcina; devido à estabilização por conjugação das duplas-ligações, o último
equilíbrio desta síntese é largamente deslocado para o lado do enol.
330
Dimedona tem certa importância como complexante e catalisador na formação de complexos com metais
de transição e tem diversas aplicações na coulometria, cristalografia, luminescência e análise
espectrofotométrica. Sua aplicação mais conhecida é na determinação gravimétrica do formaldeído, pela
facilidade da seguinte reação:
O
O
2
H3C
H3C
+
O
CH3
CH2
O CH2
O
O
+ H2O
CH3
H3C
H3C
O
Não só a sua predileção para o formaldeído, mas também para os demais aldeídos, abre caminho para um
método químico de distinguir e separar aldeídos e cetonas. A partir de uma mistura, a dimedona reage apenas
com o aldeído formando
525
A. Isenmann
Princípios da Síntese Orgânica
Anelação de Robinson
A anelação de Robinson é considerada sendo uma das técnicas mais importantes para
produzir anéis de 6 membros. O princípio do seu funcionamento é a condensação
carbonílica.
Essa reação é, ao mesmo tempo, exemplo para ciclização (ver p. 469), condensação de
Mannich e adição de Michael. Resulta uma ciclohexenona que pode fazer parte em um
sistema maior de anéis “condensados”. No exemplo a seguir isto é claramente mostrado,
pelo uso de uma cetona cíclica como substrato.
A partir da acetona, do formaldeído e da dimetilamina pode-se preparar a seguinte base de
Mannich:
+ Base(LDA)
O
H2C O + (CH3)2NH + CH3 C CH3
O
(CH3)2N
CH2 CH2 C
Base de Mannich
CH3
+ Base-
∆
O
O
O
H2C CH C
CH3
Sistema "Michael"
condensação de Michael
CH2
H2 C
O
C
CH3
O
condensação
"comum" - H2O
O
Com apoio de uma base forte não-nucleofílica, por exemplo t-butóxido ou LDA, pode ser
liberado um sistema Michael no qual o enolato da ciclohexanona pode efetuar um ataque
nucleofílico. Na última etapa se fecha o novo anel de 6 membros. Isto é mais uma
condensação comum que ocorre sob catálise básica (p. 456), seguida pela eliminação β de
O
O
H
H 3C
C
H3C
CH3
R
O
OH
CH3
O
O
H
H 3C
CH3
C
H 3C
CH3
R
O
HO
A alta seletividade se deve ao impedimento estérico que é maior, no caso da cetona.
526
A. Isenmann
Princípios da Síntese Orgânica
água. Este tipo de criação de anéis condensados é de suma importância na síntese de
esteróides e hormônios.
Condensação de Michael via enamina
Uma alquilação de compostos carbonílicos (a partir da sua enamina, ver p. 496)
infelizmente fornece muito produto N-alquilado, em vez da esperada formação de nova
ligação C-C. Como pode ser visto no esquema abaixo, o produto principal é um sal
quaternário de alquil-vinil-amônio. Neste sal podemos constatar que a polarização da dupla
ligação se inverteu, em comparação à da enamina de partida. Isso abre caminho para uma
série de reações consecutivas. Como a N-alquilação não pode ser revertida, a enamina não é
a forma adequada para efetuar alquilações em posição β do composto carbonílico (neste
exemplo: na ciclohexanona):
irreversível
δ−
N
X
δ+
δ+
N
reações consecutivas
R
-
R X
δ−
N
O
R
Hidrólise
R
Rendimento insatisfatório!
Por outro lado, as enaminas reagem prontamente com sistemas Michael, levando a uma
nova ligação C-C.
527
A. Isenmann
Princípios da Síntese Orgânica
O
δ−
N
C
O
CH C
δ−
C
N
CH C
O
C
CH C
Sistema Michael
O
O
C
Hidrólise
CH2
C
1,5-Dicetona
A explicação para a seletividade observada pode ser dada em termos do conceito de
Pearson: A adição no sentido de Michael requer um nucleófilo macio. Dentro da enamina o
β-C carbanóide é mais macio do que o nitrogênio.
Reação de Baylis-Hillman
Nem sempre um composto carbonílico α,β-insaturado (= sistema Michael) reage no
sentido, apresentado nestas últimas páginas, ou seja, ataque nucleofílico ao carbono β. A
reação apresentada a seguir mostra que também existe a possibilidade de reação eletrofílica,
no carbono α.
A reação de Baylis-Hillman é uma condensação entre um aldeído e um composto α,β insaturado com grupo retirador de elétrons (= sistema Michael; no exemplo a seguir uma
cetona α,β -insaturada). O catalisador de preferência é a base, DABCO (1,4diazabiciclo[2.2.2]octano); no produto se nota a unidade estrutural de álcool alílico.
Aprovaram-se também como catalisadores as fosfinas, P(OR)3, e outras amina
nucleofílicas, tal como DMPA ou DBU. Até a presença de solventes próticos, tais como
metanol, trietanolamina ou água, têm um efeito catlítico à reação de Baylis-Hillman.
O
R1
δ−
δ+
H
O
+
α
R2
OH
DABCO
β
Sistema Michael
R1
O
R2
528
A. Isenmann
Princípios da Síntese Orgânica
Em casos favoráveis podem ser usados até haletos de alquila como aceitador; nestes casos
aminas não servem como catalisadores, mas devem ser substituídas pelas fosfinas que não
reagem diretamente com o haleto de alquila 331.
O
Br
O
δ+
O
δ−
O
PBu3
OEt
OEt
OEt
OEt
O
O
Além destas, é conhecida como reação aza-Baylis-Hillman, onde o eletrófilo é o carbono
sp² de uma imina 332.
Mecanismo simplificado 333:
A adição do DABCO no composto α,β-insaturado leva a um intermediário carregado do
tipo betaina (definição ver nota de rodapé 437, p. 779). Este se adiciona ao aldeído
eletrofílico o que leva ao 1,3-cetoálcool. A expulsão do DABCO, finalmente, fornece o
álcool alílico.
O-
O
R2
+
N
O
O
N
H
R1
N
N
R2
R1
OOH
R2
H
- DABCO
N
O
R2
R1
N
Muito semelhante a esta síntese é a mais antiga reação de Rauhut-Currier (1963). O
catalisador, por exemplo, é o mesmo. A sua aplicação, por outro lado, tem muitas restrições
por ser pouco regiosseletiva. Portanto, é acompanhada por uma série de reações paralelas
quando usada em condensações cruzadas. Porém, dois casos especiais sejam apresentados
onde o rendimento foi satisfatório: uma dimerização e uma condensação intramolecular.
331
M.E. Krafft, K.A. Seibert, T.F.N. Haxell, C. Hirosawa, Unprecedented reactivity in the Morita-BaylisHillman reaction; intramolecular alkylation of enones using saturated alkyl halides. Chem.Commun. 46
(2005) 5772.
332
V.D.B. Bonifacio, Enancioselective aza-Baylis Hillman reaction. Org. Chem. Highlights 2006 (acessível
por: http://www.organic-chemistry.org/highlights/2006/30januaryA.shtm)
333
F. Coelho, W.P.Almeida, Reação de Baylis Hillman: uma estratégia para a preparação de intermediários
multifuncionalizados para a síntese orgânica, Quim. Nova 23 (2000) 98.
529
A. Isenmann
Princípios da Síntese Orgânica
1) Dimerização
O
O
+
EtO
O
1 eq. Bu3P
OEt
MeCN, 40°C, 3h
O
EtO
OEt
Acrilato de etila
46%
2) Condensação intramolecular
O
O
O
0,2 eq. Bu3P
O
EtOAc, 50 °C
86%
Note que este tipo de catálise é um novo campo da química dos organofosforados (compare
p. 791).
6.6.3 Resumo: regioseletividade na adição em compostos carbonílicos
α,β
β-insaturados
Bem mais importante do que a reação no carbono α, conforme descrito logo acima, é a
adição de nucleófilos ao carbono β, em sistemas conjugados C=C-C=O. Nas páginas 519 a
527, essa adição foi denominado de "Adição de Michael", ou "adição 1,4". Também foi
mencionada nesta discussão a concorrência em forma da "adição 1,2" que, na maioria dos
propósitos preparativos, leva a um produto paralelo indesejado.
Como estes caminhos são concorrentes inseparáveis, a seguinte tabela resume os fatores
que direcionam a adição do nucleófilo, para o sentido 1,2 e 1,4, respectivamente.
530
A. Isenmann
Princípios da Síntese Orgânica
Tabela 29.
Fatores que direcionam a adição de nucleófilos em compostos
carbonilados α,β-insaturados.
Critério
Adição direta
Adição de Michael
Estabilidade termodinâmica menor (perdeu a ligação maior (perdeu somente C=C)
do produto
estável C=O)
Força da dupla ligação
C=O : maior
C=C : menor
Reversível?
sim
não
Velocidade
maior
menor
Polarizabilidade do aceitador menor (carbonila = centro maior (β-carbono = centro
duro)
mole)
Aceitadores típicos
O
C C C
O
C C C
Cl
O
C C C
OR
O
C C C
H
C
Qualidades do nucleófilo:
alta basicidade
promove
atrapalha
alta polarizabilidade
atrapalha
promove
Nucleófilo estável (= pouco atrapalha
reativo)
promove
Nucleófilos típicos
são de natureza dura:
são de natureza mole:
RLi, NH2-, RO-, H-
R2CuLi (p. 520), melhor do
que RMgBr, aminas neutras,
carbânions
mais
RS-,
estáveis, enaminas
Redução seletiva (p. 590) do C=O com NaBH4 (aldeídos, H2/Catalisador
sistema C=C-C=O
cetonas); LiAlH4 (ésteres,
Esta redução não é iônica,
cloretos de acilas)
então procura reduzir a dupla
ligação que é mais simétrica
e mais fraca.
531
A. Isenmann
6.7
Princípios da Síntese Orgânica
Condensações com pseudo-ácidos sem grupo carbonila
Pseudo-ácidos são compostos com o elemento C-H que funciona como ácido de Brønsted,
quer dizer, que pode soltar o próton. A acidez C-H em alcanos e alquenos é extremamente
baixa, portanto não podem ser considerados pseudo-ácidos. Todos eles têm em comum a
propriedade de não liberar um próton ao serem tratados com bases. Isto se reflete nos
valores muito altos de pKa, que são acima de 35 para todos estes compostos (ver p.
186).Contudo, o carbono deve fazer parte de uma estrutura especial que distribui e
estabiliza a carga negativa que remanesce no carbânion da base conjugada.
Condensação com acetileno
O acetileno, como elucidado no item 2.1.2, tem um pKa notavelmente mais baixo do que
alquenos e alcanos. Isto se deve às particularidades do orbital híbrido sp usado para fazer a
ligação com o hidrogênio: em comparação ao orbital sp² e sp³ ele é menor, mais
arredondado em volta do C - resumindo, o orbital sp proporciona ao carbono maior
eletronegatividade. A acomodação de dois elétrons não ligantes neste orbital, portanto,
custa menos energia, do que a ocupação de sp²ou sp³ com dois elétrons. O carbânion sp é
termodinamicamente mais estável. A condensação com uma unidade do acetileno se chama
“etinilação” (ver também p. 186).
CH
HC
CH
KOH
HC
C-
+ Acetona
H3 C
H3 C
1) H2 / Catalisador de Lindlar
2) + H+ ; - H2O
C
C
O-
+
H3O
CH
H3 C
H3 C
C
C
OH
CH3
H2 C
C CH CH2
Isopreno
O produto desta síntese é isopreno, um importante monômero em reações de polimerização
334
.
O acoplamento com o ânion acetilídeo funciona até com moléculas de acetileno entre si. Ao
acoplar um grande número de acetilenos se consegue, sob condições especiais, um
polímero com propriedades elétricas extraordinárias (ver p. 183).
O ânion acetilídeo é de alto valor estratégico na síntese orgânica, devido à sua facilidade de
ser transformado em um grupo carbonila 335, após o acoplamento com um aceitador (a
terminologia de aceitador, ver p. 482).
334
Esses polímeros têm qualidades mecânicas excepcionais (borracha natural, copolímeros elastoméricos,
guttapercha). Também é usado na (bio-)síntese de oligômeros, tais como terpenos, sesquiterpenos,
diterpenos,....
335
M.F. Ansell, W.J.Hickinbottom, A.A. Hyatt, J.Chem.Soc. 1955, 1592.
532
A. Isenmann
Princípios da Síntese Orgânica
O
H C
C
H
OH
NaNH2 em NH3(liq)
H C
C
Ânion acetilido
= nucleófilo
OH
C CH
C CH
OH
Hg(II)
C
H+, H2O
O
Pode-se afirmar que o ânion acetilídeo é um equivalente estrutural do “ânion acila”, um
sinton (definição, ver nota de rodapé na p. 389) que em si não existe. Observe que uma
polarização negativa do carbono no grupo acila ou carbonila é contra a sua natureza!
Assim, abre-se um importante caminho para produzir compostos 1,2-difuncionais, via
condensação. Existem relativamente poucas sínteses confiáveis para compostos 1,2dicarbonilas - ao contrário de produtos 1,3-dicarbonilas e 1,5-dicarbonilas. Outras rotas
sintéticas para compostos 1,2-dicarbonilas foram apresentadas com a condensação de
aciloína e benzoína, seguido por oxidação, p. 465 (nestas tem-se uma severa restrição
referente ao esqueleto carbônico: o produto sempre é simétrico).
Sintons no planejamento da síntese (note que somente o primeiro pode ser facilmente feito,
mas não o segundo):
H C
C
=
C
O
CH3
ânion acetilida
ânion acila
(não-natural)
Condensações com nitroalcanos
O grupo nitro, mais ainda do que o grupo carbonila, é retirador de elétrons. Ele polariza as
ligações vizinhas de tal maneira que resulta uma acidez C-H notável em posição α (pKa =
10; ver tabela na p. 505). Então não é difícil desprotonar o nitroalcano e a sua base pode
preencher o papel do componente nucleofílico no sentido de uma condensação, também
conhecida como condensação de Henry. Com apoio de microondas essa condensação
demorada ocorre em apenas 7 minutos 336:
336
R.S. Varma, R. Dahiya, S. Kumar, Tetrahedron Letters 38 (1997) 5131.
533
A. Isenmann
Princípios da Síntese Orgânica
O
CH3NO2
OH
Equil.
ácido-base
H2C N
H2C N
Equil.
ácido-base
+ H2O ; - OH-
O
O
-
O
O
O-
C6H5 C
H
C6H5 CH CH2 NO2
Condensação
Nitronato
∆
OH
C6H5 CH CH2 NO2
[ OH- ]
- H2O
C6H5 CH CH NO2
Elimin. β
A primeira etapa é a desprotonação em posição α que leva ao ânion nitronato, estabilizado
por mesomeria, como pode ser verificado nas duas fórmulas de ressonância. O próprio
composto nitro não aproveita de uma estabilização deste tipo. Ao invés desta, ele somente
está em equilíbrio com a forma aci (tautomeria aci-nitro, ver pp. 665 e 400) que não traz
vantagens energéticos.
Lembre-se: uma mesomeria não deve ser confundida com um equilíbrio entre dois
isômeros! Mesomeria realmente abaixa a energia interna, então aumenta a estabilidade
termodinâmica do composto. Por outro lado, um equilíbrio químico implica a
transformação de um composto em outro, mas não melhora a sua situação energética.
É assim, no caso do composto nitro: ele está em equilíbrio com a forma aci,
O
H3C N
nitro
O
O
H2C N
aci
O
, enquanto as duas formas têm energias internas altas.
Vantagens energêticos, no caso do nitronato, significam: desprotonação fácil do
nitrometano. A partir deste exemplo pode-se generalizar que sempre se espera alguma
acidez C-H quando a base conjugada for estabilizada por mesomeria ou quando se forma
um sistema conjugado, formado por duplas ligações e/ou pares de elétrons livres.
Na segunda etapa ocorre o ataque nucleofílico do nitronato ao carbono do benzaldeído.
Nesta etapa é realmente o carbono negativado e não o oxigênio que se liga ao carbono do
grupo carbonila (ver também questão No. 15, na p. 539). Pode-se perguntar: por que o
carbono que efetua o ataque nucleofílico ao benzaldeído e não o oxigênio - que é
igualmente com carga negativa? A resposta pode ser dada em duas partes:
1) O produto que resulta do ataque do oxigênio, não é estável, então pode-se esperar
reversibilidade desta reação.
2) Um contato entre dois carbonos é favorável, no conceito de Pearson: é um contato molemole (ver cap. 10.1.3), enquanto o oxigênio representa um centro duro (= pouco
polarizável). Portanto, é menos adequado para estabelecer um contato ligante.
Comparando essa reação com as condensações apresentadas no início deste capítulo,
verifica-se que o nitronato tem seu equivalente no enolato de um composto com grupo
534
A. Isenmann
Princípios da Síntese Orgânica
carbonila 337. Uma diferença fundamental, porém, existe entre esses reagentes: o composto
nitro sempre é o componente metilênico (= nucleófilo = pseudo-ácido; ver definições na p.
482) porque não tem centro positivado que poderia funcionar como aceitador. Isso é
claramente uma vantagem, em cima de muitas outras condensações mistas, onde a
ambivalência dos compostos pode levar a uma mistura de diferentes produtos.
A disponibilidade comercial dos nitroalcanos se restringe ao nitrometano e 2-nitropropano.
Mas isso não mingua o valor preparativo desta classe. Conforme o esquema a seguir, é
possível acessar uma grande variedade de compostos nitro no laboratório.
H3C NO2
Base, CTF
Nitronato
R X
R CH2 NO2
Esta reação não só funciona com R-X sendo haletos de alquila, mas também com outros
compostos de carbono positivado, tal como vinílicos, H2C=CH-X (X = retirador de
elétrons) 338.
Reação de Nef
O produto da condensação descrito acima faz uma reação interessante que aumenta bastante
sua versatilidade: a transformação do grupo nitro em um grupo carbonila, conhecido como
reação de Nef 339. Uma aplicação concreta deve mostrar a utilidade desta reação: a
condensação com nitrometano e a reação de Nef servem para aumentar a cadeia carbônica
em açúcares:
337
Note-se que essa síntese satisfaz a definição mais ampla de “condensação”, que é a criação de uma nova
ligação C-C, através de um carbono nucleofílico e um carbono elétrofílico.
338
Uma vista geral sobre as reações dos compostos nitro se encontra no anexo 2 deste livro. Um artigo de
revisão abrangente: D. Green, T. Johnson, "Nitroalkane chemistry", encontra-se na rede (acesso em 12/2010):
www.iptonline.com/articles/public/IPTFOUR79NP.pdf
339
R. Ballini, M. Petrini, Tetrahedron 60 (2004) 1017-1047.
535
A. Isenmann
Princípios da Síntese Orgânica
O
HC O
-
+
+ [ H2C NO2 ] Na
Na+
O
N
Na
O
+
CH
R
CH
HO
CH OH
CH
R
R
D-arabinose
N
O
Nef
R
=
HO
CH
CHO
CHO
HC OH
HO
CH OH
HC OH
CH
R
R
D-glicose
D-manose
CH2OH
Mecanismo:
A reação de Nef não é uma reação redox, mas uma hidrólise dos compostos nitro, na sua
forma aci. A reação no nitronato requer um ambiente fortemente ácido – se não se forma
uma série de produtos paralelos (oximas, compostos hidroxinitrosos e outros). Ao gotejar o
composto nitro em ácido sulfúrico 50% se consegue aldeídos e cetonas com rendimentos de
até 85%. O mecanismo é via dupla protonação do produto primário da condensação:
R
O
C N
R´
Na+
R
H+
O
R´
base conjugada
(em pH > 10)
H+
O
R
OH
C N
R´
H2O
OH
R´ OH
R
N OH
OH H
forma aci
R´ OH
R
OH
C N
R
C O +
N OH
OH H
R´
H+ + "HNO"
x2
H2O + N2O
O desprendimento de uma molécula de óxido nitroso (gás hilariante; N2O), a cada dois
compostos nitro, faz com que a reação fique irreversível.
Infelizmente, as condições rigorosas aplicadas na reação de Nef podem causar danos em
outros grupos funcionais. Para estes casos foram desenvolvidos métodos de transformar o
536
A. Isenmann
Princípios da Síntese Orgânica
grupo nitro em carbonila sob condições mais brandas. O seguinte método oxidativo, por
exemplo, anda até em solução tamponada de acetato 340:
C N
CH NO2
C N
O
-
- HSO4-
O
O SO3H
R
-
N O
C
R´
O
O-
R
O-
R
+ HSO5-
-
R´
R´
R´
O-
R
[OH-]
R
+ NO2-
O
R´
O
Mas também métodos redutivos foram desenvolvidos. O reagente de McMurry, TiCl3 (ver
p. 468) se liga sob alta exotermia ao oxigênio do nitronato e o leva embora na segunda
etapa 341. Dois fatos promovem esta reação: a alta afinidade do titânio para o oxigênio e sua
preferência de ficar no NOX +4. Resulta então uma oxima que hidrolisa facilmente, em
ambiente aquoso ligeiramente ácido, liberando a cetona:
O-
R
C N
R´
O
-
TiCl3
R
OTiCl2
C N
R´
O
Nitronato
-
R
(- TiO2)
H2O / H+
R
O
N
R´
OH
R´
Oxima
O valor preparativo de compostos nitro é o mesmo do que do acetileno discutido na p. 532.
Os dois podem ser desprotonados (enquanto o composto nitro com muito mais facilidade!)
e os carbânions que resultam podem condensar com outra componente que possui um
carbono positivado. Depois, os produtos de condensação podem ser hidrolisados formando
um novo grupo carbonila - sem necessidade de trocar o NOX dos carbonos. Por esta razão
podemos considerar os compostos nitro e acetileno sendo "sintons latentes" que introduzem
o grupo carbonila e aumentam a cadeia carbônica.
Condensação no carbono benzílico
Embora os hidrogênios do tolueno não serem ácidos, a posição benzílica tem elevada
reatividade, como elucidado no capítulo 1 (p. 46). A facilidade de trocar seus hidrogênios
ainda aumenta caso presentes retiradores de elétrons no anel, especialmente em posição
orto ou para. Aqueles anéis aromáticos que facilmente reagem no sentido SN (ver p. 325),
340
341
P. Ceccherelli, Synth.Commun. 28 (1998) 3057.
J.E. McMurry, Acc.Chem.Res. 7 (1974) 281; J.E. McMurry, J. Melton, J.Org.Chem. 38 (1973) 4367.
537
A. Isenmann
Princípios da Síntese Orgânica
se tiverem um grupo -CH3 em posição benzílica, são candidatos potenciais de funcionar
como componente pseudo-ácido em condensações.
Assim se explica também porque o nucleófilo que deve atacar o anel no sentido de uma SN,
está concorrendo a entrada em posição benzílica. É a sua basicidade que acompanha
naturalmente a maioria dos nucleófilos, que o deixa entrar ("condensar") na cadeia lateral
(ver item 4.8). Dependendo do propósito preparativo, deve-se usar um reagente de alta
nucleofilia e baixa basicidade (por exemplo, bases de N prim. e sec., S, P, As,...) ou de
baixa nucleofilia e alta basicidade (por exemplo, t-butóxido ou LDA), para direcionar o
caminho da síntese.
Os dois substratos mostrados a seguir, são apropriados para reações de condensação após a
sua desprotonação em posição benzílica:
CH3
NO2
N
NO2
2,4-dinitrotolueno
CH3
o-metilpiridina
Segue um exemplo que mostra a facilidade com que ocorre essa condensação.
Trata-se da síntese de Ladenburg, fornecendo o alcalóide coniina que é de elevada
toxicidade e por sua vez material de partida para outros alcalóides.
H
C CH3
H2 / Cat.
O
N
CH3
Base
- H2O
N
N
CH CH CH3
D,L-coniina
Condensação com ciclopentadieno
A desprotonação do ciclopentadieno é extraordinariamente fácil (pKa = 14,5; compare com
alquenos “comuns”: pKa ≈ 37). Isto se deve à estabilidade aromática que se aplica somente
à forma desprotonada, o ânion ciclopentadienila (Cp-). A vantagem energética devido à
aromaticidade foi constatada com ≈ 150 kJ mol-1 (ver p. 284).
CH2 + OH-
+ H2O
CH
aromático!
538
A. Isenmann
Princípios da Síntese Orgânica
(Note-se que CpH e água têm aproximadamente a mesma acidez, então espera-se a posição
deste equilíbrio bem no meio.)
Com compostos carbonílicos o Cp- reage formando fulvenos.
O
H
C
+ R C
H
6.8
H
OH
C
CH O-
CH
R
C C
- OH-
R
Fulveno
R
Exercícios de Condensação de Compostos com grupo
Carbonila
1) Compare as autocondensações de aldeídos, cetonas e ésteres, em termos de
a) catálise,
b) facilidade de execução e
c) rendimentos.
(pp. 454 a 459)
2) Reações consecutivas dos produtos de condensação
(p. 462)
a) Explique as expressões "quebra de ácido" e "quebra de cetona".
b) Denomine as precondições estruturais do substrato que faz essas quebras.
c) Quais são as condições reacionais e reagentes, para efetuar essas quebras?
3) Descreve detalhadamente as etapas da síntese, do composto 1,3-dicetona, usado na p.
463 (Síntese de Hüning).
4) Na p. 465, foi alegado que uma condensação de benzoina com 2-fenilacetaldeído não
leva ao produto desejado:
Ph
CH2
CHO
CN(H2O / EtOH / pH 8)
O OH
Ph
CH2 C CH CH2 Ph
Não funciona!
a) Procure um argumento por que o benzaldeído faz essa autocondensação, mas não o 2fenilacetaldeído, sob as mesmas condições reacionais.
b) Qual é realmente o produto desta condensação?
c) Como poderia ser realizada a síntese da aciloína do esquema acima?
5) Acoplamento redutivo de pinacol
(p. 467)
539
A. Isenmann
Princípios da Síntese Orgânica
a) Qual é o produto do acoplamento de pinacol, a partir da butanona?
b) Qual é o produto consecutivo do rearranjo de pinacolona?
6) Condensações cruzadas
(p. 482)
a) Explique, no exemplo do acetato de etila com acetona, quais produtos (paralelos) podem
esperar-se numa condensação cruzada.
b) Quais são as estratégias, para direcionar uma condensação cruzada?
7) Condensação de Mannich
(p. 491)
a) Formule a condensação de Mannich, a partir de acetofenona, formaldeído e cloreto de
dimetilamônio.
b) A amina aplicada nesta síntese multi-componente, tem um papel ativador e um papel
desativador sobre o formaldeído, ao mesmo tempo. Explique.
c) Quais problemas poderíamos esperar, ao deixar a amina secundária (geralmente
dietilamina) embora?
d) Quais problemas poderíamos esperar, ao substituir a dietilamina por uma amina
primária?
e) Indique uma utilidade sintética da base de Mannich sendo o produto da reação do item a.
8) Condensação de Mannich ampliada
(p. 491)
A síntese de Mannich geralmente se restringe ao formaldeído sendo componente
carbonílico. Em uns casos especiais esta síntese pode ser ampliada a outros aldeídos, um
dos quais é o aldeído succínico. Com este aldeído se abre um caminho interessante que leva
a uma variedade de alcalóides.
Uma série de alcalóides pode ser produzida onde a reação chave é a condensação de
Mannich. Assim, obtém-se a tropinona, um composto intermediário na produção de
atropina, pela dupla condensação de Mannich entre aldeído succínico, metilamina e o
diácido carboxílico de acetona. Também esta síntese é possível sob "condições
fisiológicos". O diácido carboxílico de acetona pode ser obtido facilmente, ao tratar ácido
cítrico, HOOC-CH2-C(COOH)(OH)-CH2-COOH, com ácido sulfúrico concentrado.
O
HOOC
CH2
CH2
O
COOH
HOOC
HC
H
C
CH2 CH2
O
O
NH2
CH3
C
CH
COOH
O
H
- 2 H2O
HC
CH
- 2 CO2
N
N
CH3
a) O aldeído succínico, usado como material de partida, é um componente metilênico
duplamente ativado? Justifique.
540
CH3
A. Isenmann
Princípios da Síntese Orgânica
b) O diácido carboxílico de cetona, usado como material de partida, é um componente
metilênico duplamente ativado? Justifique.
c) Formule o mecanismo desta condensação.
9) Alquilação de compostos carbonílicos duplamente ativados
(p. 510)
a) Anote a reação de condensação entre acetoacetato de etila e cloreto de benzila.
b) Destaque quais os perigos e produtos paralelos esperados.
c) Descreva as providências que devem ser tomadas para reprimir os produtos paralelos
(orienta-se na lista negativa na p. 510).
10) (Questão especial)
(p. 510 e 52)
a) Explique por que o 2-fenil malonato de dietila não é acessível via condensação,
conforme descrita na última questão, a partir de malonato de dietila, base e clorobenzeno.
COOEt
Ph
HC
COOEt
2-Fenilmaloato de dietila
b) Procure uma estratégia alternativa para este produto. Dica: procure a solução num
derivado do ácido carbônico (ver p. 490).
11) Compostos carbonílicos duplamente ativados
(p. 513)
a) O que se entende por condensação de Knoevenagel?
b) Formule a condensação de Knoevenagel, entre acetoacetato de etila e acetaldeído.
12) Regiosseletividade em compostos carbonílicos duplamente ativados
(p. 514)
a) Formule a condensação, acetilacetonitrila com cloreto de acila. Indique todas as etapas
relevantes, na sequência certa.
b) Em certo momento da condensação se abre a questão: onde o carbono eletrofílico do
cloreto de acila procura os elétrons: no oxigênio ou no carbono, do componente metilênico.
Como poderiamos influenciar esta regiosseletividade? (p. 515)
13) Acilação de compostos duplamente ativados
(p. 515)
Formule a condensação entre acetilacetona e o cloreto de benzoíla. Indique a base
preferida, usada para a desprotonação da acetilacetona.
14) Anelação de Robinson
(p. 526)
a) Explique em palavras (sem fórmulas químicas) o funcionamento/estratégia, da anelação
de Robinson.
b) Qual é o valor preparativo da anelação de Robinson?
541
A. Isenmann
Princípios da Síntese Orgânica
15) Na p. 533 foi mencionado um ataque nucleofílico do ânion nitronato, ao carbono do
grupo carbonila do benzaldeído. Desenhe a(s) fórmula(s), do produto hipotético que
resultará do ataque pelo oxigênio negativado do nitronato.
16) Questão mais complexa, incluíndo a condensação de Mannich, condensação de Michael
e uma quebra de cetona:
Exemplo de síntese de um composto natural: o aminoácido triptofano
O aminoácido triptofano (composto 8 no esquema abaixo) pode ser sintetizado a partir de
indol (composto 1), por uma reação de Mannich. Note-se que para o indol existem duas
formas tautoméricas:
4
5
H
3
H
2
6
7
N
Enamina
H
Tautomeria
N
Imina
Podemos esperar deste equilíbrio que a forma reativa do indol é a enamina que se destaca
por dispor de uma dupla-ligação C=C especialmente rica em elétrons. Sendo assim, a
reatividade frente eletrófilos é mais alta em posição 3 (ver p. 322).
Forma-se a gramina (composto 4) que representa em seguida a base de Mannich. Ela é
desprotonada no nitrogênio por uma base forte e elimina a dimetilamina formando 3metilenoindol (composto 5). Esta etapa reacional é uma eliminação β, que, na verdade, é
uma eliminação forçada de Hoffmann (ver p. 142).
O composto com grupo metileno, composto 5, representa um sistema Michael! Então pode
reagir com um componente metilênico, o N-acetamido-malonato de dietila (composto 6),
que está completamente desprotonado nas condições aplicadas (NaOH).
Resulta o escatilacetaminomalonato de dietila (composto 7) cuja hidrólise e
descarboxilação térmica fornece triptofano.
542
A. Isenmann
Princípios da Síntese Orgânica
H
CH3
N
1
Indol
CH2 N(CH3)2
Mannich
+ H2 C N
CH3
H
N
2
3
CH2 N(CH3)2
1) CH3CONH C(COOEt)2
2) H+
N
N
H
Base
6
CH2
Hofmann
- H+
H
5
4
Gramina
COOEt
O
CH2 CH COOH
CH2 C NH C
COOEt
N
7
CH3
NH2
1) H+ / H2O
2) 80 °C : - CO2
H
N
H
8
Triptofano
Questões:
a) o composto 5 é um melhor ou um pior aceitador de Michael do que etilvinicetona?
Argumente em termos de efeitos I e M.
b) Como foi preparado o composto 6?
c) Escreva detalhadamente o que acontece na etapa 7 → 8.
6.9
Respostas aos exercícios de Condensação de Compostos
com grupo Carbonila
1) Comparação das autocondensações:
Critério
Aldeídos
Catálise
É catalisada por bases e
por ácidos; catalisadores
em baixas concentrações
(pH entre 2 e 11).
Ambiente pode ser aquoso.
Cetonas
Bases: a
condensação é
reversível.
Ácidos: irreversível.
Ambiente pode ser
aquoso.
Ésteres
Somente por bases,
de preferência um
alcóxido, para não
saponificar o éster.
São catalisadores
especiais.
543
A. Isenmann
Princípios da Síntese Orgânica
Facilidade
da Muito fácil; sob catálise
autocondensação básica: reversível.
Média.
Rendimento da Rendimentos altos (até
autocondensação 80%).
Na catálise básica é
necessário separar ou
sequestrar o produto,
para obter
rendimentos
satisfatórios
(Ba(OH)2 e Soxhlet).
Só em ambiente
anidro.
Pouco reativo (pouca
acidez em posição
α).
até 50%.
2) Reações seguidas dos produtos de condensação carbonílica
a) As expressões "quebra de ácido" e "quebra de cetona" não se referem às condições
(catalisadores) sob quais estas reações andam, mas sím, à natureza química do produto.
Sendo assim, o produto da quebra de ácido é um ácido carboxílico, pois essa reação é a
reversa da condensação de Claisen. Ela anda sob catálise básica, em condições que
geralmente provocam a hidrólise de ésteres. Portanto, não é exatamente a reversa, mas as
etapas mecanísticas são idênticas à da condensação.
Já a quebra de cetona fornece uma cetona simples. O composto 1,3-bifuncional, produto da
condensação de Claisen, descarboxila, logo tiver uma função de ácido carboxílico livre (COOH).
b) Para a quebra de ácido é vantajoso usar um produto 1,3-bifuncional que não contém
grupos -COO- livres, para não favorizar a concorrência da clivagen de cetona.
Já a condição para uma fácil descarboxilação é a disposição de um grupo carbonila ou
carboxila em posição γ, ao grupo -COOH clivável.
c) Quebra de ácido: ambiente fortemente básico. Pode ser em ambiente aquoso; para evitar
a liberação do carboxilato livre é melhor trabalhar em ambiente alcoólico.
Quebra de cetona: ambiente neutro a ácido (aquoso) e calor (100 °C).
