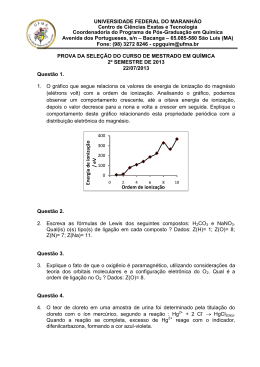
RENATA PINHEIRO BARRETO IMPLEMENTAÇÃO DE METODOLOGIA ANALÍTICA PARA A DETERMINAÇÃO DE HIDROCARBONETOS POLICÍCLICOS AROMÁTICOS NITRADOS (NHPAs) EM FULIGEM DE DIESEL Dissertação apresentada ao Curso de Pós-Graduação em Química da Universidade Federal Fluminense, como requisito parcial para a obtenção do Grau de Mestre. Área de Concentração: Química Analítica Orientador Prof. Dr. Annibal Duarte Pereira Netto Niterói Junho de 2006 B 273 Barreto, Renata Pinheiro Implementação da metodologia analítica para a determinação de hidrocarbonetos policíclicos aromáticos nitrados (NHPAs) em fuligem de diesel/ Renata Pinheiro Barreto. – Niterói: [s. n.], 2006. 112f. Dissertação – (Mestrado em Química) – Universidade Federal Fluminense, 2006. 1. Hidrocarboneto. 2. Hidrocarboneto policíclico aromático nitrado. 3. Cromatografia líquida. 4. Química analítica. 5. Diesel. I. Título. CDD.: 547.01046 ii RENATA PINHEIRO BARRETO IMPLEMENTAÇÃO DE METODOLOGIA ANALÍTICA PARA A DETERMINAÇÃO DE HIDROCARBONETOS POLICÍCLICOS AROMÁTICOS NITRADOS (NHPAs) EM FULIGEM DE DIESEL Dissertação apresentada ao Curso de Pós-Graduação em Química da Universidade Federal Fluminense, como requisito parcial para a obtenção do Grau de Mestre. Área de Concentração: Química Analítica Aprovada em Junho de 2006 Prof. Dr. Annibal Duarte Pereira Netto - orientador Universidade Federal Fluminense Prof. Dr. Ricardo J. Cassela Universidade Federal Fluminense Prof. Dr. Thomas Krauss Fundação Oswaldo Cruz Dr. Celso Blatt Agilent Technologies Niterói 2006 Dedico este trabalho à minha avó, Heloísa Bello Barreto (In Memorian) Pelo Kossen Rufo, NAM MYOHO RENGUÊ KYO iv SUMÁRIO AGRADECIMENTOS vii RESUMO viii ABSTRACT ix LISTA DE FIGURAS x LISTA DE TABELAS xii LISTA DE ABREVIATURAS xiv CAPÍTULO 1: Introdução p.16 CAPÍTULO 2: Fundamentação Teórica p.20 2.1) HPAs e NHPAs: Aspectos Gerais p.20 2.2) Determinação de HPAs e NHPAs p.26 2.3) Cromatografia a Líquido de Alta Eficiência acoplada a espectrometria de massas (CLAE-EM) p.31 2.4) Ionização Química à Pressão Atmosférica (APCI) p.36 2.5) Comparação das Interfaces APCI e ESI p.41 2.6) Espectrometria de massas acoplada à espectrometria de massas (EM-EM) p.44 2.7) Determinação de NHPAs por CLAE-EM-EM p.46 CAPÍTULO 3: Objetivos p.50 3.1) Objetivo Geral p.50 3.2) Objetivos Específicos p.50 CAPÍTULO 4: Materiais & Métodos p.51 4.1) p.51 Solventes v 4.2) Padrões p.52 4.3) Outros Materiais p.52 4.4) Instrumentação p.53 4.5) Limpeza e descontaminação de vidraria p.55 CAPÍTULO 5: Resultados & Discussão p.56 5.1) p.56 Estudos de ionização de NHPAs em interface APCI-EM 5.1.1) Avaliação dos padrões de fragmentação de 1-NPi com detecção por varredura 5.1.2) p.56 Avaliação dos padrões de fragmentação de outros NHPAs com detecção por varredura 5.1.3) p.61 Avaliação da detecção de NHPAs por monitoramento de reações múltiplas (MRM) 5.2) p.65 Otimização do sistema de eluição para determinação de NHPAs por CLAE-EM 5.3) p.66 Comparação dos fatores de resposta de soluções-padrão de 1-NPi preparadas em diferentes solventes, por injeção direta (sem coluna) p.68 5.4) Otimização da vazão do sistema de eluição p.69 5.5) Otimização de parâmetros eletrônicos do espectrômetro de massas para determinação de 1-NPi p.69 5.6) Otimização de parâmetros da interface APCI para determinação de 1-NPi p.71 5.7) Estudos de Extração (I): Avaliação da extração de 1-NPi com detecção por injeção direta p.75 5.7.1) p.76 Extração de SRM 2975 com ultra-som e diclorometano 5.8) Avaliação do efeito do DCM na detecção de 1NPi por CLAE-EM-EM p.78 5.9) Figuras de Mérito do método desenvolvido p.82 5.10) Estudos de Extração (II): Avaliação da extração de 1-NPi por CLAE-EM p.83 5.11) Avaliação da recuperação de 1-NPi em papel de filtro p.87 5.12) Determinação de 1-nitropireno em amostras reais de FD p.88 5.12.1) Análise de FD depositada nos filtros BR1 e BR2 p.88 5.12.2) Extração de Fuligem de Diesel de automóvel p.92 5.13) Avaliação de outros NHPAs em FD p.94 5.13.1) Otimização da separação de 1-nitropireno e outros NHPAs p.94 5.13.2) Avaliação preliminar de outros NHPAs no material certificado SRM 2975 p.96 CAPÍTULO 6: Conclusões p.98 REFERÊNCIAS BIBLIOGRÁFICAS p.100 vi AGRADECIMENTOS À minha mãe, sem a qual eu jamais teria chegado aqui, pelo seu amor e pelo apoio incondicional em todos os momentos de minha vida. À minha família, pelo enorme carinho e pelo exemplo constante de união e força. Aos amigos mais próximos (Raq, Walli, Dri, Suy, Bruno, Tigra, Cau, Fabs e Ti), em especial à Raq, pela força de sempre, amizade verdadeira e por me aturarem em casa durante a execução deste trabalho. Ao Annibal, por tantos anos de orientação e amizade, por sua paciência comigo, por ter me ensinado muita química e principalmente por ter me ensinado a lidar com os números. Aos amigos do laboratório, especialmente ao Daniel e à Soraia (meus fiéis escudeiros), pela ajuda de sempre e paciência comigo. Aos professores da UFF que sempre me ajudaram em diversos momentos do meu trabalho. Ao Budismo, por ter entrado em minha vida. Nam Myoho Renguê Kyo! vii RESUMO Metodologia analítica por Cromatografia a Líquido de Alta Eficiência (CLAE) acoplada a um Espectrômetro de Massas do tipo Ion Trap (EM-IT) e interface de Ionização Química a Pressão Atmosférica (APCI), com detecção por monitoramento de reações múltiplas (MRM), em modo de ionização negativo foi desenvolvida e empregada na determinação de hidrocarbonetos policíclicos aromáticos nitrados (NHPAs) em fuligem de diesel. Na etapa inicial do trabalho, os padrões de fragmentação destas substâncias foram avaliados. Ionização negativa predomina em relação a ionização positiva e o íon [M-30]- foi o fragmento predominante na maior parte dos espectros de NHPAs. A identificação foi feita através da comparação com tempos de retenção de soluções padrão e pela análise dos respectivos espectros de massas. A eficiência da metodologia desenvolvida (75-100%) foi avaliada pela análise do padrão de fuligem de diesel certificado para 1-nitropireno (NIST- SRM 2975). Limites de detecção (1,5 mg/Kg) e de quantificação (5,0 mg/Kg) foram determinados. Amostras de fuligem de diesel geradas em motores de bancada analisadas através da metodologia desenvolvida, apresentaram concentrações de 1nitropireno abaixo dos limites de detecção. Amostras de fuligem de cano de descarga de veículo diesel também apresentaram concentrações de 1-nitropireno comparáveis ao LD do método. O único NHPA detectado nas amostras estudadas foi o 1-nitropireno. O método desenvolvido tem potencial para aplicação em amostras de fuligem de diesel. viii ABSTRACT An analytical procedure using High Performance Liquid Chromatography (HPLC) coupled with an Ion Trap Mass Spectrometer (IT-MS) and Atmospheric Pressure Chemical Ionization (APCI) interface with multiple reactions monitoring (MRM) detection and negative ionization was developed and applied in the determination of nitrated polcycyclic aromatic hydrocarbons (NPAHs) in diesel soot. The fragmentation patterns were evaluated. Negative ionization predominates over positive ionization and [M-30]- ion was shown to the predominating fragment of most NPAHs. NPAHs identification was performed retention time comparison and mass spectra. A high recevery efficiency (75-100%) was obtained for 1-nitropyrene in the NIST- SRM 2975. Detection limits of 1,5 mg/Kg and quantification limits of 5,0 mg/Kg were found. Diesel soot samples from bench motors were analyzed and 1nitropyrene levels were bellow the detection limits. A sample taken from a diesel vehicle showed levels of 1-nitropyrene comparable to the detection limit of the method. The only NPAH detected in the samples detected in the samples was 1nitropyrene. The method showed potential for further application in the study of NPAHs in diesel soot samples. ix LISTA DE FIGURAS Figura 1: Estruturas dos NHPAs estudados p.23 Figura 2: Principais componentes de um sistema de CLAE-EM-IT com interface de ionização à pressão atmosférica p.34 Figura 3: Comparação das principais interfaces comerciais de CLAE-EM em função de sua aplicação (polaridade e peso molecular) p.36 Figura 4: Esquema típico de interface APCI p.37 Figura 5: detector de massas do tipo Ion Trap p.45 Figura 6: Comparação dos espectros de 1NPi por CLAE-EM-APCI, com detecção por varredura, em modo de ionização negativa (a) e positiva (b) p.59 Figura 7: estrutura de ressonância eletrônica do grupo nitro (NO2) na molécula de 1NPi p.61 Figura 8: diferença de intensidades dos íons de m/z 211 e 210 no espectro de varredura de 2NFl adquirido por CLAE-EM-APCI em modo de ionização negativo p.64 Figura 9: Cromatograma e espectro de massas de 1NPi (15 ppb) obtido por CLAE-EM-APCI, com detecção por MRM em modo de ionização negativo x p.68 Figura 10: Variação do sinal de soluções de 1-NPi em função da vazão do sistema de Eluição p.69 Figura 11: Variação da área de sinal de soluções de 1-NPi em função da temperatura de vaporização: (a) 1-NPi 5 ppb; (b) 1-NPi 100 ppb p.73 Figura 12: Variação da área de sinal de soluções de 1-NPi em função da temperatura de secagem. (a) 1-NPi 5 µg/L; (b) 1-NPi 100 µg/L p.74 Figura 13: Avaliação da detecção de 1-NPi após procedimento de extração de material certificado (NIST, SRM 2975) com ultra-som e DCM p.77 Figura 14: Variação do sinal de 1-NPi em função da concentração de DCM na solução p.80 Figura 15: Procedimento de extração de cerca de 20 mg SRM 2975 em condições otimizadas p.84 Figura 16: Procedimento de extração de cerca de 2 mg SRM 2975 em condições otimizadas, após 4 etapas de extração p.85 Figura 17: Procedimento de extração de cerca de 2 mg SRM 2975 em condições otimizadas, após 6 etapas de extração p.86 Figura 18: Procedimento de extração e tratamento das amostras de Fuligem de Diesel da BR p.90 Figura 19: Cromatogramas de 1NPi obtidos por CLAE-EM-APCI, com detecção por MRM em modo de ionização negativo. a) extrato de fuligem de diesel depositada no filtro BR1 após reconcentração e com injeção de 100 uL; b) padrão 1µg/L (20 uL) p.91 Figura 20: Procedimento de extração alternativo empregado no tratamento da amostra de fuligem de cano de descarga de automóvel movido a diesel p.93 Figura 21: Cromatogramas em CLAE-EM-APCI-MRM de 9-NA e 1-NPi em extrato de SRM 2975 p.96 xi LISTA DE TABELAS Tabela 1: Especificação de solventes orgânicos utilizados p.51 Tabela 2: Especificação de outros materiais utilizados p.53 Tabela 3: Especificação da Instrumentação utilizada p.54 Tabela 4: Principais íons observados nos espectros de varredura de 1NPi, obtidos por CLAE-EM-APCI, em modo de ionização positivo e negativo p.58 Tabela 5: Caracterização da fragmentação de NHPAs selecionados por CLAE-EM-APCI, em modo negativo, com detecção por varredura p.63 Tabela 6: detecção de 1NPi por MRM em modo de ionização positivo e negativo; sistema de eluição MeOH/H2O (20:80) p.66 Tabela 7: Valores dos parâmetros do espectrômetro de massas otimizados para a detecção de 1NPi p.70 Tabela 8: Colunas cromatográficas estudadas na separação de DCM e 1-NPi p.78 Tabela 9: Parâmetros analíticos otimizados para a determinação de 1-NPi em FD p.81 Tabela 10: Figuras de mérito do método para detecção de 1NPi em FD p.82 xii Tabela 11: Avaliação da recuperação de 1-NPi em SRM 2975 p.86 Tabela 12: Avaliação da recuperação de 1-NPi em solução p.88 Tabela 13: Tempos de retenção (tr) de 1-NPi, 9-NA e 2-NFl em coluna Vydac p.95 xiii LISTA DE ABREVIATURAS ABREVIATURA DESCRIÇÃO 1-NPi 1-nitropireno 1,3-DNP 1,3-dinitropireno 1,6-DNP 1,6-dinitropireno 1,8-DNP 1,8-dinitropireno 1,8-NN 1,8-dinitronaftaleno 2-NFl 2-nitrofluoreno 9-NA 9-nitroantraceno AHPAs Hidrocarbonetos Amino-Policíclicos Aromáticos APCI Ionização Química em Pressão Atmosférica API Ionização a Pressão Atmosférica APPI Ionização Fotoquímica a Pressão Atmosférica CCF Cromatografia em Camada Fina CGAR Cromatografia a Gás de Alta Resolução CI Ionização Química CID Dissociação Induzida por Colisão CLAE Cromatografia a Líquido de Alta Eficiência DCM Diclorometano EFS Extração em Fase Sólida EI Ionização por Impacto de Elétrons EM Espectrometria de Massas ESI Interface Eletrospray FLUO Detector de Fluorescência xiv FD Fuligem de Diesel FM Fase Móvel HPAs Hidrocarbonetos Policíclicos Aromáticos IT Ion Trap MB Interface Moving Belt MeOH Metanol MPA Material Particulado Atmosférico MRM Monitoramento de Reações Múltiplas NHPAs Hidrocarbonetos Policíclicos Nitro-Aromáticos NOx Óxidos de Nitrogênio NO2 Grupamento Nitro PB Interface Particle Beam PCBs Bifenilas Policloradas PPB Partes por Bilhão PPM Partes por Milhão SCAN Detecção por varredura SIM Monitoramento Seletivo de Íons SiO2 Sílica SPAs Substâncias Policíclicas Aromáticas TSP Interface Termospray UV Detector de Ultra Violeta xv CAPÍTULO 1 Introdução Durante muitos séculos a humanidade utilizou da ajuda de animais para realizar trabalhos pesados e como forma de transportar cargas e pessoas. Porém, com o advento da revolução industrial no século XVIII, a força animal tornou-se em grande parte obsoleta devido à introdução de máquinas a vapor, cujo combustível era o carvão. No final do século XIX, as máquinas a vapor foram gradativamente substituídas por outras movidas a combustíveis fósseis e derivados de petróleo. Desde então, devido aos preços acessíveis e à robustez de motores movidos a diesel, o emprego deste combustível foi favorecido e amplamente disseminado, caracterizando o início do uso de diesel como principal fonte de energia para o setor industrial, na agricultura e nos transportes urbanos. Entretanto, ao serem queimados, óleo diesel e outros combustíveis fósseis liberam na atmosfera partículas de fuligem às quais estão associadas substâncias genotóxicas, notadamente substâncias policíclicas aromáticas (SPAs), que se distribuem entre a fase gasosa e o material particulado atmosférico (MPA) e tendem a contaminar os compartimentos ambientais em proporções que dependem das características de cada substância e também das características de cada compartimento ambiental. Ademais, outras substâncias que podem ser nocivas à saúde e ao meio ambiente também são 16 emitidas em fase gasosa devido ao processo de combustão incompleta, contribuindo de forma significativa ao incremento dos níveis de poluição ambiental. A fuligem de diesel é composta por partículas de carbono e matéria orgânica geralmente pequenas (com diâmetros < 1 µm), que podem penetrar até às porções mais inferiores dos pulmões e a presença de SPAs no material particulado atmosférico (MPA) e