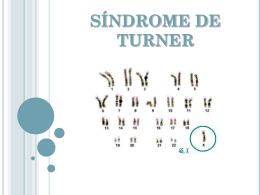
Abordagem Clínica, Epidemiológica e Laboratorial das Anomalias Congênitas Marcial Francis Galera DOENÇAS Sarampo, Caxumba, Rubéola, Varicela Causas ambientais Luxação Congênita do Quadril, Anencefalia Fibrose Cística, Hemofilia A, Acondroplasia, Distrofia Muscular Causas genéticas ambiente X genótipo Saúde D. ambiental D. genética D. multifatorial DOENÇAS GENÉTICAS DISTÚRBIOS CROMOSSÔMICOS; DISTÚRBIOS MONOGÊNICOS; DISTÚRBIOS MULTIFATORIAIS; DISTÚRBIOS MITOCONDRIAIS. Cariótipo Feminino Normal (46,XX) DOENÇAS GENÉTICAS ALTERAÇÕES CROMOSSÔMICAS Numéricas Número total de cromossomos diferente de 46. Estruturais Alteração na estrutura de algum cromossomo. DOENÇAS GENÉTICAS ABERRAÇÕES CROMOSSÔMICAS Numéricas Síndrome de Down Cariótipo: 47,XX,+21 / 47,XY,+21 DOENÇAS GENÉTICAS ABERRAÇÕES CROMOSSÔMICAS Numéricas Síndrome de Turner Cariótipo: 45,X DOENÇAS GENÉTICAS ABERRAÇÕES CROMOSSÔMICAS Estruturais - Síndrome do miado do gato (5p-) Cariótipo: 46,XX,del(5p) DOENÇAS GENÉTICAS ABERRAÇÕES CROMOSSÔMICAS Estruturais - Síndrome de Wolf-Hirschhorn (4p-) Cariótipo: 46,XX,del(4p) DOENÇAS GENÉTICAS DOENÇAS MONOGÊNICAS Autossômicas Dominantes Acondroplasia Sd. Treacher Collins DOENÇAS GENÉTICAS DOENÇAS MONOGÊNICAS Autossômicas Dominantes Neurofibromatose tipo I DOENÇAS GENÉTICAS DOENÇAS MONOGÊNICAS Autossômicas Dominantes Síndrome de Waardenburg DOENÇAS GENÉTICAS DOENÇAS MONOGÊNICAS Autossômicas Dominantes DOENÇAS GENÉTICAS DOENÇAS MONOGÊNICAS Autossômicas Recessivas Síndrome de Hurler Anemia Falciforme DOENÇAS GENÉTICAS DOENÇAS MONOGÊNICAS Autossômicas Recessivas DOENÇAS GENÉTICAS DOENÇAS MONOGÊNICAS Autossômicas Recessivas DOENÇAS GENÉTICAS DOENÇAS MONOGÊNICAS Ligadas ao X Dominante Raquitismo resistente à Vitamina D DOENÇAS GENÉTICAS DOENÇAS MONOGÊNICAS Ligadas ao X Dominante DOENÇAS GENÉTICAS DOENÇAS MONOGÊNICAS Ligadas ao X Recessiva Distrofia Muscular de Duchenne DOENÇAS GENÉTICAS DOENÇAS MONOGÊNICAS Ligadas ao X Recessiva DOENÇAS GENÉTICAS DOENÇAS MITOCONDRIAIS Oftalmopatia de Leber DOENÇAS GENÉTICAS DOENÇAS MULTIFATORIAIS Fatores ambientais e genéticos Defeito de Fechamento do Tubo Neural; Cardiopatias Congênitas; Estenose Hipertrófica do Piloro. DOENÇAS GENÉTICAS DOENÇAS MULTIFATORIAIS Defeito de Fechamento do Tubo Neural Por que estudar doenças genéticas / anomalias congênitas? Declínio da importância dos fatores ambientais (agentes infecciosos) Repercussão importante nas taxas de mortalidade infantil (< 1 ano) Maior concentração dos óbitos no período perinatal MORBIDADE • A.C. – 1 em cada 3 principais diagnósticos • 37% das admissões hospitalares pediátricas • Mortalidade hospitalar – 9,8% (dobro das outras causas) • Maior permanência hospitalar – 5,7 dias (M: 4,5) • Maior custo de internação Anomalias Genéticas / Congênitas X Hospitais Pediátricos Autor ano país % sobre total de internações Clow 1973 Canadá 33 Hall 1978 U.S.A 40 Day 1973 U.S.A 49 Barreiro 1976 Argentina 24 Carnevale 1985 México 25 Penchaszadech 1979 Venezuela 10 Pinto 1994 