3) Condensações cruzadas, estratégia da desprotonação quantitativa
O produto da síntese de Hüning, do composto 1,3-dicetona, é resultado de uma
condensação cruzada. Foi feito a partir da ciclohexanona e um cloreto de acila. Como a
cetona não é duplamente ativada e mostra a tendência para autocondensação, então
oferecem-se duas estratégias:
1) Estabilização da forma enólica - geralmente feita via enamina (p. 495);
2) Desprotonação rápida e quantitativa da cetona, numa etapa prévia (p. 502).
A mais utilizada é a segunda estratégia porque envolve uma etapa preparativa (desproteção
da cetona) e duas etapas de purificação (destilação da amina e da enamina) a menos:
544
A. Isenmann
Princípios da Síntese Orgânica
O-
O
O
O
LDA
R
O
C
COCl
R
- H+
Enolato quantitativo
4) Condensação de aciloina
a) Na cianidrina a partir do benzaldeído, são dois vizinhos ao carbono em destaque que
contribuem à estabilização da sua forma desprotonada: o grupo ciano (efeitos -I e -M) e o
grupo fenila(efeito -M). Já na cianidrina, a partir do 2-fenilacetaldeído, falta o apoio pelo
grupo fenila: aqui os elétrons π do anel de fenila e o par de elétrons do carbânion são
separados, um do outro, por um grupo -CH2-. Assim, não se tem a conjugação nem a
estabilização necessária, para produzir o carbânion em concentrações suficientemente altas.
A autocondensação, sob as condições brandas típicas (pH < 10), não funicona.
b) É uma condensação aldólica comum (p. 454). O rendimento seria baixo, devido à
reversibilidade.
O
O
Ph
CH2
C
H
+ OH
+ H2O
O
PhCH2
CH CH C
O-
Ph
H
PhCH C
- OH-
H
PhCH C
H
+ H2O
C CH2 Ph
O-
O
-
H
O
PhCH2
CH CH C
OH
Ph
H
c) Através da cianidrina protegida: por etilviniléter (enoléter, p. 499) ou por cloreto de
trimetilsilila (p. 501).
5) Acoplamento redutivo de pinacol
a) A butanona é uma cetona assimétrica, portanto se obtêm três produtos isômeros
diferentes, dois enanciômeros e uma forma meso. A redução é geralmente feita por metais
alcalinos, o mecanismo é via SET:
545
A. Isenmann
H5C2
C O
Princípios da Síntese Orgânica
Na / Xileno
1) Dimerização
dos radical-ânions
2) Neutralização
H3C
Na+
H3C
Radical-ânion cetila
C2H5
C2H5
C2H5
HO
C O
C O
H3C
H3C
H5C2
H5C2
C CH3 H3C
C OH
C OH HO C CH3
C2H5
C2H5
Enanciômeros (D,L)
H3C
C OH
H3C
C OH
C2H5
meso
b) Este pinacol, submetido a um ácido mineral forte, sofre rearranjo no esqueleto
carbônico. Espera-se uma migração preferencial do grupo etila. Devido à alta simetria do
pinacol formam-se somente duas pinacolonas (não tem mais isômeros ópticos):
O
C
C
O
CH3
e
C
C
Produto principal
6) Condensações cruzadas
a) Uma "Condensação de Claisen cruzada”, com um éster e uma cetona, é possível sob
catálise de uma base. Para não provocar uma saponificação do éster se dá preferência a um
alcóxido (em vez de hidróxido), sem solvente adicional. Isso funciona porque o éster é
menos α-ácido e menos enolizável do que a cetona.
546
A. Isenmann
Princípios da Síntese Orgânica
O
O
H3C
OEt
CH3
O
H3C
O
-
H3C
O
CH
CH2
O
- H+
CH3
Ph
+ H3C C
O
H3C
OEt
CH2
O
CH
H
pKa < 10
Acetilacetona, "acac"
Depositado em
forma de sal
Como todas etapas são reversíveis, o produto da condensação deve ser sequestrado em
forma do enaolato, um sal que se deposita. O produto acac, aliás, é um importante ligante
quelatizante para os metais de transição.
Quando se tem, por outro lado, uma cetona (A) e um éster (B) que têm propriedades
semelhantes, tanto na enolizabilidade quanto na qualidade como aceitador, temos que
contar com uma mistura de quatro produtos de condensação: dois produtos de
autocondensação, A-A e B-B, e dois condensados cruzados, A-B e B-A.
b) As estratégias gerais, para direcionar uma condensação cruzada, são:
1) Ativação do pseudo-ácido pela estabilização da forma enólica de um participante,
seja na forma da enamina, do enoléter ou do sililenoléter.
2) Com bases muito fortes se consegue desprotonar o pseudo-acido quantitativamente,
em uma etapa preliminar. Somente depois se acrescenta o outro componente.
3) Emprego de um pseudo-ácido duplamente ativado que enoliza e desprotona muito
mais fácil do que o componente carbonílico. O produto desta condensação de
Knoevenagel pode ser hidrolisado e o grupo carboxílico, -COOH, pode ser
desprendido em forma de CO2 (descarboxilação). Assim, o grupo do éster tinha o
papel de um "ativador" do componente pseudo-ácido.
7) Condensação de Mannich
a) A forma enólica da acetofenona representa o componente nucleofílico, o íon imínio
formado a partir de um formaldeído e uma molécula de dimetilamina providencia o
carbono eletrofílico.
547
A. Isenmann
Princípios da Síntese Orgânica
O
Ph
C CH3 + H2C O + H2N(CH3)2
O
- H2O
Ph
H
C CH2 CH2 N(CH3)2
Cl-
Base de Mannich protonada
Hidrocloreto da N,N-dimetilamino-propiofenona
b) Papel ativador da amina:
o A carga positiva, no produto da reação entre amina e formaldeído, já indica
que essa espécie fica mais eletrofílica frente o componente metilênico, do
que o formaldeído livre.
Papel desativador sobre o formaldeído:
o Reduz a tendência do formaldeído de entrar em autocondensação, formando
poli-oximetileno, (-O-CH2-)x.
o Diminui o risco de condensar o formaldeído repetitivamente em compostos
metilênicos que têm mais de um α-H. Além de fatores eletrônicos, é o
tamanho pequeno do formaldeído livre que facilita uma dupla-condensação
com um grupo metileno.
o Reduz a tendência do formaldeído fazer desproporcionamento de
Cannizzaro ou até reduzir algum componente aldeídico na mistura, através
de uma reação de Cannizzaro cruzada (p. 423).
c) Além de todos os perigos citados no item b, uma eletrofilia inferior do formaldeído
levaria à autocondensação do outro componente carbonílico (aldeído ou cetona).Quer dizer,
em nosso exemplo, uma segunda molécula da própria acetofenona entraria como aceitador.
d) Uma amina primária ou até amônia, NH3, têm mais de um hidrogênio a ser trocado pelo
formaldeído. Assim, formam-se produtos diferentes. Além do mais, pode-se formar uma
base de Schiff, da estrutura R-N=CH2 que seria um eletrófilo muito fraco:
548
A. Isenmann
Princípios da Síntese Orgânica
R NH2 + H2C O
R NH2 CH2 O-
R NH CH2 OH
- H2O
R N CH2
Base de Schiff
+ H+
+ H2C O
CH2 OH
R NH CH2
R NH CH2
R N CH2
Forma protonada da base de Schiff
(eletrófilo fraco)
produto paralelo
e) A utilidade principal da base de Mannich, produto direto da aminometilação, é a sua fácil
transformação em um composto carbonílico α,β -insaturado, R-CO-CH=CH2, que, por sua
vez, é muito reativo e sensível à oxidação e polimerização. Outro exemplo de reação
consecutiva é a dimerização da vinilcetona: a reação decorre com bastante facilidade e pode
ser identificada como hetero-Diels-Alder (p. 252):
O
O
O
O
Então a base de Mannich é uma forma de proteger o grupo vinila que pode ser liberada
somente na hora da sua aplicação.
A base de Mannich (β-aminocetona) também pode ser facilmente reduzida, fornecendo um
β-aminoálcool, substância que mostra atividade biológica.
8) Condensação de Mannich ampliada
a) O aldeído succínico não é um componente metilênico duplamente ativado porque não
tem nenhum grupo metileno, -CH2-, com dois vizinhos retiradores de elétrons.
b) O diácido carboxílico de cetona é um componente metilênico duplamente ativado, sim.
Têm-se até dois grupos metilenos que podem ser facilmente desprotonados. Este composto
apresenta alta porcentagem de forma enólica.
c) A amina usada nesta condensação é uma amina primária, então tem dois hidrogênios que
podem ser trocados por carbonos. Certa quantia da base iria desprotonar o diácido
carboxílico de cetona. Uma carga negativa, por outro lado, repele nucleófilos e atrai
549
A. Isenmann
Princípios da Síntese Orgânica
eletrófilos. O eletrófilo formar-se-á a partir do succinaldeído, dando um íon imínio onde o
carbono fica mais positivado do que no aldeído da partida. Ocorrerá a condensação entre o
enol/enolato do diácido carboxílico de cetona e o carbono proveniente do succinaldeído.
Como as distâncias intramoleculares são favoráveis, esta etapa de condensação se repete,
finalizando em um composto alcalóide bicíclico.
9) Alquilação de compostos carbonílicos duplamente ativados
a) A partir de acetoacetato de etila e cloreto de benzila forma-se com rendimentos médios o
β-cetoéster benzilado. A condensação requer da catálise básica, que é o etóxido no exemplo
a seguir. Esta reação, do ponto de vista do cloreto de benzila (grupo funcional num carbono
primário), é uma SN2.
O
H3C
O
O
C
C
CH2
ClCH2
OC2H5
+
[ C2H5O Na ]
H3C
C
O
CH
C
OC2H5
CH2
Acetoacetato de etila
b) Podem-se esperar produtos paralelos de autocondensação do acetoacetato de etila: ele
tem carbonos aceitadores (no grupo carbonila e no grupo éster) e também centros
nucleofílicos em posições α ao grupo carbonila. A desprotonação para o enolato é muito
fácil, devido à presença de um gupo metileno duplamente ativado.
O grupo metileno duplamente ativado tem mais um hidrogênio que pode ser substituído por
um carbono, proveniente de outra molécula de cloreto de benzila.
A presença de uma base forte pode provocar uma reação SN2 no cloreto de benzila, levando
ao éter (isto é, a síntese clássica de Williamson,1850, ver p. 136). Realmente, observamos
esta reação paralela - especialmente ao usar haletos orgânicos com grupos altamente
ramificados.
Uma eliminação de HCl, por outro lado, levando a um alqueno a partir do haleto orgânico,
não é possível neste caso (essa reação somente se observa em haletos terciários que são,
então, substratos inadequados para este tipo de condensação).
c) É de suma importância desprotonar o substrato acetoacetato de etila quantitativamente,
quer dizer, o tratamento com o etóxido deve ser feito numa etapa prévia, acrescentando o
éster aos poucos à base que se encontra no balão do fundo. Por este procedimento reprimese a autocondensação do mesmo.
Ao mesmo tempo não deixar sobrar nenhum excesso da base. Caso contrário a reação de
Williamson ocorrerá. Na prática a base desprotonar na primeira etapa o acetoacetato, aos
poucos mas não muito devagar, até chegar no ponto de equivalência (uso de indicador).
Neste ponto a desprotonação do acetoacetato de etila para o seu enolato é completa:
550
A. Isenmann
Princípios da Síntese Orgânica
lembre-se que seu pKa = 10,7, enquanto o ácido correspondente à base EtO- mostra uma
acidez em torno de pKa(EtOH) = 19.
O cloreto deve então ser acrescentado em segunda etapa, onde pode condensar com o
enolato do éster. Também o cloreto não deve ser acrescentado em excesso, para evitar uma
condensação excessiva, no grupo metileno duplamente ativado do éster.
Resumindo: os reagentes acetoacetato de etila, base e cloreto de benzila devem ser usados
nas proporções 1 : 1 : 1.
Embora todas essas providências, os rendimentos máximos desta síntese são limitados a
50%.
10) (Questão especial)
(p. 510 e 52)
a) Muito pelo contrário ao cloreto de benzila usado na última condensação (Questão 9), o
cloreto de fenila (= clorobenzeno) não serve para reações SN2, conforme discutidas no cap.
1.2.2. Apesar de uma produção completa do enolato do malonato de dietila, a condensação
não vai funcionar porque a eletrofilia do carbono do clorobenzeno não é suficiente.
Portanto, não ocorrerá uma SN2 no carbono fenílico.
b) Por condensação cruzada com um componente aceitador que não enoliza e, além disso,
fornece o carbono no NOX certo. Para este fim serve um derivado do ácido carbônico, por
exemplo o dietilcarbonato (ver estratégia usada na p. 490 e exemplo reacional apresentado
na questão 14, logo abaixo):
COOEt
Ph
Br
+
H2C
COOEt
COOEt
Ph
COOEt
O
Ph
CH2
+ base + C(OEt)2
HC
- EtO-
Fenilmalonato de dietila
COOEt
11) Compostos carbonílicos duplamente ativados
a) A expressão "Condensação de Knoevenagel" se refere, em quase todas as bibliografias, a
uma condensação carbonílica onde um dos componentes, o pseudo-ácido, é um composto
duplamente ativado, quer dizer, tem um grupo metileno (-CH2-), rodeado por dois grupos
retiradores de elétrons.
b) De maneira análoga à condensação de Knoevenagel com o éster malônico (ver p. 513)
funciona a condensação entre acetoacetato de etila e aldeídos:
551
A. Isenmann
Princípios da Síntese Orgânica
COOC2H5
H3C CHO + H2C
Acetaldeído
H3 C
- H2O
C CH3
CH C
C
CH C
C
[ H+ ]
CH3
O
O
Acetoacetato de etila
COOC2H5
H3C
COOC2H5
[Base suave]
COOH
H3 C
CH C
C CH3
CH3
∆
- CO2
O
O
O
H3 C
CH CH C CH3
Cetonas α,β− insaturadas
12)a) Condensação entre acetilacetonitrila e cloreto de acila.
O
H3C
O
H3C
C
CH2
CN
Acetilacetonitrila
(duplo-ativado)
Mg(OH)2
C
CH
CN
H3C
C
O
AcOCl
O
CH
Enolato
CN
H3C
C
CH2
CN
O
b) Para bloquear o oxigênio nucleofílico dentro do enolato, usa-se uma base que tem
poderes de complexar o oxigênio, mas deixa o carbono negativado livre. Uma base
favorável seria um hidróxido cujo metalcátion tem duas ou mais cargas. Estes íons, M2+ ou
M3+, têm a qualidade desejada. Compare também a próxima questão.
13) Acilação de compostos duplamente ativados
A acetilacetona é um composto com predileção para a forma enólica. Então um ótimo
componente metilênico. Só que é bom demais, quer dizer, tem dois centros nucleofílicos
que ambos podem condensar com o carbono positivado do cloreto de benzoíla. Portanto, a
escolha da base cai num hidróxido cujo cátion tem facilidade de formar complexos
quelatos, desta maneira bloquear o oxigênio negativado. O magnésio é um dos metais
adequados, Fe(II), Mn(II) e Al(III) são outros. Não são indicados os hidróxidos dos metais
alcalinos porque não mostram grande preferência de complexar os oxigênios - centros
nucleofílicos duros da acetilacetona.
552
A. Isenmann
O
O
Acetilacetona
Princípios da Síntese Orgânica
OH
O
Mg(OH)2
-
O
Mg
O
Complexo
quelato
Enol
O
H5C6
C
Cl
O
O
H5C6
O
Além destas bases, podem ser usadas bases orgânicas sendo catalisadores especiais,
conforme descrito na nota de rodapé 322, na p. 509.
14) Anelação de Robinson
(p. 526)
A anelação de Robinson é uma extensão da condensação de Michael e representa uma
importante estratégia para fazer anéis de 6 membros. Especialmente a química dos
esteróides aproveitou desta sequência reacional.
É altamente favorável liberar o sistema Michael, que se precisa para esta anelação, in situ,
devido sua alta reatividade. Para este propósito se oferece a base de Mannich (ver p. 495).
O nucleófilo que ataca o carbono β deste sistema Michael, é o ânion de um composto
carbonílico duplamente ativado (deve ser duplo ativado, por causa da fácil desprotonação,
sem necessidade de aplicar bases muito fortes).
Característica mais marcante do produto da anelação de Robinson é uma dupla-ligação
C=C, dentro do novo anel e uma função de cetona neste anel. As duas duplas-ligações
formam um sistema conjugado, C=C-C=O (que representa um novo sistema de Michael com todas as consequências reacionais).
Um exemplo:
553
A. Isenmann
Princípios da Síntese Orgânica
O
1) H2C=O; HNEt2
2) CH3I
O
O
NEt2
O
CH3
Base de Mannich metilada
C
EtO
/ EtOOEt
O
Elim. de Hofmann
O
O
C
OEt
Composto duplo-ativado
Aceitador de Michael
Anelação de Robinson
EtO- / EtOH
O
O
O
H2SO4
AcOH
O
C
OEt
(hidrólise do éster, descarboxilação
e condensação, em uma etapa)
Resumindo: a anelação de Robinson é uma condensação de Michael onde o nucleófilo é
feito por um composto carbonílico duplamente ativado. Resulta um composto 1,5dicarbonilado que pode ser mais uma vez condensado, levando finalmente a um ciclo de 6
membros, com uma dupla-ligação endocíclica.
15) Um ataque do oxigênio negativo ao carbono eletrofílico do benzaldeído levará a um
composto zwitteriônico (isto é, com cargas em átomos, sem jeito de neutralização). Isto
seria termodinamicamente desfavorável. Essa etapa - se ocorrer - seria certamente
reversível, então qualquer caminho paralelo iria ganhar preferência.
554
A. Isenmann
Princípios da Síntese Orgânica
O
O
H2C
C6H5 C
O
N
H
H2C N
O
O-
O-
C6H5 CH O N CH2
O
Zwitteríon
Nitronato
16) (Questão mais complexa, incluíndo a condensação de Mannich, condensação de
Michael e uma quebra de cetona)
Exemplo de síntese de um composto natural: o aminoácido triptofano
a) O composto 5 representa um sistema Michael, como já dito. Porém, o carbono em
posição exo-cíclico não é tão pobre em elétrons (eletrofílico) quanto, por exemplo, o
carbono em uma cetona α,β -insaturada. Isto se dá da maior eletronegatividade do O, em
comparação ao N.
Do efeito doador de elétrons pelo anel aromático; note-se que os elétrons π do anel e os
elétrons π exocíclicos formam um sistema conjugado de elétrons.
Comparação dos sistemas de Michael:
δ−
O
Cδ+
δ−
δ−
N
δ−
δ+
Etilvinilcetona
δ+ mais pronunciado
melhor aceitador de Michael
δ+
δ+
CH2
5
b) O composto 6 é o acetaminomalonato de dietila (também chamado de
acetamidomalonato de dietila). Pode ser feito a partir do malonato de dietila com nitrito de
sódio em ácido acético glacial, a 0 °C. Com o nitrito (ou nitrosil cátion) se tem um reagente
com nitrogênio eletrofílico!
Resulta o isonitrosomalonato de dietila (isonitroso também é chamado de oxima, ver p.
400) que, devido à presença do acetato de sódio em alta condentração, está presente como
aduto, NaOAc . 3 HO-N=C(COOEt)2 342. Este pode ser reduzido com zinco em pó em ácido
acético/acetanidrido, para o acetaminomalonato 343.
342
343
K.N.F. Shaw, Ch. Nolan, J.Org.Chem. 22 (1957) 1668.
Etapa da redução, ver M. Vignau, Bull.Soc.Chim. France 1952, 638.
555
A. Isenmann
Princípios da Síntese Orgânica
COOEt
COOEt
Tautomeria
CH2
COOEt
CH
C
HO
NaNO2
AcOH
COOEt
HO N C
HO N C
O N CH
COOEt
COOEt
OEt
Oxima
(= isonitroso)
Nitroso
Dietilmaloato
COOEt
COOEt
Tautomeria
O
Zn / AcOH
H3C
COOEt
C NH CH
OH-
CH3CONH C(COOEt)2
6
COOEt
COOEt
Note-se que acetamidomalonato é um sínton versátil na síntese orgânica.
c) Uma descarboxilação de compostos 1,3-bifuncionais (= "quebra de cetona", p. 462),
requer em primeira linha de um grupo carboxila livre que tenha em posição β um grupo
carbonila ou carboxila. O processo da descarboxilação mesmo pode ser visto como
processo eletrocíclico, portanto especialmente fácil. Na etapa 7 → 8 têm-se duas sequências
de hidrólise/descarboxilação.
COOEt
Ar
O
CH2 C NH C
COOEt
7
CH3
H+ / H2O
Hidrólise
COOH
Ar
H
O
O
CH2 C NH C
COOEt
=
CH3
EtO
O
C
C
C
ArCH2
"Clivagem de cetona"
O
NHAc
- CO2
COOEt
Ar
O
CH2 CH NH C
CH3
COOEt
Ar
O
CH2 CH NH C
CH3
H+ / H2O
Hidrólise
COOH
Ar
CH2 CH NH C
OH
O Tautomeria O H C CH
3
CH3
O
C
CH
N
CH2Ar
"Clivagem de cetona"
Ar
- CO2
CH2 CH COOH
NH2
8
556
A. Isenmann
7
Princípios da Síntese Orgânica
Planejamento, estratégias preparativas e
retrossíntese
Este capítulo é uma apresentação breve de um assunto importante, conhecimento
indispensável para o químico pro-ativo. Em todos os capítulos anteriores o foco foi nas
técnicas preparativas: como efetuar certa transformação química e como aumentar o
rendimento. Aqui, neste capítulo, o objetivo é outro: a escolha do caminho preparativo.
Embora os dois assuntos são de importância para o sucesso do produto final, temos que
admitir que, na hierarquia empresarial, o propósito estratégico deste capítulo fica acima da
tarefa executiva. Inegavelmente, o planejamento e questões estratégicas são assunto do
gerente, a não ser da chefia. Por outro lado, o que marca um bom laboratorista não é
somente a capacidade de executar reações, mas também mostrar uma visão mais generalista
do que ele faz no dia a dia. Portanto, o assunto deste capítulo é igualmente importante, para
todos que se dedicam à síntese orgânica.
Porém, a forma resumida desta apresentação é certamente insuficiente para dominar os
conceitos estratégicos e a ferramenta da retrossíntese. Uma discussão profunda, inclusive os
exercícios necessários, se encontram na literatura indicada 344. Este texto deve apenas dar
uma introdução e despertar o interesse, incentivando o leitor para abrir os livros indicados.
7.1
Planejamento da síntese
Antes de pensar em planejamento de síntese, procuramos informações em basicamente 5
assuntos:
1. Um entendimento dos mecanismos das reações a serem aplicadas.
2. Um consolidado conhecimento das reações clássicas; é bastante vantajoso conhecer
também os refinamentos destas reações que foram elaborados nos últimos anos,
porém não é essencial para conseguir o planejamento.
3. Uma noção da estereoquímica das reações a serem aplicadas.
4. Noções sobre os métodos de purificação que se oferecem a certo composto
(intermediário).
5. Uma noção dos compostos que são prontamente disponíveis.
Os itens 1 a 3, espera-se de coração, o leitor adquire ao longo da leitura deste livro. O item
4 se obtém, aos poucos e com muita paciência, no laboratório (enquanto esperar que uma
amostra de mistura desça em uma coluna preparativa de sílica....).
O item 5 será comentado a seguir.
344
S. Warren, Organic Synthesis - The Disconnection Approach, Wiley Chichester 1992 (texto introdutório
altamente recomendado; com livro de exercícios);
Suplemento: S. Warren, Designing Organic Synthesis - A Programmed Introduction to the Synthon
Approach, Wiley Chichester 1992.
Livro de exercícios: S. Warren, Workbook for Organic Synthesis: The Disconnection Approach, Wiley
Chichester 1992
Leitura mais aprofundada sobre o conceito da retrossíntese:
E.J. Corey, X.M. Chelg, The Logic of Chemical Synthesis, Wiley New York 2003 (obra bastante detalhada).
557
A. Isenmann
Princípios da Síntese Orgânica
Em geral o químico preparativo tem a tarefa de preparar um definido produto, enquanto ele
tem escolha "livre" no material de partida e também no caminho como chegar à moléculaalvo. A palavra livre está em aspas porque depende muito da disponibilidade e do preço das
substâncias básicas. Para termos uma noção do que é considerado básico e relativamente
barato, recomenda-se usar um catálogo de fornecedor de química fina. Especialmente o da
Merck® e da Sigma-Aldrich® fornecem informações altamente importantes: além do valor e
da embalagem, contém os dados físicos mais importantes da substância, tais como ponto de
fusão (m.p.), ponto de ebulição (b.p.), índice de refração ( nD20 ), além de recomendações
(frases R e S) do manuseio e toxicidade da substância.
Segue uma lista com alguns materiais de partida que podem ser levados em consideração.
Logicamente, está longe de ser completa.
Todos os alifáticos de cadeia carbônica linear, de C1 a C8. O grupo funcional, situado no
carbono C1, pode ser um álcool, haleto, ácido carboxílico, aldeído ou uma amina.
Compostos alifáticos de cadeia ramificada, com os grupos funcionais mencionados acima, é
possível achar com os seguintes esqueletos carbônicos:
X
X
X
X
X
X
X
As seguintes cetonas:
O
O
O
O
Compostos vinílicos, principalmente usados em polimerizações:
H5C6
Butadieno
Isopreno
Cl
Estireno
Cloreto de vinila
Acrilatos (R = H) e metacrilatos (R = CH3), isto é, os derivados do ácido acrílico e
metacrílico:
R
O
C
R
R
O
CN
OR´
C
H
558
A. Isenmann
Princípios da Síntese Orgânica
Compostos aromáticos: todos os monofuncionais.
Os seguintes aromáticos bifuncionais:
O
OH
NH2
OH
COOH
Ácido salicílico
COOH
Ácido antranílico
CHO
Aldeído salicílico
OH
HO
O
O
Anidrido ftálico
OH
HO
orto: catecol
meta: resorcina
para: hidroquinona
o, m e p-cresol
Bifenileno
Mesitileno
Heteroaromáticos:
N
Anilina
X
O
CHO
S: tiofeno Furfural
O: furano
HN: pirrol
Outros compostos, multifuncionais ou alicíclicos:
Todos os mono-alcoóis e cetonas alicíclicos, de C4 a C8. Além destes, são disponíveis:
559
A. Isenmann
Princípios da Síntese Orgânica
OH
O
HO
OH HO
Butinodiol
Corilona
OH
cis-butenodiol
O
CO2Et
O
O
Cl
Epiclorohidrina
CO2Et
Cicloocta-1,5-dieno
Éster de Hagemann
Esses poucos exemplos devem ilustrar que nem sempre suspeitamos um composto ser
material de partida, barato e acessível. Alguns destes são produzidos em toneladas, com
destino em uma síntese industrial, outros são obtidos em somente uma etapa, às vezes com
ajuda de catalisadores especiais, a partir do petróleo ou carvão. Mais outros, por exemplo, o
furfural, é um produto da pirólise de madeira, então um produto barato a partir de material
biológico. Portanto, sempre vale a pena buscar um possível material de partida em um dos
catálogos conforme indicada acima ou na revista "Química e Derivados" e suas edições
especiais que contêm as listas dos fornecedores.
Definições úteis no planejamento de síntese.
Tabela 30.
Resumo das expressões utilizadas na retrossíntese e no planejamento.
Expressão em
Expressão em inglês
Explicação
português
Molécula alvo
Target
Molecule Molécula que se pretende obter.
(TM)
Retrossíntese ou
Disconnection
Processo de desfazer a TM, quebrando-a
Análise retrossintética approach ou
em partes menores até alcançar um
retro-synthesis
material de partida reconhecível.
(símbolo: ⇐)
Substância química, disponível no seu
Material de partida
Starting Material
almoxarifado em quantidades suficientes,
(SM) ou
Reagent
com grau de pureza satisfatório. É o
produto da análise retrossintética.
Desconexão
Disconnection
Desfazer uma ligação carbono-carbono na
TM, para verificar se ela fornece um par
de síntons e, finalmente, reagentes
(símbolo: )
acessíveis.
560
A. Isenmann
Princípios da Síntese Orgânica
Interconversão de
grupos funcionais
Reconexão
Síntons
Equivalente sintético
ou simplesmente
Reagente.
Functional Group
Transformação de um grupo funcional da
Interconversion (FGI) TM em um outro grupo, sob a condição
345
de fornecer um intermediário acessível.
Reconnection ou
Introdução de um grupo funcional
Functional Group
adicional que promove a reatividade de
Addition (FGA)
um carbono ou fragmento, mas que não
consta da TM. Introdução de um grupo
ativador (que é oposto do grupo protetor,
compare p. 509).
Synthons
Fragmentos conceituais que se originam
da desconexão da TM; síntons são
partículas hipotéticas que não existem
nesta forma, pois geralmente carregam
cargas 346.
Synthetic equivalent
Composto (real) que reage no mesmo
sentido de um sínton (hipotético).
Para deixar mais claro qual a diferença entre sínton e equivalente sintético, sejam
referidas algumas desconexões bastante comuns na retrossíntese, exemplificadas em alcoóis
e cetonas. Note-se que todos os síntons têm cargas, enquanto os equivalentes sintéticos
apenas têm ligações polarizadas (maiores explicações sobre as polarizações, ver p. 566):
Tabela 31.
Os mais importantes síntons e seus equivalentes sintéticos.
No.
Síntons
Equivalentes sintéticos
TM ⇒
1
R
;
R
=
Alquila
RMgX,
RLi, R2CuLi; R2Cd, todos
OH
os organometálicos (cap. 10.3)
R´´
R
OH
δ−
R´
O
Álcool terciário
2
OH
R
R´´
R´
Álcool secundário
3
O
R´´
R´
(sínton natural)
R- ; R = Alquila
OH
R´
(sínton não-natural)
R- ; R = Alquila
R´´ δ+ R´
RMgX, RLi, R2CuLi; R2Cd, todos
os organometálicos
O
R´
Epóxido
RMgX, RLi, R2CuLi; R2Cd.
345
J.J. Li, E.J. Corey, Name reactions fo functional group transformations, Wiley New York 2007.
Os FGIs mais convenientes, usados na síntese de aromáticos, são apresentados na Tabela 17, na p. 300.
346
Existem também síntons radicais, porém esta consideração é avançada e deve ser estudada na literatura
especial.
561
A. Isenmann
Princípios da Síntese Orgânica
O
O
4
R´
(sínton natural)
R- ; R = Alquila
O
R
R´
O
Cetona
R´
(sínton não-natural)
R+ ; R = Alquila
Alternativa:
O
R- ; R = Alquila
O
R´
Cetona
Br
R´
β-bromohidrina (Reformatzky p.
394; reação halofórmio p. 424);
produção/reatividade problemática.
R-X, com X = Cl, Br, I, OTs, OTf,
OMs (p. 35)
R
O
R´
(sínton natural)
O
O
R´
(sínton natural)
Cetona
5
X , com X = Cl, OEt,
OAc,... (p. 440)
RMgX, RLi, R2CuLi; R2Cd, todos
os organometálicos
O
O
R
R´
R´
R´
R´
Enolato (p. 400); composto com
grupo metileno duplamente ativado
(p. 505)
RMgX, RLi, R2CuLi; R2Cd, todos
os organometálicos.
O
R´
Sistema Michael (p.
518)
A primeira desconexão é feita no mesmo carbono onde se tem o grupo funcional, no caso o
grupo hidroxila. Portanto, este tipo de desconexão é referido como (1,1). Já no segundo
exemplo o álcool é quebrado em posição 2 ao grupo hidroxila, então temos uma
desconexão (1,2). A cetona do terceiro exemplo é quebrada no carbono 1, então temos uma
desconexão (1,1). No último exemplo temos uma desconexão de uma cetona entre C3 e C4,
então é denominada de desconexão (1,3).
Para as expressões "natural" e "não-natural", ver mais embaixo (p. 566).
Da tabela acima podemos ver que não há um único jeito de desconectar. Além de escolher a
posição onde quebrar o esqueleto carbônico, o exemplo 4 demonstrou que até na
distribuição das cargas nos síntons podemos variar. Note, porém, que nem todo sínton tem
um equivalente sintético praticamente viável.
Quase sempre se oferecem alternativas na desconexão de uma TM que levam a síntons e,
finalmente, a equivalentes sintéticos totalmente diferentes. Além disso, o grupo funcional
562
A. Isenmann
Princípios da Síntese Orgânica
pode mudar durante a retrossíntese - até o estado de oxidação dos carbonos envolvidos
pode mudar, desde que se conhece uma reação redox de fácil execução. O que não pode ser
diferente, em sínton e equivalente sintético, é o número de carbonos, a posição do grupo
funcional e a estrutura do esqueleto carbônico.
7.2
Estratégias de síntese
Quais são os critérios, na hora de escolher o local da desconexção dentro da TM?
Em princípio podemos resumir em seis pontos:
1. A desconexão deve fornecer a maior simplificação.
2. Aproveitar da simetria, observada dentro da TM.
3. Aplicar somente etapas confiáveis, sínteses com altos rendimentos.
4. Respeitar as polaridades dos síntons que se escondem nos equivalentes sintéticos
como polaridades latentes.
5. Em sínteses que requerem várias etapas, o grupo funcional mais reativo deve ser
introduzido pelo último.
6. Produzir material de partida disponível em quantidade e qualidade suficiente, além
de ser barato.
O último ponto já foi esclarecido no cap. 7.1, os demais pontos sejam explicados, através
de exemplos, a seguir.
Simplificação por desconexão
Isto geralmente se alcança ao quebrar bem no meio da molécula; desta maneira resultam
dois fragmentos de tamanhos parecidos e de tamanhos médios.
Também se obtém esqueletos mais simples ao desconectar em pontos de maior ramificação,
isto é, perto de carbonos terciários ou até quaternários.
Quando a TM tem uma parte aromática, então é sempre uma boa idéia pensar em
desconectar o anel da cadeia lateral.
Um fato que pareçe estranho para o iniciante da técnica de desconexão, é que uma duplaligação C=C é mais fácil de desconectar do que uma ligação C-C simples (ao contrário que
nos sabemos das estabilidades termodinâmicas destas ligações).
Portanto, lembre-se: a retrossíntese (um procedimento puramente teórico) não tem nada a
ver com a reação reversa (que realmente existe)!
Um outro argumento para a desconexão no meio da TM é a facilidade de planejar uma rota
convergente (ver p. 579).
Exemplos:
Onde desconectar o seguinte álcool? Todas as cinco desconexões indicadas são realizáveis,
mas nem todas trazem uma grande simplificação.
563
A. Isenmann
Princípios da Síntese Orgânica
OH
Neste caso é recomendado desconectar a ligação entre os carbonos mais ramificados. Além
disso, deve ser uma ligação que separa a TM mais ou menos no meio - e não picar um
pedacinho na extremidade. Então:
C-C
(1,1)
+
OH
OH
+
CHO
Síntons
Eq. sintéticos
BrMg
A síntese é uma reação de Grignard em uma etapa.