nas fuligens é particularmente importante devido ao seu impacto biológico e atividade mutagênica, que podem ser associados à ocorrência de câncer (IARC, 1989). Substâncias Policíclicas Aromáticas (SPAs), são encontradas em grande variedade na natureza e constituem uma grande classe de substâncias orgânicas cuja estrutura molecular central é mantida por estáveis sistemas de ligações π. Compreendem tanto sistemas orgânicos heterocíclicos quanto homocíclicos. O estudo destas substâncias é de grande interesse, pois apesar de algumas delas serem pigmentos animais e vegetais de notável importância biológica e intensa coloração, outras são importantes poluentes ambientais que, em alguns casos, induzem o desenvolvimento de neoplasias e/ou provocam mutações (Pereira Netto et al., 2000; Blumer, 1976). Fontes antropogênicas encontram-se intimamente ligadas à questão da poluição e contaminação ambiental em grandes centros urbanos, principalmente devido à emissão por veículos automotores e por processos industriais (Menzie et al., 1992). Neste caso, há formação de SPAs por síntese de novo, através de mecanismos via radicais livres que são bem conhecidos (Badger e Kimber, 1960; Badger et al., 1960). A importância relativa de cada tipo de fonte depende das características industriais e econômicas de cada região, mas as estimativas de contribuição relativa de cada uma variam de autor para autor (Baek et al., 1991). Entretanto, a combustão incompleta por fontes móveis ou estacionárias é uma das mais importantes delas, tanto pela quantidade emitida quanto por sua ampla distribuição. Emissões veiculares são, portanto, consideradas uma das principais fontes primárias de substâncias mutagênicas em atmosferas urbanas (Alfheim et al., 1983), especialmente devido aos motores a diesel, que emitem alta 17 carga de partículas ao serem comparados, por exemplo, aos motores a gasolina (Rappaport et al., 1980). Devido ao crescente número de veículos que trafegam diariamente nos grandes centros urbanos e da multiplicação de indústrias ao redor das metrópoles e em áreas anteriormente rurais, a poluição ambiental decorrente da queima de óleo diesel e de combustíveis fósseis tem aumentado gradativamente e constitui importante fator de risco à saúde das populações em geral. A inalação de ar poluído por indivíduos que trabalham em locais de grande exposição ou que lidam diretamente com máquinas movidas a combustíveis fósseis aumenta fortemente a exposição ocupacional. Os efeitos da poluição atmosférica podem ser sentidos a curtos e longos prazos e se apresentam de forma bastante variada e profunda. Chuva ácida, aquecimento global, redução da visibilidade em áreas urbanas e/ou agrícolas e degradação da camada de ozônio são apenas alguns dos mais conhecidos, porém a poluição atmosférica também está associada a problemas de saúde das populações expostas, gerando aumento na demanda por atendimento médico e podendo contribuir para o congestionamento do sistema de saúde público. Além disso, o MPA (fuligens, sujeiras, fumaça, poeira e gotículas líquidas) se deposita sobre a superfície de solos e corpos d’água, proporcionando alterações bioquímicas nestes ecossistemas e seu depósito sobre estruturas de importância histórica e cultural, causa erosão e deixa mancha nas mesmas. Diante deste cenário, uma das principais questões que se apresentam é: em que extensão a queima de óleo diesel constitui uma fonte significativa de exposição humana à SPAs e o quanto contribui à concentração atmosférica total das mesmas, com interesse particular em relação à presença de hidrocarbonetos policíclicos aromáticos (HPAs) e seus derivados nitrados (NHPAs), duas das principais classes de SPAs de grande importância ambiental. Neste contexto, este trabalho apresenta o desenvolvimento e a implementação de metodologia para a determinação de NHPAs em fuligem de diesel por cromatografia a líquido de alta eficiência acoplada a espectrometria 18 de massas e os resultados obtidos com esta metodologia no estudo da emissão destas substâncias por combustão de óleo diesel nacional. 19 CAPÍTULO 2 Fundamentação Teórica 2.1) HPAs e NHPAs: Aspectos Gerais Hidrocarbonetos Policíclicos Aromáticos (HPAs) e seus derivados nitrados (NHPAs) são substâncias tóxicas persistentes (STPs), formadas durante processos de combustão incompleta envolvendo combustíveis fósseis ou qualquer outro tipo de matéria orgânica e são consideradas umas das principais famílias de SPAs. Muitos HPAs e NHPAs representam risco à saúde por serem potencialmente carcinogênicos, mutagênicos e/ou teratogênicos, mesmo em baixas concentrações (Pereira Netto et al., 2000; Boffetta et al., 1997). Sabe-se também que alguns NHPAs, como 1-nitropireno e dinitro-pirenos, estão entre as substâncias mais potencialmente mutagênicas já testadas (Rosenkranz, 1982; Rosenkranz e Mermelstein, 1983). Os HPAs possuem tempos de vida diferentes na atmosfera (Butler, 1981) pois estão susceptíveis a reações fotoquímicas que vêm sendo estudadas há pelo menos quatro décadas (Grosjean, 1983). No entanto, o interesse pelo estudo destas substâncias aumentou no final da década de 70, quando Jager e Pitts Jr demonstraram, independentemente, que HPAs podem reagir com óxidos de nitrogênio (NOx) na atmosfera, formando derivados nitrados (NHPAs). 20 Jager (1978) reportou a presença de 3-nitrofluoranteno em MPA coletado em Praga e Pitts Jr (1978, 1979) demonstrou que benzo(a)pireno, perileno e pireno eram convertidos nos respectivos nitro-derivados após serem expostos a atmosferas (simuladas) contendo NO2 e traços de HNO3 em fase gasosa. A formação de NHPAs é catalisada por baixos valores de pH e como os óxidos de nitrogênio (NOx) são responsáveis pela acidificação do meio, catalisam sua própria reação com os HPAs precursores para sua formação (Moller et al.; 1983). NHPAs podem ser emitidos diretamente na atmosfera, após serem formados em processos de combustão incompleta em motores movidos a combustíveis fósseis, principalmente a óleo diesel (Shuetzle et al., 1982, Librando et al., 1993), e também a partir de processos industriais (Nielsen et al., 1984; Nielsen e Ramdahl, 1986; Pitts Jr et al., 1985a; Ramdahl et al., 1986). Entretanto, as reações atmosféricas são as principais fontes dos NHPAs presentes no MPA (Arey et al.; 1986; Atkinson et al.; 1990; Finlayson-Pitts & Pitts Jr, 1986; Pitts Jr et al., 1985 b; Sweetman et al., 1986; Kamens et al., 1994) pois a maioria dos NHPAs é formada na atmosfera a partir de reações de HPAs da fase gasosa (Schuetzle et al., 1986; Atkinson, 1988; Nishioka et al., 1988; Lewtas et al., 1990). Diversos NHPAs foram detectados em MPA, em estudos conduzidos na cidade de Barcelona, Espanha (Bayona et al.; 1994), nos quais a ocorrência de 2-nitrofluoranteno e de 2-nitropireno está relacionada a processos de nitração de fluoranteno e pireno em fase gasosa, na presença de radicais OH ou N2O5. Por outro lado, a ocorrência de 9-nitroantraceno e de 1-nitropireno está relacionada a reações eletrofílicas com os HPAs precursores durante processos de combustão, refletindo a contribuição de fontes móveis de poluição. Assim como acontece com os HPAs precurssores, NHPAs de baixo peso molecular (por exemplo, nitrobifenilas e nitronaftalenos) predominam na fase gasosa do ar enquanto os mais pesados (por exemplo, nitropirenos, nitrofluorantenos, nitroantracenos e nitrocrisenos) encontram-se associados ao MPA, de acordo com suas pressões de vapor. NHPAs constituem um grupo de mais de 200 substâncias diferentes. São encontrados no meio ambiente junto aos HPAs precursores e outras 21 centenas de substâncias orgânicas e inorgânicas, porém em concentrações inferiores àquelas dos HPAs. Os NHPAs encontrados em diferentes matrizes ambientais possuem de 2 a 5 anéis aromáticos que podem conter 1, 2 ou 3 grupamentos nitro (NO2). No entanto, a genotoxicidade dos NHPAs depende tanto da estrutura do HPA de origem como do número e da posição do grupamento nitro (NO2). NHPAs também têm sido encontrados em diversos materiais como toners de fotocopiadoras, produtos da combustão de carvão, sedimentos e fumaça de cigarro (Moreira e Barek, 1995). As estruturas moleculares dos NHPAs estudados neste trabalho encontram-se na Figura 1. 22 NO2 NO2 NO2 1-Nitropireno (1-NPi) 9-Nitroantraceno (9-NA) 2-Nitrofluoreno (2-NFl) NO2 NO2 1-Nitronaftaleno 2-Nitronaftaleno (1-NN) (2-NN) NO2 O2 N NO2 NO2 2,5-diNitrofluoreno (2,5-diNFl) 2,7-diNitrofluoreno (2,7-diNFl) NO2 NO2 NO2 NO2 NO2 1,3-diNitronaftaleno (1,3-diNN) NO2 1,5-diNitronaftaleno (1,5-diNN) 1,8-diNitronaftaleno (1,8-diNN) Figura 1: Estruturas moleculares dos NHPAs estudados neste trabalho. 23 HPAs e NHPAs podem ser absorvidos por inalação, ingestão ou por contato dérmico e se distribuem amplamente em todo o organismo, sendo encontrados principalmente em tecidos ricos em lipídios. O metabolismo de ambos é complexo. No caso dos HPAs, epóxidos são os intermediários metabólicos e precursores de intermediários como dihidrodiois e diol-epóxidos, que são as substâncias responsáveis pelos processos iniciais de carcinogênese. A formação destes intermediários reativos explica a atividade biológica dos HPAs, que seriam naturalmente inativos (Josephy, 1997; Glusker, 1985). NHPAs são metabolizados no fígado por NADPH-citocromo P-450 redutases, formando intermediários que são reduzidos às substâncias aminoaromáticas correspondentes, que são excretadas na urina sob a forma livre ou após acetilação (Moreira e Barek, 1995). Seus efeitos tóxicos estão relacionados à formação de intermediários reativos, os quais podem reagir com macromoléculas endógenas, sobretudo com proteínas (Pryor, 1982) e ácidos nucléicos (Varghese e Withmore, 1980). No entanto, muitos NHPAs também apresentam atividade mutagênica mesmo na ausência de ativação metabólica (Moreira e Barek, 1995) e muitos NHPAs são mais mutagênicos de que os HPAs precursores em testes de mutagenicidade do tipo Ames, com Salmonella typhimurium (Pitts Jr, 1987). Muitos também demonstram ter potencial carcinogênico em animais (Ohgaki et al., 1984; Rosenkranz, 1984; Salmeen et al., 1982; Tokiwa et al., 1986). Concentrações de NHPAs normalmente encontradas na atmosfera são da ordem de ng/m3 (OMS, 2003; Librando e Fazzino, 1993), enquanto partículas de fuligem de diesel apresentam concentrações de NHPAs da ordem de µg/kg a mg/kg (IARC, 1989; Moller et al.; 1993). As concentrações de dinitro-HPAs são aproximadamente 10 vezes menores do que os NHPAs, tanto em amostras ambientais como em amostras biológicas (Cvacka et al., 1998). Até 1987, cerca de 100 diferentes mono e di-NHPAs já haviam sido relatados em fuligem de diesel (Shuetzle et al., 1982, Shuetzle, 1983; PaputaPeck et al., 1983; Pitts Jr, 1987). Dentre os principais NHPAs encontrados em fuligem de diesel destacam-se nitropirenos, nitroantracenos e nitrofenantrenos, 24 nitrofluorenos, metilnitropirenos, metilnitrofluorantenos e nitrofluorantenos (Schuetzle, 1983). No entanto, as concentrações de NHPAs em fuligem de diesel variam substancialmente de amostra para amostra, sendo difícil comparar os diferentes perfis. Geralmente, 1-nitropireno é o NHPA predominante, com concentrações de 7-165 mg/Kg (Levsen, 1988), sendo comum encontrar concentrações de 2-nitrofluoreno de cerca de 15% em relação ao 1-nitropireno (Beije e Moller, 1988). Entretanto, nem sempre 1-nitropireno predomina, especialmente em fuligens oriundas de veículos pesados, nas quais há predomínio de 2-nitrofluoreno (IPCS, 2003). Até o presente momento, não existem estudos epidemiológicos detalhados sobre exposição humana às emissões de motores a diesel, que por serem misturas complexas demandam substâncias marcadoras que sejam específicas e sensíveis para a determinação de efeitos biológicos. Neste contexto, 1-nitropireno é considerado o principal biomarcador da queima de diesel para a classe dos NHPAs e pode fornecer informação acerca da exposição a esta classe de substâncias tóxicas (Gallagher et al., 1994; Raat et al., 1994), embora certos autores também atribuam este papel ao 2nitrofluoreno (Zwirner-Baier et al., 1999). Straube e colaboradores (2004), consideraram que a única fonte relevante de exposição humana a dinitropirenos é a emissão por motores a diesel e devido a esta especificidade sugerem a utilização dos mesmos como biomarcadores. Extratos de fuligem de diesel produziram mutações em testes do tipo Ames, com Salmonella typhimurium e segundo Schuetzle e colaboradores (1981), 1-nitropireno está relacionado à cerca de 24% da atividade mutagênica destes extratos. Há estimativas de que nitro-arenos contribuem com cerca de 15-20% da atividade mutagênica direta em atmosferas urbanas (Arey et al., 1988). Deve ser considerado ainda que as concentrações de HPAs em combustíveis a base de petróleo, diesel e em óleos são da ordem de partes por milhão (ppm) (IPCS, 1998), que podem, em parte, ser liberados diretamente para a atmosfera durante a queima, bem como originar NHPAs em motores de veículos automotivos, a partir da reação com óxidos de nitrogênio que são emitidos pela 25 conversão de nitrogênio e oxigênio em altas temperaturas nas câmaras de combustão (Scheepers e Bos, 1992). Neste contexto, muitos esforços têm sido feitos no intuito de avaliar estas classes de substâncias em matrizes ambientais, sobretudo aqueles NHPAs de maior importância carcinogênica, como 1-nitropireno, 2- nitrofluoreno, 1,3-, 1,6- e 1,8- dinitropirenos. Entretanto, há apenas dois trabalhos sobre NHPAs no meio ambiente do Brasil (Vasconcellos et al., 1998; Ciccioli et al. 1996) e não foram encontrados dados sobre NHPAs em fuligens de combustível no Brasil. 2.2) Determinação de HPAs e NHPAs Devido à complexidade das misturas de HPAs e/ou de NHPAs encontradas em amostras ambientais a determinação de HPAs e NHPAs requer métodos que permitam alta resolução e alta seletividade simultaneamente. Portanto, não é possível realizar análise direta de HPAs e NHPAs em amostras ambientais, uma vez que as mesmas contêm ampla variedade de outros produtos de combustão (carbono elementar [fuligem], espécies inorgânicas, aldeídos, cetonas e ácidos carboxílicos), que tendem a co-eluir com as substâncias de interesse nas técnicas cromatográficas geralmente empregadas. Em amostras provenientes de combustão, hidrocarbonetos alifáticos tendem a predominar com concentrações 100 a 1000 vezes maiores que as dos HPAs e 10.000 a 100.000 vezes maiores que as dos NHPAs. Os extratos de HPAs podem conter também isômeros destas substâncias e no caso de NHPAs derivados contendo mais de um grupo nitro ou funções mistas. Muitas destas famílias de substâncias apresentam comportamento físico-químico semelhante ao dos analitos de interesse e por isso sua remoção completa através de processos de extração e purificação se torna muito difícil, o que aponta na direção do uso de técnicas de detecção altamente seletivas (Niessen, 2000; Cvacka et al., 1998; Anacleto et al., 1995; Moreira e Barek, 1995, Castello e Gerbino, 1993; IPCS, 1998). 