Brasil 23 MORTALIDADE INFANTIL ANO A.C. CAUSA 1980 1990 2000 5% 8% 13% 5ª 4ª 2ª DATASUS DEFEITOS CONGÊNITOS EPIDEMIOLOGIA Mortalidade infantil atribuída aos defeitos congênitos – 20% Causas das anomalias congênitas Anomalias cromossômicas: •6 a 7% dos zigotos; •50% dos abortos; •5% dos natimortos; •0,5 a 0,6% dos nascidos vivos. INCIDÊNCIAS DE PATOLOGIAS GENÉTICAS EM RECÉM-NASCIDOS VIVOS. Anomalias Cromossômicas 0,50 % Gene Autossômico Dominante 0,70 % Gene Autossômico Recessivo 0,25 % Gene Ligado ao Sexo (X) 0,07 % Herança Multifatorial 2,00 % EPIDEMIOLOGIA DAS ANOMALIAS CONGÊNITAS • É o estudo da distribuição e determinantes de doença entre populações; • Estudo das manifestações da doença e suas associações. ECLAMC (ESTUDO COLABORATIVO LATINOAMERICANO DE MALFORMAÇÕES CONGÊNITAS) Programa de investigação clínica e epidemiológica das anomalias do desenvolvimento que opera com nascimentos hospitalares em países latino-americanos. • Início – 01 de julho de 1967. • Programado como investigação dos fatores de risco na causalidade das malformações, com metodologia casocontrole. ECLAMC (1987) INCIDÊNCIA X VIGILÂNCIA 2,44% de MF em RN vivos 3,79% de MF em NATIMORTOS NO BRASIL • Política visando o atendimento às anomalias congênitas é urgente por sua crescente importância. • OMS – modificações nos serviços de saúde necessários ao atendimento da população, desviando o foco de doenças agudas para o manejo dos problemas crônicos. AÇÕES GOVERNAMENTAIS • Programa Nacional de Imunização. • Fortificação das farinhas com ácido fólico. • Campo 34 da DNV – permite que sejam registradas, de forma sistemática, as anomalias congênitas ao nascimento. CAMPO 34 CAMPO 34 QUAL A MAGNITUDE DO PROBLEMA?? Uma vez conseguido isso, o próximo passo será estudar as medidas de prevenção. PREVENÇÃO Primária - pré-concepcional e evita a ocorrência do defeito congênito; Secundária - pré-natal e evita o nascimento de um feto com defeito congênito; Terciária - pós-natal e evita as complicações dos defeitos congênitos melhorando suas possibilidades de sobrevida assim como sua qualidade de vida. OUTRO ENTRAVE: Implantação da Política de Genética Médica no SUS ainda não foi implementado! OBJETIVO DE UMA AVALIAÇÃO GENÉTICO-CLÍNICA Determinação do diagnóstico e conseqüente definição do prognóstico; Apoio aos pais e familiares; Estabelecer o prognóstico familial (Aconselhamento Genético). SUSPEITAR DIAGNÓSTICO DA DOENÇA GENÉTICA ACONSELHAMENTO GENÉTICO Doença Genética QUANDO SUSPEITAR ? HISTÓRIA FAMILIAR E PRÉ-NATAL Consangüinidade; Outros afetados, por doença genética, na família; Fatores de risco maternos (exposição a teratógenos, patologias crônicas) Doença Genética QUANDO SUSPEITAR ? HISTÓRIA FAMILIAR E PRÉ-NATAL Atividade fetal; Apresentação e tamanho do feto; Alteração no volume do líquido amniótico. QUANDO SUSPEITAR ? DADOS DE NASCIMENTO Condições gerais; Boletim de Apgar; Anormalidades de tamanho; Anormalidades do tônus; Sinais/sintomas neurológicos metabólicos inexplicados; Presença de anomalias congênitas. e/ou QUANDO SUSPEITAR ? EXAME FÍSICO Geral Morfológico (antropometria e descrição detalhada do fenótipo). ANOMALIAS CONGÊNITAS ANOMALIAS CONGÊNITAS Alterações estruturais ou metabólicas presentes ao nascimento que podem ser geneticamente determinadas ou resultado da influência ambiental durante a vida fetal ou embrionária (Apgar & Stickle, 1986). 