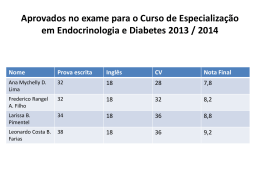
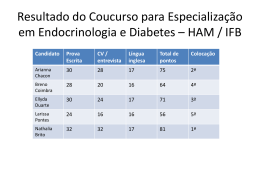
CONSERVAÇÃO DA ESTATURA FINAL EM PACIENTE COM CRANIOFARINGIOMA DE GRANDES PROPORÇÕES E PANHIPOPITUITARISMO Gondim, F; Saldanha, L; Poncell, M; Bandeira, E e Bandeira, F. Unidade de Endocrinologia e Diabetes - Hospital Agamenon Magalhães – MS / SES / UPE, Recife - PE. INTRODUÇÃO Craniofaringiomas são tumores supra-selares benignos que surgem a partir do endotélio remanescente da bolsa de Rathke com incidência de 0,5 a 2 casos por milhão de pessoas. Esta neoplasia está associada a uma significante disfunção pituitária havendo 88% de probabilidade cumulativa de deficiência de GH em 10 anos. Tal redução acarreta importante prejuízo na obtenção da altura final nesses pacientes. Apresentaremos o caso de uma paciente que apesar da deficiência de GH, conseguiu atingir o alvo esperado para a estatura familiar. RELATO DE CASO ICFS, sexo feminino, 21 anos, com diagnóstico de Craniofaringioma aos 7 anos de idade após apresentar quadro de hipotiroidismo associado a Diabetes Insípidus. Foi submetida a 3 cirurgias para ressecção tumoral aos 9, 12 e 17 anos e radioterapia aos 16 anos. Em seu internamento na enfermaria de endocrinologia, exame físico evidenciava infantilismo sexual (Estadio Puberal:M1P1) e sua estatura de 1,55m já havia atingido alvo familiar(1,56m). Exames laboratoriais demonstraram pan-hipopituitarismo (T4Livre: 0,73ng/dl; Estradiol: 27,8pg/ml; LH: 0,13miU/ml; FSH< 0,1miU/ml; PRL: 0,861ng/ml; Cortisol basal<1,00ug/dl). Dosagem sérica de GH pós estímulo com glucagon demostrou-se suprimida(<0,02mcg/l). Densitometria óssea(DXA) constatou osteoporose (T-score de -3,6 em coluna lombar) e RNM demonstrou lesão de 3,5x3,0x2,5cm com extensão até assoalho do 3° Ventrículo. Foi iniciada reposição hormonal estrogênica e a paciente recebeu alta para acompanhamento ambulatorial. CONCLUSÃO Este caso demonstra uma apresentação incomum de pan-hipopituitarismo na infância e adolescência
Baixar