26 A extração das amostras de fuligem de diesel (FD), bem como de outras amostras ambientais como MPA, solos, sedimentos e poeira de túnel, podem ser realizadas através de diversos procedimentos. Os mais empregados são extração ultra-sônica, soxhlet e agitação mecânica (Ryno et al., 2006). A extração e o preparo de amostras constituem, na maioria dos casos, as etapas mais laboriosas e lentas dos procedimentos analíticos e o consumo de tempo é maior (12 a 24 horas) quando extração em Soxhlet é realizada, que ademais, em algumas situações, pode demandar supervisão humana. Por outro lado, a extração em banho de ultra-som é mais rápida e consome menor tempo. Outra vantagem da extração em ultra-som é o fácil manuseio. De modo geral, ambas permitem altas recuperações de NHPAs (> 90 %) (Cvacka et al., 1998). Os solventes orgânicos mais utilizados são diclorometano, acetonitrila, metanol, acetona, benzeno, tolueno e hexano (Guillén, 1994). Muitos procedimentos realizam a extração com um único solvente (Cvacka et al., 1998) enquanto outros utilizam misturas dos mesmos (Moriwaki et al., 2000; Schauer et al., 2004). Devido à alta complexidade das matrizes ambientais, procedimentos de limpeza e fracionamento das amostras são geralmente usados e permitem obter extratos com menos interferentes provenientes da matriz e de outras classes ou famílias de substâncias químicas. Estes procedimentos podem ser realizados, por exemplo, por extração líquido–líquido, extração em fase sólida (EFS), cromatografia em camada fina (CCF), cromatografia líquida em coluna (CL) ou CLAE preparativa. EFS é um procedimento de limpeza amplamente utilizado devido à facilidade de operação e ao baixo consumo de tempo (Kootstra et al., 1995; Barranco et al., 2003). O emprego de EFS com cartuchos descartáveis de sílica é um procedimento usado na determinação de HPAs (Pereira Netto, 1999). Estudos comparativos de procedimentos de fracionamento mostraram que altas taxas de recuperação de NHPAs podem ser obtidas com EFS (87-91%) ou CLAE preparativa (83-92%) contra menores taxas de recuperação obtidas por CCF (55-60%) (Cvacka et al., 1998). Devido à complexidade das amostras ambientais e também à necessidade de identificação de diferentes isômeros, técnicas cromatográficas 27 têm sido amplamente usadas na determinação de HPAs e NHPAs. Cromatografia a gás de alta resolução (CGAR) e cromatografia a líquido de alta eficiência (CLAE) são empregadas freqüentemente e a principal vantagem das técnicas cromatográficas sobre as demais é possibilitar a separação (resolução) de misturas complexas. O uso ou acoplamento a detectores de alta seletividade e alta sensibilidade permite identificar e quantificar baixas concentrações de HPAs e de NHPAs, mesmo em misturas ou frações contendo outras classes ou famílias de substâncias químicas. Neste contexto, a técnica ideal deve possibilitar boa resolução de todas as substâncias de interesse entre si e em relação às interferentes, de forma a permitir simultaneamente baixos limites de detecção e reprodutibilidade de tempos de retenção para auxiliar a identificação. Durante muito tempo CGAR foi utilizada para a análise de HPAs e NHPAs, porém a principal desvantagem na utilização desta técnica na separação de HPAs e NHPAs está relacionada com a baixa volatilidade dos membros mais pesados das séries. A baixa estabilidade térmica de certos NHPAs também pode ser uma desvantagem pois pode haver perda por decomposição no injetor, na coluna e na interface CGAR-EM (Sweetman, 1982), o que pode ser problemático para amostras ambientais onde encontramse em níveis bastante baixos (ng-pg/g) destas substâncias. Uma forma de evitar a perda por decomposição de NHPAs nas regiões aquecidas do cromatógrafo à gás é reduzi-los à amino-HPAs (AHPAs) e, em seguida, derivatizá-los (Scheepers et al., 1992). Como este processo envolve duas etapas de reação, implica em maior consumo de tempo no preparo das amostras e na possibilidade de perda parcial dos NHPAs nas duas etapas de reação. Algumas das limitações da análise de HPAs e NHPAs por CGAR podem ser superadas pela utilização de CLAE, que, de modo geral, apresenta muitas vantagens na determinação de compostos termolábeis, uma vez que as análises são feitas em temperaturas próximas à ambiente, evitando decomposição. Além disso, derivatização normalmente não é necessária e como a volatilidade não é pré-requisito à determinação, substâncias de alto peso molecular podem ser determinadas. Outro fator que pode ser considerado 28 é o fato de não haver volatilização da amostra nas análises por CLAE, o que reduz as perdas na transferência para a coluna e a discriminação por peso molecular. HPAs e NHPAs presentes em diversos tipos de amostras (como MPA, emissões de veículos, materiais biológicos, extratos de alimentos, águas, plantas, óleos, poeira e fuligens) são habitualmente analisados por CLAE em fase reversa, que é a técnica mais amplamente usada para a separação dos mesmos (Wise et al, 1993; Furton et al., 1993) que, por serem substâncias de baixa polaridade, são bem retidos em octadecilsílica (ODS). Schauer e colaboradores (2004) apresentaram cromatogramas de alguns NHPAs obtidos por CLAE em fase reversa, com boa resolução em gradientes de eluição de MeOH/H2O contendo até 50% de cada solvente. Entretanto o tempo total de análise foi de cerca de 40 minutos e os picos cromatográficos obtidos apresentavam largura maior que 3 minutos. Várias técnicas de detecção têm sido usadas para a determinação de HPAs e NHPAs (Cvazka et al., 1998). Para a análise de amostras reais, a sensibilidade do detector a ser utilizado é fator determinante à escolha do mesmo, uma vez que as amostras normalmente estudadas contêm apenas traços das substâncias de interesse. Espectrofotometria de absorção no ultravioleta (UV) e fluorescência (FLUO) são as técnicas de detecção mais utilizadas para a determinação de HPAs. A detecção por UV é praticamente universal para HPAs. Entretanto, na quantificação de amostras complexas, o detector de fluorescência oferece maior sensibilidade e seletividade já que estas substâncias apresentam fluorescência natural devido ao seu sistema de elétrons π (ver, por exemplo, Wise et al., 1993). A sensibilidade e a seletividade da técnica de detecção por FLUO podem ser aumentadas utilizando-se uma programação de detecção na análise cromatográfica que permita variar os comprimentos de onda de excitação e de emissão dos compostos de interesse, de forma adequá-los aos valores máximos para cada HPA ou grupos de HPAs. Dados de literatura apresentam espectros de excitação e de emissão para vários HPAs ou tabelas de comprimentos de onda máximos de excitação e de emissão, que podem ser 29 usados como base para a otimização dos comprimentos de onda analíticos ideais (ver por exemplo, Wise et al., 1993 e Barranco et al., 2003). No caso de NHPAs, além de detecção fluorimétrica, detecção eletroquímica, detecção por quimioluminescência e por espectrometria de massas têm sido empregadas (Cvacka et al; 1998). Como NHPAs são pouco fluorescentes ou mesmo não-fluorescentes devido ao efeito desativador do grupo nitro (NO2), para a determinação fluorimétrica destas substâncias é necessário convertê-las previamente em espécies fluorescentes, o que pode ser feito por redução do grupamento nitro (NO2) em um grupo amino (NH2), gerando amino-HPAs (AHPAs), que são extremamente fluorescentes. A redução de NHPAs pode ser feita antes da introdução no sistema cromatográfico (off line) ou após a separação, previamente à detecção ou diretamente (on line) no sistema cromatográfico e ambas as metodologias tem sido empregadas na determinação de NHPAs por CLAE (Cvacka et al., 1998). Os métodos de redução on line requerem a utilização de uma coluna de redução em linha com a coluna analítica. Os métodos off line tendem a ser mais laboriosos e a consumir mais tempo de pré-tratamento das amostras. Para fins quantitativos, a etapa de redução também pode levar a perdas na determinação de traços de NHPAs. Os métodos quimioluminescentes envolvem também redução e reação, enquanto a detecção eletroquímica pode ser afetada por pequenas concentrações de O2 residual do ar (Cvacka et al., 1998). A detecção por espectrometria de massas (EM) é, possivelmente, a principal ferramenta utilizada na determinação de HPAs e NHPAs. Este detector apresenta como principal vantagem a possibilidade de elucidação de estruturas moleculares por comparação dos espectros de massas com bibliotecas de espectros e/ou através da interpretação destes espectros. Outra vantagem que se apresenta na determinação de NHPAs por CLAE com detecção por espectrometria de massas (CLAE-EM) é que não há necessidade da etapa de redução (como acontece na detecção fluorimétrica) e a determinação pode ser feita diretamente nos extratos das amostras, devido à seletividade do detector. 30 2.3) Cromatografia a Líquido de Alta Eficiência acoplada a Espectrometria de Massas (CLAE-EM) O acoplamento de cromatografia a líquido de alta eficiência com espectrometria de massas (CLAE-EM) vem sendo estudado há cerca de 30 anos, desde o início da década de 1970. Um histórico dos grupos pioneiros e uma discussão sobre o tema pode ser encontrada na revisão de Niessen e colaboradores (1995). A combinação destas técnicas (CLAE - separação; EM - detecção e informação estrutural) disponibiliza uma ferramenta analítica bastante versátil para a análise de misturas complexas contendo compostos desconhecidos e/ou analitos em baixa concentração. Dentre as áreas de aplicação, destacamse as ciências bioquímicas e biomédicas (Gelpi, 1995; Madl et al., 2006), farmacêuticas (Smyth et al., 2006) e aplicações ambientais (Amorisco et al., 2006). Três maiores dificuldades podem ser citadas em relação ao acoplamento CLAE-EM: (a) a necessidade de adaptar vazões de aproximadamente 1 mL/min do efluente oriundo do sistema cromatográfico na região de alto vácuo do espectrômetro de massas; (b) a ionização de analitos termolábeis ou não voláteis e (c) a incompatibilidade na composição dos eluentes, caso haja uso de fases móveis não voláteis no desenvolvimento de metodologia de separação cromatográfica. A questão da ionização gerava maior problemática nos primeiros anos de desenvolvimento da técnica, quando apenas tecnologias baseadas na ionização por impacto de elétrons (EI) e ionização química (CI) (que requeriam amostras no estado gasoso) eram consideradas, em contraste com as técnicas de CLAE e CLAE-EM que lidavam com substâncias não voláteis. Entretanto, considerável progresso foi alcançado no desenvolvimento de métodos de ionização mais suaves e, atualmente, a ionização de analitos em CLAE-EM não é mais considerada um problema. Isto foi obtido considerando-se também o papel da fase móvel em CLAEEM. O paradigma anterior considerava que a fase móvel estava mais relacionada ao processo cromatográfico e devia ser removida de forma mais rápida e completa possível. Atualmente, com as atuais interfaces empregadas 31 em CLAE-EM, o processo de ionização da substância de interesse é considerado na escolha da fase móvel a ser utilizada, o que por outro lado pode levar a “conflitos de interesses”, uma vez que uma determinada composição de fase móvel pode ser excelente do ponto de vista da espectrometria de massas, mas pode não ser útil em relação ao processo cromatográfico e vice-versa. O uso rotineiro de fases móveis contendo tampões salinos (fosfato, por exemplo) ou outros aditivos, não é indicado em interfaces de CLAE-EM, pois os mesmos costumam ser geralmente não-voláteis e tendem a acumular-se nas interfaces. Entretanto, sistemas que dispõem de fluxo de gás em contra corrente podem empregar tais eluentes com maior tolerância. Para resolver a questão das incompatibilidades relacionadas à fase móvel, muitas vezes o mais indicado é a substituição de seus componentes. Entretanto, continua havendo restrições na instrumentação, especialmente no que se refere ao fluxo de fase móvel e aos dispositivos de vácuo, pois, por exemplo, uma vazão de 1 mL/min de água é convertida em volumes muito maiores de vapor d’água (~1,2 L/min) à pressão atmosférica, gerando um aporte excessivo de material aos sistemas de vácuo empregados em espectrometria de massas. Visando resolver esta incompatibilidade, várias técnicas de acoplamento e várias interfaces de CLAE-EM tornaram-se disponíveis comercialmente, tendo em comum o fato de operarem em baixas e/ou moderadas pressões (Niessen et al., 1995). O acoplamento CLAE-EM é considerado um dos mais importantes avanços na área de Química Analítica (Niessen, 2000) e foi viabilizado pelo desenvolvimento de interfaces que permitem a introdução adequada do fluxo oriundo do sistema cromatográfico (à pressão atmosférica) ao ambiente sob alto vácuo exigido pelo detector de massas. Várias interfaces com estas características foram introduzidas no mercado nos últimos anos (Niessen, 2000). As primeiras interfaces comerciais de CLAE-EM apresentavam muitas dificuldades em sua manipulação, sensibilidade limitada e falta de robustez. Alguns exemplos podem ser citados: interfaces dos tipos moving belt (MB) e particle beam (PB), que se baseiam na remoção da fase móvel antes da entrada no sistema de espectrometria de massas; interfaces de introdução 32 direta de líquido e de fluxo contínuo, que diminuem a vazão de entrada com dispositivos de divisão, porém geram perda de sensibilidade de acordo com a razão de divisão e interface termospray (TSP), baseadas na vaporização do efluente da coluna. O grande aumento na aplicação de técnicas de CLAE-EM se deu em função do desenvolvimento de técnicas de ionização à pressão atmosférica (API), que apresentavam sensibilidade e robustez adequadas (Niessen, 2000). Esta denominação - API - engloba todas as técnicas de ionização nas quais íons são formados à pressão atmosférica. A aplicação destes processos de ionização à espectrometria de massas vem sendo estudada há muitos anos (Knewstubb et al., 1966; Shahin, 1966; Horning et al., 1974). Recentemente, foram desenvolvidas interfaces de ionização à pressão atmosférica (API), que diferem principalmente quanto ao princípio de nebulização do efluente cromatográfico e quanto à faixa de aplicação que cobrem. Há variações de desenho nos modelos de espectrômetros de massas disponíveis comercialmente, mas de modo geral, os íons formados na região da API são transportados para a região sob alto vácuo no interior do espectrômetro de massas por intermédio da aplicação de potenciais elétricos e de um ou mais estágios de bombeamento consecutivos. Um esquema genérico de instrumentação CLAE-EM com sistema de ionização à pressão atmosférica é apresentado na Figura 2. 