Igualmente evidente é o local da desconexão na seguinte molécula:
OH
C-C
(1,1)
ClMg
O
+
OCH3
OCH3
A síntese é de quatro etapas:
1) Cl2 [FeCl 3]
2) (CH3)2SO4; base
OH
Cl
3) Mg [Et2O]
OCH3
4)
TM
O
Vantagem de produzir um produto simétrico
A simetria observada dentro da TM não deve ser reduzida pela desconexão. Quer dizer que
deveríamos produzir fragmentos igualmente simétricos, a não ser idênticos. Isto tem a
vantagem de não correr perigo de desvios reacionais, porque nestes casos sempre se tem
pelo menos um dos reagentes que seja bifuncional.
Exemplos:
O seguinte álcool é altamente simétrico. Portanto, tentamos desconectar e manter essa alta
simetria:
564
A. Isenmann
Princípios da Síntese Orgânica
C-C
(1,1)
AcOEt + 2
MgBr
OH
.
O reagente de Grignard que resulta desta desconexão não é comercial, portanto temos que
desconectar mais uma vez. Proposta: reduzir o cloreto ao álcool, depois desconectar no
carbono mais ramificado. Como isto é uma desconexão em posição 2 ao grupo funcional,
então aproveitamos da reatividade especial do oxirano.
C-C
(1,2)
FGI
MgBr
OH
MgBr
O
+
A síntese pode então ser feita em 5 etapas 347:
1) Mg; Et2O
Br
2)
PBr3
O
OH
1) Mg; Et2O
Br 2)
EtOAc
TM
(45%)
Um outro exemplo é uma TM alicíclica, derivado do THF. Trata-se de um éter que pode ser
o produto da condensação de um diol altamente simétrico:
Ph
Ph
O
Ph C-O
éter
Ph
Ph
C-C
Ph (1,1)
OH HO
Ph
Ph
EtO2C
CO2Et
+ 4 PhMgBr
A síntese requer apenas duas etapas:
EtO2C
CO2Et
1) PhMgBr
2) H+
TM
Visar etapas de síntese de altos rendimentos.
Este ponto é certamente o mais difícil a ser previsto. É bastante trabalhoso, a não ser
impossível, achar todas as condições ótimas para a reação na literatura. Além disso,
podemos ver os rendimentos indicados lá como limite superior. Quem pensa em superar um
rendimento, indicado na literatura por um pesquisador especialista com profunda
experiência prática nesta reação, está sonhando. Além disso, temos inúmeros exemplos de
347
V.Grignard, Ann.Chim. (Paris) 24 (1901) 475.
565
A. Isenmann
Princípios da Síntese Orgânica
reações que funcionam no papel, mas na prática fornecem um produto totalmente diferente
ou uma mistura feia de produtos paralelos (ver último exemplo dado no parágrafo 2.2.5).
Afinal, temos que encorajar e tentar.
Certa segurança ganhamos, todavia, ao elaborar duas rotas diferentes. Desta maneira
podemos descartar aquela rota cuja tentativa não funcionou.
Polaridade latente natural e inversão da polarização.
Na discussão dos sistemas com elétrons π conjugados (gráficos na p. 318 e p. 518) se
evidenciou uma alteração bem regular entre carbonos positivados e negativados. Isto foi
explicado com o longo alcance do efeito mesomérico (efeito M) que se propaga ao longo
de todo o sistema conjugado. Nos seguintes exemplos este fato seja repetido:
δ−
O
δ+
δ+
δ−
δ−
δ+
δ+
δ−
NR
δ+
δ+
δ+
δ−
δ−
X
δ−
δ+
δ−
δ+
δ+
δ+
δ−
δ−
δ−
δ+
δ−
X = OH, Br, NHR,...
δ−
Cl
δ−
δ−
mas:
δ+
δ+
MgCl
Podemos ampliar esta observação e incluir carbonos sp³, com a finalidade de explicar a
facilidade de produzir TMs, a partir de fragmentos que já tenham as polaridades favoráveis,
a serem simplesmente transcritas na TM. Esse conceito é denominado polaridade latente.
No capítulo 6 pode-se verificar que a maioria das condensações forneceu moléculas 1,3bifuncionais, muitas destes são 1,3-dicarboniladas. Significa que, quando acharmos os
grupos funcionais na relação 1,3 dentro da TM, a química das condensações carbonílicas
disponibiliza um prato cheio para o químico preparativo.
Um exemplo:
O
δ−
Análise da polaridade latente na TM:
a partir da cetona
O
OH
δ+
Ph
OH
δ−
Ph
δ−
O
OH
a partir do álcool
δ+
δ+
δ−
δ+
Ph
Essa análise da polaridade levou ao mesmo resultado, não importa qual grupo funcional foi
escolhido como ponto de partida. Análises com essa evidência são conhecidas como
sintonia da polaridade.
566
A. Isenmann
Princípios da Síntese Orgânica
Igualmente consoante é a polaridade dentro do TM 1,5-bifuncional.
Um exemplo:
δ−
δ−
O
O
O
δ+
δ−
δ+
δ−
δ−
δ+
O
R
R
Identificamos um sistema Michael que é atacado por um carbono nucleofílico (p. 523).
O
O
NaOH
O
O
O
R
O
R
Enolato
A desconexão de compostos 1,3 e 1,5-bifuncionais é bem óbvia, porque podemos formular
dois síntons na sua polaridade natural. Raros, por outro lado, são os métodos que levam a
compostos 1,2 e 1,4-bifuncionais, porque nestes casos produzimos pelo menos um sínton
de polaridade não-natural. A polaridade natural de equivalentes sintéticos já foi apresentada
em diferentes lugares: em haletos, alcoóis, aminas (p. 14), sais de amônio (p. 365),
compostos com grupo carbonila (p. 456) e finalmente em compostos com sistema de
elétrons π conjugados, onde uma das duplas-ligações é uma ligação de polaridade
permanente, conforme mostrado no início deste parágrafo.
Exemplo:
δ−
Polaridade latente, a partir do:
O
O
δ+
grupo carbonila esquerdo
δ−
O
1,4-dicetona
grupo carbonila direito
δ+
O
O
δ−
δ−
δ−
δ−
δ+
δ+
O
δ−
É evidente que aqui se sobrepõem polaridades dissonantes. Qualquer desconexão finaliza
em um sínton de polaridade errada - independente do local da desconexão.
Desconectamos, por exemplo, no meio:
567
A. Isenmann
Princípios da Síntese Orgânica
O
O
Síntons
O
O
?
Enolato
Equivalentes
Fazer o quê, quando não achamos equivalente de polaridade natural para o sínton da nossa
desconexão? A resposta é: manipular a polarização latente , através de um grupo funcional
auxiliar que altamente polariza a ligação com o carbono cuja polaridade deu errada. Este
grupo pode ser, de um lado, um elemento bastante eletronegativo, então retirador de
elétrons (ingês: electron withdrawing group, EWG). Por outro lado, podemos pensar em
metais que todos são mais eletropositivos do que o carbono - entre eles especialmente os
elementos Li, Na, Mg e Al, para mencionar os mais utilizados. No primeiro caso, o carbono
da ligação C-X fica positivado, no segundo onde se tem uma ligação C-M, o carbono fica
negativado. Outros equivalentes sintéticos importantes, portadores de polarizações nãonaturais, são o acetileno (ver as reações no cap. 2.5) e os epóxidos (exemplo 2 dado na
Tabela 31 na p. 560).
Um papel estratégico bastante aplicado é a inversão da polarização (pp. 165, 431 e 700),
também conhecido como "Umpolung" (do Alemão). Quer dizer que um carbono de
polarização positiva ganha, através de uma reação estabelecida, um vizinho eletropositivo e
muda assim sua polarização para negativado. O exemplo mais famoso é a metalação, a
reação-base de preparo do reagente de Grignard (p. 431).
Mas também a polarização δ+ no carbono do grupo carbonila pode ser transformada em um
carbono negativado. A estratégia foi estabelecida por Corey e Seebach (observe a
semelhança com a redução de Mozingo, p. 611):
O
R
HS
H
SH
Catalisador:
ácido de Lewis
BF3.Et 2O
reage com nucleófilos
S
S
R
H
Acidez:
pKa ~32
n-BuLi
S
R
S
Li+
reage com eletrófilos
Este procedimento parece bastante forçado - e na verdade é forçado. Portanto, podemos
falar de sorte que temos reagentes (equivalentes) especiais que já têm a polaridade desejada
ou aqueles que podem ser transformados, via FGI, em síntons da polaridade necessária. Na
tabela a seguir são listados os equivalentes sintéticos não-naturais mais frequentemente
usados.
568
A. Isenmann
Princípios da Síntese Orgânica
Tabela 32.
Equivalentes para síntons de polaridade não-natural:
Sínton nãonatural
Equivalente sintético
OH
R
OH
O
Epóxido ou β-bromoidrina
(p. 224)
O
Br
R´
R´
Acesso ao equivalente
/comentários
O epóxido é mais usado porque é
o equivalente mais reativo. Sua
preparação é fácil e segura, a
partir do alqueno.
(p. 163)
O primeiro equivalente é
adequado para reações em
O
O
Br ou
ambiente ácido, o segundo em
Br
R
R
ambiente básico.
As propriedades e a síntese do
OEt
OEt
viniléter, ver pp. 189, 405 e 499.
OEt
E+
Uma propriedade que importa
R
H
neste contexto é a sua acidez que
R
Li
R
E
é ligeiramente acima dos
+ sec-BuLi
Etoxivinilítio
+
H /H2O hidrocarbonetos, então pode ser
desprotonado com butilítio ou
fenilítio. Já a desprotonação do
O
ou:
acetileno (pKa ≈ 25) não requer
bases tão fortes, então pode-se
E
usar LDA, por exemplo (nota de
H+/H2O rodapé 307).
O
R
O
H3C
1) base
2) E+
OH
E H+/H2O
HgO
O
R
O
S
Estratégia de Corey-Seebach (ver
texto acima).
+ n-BuLi
R
S
S
S
H
E
A primeira alternativa é uma
variação de Corey-Seebach;
+ n-BuLi
S
a segunda é uma ampliação da
reação de Nef (p. 535)
ou:
H3C NO2 + base
O
Ar
O
Ar
H
1) (CH3)3Si CN
2) LDA
OSi(CH3)3
Ar
C
CN
A cianidrina livre é bastante
frágil, portanto é melhor protegêla via sililação antes da
desprotonação.
Aplicação: ver condensação de
benzoina (p. 465).
569
A. Isenmann
Princípios da Síntese Orgânica
CN-
O
NaCN (pp. 31 e 377) em solvente
polar aprótico.
HO
Escolha da ordem das etapas, em sínteses multi-etapas
Em sínteses sobre várias etapas, o grupo funcional mais reativo deve ser introduzido pelo
último, conforme ponto 5 da lista acima. Caso diferente, as demais etapas operacionais
(inclusive as de purificação!) podem danificar este grupo. Isto significa que, na
retrossíntese, este grupo reativo deve ser desconectado em primeiro lugar.
Grupos desta categoria são nitro, nitroso, haletos, epóxidos, cloretos de acila, aminas afinal, a lista é tão extensa quanto a lista daqueles grupos que são considerados robustos.
Uma análise profunda das condições, aplicadas em cada uma das etapas subsequentes, é
indispensável.
Um exemplo:
N
N
N
(Bu)2AlH
SM
Perácido
O
OH
OH
O
Caso contrário:
N
Perácido
N
(Bu)2AlH
O
+
HO
O
OH
HO
N
OH
Mas vamos pegar um caso especialmente desfavorável: um grupo sensível esteja presente,
já no início da síntese e não queremos perdê-lo 348. Então precisamos de um método para
contornar quaisquer ataques perigosos nele; temos que mascarar ou proteger este grupo
funcional. Significa que, através de uma transformação química reversível, o grupo se torna
resistente às condições reacionais subsequentes (reagentes, solventes, temperatura alta,
pH,...). Como esta estratégia requer pelo menos três etapas operacionais extras (proteção,
remoção do excesso de reagente protetor e desproteção), então fica na segunda linha das
nossas opções. Sempre quando pudermos, resolvemos o problema de reações num grupo
funcional através da sequência certa dos eventos.
348
Essa necessidade pode surgir, por exemplo, nas SE aromáticos, onde o primeiro substituinte tem certo
efeito dirigente ao segundo substituinte (ver p. 314).
570
A. Isenmann
Princípios da Síntese Orgânica
Exemplo para proteção de uma cetona, frente um agente redutor (descrição na p. 403):
Proteção
etilenoglicol
O
Ph
CO2CH3
Catalisador:
TsOH
O
O
Ph
CO2CH3
Cetal
(estável frente bases
e nucleófilos)
Etapa necessária LiAlH4; Et2O
O
O
Ph
7.2.1
Desproteção
H+ / H2O
OH
O
Ph
OH
A estratégia da reconexão (FGA)
A reconexão, conforme a tabela na p. 560, tem a finalidade de ativação temporária de um
dos equivalentes sintéticos. Esse grupo não consta da TM, então é abstraída durante a rota
sintética. Um dos mais famosos exemplos é a dupla ativação do grupo metileno em ácido
malônico e seus derivados (p. 509). Logo após ter efetuado a condensação, um dos grupos COOH pode ser facilmente abstraído - uma simples reação térmica chamada de
descarboxilação (p. 462).
A facilidade com que se removem halogênios de uma molécula (p. 594), também abrem
caminho para uma FGA. A desalogenação→hidrogenação seletiva, com catalisador
CuCr2O4 (p. 598), por exemplo, pode ser feita após ter superada a etapa mais problemática
da síntese.
Na discussão das condensações cruzadas (cap. 6.4) foi discutida extensamente a estratégia
de estabilizar a forma enólica em um dos participantes, provocando assim uma reatividade
elevada como componente pseudo-ácido (= nucleofílico, ver estratégia No. 3, na lita da p.
482). Só para alcançar este propósito foram apresentados 3 métodos, enamina, enoléter e
sililenoléter, pp. 495 em seguida).
Uma maneira mais complexa de ativação (e desativação), no caso do formaldeído, foi
apresentada com a condensação de Mannich (ver p. 491 e questão 7 na p. 539).
Resumindo: o conceito da FGA é aplicar um grupo funcional auxiliar que tem o efeito
oposto de um grupo protetor, mencionado no item anterior: ele é ativador.
Dois exemplos concretos que devem ilustrar o valor estratégico da reconexão. Admita-se
que esta metodologia requer um elevado grau de intuição. A seguinte cetona α,β -insaturada
pode ser transformada em um composto 1,3-bifuncional que pode ser desconectado de
maneira padrão:
571
A. Isenmann
Princípios da Síntese Orgânica
O
TM
Ph
Ph
FGA
+ H2O
O
Ph
Ph
O
OH
Ph
O
Ph
OH
Polaridade consonante
Polaridade dissonante
não leva a síntons simples
Ph
OH
Ph
Equivalentes:
O
+
CHO
Ph
Ph
A FGA desta análise revela a necessidade de eliminar água a partir de um aldol. Iso pode
ser feito, de maneira especialmente elegante ao aplicar um catalisador ácido, em vez de
uma base (o que seria padrão nas condensações). Daí a síntese pode ser feita em apenas
uma etapa (ver p. 455).
A estratégia da FGA torna-se até essencial quando a TM não tem grupos funcionais ou
apenas grupos aparentemente pouco reativos.
Um exemplo:
HO
FGA
HO
+
(1,1)
Oferece-se a síntese em 4 etapas:
Br
HO
1) Mg [Et 2O]
2)
H+
O
H2 [Pd/C]
Podemos pensar em até uma alternativa, baseada na reatividade de sistemas Michael:
TM
FGA
O
O
Na execução desta síntese temos que conferir um caráter mais mole possível ao nucleófilo
(estratégia de Corey-House, ver p. 520):
572
A. Isenmann
O
Princípios da Síntese Orgânica
CuLi
2
O
Clemmensen
ou Wolff-Kishner
7.2.2
TM
Resumo das etapas do planejamento por retrossíntese
De forma mais resumida, podemos dividir a retrossíntese em cinco etapas:
1. Identificar todos os grupos funcionais na TM, assim que as posições relativas entre eles.
2. Se houver dificuldades em achar síntons realizáveis, aplicam-se transformações dos
grupos funcionais da TM (chamadas de FGI), até que uma desconexão for plausível. Este
item inclui oxidação e redução. Em casos excepcionais pode-se acrescentar um grupo
funcional auxiliar, para gerar a bifuncionalidade desejada (FGA).
Geralmente facilita a elaboração da estratégia quando todos os grupos funcionais da TM
forem convertidos em funções que contenham somente C, H e O.
3. Desconectar o esqueleto carbônico, pensando somente em reações de condensação
clássicas (inclusive as reações organometálicas, tipo Grignard; p. 436).
Siga as seguintes regras gerais:
• Pense em desconectar as ligações diretamente nos anéis aromáticos - sejam Ar-C ou
Ar-X.
• Desconectar as ligações C-X é geralmente fácil, especialmente quando a ligação é
diretamente num grupo carbonila, isto é, RCO-X.
• Desconectar C-C da TM 1,2-, 1,3- e 1,4-bifuncional, de modo que resultem dois
fragmentos com um grupo funcional cada.
• Abrir um anel não-aromático (alicíclico) é vantajoso porque as ciclizações são
etapas cuja termodinâmica, dentro desta a entropia, sempre é favorável (p. 469); podese pensar em ciclização por reação eletrocíclica (tipo Diels-Alder, p. 252) ou em
condensações carbonílicas intramoleculares (p. 471).
4. Repita a desconexão, se for necessário, até que chegue em material de partida (SM)
disponível.
5. Evite etapas das quais já se sabe que o produto somente pode ser obtido em forma de
mistura e/ou de baixa porcentagem. Pois, lembra-se de que o químico preparativo gasta
apenas uns 30% do seu tempo com a síntese, mas 70% com a purificação!
7.2.3
Resumo da execução do trabalho prático
A síntese da TM pode ser resumida igualmente em cinco pontos:
1. Anote (com papel e lápis!) o plano da síntese, simplesmente invertendo a análise
retrossintética. Adicione todos os reagentes, temperaturas, catalisadores e tempos de
reação, de acordo com o método clássico.
573
A. Isenmann
Princípios da Síntese Orgânica
2. Verifique se a ordem das etapas pode ainda ser otimizada; este ponto é especialmente
importante quando se têm terceiros grupos funcionais que podem ser danificados pelas
condições aplicadas em seguida.
3. Um critério importante em toda síntese multi-etapas é o caráter convergente (p. 579)
que pode ser estabelecido pela sequência certa dos eventos. Às vezes é inevitável incluir
etapas difíceis; elas devem ser colocadas o mais cedo possível, na cadência da síntese
multi-etapas. O oposto vale para etapas que requerem reagentes caros: essas devem ser
aplicadas no final da sequência reacional.
4. Verifique se todos os aspectos de quimosseletividade foram levados em consideração.
Temos que excluir a possibilidade de reação em outro local do reagente do que foi
intencionada. Caso não houver solução alternativa, podemos pensar em introduzir um
grupo protetor.
5. Modificar e otimizar as etapas em casos de falhas inesperadas - que sempre acontecem
na prática (informe-se sobre as ´Leis de Murphy´ que, sem dúvida alguma, regem no
laboratório de síntese orgânica!).
Os aspectos estereoquímicos, especialmente a formação do enanciômero certo a partir de
um centro pro-quiral, é um assunto avançado que deve ficar reservado à literatura indicada
na nota de rodapé 344 na p. 557 349.
7.3
Aplicando o conceito da retrossíntese
Para documentar um pensamento retrossintético, usa-se uma flecha da forma "⇒";
desconexões são indicadas por uma linha ondulada que corta a TM em dois pedaços.
O "coração" de cada síntese é a construção do esqueleto carbônico da TM, a partir de SM
cuja estrutura carbônica é bem mais simples. Mais simples geralmente significa: menor
(menos carbonos em total) e menos ramificado (menos carbonos terciários e secundários,
em relação aos primários). As condensações carbonílicas e as reações organometálicas do
tipo Grignard são consideradas as estratégias mais importantes para este fim.
7.3.1
Exemplo de retrossíntese, desenvolvendo caminhos opcionais via
condensações
A molécula alvo seja um álcool, da seguinte estrutura:
OH
OH
abreviado: Ph
t-Bu
TM
349
As ferramentas da síntese estereocontrolada foram apresentadas, de forma resumida, no excurso do cap. 3,
na p. 225 em diante.
574
A. Isenmann
Princípios da Síntese Orgânica
A primeira idéia é desconectar o anel aromático do resto alifático, uma estratégia que quase
sempre leva a alguma simplificação razoável e, finalmente, a uma reação química
estabelecida.
OH
Ph
t-Bu
A
B
OH
OH
Síntons:
t-Bu
t-Bu
O
Equivalentes: PhMgBr
C
t-Bu
H
?
?
Dos dois possíveis pares de síntons, somente a opção A leva a equivalentes sintéticos cuja
polaridade é natural. Opção B, por outro lado, é difícil de realizar, já que a polaridade dos
síntons é contra a natureza dos possíveis equivalentes (o carbono do clorobenzeno, por
exemplo, não é suficientemente positivado, para receber um carbânion em virtude de uma
SN2).
Assim, podemos pensar na seguinte síntese:
Primeira opção:
PhBr
Mg [THF]
O
PhMgBr
H
C
OH
t-Bu
Ph
t-Bu
.
Essa certamente não é a única maneira de desconectar. A segunda, menos óbvia, é a
desconexão para um fragmento benzílico:
575
A. Isenmann
Princípios da Síntese Orgânica
OH
Ph
t-Bu
A
B
OH
OH
Síntons:
t-Bu
t-Bu
Equivalentes: PhCHO
BrMg
t-Bu
Br
?
t-Bu
Por meio da distribuição das cargas conforme caminho B, temos que lidar com o problema
sintético de criar um carbono negativado em posição benzílica - além do mais na
vizinhança do grupo hidroxila.
O caminho A é realizável, novamente por meio de uma condensação via composto de
Grignard:
Segunda opção:
t-Bu
OH
Br
Mg [THF]
t-Bu
MgBr
PhCHO
Ph
t-Bu
E vamos pensar em mais uma possibilidade de desconectar:
OH
Ph
t-Bu
Embora essa desconexão forneça fragmentos bastante desiguais, os reagentes equivalentes
são de acesso surpreendentemente fácil:
576
A. Isenmann
Princípios da Síntese Orgânica
OH
Síntons:
TM
t-Bu
Ph
O
Equivalentes:
BrMg
Ph
Então se oferece a síntese número 3:
Grignard
Ph
BrMg
O
Perácido
TM
Ph
Estireno
.
Incluímos também a possibilidade de transformar os grupos funcionais da TM ("FGI"). Daí,
chegamos à cetona que evidencia uma nova desconexão razoável: a alquilação de cetonas
em posição α (pp. 496, 498 e 502).
OH
Ph
O
FGI
t-Bu
Ph
t-Bu
O
Síntons:
Ph
t-Bu
O
Equivalentes:
Ph
Br
t-Bu
Síntese número 4:
O
Ph
base forte
quantitativo
O
Ph
Enolato
Br
O
t-Bu
Alquilação
Ph
LiAlH4
t-Bu Redução
TM
.
577
A. Isenmann
Princípios da Síntese Orgânica
Incluímos a possibilidade de condensação do tipo Michael (p. 523), então desconectamos
diretamente o grupo t-butila. Este grupo tem que atacar como nucleófilo, então deve ter um
carbono negativado. Para evitar um ataque no centro duro de oxigênio, então escolhemos o
reagente de Corey-House, por sua vez mole e afim à posição β da etilfenilcetona.
OH
Ph
O
FGI
t-Bu
Ph
t-Bu
O
Síntons:
t-Bu
Ph
O
Equivalentes:
LiCu(t-Bu)2
Ph
Resulta a síntese da opção número 5:
O
O
t-Bu2CuLi
Ph
NaBH4
Ph
t-Bu Redução
TM
Uma sexta possibilidade fica em segunda linha porque não fornece um SM de acesso
barato. Mesmo assim, consideramos a desconexão do heteroátomo como possível caminho:
OH
Ph
t-Bu
Síntons:
Ph
t-Bu OH
Equivalentes:
Ph
t-Bu
"H2O"
A adição de água à dupla-ligação C=C, para fornecer um álcool no sentido Markownikov,
requer da catálise de mercúrio (ver p. 160):
578
A. Isenmann
Princípios da Síntese Orgânica
Opção 6:
OH
Hg(OAc)2
Ph
t-Bu
H2O
Ph
t-Bu
NaBH4
TM
HgOAc
Pode-se ver que vale a pena refletir sobre alternativas desconexões. Caso a primeira
tentativa não der o resultado esperado, podemos descartar a estratégia completa e tentar
outra. Os critérios para a nossa escolha são bastante subjetivos. Afinal é uma escolha
pessoal, qual é o caminho da síntese. Cada químico tem reagentes preferidos (quer dizer,
com quais ele já obteve resultados bons) e métodos e aparelhos que lhe prometem sucesso e
manuseio confortável. A nova tarefa pode ter alguma semelhança com uma antiga síntese
que foi feita com grande sucesso. Por outro lado, o químico preparativo também não
esquece os dias pretos, onde uma reação não forneceu o resultado esperado, uma técnica embora de ter feito várias tentativas de melhoramento - finalizou em um fracasso. É claro
que isso causa uma aversão contra certos reagentes e técnicas. Tudo isso afeta a escolha
final do nosso método.
Outros pontos que sempre devem influenciar nossa escolha são:
• Os rendimentos, conhecidos da literatura, devem ser altos. Esses valores, indicados nos
artigos, a gente geralmente não atinge (às vezes podemos ser felizes obter a metade do
rendimento indicado!).
• O perigo de reações paralelas e caminhos de desvio. (Observe o perigo da
autocondensação da cetona na opção 4, ao não efetuar sua desprotonação rapida e
quantitativamente!)
• A síntese deve conter menos etapas possíveis, pois cada etapa e seu processo de
purificação, significa perda em material. Este efeito é especialmente grave em sínteses
lineares. Menos perda geralmente se espera em sínteses convergentes, conforme
explicado a seguir.
7.3.2
O que significa sequência linear, o que é síntese convergente?
Visando uma redução de etapas consecutivas, a síntese convergente é uma ferramenta
valente, como mostrado no seguinte exemplo, uma conexão de 6 unidades estruturais
fictício (A, B, C, D, E, F). Note-se que cada etapa vertical causa perdas com rendimento.
A retrossíntese pode ser formulada de duas maneiras:
579
A. Isenmann
Princípios da Síntese Orgânica
Convergente (ideal):
Linear:
ABCDEF
ABCD
AB +
+
CD;
ABCDEF
EF
ABCDE
E + F
+ F
ABCD
A + B; C + D
+ E
ABC + D
AB +
C
+
B
A
A economia de duas etapas verticais tem um efeito drástico ao rendimento global da
síntese. Escrita por extenso, pressupondo rendimentos típicos de 80% em cada etapa
vertical:
Convergente:
A
C
E
Rendimento global:
Linear:
A
Rendimento global:
rend. 80%
rend. 80%
rend. 80%
AB
rend. 80%
CD
80%
rend. 80%
TM
EF
80%
Etapa 1:
rend. 80%
ABCD
64%
AB
Etapa 1:
rend. 80%
64%
ABC
51%
Etapa 1:
rend. 80%
51%
ABCD
Etapa 1:
rend. 80%
41%
ABCDE
Etapa 1:
rend. 80%
33%
As etapas verticais são indispensáveis, em qualquer síntese; cada síntese é linear de certo
grau. Todavia, vale a pena buscar por possibilidades de incluir uma etapa convergente e,
desta maneira, economizar uma etapa vertical - inclusive as perdas na purificação da
mesma.
Este exemplo, ao mesmo tempo, sublinha a vantagem de desconectar a TM bem no meio,
em vez de picar pedacinhos da sua extremidade, conforme indicado na p. 563.
580
TM
A. Isenmann
Princípios da Síntese Orgânica
581
A. Isenmann
8
Redução
8.1
Número de oxidação
Princípios da Síntese Orgânica
O elemento carbono, essencial em todos compostos orgânicos, pode ser caracterizado pelo
número de oxidação, NOX. Este número serve, em primeira linha, para elucidar
parentescos entre duas substâncias diferentes. Muitas vezes (mas nem sempre) pode-se
esperar reatividade química semelhante, quando se têm substâncias da mesma família.
Igualmente útil é a elaboração do NOX para atribuir o caráter de uma reação química:
• NOX diminui ao decorrer de uma reação, isto é então uma Redução, assunto deste
capítulo.
• NOX aumenta ao decorrer de uma reação, isto é então uma Oxidação, assunto do
capítulo 9.
O balanceamento final da reação redox, isto é, igualar o número de elétrons que se perdem
na oxidação e se ganham na redução, está descrita na segunda parte introdutória, p. 644.
O uso do NOX na química orgânica é muito disseminado, e realmente aproveitamos em
muitas situações da sua utilidade, especialmente nos capítulos 8 e 9. Embora que o NOX
não tem justificativa rigorosa na fisico-química.
No caso dos metais de transição e compostos de elevado caráter iônico, especialmente
aqueles aplicados em reações inorgânicas, o NOX representa o critério principal para sua
reatividade. Lá podemos afirmar que o número de oxidação é em grande parte idêntico com
a carga de um elemento (= íon). Isto implica: oxidação é perder elétrons e redução é ganhar
elétrons.
Em compostos orgânicos a relação entre NOX e reatividade é mais sútil. Geralmente não é
possível concluir à reatividade de um grupo funcional, sem levar em consideração as
demais particularidades da molécula. Este fato ficará especialmente claro na discussão dos
compostos azo (cap. 11), onde os aromáticos são um outro mundo do que os alifáticos,
enquanto o grupo -N=N- / -N2+ é o mesmo. Vamos lembrar-nos que compostos orgânicos
são em grande parte moléculas com ligações covalentes de pouca polarização, a não serem
apolares. Então não podemos identificar a troca de NOX de certo átomo em uma molécula
orgânica, com a perda ou ganho de uma carga. Uma molécula orgânica geralmente não
deixa de ser covalente/apolar, mesmo que um dos seus átomos troque o NOX. A criação de
uma carga, conforme a identificação acima, significaria que em cada redução e oxidação
seria envolvido pelo menos um sal - o que evidentemente não é o caso. Vamos procurar por
uma definição para o NOX, mais cabível às moléculas orgânicas.
A elaboração do NOX é bastante simples, pois segue algumas poucas regras. O critério
central a ser aplicado em todos os átomos da molécula é a eletronegatividade atômica EN
(ver tabela no anexo 2 deste livro), uma grandeza que somente influencia o(s) átomo(s)
vizinho(s), isto é, não se propaga a distâncias maiores do que 2,0 Å. Os valores absolutos
não são de interesse, sempre se considera a diferença ∆EN de átomos vizinhos. Agora a
seguinte frase contém a não-verdade da qual o químico orgânico aproveita tanto: o átomo
mais eletronegativo ganha um elétron, o mais eletropositivo o perde - até mesmo se ∆EN
for um valor bastante pequeno. Essa consideração ser faz à cada ligação química dentro da
582
A. Isenmann
Princípios da Síntese Orgânica
molécula. Duplas ligações contam duas vezes, triplas três vezes, no sentido indicado pela
∆EN.
Alguns exemplos:
•
Dos vizinhos "C-N" o carbono perde um elétron para o nitrogênio, portanto seu
NOX aumenta por unidade;
•
Na unidade "C=O" o carbono perde dois elétrons ao oxigênio, portanto seu NOX
aumenta duas unidades..
•
Na unidade "H3C" o carbono ganha um elétron de cada hidrogênio, portanto seu
NOX aumenta por 3.
•
No segmento C-C-C os vizinhos são igualmente carbonos. Neste caso e só neste
caso o NOX fica inalterado (isto implica que um carbono quaternário tem NOX 0).
Agora fica claro porque foi necessário alertar de uma sobre-interpretação do NOX: estes
números têm uma anotação parecida à das cargas em compostos iônicos, mas jamais devem
ser interpretados como tais. Uma ligação covalente não deixa de ser covalente, só por causa
deste formalismo!
As regras para a elaboração do NOX de certo átomo são:
1. O NOX aumenta por unidade quando ligado a um elemento mais eletronegativo,
através de uma ligação simples. Os mais importantes átomos vizinhos do carbono
que aumentam seu NOX são, portanto: halogênios, N, O, S, P 350.
2. O NOX cai por unidade quando o átomo em destaque for ligado a um elemento
mais eletropositivo. Os átomos menos eletronegativos, frequentemente encontrados
na vizinhança do carbono são: H, B, Si, e todos os metais.
3. Átomos vizinhos iguais ao átomo em destaque não provocam mudança no seu
NOX. Conclusão lógica é que todos os elementos, na sua forma elementar, têm
NOX = 0.
4. Em caso de ligação múltipla, os efeitos dos itens 1 e 2 se multiplicam. Um carbono
no grupo nitrila (...C-C≡N), por exemplo perde formalmente três elétrons para o
nitrogênio; o carbono do grupo carbonila perde formalmente dois elétrons para o
oxigênio.
5. Quando uma reação química leva ao aumento do NOX, então o respectivo átomo foi
oxidado; quando uma reação leva à diminuição do NOX, então se trata de uma
redução. "Oxidação" e "Redução" se refere corretamente a um discreto átomo, mas
o químico orgânico custuma atribuir essa expressão à molécula inteira. Há duas
exceções: quando dois átomos da mesma molécula mudam de NOX em sentido
contrário. Fala-se de comproporcionamento caso os NOX se aproximam, e de
disproporcionamento quando os NOX se afastam.
Importante, neste aspecto, é uma comparação cuidadosa, isto é, átomo por átomo, entre
reagente e produto da semi-reação.
Mais uma vez: o NOX não permite conclusões sobre a reatividade e o comportamento
químico da molécula inteira!
350
Note-se que os elementos S e P, neste formalismo, retiram um elétron do carbono e H doa um elétron ao
carbono - embora de terem eletronegatividades muito pouco diferentes ao carbono. Portanto: este NOX é um
número formal, sem significado físico - muito pelo contrário de uma carga!
583
A. Isenmann
Princípios da Síntese Orgânica
Importante no contexto deste capítulo, “Redução”, são dois casos:
1. O carbono aumenta o número de hidrogênios vizinhos.
2. O carbono é metalizado.
8.2
Classes de redutores
Os redutores de importância preparativa podem ser classificados como:
1. H2 mais um catalisador metálico (Ni, Pd, Pt);
2. Metais não-nobres;
3. Doadores de hidreto;
4. Sistemas redox e sais de metais pesados;
5. Reação no cátodo.
Sobre os redutores de importância preventiva, denominados de anti-oxidantes, foi relatado
no cap. 1.5.1.
A organização dos capítulos a seguir (o mesmo vale para os sub-capítulos da “Oxidação”)
foi feita em relação ao substrato orgânico a ser reduzido, enquanto o agrupamento por
agentes redutores fica em segunda linha (exceção: capítulo 8.13).
8.3
Redução de Alquenos
8.3.1
A hidrogenação catalítica
A hidrogenação 351 (a não ser confundida com a hidratação, que significa reação com
água) geralmente é uma adição cis de hidrogênio, no lado com menor impedimento espacial
do substrato insaturado. Isto vale especialmente ao se aplicar H2 a alta pressão e pequena
quantidade de catalisador. Porém, as hidrogenações mais difíceis podem mostrar desvios
desta adição cis.