3 - 5% dos recém-nascidos apresentam pelo menos uma anomalia menor presente ao nascimento. ANOMALIAS CONGÊNITAS MENORES MAIORES ANOMALIAS MENORES São alterações estruturais freqüentes que não produzem alterações significativas na saúde ou comprometimento social. ANOMALIAS MENORES Prega palmar transversal única Clinodactilia Causa desconhecida – 50 a 60% Anomalias menores Presentes em 14% dos recém-nascidos 90% dos recém-nascidos com três ou mais anomalias menores - têm anomalia maior. ANOMALIAS MAIORES Têm conseqüências médicas e sociais; Requerem atenção médica; Geralmente deixam seqüelas. ANOMALIAS MAIORES Fenda labial Hemangioma Atresia de duodeno e esôfago METODOLOGIA História familiar Exame genético-clínico Exame físico geral; Descrição detalhada do fenótipo; Exame antropométrico; Análise dos padrões dermo-papilares; Documentação fotográfica. Exames complementares Criança com Dismorfias múltiplas Criança com Dismorfias múltiplas Obter anamnese Criança com Dismorfias múltiplas Obter anamnese Realizar exame morfológico Criança com Dismorfias múltiplas Obter anamnese Realizar exame morfológico Reconhece uma síndrome? Criança com Dismorfias múltiplas Obter anamnese Realizar exame morfológico Reconhece uma síndrome? SIM Criança com Dismorfias múltiplas Obter anamnese Realizar exame morfológico Reconhece uma síndrome? SIM Confirmação laboratorial, quando possível Criança com Dismorfias múltiplas Obter anamnese Realizar exame morfológico Reconhece uma síndrome? SIM Confirmação laboratorial, quando possível ORIENTAÇÃO, PROGNÓSTICO E ACONSELHAMENTO GENÉTICO Criança com Dismorfias múltiplas Obter anamnese Realizar exame morfológico Reconhece uma síndrome? NÃO Criança com Dismorfias múltiplas Obter anamnese Realizar exame morfológico Reconhece uma síndrome? NÃO Escolher sinais maiores (ganchos) Criança com Dismorfias múltiplas Obter anamnese Realizar exame morfológico Reconhece uma síndrome? NÃO Escolher sinais maiores (ganchos) Pesquisa na literatura Criança com Dismorfias múltiplas Obter anamnese Realizar exame morfológico Reconhece uma síndrome? Escolher sinais maiores (ganchos) Pesquisa na literatura NÃO Reconhece uma síndrome? Criança com Dismorfias múltiplas Obter anamnese Realizar exame morfológico Reconhece uma síndrome? SIM Escolher sinais maiores (ganchos) Pesquisa na literatura NÃO Reconhece uma síndrome? Confirmação laboratorial, quando possível Criança com Dismorfias múltiplas Obter anamnese Escolher sinais maiores (ganchos) Realizar exame morfológico Reconhece uma síndrome? SIM Pesquisa na literatura NÃO Reconhece uma síndrome? Confirmação laboratorial, quando possível ORIENTAÇÃO, PROGNÓSTICO E ACONSELHAMENTO GENÉTICO Criança com