33 Ion Trap Detector Câmara de nebulização Figura 2: Principais componentes de um sistema de CLAE-EM-IT com interface de ionização à pressão atmosférica. A câmara de ionização em pressão atmosférica termina em um capilar de transferência, que atua como um “nariz” atraindo os íons formados nesta região. Este capilar já se encontra na face de baixa pressão do espectrômetro de massas e apenas um orifício encontra-se aberto para região em pressão atmosférica. No entanto, a passagem do gás da região de pressão atmosférica para a região de vácuo é acompanhada por um processo de expansão, onde as partículas com as menores massas tendem a se difundir mais, afastando-se daquelas de maior massa, e o resultado deste fenômeno é o enriquecimento das espécies de maior massa. Tanto em CGAR-EM quanto em CLAE-EM, como as moléculas dos analitos têm massas moleculares consideravelmente maiores do que os constituintes da fase móvel, a conseqüente separação que ocorre durante o processo de expansão leva ao enriquecimento das espécies do analito em fase gasosa (Niessen, 1995). Após o processo de vaporização, o fluxo oriundo do sistema cromatográfico, só pode ser introduzido na região de vácuo do EM por intermédio da aplicação de potenciais elétricos e de estágios diferenciados de bombeamento (vácuo). 34 Existem sistemas que dispõem de um único estágio de bombeamento ou expansão, mas na prática há duas razões para que seja empregado um segundo estágio: a evaporação incompleta de líquido e a transmissão das espécies do analito até o analisador de massas. A evaporação das gotículas geralmente é incompleta, resultando na entrada de uma grande fração de líquido no espectrômetro de massas. Visando a prevenção destes problemas, sistemas com dois estágios de bombeamento diferenciados são empregados. Além disso, modelos com dois estágios permitem o uso de skimmers (separadores de massas) mais largos, que favorecem a transmissão das espécies do analito até a região de alto vácuo no interior do espectrômetro de massas. Os íons são focalizados e guiados até o analisador de massas através dos skimmers e pela aplicação de campos elétricos apropriados em dispositivos do tipo octapolo. A primeira região de vácuo é produzida por intermédio de bombas turbo-moleculares e encontra-se entre o capilar de transferência e o primeiro skimmer. Alguns sistemas empregam um segundo skimmer, que conduz a um segundo estágio de vácuo. Geralmente, o vácuo a partir desta região é produzido por bombas do tipo turbo-molecular. Em equipamentos mais modernos, ao invés de sistemas convencionais de lentes, são empregados quadrupolos, hexapolos ou octapolos como dispositivos de focalização, através da aplicação de rádio-freqüência apenas, para préconcentrar os íons do analito e direcioná-los ao analisador de massas. Esta aplicação produz melhora na relação sinal-ruído devido ao melhor efeito de focalização alcançado. Atualmente, as principais interfaces comerciais baseiam-se em ionização à pressão atmosférica (API), compreendendo electrospray (ESI), ionização química à pressão atmosférica (APCI) e ionização fotoquímica à pressão atmosférica (APPI). Esta recente introdução de novas interfaces comerciais abriu novas perspectivas e impulsionou a aplicação da técnica na análise de misturas orgânicas complexas contendo substâncias instáveis e/ou de alto peso molecular, proporcionando limites de detecção da ordem de pg ou sub-pg. Entretanto, as interfaces não devem ser entendidas apenas como dispositivos para introdução de amostras em sistemas de espectrometria de massas. Uma interface é, além disto, o que determina ou permite a ionização 35 dos analitos de interesse e consequentemente, sua escolha influencia fortemente os resultados obtidos. O diagrama esquemático abaixo (Figura 3) indica as áreas de aplicação das três principais interfaces comerciais de CLAE-EM. Figura 3: Comparação das principais interfaces comerciais de CLAE-EM em função de sua aplicação (polaridade e peso molecular). 2.4) Ionização Química à Pressão Atmosférica (APCI) O desenvolvimento de interfaces APCI para sistemas CLAE-EM reporta- se ao trabalho de Horning e colaboradores (Horning et al., 1974), porém devido à falta de instrumentação adequada, a implementação desta técnica em larga escala se consolidou apenas no início da década de 1990. O processo de APCI é iniciado a partir da descarga de elétrons proveniente de uma agulha do tipo corona e os íons gerados na interface, em fase gasosa. Atualmente, o uso de APCI para ionização de analitos é outra das estratégias mais utilizadas. Em APCI, a ionização das moléculas do analito ocorre por descarga de elétrons provenientes de uma agulha do tipo corona (Niessen, 1995). Esta descarga ioniza não apenas as moléculas do analito, pois moléculas do eluente também são ionizadas e reagem com as moléculas do analito, em fase gasosa. 36 Os mecanismos de ionização em APCI são os mesmos observados em procedimentos de ionização química em pressões moderadas, porém quando comparada aos mesmos verifica-se que a ionização é mais eficiente, uma vez que a frequência de colisões é maior em pressão mais alta (atmosférica). A formação de íons positivos ocorre por transferência de prótons, formação de adutos ou reações de transferência de cargas, enquanto a formação de íons negativos acontece devido à abstração de prótons, acoplamento de ânion ou por reações de captura de elétrons (Lemière, 2001). As interfaces APCI geralmente consistem dos seguintes componentes: (a) um sistema de aporte do efluente do sistema cromatográfico, geralmente em vazões na faixa de 0,5 a 2,0 mL/min.; (b) um dispositivo de nebulização e vaporização deste efluente e (c) uma fonte de íons a pressão atmosférica, contendo uma agulha do tipo corona. A Figura 4 apresenta um esquema de interface APCI. Nebulizer Pressure Corona current Heater Vcap Drying gas Temperature and Flow Fragmentor Figura 4: Esquema típico de interface APCI Para o acoplamento do sistema de CLAE ao sistema APCI, o efluente líquido da coluna cromatográfica é transferido por meio de tubos capilares, que podem ser de metal ou sintéticos (tubos peek). A distância entre os dois 37 equipamentos e o diâmetro do tubo conector utilizados devem ser avaliados para evitar a criação de volume morto excessivo no sistema. O emprego de altas vazões é fundamental para a performance desta técnica e o uso de colunas de CLAE com 4.6mm e grandes volumes de amostra são relatados constantemente na literatura (Lemière, 2001). Como o processo de APCI ocorre em fase gasosa após aquecimento do efluente nebulizado, diversas montagens de nebulizadores e vaporizadores estão disponíveis no mercado, fabricados por diferentes fornecedores. Os nebulizadores mais utilizados são do tipo pneumático e compreendem três tubos concêntricos. O primeiro deles é um tubo capilar através do qual é carreado o eluente oriundo do sistema cromatográfico; o segundo e o terceiro têm a função de carrear o gás de nebulização e o gás de secagem auxiliar, respectivamente. O tubo através do qual passa o gás de nebulização possui um dispositivo aquecedor em seu interior e o aerossol préaquecido chega à câmara de vaporização, localizada diretamente em frente ao nebulizador, para completar o processo de vaporização. A agulha corona localiza-se em posição axial, próxima ou até mesmo dentro da câmara de vaporização. A vazão empregada nos processos de nebulização-vaporizaçãosecagem varia de acordo com o fabricante de cada equipamento e com as características químicas dos compostos de interesse e do sistema de eluição empregado. O gás mais utilizado é o nitrogênio (N2) e sistemas APCI requerem grande suprimento do mesmo, podendo até mesmo ser maior que 600 L/h em alguns instrumentos. Consumos tão altos muitas vezes requerem aporte de N2 a partir de cilindros de N2 líquido ou até mesmo de unidades geradoras de N2 a partir de ar comprimido. Os produtos do processo de ionização em interface APCI são íons em fase gasosa oriundos do eluente e do analito, além de íons e moléculas dos gases envolvidos no processo de secagem. Além disto, ocorre formação de agregados com água (cluster ions) e com íons provenientes do sistema de eluição que não contenham íons do analito. Estes agregados devem ser destruídos antes de passar à região de alto vácuo do espectrômetro de massas a fim de reduzir o ruído na linha de base, que pode mascarar o sinal do analito. O aumento na temperatura da interface, bem como do fluxo do gás de 38 secagem podem reduzir a formação destes agregados, porém se o fluxo de gás for aumentado excessivamente, poderá causar atenuação no sinal do analito, por perda na interface. Os íons dos analitos formados estão solvatados por moléculas de água, misturados aos íons do sistema de eluição e por moléculas dos gases utilizados no processo de vaporização. A desagregação destes clusters pode ser conseguida com o auxílio de um fluxo de gás em contra corrente, localizado no mesmo patamar onde se encontra o orifício do capilar de transferência ou pelo uso de um capilar de transferência aquecido ou pela combinação de ambos. Outro artifício que pode ser empregado é a aplicação de um suave deslocamento potencial entre o capilar de transferência e o skimmer, visando promover colisões entre os agregados e as moléculas de gás que estiverem presentes. Este artifício é conhecido como dissociação induzida por colisão – CID. As colisões dos clusters com superfícies aquecidas dentro do espectrômetro de massas provocam transferência de calor e, consequentemente, evaporação. Sendo assim, o fluxo de gás efetivo dentro do espectrômetro de massas aumenta e pode exceder a capacidade da bomba de vácuo. Por esta razão, também são empregados sistemas com dois estágios de bombeamento diferenciados. As reações de ionização química são iniciadas após descarga de elétrons emitidos a partir de uma agulha do tipo corona (5-10kV) e os elétrons emitidos deflagram uma série de reações. O processo de ionização em interfaces do tipo APCI pode ocorrer de diferentes maneiras. Em modo positivo, ocorre por transferência de prótons ou por reações de transferência de cargas. A transferência de cargas entre íons positivos ocorre entre um íon-reagente (gerado a partir de uma molécula com alto potencial de ionização) e uma molécula em fase gasosa com baixo potencial de ionização. Em modo negativo, reações de captura de elétrons ocorrem quando moléculas susceptíveis a tais processos são ionizadas pela fonte de API, produzindo ânions de peso molecular ou por captura de elétrons após dissociação, produzindo fragmentos de ânions negativos. Nas interfaces APCI, o efluente da coluna é nebulizado pneumaticamente em um tubo aquecido (geralmente de quartzo ou aço inoxidável), impulsionado por um fluxo de gás adicional, que circunda o 39 nebulizador. Uma vez vaporizado, o aerossol formado pela mistura de vapor e solvente é transferido para a interface, onde uma descarga de elétrons proveniente da agulha corona inicia o processo de ionização. As moléculas do eluente estão em excesso em relação às moléculas do analito, sendo ionizadas preferencialmente. Em um segundo momento, as moléculas do analito são ionizadas (preferencialmente) a partir das moléculas do eluente. Altas temperaturas de vaporização e secagem na interface APCI podem levar à degradação de alguns analitos, mas não há sérios relatos de decomposição térmica na literatura (Niessen, 1995), uma vez que as altas vazões empregadas e o fluxo de N2 auxiliar previnem a quebra das moléculas (Lemière, 2001). A interface APCI é fácil de ser operada e não requer complexos procedimentos de otimização de temperatura. Nos sistemas que dispõem de gás de secagem em contra corrente, podem ser usados tanto tampões voláteis quanto não-voláteis, pois qualquer material ou contaminação não volátil depositada na câmara de ionização pode ser facilmente removida com procedimentos de limpeza que não requerem o desligamento do sistema de vácuo do espectrômetro de massas. A interface APCI é, de modo geral, menos sensível a interferências químicas do que a interface ESI, pois o processo de ionização na APCI é muito eficiente, sendo o método de escolha para muitas drogas e metabólitos e utilizada em larga escala na indústria farmacêutica. Triglicerídios, PCBs, HPAs, ftalatos e ácidos graxos são alguns exemplos de analitos típicos para análise em APCI (Niessen, 1995). Amostras que contém hetero-átomos como benzodiazepinas e carbamatos também são passíveis de análise nesta interface. Amostras termolábeis e/ou fotossensíveis que possam se decompor durante o processo de nebulização, que ocorre sob aquecimento, devem ser evitadas assim como analitos que se encontrem carregados em solução, como peptídeos, proteínas, aminoácidos e nucleotídeos, pois a sensibilidade na detecção será reduzida. Ademais estas espécies tendem a ser termolábeis e, devido ao alto peso molecular, são menos voláteis. A aplicação de CLAE-EMAPCI para a análise de NHPAs foi relatada inicialmente na década de 80 (Henderson et al.; 1983), mas devido à limitada sensibilidade deste método 40 naquela época, poucas aplicações são reportadas na literatura, como a determinação de nitropirenos (Moriwaki et al.; 2000). Substâncias contendo grupos funcionais eletronegativos (NO2, halogênios) ou que contenham carbonilas conjugadas, são passíveis de ionização em APCI negativo, uma vez que a descarga de elétrons em APCI é feita por agulha corona. A maioria das reações íon-molécula em APCI é do tipo ácido-base em fase gasosa. Para operação em modo positivo, os íonsreagentes são ácidos e para operação em modo negativo, os íons reagentes são bases. No entanto, o mecanismo de ionização predominante depende das características químicas da fase móvel e da substância considerada. O maior benefício da ionização a pressão atmosférica é que todos os íons em fase gasosa sofrem colisões com as demais moléculas, resultando em interações com todos os gases que se encontram nesta região no momento da ionização. Os principais íons formados são N2+, O2+, H2O+e NO+ e na presença de traços de água uma série de reações em cascata se inicia, resultando principalmente na formação de íons agregados com água (clusters). Uma vez que estes agregados com água são a principal fonte de íons reagentes, a afinidade protônica dos mesmos em relação à afinidade protônica dos íons do analito e dos íons do eluente possui grande efeito na sensibilidade do sistema de detecção. 