A figura a seguir rascunha o mecanismo da hidrogenação catalítica. Acredita-se na
existência de "sítios ativados" na superfície do metal. Isto são valências (= orbitais
direcionados, não ligantes) de certos átomos superficiais que mostram para fora do cristal.
Alguns poucos destes átomos são posicionados em uma distância que coincide com a
distância interatômica da molécula H-H (isto é, 74 pm). Isto facilita a reação entre o metal e
H2. Na mesma medida que se formam as novas ligações entre metal e hidrogênio, a velha
ligação de H-H enfraquece e o hidrogênio torna-se mais reativo. Como os dois participantes
são apolares, então é provável que tudo isso ocorre via um processo sincronizado; os
estados intermediários certamente têm caráter radicalar.
Os pormenores e a cinética das etapas especificadas não são bem entendidos. Isto é uma
dificuldade geral de reações sob catálise heterogênea, que fogem da maioria dos métodos
351
Paul N. Rylander, Hydrogenation Methods, Academic Press London 1985
584
A. Isenmann
Princípios da Síntese Orgânica
espectroscópicos 352, portanto se sabe pouco sobre intermediários e etapas mecanísticas. Por
outro lado, os catalisadores heterogêneos têm uma grande vantagem prática: após a reação
eles são facilmente removidos da mistura reacional (decantação, filtração, centrifugação,...)
e recuperados para outras aplicações.
R
H
H
+
H
H
C
C
C H
H
R
C
H
C
C
R
C
C
C
R
H
C
H
C H
H
C
H
R C
H
C
H
H +
H
C
H
Produto principal
da hidrogenação
Complexo π
R C
H
C
H
C
H
Reações paralelas:
R
C
= Superfície do catalisador metálico
H
R C
H
C H
C
H
C H
C
H
C
R C H
C
H
C
H
Mudança de hidreto
R
C
+
C H
Isomerização cis-trans
(menos frequente)
Figura 34. Esquema geral do mecanismo da hidrogenação catalítica, sendo os
catalisadores os metais Ni, Pd ou Pt.
Neste esquema incluído, a adsorção específica do H2 e a complexação do alqueno, na
superfície do catalisador. Estes processos leva a ligações entre reagentes e catalisador que
são muito mais intenso do que uma adsorção simples, por exemplo, a adsorção de
substâncias apolares na superfície de carvão ativado. Lá têm-se interações fracas entre o
adsorbato e a superfície do sólido, chamada fisissorção, então ligações com energias na
ordem das ligações Van der Waals (< 20 kJ mol-1). O que ocorre na superfície do
352
Exceções são ESCA (Electron Spectroscopy for Chemical Analysis) e LAMMA (Laser microprobe
analysis), métodos avançados de exame superficial e pontual, respectivamente.
585
A. Isenmann
Princípios da Síntese Orgânica
catalisador da hidrogenação, por outro lado, é uma quimissorção. Essas forças são muito
mais intensas, que chegam na ordem de ligações covalentes (até 200 kJ mol-1).
Não só a adsorção firme dos reagentes na superfície metálica é de importância para o
funcionamento desta hidrogenação, mas também a facilidade de desprender o produto - que
é um alcano.
O esquema já indica que existem reações paralelas na hidrogenação catalítica:
1. Mudança de hidreto (também podemos chamar de mudança da dupla ligação C=C);
2. Isomerização cis-trans;
3. Além disto pode ocorrer troca de H por D, em alquenos parcialmente deuterados.
Estas reações paralelas se devem ao fato de que tanto o hidrogênio quanto o alqueno, uma
vez estiverem quimissorvidos, estão bastante reativos. As suas ligações intramoleculares
são mais fracas (a energia que for precisa para soltar as ligações é compensada pela
exotermia das novas "ligações", reagentes - metal). Como já foi mencionado acima, os
reagentes neste estado certamente têm caráter radicalar - o que sublinha sua reatividade.
Catalisadores metálicos da hidrogenação
Os catalisadores mais comumente usados são os metais de transição do grupo 10 (segundo
a nova recomendação da IUPAC) da tabela periódica, Ni, Pd e Pt. Eles apresentam a maior
afinidade de todos os metais para o hidrogênio 353. Menos usados, mas também com
atividade catalítica para a hidrogenação são Ru e Rh. As morfologias com as quais se
aplicam estes metais, devem conferir a maior superfície catalítica, em relação à quantidade
de metal empregado. Isto é necessário porque o preço destes metais é consideravelmente
alto. Muitos destes catalisadores estão comercializados na forma de depósito em cima de
um suporte com alta área superficial. Como exemplos têm-se níquel de Raney e paládio,
depositados sobre carvão ativado.
A forma mais ativa de todos os catalisadores é produzida in situ, a partir de PtO2. O
hidrogênio reduz numa etapa preliminar o dióxido de platina, formando um pó finíssimo de
Pt. Este catalisador é tão ativo que até duplas ligações estáveis em sistemas aromáticos são
reduzidas - à temperatura ambiente! Já o níquel de Raney e Pd/C são bem menos ativos e,
portanto, mais seletivos.
Um exemplo para ilustrar esta seletividade é dado abaixo:
353
O extremo oposto é o mercúrio que é famoso por sua repelência frente o H2. Esta propriedade torna o Hg o
melhor material de eletrodo na polarografia, onde é necessária uma alta sobretensão com H2 (e O2, também).
Mediante esta propriedade é possível reduzir metais não nobres (ε « 0 V) em ambiente aquoso (ε = 0 V), sem
decomsição antecipada da água. Note que este fenômeno não pode ser descrito pela termodinâmica (Equação
de Nernst etc.), pois se deve a inibições cinéticas.
586
A. Isenmann
Princípios da Síntese Orgânica
CH2 CH3
H2
CH2
CH
Etilbenzeno
Ni ou Pd/C
H2
"PtO2"
Estireno
CH2 CH3
Etilcicloexano
Todos os catalisadores mencionados acima entram em contato com o substrato dissolvido,
de maneira heterogênea. Isto tem vantagens e desvantagens, como já foi mencionado no
item anterior: a vantagem é a fácil separação do catalisador da mistura reacional. Filtração,
centrifugação, mas também a fixação do catalisador num suporte polimérico facilitam a
vida do químico preparativo. As desvantagens são o mau entendimento mecanístico e, em
consequência, a dificuldade da previsão de produtos em caso de isomerizações além das
dificuldades na otimização sistemática, da eficácia do sistema catalítico.
Este último ponto pode ser superado ao empregar complexos metálicos do tipo Wilkinson.
Este catalisador, (Ph3P)3RhCl, foi o primeiro sistema para hidrogenações em fase
homogênea que permite a redução de alquenos e alquinos em alta velocidade, à temperatura
ambiente e 1 atm de H2.
Ph3P
Ph3P
Rh
Cl
PPh3
Catalisador de Wilkinson
(solução homogênea em hidrocarbonetos)
8.3.2
Redução com diimida
A diimida, N2H2, é redutor específico para insaturações simétricas, tais como:
C C
,
C C
,
N N
.
Duplas ligações polares ou olefinas assimétricas (tais como nitrilas, grupos carbonilas ou
compostos vinílicos) são reduzidas muito mais lentamente.
A diimida deve ser preparada in situ, pois existe apenas a –195 °C e por curto tempo. Ela é
obtida pela oxidação (!) da hidrazina, N2H4, conforme esquema abaixo. Note-se que a
diimida é uma molécula de geometria angulada, devido aos pares de elétrons não ligantes
nos nitrogênios. O isômero ativa no sentido desta redução é a diimida cis.
Reação preliminar:
587
A. Isenmann
Princípios da Síntese Orgânica
H2N NH2
Sais de Fe3+
[ Catal.: Cu2+ ]
Hidrazina
Oxidação
H
H
N N
Diimida cis
Observamos uma coisa estranha neste procedimento: o papel da diimida é redutor. Isto é, a
própria diimida está sendo oxidada, ao decorrer da reação principal. Ao mesmo tempo,
temos uma reação preliminar que causa a oxidação da hidrazina! Observe as paralelas com
o redutor específico, ácido fórmico (ver reduções de Leuckert-Wallach e EschweilerClarke, pp. 605 e 606, respectivamente), que em si já tem um carbono de NOX alto (+2),
mas o aumenta ainda voluntariamente para +4. A explicação aqui e lá é a mesma: a
altíssima estabilidade do produto final: N2 (CO2, respectivamente).
A sequência toda desta síntese, do ponto de vista do reagente (= oxidações):
N2H4 (estabilidade média) → N2H2 (muito reativo) → N2 (extremamente estável).
Reação principal:
N
N
H
+
H
C
N
C
N
H
C
N
C
N
+
H C
H C
H
Hidrogenação
cis
A redução com diimida percorre um estado intermediário cíclico onde 6 elétrons se
movimentam de maneira sincronizada. Por isso esta redução é altamente estereoespecífica,
cis (também chamada sin).
Somente recentemente 354 foi mostrado que a diimida também é um redutor eficaz para
compostos nitro aromáticos, percorrendo todos os estados de oxidação descritos no cap.
8.10, até chegar à amina, com altos rendimentos.
O maior valor preparativo da redução com hidrazina via diimida são consideradas as
condições suaves (em comparação à redução de Wolff-Kishner, p. 610, por exemplo) e a
indiferença frente ao grupo carbonila, ou seja, à hidrazona que se forma imediatamente
quando hidrazina e C=O entrem em contato.
Hidroboração
A adição de BH3 a alquenos é uma redução reversível, a 150 °C em “diglime”
(dietilenoglicol-dimetiléter, um solvente polar aprótico de alto ponto de ebulição, ver p.
610).
Esta reação seria apresentada em pormenores no cap. 10.7.1. Com o boro e o hidreto foram
introduzidos elementos mais eletropositivos do que o carbono, portanto se trata de uma
354
M. Kumarraja, Applied Catalysis A: General 265 (2004) 135-9.
588
A. Isenmann
Princípios da Síntese Orgânica
redução. Acrescentamos aqui a possibilidade de ocorrer isomerização, quer dizer,
mudanças da dupla-ligação C=C (por ser a reversa da hidroboração, uma etapa oxidativa).
+ H
b
R CH CH CH3
- H
R CH CH CH2
150 °C
[ diglima ]
b
R CH2 CH CH2
H
b
H
"Anti-Markovnikow"
b = 1/3 B (= boro no borano)
Com este procedimento pode-se então deslocar uma insaturação, C=C, do meio da cadeia
carbônica para a extremidade do alqueno – quase sempre uma reação paralela indesejada.
8.3.3
Redução de dienos
Dienos podem ser reduzidos com sódio em amônia líquida, na presença de etanol (uma
variação da redução de Birch, ver também p. 592). O esquema abaixo mostra o mecanismo
desta reação, uma transferência de elétrons desemparelhados (“SET”):
H 2C
CH CH CH2
+ Na
- Na+
H 2C
H 2C
+ Na
- Na
+
H 3C
CH CH CH2
CH CH CH2
+ C2H5OH
- C2H5O-
H 3C
CH CH CH2
CH CH CH2
+ C2H5OH
+
- C2H5O-
+
Produto
principal
A redução de olefinas simples não ocorre com este reagente, porque o radical-ânion que se
formaria como intermediário, não é estável o suficiente. No caso de dienos conjugados,
tanto o radical-ânion- quanto os intermediários subsequentes são estabilizados por serem
sistemas de elétrons π conjugados.
8.3.4
Compostos carbonílicos α,β-insaturados
Redução com LiAlH4
O hidreto de lítio e alumínio não reduz alquenos simples e apolares. Entretanto, este
reagente pode ser usado na redução de duplas ligações C=C conjugadas à carbonila (=
sistema Michael, ver cap. 6.6 e nota de rodapé 108 na p. 183):
589
A. Isenmann
Princípios da Síntese Orgânica
δ−
δ+
δ− δ+
O
C C C
OH
LiAlH4
R
R
Explicação: o grupo carbonila induz uma polarização na dupla ligação C=C que gera um
carbono eletrofílico, em posição β ao grupo carbonila.
Observação: LiAlH4 reduz todas as ligações π polares.
Redução específica de somente um grupo funcional
O
H2 / Pd (C)
C
ou Li / NH3
C C
R
H H
O
C
C C
R
OH
NaBH4
ou H2 / CuCr 2O4
C
C C
R
A olefina é reduzida muito lentamente, na presença do catalisador CuCr2O4. Igualmente, o
boridreto de sódio reduz especificamente o grupo carbonila (explicação ver p. 160).
8.4
Redução de Alquinos
8.4.1
Redução para olefinas cis
A comparação energética dos isômeros cis e trans, do mesmo alqueno, revela maior
estabilidade termodinâmica do isômero trans. Mesmo assim, os métodos para olefinas cis, a
partir de alquinos, não são raros. Especialmente as hidrogenações catalíticas, como
mostrado no exemplo a seguir, representam uma adição cis seletiva à tripla-ligação.
Redução com H2 e catalisador de Lindlar
355
, Pd/CaCO3/PbO
Neste catalisador o metal ativo, Pd, é depositado sobre carbonato de cálcio e posteriormente
“envenenado” pelo chumbo. Isto quer dizer, atenuada sua atividade catalítica, de forma que
não catalise a hidrogenação de duplas ligações apolares. A redução é regio e
estereosseletiva, de alquinos para olefinas cis:
355
H. Lindlar, R.. Dubuis Organic Synthesis V, 880.
590
A. Isenmann
Princípios da Síntese Orgânica
H2
+
Pd / CaCO3 / PbO
Hexa(2 cis,4 trans)dieno
Redução com B2H6/CH3COOH a 25 °C.
A hidroboração pode ser conduzida seletivamente até o estado da olefina quando o
diborano for hidrolisado in situ. Resulta a olefina cis em boa seletividade 356.
Um redutor alternativo que fornece igualmente o alqueno cis com bom rendimento é
DIBAL-H (= hidreto de diisobutilalumínio) 357.
Redução com diimida
A redução com diimida funciona bem, porém não se consegue parar completamente no
estado da olefina cis. Parte do substrato sempre será reduzida totalmente, até o alcano
correspondente 358.
8.4.2
Redução para olefinas trans
As olefinas trans se formam quando a redução não decorre sob controle cinético, pois o
isômero trans é o produto mais estável. Especialmente quando o mecanismo da redução é
em etapas, geralmente via SET, podemos esperar o isômero trans.
Redução com sódio em amônia líquida
(Para o mecanismo da redução de Birch, recorra a p. 592). O grupo acetileno é atacado
mais rapidamente do que um sistema aromático. Assim, é possível reduzir seletivamente o
alquino, ao trabalhar com quantidade estequiométrica de sódio:
Na, NH3(l)
trans-estilbeno
Um outro sistema redutor, aplicado em alquinos altamente hidrofílicos, é o sal do Cr(II) em
H2O ou DMF (dimetilformamida). Esse íon é um redutor eficaz, mas não muito forte (e
portanto específico):
ε0 = -0,408 V
Cr2+
Cr3+ + e
É uma síntese especialmente branda que funciona à temperatura ambiente.
356
H.C. Brown; G. Zweifel, J.Am.Chem.Soc. 1959, 81, 1512
J.G. Ulan, W.F. Maier, D.A. Smith, J.Org.Chem. 52 (1987) 3132.
358
Artigo de revisão sobre o redutor diimida: D.J.Pasto, R.T. Taylor, Org.React. 40 (1991) 91
357
591
A. Isenmann
Princípios da Síntese Orgânica
8.5
Redução de hidrocarbonetos aromáticos
8.5.1
Hidrogenação catalítica
A redução total, de aromáticos para cicloalcanos, se consegue com H2 de altas pressões e
um dos seguintes sistemas catalíticos:
1. Rh (bem seletivo para sistemas aromáticos)
2. Pt/AcOH, a 25 °C
3. Ni, sob alta temperatura e pressão de H2
Problemas ao ajustar a atividade do catalisador e a pressão necessitada impedem muitas
vezes estas reduções no laboratório, porém elas tem alta importância industrial.
8.5.2
Redução de Birch
Metais apropriados para a redução de Birch 359 são Li, Na e K. Entre eles o lítio geralmente
ganha preferência porque desprende hidrogênio muito lentamente, enquanto Na e K
decompõem o solvente (amônia ou etilamina) e o etanol com bastante rapidez. Desta
maneira se perde o reagente redutor destinado ao substrato aromático.
Dependendo das condições reacionais a redução prossegue até o 1,4-dieno ou até a monoolefina. Uma dupla ligação isolada não é reduzida por este método.
Redução com metal/NH3/EtOH:
Em uma reação rápida forma-se, sob controle cinético, o dieno não conjugado (para a
definição de conjugado, ver p. 131).
H
+ Li
- Li+
+ EtOH
- EtO(rápido)
H
H
H
+ Li
+ EtOH
- Li+
- EtO(rápido)
H
H
H
H
Redução com sódio em amônia líquida, sem adição de etanol:
O produto é a olefina simples.
359
Monografia: A.A.Akhrem, I.G. Reshotova, Yu.A. Titov, Birch Reduction of aromatic compounds, Plenum
Press New York 1972.
Artigos de revisão:
A.J.Birch, Pure Appl.Chem. 68 (1996) 553.
P.W. Rabideau, Tetrahedron 45 (1989) 1579.
592
A. Isenmann
Princípios da Síntese Orgânica
H
+ Li
- Li
H
H
- Li
H
H
H
H
+ NH3
+ Li
+ NH3
- NH2(lento)
+
H
+
- NH2(reversível)
+ NH3
H
H
H H
H
H
H
H
H
H H
H
H
H H
+ NH3
- NH2(lento)
H
+ Li
H
H H
- Li+
H H
H
H
H
+ Li
- Li+
+ NH3
- NH2-
O etanol é um donor de prótons fraco (pKa ≈ 18), certamente um composto de maior acidez
que a amônia (pKa > 30).
Ao trabalhar na presença de etanol se forma o 1,4-dieno em uma reação cineticamente
controlada (para a explicação recorra à Figura 12, p. 131). Se não estiver presente o álcool,
a etapa da protonação que precede a formação do 1,4-dieno é lenta e reversível. Isto abre o
caminho para a isomerização que fornece o produto de maior estabilidade termodinâmica.
Então pode se formar sob estas condições em uma etapa bem lenta, o 1,3-ciclohexadieno.
Como já foi discutido no item 2.1.1, o dieno conjugado é mais estável do que o 1,4-dieno
por aproximadamente 17 kJ.mol-1. Em seguida o 1,3-dieno pode ser mais uma vez reduzido
conforme enunciado acima (p. 589), então fornecendo 100% da olefina simples.
A redução sem o aditivo etanol é uma reação bastante demorada que ocorre via SET, do
metal para o substrato. Isto pode gerar um problema preparativo porque outros grupos
funcionais podem ser reduzidos antes do sistema aromático. Sendo assim, grupos -CH2OH
podem sofrer hidrogenólise para -CH3; qualquer halogênio se perde no substrato, também.
8.5.3
Redução de heteroaromáticos para heterocíclicos saturados
Em alguns casos a reação catalítica com H2 pode ser usada na redução de compostos
heteroaromáticos. Porém, qualquer composto contendo enxofre envenena o catalisador.
593
A. Isenmann
Princípios da Síntese Orgânica
Nesses casos, deve-se usar um metal alcalino sendo fonte de elétrons, mais um solvente que
fornece os hidrogênios em falta.
Exemplo:
Na / EtOH
ou H2 / Raney-Ni
N
N
H
Piridina
Piperidina
Mas:
H2 / Pd/C
N
S
Na / EtOH
N
S
H
Igualmente foram reduzidos pirol para pirolidina
sistema redutor de Birch.
8.6
Redução de haletos orgânicos
8.6.1
Redução com estanano
360
e furano para THF
361
, usando o
A remoção de um halogênio e sua substituição por hidrogênio geralmente não é uma tarefa
difícil. Portanto, é indicado usar um redutor menos poderoso, para não correr o perigo de
reduzir outros grupos funcionais, sem querer. Também pede, numa síntese sobre várias
etapas, fazer primeiro as reduções necessárias, e só depois a halogenação. O hidreto de
tributilestanho, Bu3SnH, é um reagente redutor muito seletivo 362. Na maioria dos casos
representa a melhor opção para reduzir compostos halogenados.
A reação é bastante rápida, pois ocorre em cadeia radicalar (ver também cap. 10.4.1). Ela é
iniciada, por exemplo, por AIBN (ver p. 54), adicionado em quantidades catalíticas.
360
T.J. Donohoe, P.M. Guyo, J.Org.Chem. 61 (1996) 7664.
T.Kinoshita, D. Ichinari, J. Sinja, J.Heterocyclic Chem. 33 (1996) 1313.
362
H.G.KuiViala, Synthesis 1979, 499.
361
594
A. Isenmann
Princípios da Síntese Orgânica
Propagação: 1) R
+
2) Bu3Sn
Bu3SnH
+
R
R H
+ Bu3Sn
Bu3SnBr
Br
+
R
Os radicais trialquilestanil são apropriados para a transferir seletivamente o hidrogênio na
posição do halogênio. O fato de que a ligação Sn-Br que se formou no subproduto Bu3SnBr
ser bastante estável (contato macio-macio) faz com que as posições dos hidrogênios dentro
do substrato fiquem inalteradas. Isto é uma vantagem preparativa porque desta forma
quaisquer isomerizações ficam ausentes. Embora que hoje ser possível comprar Bu3SnH,
ele pode ser substituído por reagentes mais convencionais. A alternativa é então a redução
do composto halogenado usando 1 equivalente de LiAlH4 em THF, na presença de
quantidades catalíticas de cloreto de trialquilestanila, por exemplo Bu3SnCl. Neste caso a
restituição do estanano se provou ser a reação mais rápida, o que confirma o papel catalítico
do Sn. Essa, aliás, é a variação mais econômica desta síntese.
Reações paralelas podem ser esperadas em substratos que têm grupos ceto, alcoóis
primários 363 ou alcoóis secundários 364 que, embora de menos rapidamente, são reduzidos
pelo Bu3SnH, também.
8.6.2
C
Redução de haletos orgânicos com metais alcalinos e alcalinos-terrosos
X
+ Mg
C MgX
+ H2O
Hidrólise
C H
+
Mg2+
+
OH- + X-
Reagente de Grignard
A hidrólise na segunda etapa pode ser substituída por uma alcoólise. Os seguintes sistemas
redutores representam alternativas ao reagente de Grignard:
Li/t-BuOH ; Na/C2H5OH ; Zn/HCl/H2O.
Atenção: há perigo de acoplamento C-C entre as duas unidades estruturais, C-MgX e C-X,
já que esses carbonos têm polarizações opostas (reação de Wurtz, ver p. 750).
8.6.3
Hidrogenólise de haletos orgânicos
C6H5 CH2 Cl
1 mol H2
catalisador
C6H5 CH3
+ HCl
Cloreto de benzila
O mecanismo corresponde em partes ao da hidrogenação de alquenos (p. 584):
363
364
D.L.J. Clive, J.Chem.Soc. Chem. Commun.1978, 41.
E.J. Brisbe, J.C. Martin, Synth. Commun. 15 (1985) 401.
595
A. Isenmann
Princípios da Síntese Orgânica
H
C
H
C
X
C
H
H
H
C H
C H
C
C H
C
H
C H
C H
C
H
C
- HX
H
H
H
= Superfície do catalisador metálico
Pode-se afirmar que os haletos alifáticos saturados são reduzidos mais lentamente do que os
haletos nas posições benzila ou alila – o que pode ser entendido em termos de estabilidade
dos radicais (ver p. 65).
Facilidade da substituição (= reatividade relativa):
Haleto de alila > haleto de benzila > haleto de arila > haleto de alquila
R-I > R-Br > R-Cl > R-F
Pt > Ni, Pd
A adição de bases ou o uso de solvente contendo grupo(s) hidroxila(s) promovem a
desalogenação.
Não pode ser negado que a desalogenação catalítica costuma ser uma reação paralela
indesejável que ocorre durante hidrogenações de compostos insaturados contendo haletos.
Nem sempre as duplas ligações são reduzidas mais rapidamente. Sob este aspecto pode ser
vista como vantagem preparativa que os cloretos de alquilas primários e secundários (que
se encontram no final da escala de reatividade dada acima) são mais resistentes à
hidrogenólise. Eles geralmente não são afetados quando se trabalha em ambiente neutro, a
25 °C.
Além da atividade ante a ligação C-X, a hidrogenólise provoca também a quebra de
diversas ligações simples e fracas, tais como:
O-O , N-N , S-S , C-S.
8.6.4
Redução com SnCl2 ou CrCl2 em HCl ou DMF
O
Cl
CH2
O
C
O
H
+ SnCl2 +
HCl
H2O
H 3C
O
C
H
+
SnCl4
596
A. Isenmann
Princípios da Síntese Orgânica
A redução seletiva pode ser feita alternativamente no solvente polar aprótico,
dimetilformamida (DMF). A ordem de reatividade dos haletos neste solvente é
Benzila > alila > alquila.
Também as cetonas halogenadas em posição α, Ar-CO-CH2X, podem ser reduzidas,
formando Ar-CO-CH3.
Os haletos aromáticos, Ar-X, não são reduzidos por este método.
8.6.5
Redução com LiAlH4
Aqueles grupos que têm grande facilidade de sair pelo mecanismo SN2, tais como tosilato,
sulfato ou haleto, podem ser substituídos pelo hidreto H- , usando-se LiAlH4. Lembre-se
que H- é um dos melhores nucleófilos (ver caps. 1.3.7 e 5.2.3):
O
H5C2
O
S
O C2H5
O
1) LiAlH4
2 C2H6 + SO42-
2) H2O
Como podemos esperar de uma SN2 (ver p. 17), qualquer impedimento espacial, isto é,
maior grau de substituição no carbono, dificulta a redução.
Reatividade dos haletos de alquila:
•
primário > secundário > terciário
•
R-I > R-Br > R-Cl
Os haletos aromáticos, Ar-X, não são reduzidos por este método.
8.6.6
Redução de iodetos com HI
R
+
I
HI
R H
+
I2
Como já a desbromação com iodeto (ver p. 152) nos surpreendeu, esta reação também
parece fora do padrão. Porém, ela ocorre seletiva e suavemente.
8.7
Redução de alcoóis, éteres e fenóis
8.7.1
Redução de alcoóis para hidrocarbonetos
O grupo hidroxila em compostos alifáticos mostra-se bastante resistente à redução. Porém,
existem dois métodos dos quais o segundo se processa sob condições mais drásticas.
O processo radicalar com que é possível reduzir alcoóis, usando Bu3SnH, já foi
mencionado (p. 594).
597
A. Isenmann
Princípios da Síntese Orgânica
Redução indireta, via haleto
A transformação do álcool em um haleto é uma reação de SN simples (usando SOCl2, PCl3,
ver p. 24; Figura 4 na p. 38). A redução em seguida ocorre conforme descrito no item 8.6.
R OH
+ H X
R X
- H2O
R H
Destilação com zinco em pó
A força para esta reação se explica com a alta afinidade do zinco para oxigênio: forma-se
como subproduto o óxido de zinco 365.
8.7.2
Quebra da ligação O-C benzílico
Hidrogenólise com H2/CuCr2O4
A cromita de cobre, CuCr2O4, é um espinelo (estrutura cúbica; típica em diversas pedras
preciosas) que é um catalisador de atividade moderada em hidrogenações 366 e também em
desidrogenações (= oxidação, ver p. 645). Sua vantagem principal é o preço: é bem mais
barato que os catalisadores de Pd e Pt.
A seguir algumas hidrogenações viáveis, usando esse catalisador; algumas destas requerem
elevadas pressões de hidrogênio e temperaturas de até 300 °C.
C6H5 CH2 OH
H2
CuCr 2O4
C6H5 CH3 + H2O
Álcool benzílico
C6H5 CH2 OR
H2
CuCr 2O4
C6H5 CH3 + ROH
Éter benzílico
O
C6H5 CH2 OC
CH3
H2
CuCr 2O4
C6H5 CH3 + H3C
COOH
Éster benzílico
Note-se que a hidrogenólise da ligação C-O se restringe à posição benzílica, enquanto em
outros alcoóis, éteres e ésteres esta redução não funciona.
365
Esta redução é historicamente importante, porque contribuiu na descoberta da constituição da alizarina.
Sua síntese finalmente, a partir do antraceno, removeu as últimas dúvidas (A.v.Baeyer, Graebe, Liebermann,
1862).
366
H.Adkins, E.E. Burgoyne, H.J. Schneider, The copper-chromium oxide catalyst for hydrogenation,
J.Am.Chem.Soc. 72 (1950) 2626
598
A. Isenmann
Princípios da Síntese Orgânica
Aplicação desta redução na síntese de (oligo-)peptídeos
Existe um derivado do ácido carbônico, o cloreto de carbobenziloxi, CBO (Bergmann
1932), usado para proteger seletivamente o grupo –NH2 do primeiro aminoácido da cadeia
em uma série de condensações sucessivas que levam ao oligopeptídeo. Sem efetuar essa
proteção, o peptídeo pode crescer em ambos os lados, tanto no final da amina quanto no
final do ácido carboxílico, já que cada monômero dispõe dos dois grupos funcionais. Após
de terminar a formação do peptídeo por policondensação, o grupo protetor CBO pode ser
removido redutivamente, sob condições que não interferem nas ligações de amidas entre as
unidades de aminoácidos, nem nos grupos específicos dos aminoácidos.
O procedimento de proteger seletivamente um grupo funcional do primeiro aminoácido,
fazer sucessivamente as condensações com outros e finalmente soltar o grupo protetor é
uma sequência reacional bastante complexa, mas pode ser automatizado por fixação do
grupo CBO num suporte insolúvel. Para este fim emprega-se um poliestireno
funcionalizado onde as cadeias laterais são grupos CBOs. Para a imobilização do reagente
em polímeros insolúveis se estabeleceu a expressão “Síntese em fase sólida” 367. Este
exemplo em particular é conhecido como síntese de Merryfield:
O
C6H5 CH2 OC
H
Cl
+
O
H2N C COOH
R
CBO
O
H O
R
+ HO
R
C
OH
H2 / CuCr 2O4
n
H O
C NH C
COOH
R
α-aminoácido
com grupo amino
protegido
C6H5 CH2 O C NH C C
O
C6H5 CH3
C6H5 CH2 O C NH C
- HCl
α-aminoácido
Síntese de
peptídeos
H
H O
OH
n
- CO2
H NH C
R
C
OH
n
Um protetor igualmente bom é o éter benzílico: ele serve para proteger alcoóis, aminas e
até ácidos carboxílicos. Sua remoção pode ser efetuada, em analogia ao grupo CBO, por
redução seletiva e suave.
Procedimentos:
367
Marquardt, M., Eifler-Lima, V.L. A síntese orgânica em fase sólida e seus suportes poliméricos mais
empregados. Quím. Nova 24 (2001) 846-855.
599
A. Isenmann
Princípios da Síntese Orgânica
Substrato
protegido
PhCH2Br
R OH
Base
"Williamson"
H2 / cat.
R O CH2Ph
HBr
Éter
H2 / cat.
2 PhCH2Br
R NH2
Base
R N(CH2Ph)2
HBr
Amina terc.
R COOH
PhCH2OH / H+
O
H2 / cat.
R C
O CH2Ph
HBr
reconstituição
R OH + Ph
CH3
R OH + Ph
CH2Br
R NH2 + Ph
CH3
R NH2 + Ph
CH2Br
R COOH + Ph
CH3
R COOH + Ph
CH2Br
Éster
Redução com cloreto de cromo (II)
O NOX +2 é incomum para o cromo, os estados bem mais estáveis são +3 e +6. Portanto,
este sal de Cr(II) é redutor (ver p. 591).
C6H5 CH2 OH
Cr2+ / HCl
C6H5 CH3
+ C6H5 CH2 CH2 C6H5
O subproduto de dimerização nesta síntese se deve a um mecanismo radicalar.
Redução com sódio em amônia líquida
A redução com o sistema de Birch percorre, igual a síntese acima, etapas de SET.
8.7.3
Remoção de grupos -OH fenólicos
Tanto a remoção quanto a introdução de grupos fenólicos (em geral: grupos alcoólicos) é
uma tarefa preparativa bastante delicada. Existem poucos métodos específicos, seletivos e
de alto rendimento. Portanto, essas etapas devem ser contornadas onde for possível
(exceção: dióis a partir de olefinas, ver p. 628).
Destilação com zinco em pó
A destilação com zinco em pó tem relativamente pouca importância preparativa neste
sentido. Historicamente, porém, foi de grande utilidade para elucidar a constituição de
substâncias naturais, através da formação de derivados menos reativos, mais voláteis e de
análise simplificada.
Formação do tosilato e redução do mesmo com hidrogênio e catalisador.
600
A. Isenmann
Ar
Princípios da Síntese Orgânica
+ TsCl
- HCl
OH
Ar
O
H2 / Ni
SO2 Ar´
Ar
H
Tosilato
Redução com 2,4-dinitrofluorbenzeno
Este reagente foi usado por Sanger (1954), em uma das primeiras análises de sequência de
peptídeos (Método dinitrofenil ou DNP). Como variação desta podem-se também reduzir
aminas aromáticas em vez de fenóis. Vale ressaltar que não se formam compostos azo, nem
hidrazo, durante esta redução, enquanto com outros sistemas redutores consegue-se parar
nestes estados oxidativos (ver p. 616).
O2N
O2N
(-NH2)
O- + F
NO2
(-NH-)
O
-
- F
NO2
H2 / Ni
SN no anel
H2N
H2N
(-NH-)
O
NH2
Na / NH3
H
+
(H2N-)
HO
NH2
Redução de Kenner e Pelletier
A primeira etapa é a esterificação do fenol com o derivado do ácido fosfórico, em ambiente
levemente ácida. O éster produzido pode ser removido do equilíbrio por ser bem solúvel em
solventes apolares (operação em sistema bifásico). Tanto a esterificação quanto a redução
se processam sob condições suaves. Porém, esta síntese somente é útil em escala semimicro.
O
Ar
OH
+
HO
P (OC2H5)2
[H+]
H2O / hexano
O
Ar
O
P (OC2H5)2
O
O
Ar
8.7.4
O
P (OC2H5)2 + 2 Na +
NH3
Ar
H + NaNH2 + Na+ -O
P (OC2H5)2
Quebra redutiva de feniléteres
Redução com sódio em etanol
601
A. Isenmann
Princípios da Síntese Orgânica
Um éter com uma parte aromática e outra alifática, pode ser reduzido por elétrons livres,
disponibilizados por sódio. Note-se que somente a parte alifática sofre redução, enquanto a
parte aromática permanece em forma de fenol.
C6H5 O CH3
Na
EtOH
C6H5 OH
+ CH4
Como meio redutivo podem também ser usados, com o mesmo resultado:
Na/NH3 ; Na/diglime (= dietilenoglicoldimetiléter) ; Na/piridina.
O grupo fenoxi ganha, assim, certo significado como grupo protetor (o benziléter, porém, é
bem mais utilizado para este fim, ver p. 598).
