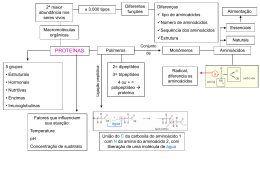
Introdução Moléculas de proteínas se dobram formando estruturas tridimensionais específicas A função de uma proteína está diretamente ligada à sua estrutura 3D Como resultado, há um grande esforço, tanto experimental como computacional, em determinar as estruturas de uma proteína Protein Folding Introdução A estrutura de uma proteína pode ser determinada experimentalmente por: Cristolografia de raios x NMR (nuclear magnetic resonance) spectroscopy Esses métodos porém, nem sempre podem ser aplicados: Cristolografia é limitada pela dificuldade de fazer algumas proteínas formarem cristais NMR só pode ser aplicado em moléculas de proteínas relativamente pequenas Introdução Além disso, apesar de décadas de trabalho, o problema da predição da estrutura 3D de uma proteína, dada sua sequência de aminoácidos, ainda continua não resolvido Métodos computacionais no entanto podem fornecer uma boa previsão e são amplamente utilizados Carbono α Grupo Amina Side Chain Grupo Carboxila Aminoácidos Há 20 side chains diferentes especificados pelo código genético, cada um com diferentes átomos e propriedades químicas: (hidrofóbico, polar, positively charged, etc) É devido a essas diferenças nas propriedades que existem uma enorme variedade de ‘foldings’ de proteínas na natureza Aminoácidos Várias forças atuam provocando o folding da proteína. Uma dessas forças é o efeito hidrofóbico, que acaba fazendo com que proteínas solúveis em agua formem um núcleo hidrofóbico No entanto o backbone dessas proteínas são altamente polares, o que é indesejado nesse ambiente do núcleo hidrofóbico Aminoácidos Para neutralizar esse grupos polares, são formados várias ligações de hidrogênio entre os átomos do backbone Estrutura secundária são essas estruturas formadas devidos a essas ligações de hidrogênio alpha-helix, beta-sheets, etc... Alpha Helix Formado através de sequências contínuas de aminoácidos, através de ligações de hidrogênio entre átomos nas posições i e i+4 Tamanho pode variar, de 4 a até centenas de aminoácidos Beta Strands Beta Sheet Beta Strands interagem com outros Beta Strands através de pontos de hidrogênios, formando um Beta Sheet Em sheets paralelos, os Strands correm na mesma direção. Em antiparalelos correm em direções contrárias. Há também sheets mistos ... A sequencia de aminoácidos Ligacões de Hidrogênio nas grupos amina e carboxila dos aminoácidos formam estruturas secundárias Estruturas Super Secundarias são formadas por combinações de estruturas secundarias Estruturas Terciarias são formadas por Estruturas Secundarias e SuperSecundarias combinadas e definem o dobramento em 3 dimensões da proteína Estruturas Quaternarias definem o arranjo espacial de mais de uma proteína numa cadeia de proteínas Chou-Fasman Method [2] Uma das primeiras abordagens para predição de estruturas secundárias Taxa de acerto de 50% a 60% dependendo da proteína Usa uma combinação de regras estatísticas e heurísticas Conjunto de Sequências de Proteínas com estruturas secundárias já conhecidas (através de cristolografia de raio X) Calcula a frequência com que cada aminoácido aparece em um tipo particular de estrutura secundária, utilizando o conjunto de sequências com estruturas já conhecidas Idéia: Diferentes aminoácidos ocorrem preferencialmente em diferentes elementos de estruturas secundárias Atribui 3 parâmetros para cada aminoácido, baseado nas frequências observadas P(a): Tendência de formar um alpha helix P(b): Tendência de formar um beta sheet P(turn): Tendência de formar um beta turn ... Além disso, atribui 4 parâmetrs baseado na frequência em que foram observados na 1ª, 2ª, 3ª ou 4ª posições de um beta turn 1. Algoritmo recebe a entrada (sequência de aminoácidos) 2. Varre essa