2.5) Comparação das Interfaces APCI e ESI Nas aplicações de CLAE-EM, podem ser destacadas principalmente duas técnicas de ionização, APCI e ESI. ESI é o método de ionização à pressão atmosférica mais usado em espectrometria de massas atualmente, embora seja um processo conhecido há muitos anos. O primeiro relato sobre esta técnica de ionização foi feito por Zeleny (1917) que descreveu como a aplicação de um potencial elétrico sobre um capilar contendo solvente produzira a quebra do mesmo em finas gotículas. Este processo é ainda hoje a base de funcionamento da maioria dos modelos de interface ESI disponíveis. Posteriormente, o primeiro relato do uso de electrospray em uma interface foi feito por Dole e colaboradores (Dole et al., 1968) no final da 41 década de 1960. Eles investigaram a possibilidade de produzir íons em fase gasosa a partir de macromoléculas em solução utilizando um spray eletrostático à pressão atmosférica e demonstraram que a técnica proposta era capaz de produzir íons em fase gasosa. Após este primeiro trabalho de sucesso, apenas no final da década de 1970, deu-se continuidade às pesquisas sobre o tema com os trabalhos de Thomson e Iribarne (Thomson, 1979; Iribarne, 1976). O acoplamento CLAE-EM-ESI foi reportado em seguida, quase simultaneamente, por Yamashita e Whitehouse (Yamashita et al., 1984; Whitehouse et al., 1985) e desde então, encontram-se na literatura inúmeras aplicações sobre o tema. Devido à grande semelhança entre as interfaces APCI e ESI, os sistemas de CLAE-EM-API de última geração geralmente são equipados para ambos os processos de ionização, requerendo um mínimo de interação do operador ou modificação no equipamento. As interfaces APCI diferem das interfaces ESI em dois aspectos principais. O primeiro deles refere-se à inclusão de uma região aquecida que vai além da extremidade do capilar de nebulização, através do qual passa um fluxo de gás para vaporizar eluente(s) e analito(s). O segundo aspecto refere-se à inclusão da agulha corona, para produzir a descarga de elétrons que deflagra o processo de ionização. Portanto, em contraste com a interface ESI, os processos de evaporação do solvente e de formação de íons ocorrem separadamente e, desta forma, a ionização em APCI ocorre na fase gasosa resultante do aquecimento do efluente nebulizado. Por outro lado, na interface ESI, o efluente da coluna é nebulizado em uma câmara de ionização a pressão atmosférica e a ionização ocorre como resultado da ação de um campo elétrico. Em alguns sistemas a nebulização em ESI é feita por um nebulizador pneumático (ES-pneumaticamente assistida), que difere principalmente no processo de formação do spray. Outra diferença muito importante entre as duas técnicas refere-se às vazões de fase móvel empregadas. A interface ESI opera em vazões baixas, na faixa de até nL/min., enquanto a interface APCI tem melhor performance em vazões mais altas (mL/min). Vazões mais baixas até podem ser usadas em APCI, mas neste caso a estabilidade da descarga da agulha corona pode apresentar problemas. Enquanto ESI é uma técnica que favorece a 42 miniaturização, a redução das dimensões em APCI seria muito trabalhosa e pouco proveitosa (Lemière, 2001). Pelo fato do processo de ionização ocorrer em solução, de modo geral a interface ESI é mais sensível aos efeitos de matriz e à composição de fase móvel que a interface APCI. Em ESI, o pH dos solventes afeta diretamente o processo de ionização uma vez que os analitos são íons em solução e pode haver formação de adutos com cátions (Na+, K+) e ânions (Cl-, CF3 e COO-). Logo, a otimização do pH pode afetar positiva ou negativamente a performance desta interface. Em APCI, a escolha do solvente afeta diretamente o processo de ionização, porém o efeito de pH é pouco pronunciado. Outra forma de se comparar interfaces CLAE-EM é a avaliação de suas figuras de mérito, ou seja, comparar seus limites de detecção e de quantificação e/ou ainda, comparar o conteúdo de informação estrutural fornecida. É importante ressaltar que o tipo de informação que se deseja adquirir depende da finalidade da aplicação: Em uma análise qualitativa, por exemplo, deve-se adquirir consistente informação acerca da estrutura química dos íons e substâncias, enquanto em análise quantitativa, a presença de 1-3 picos distintos no espectro de massas é suficiente. Em relação às figuras de mérito, torna-se difícil fazer uma comparação das interfaces CLAE-EM, uma vez que é complicado extrair dados comparáveis da literatura e isto só seria possível caso uma mesma substância fosse analisada por diferentes interfaces (Niessen, 1995). Ademais, as diferenças em sistemas de detecção têm papel importante nos valores obtidos. Uma característica marcante da técnica de ESI é que a geração de íons em fase gasosa é um processo suave, que permite a análise de substâncias lábeis sem que haja comprometimento estrutural das mesmas. Incluem-se entre as amostras mais indicadas para ESI aquelas carregadas em solução, como proteínas, peptídeos, oligonucleotídeos e aminas quaternárias (em geral macro moléculas), por exemplo, enquanto devem ser evitados compostos de baixa polaridade, como HPAs e PCBs. Já em APCI, são indicados compostos de menor peso molecular (geralmente abaixo de 1000 Da) e de polaridade baixa a intermediária. Em geral, substâncias não-voláteis devem ser evitadas, pois os analitos precisam ter certa volatilidade, devido aos 43 mecanismos envolvidos. Espécies previamente carregadas em solução e substâncias termolábeis devem ser evitadas. Analitos típicos para APCI são HPAs, NHPAs, PCBs, ácidos graxos, ftalatos e esteróides (Perez e Barceló, 2001; Ingelse et al., 2001). Uma comparação sistemática de três diferentes técnicas de ionização (ESI, APCI e APPI) foi feita por Straube e colaboradores (2004), que utilizaram interfaces comerciais para a análise de aminas poliaromáticas e de substâncias nitroaromáticas por CLAE-EM-EM. 2.6) Espectrometria de Massas acoplada à Espectrometria de Massas (EM-EM) Técnicas de detecção que empregam múltiplas etapas de espectometria de massas (EMn) oferecem grande especificidade para análises qualitativas. Do ponto de vista quantitativo, a detecção por EMn apresenta sensibilidade e precisão adequadas para muitas das determinações de interesse em análise ambiental, pois permitem detectar as substâncias de interesse em misturas complexas, com alta seletividade e com eficiente redução nos níveis de ruído (background). Além disto, a seletividade e especificidade de duas ou mais etapas de EM (CLAE-EM-EM) tende a reduzir a complexidade dos procedimentos de preparo de amostras, com baixos limites de detecção e com exatidão e precisão adequadas. Há sistemas de detecção que permitem várias etapas de isolamento e fragmentação, dando origem a uma determinação extremamente seletiva e de um modo geral, qualquer um dos íons gerados pode ser usado em análise quantitativa, embora o sinal gerado caia com o aumento do número de etapas de fragmentação e isolamento consecutivas. Atualmente, espectrômetros de massas do tipo triplo quadrupolo (QqQ), Ion Trap (IT) e quadrupolo associado a detecção por tempo de vôo (Q-TOF) são exemplos de sistemas disponíveis comercialmente que podem empregar a detecção por EMn. Além do alto custo e de serem mais complicados no que se refere a sua operação e manutenção do que os sistemas IT, os sistemas de QqQ e Q-TOF são limitados a apenas duas etapas de espectrometria de massas (EM-EM). 44 Além de possibilitar a realização de EMn, que oferece vantagens em relação à obtenção de informação qualitativa adicional, sistemas IT permitem adquirir espectros de varredura em alta velocidade, com boa resolução e com maior sensibilidade do que sistemas QqQ. Ademais, os espectros de massas gerados por IT no modo EM-EM são fáceis de ser interpretados, pois os mecanismos de fragmentação são bem definidos, além de haver menor fragmentação do que nos sistemas QqQ (Agilent Technologies, sem data). As desvantagens dos sitemas IT estão relacionadas à menor precisão na informação das massas (m/z) adquiridas (em relação ao Q-TOF), bem como à menor sensibilidade para análises quantitativas do que os sistemas QqQ devido ao mecanismo de funcionamento do IT, que será explicado detalhadamente a seguir. Entretanto, levando-se em consideração o custo dos equipamentos, a vantagem de um sistema IT está relacionada à flexibilidade de análises que o mesmo oferece. Um detector de massas do tipo IT (Figura 5) é composto por dois eletrodos em forma de anel, ,que estão relacionados com a análise das massas e por dois eletrodos do tipo “end caps”, que estão relacionados com o isolamento das massas adquiridas, com o processo de dissociação induzida por colisões (CID) e com o processo de ejeção por ressonância não linear. Há também um sistema de aporte de gás (He), o qual está relacionado à mobilidade dos íons (m/z) dentro do detector, bem como ao processo de CID e à resolução de massas. Figura 5: Detector de massas do tipo Ion Trap. 45 Inicialmente, ocorre acumulação dos íons que são formados na câmara de ionização em pressão atmosférica. Em seguida, o Trap opera em ressonância não linear, de forma a ejetar os íons de modo seqüencial, de acordo com os interesses de cada análise. Desta forma, é possível empregar a técnica de detecção por monitoramento de reações múltiplas (MRM), que permite isolar um ou mais íons e fragmentá-los seletivamente, gerando vários “íons filhos”, dos quais pode-se novamente isolar aqueles de interesse, reiniciando assim o processo de fragmentação seletiva. 2.7) Determinação de NHPAs por CLAE-EM-EM Há relativamente poucos dados disponíveis na literatura sobre o comportamento de NHPAs por CLAE-EM-EM (Bonfanti et al., 1996; Moriwaki et al., 2000; Williams e Perreault, 2000; Schauer et al., 2004; Straube et al., 2004). Entretanto, nos últimos anos, o aumento no número de publicações sobre aplicações desta técnica na determinação de substâncias poliaromáticas é indicativo de sua viabilidade na determinação de NHPAs. No trabalho de Bonfanti e colaboradores (1996), o estudo de NHPAs por CLAE com interface do tipo Particle Beam (PB) gerou espectros de massas de baixa qualidade tanto em modo de ionização negativo quanto positivo quando foi empregada ionização por impacto de elétrons. Quando ionização química com detecção em modo negativo foi empregada, melhores respostas em termos foram obtidas e íons [M]- foram detectados predominantemente para a maioria dos NHPAs estudados. Moriwaki e colaboradores (2000) realizaram estudos com 1-nitropireno e dinitro-pirenos. A formação de picos correspondentes aos íons de peso molecular não foi observada por CLAE-EM-ESI, em modo negativo e em modo positivo. Com interface APCI, em modo de ionização negativo, apenas íons [M]foram observados. Straube e colaboradores (2004) compararam diferentes interfaces de CLAE-EM (ESI e APCI) para a análise de 1,6-dinitropireno e observaram que não houve ionização consistente em interface ESI. Por outro lado, observaram 46 a formação de íons negativamente carregados ([M]-) em interface APCI e atribuíram este fato à capacidade aceptora de elétrons dos nitroaromáticos. Schauer e colaboradores (2004) investigaram o comportamento de alguns NHPAs (1- e 2-nitronaftalenos; 2-nitrofluoreno; 9-nitroantraceno e 1nitropireno, dentre outros) através de CLAE-EM-APCI com detecção por TOF. Em modo de ionização positivo observaram predominantemente a formação de íons do tipo [M+H]+, exceto no caso dos nitronaftalenos, onde observaram predomínio dos íons do tipo [M+H-30]+. Em modo de ionização negativo, observaram predominância do íon [M]- e do fragmento correspondente à perda de 30 unidades de massa [M-30]-. No estudo de NHPAs por CLAE-EM-ESI realizado por Williams e Perreault (2000), não foram obtidas condições consistentes para gerar íons positivos estáveis que permitissem o desenvolvimento de metodologia sistemática para a detecção de NHPAs em modo de ionização positivo. No entanto, em modo de ionização negativo foi observado que apenas os compostos contendo grupos “puxadores” de elétrons foram prontamente ionizados em modo negativo e também que, dentre os compostos estudados, apenas os NHPAs (1-nitronaftaleno, 9-nitroantraceno e 1-nitropireno) produziram fragmentação significativa, gerando íons do tipo [M]-, [M-H]- e [MH+16]-, que posteriormente sofreram perda de 30 u.m.a., gerando fragmentos do tipo [M-30]-, [M-H-30]- e [M-H+16-30]-. A perda de 30 unidades de massa a partir a partir do íon precursor de NHPAs já foi observada anteriormente com outras técnicas de ionização, como por exemplo impacto de elétrons com detecção de íons positivos, ionização química em modo negativo e por MALDI em modo positivo e tem sido interpretada por alguns autores como decorrente da perda de NO ([M-NO]- ou [M-NO+H]+) (Korfmacher et al., 1987; Schauer et al., 2004). No entanto, a redução na fonte de ionização que leva à formação de uma amina ([M - 2O + 2H]- ou [M - 2O + 3H]+) também tem sido considerada como hipótese à formação do fragmento correspondente à perda de 30 unidades de massa (Straube et al., 2004; Williams e Perreault, 2000; Bonfanti et al., 1996). Esta hipótese é corroborada por estudo realizado em interface APCI com substâncias nitradas e solventes deuterados, onde a perda de 28 unidades de massa foi observada e considerada conseqüência da formação de 47 amina contendo dois átomos de deutério (Karancsi e Slegel, 1999). Bonfanti e colaboradores (1996) observaram a perda de 30 u.m.a. nos espectros da maioria dos compostos nitrados estudados em modo de ionização negativo, exceto no espectro do 1NPi, no qual o produto de redução não foi identificado. Eles atribuem este fato à hipótese de que a reação de redução seja isômeroespecífica. No trabalho de Moriwaki e colaboradores (2000), também não foi observada a formação de produtos de redução ou de eliminação do grupo nitro (NO2) nos espectros de massas obtidos em interface APCI dos nitropirenos estudados (1-nitropireno; 1,3-, 1,6- e 1,8-dinitropirenos) em modo de ionização positiva. No entanto, como aminopirenos (1-aminopireno; 1,3; 1,6 e 1,8diaminopirenos) formaram íons do tipo [M+H]+, foi sugerido que nitropirenos não formam amino-derivados, ou seja, não sofrem redução em interfaces CLAE-EM-APCI. Por exemplo, Bezabeh e colaboradores (2003) observaram que a técnica de ionização química em modo negativo apresenta maior seletividade na ionização de substâncias contendo grupos eletronegativos, inclusive NHPAs, em matrizes ambientais complexas. Outra abordagem feita em relação à ionização de substâncias policíclicas aromáticas considerando a afinidade protônica das espécies (Takáts e Vékey, 1998). Eles sugerem que a ionização positiva em sistemas APCI ou ESI não ocorre de forma satisfatória devido à afinidade protônica de NHPAs ser geralmente menor do que a dos eluentes comumente usados em CLAE em fase reversa, como acetonitrila e metanol. Isto é explicado devido ao efeito de supressão provocado na presença destes eluentes, pois compostos aromáticos de baixa polaridade possuem afinidade protônica menor ou igual do que metanol ou acetonitrila e não são protonados na presença dos mesmos. A presença destes íons em espectros de NHPAs obtidos por CLAE-EMESI em modo de ionização negativo, foi descrita por Williams e Perreault (2000), que sugeriram que a natureza deste fragmento está relacionada à perda de um próton e ao ganho de um átomo de oxigênio ([M-H+O]-) num mecanismo de captura de elétrons via oxidação. Entretanto, esta oxidação ou adição de 16 u.m.a. não foi observada em MALDI em modos positivo e 48 negativo, nem por ionização química em modo negativo. (Williams e Perreault, 2000). 