8.7.5
Redução de epóxidos
Um epóxido é formalmente no mesmo nível de oxidação do que um álcool ou um éter. Isto
pode-se verificar para ambos os carbonos envolvidos no anel. A tensão de pequenos anéis,
porém, o deixa mais reativo, em todos os sentidos, que os seus parentes.
A partir de um epóxido assimétrico se obtém entre 95% e 100% do álcool mais substituído
ao reduzi-lo com LiAlH4 ou (t-BuO)3LiAlH: estes hidretos entram com maior facilidade no
carbono menos substituído (= mais positivado). Isto não é somente uma particularidade do
hidreto, mas vale para todos os nucleófilos (geralmente são nucleófilos fortes, tais como
reagente de Grignard, p. 438, alcóxidos ou haletos). O mecanismo é SN2, implica que com
a entrada do hidreto a configuração no carbono se inverte (ataque de trás).
δ−
+I (δ+)
O
δ+
Redução
R CH CH2
" H- "
OH
R CH CH3
+
R CH2 CH2OH
Produto principal
A redução com H2/Pd, por outro lado, fornece o álcool menos substituído com altos
rendimentos. A redução catalítica não segue um mecanismo iônico, mas é sincronizada ou
envolve etapas com caráter radicalar (p. 584). A reatividade depende particularmente dos
contatos H2↔catalisador e substrato↔catalisador. Mais intenso o contato com a superfície
do catalisador, mais reativos são os reagentes. Um impedimento espacial deixa então um
lado do substrato mais distante do catalisador, portanto fica menos reativo. O produto da
redução catalítica é, portanto, o álcool primário 368.
368
M. Oshima, H. Yamazaki, I. Shimizu, M. Nizar, J.Am.Chem.Soc. 111 (1989) 6280.
S. Krishnamurthy, R.M. Schubert, H.C.Brown, J.Am.Chem.Soc. 95 (1973) 8486.
602
A. Isenmann
8.7.6
Princípios da Síntese Orgânica
Redução de α-hidroxicetonas (aciloínas)
Sobre o acesso das aciloínas ver pp. 465 e 612.
O OH
R C CH
R
O
Zn / HCl / AcOH
R C CH2 R
∆
8.8
Redução de aldeídos e cetonas
8.8.1
Redução de aldeídos e cetonas para alcoóis
Redução com hidretos metálicos
Para obter uma vista geral sobre os poderes redutores dos hidretos metálicos, recorra ao
quadro geral, na p. 621. A facilidade desta reação não só se explica pela alta reatividade e
disponibilidade do hidreto, mas também pela afinidade extraordinária do alumínio pelo
oxigênio. Um bom funcionamento da redução com LiAlH4 requer a rigorosa ausência de
umidade; solvente bastante utilizado é THF, no qual a redução se torna mais seletiva.
AlH4 Li
+
AlH3 +
C O
H
C O- +
Li+
+ 3 O C
Li+ Al O CH
+
H C O AlH3 Li
4
Complexo estável
Do ponto de vista do LiAlH4, temos uma inserção do grupo carbonila, entre a ligação Al-H.
O alcóxido de alumínio pode ser hidrolisado posteriormente usando álcool ou água:
Li+ Al O CH
+ 2 H2O
4
CH OH
+
LiAlO2
4
Para a redução da maioria dos aldeídos/cetonas geralmente basta usar o NaBH4, redutor
mais suave e seletivo. Este hidreto tem a grande vantagem de funcionar até na presença de
grupos hidroxilas no substrato, enquanto LiAlH4 desprotona estes grupos imediatamente.
Então, o redutor se perde em forma de H2 e a reatividade do substrato é modificada. Ao
usar NaBH4 pode-se até operar em solução alcalina aquosa.
Em moléculas onde o grupo carbonila é um centro pro-quiral e, além deste, existe na sua
direta vizinhança um carbono assimétrico, a redução ocorre com diferentes facilidades
pelas duas faces do grupo carbonila. Sob condições brandas pode-se obter um dos
603
A. Isenmann
Princípios da Síntese Orgânica
enanciômeros em excesso (até 75% ee, segundo a regra de Cram da indução assimétrica 369;
ver também p. 229). A D-frutose é um bom exemplo onde a redução fornece dois alcoóis
(os hexóis diastereoisômeros, D-sorbita e D-manita), em partes desiguais.
Exemplo:
R2
R1
O
C*
C
LiAlH4
R4
R1
R2
R1
H
+
C* C* R4
C* C* R4
R3
R3 OH
Produto principal
(ee de até 75%)
R3
R2
OH
H
Redução de Meerwein-Ponndorf-Verley (MPV)
Por redução MPV se entende a redução de aldeídos e cetonas a alcoóis usando isopropóxido
de alumínio em isopropanol. Interessante é que o grupo isopropóxido é oxidado nesta
reação, fornecendo acetona. Consequentemente se trata de uma óxido-redução reversível
cujo equilíbrio deve ser deslocado em direção aos produtos. Para este fim a acetona é
removida continuamente por destilação. A reversa desta síntese também tem aplicação
prática: a oxidação de alcoóis usando-se acetona em ambiente fortemente alcalino é
conhecida como oxidação de Oppenauer (ver p. 650).
H3 C
O
(CH3)2CH O Al
2
C
O
CH3
H3 C
C
CH3
+ (CH3)2 CH OH
O
H
+
CR2
(CH3)2CH O Al
2
H
R2CH OH
+
- (CH3)2CH O-
(CH3)2CH O Al
O CR2
3
Redução com sódio em xileno: síntese de pinacol
Este acoplamento redutivo já foi apresentado na p. 467.
O
2 H3C
C CH3
+ 2 Na
- 2 Na+
O- O-
O2 H3C
C CH3
H3C
C C CH3
CH3CH3
Importância particular tem esta reação quando o substrato tem dois grupos cetonas,
separados por uma cadeia hidrocarbônica de até 8 carbonos. Neste caso um anel está sendo
fechado, conhecido como condensação de aciloína segundo Prelog e Stoll (ver p. 473).
369
P.A. Bartlett, Stereocontrol in the Synthesis of Acyclic Systems, Tetrahedron 36, 3 (1980).
604
A. Isenmann
Princípios da Síntese Orgânica
Pré-condição para o sucesso desta reação é o trabalho em solvente aprótico. Na presença de
hidrogênios ácidos o íon-radical cetila que se forma na primeira etapa é protonado e,
recebendo mais um elétron, então é novamente reduzido. Forma-se então um álcool.
OH 3C
C
+ ROH
CH3
- RO-
OH
H3 C
C CH3
OH
+ Na
- Na+
H3 C
C
+ ROH
CH3 - RO-
OH
H3C
C CH3
H
Este processo, realizado desde o início em solvente prótico, é conhecido como redução de
Bouveault-Blanc. O sistema redutor mais aplicado é o sódio em etanol. Na sua execução
deve-se considerar a perda de qualquer halogênio em posição α ao grupo carbonila:
O X
R C CR2
O
- X-
R C CR2
+ Na
- Na+
O
H+
R C CR2
O
R C CHR2
Redução
até o álcool
Enolato
8.8.2
....
Redução de aldeídos e cetonas para aminas
Aminação redutiva
R
C O
NH3
R
R
C NH
R
H2 / PtO2
R
H
C
R
NH2
Redução via oxima
H2NOH
R
C O
R
Hidroxilamina
R
C NOH
R
H2 / Cat
ou LiAlH4
R
ou Na / EtOH
R
H
C
NH2
Redução de Leuckart-Wallach
Este é um bom método de preparo de aminas, especialmente de aminas terciárias. Neste
caso os reagentes são aminas secundárias e ácido fórmico em excesso. Seguem dois
exemplos típicos desta redução, dos quais o segundo mostra claramente a vantagem desta
síntese: produzindo uma amina terciária que é de acesso desconfortável por outros métodos.
Exemplo 1:
605
A. Isenmann
Princípios da Síntese Orgânica
O
R
R
C O +
OH
C
NH3
R
R
NH2
H C
R
+ H+
- H2O
C NH2
OH
R
H
C
R
R
Cátion imônio
+ CO2 + H+
NH2
Exemplo 2:
O
R(H)
R(H)
C O
+
OH
C
HN
R(H)
R(H)
N
+ H+
- H2O
H C
R2(H2)C N
OH
H
+ CO2 + H+
R2(H2)C N
A primeira etapa em cada exemplo é a formação do cátion imônio (este também é o
princípio da ativação do componente “carbonílico”, na condensação de Mannich, ver p.
491). Já na segunda etapa ocorre o ataque do ácido fórmico que é, neste caso, o portador do
hidreto. O próprio ácido fórmico é oxidado a CO2, por sua vez um produto de alta
estabilidade termodinâmica.
Devem ser mencionadas duas variações da reação de Leuckart-Wallach que têm relevância
prática:
1. Redução da cetona com formiato de amônio, em altas temperaturas (180 °C)
2. Reação com DMF (dimetilformamida), sob refluxo (150 °C)
A última possibilidade mostra que nem sempre a DMF tem o papel de solvente polar
aprótico, mas também pode reagir com substratos contendo grupos carbonilas (outra reação
onde o DMF entra como reagente, ver formilação de Vilsmeyer, pp. 304 e 796).
Redução de Eschweiler-Clarke
Embora não ser um método para aldeídos/cetonas em geral, a síntese a seguir deve ser
discutida próxima à redução de Leuckart-Wallach, devido às semelhança em reagentes e
mecanismo.
A reação de Eschweiler-Clarke pode ser descrita como alquilação redutiva de aminas. O
reagente é formaldeído contendo ácido fórmico (que, aliás, sempre está presente em traços,
na formalina e no paraformaldeído comercial).
R
H
N H
R´
+
O
H
HCOOH
- CO2
R
N CH3
R´
606
A. Isenmann
Princípios da Síntese Orgânica
Do ponto de vista do formaldeído temos então uma redução do aldeído à amina, portanto o
assunto encaixa neste capítulo. Em analogia à redução de Leuckart-Wallach, o agente
redutor é o ácido fórmico (ou formiato) que está sendo reduzido ao gás carbônico. O
esquema reacional mostra uma amina secundária como material de partida, mas a reação
também funciona com alquilaminas primárias, onde resultará uma dimetilalquilamina (ver
também questão 7, na p. 671).
R
H
N H
+
R´
O
H
R
HCOOH
- CO2
N CH3
R´
Ao contrário da maioria dos métodos clássicos de metilação (por exemplo, com iodeto de
metila ou sulfato de metila), aqui não se corre o perigo da "superalquilação" da amina, quer
dizer, no método de Eschweiler-Clarke se formam exclusivamente aminas terciárias, mas
não sais quaternários de amônio. As outras vantagens desta síntese são os reagentes baratos
e a facilidade do scale-up, quer dizer, sua aplicação em escalas maiores.
Mecanismo:
A amina e o formaldeído reagem formando uma imina (ou um íon imônio, conforme visto
na condensação de Mannich, p. 491). Já na segunda etapa o ácido fórmico transfere um
hidreto à imina, liberando irreversivelmente CO2. Em caso de amina primária estas etapas
se repetem, com leves mudanças nas cargas (questão 7, na p. 671).
H
H
O
H
1)
R NH2
CH2
2)
N
R
O
H
R
H
H
N
H
+H
H
R
O
O
O
+
N
H
CH2
H
- H2O
- H+
N
R
Imina
CH3
- CO2
R
N
H
Forma-se um sal, o formiato de amônio (terciário), que pode ser separado da mistura
reacional, facilmente por decantação.
Sob as condições aplicadas se justifica a formulação do estado de transição como ciclo de 6
membros, conforme feito no esquema acima. Traços de umidade e a basicidade da própria
amina, por outro lado, levam à desprotonação do ácido fórmico, e com o formiato podemos
formular um estado de transição não cíclico:
607
A. Isenmann
R
Princípios da Síntese Orgânica
O
H
N
R´
N CH3
O-
H
O
R
R´
H
+
C
O
Formiato
Produtos paralelos:
Em alguns casos o rendimento em amina terciária é diminuido devido à formação de
compostos carbonilados que se formam a partir da imina (= base de Schiff, ver p. 411) por
hidrólise. Isso acontece especialmente fácil quando R contém um grupo CH2 em posição α
ao nitrogênio:
R
R
N
N
H2O
R
O
+ NH2
Também formamidas podem resultar como produtos paralelos em menor porcentagem.
Variações da reação de Eschweiler-Clarke:
1) O poder redutor do formiato é suficientemente alto para reduzir também compostos
nitrosos (mas não nitro!). Após ter efetuada a redução do grupo nitroso para amina, segue a
metilação no N e resulta, conforme esperado, uma N,N-dimetilamina. Essa síntese é
especialmente valiosa na a química dos aromáticos. Como discutido lá (p. 290), a
introdução do grupo nitroso no sistema aromático rico em elétrons é uma reação bastante
fácil.
R NO
HCHO
HCOOH
R N
CH3
CH3
2) Um procedimento mais recente 370 evita o uso do tóxico formaldeído e ácido fórmico
concentrado. Em vez destes pode-se usar paraformaldeído (sólido) e ácido oxálico. O
último tem aproximadamente o mesmo poder redutor do que o ácido fórmico, porém seu
manuseio é mais fácil (menos volátil). Desta forma a metilação da amina (muitas vezes um
líquido) não requer de solvente adicional, um avanço em direção à "green chemistry"; basta
aquecer a mistura a 100 °C por 1 hora e não se requerem etapas demoradas de purificação
do produto.
Exemplo:
370
T. Rosenau, A. Potthast, J. Röhrling, A. Hofinger, H. Sixta, P. Kosma, Synth. Comm. 32 (2002) 457-65.
608
A. Isenmann
Princípios da Síntese Orgânica
HO
O
O
.
O
N
H
Morfolina
OH
CH2 O
n
O
2 H2O
100 °C, 1 h.
N
100%
3) Segundo Fache 371 a reação de Eschweiler-Clarke pode ser estendida à N-alquilação de
amidas. A maioria dos métodos padrões para essa tarefa falha ou leva a baixo rendimento,
em caso de amidas secundárias ou até cíclicas - em geral com amidas onde a acidez N-H é
especialmente baixa. Além disso, este método pode ser aplicado para conectar o nitrogênio
a um grupo alquil secundário - uma reação que se impede por outras estratégias, devido à
eliminação sempre concorrente no carbono secundário 372. O redutor desta vez deve ser
H2/Pd, em vez de ácido fórmico, o solvente é aprótico e seco.
Exemplos:
R
NH2
R
Pd/C/Na2SO4
H2, 40 bar
O
O
ou
+
R1
R2
[EtOAc], 100 °C
NH
O
R2
R1
ou
O
O
N
H
N
R1
8.8.3
81 a 95%
R2
Redução de aldeídos e cetonas a hidrocarbonetos
Redução de Clemmensen
A cetona à qual se aplica esta redução muitas vezes é o produto da acilação de FriedelCrafts (ver p. 297). Sendo esta uma reação bem controlável, a combinação com a redução
de Clemmensen é a melhor opção para alquilar um anel aromático. Para verificar as
desvantagens do procedimento alternativo, a "alquilação de Friedel-Crafts", recorra à
Tabela 16, na p. 299.
371
372
F. Fache, L. Jaquot, M. Lemaire, Tetrahedron Lett. 20 (1994) 3313.
T. Gajdo, A. Zwierak, Synthesis 1981 1005.
609
A. Isenmann
Princípios da Síntese Orgânica
O
C CH3
Zn / Hg / HCl
CH2 CH2 CH3
∆
Um solvente frequentemente usado é o tolueno, desde que não seja muito semelhante ao
produto desta redução - se não a purificação torna-se muito difícil. Este método não pode
ser aplicado em substratos com outros grupos funcionais que são sensíveis ao ácido. Nesses
casos deve-se aplicar a redução de Wolff-Kishner.
Redução de Wolff-Kishner
Na redução de Wolff-Kishner, o meio redutivo é hidrazina e KOH, então um ambiente
fortemente alcalino.
O solvente preferido nesta reação é "diglime”, dietilenoglicol dimetiléter, H3CO-CH2-CH2O-CH2-CH2-OCH3. Este solvente é polar e se destaca por ter um ponto de ebulição muito
alto. Assim, consegue-se conduzir a reação entre 200 e 300 °C.
R
C N NH
R
R
C O
+
-
C N NH2
H2N NH2
+ OH
R
R
R
R
C N NH
R
+ H2O
- OH-
R
CH N NH
+ OH-
R
R
R
CH N N
R
- N2
CH
R
+ H2O
- OH-
R
CH2
R
Para muitos substratos ambas reduções, Clemmensen e Wolff-Kishner, são possíveis. Neste
caso deve–se lembrar da desvantagem do procedimento de Wolff-Kishner de operar a
temperaturas muito altas. A sua vantagem, por outro lado, é a melhor adaptação a
quantidades maiores de substrato, porque produz menos resíduos de impacto ambiental.
Redução de tosilidrazonas com NaBH4
Utilizada com finalidade de redução, a tosilidrazina também é conhecida como reagente de
Bamford-Stevens (p. 873).
610
A. Isenmann
Princípios da Síntese Orgânica
R
R
C O
+
H2N NH
R
SO2
CH3
- H2O
Tosilidrazina
C N NH
R
SO2
CH3
Tosilidrazona
R
NaBH4
CH2
+
N2
+
SO2
CH3
R
Remoção de enxofre em tiocetais segundo Mozingo
O sulfeto é um veneno para catalisadores de metais de transição. Por isso deve-se empregar
quantidades maiores de níquel, para efetuar a seguinte redução do tiocetal 373:
R
C O
+
R
HS
CH2
HS
CH2
H+
R
R
S
S
CH2
CH2
H2 / Ni
R
CH2 + 2 H2S +
H3C CH 3
R
Este método é especialmente valioso por se consegue a remoção total do grupo funcional, o
grupo carbonila sob condições suaves. Desta maneira o grupo carbonila pode ser usado
como grupo ativador, dentro de uma síntese multi-etapas, para ser removido segundo
Mozingo no final da síntese ("FGA", ver p. 571).
8.9
Redução de ácidos carboxílicos e seus derivados
8.9.1
Redução para alcoóis e aminas
Redução com hidretos metálicos
Para redução dos derivados do ácido carboxílico por complexos metálicos de hidreto,
recorra à vista geral no item 8.13. Observa-se que o ácido carboxílico livre não pode ser
reduzido por hidretos metálicos porque seu próton ácido reage rapidamente com o hidreto,
liberando H2 e tanta energia que pode levar ao incêndio. O produto desta reação ácido-base
é o carboxilato cuja carga negativa dificulta ou até impede a reação no sentido desejado
com o hidreto. Além do mais, o sal R-COO-Li+ é de baixa solubilidade, portanto recusa-se à
redução. Assim, perdemos grande quantidade de reagente hidreto metálico, sem efeito
redutor no substrato.
R COOH
+ "H-"
- H2
R COO-
"H-"
não há redução
A solução neste caso é uma derivatização do ácido carboxílico para o éster, usando metanol
(ou etanol) e um catalisador ácido (ver cap. 5.2.3). Com os ésteres não se espera tais
dificuldades, o carbóxido de metila (ou etila) pode ser abandonado, sem impedimentos e
373
H.O.House, Modern Synthetic Reactions, Benjamin, Menlo Park 1972, p. 15-16.
611
A. Isenmann
Princípios da Síntese Orgânica
reações consecutivas indesejadas. As demais etapas redutivas no substrato são idênticas
com as da redução de aldeídos (ver p. 603).
Observação: Enquanto com LiAlH4 se consegue reduzir qualquer grupo carbonila e
carboxila, a força redutora do NaBH4 (isto é, mais corretamente, a força nucleofílica do
hidreto) basta para reduzir aldeídos e cetonas, mas não os derivados do ácido carboxílico nem o grupo nitro. Então, NaBH4 não serve para a redução descrita aqui!
Redução de ésteres segundo Bouveault-Blanc
Como já foi apresentado para as cetonas (p. 604), a redução de Bouveault-Blanc também
pode ser aplicada em ésteres. Aplica-se sódio em etanol. Esta reação é muito apropriada,
também para quantidades maiores de substrato.
Mecanismo:
O
R C OC2H5
O-
+ Na
+
- Na
O
- EtO
C2H5O-
+
R C
+ EtOH
R C OC2H5
-
O-
O
+ Na
- Na+
R C OC2H5
H
R C OC2H5
H
redução até o álcool
H
O aldeído, conforme descrito acima (p. 603), é reduzido facilmente ao álcool primário.
Com o reagente de Bouveault-Blanc também podem ser reduzidas nitrilas para aminas
primárias:
R CN
Na
EtOH
R CH2 NH2
Condensação de aciloina
Em analogia à síntese de pinacol (p. 604) esta mecanismo passa pela formação do ânionradical intermediário. De maneira semelhante, se trabalha com sódio em solvente aprótico e
inerte, de preferência em xileno.
O-
-
O
2 R C OC2H5
+ 2 Na
- 2 Na+
O
R C OC2H5
2 R C OC2H5
R C OC2H5
O-
R
- 2 C2H5OR
O
C
+ 2 Na
C
- 2 Na+
O
R
OC
C
R
O-
+ 2 H+
R
O
C
R CH OH
Aciloina
612
A. Isenmann
Princípios da Síntese Orgânica
O nome genérico do produto é aciloína; caso os dois grupos R sejam aromáticos então é
chamado de benzoína. A aciloína é formalmente o produto de oxidação do pinacol (ver p.
603); mesmo assim, é mais fácil sintetizar a aciloína a partir de ésteres do que oxidar o
pinacol, porque a oxidação dificilmente pára nesta etapa oxidativa.
Note que existe um acesso alternativo à aciloína: uma condensação de mecanismo aniônico
(ver p. 473).
Vale salientar a diferença entre esta reação e a redução de Bouveault-Blanc: somente na
ausência de prótons o ânion-radical tem tempo suficiente para procurar um outro ânionradical, para se dimerizar. O solvente usado para efetuar a condensação de aciloina deve ser
bem seco e é aconselhado o trabalho sob atmosfera inerte. No caso de Bouveault-Blanc, por
outro lado, tem-se acesso ilimitado de hidrogênios ácidos, pois se processa em etanol (pKa
= 18).
Redução com H2/CuCr2O4
Este sistema redutor reduz cataliticamente ésteres a alcoóis 374.
Método de Weygand
Aminas secundárias reagem com o cloreto de acila formando uma amida terciária que pode
ser reduzida em seguida por hidretos metálicos. Os rendimentos desta síntese são bons (de
80 a 90%).
Dois exemplos:
O
R C
H3C NH C6H5
- HCl
Cl
O
R C
LiAlH4
CH3
R CH2 OH + HN
N C6H5
C6H5
CH3
O
R C
Cl
NH(CH3)2
- HCl
O
R C
N CH3
LiAlH(OEt)3
CH3
R CH2 OH + HN
CH3
CH3
Redução de nitrilas para aminas
Existem várias opções para reduzir uma nitrila. Uma delas, a redução de Bouveault-Blanc
(Na em EtOH), já foi mencionada acima. As outras que fornecem aminas primárias são:
•
374
com os hidretos LiAlH4 (ver também p. 379), AlH3 ou B2H6
R.B.C. Pillai, A study of preactivation of a copper chromite catalyst, Catalysis Letters 26 (1994) 365.
613
A. Isenmann
•
Princípios da Síntese Orgânica
com H2/Pt em ácido acético glacial ou etanol. Ao trabalhar em ambiente neutro
podem se formar aminas secundáras (isomerizações!).
8.9.2
Redução para aldeídos e cetonas
Na redução de ácidos carboxílicos para alcoóis primários, conforme apresentado na secção
anterior, não há necessidade de controlar o poder redutivo do redutor porque o álcool
representa o composto com número de oxidação mais baixo, na sequência: ácido
carboxílico → aldeído → álcool. Uma redução do álcool, por sua vez, exige de condições
bem mais drásticas, conforme alegado na p. 597.
Quando se pretende reduzir o derivado do ácido carboxílico para o aldeído, deve-se
escolher um agente redutor mais seletivo, ou seja, o mais suave possível, para que o
produto da redução, o aldeído, não seja reduzido ao álcool. Também a gama de substratos é
mais restrita: essencialmente as nitrilas e os cloretos do ácido carboxílico que representam
os derivados mais apropriados neste sentido, quer dizer, podem ser reduzidos seletivamente
para aldeídos.
Redução de Rosenmund
Esta redução funciona com H2 e um catalisador especial - a não dizer especialmente fraco.
A redução pára seletivamente na etapa do aldeído e os rendimentos são surpreendentemente
bons:
O
R C
Cl
H2 / Pd (BaSO4)
∆
O
R C
[ Xileno ]
H
A atenuação da atividade catalítica do Pd, como já foi mostrado na redução parcial de
alquinos (p. 590), pode ser alcançado com metais pesados.
Observação: o catalisador da redução de Rosenmund funciona pelo mesmo princípio que o
catalisador de Lindlar (p. 590).
Redução por hidretos complexados de reatividade moderada
A aplicação de (t-BuO)3LiAlH em cloretos de ácidos carboxílicos fornece o aldeído, porém
exige condições reacionais mais brandas possíveis. Trabalha-se preferencialmente em
“diglime” a –70 °C. Mais novo é um redutor seletivo conhecido como “Red-Al” 375 que já
vem estabilizado em diglime: NaAlH2(OCH2CH2OMe)2. Com este redutor se consegue
seletivamente aldeídos, a partir de ésteres e a partir de nitrilas, alquenos a partir alquinos e
outros. A vantagem do Red-Al, sobre o LiAlH4, é a sua insensibilidade ao oxigênio do ar e
sua boa solubilidade em solventes orgânicos.
375
C.T. Meta, K.Koide, Org.Lett. 6 (2004) 1785.
614
A. Isenmann
Princípios da Síntese Orgânica
Reação com compostos organo-cádmio
Os compostos organo-cádmio podem ser preparados em uma etapa prévia, a partir do
reagente de Grignard e CdCl2, em solução benzênica (princípio de transmetalação e
metatese, ver pp. 699 e 700, respectivamente). Eles têm uma polaridade baixa, ou seja, um
caráter covalente bastante alto. Portanto, reagem apenas com o cloreto do ácido carboxílico
(derivado mais reativo do ácido carboxílico), formando uma cetona (ver também p. 441).
O
O
1/2 CdR´2
R C
R
Cl
C
+ 1/2 CdCl2
R´
Ao contrário do reagente de Grignard, os compostos CdR2 não reagem com a cetona.
Portanto, isto representa um bom método para preparar cetonas. A exotermia desta síntese
pede a sua execução à temperatura baixa (-18 °C ou abaixo).
Neste lugar pode-se também lembrar da reação do ortoéster do ácido fórmico, com reagente
de Grignard, fornecendo um aldeído (via acetal, p. 436).
Redução de nitrilas para aldeídos
Aparentemente a redução para aldeídos é menos difícil a partir das nitrilas do que a partir
dos cloretos do ácido carboxílico. Já a redução com (t-BuO)3LiAlH (p. 614) ou Red-Al (p.
614) não precisa ser executada em temperaturas tão baixas.
Uma outra opção é a redução de Stephen que funciona com quantidades estequiométricas
de SnCl2, em ambiente ácido.
R CN
Cl
+ HCl
R C
NH
SnCl2 + HCl
- SnCl4
R CH NH
+ H2O
- NH3
R CH O
Redução de nitrilas para cetonas
δ−
R MgBr
+
δ+
R´ CN
NMgBr+
R´ C
+ R MgBr
R
Ânion imeno
R2C N2- Mg2+
R´
não ocorre
+
+ H3O
O
R
C
R´
+ NH3
A nitrila somente adiciona um carbânion, providenciado pelo reagente de Grignard. O
ataque de um segundo carbânion é impedido pela carga negativa do complexo
615
A. Isenmann
Princípios da Síntese Orgânica
intermediário (ânion imeno). A nucleofilia do carbânion de R evidentemente não é
suficiente para atacar - ao contrário dos dois hidretos que entram na nitrila, reduzindo-a à
amina, ver pp. 613 e 621. Além de obstáculos cinéticos pode-se argumentar também com a
estabilidade do produto que seria, no caso da reação com dois reagentes de Grignard,
desfavorável devido às cargas negativas acumuladas no –N2-.
A hidrólise do ânion imeno é quantitativa e rápida, fornecendo a cetona e amônia.
A reação funciona igualmente bem com o reagente organo-lítio, em vez de Grignard.
8.10 Redução de compostos nitro
8.10.1 Redução de compostos nitro-alifáticos
A redução de compostos nitro alifáticos para aminas primárias, geralmente não é difícil. Ela
pode ser efetuada com bons rendimentos, tanto com hidrogênio e catalisador de níquel,
quanto com LiAlH4.
8.10.2 Redução de compostos nitro-aromáticos
Esta redução é uma operação muito aplicada em síntese orgânica, porque a nitração de
aromáticos é uma reação tranquila.
No caso geral a redução dos compostos nitro leva a aminas (= anilinas). Sob certos
cuidados, porém, a redução pode ser parada a níveis de oxidação mais altos, antes de chegar
ao nível da anilina.
São apropriados os seguintes agentes redutores:
•
H2/Ni 376
•
SnCl2/HCl
•
Zn/HCl
•
Fe/HCl
O primeiro sistema redutor fornece diretamente a anilina. Para os demais sistemas, todos
eles contendo metais em baixo nível de oxidação e operando em meio ácido, o seguinte
mecanismo é plausível:
376
Ao usar o catalisador mais poderoso, Pd sobre carbono, existe uma variação interessante dessa
hidrogenação: na presença de formaldeído forma-se a partir do Ar-NO2 diretamente a N,N-dimetilanilina.
616
A. Isenmann
Ar
NO2
Princípios da Síntese Orgânica
O
+ e-
Ar
OH
+ H+
Ar
N
Nitro
N
Ar
N O
+ H+
Ar
N
Ar
N OH
- OH-
O
O
O
+ e-
OH
+ e-
1) + e-
Ar
2) + H+
Ar
N O
Nitroso
Ar
NH OH
NH2
Amina
Hidroxilamina
Trata-se de uma transferência de elétrons não emparelhados (SET); a fonte de elétrons é o
respectivo metal não-nobre. Em ambiente alcalino a redução ocorre bem mais lentamente.
Especialmente a etapa da protonação do ânion-radical é lenta por causa da baixa
concentração em H+. Isto abre caminhos para formação de azoxibenzeno, azobenzeno e
hidrazobenzeno:
Ar
N O
+
Ar
N OH
O-
Dimerização
Ar
N N Ar
- OH-
OH
Ar
N N Ar
redução
Ar
N N Ar
redução
Ar
NH NH Ar
-
O
Azoxibenzeno
Hidrazobenzeno
Azobenzeno
A redução pode ser conduzida seletivamente até qualquer um dos produtos (intermediários)
da sequência reacional acima. Para isto, usam-se redutores seletivos:
•
com As2O3 em NaOH diluído
→
Azoxibenzeno 377
•
com Zn em NaOH diluído
→
Azobenzeno
•
alternativa: com SnCl2/NaOH diluído
→
Azobenzeno
•
com N2H4 e catalisador de Ru (C)
→
Hidrazobenzeno
Atenção: o hidrazobenzeno, ao ser exposto ao ambiente ácido, é sujeito ao rearranjo de
benzidina (Ingold, 1933):
377
Acesso ao azoxibenzeno via oxidação: ver p. 669.
617
A. Isenmann
Princípios da Síntese Orgânica
NH NH
NH2 NH2
+ 2 H+
o
Hidrazobenzeno
NH2
NH2
- 2 H+
H
H2N
NH2
Benzidina
H
Um outro exemplo onde se alcança uma redução parcial, aproveita da alta seletividade
redutora do enxofre de baixo nível de oxidação. Por este meio se consegue reduzir
aromáticos dinitro, para nitroanilinas:
O2N
O2N
NO2
1,3-dinitrobenzeno
H2S
NH2
ou S / NH3
3-nitroanilina
O H2S representa um agente redutor fraco e, portanto, bastante seletivo. O enxofre (NOX 2) consegue reduzir o composto dinitro que tem uma densidade eletrônica extremamente
baixa no nitrogênio. Porém, uma vez tendo reduzido o primeiro grupo nitro para amina, o
sistema aromático torna-se mais rico em elétrons (forte efeito +M pelo grupo -NH2) e
exerce um efeito doador de elétrons ao segundo grupo nitro, desta maneira atenua sua falta
de elétrons. Consequentemente, o ataque do H2S ao segundo grupo nitro é impedido mesmo se o agente redutor for colocado em grande excesso.
8.11 Redução de outros compostos contendo nitrogênio
8.11.1 Redução de azidas segundo Staudinger
Azidas, na maioria das vezes acessíveis por substituição nucleofílica SN2 sob condições de
CTF (ver p. 32), podem ser seletivamente reduzidas para aminas, usando fosfinas como
agente redutor.
Exemplo:
618
A. Isenmann
Princípios da Síntese Orgânica
N3
NH2
PPh3
N3
N3
THF, H2O
H2N
NH2
Mecanismo:
O ponto central do mecanismo desta redução é o ataque nucleofílico da fosfina, ao
nitrogênio terminal da formação -N3, levando ao iminofosforano. Segue uma expulsão de
nitrogênio N2, sempre um processo facilitado pela alta estabilidade deste subproduto.
Afinal, o intermediário iminofosforano é hidrolisado fornecendo o fosfinóxido e a amina.
R R
R´
R
R
R´
P
R + N N N
R
R
R
N
P
R´
R
N N
P
R
N
R
- N2
N N
R´
P N
R
Iminofosforano
H 2O
R
R
R´
P O + N 2N
R
378
Uma variação interessante desta reação é a "ligação de Staudinger" . Nesta reação de
acoplamento aproveita-se da presença de um ligante na fosfina, geralmente um metiléster,
que exerce o papel de sequestrador eletrofílico. Forma-se um intermediário que podemos
chamar de aza-ilídeo que sofre um rearranjo de tal forma que o N-, altamente nucleofílico,
repõe o grupo metóxido do éster. O ataque nucleofílico é facilitado neste complexo, pela
proximidade entre N- e o carbono positivado do grupo éster (formação de um ciclo de 5
membros). Em ambiente aquoso ocorre prontamente hidrólise, formando uma nova ligação
de amida e um fosfinóxido, desta vez não em forma de compostos separados, mas na
mesma molécula. Este método brando de formar uma nova ligação de amida é bastante
versátil, aplicado com sucesso na área da química biológica. Sendo assim, foi acoplado um
marcador fluorescente num nucleosídeo in vivo 379.
O
O
OCH3
R
PPh2
R´N3
- N2
O
OCH3
R
P
Ph
Ph
R´
- H3COH
N
R´
NH
H2O
R
P
Ph
O
Ph
"Aza-ilídeo"
378
E. Saxon, C.R. Bertozzi, Cell suface engineering by a modified Staudinger reaction. Science 287 (2000)
2007-10.
379
I. Kosiova, A. Janikova, P. Kois, Synthesis of coumarin or ferrocen labeled nucleosides via Staudinger
ligation.Beilstein Journal of Organic Chemistry 2 (2006) 23.