sequência em busca de subsequências (núcleos) com alta concentração de aminoácidos com tendência a formar helix ou sheet 3. Verifica através de heurísticas se essas regiões podem ser classificadas em alpha-helix ou betasheets [2] [3] Chau-Fasman Method Há regras para classificar a subsequência em beta-sheets ou betaturns também Predições conflitantes também são resolvidas através de heurísticas Exemplo: Exemplo: A R E Q T S P C P(a) 142 98 151 111 83 77 57 70 P(b) 83 93 37 110 119 75 55 119 4 de 6 aminoácidos com P(a) > 100 ... T S P C E Q A R E Q A Q R T S P C ... Total P(a) = 1115 Total P(b) = 756 Maior, logo prediz região como alpha-helix GOR Method Idéia: Experimentos mostram que cada aminoácido tem um efeito significante na estrutura de aminoácidos em posições até 8 a frente ou atrás dele Similar ao método de Chau-Fasman, porém ao invés de considerar apenas a tendência de um determinado aminoácido formar uma certa estrutura secundária... Ele também considera a probabilidade condicional desse aminoácido formar essa estrutura dado que seus vizinhos já o fizeram GOR METHOD 25 proteínas com estruturas conhecidas foram analisadas, e a frequência com que cada aminoácido foi encontrado em um helix, sheet, turn or coil dentro de uma janela de 17 posições foi determinada Criando uma matriz 17 * 20 usada para calcular a estrutura mais provável para cada aminoácido dentro da janela de 17 posições A janela percorre a sequência primária, calculando a estrutura mais provável para cada aminoácido, baseado nos aminoácidos vizinhos Taxa de acerto de aproximadamente 65% Dependências Locais As técnicas vistas até agora prediziam estruturas secundárias examinando apenas cada aminoácido individualmente Abordagens posteriores passaram a considerar interações de alta ordem entre os resíduos das seqüências, melhorando a taxa de acerto. Dependências Locais Uma forma de fazer isso é uma extensão do GOR que leva em conta o tipo dos resíduos vizinhos na janela Outras técnicas incluem métodos de aprendizagem de máquina como: Nearest-Neighbor Neural Networks K-Nearest Neighbors Ponto preto está sendo classificado K=9 Dos 9 vizinhos mais próximos, 6 são da classe azul e 3 da vermelha O classificador irá então prever a classe do ponto preto como azul Nearest Neighbors aplicado a predição de estruturas Secundárias Predizer a estrutura secundária de um resíduo considerando uma janela de resíduos ao redor dele, e encontrando alinhamentos similares nas sequências com estruturas conhecidas Idéia: Pequenas sequências de aminoácidos muito similares entre si possuem estruturas secundárias similares, mesmo que estejam não homólogas Redes Neurais Tenta predizer a estrutura de um resíduo considerando os resíduos rj-8, ... , rj, ... , rj+8 Cada resíduo é representado por 21 bits (1 bit pra cada tipo de aminoácido +1 bit extra). Portando 17x21 bits de entrada Treinamento: Se estrutura é helix, output = 1 p/ helix e 0 p/ sheet Nova Sequência: Classifica como helix quando 4 ou mais resíduos onde o output helix é maior que tanto o output sheet e um certo threshold Explorando informação evolucionária Fato: A estrutura de uma proteína é mais conservada que a sequência da proteína. Se duas proteínas compartilham mais que 30% da sequência então provavelmente possuem estruturas similares Idéia: Quando predizendo a estrutura secundária de uma proteína em particular, predições das proteínas homólogas podem ser úteis Métodos de previsão tem alcançado melhores resultados usando proteínas homológas também como entradas Tight Turns Estruturas secundarias. Formadas por poucos residuos (no máximo 6) Ligação de dois resíduos formando uma ponte de hidrogênio Distancia entre os Cα dos resíduos que formam a ponte é menor que 7Å Tight Turns Tipos de Tight