49 CAPÍTULO 3 Objetivos 3.1) Objetivo Geral Desenvolvimento e implementação de metodologia para a determinação de Hidrocarbonetos Policíclicos Aromáticos Nitrados (NHPAs) em fuligem de diesel. 3.2) Objetivos Específicos Otimização de metodologia para a detecção de NHPAs por EM-EM; Desenvolvimento de sistema de eluição para a separação de NHPAs Cromatografia a Líquido de Alta Eficiência (CLAE); Validação da metodologia desenvolvida (CLAE-EM-EM); Determinação de HPAs e NHPAs em amostras de fuligem de diesel fornecidas pela Petrobrás-BR, através da metodologia validada. 50 CAPÍTULO 4 Materiais & Métodos 4.1) Solventes Metanol, acetonitrila, diclorometano e tolueno grau HPLC ou espectrocópico (Tedia Brazil) foram usados (Tabela 1). Tabela 1: Especificação de solventes orgânicos utilizados SOLVENTE ESPECIFICAÇÃO FABRICANTE Metanol (CH3OH) CLAE-ESPECTRO Tedia Company, EUA Acetonitrila (CH3CN) CLAE-ESPECTRO Tedia Company, EUA Diclorometano (CH2Cl2) UV/CLAE Tedia Company, EUA Tolueno (C6H5CH3) UV-Análise de Resíduos EM Science, EUA 51 4.2) Padrões NHPAs sólidos com purezas maiores que 98% (Sigma; Aldrich - EUA) foram adquiridos e utilizados sem purificação adicional. As soluções de todos os NHPAs utilizados neste trabalho foram preparadas a partir da dissolução dos respectivos sólidos. As pesagens foram feitas em balança analítica com precisão de 0,01 mg. 4.3) Outros materiais O material certificado de referência Fuligem de Diesel, SRM 2975 (NIST, EUA) foi empregado para validação de metodologia analítica. Nitrogênio líquido e hélio de alta pureza (PRAXAIR-White Martins, Brasil) foram empregados no sistema de espectrometria de massas. Colunas e pré-colunas (Vydac 201TP54 e Zorbax Eclipse XDB) de fase reversa (C8, C18) foram empregadas no sistema CLAE. Micropipetas de volume fixo e/ou variável e ponteiras descartáveis foram utilizadas no preparo de soluções padrão e na manipulação dos extratos de fuligem. Filtros descartáveis de seringa foram empregados na filtração dos extratos antes de análise por CLAE-EM (Tabela 2). 52 Tabela 2: Especificação de outros materiais utilizados MATERIAL ESPECIFICAÇÃO FABRICANTE Material certificado Fuligem de Diesel NIST, EUA de referência (SRM 2975) Gases N2 líquido He (Pureza > White Martins 99,995%) PRAXAIR, Brasil Colunas e pré-colunas Zorbax Eclipse XDB para CLAE C8 (2.1 x 5 x 50) 201TP54 Agilent, EUA Vydac, EUA C18 (4.6 x 5 x 250) Micropipetas e ponteiras de (50, 100, 250, 500 volume fixo e/ou variável e 1000 µL) Filtros descartáveis de (PTFE; 0,45 µm; 13 mm) Gilson, França Agilent, EUA seringa 4.4) Instrumentação Balança analítica (Série GR, modelo GR-202 – AND, Japão), com precisão de 0,01 mg, foi utilizada para pesagem de NHPAs sólidos e das amostras de fuligem. Com base nas altas taxas de recuperação relatadas na literatura (Schauer et al., 2004; Cvacka et al., 1998) e visando diminuir o tempo total gasto no procedimento analítico, o procedimento de extração de fuligem de diesel desenvolvido neste trabalho foi realizado por banho de ultra-som (USC1800, Unique, Brasil). Uma centrífuga (Excelsa II 206 BL – Fanem, Brasil) e um agitador Vortex (AP 56 – Phoenix, Brasil) também foram usados no procedimento de extração de amostras de fuligem. 53 Para separação e detecção das substâncias de interesse foram empregados, um sistema de cromatografia a líquido de alta eficiência acoplado a um sistema de espectrometria de massas (CLAE-EM) Agilent série 1100, dotado de bomba quaternária, degaseificador, injetor automático, forno de coluna e detector por espectrometria de massas (Agilent SL), com analisador do tipo Ion Trap e interface por ionização a pressão atmosférica por ionização química (APCI). Uma estação de trabalho Agilent foi usada para aquisição de dados e controle do sistema CLAE-EM. Água ultrapura foi obtida por destilação seguida de tratamento em sistema Simplicty (Millipore, EUA). Estufa (Icamo, modelo 3; Brasil) foi utilizada para secagem da vidraria utilizada. Todos os instrumentos utilizados neste trabalho são apresentados na Tabela 3. Tabela 3: Especificação da Instrumentação utilizada INSTRUMENTO ESPECIFICAÇÃO FABRICANTE Série 1100 Agilent, EUA Série 1100 Agilent, EUA Ionização Química à Pressão Agilent, EUA Cromatógrafo a Líquido de Alta Eficiência Espectrômetro de Massas por captura de íons (Ion Trap) Interface CLAE-EM Atmosférica (APCI) Programa de tratamento e aquisição de Versão 4.2 Agilent, EUA Simplicity 185 Millipore, EUA USC-1800 Unique, Brasil AP 56 Phoenix, Brasil Excelsa II 206 BL Fanem, Brasil Série GR, modelo GR-202 AND, Japão Modelo 3 Icamo, Brasil dados (ChemStation) Sistema de purificação de água Banho de ultra-som Agitador Vortex Centrífuga Balança analítica Estufa 54 4.5) Limpeza e descontaminação de vidraria Toda a vidraria empregada no trabalho foi descontaminada através de várias etapas de limpeza. Recipientes e frascos usados na extração e manipulação de amostras eram previamente rinsados com solvente orgânicos (PA) para retirada de resíduos visíveis. Após esta etapa, a vidraria era imersa por 12 horas em banho contendo detergente neutro, específico para limpeza de vidraria (Extran Merck-Brasil ou Detertec, Vetec-Brasil). A vidraria era lavada com água potável, depois com água ultra-pura e em seguida aquecida por 6 a 8 horas na temperatura de 250oC. 55 CAPÍTULO 5 Resultados & Discussão 5.1) Estudos de ionização de NHPAs em interface APCI-EM Nos estudos de ionização, exceto quando mencionado no texto, 20 uL de soluções contendo diferentes NHPAs foram injetados diretamente na interface de ionização química à pressão atmosférica (APCI), sem uso de coluna cromatográfica, na vazão de 0,3 mL/min. Os estudos de ionização realizados neste trabalho utilizaram interface de ionização química à pressão atmosférica (APCI), que foi escolhida por apresentar melhor resposta para substâncias não-iônicas, como no caso das substâncias de interesse. As características de interesse deste tipo de interface foram discutidas no item 2.4. 5.1.1) Avaliação dos padrões de fragmentação de 1-NPi com detecção por varredura Espectros de massas foram adquiridos, em ionização positiva e negativa, por varredura, na faixa de m/z = 100-500 para soluções de 1-NPi 1000, 500 e 100 µg/L, preparadas em ACN. O sistema de eluição utilizado foi MeOH/H2O (20:80). O padrão de fragmentação de 1-NPi foi avaliado 56 comparando-se as massas dos fragmentos e as áreas de cada um deles nos dois modos de ionização. O programa de integração do EM foi utilizado com esta finalidade. Em modo de ionização negativo foi observada principalmente a formação do íon de peso molecular m/z 247 ([M]-), que corresponde ao ganho de 1 elétron. O principal fragmento observado em modo negativo foi o íon de m/z 217 ([M-30]-) que corresponde à perda de 30 unidades de massa. Outros fragmentos observados em menor intensidade foram os íons de m/z 262 e 231. O íon de m/z 262 corresponde a um mecanismo de oxidação e desprotonação ([M-H+16]-) (Williams e Perrault, 2000), enquanto o íon de m/z 231 corresponde à perda de um átomo de oxigênio ([M-16]-) (Bonfanti et al., 1996). Traços do fragmento de m/z 201, correspondente à perda de NO2 ([M-46]-) também foram observados. Em modo de ionização positivo, o principal íon observado foi o fragmento correspondente à protonação da molécula original seguida de perda de um átomo de oxigênio de (m/z 232; [M+H-O]+). Nos espectros adquiridos em modo positivo, foi observada em menor intensidade a formação do íon de pseudo-peso molecular (m/z 248; [M+H]+), que corresponde à protonação da molécula original. Também foram observados os fragmentos de m/z 218 ([M-30+H]+), que corresponde à perda de 30 unidades de massa em decorrência da fragmentação do íon de m/z 248 ([M+H]+) e traços do fragmento de m/z 202, que corresponde à protonação da molécula original seguida de perda de NO2 ([M+H-46]+). Os resultados obtidos em ambos os modos de ionização estão apresentados na Tabela 4 e na Figura 6. 57 Tabela 4: Principais íons observados nos espectros de varredura de 1NPi, obtidos por CLAE-EM-APCI, em modo de ionização positivo e negativo. Modo de Íons Observados Fragmentos Possível Ionização (m/z) observados Explicação APCI 247 [M]- Captura de 1 elétron Negativo 217 [M-30]- Redução e/ou perda de NO 262 [M-H+16]- Oxidação e desprotonação 231 [M-16]- Perda de um átomo de oxigênio 201 [M-46]- Perda de NO2 232 [M+H-O]+ Protonação seguida de perda APCI Positivo de oxigênio 248 [M+H]+ Protonação 218 [M+H-30]- Redução e/ou perda de NO 202 [M+H-46]- Protonação seguida de perda de NO2 Em nossos experimentos, as melhores condições de ionização foram obtidas com fase móvel contendo os solventes próticos MeOH e H2O, o que de acordo com a hipótese de Karancsi e Slegel (1999) favorece a ionização de NHPAs, indicando que a redução dos mesmos é uma hipótese plausível. Os espectros de massa da Figura 6 ilustram a diferença de intensidade de sinal em ionização positiva e em ionização negativa, bem como os principais íons observados em cada situação. Em modo negativo, os sinais obtidos para o íon de m/z 247 [M]- foram sempre maiores do que os obtidos em modo positivo para o íon de m/z 248 ([M+H]+). 58 a) b) Figura 6: Comparação dos espectros de 1NPi por CLAE-EM-APCI, com detecção por varredura, em modo de ionização negativa (a) e positiva (b). 59 Como é possível observar na comparação das Figuras 6a e 6b, o sinal em modo positivo tem intensidade que é, pelo menos, uma ordem de grandeza inferior a obtida em modo positivo. Ademais, o espectro obtido em modo positivo apresenta maior contribuição do background do sistema, com vários íons inespecíficos. A maior sensibilidade observada em modo de ionização negativo está certamente relacionada com o caráter eletrofílico dos NHPAs, que podem estabilizar uma carga negativa adicional obtida por um mecanismo de captura de elétrons, fato também observado por outros autores (Bonfanti et al., 1996; Moriwaki et al., 2000; Williams & Perreault, 2000; Bezabeh et al., 2003; Schauer et al., 2004; Straube et al., 2004). Este fato pode ser interpretado considerando-se, nas moléculas estudadas, as estruturas de ressonância eletrônica do grupo nitro (NO2), que, por ser eletronegativo, é considerado é um forte retirador de elétrons do sistema aromático. Independente do posicionamento dos elétrons nas estruturas de Lewis (Figura 7), o átomo de nitrogênio sempre possui uma carga formal positiva e por efeito de indução diminui a densidade eletrônica do anel aromático e aumenta seu caráter eletrofílico, favorecendo a captura de elétrons disponíveis no sistema de ionização. Em modo de ionização negativo, a conseqüência da descarga de elétrons emitida pela agulha corona sobre as moléculas dos NHPAs na interface APCI é a captura de um elétron, formando predominantemente um ânion que tem o peso molecular da molécula original. A aromaticidade do anel é mantida e por este motivo a carga negativa é bem estabilizada nos sistemas aromáticos dos NHPAs. 60 O N + _ O Figura 7: estrutura de ressonância eletrônica do grupo nitro (NO2) na molécula de 1NPi. Em modo de ionização positivo, a formação de íons positivamente carregados pode ocorrer por um mecanismo de captura de um próton ou protonação, decorrente de um ataque eletrofílico. A conseqüência da entrada de um eletrófilo no sistema aromático (neste caso, de um próton oriundo do sistema de eluição) é a quebra da aromaticidade do sistema, gerando espécies iônicas altamente instáveis. No sentido de reaver a aromaticidade e conseqüentemente diminuir a energia potencial do sistema, pode ocorrer uma reação de substituição eletrofílica aromática, pela abstração de um próton ou pode haver a reversão da reação. Por este motivo, a ionização de NHPAs em modo positivo não é favorecida, uma vez que os sistemas aromáticos dos NHPAs não são capazes de estabilizar a carga positiva gerada pela captura de um próton tão bem quanto são capazes de estabilizar a adição de uma carga negativa. Deste modo, a ionização negativa é favorecida em detrimento da positiva. 5.1.2) Avaliação dos padrões de fragmentação de outros NHPAs com detecção por varredura O interesse no estudo dos padrões de fragmentação de outros NHPAs visou o posterior desenvolvimento de metodologia para sua determinação em fuligem de diesel e devido aos resultados obtidos anteriormente com 1-NPi (ver ítem 5.1.1), apenas o modo de ionização negativo foi considerado. 