619
A. Isenmann
Princípios da Síntese Orgânica
8.12 Redução de compostos Organo-Enxofre
Ainda mais diversificados do que os compostos contendo nitrogênio, como visto acima, são
os produtos de redução que contêm enxofre. A primeira etapa de uma síntese de compostos
organo-enxofre é, na maioria dos casos, uma sulfonação, usando ácido sulfúrico ou óleum.
Este processo introduz um grupo funcional onde o enxofre tem alto NOX. Em segunda
etapa então se aplica a redução para os demais compostos organo-enxofre, de NOX +4, +2,
0, -1 e -2.
Os seguintes sistemas redutores podem ser aplicados para abaixar o NOX do enxofre
seletivamente.
Substrato
Produto
Comentário
Ar-SO2Cl
e
R-SO2Cl
Ar-SO2H
e
R-SO2H
Os derivados dos ácidos sulfônicos, R-SO2-OH e Ar-SO2OH, mais comumente usados são o cloreto de tosila e os
cloretos de alquil-sulfonila, respectivamente. Eles podem
ser reduzidos para o ácido sulfínico, Ar-SO2H e R-SO2H, ao
serem tratados com zinco em pó em éter. O produto
também pode ser o mercaptano, Ar-SH e R-SH. Este é o
caso quando se aplica um excesso do agente redutor.
Essa reação é bastante aplicada porque o acesso aos
substratos, via sulfocloração, é barato (ver p. 74).
ou
Ar-SH
e
R-SH
R-SO2-R
Sulfonas podem ser reduzidas da mesma maneira que os
sulfóxidos. Essa redução passa pelo sulfóxido, mas
dificilmente pode ser interrompida nesta etapa. Significa
que o sulfóxido não pode ser obtido com bom rendimento
por este caminho. Porém, uma reação oxidativa, a partir de
tioéteres, pode ser conduzida seletivamente até o estado do
sulfóxido e também à sulfona.
R-SO-R
Sulfóxidos podem ser reduzidos para o tioéter, R-S-R, com
LiAlH4 em etilbutiléter, sob refluxo.
R-S-S-R
C-S
(em geral)
R-SH
A redução de dissulfetos para R-SH é muito fácil. Bons
rendimentos se obtêm com SnCl2/HCl. Aliás, pontes de
dissulfeto ou polissulfeto R-(S)x-R, com x > 2, podem ser
facilmente introduzidos, ao tratar hidrocarbonetos
insaturados (alquenos alifáticos) com enxofre elementar. O
processo desta “vulcanização” é aplicado em larga escala,
na produção de artefatos de borracha, a partir do látex
natural ou outros pré-polímeros lineares com insaturações.
A hidrogenólise, com bastante níquel de Raney, é
geralmente viável. Isto já foi mostrado na reação da retirada
620
A. Isenmann
Princípios da Síntese Orgânica
de enxofre de tiocetais (ver p. 605). Porém, lembre-se que o
enxofre é veneno para o catalisador metálico que, portanto,
deve ser aplicado em quantidades maiores.
8.13 Redução com Hidretos complexos – Vista Geral
Uma descrição detalhada dos hidretos metálicos como reagentes de redução, se encontra na
monografia de Seyden-Penne 380. Um excelente resumo, onde para cada sistema de hidreto
metálico é indicada sua eficiência para uma série de reações, listadas em ordem da
facilidade da redução, se encontra na monografia de March 381.
380
J.Seyden-Penne, Reductions by the Alumino- and Borohydrides in Organic Synthesis, Wiley-VCH New
York 1997
381
J.March, M.B.Smith, Advanced Organic Chemistry, Ed. 5, Wiley New York 2001, Cap. 19: Oxidations
and Reductions.
621
B2H6 em THF
Diisoamil-borano em THF
(t-BuO)3LiAlH em THF
(H3C-O)3LiAlH em THF
LiAlH4 em Dietiléter
LiAlH4 em THF, refluxo
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
-
-
+ 382
+
+
+
+
+
+
+
+
+
+
-
+
+
-
+
+
+
-
+
+ 383
-
-
+
+
+
-
-
+
-
-
+
+
+
-
-
-
--
-
-
+
+
-
-
+
+
+
+
+
+
-
-
-
+
-
+
-
-
-
+
+
-
-
+
382
383
-
Sistema redutor
NaBH4 + LiCl, em diglime
Princípios da Síntese Orgânica
NaBH4 em EtOH ou glime
A. Isenmann
Substrato
Aldeído
Cetona
Cloreto de ácido
carboxílico
Lactona
Epóxido
Éster
Ácido carboxílico
Sal do ácido
carboxílico
Nitrila
Composto nitro
Olefinas
fornece aldeídos
muito devagar
622
A. Isenmann
Princípios da Síntese Orgânica
9
Oxidação
9.1
Significado de Oxidação em química orgânica.
Aplicando os princípios, introduzidos com o número de oxidação "NOX" (ver p. 582), as
oxidações mais comumente encontradas são:
Remoção de hidrogênio = desidrogenação
Exemplos:
a)
Ciclohexano
b) H3C
- [H]
CH2 OH
Cicloexeno
- [H]
H 3C
+ H2
CHO + H2
Introdução de substituintes que são mais eletronegativos do que o hidrogênio ou carbono:
As eletronegatividades (EN), dos elementos mais espalhados em química orgânica, são:
EN (C) = 2,5 e
EN (H) = 2,1;
conforme a escala de Pauling (ver tabela no anexo 2 deste livro).
Sendo assim, a introdução de oxigênio, EN = 3,5, representa uma oxidação.
H3C
CH O
+ [O]
H3C
COOH
Neste exemplo foi substituído –H por –OH.
A esta categoria pertencem também as substituições radicalares, R-H → R-X , que já foram
discutidas no item 1.4.
9.2
Oxidação de alcanos
9.2.1
Oxidação com ar (= auto-oxidação)
1) Motores de combustão
2) Parafinas
O2 ; 100 - 130°C
0,3% KMnO4
R COOH
+ HOOC
R´
cisão da cadeia
carbônica da parafina
623
A. Isenmann
Princípios da Síntese Orgânica
Qual a diferença entre a primeira e a segunda auto-oxidação?
A primeira é uma “combustão completa”, quer dizer, em qualquer momento da reação se
tem um excesso de oxidante que é, no caso, o oxigênio do ar. Os produtos são CO2 e H2O
(somente a temperaturas extremamente altas e prolongadas, o monóxido de carbono, CO,
composto menos exotérmico, é produzido a custo do gás carbônico CO2). Como o CO2 não
tem valor preparativo, então esse tipo de oxidação somente tem aplicação na geração de
calor e volume, como é o caso no motor do carro.
Muito pelo contrário desta, temos no segundo exemplo uma quantidade limitada e
controlada de oxigênio, portanto não é uma combustão completa. Isto quer dizer, o carbono
não é oxidado ao NOX +4, como está o caso no CO2, mas apenas a +3, NOX típico em
ácidos carboxílicos. Isto é então uma reação de alto valor preparativo.
Um outro exemplo para uma auto-oxidação que não seja uma combustão completa, é a
quebra oxidativa e controlada de butano. No catalisador de contato ácido é produzido
exclusivamente ácido acético:
OH
O
+ O2
H
°
H3C
CHO
+
O
O
+ O2
- H2O
Eliminação β
O
OH
H3C COOH
+ 1/2 O2 (rápido)
H3C
COOH
Atenção: caso estiver presente uma dupla-ligação C=C, a oxidação ocorre de preferência e
com muito mais facilidade, em posição alílica (ver pp. 95 e 632); caso tiver uma unidade CO-C (= éter), então a oxidação ocorrerá com seletividade em posição α ao oxigênio (ver pp.
96 e 664).
9.2.2
Oxidação com CrO3 ou KMnO4/OH-
Estes reagentes têm grande importância na síntese. São os meios oxidantes mais usados em
escala de laboratório porque são baratos e de fácil acesso. Em preparações maiores, porém,
deve-se considerar o impacto ambiental que os resíduos de metais pesados podem gerar.
Os óxidos de metais de transição com alto número de oxidação podem ser favoravelmente
aplicados em alcanos onde seletivamente oxidam as ligações C-H de carbonos terciários.
Como já foi mencionado antes (p. 252) a força oxidante dos óxidos dos metais de transição
depende fortemente do pH do meio. Como regra geral o agente oxidante é mais forte, isto é,
624
A. Isenmann
Princípios da Síntese Orgânica
o potencial de oxidação ε é mais positivo, em ambiente ácido do que em ambiente básico.
Isto se deve à complexação (= estabilização) do metal de alto NOX, que é melhor com OHdo que com H2O. Além da maior estabilidade do complexo com hidróxido, tem-se a
compensação da (alta) carga positiva do metal. Assim, o reagente torna-se menos solúvel e
pode precipitar-se.
Na equação abaixo este efeito se efetua como uma diminuição de [Ox]. Quantitativamente
pode-se verificar o efeito das concentrações sobre o potencial eletroquímico real, na
equação de Nernst simplificada, onde a concentração do íon metálico de alto estado de
oxidação, [Ox], contribui positivamente ao valor do potencial eletroquímico, ε:
ε = ε0 +
[Ox] ,
0,059
log
[Re d ]
n
Equação de Nernst simplificada.
ε: potencial redox real
ε0 : potencial redox padrão (atividades de 1 mol l-1, à temperatura e pressão ambiente)
n: número de elétrons transferidos na semi-equação do metal
[Ox]: concentração 384 da forma oxidada, por exemplo Cr6+
[Red]: concentração da forma reduzida, por exemplo Cr3+
O fator 0,059 tem validade para sistemas à temperatura ambiente.
Para o permanganato, o potencial ε a pH 0, mantendo as demais grandezas dentro do
padrão, toma o valor +1,51 V, enquanto a pH14 se tem ε0 de apenas +0,59 V. Os demais
valores de ε, para quaisquer outras concentrações iônicas, se obtêm através da equação
ε = 1,51 +
[ MnO4− ][ H + ]8
0,059
.
⋅ lg
5
[ Mn 2 + ]
A equação de Nernst deve-se aplicar, tanto para o agente oxidante quanto para o substrato a
ser oxidado. Somente depois de ter elaborado os dois valores de ε (potencial real) para
ambas as semi-equações, oxidação e redução, a força termodinâmica da reação redox pode
ser indicada, a base da diferença dos potenciais ∆ε = εox - εred:
∆G = −n ⋅ F ⋅ ln ∆ε .
∆G: Entalpia livre de Gibbs = força propulsora da reação
n: número de elétrons abandonados (recebidos) no processo da oxidação (redução)
F: constante de Faraday = carga de 1 mol de elétrons = 96.485 C·mol-1
∆ε = εOx - εRed = “força eletromotriz”
Assim se explica a relativamente baixa reatividade e, portanto, alta seletividade dos
sistemas CrO3 ou KMnO4, em ambiente básico.
384
Corretamente deve-se usar a atividade; ela pode ser considerada igual a concentração em casos de alta
diluição.
625
A. Isenmann
Princípios da Síntese Orgânica
Os eventuais efeitos cinéticos que podem inibir uma reação redox (sobre-tensão, períodos
de indução,...), não serão discutidos aqui 385. Somente vale ressaltar que mesmo quando
deve ocorrer a reação redox - isto é o caso com valores ∆G negativos – a reação pode
demorar muito ou até mesmo não acontecer. Neste caso, a tensão propulsora não pode se
aliviar, o sistema fica fora do seu equilíbrio.
Igualmente problemático é o trabalho em sistemas bifásicos: os oxidantes são tipicamente
sais dissolvidos na fase aquosa (os sais do nosso exemplo são solubilizados em forma de
CrO42- e MnO4- caso o ambiente for alcalino, e H2Cr2O7 e HMnO4 em ambiente ácido). O
substrato orgânico, por outro lado, é uma molécula hidrofóbica que tem pequena área de
contato com o agente oxidante. A consequência é uma reação demorada.
Este último problema pode ser contornado, por uma das seguintes providências:
1. Trabalhar à temperatura alta: daí, a constante dielétrica da água fica mais baixa (=
momento dipolar mais baixo) e se mistura com a fase orgânica com maior
facilidade.
2. A adição de um catalisador de transferência das fases (do tipo “sabão” ou sal
quaternário de amônio, ver p. 32)
3. Uma agitação intensa da mistura reacional sempre é de vantagem.
R 3C
H
[O]
R C
OH
3
é via SET. As etapas
O mecanismo de reações do tipo
intermediárias podem até ser comprovadas por reagentes que apanham os radicais (=
sequestradores).
Para o caso do permanganato podemos formular as seguintes etapas mecanísticas:
R3C
H
+
+7
MnO4-
R3C
+5
+6
R3C O MnO2-
HO MnO3-
+
OH
+ H3O+
R3C OH
+5
+ H3MnO4
O óxido de manganês(V) é instável e sofre disproporcionamento (quer dizer, reage com
igual e fica em estados de oxidação, um maior outro menor que V; no sentido do falado no
item 5 na p. 583). Em ambiente ácido o mais estável é Mn(II) (incolor), enquanto em
ambiente neutro e básico o dióxido de manganês (marrom) é o mais produzido.
Para o cromato podemos formular, em toda analogia:
R3C
H
+ H2O
+
+6
CrO42-
R3C OH
R3C
+
+5
CrO4H2-
+ 2 H+
+4
R3C O Cr(OH)3
+4
+ H4CrO4
385
leia sobre sistemas fora do equilíbrio em livros da físico-química, por exemplo P. Atkins, Físico-Química
Vol. 3 (cinética de reações eletroquímicas); LTC Rio de Janeiro 2003.
626
A. Isenmann
Princípios da Síntese Orgânica
O cromo prefere o estado de Cr(III), verde, que pode ser alcançado pelo mesmo princípio
do que o Mn(II).
9.2.3
Desidrogenação e ciclodesidrogenação
∆
Cat.
H3C
CH CH3
H3C
C
H3C
CH2
+
H2
H3C
Todas essas reações heterolíticas, que ocorrem em fase gasosa a altas temperaturas, na
superfície de catalisadores sólidos, são reversíveis. As condições típicas são 500 °C e baixa
pressão (por que?); os catalisadores podem ser ácidos (Cr2O3, Al2O3) ou metais (Pd ou Pt).
A temperaturas altas prevalece a desidrogenação, a temperaturas mais baixas e excesso de
H2 a hidrogenação (mecanismo discutido no item 8.3.1).
A desidrogenação puramente térmica, sem catalisador, não tem valor preparativo porque a
fragmentação dos hidrocarbonetos (inglês: Cracking, ver p. 76) predomina, por exemplo:
H3C
∆
CH2 CH3
Pirólise
H2C
CH2
+
CH4
Exemplos para desidrogenações de importância industrial:
1) Obtenção de aromáticos, a partir da fração de parafinas cíclicas do petróleo:
Ciclohexano
∆
[ Pt ]
Benzeno
2) Ciclodesidrogenação = ciclização acompanhada de desidrogenação:
Cr2O3 , Pt
H+ / H2O
900 °C
450 °C
+ CH3OH
- 3 H2
A ciclodesidrogenação direta, levando à molécula-alvo em somente uma etapa a partir do
hexano, não funciona. Por outro lado, o tolueno que se consegue a partir do heptano, pode
ser submetido a uma segunda etapa: o tratamento dos vapores do tolueno com HCl e água,
daí a transformação em benzeno e metanol.
Observação: a segunda etapa desta síntese é o reverso da alquilação de Friedel-Crafts (ver
p. 296)!
627
A. Isenmann
Princípios da Síntese Orgânica
9.3
Oxidação de alquenos
9.3.1
Oxidação com oxigênio molecular
Síntese de óxido de etileno (= oxirano)
H2C
CH2
O2
[ Ag ]
H2C
O
CH2
Esta reação só tem sucesso com etileno e estireno porque todos os demais alquenos serão
preferencialmente oxidados em posição alílica. Além disso, isomerizações podem ocorrer
no esqueleto carbônico:
R CH2 CH CH2
H
+ O2
R C CH CH2
O OH
- H2O
Eliminação β
R C CH CH2
O
Formalmente foi então oxidada em posição alílica. Como será ilustrado mais
detalhadamente na p. 632, isto não é um caso raro, mas sim, a posição mais regularmente
atacada. No entanto, uma oxidação diretamente na C=C, sob conservação da dupla-ligação,
é um evento raro (p. 635, item a).
Um exemplo de relevância técnica é a dupla oxidação alílica de 2-buteno fornecendo o
anidrido maléico, um importante monômero para copolímeros (a oxidação funciona
igualmente com 1-buteno, devido à isomerização para 2-buteno sob as condições
aplicadas):
O
+ O2
450 °C
[V2O5]
O
+ 3 H2O
O
Anídrido maléico
9.3.2
Formação de cis-dióis
A oxidação de alquenos com KMnO4 e/ou OsO4, à temperatura ambiente, fornece dióis
(também chamados glicóis) de conformação cis (compare p. 252).
KMnO4
H2O / H+
OH
OH
628
A. Isenmann
9.3.3
Princípios da Síntese Orgânica
Formação de dióis trans
Oxidação do alqueno com perácido
Sob a influência de perácidos pode formar-se o epóxido que posteriormente pode ser aberto
por hidrólise ácida ou básica (ver p. 224). Geralmente a hidrólise ácida ocorre com maior
facilidade:
O
HO
R C
H2O
OOH
+
O
[H ]
OH
HO
HO
SN2
H2O
HO
HO
HO
Dentro do alqueno do nosso exemplo se escondem dois centros proquirais. Observa-se, em
casos de esqueletos assimétricos, a formação de dois diastereoisômeros, onde os dois
carbonos do anel oxirano têm configurações invertidas.
Reação de Prevost, com I2 e C6H5COOAg
A oxidação de alquenos pode ser efetuada, segundo Prevost, com iodo e benzoato ou
acetato de prata, sob condições especialmente brandas. Os rendimentos são excelentes,
porém o alto custo do reagente somente permite a aplicação deste método em pequena
escala.
629
A. Isenmann
C
+ I2 + Ag+
C
Princípios da Síntese Orgânica
C
- AgI
C
O
I
C6H5
C
I
C O C
O
+ Ag+
- AgI
C6H 5
C
O
C6H5 COOO
O
+ C6H5 COO-
C6H5
C O C
C6H5 + 2 H2O
C O C
2 C6H5 COOH
+
C OH
HO
C
Diol
trans
O papel da prata é a fixação de iodeto e então a disponibilização do eletrófilo, I+. Isso
implica que a oxidação de Prevost não é um processo radicalar, mas ocorre por um
mecanismo iônico iniciado pela quebra heterolítica do iodo.
O iodo é o único halogênio com que a reação de Prevost sucede. Tentativas com bromo não
fornecem o diol desejado. Em vez da oxidação do substrato alqueno ocorre uma reação dos
dois reagentes AgOAc e Br2 entre si: a quebra do Br2 é homolítica, fornecendo AgBr
(precipitado) e o brometo de oxacila, AcOBr. O último sofre degradação logo depois da sua
geração, fornecendo o brometo de metila e CO2 (descarboxilação de Hunsdiecker, ver p.
104).
9.3.4
Ozonólise
A reação com mecanismo foi apresentada na página 248. O resultado é a quebra da
molécula no local da dupla-ligação C=C. Quais serão os grupos funcionais dos produtos
fragmentados, isso depende do pós-tratamento do ozonóide. Na maioria das aplicações se
produzem ácidos carboxílicos.
O valor preparativo deste método é limitado porque o ozônio ataca muitos outros grupos
funcionais, também.
9.3.5
Oxidação de Lemieux com NaIO4
O reagente de Lemieux é o periodato de sódio, NaIO4, com pequena quantidade de
permanganato ou tetróxido de ósmio:
H3C
C
H3C
CH CH3
NaIO4
KMnO4 (Cat.)
e/ou
H3C
C O
+
CH3CHO
H3C
OsO4 (Cat.)
CH3 COOH
630
C
C
A. Isenmann
Princípios da Síntese Orgânica
Mesmo quando o KMnO4 (OsO4) for acrescentado em apenas quantidades catalíticas, é ele
que entra em reação com a dupla ligação do substrato. O próprio periodato é incapaz de
reagir com o substrato olefínico. Por outro lado, o cis-diol que se formou na primeira etapa
pode ser facilmente quebrado pelo periodato (reação de Malaprade, ver p. 653), fornecendo
em primeira instância aldeídos e cetonas.
Sob a influência do permanganato o aldeído é oxidado mais uma vez, fornecendo o ácido
carboxílico. No final da reação o manganês se encontra em estado oxidativo baixo. Mas o
periodato é um oxidante poderoso e consegue reoxidá-lo para MnO4- (OsO4) para que
possam entrar no próximo ciclo reacional 386. O único reagente consumido é então o
periodato - o que confirma o papel do catalisador do permanganato (OsO4).
A oxidação de Lemieux age muito seletivamente, somente para insaturações olefínicas
isoladas. Embora aplicando oxidantes poderosos, esta síntese pode ser considerada branda,
já que anda à temperatura ambiente.
9.3.6
Oxidação com ácido crômico
Existe uma concorrência entre oxidação em posição alílica e da própria dupla ligação C=C.
Ao conduzir a reação em ambiente aquoso a reação principal é a quebra da dupla ligação
(caminho 1). Ao trabalhar em ácido acético glacial a oxidação alílica prevalesce (caminho
2).
COOH
1
COOH
em H2O
CrO3
O
2
em CH3COOH
A variedade de produtos que se consegue com o oxidante ácido crômico é muito ampla
(compare nas pp. 642, 650 e 653).
9.3.7
Desidrogenação com cloranil
Esta reação somente funciona quando o substrato hidrocarboneto já possui pelo menos uma
dupla ligação. A saída do hidreto, H-, é uma etapa bastante endotérmica. Neste caso, porém,
é facilitada pela estabilização mesomérica do carbocátion remanescente (= estabilização
alílica).
386
Mais corretamente trata-se da transição do ácido orto-periôdico, H5IO6, para o iodato, IO3-. O potencial
padrão desta semi-reação em ambiente ácido é ε0 = +1,7 V, então acima dos óxidos de Mn e Os; portanto
provoca a oxidação dos mesmos.
631
A. Isenmann
Princípios da Síntese Orgânica
Cl
H
H
Cl
Cl
O
H
O
Cl
Cl
Transferência
de H(lenta)
Cl
O-
HO
+
H
Cl
H
Cl
Cloranil
H
Cl
H
Cl
Cl
Cl
H
+
-
O
OH
H
Cl
H
Cl
Transferência
de H+
(rápida)
+ HO
OH
Cl
Cl
mais um cloranil
Já a quinona dicloro diciano ("DDQ") mostra uma reativade diferenciada (ver nota de
rodapé 397, na p. 668.
9.3.8
Oxidação em posição alílica
As oxidações em posição alílica ocorrem com bastante facilidade (compare p. 72 e 79).
Portanto é aconselhado usar agentes oxidantes suaves e seletivos que não atacam outros
grupos funcionais no substrato.
Oxidação alílica com Hg(OAc)2
A precondição do funcionamento desta oxidação é a ausência de água.
632
A. Isenmann
R
Princípios da Síntese Orgânica
CH
CH
CH CH
- AcOH
H
AcO
R
CH2
Hg
CH2
Hg
AcO
OAc
rápido
R
CH
CH
- Hg0
Hg
OAc
R
CH
CH
CH CH CH2
AcO
rápido
CH2
H
AcO Hg
R
CH2
R
CH
- AcOH
OAc
CH
AcO
CH2
Hg
Podemos observar neste mecanismo que sempre se forma o acetato alílico mais substituído
porque o complexo organo-mercúrico intermediário sofre isomerização alílica. Este
deslocamento da dupla ligação ocorre mais rapidamente do que a perda do metal. O
abandono de Hg0 finalmente representa a etapa de oxidação do substrato.
Observação: na presença de água (álcool) o centro reativo não é a posição alílica, mas a
própria dupla ligação. Neste caso se perde a dupla-ligação por adição de água (álcool) no
substrato (ver p. 160, “Hidroximercuração-redução”).
Dióxido de selênio em anidrido acético
R CH2 CH CH2
SeO2
Ac2O
R CH CH CH2
OAc
O mecanismo desta oxidação ainda está incerto. Por enquanto têm-se fortes indicativos para
estados intermediares radicalares.
Informe-se sobre a toxicidade deste reagente.
Oxidação de Kharash-Sosnovsky
Com perésteres (muito usado é o perbenzoato de t-butila) e Cu(I) ou Fe(II), se consegue
oxidar compostos insaturados seletivamente, nas posições alílica e benzílica. Além destas,
podem ser oxidadas as ligações C-H em posição α de éteres e tioéteres.
Essa reação ocorre via radicais e mostra uma cinética típica para reações em cadeia (ver p.
62). A própria iniciação da cadeia radicalar acontece por uma reação redox, usando Cu(I)
como catalisador.
633
A. Isenmann
Princípios da Síntese Orgânica
Cadeia cinética:
O
1)
+1
C6H5 C O O C(CH3)3
Éster do perácido benzóico
+
CuCl
+2
(CH3)3C-O
+
COO- CuCl+
C6H5
H
2)
+
(CH3)3C O
H
(CH3)3C OH
+
H
H
3)
+
H
C6H5 COOCuCl
CuCl
+
H
+
O
O C
ou
3a)
H
+
Cu(II)
H
H
+
C6H5 COO
-
Cu(I)
O
O C
Éster do ácido benzóico
A regio-seletividade para a posição alílica é uma consequência da elevada estabilidade do
radical t-butóxido que é sustentador da cadeia cinética da reação de Kharash-Sosnovsky
(compare p. 70). Existem incertezas se as etapas 3) ou 3a) – ou até as duas – sejam
envolvidas. Para o bom funcionamento da cadeia cinética é importante que o catalisador
Cu(I) é reformado para que possa iniciar um novo ciclo, por decomposição do reagente de
peréster.
O produto da reação de Kharash-Sosnovsky pode ser saponificado posteriormente. O
produto final é um álcool alílico:
H
O
O C
H2O / OH-
H
+
COO-
OH
Álcool alílico
A reação ocorre rápida, completa e seletivamente em substratos com duplas ligações
isoladas.
Nesta síntese, como em todas as reações em posição alílica, temos que lidar com dois
possíveis produtos, devido às diferentes posições alílicas no substrato e também por causa
da isomerização no intermediário radical alílico,conforme mostrado abaixo.
634
A. Isenmann
Princípios da Síntese Orgânica
Na maioria dos substratos se encontra mais de um carbono em posição alílica, então há
mais de um hidrogênio que pode ser substituído. No exemplo dado abaixo reação pode
ocorrer nos carbonos 3 e 6 do (4-metil)cicloexeno. A partir de um substrato de baixa
simetria, como este, uma mistura de produtos é inevitável. Além disso, não pode-se
esquecer que a mesomeria do radical alílico permite também uma substituição em posição 1
ou 2. Afinal, o radical sustentador não tem nenhuma enanciosseletividade. Na oxidação do
(4-metil)cicloexeno podemos então esperar 8 diferentes produtos isoméricos!
1
Y
2
6
Y
3
1
Y
Y
5
2
6
[Ox]
3
5
1
6
4-metilciclohexeno
Y
2
3
Y
5
Y
Y
Ressonância do radical alílico
Oxipaladação com Pd(OAc)2
a) Substratos com duplas ligações finais podem reagir com acetato de paládio formando
acetatos enólicos(= acetatos de vinila; a não ser confundido com acetato de alila que se
formou na mercuração, na p. 632).
1
H2C
2
3
+ Pd(OAc) 2
CH CH2 R
1
AcOPd
H
2
CH2 C
3
CH2 R
OAc
1
2
3
H2C
C
CH2 R
+
AcOH
+ Pd
OAc
Vinilacetato
Esta oxidação é um dos raros exemplos onde se introduz um elemento eletronegativo
diretamente num carbono envolvido na dupla-ligação C=C.
635
A. Isenmann
Princípios da Síntese Orgânica
Na maioria das aplicações com acetato de paládio essa oxidação representa uma reação
paralela indesejada, enquanto se visa um acoplamento sendo reação principal (ver p. 306).
b) Duplas ligações do meio da cadeia carbônica fornecem exclusivamente acetatos de alila.
Este produto corresponde plenamente ao da mercuração:
H3C
CH CH CH3
Pd(OAc) 2
H3C
CH CH CH3
H3C
OAc PdOAc
CH CH CH2 + AcOH + Pd
OAc
Síntese de acetaldeído segundo Wacker
Semelhante à oxipaladação, sub-item a, reage etileno com cloreto de paládio(II). Como o
substrato é uma molécula bastante simples então somente há um produto possível.
O processo é igualmente simples: uma mistura de etileno e ar é borbolhado em uma solução
aquosa (ácida) de PdCl2. O reagente Pd(II) é reduzido para Pd(0) logo após de ser abstraido
do complexo com etileno. Em seguida o Pd(0) é reoxidado pelo oxigênio. A reoxidação
ainda é facilitada, pela presença do co-catalisador CuCl2.
A síntese de Wacker tem grande importância industrial, pois o acetaldeído é material de
partida de inúmeras reações (inclusive polimerizações).
H
H2C
CH2 +
PdCl2
H2C
CH2
+ H2O
- HCl
PdCl2
Complexo π
- HCl
H2C
- Pd
2 CuCl 2
CH OH
Enol
tautomeria
CH2 CH OH
Pd
Cl
H3C
CH O
Acetaldeído
PdCl2
+ 2 CuCl
+ 1/2 O2
+ HCl
- H2O
Ao se submeter um 1-alqueno a este tipo de oxidação, o resultado é uma metilcetona. E
quando se usa em vez da água, uma solução alcoólica ou ácido acético, os produtos serão
um éter ou um éster, respectivamente.
9.4
Oxidação de alquinos
O acoplamento oxidativo segundo Glaser já foi discutido na p. 191.
636
A. Isenmann
Princípios da Síntese Orgânica
Oxidação com permanganato
A oxidação branda ocorre quando adicionar o alquino a uma solução de KMnO4 em meio
neutro a levemente alcalino. Formalmente, os oxigênios são transferidos ao alquino, sem
ele perder seu caráter insaturado. Afinal, ocorre uma oxidação e, via mesomeria, formam-se
duas moléculas de glioxal.
HC
CH
H
HC
CH
Mn
O
O
KMnO4
H2O/OH-
H
C
O
C
+
MnO2
O
Glioxal
Note que, quando usarmos o permanganato em solução ácida (potencial oxidativo mais
forte), em vez de formar o aldeído forma-se o produto de oxidação total, ou seja, o gás
carbônico. Já a partir de alquinos secundários obteremos os ácidos carboxílicos como
produtos finais.
Oxidação total:
HC
KMnO4
em H2O/H+
CH
- Mn
2+
H
H
[O]
C
2
HO
OH
C
2
O
Ác. carbônico
O
Formaldeído
Oxidação de alquinos substituídos:
H3C
9.5
C
C
CH3
KMnO4
em H2O/H+
- Mn2+
2
H3C
H
C
O
Acetaldeído
[O]
H3C
2
OH
C
O
Ác. acético
Oxidação de cadeias laterais em compostos aromáticos
As oxidações nas cadeias laterais de aromáticos geralmente ocorrem em posição benzílica,
Ar-CH2-R, onde R pode ser hidrogênio, alquila ou arila. As introduções de halogênios e do
grupo hidroxila em posição benzílica já foram amplamente discutidas nas pp. 70 e 79,
respectivamente.
Os demais métodos comumente usados para oxidar cadeias laterais em aromáticos
fornecem diretamente aldeídos, cetonas ou ácidos carboxílicos. A reatividade elevada da
posição benzílica se deve à vantagem cinética. Lembre-se da estabilidade dos
intermediários, seja radical ou carbocátion: qualquer destas formas reativas recebe
estabilização, por entrar em conjugação com os elétrons π do anel (ver as energias de
dissociação homolítica, na p. 63, e estabilidade de radicais, na p. 65; também a tabela na p.
52).
637
A. Isenmann
Princípios da Síntese Orgânica
Também do ponto de vista termodinâmico uma reação em posição benzílica é favorável: o
grupo carbonila, introduzido pela oxidação, fica em conjugação com os elétrons π do anel o que abaixa a energia interna deste produto.
9.5.1
Síntese de aldeídos e cetonas
Oxidação catalítica com O2
A seguinte oxidação é bastante acelerada por catalisadores de cobre:
H
O
OOH
Elim. β
O2
- H2O
Cu-Cat.
Essa oxidação é uma reação radicalar típica e segue uma cinética em cadeia (Autooxidação). O oxigênio é um radical bastante estável, então pouco reativo. Por isso, tem alta
seletividade para a posição benzílica (e alílica). A segunda etapa, a transformação do
hidroperóxido em cetona, é uma eliminação β de água.
Halogenação radicalar e hidrólise de dihaletos geminais
Em complementação ao já dito no cap. 1.4 seja referida a dupla halogenação em posição
benzílica, fornecendo um dihaleto geminal.
A primeira etapa deve ser realizada a temperaturas elevadas e com iniciadores de radicais
(peróxidos ou luz UV). A presença de catalisadores ácidos, por outro lado, deve ser evitada,
para não promover uma substituição no anel (ver p. 295).
Ar
CH3
+ 2 Cl2
- 2 HCl
radical
Ar
CHCl2
+ H2O
- 2 HCl
Ar
CHO
SN
Após a dupla substituição radicalar segue uma substituição nucleofílica onde os
substituintes haletos estão trocados por hidroxilas. Esta SN se efetua favoravelmente em
meio fortemente básico. Finalmente forma-se o aldeído (ou a cetona) pela desidratação do
diol geminal - um equilíbrio deslocado largamente para o lado do aldeído (ver p. 402).
Atenção: um problema inerente desta síntese é acertar o grau exato da cloração em posição
benzílica: a monocloração levará ao álcool benzílico, enquanto um excesso de cloro levará
à formação do tricloreto de benzila, por sua vez derivado do ácido benzóico (p. 641).
Oxidação com ácido crômico em anidrido acético
O reagente CrO3 é suficientemente forte para oxidar a posição benzílica:
638
A. Isenmann
Ar
CH3
CrO3
Princípios da Síntese Orgânica
[ Ar
CHO ]
OAc
Ac2O
Ar
CH
OAc
1. purificação
2. H2O
Ar
CHO
Ar
COOH
sem Ac2O
Ar
COOH
O aldeído, uma vez formado, deve ser removido imediatamente da mistura reacional para
não perdê-lo por mais uma oxidação para o ácido benzóico. Isto não pode ser realizado por
destilação porque o ponto de ebulição do aldeído é próximo ao material de partida. Mas
através da reação com anidrido acético se consegue protegé-lo efetivamente. Forma-se o
diacetato do hidrato do aldeído, um derivado bastante estável à oxidação. Após terminar a
oxidação do substrato aromático, os restos do ácido crômico podem ser separados do
diacetato. Depois disso o produto purificado pode ser hidrolisado tranquilamente, sem
perigo de perder o aldeído.