Dismorfias múltiplas Obter anamnese Realizar exame morfológico Reconhece uma síndrome? Escolher sinais maiores (ganchos) Pesquisa na literatura NÃO Reconhece uma síndrome? Criança com Dismorfias múltiplas Obter anamnese Escolher sinais maiores (ganchos) Realizar exame morfológico Reconhece uma síndrome? SIM Pesquisa na literatura NÃO Reconhece uma síndrome? Confirmação laboratorial, quando possível ORIENTAÇÃO, PROGNÓSTICO E ACONSELHAMENTO GENÉTICO METODOLOGIA PESQUISA NA LITERATURA Livros Artigos científicos Atlas de síndromes Site: www.ncbi.nlm.nih.gov/entrez/query.fcgi?db= OMIM Programas: LDDB LNDB POSSUM SIMULAÇÃO SIMULAÇÃO Luxação do Cristalino SIMULAÇÃO Luxação do Cristalino Aracnodactilia SIMULAÇÃO Luxação do Cristalino Aracnodactilia Escoliose SIMULAÇÃO Luxação do Cristalino Aracnodactilia Escoliose Dilatação da Aorta LISTA DE SINAIS •Luxação do Cristalino; •Aracnodactilia; •Escoliose; •Dilatação da aorta. PROGRAMA LDDB METODOLOGIA Exames complementares Métodos de diagnóstico por imagem: Radiografia simples; Tomografia Computadorizada; Ressonância Magnética; Ultra-som. METODOLOGIA Exames complementares Laboratoriais: Hematologia; Bioquímica; Citogenética Clássica e Molecular; Biologia Molecular. ANÁLISE CROMOSSÔMICA C A R I Ó T I P O MATERIAL: •LINFÓCITOS •MEDULA ÓSSEA •FIBROBLASTOS DE PELE •VILOSIDADES CORIÔNICAS •LÍQUIDO AMNIÓTICO •TECIDO TUMORAL •RESTOS OVULARES MÉTODOS DE COLORAÇÃO: •COLORAÇÃO CONVENCIONAL •BANDAS G, Q, R. •FISH CROMOSSÔMOS HUMANOS BANDAS Cariótipo Masculino Normal (46,XY) ESTUDOS BIOQUÍMICOS •TESTES QUALITATIVOS DE URINA; •CROMATOGRAFIA DE AMINOÁCIDOS; •CROMATOGRAFIA DE OLIGOSSACARÍDEOS; •CROMATOGRAFIA DE MUCOPOLISSACÁRIDES; •DOSAGENS PLASMÁTICAS; •ENSAIOS ENZIMÁTICOS NO PLASMA; •ENSAIOS ENZIMÁTICOS EM LEUCÓCITOS. ESTRATÉGIAS DE ANÁLISE MOLECULAR Estratégia por análise direta: Gene seqüenciado e mutações restritas. Exemplo: Acondroplasia (FGFR3) Estratégia por análise indireta: Gene mapeado e não seqüenciado / gene muito extenso / sem mutações prevalentes. Exemplo: Doença policística renal tipo adulto Reação em Cadeia da Polimerase (PCR) Aplicação: •Análise qualitativa para deteção de DNA/RNA de vírus, bactérias, protozoários; •Detecção de fragmentos cromossômicos (Y); •Detecção de rearranjo gênico (LMC, abl/bcr; 9/22) •Detecção de mutações de ponto (AF - GAG - GTG) •Detecção de deleções (FC - Delta F508) •Determinação de vínculo biológico DOENÇAS INFECCIOSAS •CMV •Clamídia •EBV •Hepatite B, C •Herpes 1 e 2 •HIV •Listeriose •HPV •Parvovirose •Rubéola •Lues •Toxoplasmose •Varicela DOENÇAS GENÉTICAS •Cranioestenoses •Fibrose Cística •Displasias Esqueléticas •Anemia Falciforme •Fenilcetonúria •Acondroplasia •Síndrome do X Frágil •Distrofia muscular •Hemofilia A •EIMs •Neurofibromatose I •Coréia de Huntington •HAC •Osteogênese Imperfeita AVALIAÇÃO EM GENÉTICA CLÍNICA Determinação do diagnóstico e conseqüente definição do prognóstico; Apoio aos pais e familiares; Estabelecer o prognóstico familial (Aconselhamento Genético). OBRIGADO [email protected]
Baixar