61 Espectros de massas com detecção por varredura (SCAN) foram adquiridos na faixa de 100-500 (m/z) para estudo dos padrões de fragmentação dos seguintes NHPAs selecionados: 9-nitroantraceno (9-NA), 2-nitrofluoreno (2NFl), 1- e 2-nitronaftalenos (1-NN e 2-NN) e para os seguintes di-NHPAs: 1,3-, 1,5- e 1,8-dinitronaftalenos (1,3-, 1,5- e 1,8-diNN), 2,5- e 2,7-dinitrofluorenos (2,5- e 2,7-diNFLs). O sistema de eluição utilizado foi MeOH/H2O (20:80). Os íons predominantes na fragmentação de cada NHPA estudado estão descritos na Tabela 5. A aquisição de dados em varredura mostrou que os principais íons gerados, para a maioria dos NHPAs, foram os respectivos íons de peso molecular ([M]-), correspondentes à captura de elétron. Exceto no caso do 2NFl e dos isômeros 2,5 e 2,7-diNFL, foi observada principalmente formação de íons do tipo [M-H]-, correspondendo à captura de elétron e desprotonação, em detrimento dos íons do tipo [M]-. No caso do 2-NFl, a intensidade do íon de m/z 210 ([M-H]-) foi sempre maior do que a do íon de peso molecular de m/z = 211 ([M]-) (Figura 8). A perda de um próton permite que o carbono 9 do 2-nitrofluoreno participe do sistema aromático aumentando a estabilidade do ânion obtido. Resultado análogo é obtido na ionização de fluoreno, onde o íon de peso molecular e o íon de [M-H]- tem intensidades comparáveis. O mesmo padrão de fragmentação foi observado no caso do 2,5diNFL, onde o íon de m/z 255 ([M-H]-) predominou em relação ao íon de m/z = 256 ([M]-). A presença de íons [M-H]- foi também observada nos espectros de 1-NPi, 9-NA e 1-NN obtidos por ionização em interface ESI (Williams e Perreault, 2000), mas a razão entre as intensidades dos mesmos não foi discutida por ela. 62 Tabela 5: Caracterização da fragmentação de NHPAs selecionados por CLAEEM-APCI, em modo negativo, com detecção por varredura. NHPA 9-NA PM Íons (m/z) Fragmentos Possível (g/mol) Observados observados Explicação 223 223 [M]- captura de elétron 193 [M-30]- perda de NO ou redução na fonte 2-NFl 211 238 [M-H+16]- Oxidação e desprotonação 210 (>) [M-H]- Desprotonação 211 (<) [M]- captura de elétron 180 [M-H-30]- perda de NO ou redução na fonte e desprotonação 181 - [M-30] perda de NO ou redução na fonte 1-NN 225 [M-H+16]- Oxidação e desprotonação 173 173 [M]- captura de elétron 218 218 [M]- captura de elétron 172 [M-46]- perda de NO2 188 [M-30]- perda de NO ou redução na 2-NN 1,3diNN 1,5diNN fonte 1,8diNN 233 [M-H+16]- Oxidação e desprotonação 158 [M-60]- Perda e/ou redução de 1 e/ou 2 grupos NO 255 (>) [M-H]- Desprotonação e 256 (<) [M]- captura de elétron 2,7- 225 (>) [M-H-30]- perda de NO ou redução na 2,5- 256 diNFl diNFl fonte e desprotonação 226 (<) - [M-30] perda de NO ou redução na fonte 63 Figura 8: diferença de intensidades dos íons de m/z 211 e 210 no espectro de varredura de 2NFl adquirido por CLAE-EM-APCI em modo de ionização negativo. Assim como observado para 1-NPi, nos espectros de varredura dos demais NHPAs também foi identificada a perda de 30 unidades de massa ([M30]-) a partir do íon de peso molecular, que indica a perda de NO (Kormacher et al., 1987; Schauer et al., 2004) ou um processo de redução na fonte (Bonfanti et al., 1996; Williams & Perreault, 2000; Straube et al., 2004), conforme discutido anteriormente. No caso do 2NFl e dos isômeros 2,5 e 2,7-diNFL, os fragmentos de m/z 180 e 225 ([M-H-30]-) predominaram em relação aos fragmentos de m/z 181 e 226 ([M-30]-), respectivamente, embora estes últimos também tenham sido identificados em menor intensidade. No caso do 1,8-diNN, houve predominância do fragmento de m/z 172 ([M-46]-) em relação ao fragmento de m/z 188 ([M-30]-). Este fato sugere que a perda de um dos grupos nitro (NO2) pode ser um processo importante para diNHPAs. Na investigação espectral não foi observada formação de nenhum outro fragmento que indicasse a perda do segundo grupo NO2. Ainda no 64 espectro do 1,8-diNN, foi observada pequeno sinal do íon 158, que correspondente ao íon [M-60]- e que poderia indicar a redução de um dos grupos NO, a redução de ambos ou a perda de um deles e a redução de outro, ou mesmo uma mistura destes mecanismos. Traços do fragmento de m/z 172 ([M-NO2]-) também foram observados por Bonfanti e colaboradores (1996), em espectros de massa obtidos por ionização química em modo negativo de 1,8diNN. Na fragmentação de alguns NHPAs selecionados (9-NA; 1,3-; 1,5- e 1,8diNN; 2-NFl), foi identificada a presença de íons do tipo [M-H+16]- (Tabela 5), que correspondem à perda de um próton e ao ganho de um átomo de oxigênio ([M-H+O]-) num mecanismo de captura de elétrons via oxidação (Williams e Perreault, 2000). 5.1.3) Avaliação da detecção de NHPAs por monitoramento de reações múltiplas (MRM) Foi realizada a comparação dos sinais e da seletividade da detecção de NHPAs por monitoramento de reações múltiplas (MRM). O sistema de eluição utilizado foi MeOH/H2O (20:80) e a aquisição de dados foi feita em modo de ionização negativo, pelo monitoramento de duas etapas consecutivas de fragmentação, considerando-se os principais íons de cada substância. O modo de MRM foi escolhido para a detecção do 1-NPi devido a seletividade que esta forma de detecção permite. A detecção por monitoramento de reações múltiplas (MRM) pressupõe o isolamento de um íon e sua fragmentação após isolamento pelo sistema Ion Trap, o que torna a detecção extremamente seletiva. Algumas das características desta técnica de detecção foram discutidas anteriormente (ítem 2.6). Com base nos resultados anteriores, foi avaliado que a melhor forma de detecção de NHPAs seria o uso de MRM em modo negativo, isolando-se os íons de peso molecular e monitorando-se o íon [M-30]- na etapa seguinte de fragmentação. Exceto no caso do 2-NFl e dos isômeros 2,5 e 2,7-diNFL, foram detectados os íons de m/z 180 e 225 ([M-H-30]-) a partir do isolamento e fragmentação dos íons precurssores 210 e 255 do tipo [M-H]-, conforme discussão anterior. 65 Uma limitação desta técnica de detecção é quando o sinal gerado pelo íon proveniente do 2º estágio de fragmentação apresenta baixa intensidade. No caso dos NHPAs estudados e especificamente no caso do 1-NPi, esta relação é suficientemente elevada em modo de ionização negativo para a detecção com baixos limites de detecção, como será discutido adiante. O modo de ionização positivo foi também avaliado nestas condições, embora os próprios estudos de varredura já apontassem para menor sensibilidade de detecção neste modo. Resultados típicos são apresentados na Tabela 6, onde é possível verificar que a intensidade em MRM em modo negativo é cerca de 400 vezes maior. Tal fato é certamente decorrente da ionização menos eficiente em modo positivo. Tabela 6: detecção de 1NPi por MRM em modo de ionização positivo e negativo; sistema de eluição MeOH/H2O (20:80) Modo de EM1 EM2 Ionização (m/z) (m/z) Negativo 247 217 [M]- [M-30]- 248 218 [M+H]+ [M-30+H]+ Positivo Intensidade de sinal (u.a) 1,9. 106 4,5. 103 Como o 1-NPi é o principal NHPA de interesse em fuligem de diesel e é o único certificado em fuligem de diesel (SRM 2975 – NIST-EUA), grande parte do trabalho subseqüente foi baseado na otimização de condições para a detecção desta substância nesta matriz. 5.2) Otimização do sistema de eluição para determinação de NHPAs por CLAE-EM A resposta de 1-NPi com detecção por MRM, utilizando-se os íons descritos acima (Tabela 6), foi comparada nos dois modos de ionização (positivo e negativo) em função de três sistemas de solventes de eluição: 66 ACN/H2O, ACN/MeOH e MeOH/H2O, em proporções variáveis entre 20 a 80% de cada solvente. A ionização das moléculas do analito em interfaces APCI ocorre por descarga de elétrons provenientes de uma agulha do tipo corona. Neste momento, as moléculas do eluente encontram-se em excesso em relação às moléculas do analito e são ionizadas preferencialmente. Conseqüentemente, as moléculas do analito são ionizadas a partir das moléculas do eluente em fase gasosa e, portanto, dependem da composição do meio (Niessen, 1995). Foi observado que, em modo de ionização positivo, o íon de m/z 218 ([M-30+H]+) gerou sinais comparáveis ao ruído eletrônico nos três sistemas de eluição testados, o que, mais uma vez confirmava a inviabilidade de detecção deste NHPA por MRM neste modo. Em modo negativo (Figura 6), os sinais obtidos para o íon de m/z 217 ([M-30]-) foram sempre cerca de três ordens de grandeza maiores do que os resultados obtidos em modo positivo para o íon de m/z 218 ([M-30+H]+) (Tabela 6). A fase móvel MeOH/H2O (80:20) apresentou os sinais de maiores intensidades e melhores relações sinal/ruído em modo de ionização negativo (Figura 9). Esta composição de fase móvel foi adotada nas etapas posteriores do trabalho. O aumento de sinal com metanol está relacionado às reações de transferência de carga em CLAE-EM-APCI, pois o uso de ACN leva à redução da sensibilidade na medida. A redução da detecção de 1-NPi em sistemas de eluição contendo ACN foi anteriormente observada em interface ESI (Williams e Perreault, 2000). 67 Figura 9: Cromatograma e espectro de massas de 1NPi (15 µg/L) obtido por CLAE-EM-APCI, com detecção por MRM em modo de ionização negativo. 5.3) Comparação dos fatores de resposta de soluções-padrão de 1-NPi preparadas em diferentes solventes, por injeção direta (sem coluna) Foram preparadas soluções de 1-NPi com concentrações de 1, 10, 50, 100 e 500 µg/L em ACN, MeOH e DCM e foram injetados 20 µL de cada uma das soluções, diretamente no espectrômetro de massas, sem uso de coluna cromatográfica, na vazão de 0,3 mL/min. O sistema de eluição utilizado foi MeOH/H2O (80:20). A aquisição de dados foi feita por MRM, em interface APCI com ionização negativa. DCM, MeOH e ACN foram selecionados por serem alguns dos principais solventes orgânicos usados em extrações de substâncias orgânicas ou como fase móvel em CLAE. DCM é particularmente importante por ser um solvente de escolha na extração de HPAs e NHPAs de amostras sólidas (Guillén, 1994). A ionização em modo negativo apresentou maiores sinais em soluções preparadas em metanol, que foi, portanto, usado na preparação das soluções de NHPAs estudadas neste trabalho. No desenvolvimento do trabalho foi observada forte redução do sinal na injeção direta de soluções preparadas apenas em DCM, onde não era possível detectar 1-NPi. Este fato afetou de forma drástica o desenvolvimento do trabalho conforme será discutido adiante, já que inicialmente era pretendido 68 fazer injeção direta no sistema de CLAE-EM e tirar proveito da seletividade do sistema de detecção. 5.4) Otimização da vazão do sistema de eluição A influência da vazão do sistema de eluição de CLAE no sinal de espectrometria de massas foi avaliada utilizando-se detecção por MRM e injeção direta no espectrômetro de massas. Duas concentrações diferentes de 1-NPi (100 e 500 µg/L; MeOH) foram avaliadas em vazões de de 0,3 a 0,5 mL/min do sistema de eluição MeOH/H2O (20:80). As respostas relativas para injeção de 20 uL de soluções de 1-NPi nas diferentes vazões são apresentadas na Figura 10. Na menor vazão há a maior resposta relativa, como seria de se esperar, pois há menor diluição do 1-NPi. 4,00E+07 3,50E+07 3,00E+07 2,50E+07 área (EIC m/z = 217) 2,00E+07 1NPi 500 ppb 1NPi 100 ppb 1,50E+07 1,00E+07 5,00E+06 0,00E+00 0,3 0,4 0,5 vazão de fase móvel (mL/min) Figura 10: Variação do sinal de soluções de 1-NPi em função da vazão do sistema de eluição 5.5) Otimização de parâmetros eletrônicos do espectrômetro de massas para determinação de 1-NPi A otimização de parâmetros eletrônicos do espectrômetro de massas foi realizada com infusão de solução de 1-NPi (100 µg/L; MeOH) diretamente no 69 espectrômetro de massas, em vazão constante de 300 µL/min e monitoramento do íon de m/z = 247. Foram otimizadas (automaticamente através da estação de trabalho do equipamento) as voltagens para os seguintes parâmetros do espectrômetro de massas: capilar, skimmer, saída do capilar, octapolos, trap drive e lentes (Tabela 7). Tabela 7: Valores dos parâmetros do espectrômetro de massas otimizados para a detecção de 1-NPi PARÂMETRO VALOR OTIMIZADO CAPILAR 1000 V SKIMMER -33,1 V SAÍDA DO CAPILAR -107,4 V OCTAPOLO 1 DC -4,4 V OCTAPOLO 2 DC -1,37 V TRAP DRIVE 40,03 V OCTAPOLO RF 136 V LENTE 1 0,82 V LENTE 2 33,6 V TARGET MASS 30.000 TEMPO DE ACUMULAÇÃO MÁXIMO 50 ms 70 5.6) Otimização de parâmetros da interface APCI para determinação de 1-NPi A ionização em APCI acontece em fase vapor, logo é influenciada diretamente pela temperatura de vaporização (Tvap) no nebulizador e pela temperatura de secagem (Tsec) do spray na interface e a formação do aerossol é um parâmetro crítico no processo de ionização nesse tipo de interface. Para auxiliar o processo de secagem do aerossol na interface APCI, o equipamento utilizado neste trabalho dispõe de um mecanismo auxiliar que fornece um aporte de gás (N2) em sentido contrário ao orifício de entrada do capilar do espectrômetro de massas. Condições de CLAE-EM iguais às descritas no item 5.5 foram usadas neste experimento. Além destas, foi usada a vazão de gás de secagem de 5 L/min recomendada (Agilent Technologies, sem data) para a interface APCI na vazão de eluente empregada (0,300 mL/min) e embora a pressão do gás de nebulização também possa ser otimizada, foi usado o valor de 60 PSI. Para a otimização da temperatura de vaporização e da temperatura de secagem, foram usadas soluções de 1-NPi de diferentes concentrações (5 e 100 µg/L; MeOH) e dois experimentos foram realizados. No primeiro, variou-se a temperatura de vaporização em função da temperatura de secagem fixada em 350°C. Os melhores resultados foram obtidos na faixa de 350-400°C, para ambas as concentrações estudadas (Figura 11a-b). Em função dos resultados obtidos na avaliação da temperatura de vaporização, a temperatura de secagem foi avaliada em duas temperaturas de vaporização (350°C e em 400°C). Soluções de 1-NPi 5 e 100 µg/L foram estudadas e os resultados obtidos estão apresentados na Figura 12a-b. Para as duas concentrações estudadas, os melhores resultados, isto é os sinais de maiores intensidades, foram obtidos em temperatura de vaporização de 350°C. A melhor temperatura de secagem para a solução de 5 µg/L foi de 350°C e para a solução de 100 µg/L foi de 300°C. Certamente esta diferença está relacionada às concentrações avaliadas em cada caso, pois as respostas não são proporcionais para as duas soluções em cada temperatura. 