Reação com cloreto de cromila segundo Etard
O agente oxidante cloreto de cromila, CrO2Cl2, é um líquido vermelho escuro (Teb =
117°C) que se produz in situ, ao tratar uma mistura de dicromato de potássio e cloreto de
potássio com ácido sulfúrico concentrado. A temperaturas elevadas o CrO2Cl2 (muito
higroscópico) pode ser destilado e diretamente usado:
Cl
6+
Ar
CH3
+ 2 CrO2Cl2
[ CCl4 ]
Ar
CH2 O Cr
Cl
Cl O
O Cr
Cl OH
H3O+
- Cr3+
Ar
CHO
"Complexo de Etard"
(vermelho)
Oxidação com dióxido de selênio
O dióxido de selênio é valorizado como oxidante bastante seletivo (ver também p. 633 e
651). Porém, seu uso é restrito a pequenas quantidades, devido à toxicidade que é até mais
alta que a dos compostos do seu vizinho à esquerda, o arsênio. Isto vale especialmente para
sua forma dissolvida, o ácido selênico H2SeO4, que pode gerar nesta forma problemas
ambientais. Além do mais, o reagente não é barato.
CH3
+4
SeO2
CHO
- Se0
Esta reação percorre estados radicalares.
639
A. Isenmann
9.5.2
Princípios da Síntese Orgânica
Preparo do ácido benzóico e derivados, via oxidação.
1. Métodos padrões
Os métodos preparativos certamente mais importantes são as oxidações com
•
CrO3/H+
•
KMnO4/OH- 387
•
HNO3 concentrado
O último reagente pode levar a produtos paralelos. Como apresentado na p. 288 o ácido
nítrico concentrado também é apropriado para nitrar o anel aromático. Por isso haverá
competição entre a oxidação da cadeia lateral e a nitração do anel – especialmente ao
trabalhar a temperaturas elevadas.
Exemplos reacionais:
CH3
HNO3 conc.
CH3
CH3
o-Xilol
COOH
COOH
KMnO4 / OHou O2 / cat. de Cu
COOH
Ácido ftálico
Não apenas grupos metilas, mas também cadeias laterais mais compridas podem ser
oxidadas por estes reagentes. Nestes casos o oxidante poderoso quebra a cadeia carbônica
lateral, ao decorrer da oxidação do carbono benzílico.
Exemplo:
N
N
Nicotina
CH3
COOH
HNO3 conc.
N
Ácido nicótico
387
em casos onde não suceder oxidação deve-se somente abaixar o pH para aumentar a força oxidativa do
permanganato, conforme explicado nos capítulos 3.5.5 e 9.2.2.
640
A. Isenmann
Princípios da Síntese Orgânica
2. Oxidação do tolueno pelo oxigênio do ar (auto-oxidação)
Os óxidos de metais descritos acima não devem ser usados ao oxidar grandes quantidades.
Neste caso o método mais limpo é a oxidação catalítica com ar:
CH3
CHO
CH2 OOH
O2
COOH
O2
- H2O
Traços de íons de metais de transição catalisam esta reação. A reação não pára na etapa do
aldeído sendo o composto mais sensível à oxidação de todos os compostos envolvidos nesta
reação (ver p. 645).
3. Saponificação de benzotricloreto
Em analogia à reação descrita na p. 638, o substrato pode ser seletivamente clorado em
posição benzílica – esta vez com excesso de cloro. O mecanismo radicalar fornece o
tricloreto que pode ser hidrolisado numa etapa subsequente:
CH3
CCl3
+ 3 Cl2
H2O
COOH
/ OH-
radicalar
Benzotricloreto
9.6
Oxidação do anel aromático
9.6.1
Oxidação por ozônio
Com ozônio se consegue oxidar qualquer dupla ligação carbono-carbono. Como a
estabilidade aromática já deixa supor, as insaturações aromáticas são oxidadas mais
lentamente do que alquenos não-aromáticos. Com o propósito de oxidar seletivamente
insaturações não-aromáticas, a introdução do ozônio deve ser interrompida no momento
certo. Ao manter a reação sob controle rigoroso, isto é, fornecimento estequiométrico de
O3, é possível oxidar seletivamente cadeias laterais insaturadas em compostos aromáticos.
Caso contrário o anel aromático será destruído, conforme descrito a seguir (o mecanismo
desta ciclo-adição 1,3-dipolar está descrito na p. 247):
641
A. Isenmann
Princípios da Síntese Orgânica
O
3 O3
O
O
O
O O
H + 3 H2 O2
3 H C C
Ox.
HOOC
COOH
Ácido oxálico
O
O
O
O
9.6.2
O
H2O
H2 / Pd
O O
3 H C C H + 3 H2 O
Glioxal
Oxidação catalítica no ar (auto-oxidação)
O catalisador mais usado para oxidações com ar é óxido cúprico, Cu2O. Mas existem
também outros óxidos de metais de transição que têm atividade catalítica, desde que
tenham propriedades ácidas.
A seguinte reação é feita a altas temperaturas em fase gasosa:
O
4 O2
O
V2O5 360 °C
+
2 CO2
+ H2O
O
Anídrido ftálico
9.6.3
Oxidação com ácido crômico
O ácido crômico, CrO3, consegue oxidar compostos aromáticos de várias maneiras
diferentes. Muitas vezes uma mistura de produtos é inevitável. Nestes casos deve-se
procurar um outro método para evitar altos gastos na separação e purificação do produto
principal.
Compostos aromáticos condensados, aqui representado pelo naftaleno, podem ser
degradados por oxidação onde um dos anéis aromáticos quebra. Em aromáticos policíclicos
podem decompor-se até vários anéis; a síntese de um produto definido requer bastante
experiência. Apenas o último anel aromático do substrato policíclico certamente não está
sendo atacado pelo ácido crômico.
As seguintes reações ocorrem ao aplicar um excesso de CrO3 e temperaturas elevadas:
642
A. Isenmann
1)
Princípios da Síntese Orgânica
COOH
CrO3
COOH
O
2)
CH3
CH3
CrO3
O
Menadion
No primeiro exemplo o esqueleto carbônico é quebrado e apenas um anel aromático sobrou
.
No segundo exemplo reacional os anéis ficam intactos e apenas a aromaticidade de um dos
anéis se perde 389. Forma-se uma quinona (ver p. 668), “menadion”, que serve como
substituinte da vitamina K (importante na coagulação do sangue humano e também na
fotossíntese).
Estes dois exemplos sejam referidos simbolicamente, para ilustrar a grande variação nas
oxidações com CrO3. Por isso é recomendado de consultar sempre a literatura especial 390
para acertar as melhores condições reacionais (T, p, concentrações, catalisadores) para um
substrato específico.
388
Vermelho de Wurster
Uma síntese especial de quinonas, a partir da diamina aromática, deve ser mencionada
neste lugar. Este exemplo é excepcional porque se forma um complexo de radical cátion tão
estável, que pode até ser isolado 391! É conhecido como “Vermelho de Wurster”. Como
todos os radicais orgânicos, ele absorve a luz na região visível, então é colorido. Todas as
etapas são transferências de elétrons únicos (SET). O método físico-químico mais adequado
para estudar os radicais é a espectroscopia de spins eletrônicos (ESR ou EPR, electron
paramagnetic ressonance), parente da r.m.n.
388
L. Friedman, D.L. Fishel, H.Shechter, J.Org.Chem. 30 (1965) 1453.
B.Sket, M. Zupan, Synth. Commun. 20 (1990) 933; Ref. 128.
390
Uma lista de vários exemplos, juntos às condições reacionais, se encontra em: J. March, Advanced organic
chemistry: reaction, mechanisms, and structure. Edição 5, John Wiley New York 1992
389
O2N
Ph2N N
NO2
O2N
Outro radical estável e com vida de prateleira é o 1,1-difenil 2 picrilidrazil,
, com
elétron desemparelhado e deslocalisado sobre muitos átomos, que serve como reagente sequestrador de
radicais livres em estudos cinéticos de reações em solução. Ao recombinar-se com outros radicais (que são
muito mais reativos), o 1,1-difenil 2 picrilidrazil perde sua coloração violeta. Assim, se tem um método
indireto e elegante, para medir a quantidade dos radicais que são liberados na reação investigada; o método de
monitoramento pode ser feito por espectrofotometria UV/visível – simples, barato e comum nos laboratórios.
391
643
A. Isenmann
Princípios da Síntese Orgânica
Oxidação via SET:
NH2
NH2
NH2
NH2
+ 1/2 Br2
....
- Br-
N(CH3)2
N(CH3)2
N(CH3)2
Br-
N(CH3)2
Vermelho de Wurster
NH
+ 1/2 Br2
O
+ 2 H2O
+
- HBr
N(CH3)2
NH4+
+ HN(CH3)2
O
9.7
Oxidação de Alcoóis
9.7.1
Oxidação de alcoóis primários para ácidos carboxílicos
Esta secção pode ser vista como complemento do texto introdutório ao NOX, dado no
início do capítulo “Redução”, p. 582. Os exemplos de oxidação neste item são bastante
adequados para treinar o formalismo de balancear uma reação redox, a base dos NOX.
Antes de entrar no formalismo geral, devemos conhecer os NOX de todos os reagentes e
todos os produtos que se esperam de uma reação redox.
Procedimento padrão e regras gerais, para equilibrar uma reação redox orgânica:
1. Analisar o acontecimento geral e separar em duas "semi-equações" que são
denomindas de “Redução” e “Oxidação”, respectivamente. Na redução se
consomem elétrons, na oxidação se liberam elétrons.
2. Balancear cada semi-equação por si. É mais fácil equilibrar uma semi-equação do
que a reação redox completa, portanto é aconselhado fazer o balanceamento nesta
etapa. Deve-se procurar o número de átomos igual, em ambos os lados da semiequação. A maioria das reações redox é executada em ambiente aquoso. Neste caso,
a equação pode ser ampliada, à vontade com água e - dependendo do pH do meio com H+ ou OH-.
3. Evite o uso de “O2-“ em ambiente aquoso, pois não existe: O2- + H2O → 2 OH- ;
igualmente inadequado é escrever “H+”, ao se ter condições básicas ou “OH-“,
quando o ambiente da reação for ácido. O acréscimo adequado de H2O, H+ e OHsempre permite equilibrar o número de hidrogênios e oxigênios em cada semiequação.
644
A. Isenmann
Princípios da Síntese Orgânica
4. Considerar os NOX apenas daqueles elementos que sofrem redução/oxidação. A
comparação de substrato e produto em cada semi-reação revela um excesso de
elétrons em um dos lados. Esses devem ser acrescentados no outro lado da semiequação em forma de "e-" - como se fossem substâncias químicas. Por este
procedimento o número de elétrons recebidos/perdidos é compensado em cada
semi-equação.
5. Multiplicar ambas as semi-equações com os fatores menores possíveis, para que
resulte o mesmo número de elétrons, recebidos na redução e perdidos na oxidação.
6. Juntar as semi-equações para a equação final.
7. Cortar aquelas moléculas e íons que aparecem em ambos lados da equação final.
8. Juntar aqueles íons, na sua fórmula correta, que formam sais de baixa solubilidade.
Exemplo: Oxidação de um álcool primário usando KMnO4 em solução ácida (outros
exemplos, ver questão 5, na p. 671).
+7
Red: MnO4Ox:
+ 8 H+
5 e-
Mn2+
+3
-1
R CH2 OH
+ H2O
4 MnO4- + 12 H+
9.7.2
+
R COOH
+ 5 R CH2OH
+
4 H2O
+ 4 H+
+ 4 e-
x4
x5
4 Mn2+ + 5 R COOH + 11 H2O
Oxidação de alcoóis para aldeídos e cetonas
A oxidação de alcoóis terciários geralmente não é possível. Os agentes oxidantes comuns
provocam eliminação β de água. Daí resulta o alqueno (ver p. 46), antes de oxidar o
substrato. A reação mais típica dos alcoóis terciários é a eliminação.
A oxidação de alcoóis secundários fornece cetonas. Essa reação é fácil de executar e não
requisita condições muito bem controladas: temperatura e quantidade do agente oxidante
são bem flexíveis; geralmente se aplica o oxidante em largo excesso. O dicromato e o
permanganato em ambiente ácido oxidam estes alcoóis rapida e completamente.
Praticamente não existem alcoóis secundários onde uma oxidação à cetona seria impedida
pelos fatores indutivo ou mesomérico (efeitos eletrônicos). Único fator que dificulta o
ataque do oxidante é o impedimento espacial, por grupos vizinhos volumosos (grupos
fenilas, t-butilas, isopropilas, etc). Para este caso foi apresentado na p. 467 um método
especial, com o rearranjo da pinacolona.
Caso estejam presentes outros grupos funcionais que não podem sofrer oxidação, porém,
deve-se pensar em equipá-los com grupos protetores (um exemplo dado na p. 638), antes de
efetuar a oxidação. Também pode-se tentar um método oxidativo brando e seletivo - que
pode ser um dos 12 métodos descritos a seguir.
645
A. Isenmann
Princípios da Síntese Orgânica
Mais difícil do que a oxidação de alcoóis secundários é o preparo de aldeídos, a partir de
alcoóis primários, por causa da seguinte sequência reacional:
Álcool primário
Ox.
Aldeído
Ox.
Ácido carboxílico
A obtenção dos aldeídos por este caminho é difícil, já que o próprio aldeído é mais
susceptível à oxidação do que o próprio álcool. Por isso, o problema preparativo não pode
ser resolvido ao simplesmente aplicar quantidades estequiométricas de agente oxidante (que
resultam do balanceamento conforme descrito acima), nem por limitar o tempo reacional.
Todavia, existem três estratégias para se obter os aldeídos com rendimentos satisfatórios:
1. Remoção do aldeído da mistura reacional, imediatamente depois da sua produção.
Isto pode ser feito muitas vezes por destilação porque os pontos de ebulição
diminuem na sequência: acido carboxílico > álcool > aldeído. Deve-se trabalhar
então a temperaturas acima da ebulição do aldeído e abaixo do álcool. Todavia, os
rendimentos não excederão 50%.
2. Estabilizar o aldeído em forma de um derivado estável à oxidação, por exemplo, o
diacetato do seu hidrato (ver p. 638).
3. Uso de agente oxidante específico que se caracteriza por ter impedimentos cinéticos
frente o aldeído.
A terceira estratégia parece estar em contradição à observação de que o aldeído oxida mais
facilmente do que o álcool. Ela não pode ser explicada em termos da termodinâmica
segundo a qual sempre o composto com o menor potencial redox ε é oxidado. Em todos os
casos isto seria o aldeído. A explicação do funcionamento destas oxidações deve-se
procurar em efeitos cinéticos (ver p. 624). Existe algum fator que impede uma oxidação
rápida do aldeído, enquanto o álcool primário não é afetado por este impedimento cinético.
Em vez de discutir as cinéticas em particular é mais indicado apresentar os métodos mais
promissores. O químico preparativo tem a escolha entre os seguintes reagentes:
1) Desidrogenação catalítica usando cromita de cobre, CuCr2O4 (o uso deste
catalisador em reduções já foi apresentado na p. 598).
2) Reagente de Collins
3) PCC
4) Ferrato (VI)
5) Dioxido de manganês
6) NBS
7) Tetra acetato de chumbo em piridina
8) Oxigênio e catalisador
9) DMSO (Swern)
10) com restrição: Dicromato em acetona e ácido crômico em ácido acético glacial.
646
A. Isenmann
Princípios da Síntese Orgânica
Oxidação com CrO3 e piridina, 1 : 2; reagente de Collins
O ácido crômico e a piridina formam um complexo, CrO3 * 2 Py, também conhecido como
reagente de Collins que representa um agente oxidante suave e seletivo. Todavia, para obter
aldeídos com melhores redimentos devem ser aplicadas quantidades estequiométricas de
ácido crômico.
Rascunho do mecanismo (álcool primário ou secundário):
H
H
H
OH
+6
O
O - H2O
C OH + H2CrO4
C
Cr
O
H
H HO
Base
- H+
H
+4
C O +
H
C
+6
O
H HO
Cr
O
O
Base
HCrO3-
A base mencionada neste esquema reacional é a piridina. Tanto aldeídos quanto cetonas
podem ser obtidos por este método.
Oxidação por clorocromato de piridínio, “PCC”
A mistura oxidante, conhecida como PCC, se prepara da seguinte maneira: dissolver (até
saturação) o dicromato de potássio em ácido clorídrico de concentração 6 mol L-1, depois
adicionar a piridina em excesso. Os rendimentos com PCC são bons - comparáveis ao
reagente de Collins.
Tetraacetato de chumbo em piridina
Apropriado para síntese de aldeídos e cetonas é o sistema Pb(OAc)4/Py.
Ferrato de potássio, K2FeO4, em ambiente aquoso
Os complexos com ferro de número de oxidação +6 são relativamente raros e somente
estáveis em ambiente básico. Eles podem ser obtidos a partir de Fe(OH)3 e cloro, em soda
cáustica concentrada ou, alternativamente, por oxidação anôdica de ferro metálico,
igualmente em ambiente fortemente alcalino.
O íon de FeO42- é isômorfo ao SO42- e CrO42-, é paramagnético e tem uma bonita cor
púrpura. Já em ambiente neutro o ferrato se decompõe liberando oxigênio e Fe(III). A semiequação do ferrato é
FeO42- + 4 H2O + 3 eFe3+ + 8 OH,
com um potencial padrão de ε0 = +2,20 V (!), então o ferrato é um oxidante mais poderoso
do que MnO4-. Mesmo assim, este método é adequado para preparar aldeídos e cetonas.
647
A. Isenmann
Princípios da Síntese Orgânica
Dióxido de manganês em solvente orgânico
MnO2 em solvente orgânico polar, geralmente acetona, representa um sistema bastante
suave e seletivo, adequado para preparar aldeídos e cetonas a partir de alcoóis
polifuncionais. Os melhores rendimentos se obtêm a partir de alcoóis alílicos ou benzílicos.
Exemplo:
OH
CHO
MnO2
Acetona
Cinamaldeído
Também compostos com tripla ligação (a unidade -C≡C-C-Y tem o nome trivial
"propargila", ver nota de rodapé 58, na p. 129) podem ser oxidados com bons rendimentos:
R C C
CH R´
OH
MnO2
Acetona
R C C C
R´
O
NBS ou hipoclorito de t-butila
O hipoclorito de t-butila e alternativamente a N-bromossuccinimida (NBS), transferem o
álcool numa primeira etapa em um hipoalogenito. Este, por sua vez, pode ser submetido à
eliminação α, tratando-o com base. O resultado é um grupo carbonila.
R CH2 OH
t-BuOCl
ou NBS
R CH O Hal
R CH O + HB+
+ Hal-
H
Base
O método é adequado para preparar aldeídos.
Note-se que este processo não decorre via radicais – ao contrário do uso geral destes
reagentes, tanto do NBS (p. 70), quanto do hipoclorito de t-butila (p. 70).
Desidrogenação catalítica
O equilíbrio que se estabelece rapidamente, na presença do catalisador, é continuamente
deslocado para o lado do produto, já que um dos produtos, o hidrogênio, é removido
irreversivelmente pela oxidação com ar:
648
A. Isenmann
Princípios da Síntese Orgânica
Catalisador
R CH2 OH
R CH O
+ H2
+ 1/2 O2
H2O
Os catalisadores são:
•
Cu/O2
•
Ag/O2
•
Cu/Cr2O3
Oxidações com DMSO
A grande utilidade do dimetilsulfóxido (DMSO) é ser solvente polar e aprótico, devido à
sua inércia química. Graças ao DMSO já se contornaram muitos problemas até então
insuperados, principalmente os de solubilidade do reagente (p. 31), sensibilidade do
substrato/produto à acidez ou de insuficiência em poder nucleofílico do reagente (ver p.
29). Além destas aplicações, é usado na espectroscopia r.m.n. e em estudos mecanísticos
onde se procura reprimir a troca de hidrogênio entre grupos hidroxilas e/ou aminas (DMSO
quebra as ligações de hidrogênio, igual à uréia). Em todos estes exemplos se aproveita da
estabilidade térmica e da baixa reatividade da molécula do DMSO.
Muito menos conhecida é sua utilidade como reagente. Realmente, o DMSO tem o papel de
oxidante bastante específico, para alcoóis primários e haletos orgânicos, como os seguintes
dois exemplos mostram.
a) Dimetilsulfóxido com cloreto de tionila
R CH2 OH
δ+
R CH2
Clδ−
+
SOCl 2
R CH2 Cl
SN
δ− δ+
O
CH3
S
CH3
DMSO
SN
- Cl-
CH3
R CH O
H
S
- H+
R CH O
+ H 3C
S
CH3
CH3
Base
auxiliar
A mistura de O=SMe2 com O=SCl2 age como oxidante bastante suave, então serve para
sínteses de aldeídos e cetonas.
b) Oxidação de Swern
O sistema DMSO com cloreto de oxalila, (COCl)2, é igualmente brando e serve para
perparar aldeídos.
649
A. Isenmann
Princípios da Síntese Orgânica
Observação: a temperatura reacional não deve exceder –50 °C, menos na última etapa que
se processa à temperatura ambiente.
O
H3C
S
O
CH3
+
O
C
Cl
C
Cl
H3 C
S
[CH2Cl2]
O
H3 C
-50°C
Cl
C
O
C
H3 C
Cl-
- CO
- CO2
O
S
Cl
H3C
Cloreto de
oxalila
R CH2OH
-50°C
H3C
S
-
O CH2 R Cl
H3C
Base (NEt 3)
- H+ , - S(CH3)2
O
R C
H
Investigações profundas do mecanismo desta oxidação revelaram uma reação prévia entre o
cloreto de oxalila e o DMSO, em vez da esperada troca nucleofílica do grupo –OH pelo –Cl
no substrato orgânico, como foi constatado no exemplo do item a. Este novo complexo é
bastante instável e sofre, logo depois da sua formação, descarboxilação (- CO2) e
descarbonilação (- CO). Sobra o cátion dimetilclorossulfônico, um eletrófilo que procura os
elétrons do substrato alcoólico. Sob a influência de uma amina, finalmente, separa-se o
tioéter dimetílico, liberando o aldeído desejado.
Oxidação com Cr2O72-/acetona/ácido acético glacial (/ H2SO4)
Este sistema oxidante pode ser usado com ou sem ácido sulfúrico. O papel do H2SO4 é o
ajuste da força eletromotriz: a presença de ácido sulfúrico aumenta bastante a força
oxidativa do sistema (ver p. 252). Por este motivo, não é surpreendente que a oxidação para
aldeídos somente funciona na ausência de H2SO4. A síntese de cetonas, por outro lado, é
bastante facilitada pelo H2SO4.
Em muitos textos este método é referido como método padrão para o preparo de aldeídos.
Todavia, deve-se lembrar das suas desvantagens:
•
o gasto de solvente é alto
•
especialmente com tempos reacionais prorrogados as insaturações do substrato
podem ser oxidadas, também.
Os três métodos a seguir, certamente não servem para oxidar um álcool primário para o
aldeído.
Oxidação de Oppenauer
Essa reação funciona com t-butóxido de alumínio e uma cetona de sacrifício. É a reversa da
redução de Meerwein-Ponndorf-Verley, apresentada na p. 603. Ela é útil somente para a
síntese de cetonas.
650
A. Isenmann
Princípios da Síntese Orgânica
R H
+
C
R OH
Al(t-BuO)3
R
O
H
C O
+
R
OH
Cetona de sacrifício
Este método é bastante aplicado em compostos naturais (esteróides, alcalóides,...).
Dióxido de selênio
A força oxidativa do SeO2 é muito alta para a preparação de aldeídos: a reação sempre
procede ao ácido carboxílico. Este método então somente serve para a síntese de cetonas.
KMnO4 em ambiente básico
Com este reagente também não podem ser obtidos aldeídos, mas somente cetonas. O
mecanismo inclui etapas de SET:
R2CH OH
+ OH+7
R2C O- + MnO4-
R2C O-
+ H2O
+6
R2C O-
+ MnO42-
+ H+
Radical cetila
R2C O- + MnO43 MnO42-
+ 2 H2O
R2C O
+4
MnO2
+ MnO42+7
+ 2 MnO4-
+ 4 OH-
A última linha deste esquema não tem a ver com a reação orgânica, mas representa a reação
seguida do óxido de manganês (VI) que tem pouca estabilidade. É um
desproporcionamento para Mn(IV) e Mn(VII), as formas mais estáveis em ambiente aquoso
básico.
9.7.3
Oxidação de 1,2-dióis (= glicóis)
Pela análise dos NOX nos carbonos, o epóxido pode ser visto como 1,2-diol, também. Um
método especial para sua oxidação é referido na p. 467.
Quebra com tetraacetato de chumbo segundo Criegée
a) Quebra de cis-dióis
651
A. Isenmann
Princípios da Síntese Orgânica
+4
0
Pb(OAc) 4
C
- 2 AcOH
+2
0
C
+1
O
O
+ Pb(OAc)2
CH CH
O O
Pb
AcO
OAc
OH OH
+1
b) Quebra de trans-dióis
OH H
Pb(OAc) 4
- AcOH
+2
O HH
- AcOH
H OH
H
O
+1
+1
O
O
+ Pb(OAc)2
CH CH
Pb(OAc)3
Observa-se uma velocidade de quebra do isômero cis, 300.000 vezes mais rápida do que do
isômero trans. A causa desta diferença é a dificuldade em formar um complexo cíclico no
último caso.
Dentre o ciclo de 5 membros é plausível um movimento sincronizado dos elétrons. Ao
mesmo tempo que uma ligação quebra (= endotérmico) uma outra se forma (= exotérmico).
O balanço fornece uma energia (= energia de ativação!) bastante baixa. Consequentemente,
a reação é fácil e rápida. O resultado é uma eliminação redutora do chumbo.
Pode-se objetar que os esquemas acima não representam o substrato na sua conformação
correta. Mesmo assim, as conclusões sobre as diferentes reatividades têm validade,
conforme mostrado a seguir.
Enquanto o cis-diol mantém seus grupos em distância favorável em qualquer momento (é
sempre um grupo –OH axial e o outro equatorial), a distância entre os grupos hidroxilas no
trans-diol varia muito quando o anel alicíclico troca de conformação. Isto pode ser
verificado, de preferência nas conformações de assento - por sua vez mais provável para
ciclos de 6 membros:
652
A. Isenmann
Princípios da Síntese Orgânica
OH
OH
cis
HO
HO
OH
trans HO
HO
OH
Distância
desfavorável
Os trans-1,2-dióis de cadeia aberta (entre eles os açúcares e carboidratos!) não têm este
impedimento estérico. Portanto, mostram a mesma facilidade de ser quebrados do que o
isômero cis, já que a rotação em volta das ligações simples é rápida. Uma discussão
conformacional é desnecessária.
Quebra segundo Malaprade
O reagente da quebra de Malaprade é o periodato que já foi apresentado na oxidação de
Lemieux (ver p. 630) onde tinha duas funções, re-oxidar o permanganato e reagir com o
composto intermediário, um diol. Quem reagiu com o substrato na oxidação de Lemieux foi
o MnO4- (ou OsO4) que tinha então o papel de catalisador. No método de Malaprade não há
necessidade de um catalisador, já que o próprio substrato é um diol.
Esta vez é realmente o periodato que reage com o substrato:
+1
C OH
C OH
+ HIO4
O
O
+7
- H2O
I
O
+2
C O
+5
+
O C
+ HIO3
O
Quebra com ácido crômico anidro
Mais um exemplo para as inúmeras oxidações com ácido crômico é a quebra oxidativa de
dióis. Para quebrar a ligação σ (C-C) é necessária alta temperatura e um sistema livre de
água. Essa oxidação não pára no NOX do aldeído.
653
A. Isenmann
Princípios da Síntese Orgânica
H
H
H3C
H3C
0
+6
C OH
CrO3
H3C
C OH
H
O
Cr
O O
O
H3C
+1
2
H3C
H
CHO
+4
+
CrO3
[ CrO2 ]
H3C
COOH
Oxidação com H2O2 e Fe2+ (Reagente de Fenton)
A força oxidativa da água oxigenada é largamente conhecida (ε0 = +1,77 V, a pH 0). Ao
estiverem presentes traços de sais de Fe2+ o sistema torna-se mais agressivo ainda.
+ 2 OH
CH2 CH2
OH OH
+ 2 H2O
CH2 CHO
OH
A fraca ligação O-O do reagente é predestinada para quebrar de maneira homolítica;
consequentemente, esta oxidação ocorre via radicais. É exatamente na quebra desta ligação
onde o ferro entra como catalisador. Um dos radicais •OH entra em reação redox prévia
com o Fe2+, formando Fe3+ e OH-.
-1
2+
Fe
+
-1
HO OH
Fe3+
-2
+
OH-
-1
+
OH
Esta reação explica porque o reagente de Fenton pode também ser usado como iniciador de
reações radicalares em cadeia (ver p. 93). Uma outra aplicação recente do reagente de
Fenton é a preparação de água de abastecimento, substituindo assim o cloro (que causa mal
gosto e cheiro e promove a corrosão nas tubulações metálicas).
9.8
Oxidação de aldeídos a ácidos carboxílicos
A oxidação de aldeídos para ácidos carboxílicos ocorre com bastante facilidade, como já foi
elucidado no item 9.7. Qualquer aldeído deve ser guardado, por este motivo, em garrafas
escuras e bem fechadas, para não sofrer oxidação pelo ar (para o mecanismo da autooxidação, recorra à p. 79).
Os reagentes mais comuns para oxidar rapida e quantitativamente um aldeído são:
•
CrO3
•
KMnO4/OH-
•
HNO3 concentrado.
654
A. Isenmann
Princípios da Síntese Orgânica
O permanganato em ambiente aquoso ácido é muito referido como método preparativo
padrão. Porém, o substrato orgânico não deve conter outros grupos funcionais que podem
ser atacados também por este oxidante forte:
δ+
Ar CHO
OH
+
MnO4
-
+
+ H
rápido
Em toda analogia o ácido crômico:
Ar
CHO
+
H2CrO4
+7
C O MnO3
H
- H+
Ar
CH O CrO3H
+3
Ar
+5
COOH + [ MnO3- ]
Ar
OH
+6
+1
Ar
+1
+3
COOH
+
+4
[ CrO2 ]
Oxidações com finalidade analítica
As seguintes oxidações têm certo valor analítico. São sistemas bastante seletivos para
grupos aldeídicos, enquanto alcoóis e cetonas não são atacados.
Reagente de Fehling: Cu2+/complexante/OHEste reagente é conhecido desde o século 19. Foi usado, por exemplo, por E. Fischer nos
seus trabalhos fundamentais sobre carboidratos e açúcares, para distinguir cetosas das
aldosas. Trata-se de uma mistura de sulfato de cobre(II) com o sal de Seignette e NaOH.
O sal de Seignette é o tartarato de sódio e amônio e serve para complexar o Cu(II). O metal
precisa desta complexação para não ser precipitado em forma de Cu(OH)2 quando o NaOH
é adicionado.
COOH
HO
H
H
OH
COOH
D(-) -ácido tartárico
Os aldeídos (aldosos) são então oxidados, formando ácido carboxílico e um precipitado de
composição desconhecida contendo traços de Cu2O. A cor característica do Cu2O (entre
marrom e vermelho) indica a presença de aldeídos. Cetosas, isto são açúcares com grupo de
cetona, não reagem com reagente de Fehling; com metilcetonas este teste não fornece
resultados confiáveis.
Reagente de Tollens
Do mesmo princípio do sistema de Fehling funciona um reagente oxidante a base de prata
(= homólogo do cobre), conhecido como reagente de Tollens. Pode ser usado
alternativamente para testar a presença de grupos aldeídicos. Desta vez é a amônia que tem
o papel do complexante e ajuste do pH, ao mesmo tempo. Forma-se o complexo solúvel,
[Ag(NH3)2]+. Esta solução, ao ser aquecida brandamente num tubo de ensaio junto ao
655
A. Isenmann
Princípios da Síntese Orgânica
substrato em questão, indica a presença de aldeídos por um bonito espelho de prata,
depositado na parede do vidro. Em analogia ao teste de Fehling, as cetonas não reagem
com o reagente de Tollens - nem as metilcetonas.
9.9
Oxidação de cetonas
A discussão da oxidação de alcoóis (p. 645) podia dar erroniamente a impressão de ser
impossível oxidar cetonas – o que não é verdade.
Para oxidar uma cetona existem principalmente duas possibilidades (o funcionamento
excepcional da reação de Willgerodt-Kindler ver p. 660):
1. Quebra da ligação C-C próxima ao grupo carbonila e reposição do carbono por um
elemento mais eletronegativo (O ou F). Conhecem-se sistemas oxidantes que fazem
isso, mas requerem de condições mais drásticas, devido à alta estabilidade da
ligação C-C.
2. Oxidação da dupla ligação carbono-carbono da forma enólica. Formalmente é uma
reação no carbono em posição α ao grupo carbonila. Esta oxidação pode ser
efetuada com reagentes especiais.
Oxidação com CrO3/H+; KMnO4/OH- ; HNO3
A oxidação por estes oxidantes “comuns” ocorre somente a temperaturas mais altas e
tempos reacionais prolongados. Geralmente não é um bom método preparativo por causa da
insensibilidade das cetonas ante estes reagentes. Muitos outros grupos funcionais dentre o
substrato podem sofrer oxidação antes do grupo carbonila.
Seguem dois exemplos, ambos de importância industrial, onde se consegue preparar ácidos
carboxílicos, a partir de cetonas de estrutura bastante simples:
1)
O
Oxidação
O
R C
CH3
COOH
Ácido adípico
Ciclohexanona
2)
HOOC
Oxidação
R COOH
A primeira reação tem importância, especialmente para oxidar alcoóis e cetonas alicíclicos
(= cíclico e não-aromático), para produzir diácidos. Estes produtos são largamente usados
como monômeros em policondensados, tais como poliésteres e nylon.
656
A. Isenmann
Princípios da Síntese Orgânica
Acesso a compostos 1,2-dicarbonilados, via oxidação
Compostos 1,2-dicarbonilados são de importância estratégia na síntese orgânica, porém não
são fáceis de se obter (devido à polaridade não natural ou dissonantes dos síntons iônicos,
ver detalhes na p. 566). A condensação de aciloína/benzoína, já foi apresentada como
processo aniônico (benzaldeído + cianeto, ver p. 465), e como processo radicalar redutivo
(éster + sódio, ver p. 612). Aqui mais dois métodos específicos oxidativos, dos quais o
primeiro é mais brando e seletivo.
a) Com HNO2:
O eletrófilo que ataca a dupla ligação do enol é o cátion nitrosil, NO+. Isto é em analogia à
formação do sal de diazônio (p. 289). As condições reacionais também são bem
semelhantes: trabalha-se em ambiente moderadamente ácido. Em vez de nitrito de sódio
podem ser usados os reagentes HNO2 ou RONO ou N2O5, também.
Uma vez o NO+ se ligou ao carbono em posição α, o complexo pode sofrer uma segunda
tautomeria que fornece uma oxima. No exemplo a seguir é oxidada uma metilcetona, então
forma-se uma α-ceto-aldoxima (ver também p. 426):
O
OH
α
C6H5 C CH2
C6H5 C CH3
Tautomeria 1
OH
C6H5 C CH2 NO
Enol
O
O
- H+
NO+
C6H5 C CH N OH
C6H5 C CH2 NO
Tautomeria 2
H3O+
- H2NOH
O
C6H5 C CHO
Oxima
A partir da aldoxima/cetoxima existem várias possibilidades de reagir. Uma delas é a
hidrólise em ambiente ácido que abstrai a molécula de hidroxilamina, em troca de uma
nova função carbonila.