Turns β-turn os residuos ligados por pontes de hidrogênio são separados por 3 outros residuos γ-turn os residuos ligados por pontes de hidrogênio são separados por 2 outros residuos α-turn os residuos ligados por pontes de hidrogênio são separados por 4 outros residuos π-turn os residuos ligados por pontes de hidrogênio são separados por 5 outros residuos β-turns β-turn os residuos ligados por pontes de hidrogênio são separados por 3 outros residuos Mais comum e mais estudada São tambem classificadas de acordo com os ângulos entre os resíduos r+1 e r+2 Predição de β-turns Os primeiros métodos eram focados em identificar quais resíduos fazem parte de β-turns Métodos mais recentes têm tentado identificar o tipo de β-turn Predição de β-turns Método probabilistico Computa a probabilidade de um amino acido ai estar localizado na j-esima posição da βturn Predição de β-turns (cont) Métodos De Aprendizagem de Máquina Redes neurais Método inicial Janela de 4 resíduos como entrada (20 bits cada) 1 Camada intermediaria 4 Saidas βturn tipo 1 βturn tipo 2 Outro tipo de βturn Não é βturn Método mais recente KNN Várias camadas de redes Começa com uma janela de 9 resíduos, acaba com uma de 4 Utiliza predição de outras estruturas secundarias e SVM tambem podem ser utilizados Predição de outras turns Recentemente, existem tentativas de se predizer γ-turns e α-turns com técnicas similares Como são poucos os resíduos que fazem parte de γ-turns e α-turns, estes métodos obtiveram sucessos limitados. β-hairpins β-hairpins Estruturas super-secundarias muito simples É composta de uma β-turn ligando duas β-strands antiparalelas. Esta turn geralmente contem de 2 a 5 resíduos Predição β-hairpins Métodos de predição começaram a aparecer há pouco tempo Os 2 métodos mais recentes utilizam redes neurais Primeiro Método Identifica sequencias β-strand - β-turn – β-strand Compara com as β-hairpins ja conhecidas 14 Scores são calculados e jogados como entrada em uma rede neural treinada para diferenciar β-hairpins e não βhairpins Predição β-hairpins (cont) Segundo Obtem-se homologos utilizando o PSI-BLAST Duas redes neurais são treinadas Método A primeira rede prediz o primeiro residuo da turn Considera os 4 resíduos anteriores e os 7 posteriores A segunda rede prediz o ultimo residuo da turn Considera os 7 resíduos anteriores e os 4 posteriores Finalmente, os resultados são combinados para predizer se a turn faz parte de um hairpin ou não Coiled Coils Coiled Coils Formados por duas ou mais α-helix ligadas As helices apresentam uma sequência de 7 resíduos que se repetem chamados heptad Os resíduos “a” e “d” são hidrofóbicos, e os resíduos “e” e “g” são hidrofílicos A ligação entre as helices se dá pelos resíduos hidrofóbicos. Predição de Coiled Coils Método probabilistico Analisam as frequencias dos resíduos que fazem parte do heptad numa tabela 20x7 Similar ao Chou and Fasman Este método tambem é utilizado para predizer “Leucine Zippers” Predição de Coiled Coils Predição inter-proteínas Coiled coils são formados por duas ou mais α-helix Logo, predizendo as ligações entre α-helix é o método mais intuitivo Porém, as α-helix podem estar em sequencias diferentes São necessarios estudos de predição interproteínas Predição de Coiled Coils Predição de estruturas secundarias melhorou bastante com informações evolucionarias utilizando homologos O proximo passo é utilizar estas informações para predizer quando as α-helix fazem parte de coiled coils Porém, sequencias homologas podem demonstrar interações entre α-helix bem diferentes Os métodos podem β-Barrel Referências Wilkes University: Bioinformatics work 8 lecture (http://course.wilkes.edu/bioinformatics/stories/storyRea der$122) Handbook of Computational Molecular Biology Wikipedia
Baixar