Deve-se ressaltar que a cetoxima não corre perigo de rearranjo de Beckmann (ver p. 429).
Isto tem sua explicação na situação eletrônica: a α-cetoxima somente pode oferecer um
grupo pobre em elétrons (que é o grupo carbonila), para a migração para o N com sexteto
de elétrons de valência. Isso é energeticamente insatisfatório, então não acontece o
rearranjo de Beckmann.
b) Oxidação com SeO2 em álcool ou dioxano
Uma oxidação de cetonas com SeO2 fornece uma α-dicetona em uma etapa só. Como estes
produtos são altamente valorizados, tanto na síntese orgânica quanto na inorgânica como
complexante de metais pesados, o emprego deste oxidante se estabeleceu no últimos anos
como método padrão – apesar da sua alta toxicidade.
Este oxidante ataca a forma enólica da cetona:
657
A. Isenmann
Princípios da Síntese Orgânica
H O
O
OH
C
O
C
Se
OH
H
C O
C
- H2O
H
O
Se
C
C
OH
+
[SeO]
+
H2O
O
O subproduto, [SeO], foi anotado neste esquema em colchetes, por que sua redução
geralmente não pára neste estado, mas sim, no estado elementar, Se. Daí o SeO serve para
oxidar mais uma molécula de enol.
O reagente, SeO2, já foi introduzido como oxidante seletivo para alcoóis secundários,
posições alílicas e benzílicas (ver as pp. 651, 633, e 639, respectivamente). A facilidade
com que estas reações ocorrem, pode ser resumida na seguinte sequência de reatividade:
O
OH
CH
>
C
CH 2
>
C
C
CH 2
>
C6H5 CH2
Lembre-se: ambos os métodos oxidativos somente fornecem produtos definidos, quando a
cetona de partida é simétrica. Em casos de substratos assimétricos o lado da enolização
deve ser bem definido, se não temos que contar com uma mistura de produtos (ver tambem
p. 457).
Contração oxidativa de cetonas cíclicas
Uma reação organometálica usando Tl(III) permite a oxidação de cetonas alicíclicas,
enquanto o esqueleto carbônico do substrato sofre um rearranjo, fornecendo um anel
reduzido por um carbono 392:
O
Tl(NO3)3
COOH
AcOH
25 °C
84 %
O mecanismo proposto é a partir da forma enólica do substrato. O Tl(III) está procurando a
posição α onde tem alta densidade eletrônica. Logo em seguida ocorre a formação do diol
geminal, sua vez facilitada pela presença do retirador de elétrons. Uma eliminação redutiva
do Tl ocorre na terceira etapa enquanto no complexo reativo se fecha um segundo anel: o
produto intermediário é um epóxido cujo anel de oxirano é bastante carente em elétrons.
Em ambiente ácido a falta em elétrons se torna mais pronunciada ainda. A abertura do anel
de três membros libera um carbocátion no qual ocorre imediatamente o rearranjo dos
carbonos. Isto sempre ocorre no lado traseiro do anel de oxirano.
392
A. McKillop, J.D.Hunt, E.C.Taylor, J.Org.Chem. 37 (1972) 3381.
658
A. Isenmann
Princípios da Síntese Orgânica
O
OH
HO
Tl(NO3)3
OH
+1
Tl(NO3)2 - Tl(NO3)3
HO
O
- HNO3
HO
H O
O
+
H
O
COOH
H+
- H+
A estereoquímica deste rearranjo oxidativo é bem controlada: a partir da 4metilciclohexanona forma-se exclusivamente o isômero cis:
OH
O
H
- H+
COOH
Uma reação com o mesmo propósito preparativo, porém com mecanismo totalmente
diferente, é apresentada no contexto do rearranjo de Favorskii (p. 429).
Reação halofórmio
O benefício principal desta reação (além de ter certa importância analítica) é um produto
reduzido por uma unidade carbônica (isto é, uma alternativa à descarboxilação descrita na
p. 462).
Os reagentes são: I2/OH- ; Br2/OH- ; Cl2/OH-.
Os substratos prediletos são as metilcetonas. Mas também α-metil alcoóis (isto é, com
grupo hidroxila em posição 2) fazem esta reação, após serem oxidados in situ para as
metilcetonas.
659
A. Isenmann
O
H3C
C
R
Princípios da Síntese Orgânica
+ 3 Hal2
- 3 HHal
O
Hal3C C
-
R
+ OH
OHal3C
C
Hal3C-
R
+
HOOC
R
ânion
halofórmio
OH
Ox.
OH
H3C CH R
Hal3CH
+
-
OOC
R
Produto
= carboxilato
A α-halogenação ocorre sempre na forma do enolato do substrato cuja dupla ligação é rica
em elétrons e pode ser atacada pelo halogênio eletrofílico (para o mecanismo destas etapas,
recorra à p. 424). Após percorrer 3 vezes esta halogenação o ânion halofórmio tornou-se
estável o suficiente e pode ser abandonado. O produto da reação é o carboxilato, de cadeia
carbônica reduzida por um carbono.
Reação de Willgerodt-Kindler
Um reagente oxidante especial, adequado para oxidar aldeídos e cetonas aromáticos, é a
morfolina com polissulfeto de amônio (= enxofre solubilizado). Na primeira etapa da
reação de Willgerodt-Kindler se forma uma enamina que reage como o enxofre da seguinte
maneira:
O
O
Ar
C
N
CH3
Fenona
+
HN
O
Morfolina
- H2O
Ar
C CH2
Enamina
O
"S"
130 °C
N
Ar
C CH2 S"Betaina"
O produto primário que resulta desta sequência tem duas cargas opostas em locais distantes,
comumente chamado de "betaina".
Segue uma tautomeria especial 393. A situação eletrônica dentro do α-amino-tialdeído
facilita a migração do grupo morfolina, para o final da cadeia carbônica. Finalmente se
obtém como produto uma tioamida que é derivado do ácido carboxílico (p. 349) e que pode
ser hidrolisada para tal. Portanto, esta síntese pode ser referida como oxidação.
393
Tautomeria especial porque, além da mudança de hidrogênio, tem-se a migração da dupla-ligação para
uma posição não vizinha.
660
A. Isenmann
Princípios da Síntese Orgânica
O
N
Ar
C
CH S-
tautomeria
especial
O
O
N
N
Ar
H
C
CH
Ar
Ar
O
Ar
CH C
O
N
S-
CH CH
SCátion aziridínio
H
α−aminotialdeído
O
N
S
CH C
SH
tautomeria
comum
N
Ar
H
CH2 C
S
Tioamida
A tioamida pode ser facilmente hidrolisada formando uma amida, ao tratá-la com NaOH
meio-concentrada. A reação de Willgerodt-Kindler é então a síntese de amidas (ou seja, de
ácidos carboxílicos) a partir de fenonas (= cetonas aromáticas) e aldeídos aromáticos,
enquanto o grupo da amida pode mudar para um local diferente do grupo carbonila do
material de partida. Interessante é que esta reação funciona até com fenonas nas quais o
grupo carbonila é vizinhado por uma cadeia carbônica mais comprida. Neste caso observase a migração do grupo carbonila ao longo da cadeia toda, até chegar no seu final (ver
terceiro exemplo no esquema a seguir)!
O
Ar
C
O
H
Ar
C
NH2
O
O
Ar
C
CH3
Ar
CH2
C
NH2
O
O
Ar
C
(CH2)n CH3
Ar
(CH2)n+1
C
NH2
Tanto os aldeídos aromáticos quanto as fenonas de partida são de facil acesso (ver as
substituições com carbono eletrofílico na p. 304, e a acilação Friedel-Crafts, p. 297,
respectivamente). Portanto, a oxidação de Willgerodt-Kindler é uma síntese bastante útil e
versátil.
661
A. Isenmann
Princípios da Síntese Orgânica
Oxidação de Baeyer-Villiger
Um importante método preparativo é a quebra oxidativa de Baeyer-Villiger 394 (ver também
p. 427) que produz um éster a partir de uma cetona, em uma etapa só (inglês: one-pot
reaction). O reagente, geralmente um perácido carboxílico, é de fácil acesso.
O
CF3C
O
(CH3)3C
C CH3
O OH
OH
(CH3)3C
C
CH3
°
O C
CF3
O
OH
C O C(CH3)3
H3C
+ CF3COO-
O
O
H3C
C O C(CH3)3
+ CF3COOH
Na etapa central desta síntese se tem um rearranjo (= reorganização do esqueleto carbônico,
ver item 5.5.12) que ocorre num sexteto de elétrons no oxigênio, marcado em negrito no
esquema acima.
Ao haver dois grupos potenciais que podem mudar-se para o oxigênio com sexteto de
elétrons, o mais substituído ganha a preferência. Isto tem motivo eletrônico (efeito +I dos
substituintes), mais do que estérico. A saída da parte proveniente do reagente (no exemplo
acima: trifluoracetato) e a mudança do grupo isobutila não acontece em etapas sequenciais,
mas é um movimento sincronizado. Em consequência a configuração absoluta no grupo em
movimento se retém.
O produto principal é o éster a partir dos fragmentos da mesma molécula de substrato. Uma
transesterificação com o trifluoracetato não ocorre, sob as condições aplicadas, porque se
trabalha sob exclusão de água. Sendo assim, o éster produzido inicialmente não sofre
hidrolise ou transesterificação.
O reagente da oxidação de Baeyer-Villiger pode ser um dos seguintes perácidos, colocados
na ordem de reatividade:
CF3CO2OH
>
HCO2OH
>
CH3CO2OH ; C6H5CO2OH
Nenhum destes perácidos tem uma longa vida de prateleira, portanto é produzido logo antes
de ser aplicado na oxidação, a partir do respectivo ácido carboxílico e H2O2. O perigo que
acompanha o uso do H2O2, especialmente quando aplicado em altas concentrações
(detonações espontâneas), é bastante reduzido ao se utilizar estes perácidos. Como solvente
pode ser usado o próprio substrato (cetona) ou cloreto de metileno, CH2Cl2.
Uma reação que fornece um produto semelhante à reação de Baeyer-Villiger, mas por um
caminho diferente, é apresentada no capítulo dos rearranjos (ver rearranjo de Schmidt, p.
429).
394
Esta reação somente é uma das grandes descobertas mecanísticas e preparativas de Adolf von Baeyer
(prêmio Nobel em 1905); outros méritos são a estrutura da fluoresceina, as sínteses do índigo, a resina
fenólica e a destilação de zinco em pó.
662
A. Isenmann
Princípios da Síntese Orgânica
9.10 Oxidação de ácidos carboxílicos
Os ácidos carboxílicos são ainda mais resistentes à oxidação do que as cetonas. Única
possibilidade de aumentar o número de oxidação do carbono carboxílico é na forma de
CO2. Ao aplicar altas temperaturas, geralmente é possível provocar uma descarboxilação,
isto é, o desprendimento de CO2.
A seguir sejam referidos um exemplo químico e um eletroquímico, onde o ácido
carboxílico sofre degradação oxidativa (o método de Hunsdiecker foi referido em outros
lugares, pp. 104 e 393).
Eletrólise de Kolbe na presença de álcool
Em comparação com a química inorgânica, os métodos eletroquímicos na síntese orgânica
são raros, devido aos desvios e produtos paralelos com quais reações orgânicas são
afetadas. Um segundo motivo, certamente, é a hidrofobia, da maioria dos substratos
orgânicos 395. Aqui temos um substrato que é bem solúvel em ambiente aquoso - o
carboxilato de metais alcalinos.
Como todos os processos eletroquímicos em substâncias orgânicas a eletrólise de Kolbe é
de natureza radicalar. Uma solução de carboxilatos pode ser eletrolisada, como já foi
discutido na p. 394. Aqui seja completado o espectro sintético pela possibilidade de
produzir um éter, a partir do radical intermediário mais um álcool:
2
R COO- - 2 e-
2
R COO
-
- 2 CO2
- e , - CO2
R R
2 R
R+
+ CH3OH
R OCH3
+ H+
O produto principal, na maioria das condições aplicadas, continua o dimero do radical
descarboxilado intermediário.
Atenção: ao aplicar potenciais maiores se obtêm produtos derivados do carbocátion, em vez
do radical! Uma destas possibilidades já foi incluída no esquema acima. Na presença de um
álcool se forma um éter cuja nova parte carbônica é diminuída por unidade, em comparação
com o carboxilato da partida.
Outro exemplo de síntese radicalar, induzida por eletrólise, é a redução de cetonas no
cátodo. Quando feita em ambiente aquoso ácido se obtem o pinacol (p. 467) com bons
rendimentos.
Oxidação com tetraacetato de chumbo
395
M.E. Baizer, Recent developments in organic synthesis by electrolysis, Tetrahedron 40 (1984) 935.
663
A. Isenmann
Princípios da Síntese Orgânica
O
COOH
COOH
Pb(OAc) 4
- 2 AcOH
C O
OAc
+ Pb(OAc)2
OAc
+ 2 CO2
Pb
C O
+ H2O
O
Como se perde o subproduto CO2 em forma de gás, a reação torna-se irreversível e anda a
alto rendimento. Nota-se que o movimento dos elétrons no estado de transição é idêntico ao
da oxidação de Criegée (ver p. 651).
O mesmo produto, um alqueno, se obtém, aliás, a partir deste ácido dicarboxílico, com a
eletrólise de Kolbe (ver acima).
9.11 Oxidação de éteres
A auto-oxidação de éteres e seus produtos perigosos já foi discutida na p. 96. Ela ocorre
especialmente fácil, a temperaturas baixas e sem necessidade de catalisador.
R CH2 O CH2 R +
O2
R CH O CH2 R
O
Ramificação para peroxiéteres
poliméricos (explosivos!)
OH
Na exposição ao ar (especialmente sob incidência de luz) os éteres oxidam em posição α,
formando peróxidos explosivos. Eles podem polimerizar e formar compostos indiferentes
ao teste oxidorredutivo para hidroperóxidos, feito com iodeto/amido. Então não pode-se
excluir a presença de peróxidos (que sempre significa perigo) quando este teste deu
negativo (isto é, ausência da coloração típica azul proveniente do íon I3- complexado).
Para evitar a acumulação de peróxidos, especialmente no resíduo de uma destilação, cada
éter deve ser armazenado em garrafas escuras, a fim de excluir a iniciação radicalar pela
luz. Além disso, deve-se acrescentar KOH sólido. Assim, precipitam-se os sais pouco
solúveis dos hidroperóxidos e podem ser descartados sem perigo. Uma outra possibilidade
é a adição de quantidades catalíticas de Fe2+ que promove a decomposição dos peróxidos,
seguindo um mecanismo parecido ao do reagente de Fenton (ver p. 654).
Diversos éteres são também comercializados com aditivos antioxidantes.
Portanto: nunca deixe secar o balão na destilação de éteres! Os últimos 10% do éter cru são
descarte!
O dito vale para todos os éteres, inclusive os solventes polar-apróticos glime, diglime,
polietilenoglicol, THF, dioxano, morfolina, etc.
664
A. Isenmann
Princípios da Síntese Orgânica
9.12 Oxidação de compostos contendo nitrogênio
9.12.1 Oxidação de aminas primárias
Muitas das aminas primárias se oxidam tão facilmente que, somente ao expô-las ao ar por
certo tempo, à temperatura ambiente, elas sofrem auto-oxidação. Este caminho, porém, não
é útil sob aspectos preparativos porque se forma uma mistura complexa de produtos. Por
isso precisa-se de agentes oxidantes eficazes que levam a somente um produto com alta
seletividade.
Os oxidantes certamente mais importantes para aminas são H2O2 e perácidos.
H2O2
R CH2 NH2
R CH NOH
Aldoxima
O mecanismo mais provável é a substituição nucleofílica, no oxigênio deficitário em
elétrons. A amina, como já foi constatado no capítulo 1, é um núcleófilo muito bom.
R CH2 NH2
+
HO OH
- OH-
R CH2 NH2 OH
- H+
R CH2 NH OH
Hidroxilamina
+ H2O2
R CH2 N(OH)2
- OH- H+
Elimin. β
- H2O
R CH NOH
Observação: a estequiometria desta reação é 1 : 2. Na prática, porém, não é necessário
cuidar das quantidades exatas; em vez disso aplica-se o oxidante (barato) em largo excesso.
Ao se usar perácido trifluoracético as aldoximas são oxidadas mais uma vez para o
composto nitro 396. Isto é, o gasto em oxidante são 3 equivalentes.
R CH N OH
+ CF3CO2OH
- CF3CO2H
O
O
tautomeria
R CH2 N
R CH N
O
OH
"aci"
"nitro"
R = Alquila ou H
396
Os nitroalcanos são excelentes solventes para uma série de polímeros, tais como resinas vinílicas,
poliestireno, nitro- e acetilcelulose e poliacrilonitrila. Uma outra aplicação dos compostos nitro é sua hidrólise
ácida, chamada de reação de Nef, fornecendo aldeídos e cetonas (ver p. 535).
665
A. Isenmann
Princípios da Síntese Orgânica
O primeiro produto desta oxidação total é a forma aci que entra em equilíbrio com a forma
nitro. O mecanismo é semelhante ao da tautomeria ceto-enólica (ver p. 400).
Aliás: a própria aldoxima efetua um equilíbrio tautomérico, também:
R CH N OH
tautomeria
R CH2 N O
Aldoxima
Nitrosoalcano
9.12.2 Oxidação de aminas secundárias
A oxidação de aminas secundárias é tão fácil como a de aminas primárias. Porém, a cascata
de oxidações já pára depois a primeira etapa, fornecendo a hidroxilamina. O único próton
ligado ao nitrogênio do substrato foi então trocado pelo grupo hidroxila. Uma oxima ou até
um composto nitro não são observados:
H2O2
R2NH
R2N OH
+
H 2O
Hidroxilamina
9.12.3 Oxidação de aminas terciárias
A partir de aminas terciáras se formam aminóxidos, compostos zwitter-iônicos de
estabilidade surpreendentemente alta. Devido à contribuição de forças de Coulomb, a
ligação N-O tem apenas 136 pm. Este composto de alto momento dipolar mostra
propriedades moderadamente básicas, devido à eletronegatividade do oxigênio. Portanto, é
muito bem solúvel em água e em ácidos minerais diluídos.
R3 N
H2O2 / AcOH
R3 N O
N-óxido
Os N-óxidos (= aminóxidos terciários) representam o material de partida para a síntese
regio- e estereoespecífica de alquenos, via eliminação cis ou eliminação de Cope (ver p.
147).
9.12.4 Oxidação de aminas aromáticas
Oxidação de anilinas para compostos nitro
Ar
NH2
CF3CO2OH
Ar
NO2
666
A. Isenmann
Princípios da Síntese Orgânica
Esta reação somente tem sucesso na ausência de substituintes doadores de elétrons no anel,
devido o efeito +M. A aplicação desta síntese é rara; mais comum é o sentido reverso: o
composto nitro, de fácil acesso via nitração (p.288) é reduzido à anilina (Tabela 17 na p.
300), por sua vez de acesso difícil por outros caminhos.
Oxidação para composto nitroso
Ar
NH2
Oxidação
Agentes oxidantes:
Ar
NO
H2SO5 = perácido sulfúrico = ácido de Caro
H2S2O8 = Ácido peroxodissulfúrico
Para providenciar que o composto nitroso não sofra oxidação para o composto nitro, a
reação tem que ser conduzida abaixo de 0 °C.
Oxidação de hidroxilaminas aromáticas para compostos nitrosos
Ar
NH OH
Cr2O72H2SO4
Ar
NO
Essa oxidação tem uma aplicação interessante: os compostos nitrosos não são acessíveis, de
maneira direta, a partir de compostos nitro. Apesar de percorrer o estado nitroso, a redução
do composto nitro não pára neste estado. A solução é a redução do substrato à
hidroxilamina, na qual se aplica a oxidação seletiva para o alvo, o composto nitroso.
Ar
Ar
NO2
NO
Oxidação
Zn / NH4Cl
Ar
NH OH
Compostos nitrosos aromáticos têm diversas utilidades. Grandes quantidades são
convertidas em azometinas que são, como também os compostos azo aromáticos (p. 290 e
853), uma importante classe de corantes. Um exemplo típico é o acoplamento de γ-picolino
com N,N-dimetilamino 4-nitrosobenzeno, fornecendo uma azometina de coloração
intensiva.
667
A. Isenmann
Princípios da Síntese Orgânica
CH3
CH N
+
O N
N(CH3)2
N(CH3)2
- H2O
N
N
γ− Picolino
Azometina
Outra reação onde participam compostos nitrosos é a reação de Barton (ver p. 104).
Oxidação de diaminas aromáticas para quinonas
O
NH2
FeCl3
∆
O
NH2
As quinonas 397 têm grande importância como “tampão” em reações redox e radicalares, o
que quer dizer, elas podem inibir essas reações por reagirem mais rápida e facilmente com
os intermediários reativos. Isto se deve à grande facilidade de serem reduzidas. Portanto as
quinonas são largamente aplicadas na função de antioxidantes e “quenchers” (pp. 59 e 81),
isto é, aditivos estabilizantes em polímeros e cosméticos, por exemplo.
Uma outra aplicação de importância industrial é ilustrada no exemplo da antraquinona para
9,10-antraidroquinona:
O- Na+
O
Na2S2O4 , NaOH
reverso:
O2
O- Na+
9,10-antraidroquionona
O
Antraquinona
H2O2
H2O
O diânion da antraidroquinona é solúvel em água enquanto a antraquinona, um corante
usado na indústria têxtil, não é. Por isso o corante é tratado com ditionita de sódio para
solubilizá-lo e levá-lo a uma forma aplicável à fibra ("base de leuco"). Depois disso, deixar
a fibra / o tecido simplesmente secar no ar, enquanto o corante é restaurado.
397
com exceção da quinona dicloro-diciano, conhecida como DDQ, por sua vez um oxidante seletivo para
desidrogenações nas posições α e β de compostos carbonílicos.
668
A. Isenmann
Princípios da Síntese Orgânica
Um efeito colateral se vê nesta reação: o subproduto é água oxigenada. Então existe um
método barato de produzir H2O2 a partir de água e ar usando uma hidroquinona como
catalisador. O ponto negativo: o poder oxidante da água oxigenada, liberada por esta
reação, pode prejudicar outros corantes que são mais sensíveis à oxidação (ver próximo
item). Significa que a combinação desses corantes com o corante quinóide não serve como
receita de tintura.
O índigo, corante téxtil azul mais importante do mundo (calça jeans), é tratado desta mesma
maneira, para render tinturas uniformes, duráveis e sem manchas.
O
H
N
Redução:
H
O2 / luz
O
H
N
Na2S2O4 , NaOH
Oxidação:
N
HO
N
H
OH
Leuco-índigo
amarelo-pálido
solúvel
Índigo
azul
insolúvel
Oxidação de azobenzeno para azoxibenzeno
Já a importante classe dos corantes azo (cuja substância-mãe é o azobenzeno, ver p. 855)
reage com a água oxigenada, formando uma forma polar zwitteriônica, do tipo
azoxibenzeno. Este produto, além de perder a coloração típica que o grupo Ar-N=N-Ar´
proporcionou, é bem mais solúvel em água do que a molécula de partida. Daí, um tecido
pode ser descolorido com bastante facilidade - um efeito indesejado, na maioria dos casos.
O
N N
+ H2O2
[ AcOH glacial ]
N N
- H2O
Azobenzeno
Azoxibenzeno
O azoxibenzeno também é acessível por redução, a partir de nitrobenzeno (ver p. 616).
9.13 Oxidação de compostos com enxofre
1. A oxidação de mercaptanos, R-SH, geralmente é muito fácil. Oxidantes suaves e
seletivos atendem esta finalidade.
Exemplo 1:
669
A. Isenmann
Princípios da Síntese Orgânica
Com I2/NaOH se consegue o dissulfeto. A unidade estrutural –S-S- é bastante comum na
química orgânica: enzimas são fixadas na sua conformação, tanto quanto polímeros
sintéticos.
2 R
SH
+
I2
+
2 NaOH
R
S
S
R
+
2 NaI
+
2 H2 O
A vulcanização de látex 398 se baseia neste princípio estrutural, pontes de dissulfeto.
Somente por estes pontes se obtém um material com qualidades elastoméricas ("borracha").
Porém, o acesso nestes pontes é diferente do descrito acima - na verdade a descoberta desta
"borracha natural" foi, como tantas reações orgânicas, um produto de coincidência: C.
Goodyear (1838) simplesmente aqueceu o látex do poliisopreno junto com enxofre
elementar.
Exemplo 2:
Com O2 ou DMSO (dimetilsulfóxido) também é possível oxidar até o dissulfeto:
C2H5 SH + HS
C2H5 S
C2H5 + 1/2 O2
S
C2H5
+
H 2O
outros produtos de oxidação
Aplicando um agente oxidante mais forte, por outro lado, a oxidação prossegue até o ácido
sulfônico ou o sulfato:
R
SH
+
HNO3 conc.
R
SO3H
Oxidação de tioéteres
A partir de tioéteres, com a calculada quantidade de H2O2 ou com HNO3 diluído, se
conseguem sintetizar os sulfóxidos. Ao aplicar um excesso de agente oxidante, HNO3
concentrado ou KMnO4 se obtém facilmente a sulfona.
R
S
R
[O]
O
R
S
R
Sulfóxido
[O]
O
R
S
R
O
Sulfona
Este caminho é o mais viável para obter sulfóxidos e sulfonas, já que mercaptanos e
tioéteres são facilmente acessíveis, via substituição nucleofílica (uma variação da síntese de
Williamson, ver p. 136).
398
O látex da borracha natural, é obtido da árvore seringueira na Amazônia, em forma de líquido leitoso.
Após a separação da fase aquosa se obtém um sólido marrom e pegajoso, com pouca estabilidade
dimensional. É o poli-cis-isopreno, então um polímero de cadeias lineares sem ligações entre si, mas cada
uma com muitas duplas ligações C=C isoladas e, logico, muitas posições alílicas. As cadeias de poli-cisisopreno são prestes a serem conectadas via processos radicalares (a fragilização por exposição ao ar/luz é um
deles) ou, tecnicamente mais interessante, por processos de vulcanização.
670
A. Isenmann
Princípios da Síntese Orgânica
9.14 Auto-oxidação de compostos organometálicos
Os compostos R-M têm uma ligação bastante polarizada que justifica em alguns casos uma
anotação iônica, R-M+ (ver cap. 10.1.1). O carbono nestes compostos carbanóides é
oxidado com muita facilidade - realmente cada reação do tipo Grignard é uma oxidação
deste carbono já que sua densidade eletrônica está reduzida ao ligar-se a um carbono
positivado.
Quase todos os compostos organometálicos reagem com oxigênio atmosférico. Por isso, as
sínteses envolvendo etapas com organometálicos devem ser conduzidas sob atmosfera
inerte.
Na etapa da oxidação se forma um sal do hidroperóxido o qual somente pode ser
aproveitado quando a temperatura for mantida permanentemente abaixo de –70 °C e o
produto for refinado hidroliticamente. A temperaturas mais altas o hidroperóxido
comproporciona com mais um substrato organometálico, fornecendo finalmente o álcool.
3
Passo 1:
R MgX +
O2
Passo 2:
R O O- +MgX
Inserção
+
R O O- +MgX
R MgX
> 0 °C
-70°C
2 RO- +MgX
R O OH
+ 2 H2O
2 R OH
- 2 MgX(OH)
Rendimentos razoáveis são obtidos, somente ao conduzir a síntese de maneira bem
controlada.
Uma observação no mecanismo da auto-oxidação:
Uma reação direta tipo sincronizada com oxigênio tripleto, 3O2 (que é o estado fundamental
desta molécula, ver p. 79), é impedida pelo princípio quântico da manutenção dos spins
dentro do sistema. Conforme descrito no gráfico abaixo, a reação se processa em vez desta,
através de radicais e em várias etapas. O processo é então uma típica SET, com iniciação,
propagação para outra molécula, e término.
R-
+
O2
R O O-
SET
radicais
livres
R
+
OO-
9.15 Exercícios de Redução e Oxidação de Compostos Orgânicos
1) Indique o número de oxidação em todos os carbonos:
(p. 582)
671
A. Isenmann
Princípios da Síntese Orgânica
CH3
H3C
C
CH2 CH3
O C N
N C O
CH3
H3C
O
S
C
C
CH2
CN
H3CO
OCH3
MgBr
O
Li
O
OH
O
H3C NO2
2) Avalie se nas seguintes reações se trata de uma oxidação, redução, ambos ao mesmo
tempo (comproporcionamento/disproporcionamento) ou nenhum dos dois casos. Refira-se
sempre à fórmula em negrito.
R
COOC2H5
C
R´
+
H2N
COOC2H5
H2N
C
OH
HO
+ O
C
R
C O
R´
C O
+
2 C2H5OH
CO NH
O
C
Se
+ Se + H2O
C
OH
H
CO NH
C
O
OH
O
+ H 2O 2
+ O2
OH
O
O
C N
C N
+ H C
2
H3C C
CH3
CH3
+ CO2
H H
OH
H 2C
C
CH3
+
CH3
CH3
H3C CH
CH3
672
A. Isenmann
Princípios da Síntese Orgânica
3) Elabore os números de oxidação para todos os átomos aparentes na redução de Stephen.
(p. 615)
4) a) Formule a oxidação de Kharash-Sosnovsky, do 2-penteno.
(pp. 76 e 633)
b) Quais são os isômeros formados nesta oxidação?
c) Na p. 635 foram apresentados os isômeros da oxidação alílica, do 4-metilexeno. Procure
os pares enanciômeros. Quais são diastereoisômeros?
5) O formalismo de equilibrar uma reação redox, a base dos NOX de reagentes e produtos,
conforme introduzido na p. 644, pode ser treinado nos seguintes sistemas. Os reagentes de
cada item sejam usados, para transformar um álcool primário, em ácido carboxílico.
a) KMnO4/OH-. Dica: sob condições básicas o estado mais estável do manganês é +4,
enquanto em ambiente ácido é +2.
b) MnO2/H+
c) K2Cr2O7/H+
6)a) Formule a reação bromofórmio (p. 659), indicando reagentes e produto final, a partir
de
OH
CH3
b) Formule as etapas importantes do mecanismo.
7) Formule o mecanismo da redução de Eschweiler-Clarke, de uma amina primária. (p.
606)
673
A. Isenmann
Princípios da Síntese Orgânica
9.16 Respostas aos exercícios de Redução e Oxidação de
Compostos Orgânicos
1) Elaborar NOX:
-3
CH3
+1 -3
0
H3C
C
CH2 CH3
-3
-1
-3
+4
-2
-3
-2 +1
+1
O C N
CH3
-1
-2
-2
O
+1 -3
H 3C
S
+3 -3
+2
C
-1
-2
CH2
+1 -2 -2
CN
H3CO
+4
C
OCH3
-1
-1
-1
+2 -1
+1
-3
Li
-2
-2
O
-3
-2
MgBr
-1
-1
-3
N C O
O
+2
-3
-2 +1
-2
OH
O
+1
+2
+1 -2
+3-2
H3C NO2
-3
-1
-2
2) Indicar tipo de reação:
R
C
R´
COOC2H5
+
COOC2H5
H2N
R
C O
CO NH
C
H2N
R´
C O
+
2 C2H5OH
CO NH
Condensação
nem oxidação, nem redução
HO
OH
C
+ O
C
H
Se
OH
O
C
+ Se + H2O
C
O
Oxidação do enol para 1,2-dicetona
674
A. Isenmann
Princípios da Síntese Orgânica
OH
O
+ H 2O 2
+ O2
Oxidação da hidroquinona para quinona
OH
O
O
C N
C N
+ H C
H H
OH
H 3C
C
CH3
CH3
+ H 3C
+ CO2
Redução de Leukert-Wallach
CH3
C
H 2C
CH3
C
CH3
+
CH3
CH3
H3C CH
CH3
Desproporcionamento de radicais
(Oxidação e redução da mesma espécie,
ao mesmo tempo).
3)
+3 -3
R CN
-1
+1+1
+ HCl
+3
Cl
+2 -1
+1 -1
SnCl2 + HCl
R C -3 +1
NH
+4
+1+1
-3 +1
R CH NH
+1 -2
+ H2O
-3 +1
+1+1
-2
R CH O
- NH3
- SnCl4
4 a)
OBz
K. - S.
+
OBz
Saponificação
OH
+
OH
b)
OH
K. - S.
+
OH
675
A. Isenmann
Princípios da Síntese Orgânica
c)
Y
Enanciômeros são:
1 e 3; 2 e 4;
os outros são diastereoisômeros
Y
1
2
Y
Y
3
4
Y
Y
5
6
Y
Y
7
8
5) a) 4 MnO4- + 3 R-CH2-OH → 4 MnO2 + 3 R-COO- + 3 OH- + 2 H2O
b) 2 MnO2 + R-CH2-OH + 4 H+ → 2 Mn2+ + R-COOH + 3 H2O
c) 2 Cr2O72- + 3 R-CH2-OH + 16 H+ → 4 Cr3+ + 3 R-COOH + 11 H2O
6)a) A reação bromofórmio serve para oxidar metilcetonas, usando bromo em ambiente
alcalino. Resulta a 1,1,1-tribromocetona que pode liberar a molécula de bromofórmio.
Assim, o esqueleto carbônico do substrato é picado, quer dizer, perde um carbono. Fica
atrás um ácido carboxílico, C(n-1). Em nosso exemplo:
COOH
b)
676
A. Isenmann
O-
[OH-]
O
H3 C
Princípios da Síntese Orgânica
C
H2C
R
Br2
(eletrófilo)
O
C
H 2C
R
C
R
O
Br
H2 C
C
R
Enolato
(nucleófilo)
Têm-se mais duas etapas destas, levando à
O
Br3C
C
O-
+ OH-
R
Br3C
C
Br3C- + HOOC
R
ânion
bromofórmio
OH
1,1,1-tribromocetona
Br3CH + -OOC R
Carboxilato
Bromofórmio
R
7) Em analogia ao mecanismo discutido na p. 606, as etapas da formação da imina e sua
redução pelo ácido fórmico se repetem. É claro que os reagentes, formalina e ácido
fórmico, devem ser usados em grande excesso para que essas etapas se completem.
H
H
O
H
1)
R NH2
CH2
2)
N
R
H
H
R
O
H
N
O
H
+H
+
H
R
O
N
O
R
- CO2
R
H
N
N
R
Imina
N
H
H
O
1´)
- H2O
- H+
H
CH3
H
H
CH2
H
O
H
CH3
R
N
H
O
+
+H
CH3
R
N
CH2
H
CH3
- H2O
N
R
CH3
Íon imônio
CH2
2´)
N
R
H
CH3 H
O
O
CH3
- CO2
R
N H
CH3
- H+
R N(CH3)2
Na mistura reacional o produto se encontra em forma do sal de amônio. Logo após o
isolamento deste sal um dosamento adequado de base libera o produto final - uma amina
terciária.
677
A. Isenmann
Princípios da Síntese Orgânica
678
Download