71 Como a análise de amostras contendo baixas concentrações de NHPAs, era de interesse do método, tanto a temperatura de vaporização quanto a temperatura de secagem foram mantidas em 350°C. 72 Otimização da Temperatura de Vaporização; 1NPi 5 ug/L (MeOH) 1,80E+06 1,60E+06 1,40E+06 área EIC 217 (MRM) 1,20E+06 1,00E+06 8,00E+05 6,00E+05 4,00E+05 2,00E+05 0,00E+00 220 250 280 320 350 380 400 450 Temperatura de Vaporização (°C) Otimização da Temperatura de Vaporização; 1NPi 100 ug/L (MeOH) 3,50E+07 3,00E+07 área EIC 217 (MRM) 2,50E+07 2,00E+07 1,50E+07 1,00E+07 5,00E+06 0,00E+00 220 250 280 320 350 380 400 450 Temperatura de Vaporização (°C) Figura 11: Variação da área de sinal de soluções de 1-NPi em função da temperatura de vaporização: (a) 1-NPi 5 µg/L; (b) 1-NPi 100 µg/L 73 Otimização da Temperatura de Secagem; 1NPi 5 ug/L (MeOH) 2,00E+06 1,80E+06 1,60E+06 área EIC 217 (MRM) 1,40E+06 1,20E+06 VAP TEMP = 350°C VAP TEMP = 400°C 1,00E+06 8,00E+05 6,00E+05 4,00E+05 2,00E+05 0,00E+00 300 335 350 Temperatura de Secagem (°C) Otimização da Temperatura de Secagem; 1NPi 100 ug/L (MeOH) 3,50E+07 3,00E+07 área EIC 217 (MRM) 2,50E+07 2,00E+07 VAP TEMP = 350°C VAP TEMP = 400°C 1,50E+07 1,00E+07 5,00E+06 0,00E+00 300 335 350 Temperatura de Secagem (°C) Figura 12: Variação da área de sinal de soluções de 1-NPi em função da temperatura de secagem. (a) 1-NPi 5 µg/L; (b) 1-NPi 100 µg/L 74 5.7) Estudos de Extração (I): Avaliação da extração de 1-NPi com detecção por injeção direta Extração ultra-sônica foi escolhida para realizar a extração de NHPAs de fuligem de diesel. Esta técnica de tem sido rotineiramente utilizada em nosso laboratório para a determinação de HPAs (Pereira Netto et al., 2000, 2001, 2002a,b) e mais recentemente para a extração de metais (Soriano et al., 2006). Ela apresenta vantagens como rapidez, custo relativamente baixo e tanto reprodutibilidade quanto robustez adequadas. Substâncias policíclicas aromáticas com substituintes polares (nitro, amino e hidroxi derivados) têm sido caracterizadas em materiais certificados de referência para material particulado de diesel, tanto por CGAR-EM quanto por CLAE-EM (Bayona et al, 1988). O material de referência certificado SRM 2975 (National Institute of Standards & Technology - NIST) é um padrão de referência internacional para a avaliação de metodologias analíticas empregadas na determinação de HPAs em material particulado de diesel e matrizes similares. Dentre os NHPAs, fornece informação acerca da concentração de 1-NPi (36 mg/Kg). O material de referência (fuligem de diesel; SRM 2975; NIST, EUA), foi armazenado em geladeira, dentro de dessecador de vidro, destinado exclusivamente a esta finalidade, no qual a sílica era periodicamente seca. O dessecador e o frasco contendo tal padrão eram retirados da geladeira pelo menos 20 minutos antes da pesagem, até estabilização da temperatura. Em seguida, a fuligem era retirada do frasco com o auxílio de espátula de vidro e a pesagem do material realizada em balança analítica com precisão de 0,01 mg. O material era pesado diretamente no recipiente onde era realizada a extração. Imediatamente após a pesagem, o mesmo era coberto com papel alumínio e levado à capela de exaustão, onde o solvente de extração era adicionado. Nesta primeira parte dos estudos de extração, foram extraídas inicialmente massas de cerca de 20 mg (cerca de 720 ng de 1-NPi). As condições de extração foram avaliadas e otimizadas considerando-se a eficiência e a recuperação obtida em cada conjunto de situações avaliada. Os extratos obtidos na primeira parte dos estudos de extração foram analisados por injeção direta (sem coluna cromatográfica) e, exceto quando 75 mencionado no texto, foram injetados 20 µL de cada extrato diretamente no espectrômetro de massas, na vazão de 0,3 mL/min. É importante ressaltar que até esta etapa, o trabalho foi realizado por injeção direta das soluções de NHPAs no espectrômetro de massas, ou seja, até este momento o sistema de CLAE era utilizado apenas como sistema de injeção automático, sem coluna cromatográfica. 5.7.1) Extração de SRM 2975 com ultra-som e diclorometano Para a realização dos experimentos de extração, o solvente de escolha foi DCM, pois este é um dos solventes mais eficientes para a extração de NHPAs de amostras sólidas (Guillén, 1994). Alíquotas de cerca de 20 mg de SRM 2975 foram pesadas em tubo de ensaio e submetidas a três etapas de extração em banho de ultra-som (15 mL de DCM; 20min cada). Ao término de cada etapa de sonicação, o extrato parcial era submetido à centrifugação (5 min, 3500 RPM). Após centrifugação, o sobrenadante de cada etapa, foi retirado e transferido para balão volumétrico, cujo volume era completado com DCM. Considerando-se a concentração de 1-NPi na amostra certificada (36 mg/kg) e a massa de amostra certificada pesada, a concentração do extrato bruto resultante era da ordem de 14,4 µg/L. Uma alíquota de 10 mL deste extrato foi concentrada sob fluxo de N2 até 1mL, ([1-NPi] ~144 µg/L), filtrada e analisada. A Figura 13 representa o procedimento de extração realizado. 76 20 mg SRM 2975 15 mL DCM; U.S.; 20 min. Centrifugar (5min, 3500 RPM) Repetir 3 vezes Retirar sobrenadante e transferir Extrato bruto (DCM; 50 mL); [1-NPi]~14,4 µg/L 1) 2) alíquota 10 mL N2 (g) 3) filtração Extrato concentrado (DCM; 1 mL); [1-NPi]~ 144 µg/L CLAE-EMEM Figura 13: Avaliação da detecção de 1-NPi após procedimento de extração de material certificado (NIST, SRM 2975) com ultra-som e DCM. Os extratos obtidos em DCM não apresentaram sinal de resposta de 1NPi. Na tentativa de solucionar a questão que se apresentava, vários procedimentos foram realizados e a resposta de 1-NPi era avaliada após cada um deles: 1) troca de solvente para ACN e para MeOH após a extração com DCM; 2) extração ultra-sônica com metanol e com tolueno; 3) extração com agitação mecânica; 5) adição de solução padrão de 1-NPi aos extratos obtidos com DCM. Nenhuma destas intervenções resultou na detecção de 1-NPi e foi verificado que a interferência do DCM na ionização daquele NHPA era crítica. Desta forma, foi decidido usar uma coluna cromatográfica para a separação prévia do DCM e do 1-NPi nos extratos, antes da transferência para a interface de ionização. 77 5.8) Avaliação do efeito do DCM na detecção de 1NPi por CLAE-EM-EM Os experimentos anteriores (nos quais a detecção de 1-NPi foi realizada por injeção direta) demonstraram que há influência direta na determinação de 1NPi por CLAE-EM/APCI, em modo negativo, de acordo com a presença de DCM em solução. Diante da impossibilidade em detectar 1-NPi em soluções preparadas apenas em DCM, através de injeção direta no sistema de espectrometria de massas, foi introduzida uma etapa cromatográfica na metodologia desenvolvida, a fim de avaliar se a separação do solvente (DCM) e da substância de interesse (1-NPi) produziria resultado positivo em sua detecção por APCI em modo negativo. Dada a ampla e consagrada utilização de colunas de fase reversa para a separação de HPAs e NHPAs (Wise et al, 1993; Furton et al., 1993) e dadas as características físico-químicas dos mesmos, foram avaliadas duas colunas cromatográficas de fase reversa do tipo octadecilsílica (Tabela 8). Tabela 8: Colunas cromatográficas estudadas na separação de DCM e 1-NPi Nome comercial Diâmetro Tamanho de Comprimento Interno (mm) Partícula (um) (mm) Zorbax Eclipse XDB 2.1 5 50 Vydac 201TP54 4.6 5 250 Nos experimentos com a coluna Zorbax Eclipse XDB, foram utilizadas soluções individuais de 1-NPi, com concentração 1000 µg/L em DCM e em MeOH e suas respostas foram avaliadas em função de fases móveis contendo ACN (100%) e de composição MeOH/H2O (20:80), com vazão de 0,3 mL/min e detecção por CLAE-EM-APCI, em modo negativo (MRM, m/z 247/217). Nos experimentos com a coluna Vydac 201TP54, foram preparadas soluções individuais de 1-NPi de concentração 100 e 1000 µg/L em diferentes composições de DCM e em MeOH (0 a 100%) e suas respostas foram 78 avaliadas utilizando como fase móvel apenas MeOH, com vazão de 1 mL/min e detecção por CLAE-EM-EM-APCI, em modo negativo (MRM, m/z 247/217). Os resultados obtidos com o uso da coluna Zorbax Eclipse XDB não foram satisfatórios e seu o uso foi descartado. Dadas as características estruturais desta coluna, a mesma opera em baixas vazões, o que por sua vez prejudica a ionização dos analitos na interface APCI, vide o mecanismo de ionização que ocorre neste tipo de interface, descrito anteriormente (ver item 2.4). Por outro lado, os resultados obtidos com a coluna Vydac 201TP54 foram promissores e satisfatórios e sendo assim, todas as etapas subseqüentes deste trabalho foram realizadas com a coluna Vydac 201TP54, a qual apresenta performance otimizada em vazões mais altas do que a coluna Zorbax Eclipse XDB, favorecendo o processo de ionização na interface APCI. A fim de obter uma avaliação mais detalhada da influência de DCM na ionização de 1-NPi por APCI em modo de ionização negativo (MRM m/z 247/217) e de avaliar se havia uma condição analítica que permitisse a presença de DCM em solução sem que isto prejudicasse sua detecção por este modo de ionização, foram analisadas soluções de 1-NPi de concentração 100 e 1000 µg/L, preparadas em diferentes composições de DCM e MeOH. Ao comparar as soluções preparadas em 100% de cada solvente, foi verificado que a solução preparada apenas em DCM apresentava sinal reduzido em cerca de três ordens de grandeza em relação à solução preparada em MeOH. A Figura 14 apresenta a variação do sinal de 1-NPi em função da porcentagem de DCM em solução. 79 3,20E+07 3,10E+07 área EIC 217 (MRM) 3,00E+07 2,90E+07 2,80E+07 2,70E+07 2,60E+07 2,50E+07 0 20 30 40 50 60 % DCM Figura 14: Variação do sinal de 1-NPi em função da concentração de DCM na solução. Em virtude do demasiado efeito de solvatação exercido pelo DCM sobre as moléculas de 1-NPi (diminuindo a interação das mesmas com a coluna), foi observado alargamento dos picos cromatográficos de acordo com o aumento da % de DCM nas soluções, que ademais apresentaram aspecto “serrilhado”. Tão pouco foi possível realizar integração automática dos picos, sendo necessário proceder de forma manual. Assim, para tornar o procedimento de integração manual mais consistente, foi necessário utilizar a ferramenta de suavização de picos cromatográficos (smooth chromatogram) disponível no software de tratamento e análises de dados do equipamento, tornando possível realizar a integração de forma mais uniforme. A composição DCM/MeOH (30:70) apresentou sinal ligeiramente maior que as demais, indicando que o DCM pode, de alguma forma, facilitar o processo de transferência de cargas em interface APCI. A partir da composição de DCM/MeOH (40:60) foi observado alargamento significativo dos picos cromatográficos, resultando em diminuição de aproximadamente 10% das áreas medidas. 80 Estes resultados são importantes, pois viabilizam o uso de DCM para a extração dos NHPAs de interesse, sem que haja necessidade de eliminá-lo totalmente do extrato, visto que este solvente sempre foi nossa primeira opção para o processo de extração. A composição DCM/MeOH (30:70) foi escolhida e fixada para a composição final dos extratos de 1NPi em fuligem de diesel a serem estudados posteriormente. Com esta condição, não foi necessário introduzir uma etapa adicional de eliminação do DCM por evaporação, que poderia acarretar na perda por volatilização dos compostos de interesse (sobretudo dos mais leves), diminuição da eficiência e aumento do tempo total de análise da metodologia desenvolvida. A Tabela 9 apresenta as condições otimizadas para a determinação de NHPAs em fuligem de diesel (FD) por CLAE-EM-APCI. Os parâmetros eletrônicos do espectrômetro de massas utilizados foram aqueles apresentados na Tabela 7. Tabela 9: Parâmetros analíticos otimizados para a determinação de 1-NPi em FD PARÂMETRO OTIMIZAÇÃO Método de extração Ultra-som Solvente de extração DCM Composição do extrato de FD DCM/MeOH (30:70) Coluna cromatográfica (sistema CLAE) Vydac 201TP54 (ODS; 4.6mm; 5um; 250mm) Fase Móvel MeOH (100%) Volume de injeção 20 uL; injeção automática Vazão de fase móvel 1 mL/min Interface CLAE-EM APCI Modo de ionização Negativo Temperatura de vaporização 350°C Temperatura de secagem 350°C Vazão do gás de secagem 5 L/min Pressão de nebulização 60 PSI Modo de detecção (EM/ION TRAP)* MRM 81 5.9) Figuras de Mérito do método desenvolvido De modo geral, as calibrações foram realizadas nas faixas de 0,5 a 15 µg/L ou de 5 a 50 µg/L, em função da massa de amostra extraída e, portanto, da concentração de 1-NPi esperada. Padrões e extratos foram analisados em triplicata nas condições das Tabelas 7 e 9 para a quantificação de 1-NPi nos diferentes experimentos de extração realizados. Valores experimentais dos coeficientes de correlação e dos limites de detecção (3*S/N) e de quantificação (10*S/N) são apresentados na Tabela 10. Tabela 10: Figuras de mérito do método para detecção de 1NPi em FD Região da curva Coeficiente de Em solução Massa injetada* Em amostra** de calibração correlação (µg/L) (pg) (mg/Kg) (µg/L) LD LQ LD LQ LD LQ 0,5 a 15 > 0,999 0,2 0,5 3 10 1,5 5,0 5 a 50 > 0,999 1,2 4,2 24 84 3,0 10,0 LD = limite de detecção; LQ = limite de quantificação. *calculado considerando-se 20 µL **calculados em relação às massas de SRM extraídas em diferentes experimentos O limite de detecção calculado para o método desenvolvido (0,2 µg/L) é melhor do que o obtido por Moriwaki (2000) (25 pg em termos de massa injetada) por CLAE-EM-APCI foram calculados, embora tenham sido piores que os obtidos por Bonfanti (1996) (5 pg em termos de massa injetada) através de CLAE-EM-PB. O limite de detecção calculado também é melhor que o valor apresentado por Schauer (2004) (0,8 µg/L em solução) em análises conduzidas por CLAE-EM-APCI-TOF. Entretanto deve ser considerado que os diferentes arranjos experimentais utilizados (fontes, interfaces, sistema de espectrometria de massas etc) não permitem uma comparação direta destes resultados. Valores dos coeficientes de correlação e dos limites de detecção (LD) e de quantificação (LQ) expressos em termos do método desenvolvido e das massas estudadas (~2 mg de FD) são também apresentados na Tabela 10. Estes resultados indicam que é possível detectar até cerca de 1,5 mg de 1-NPi por Kg de FD. 82 5.10) Estudos de Extração (II): Avaliação da extração de 1-NPi por CLAEEM Após a introdução da etapa cromatográfica e da definição dos demais parâmetros analíticos do método, foi realizada nova etapa de otimização da extração, na qual todos os procedimentos avaliados foram realizados por extração em banho de ultra-som e todos os extratos obtidos tinham composição final de DCM/MeOH (30:70). Nesta etapa, também foi utilizado o material de referência certificado. Nos experimentos descritos abaixo, sempre foi utilizada coluna Vydac 201TP54 (ODS; 4.6mm; 5um; 250mm). A aquisição de dados foi feita sempre por CLAE-EM-APCI, em modo de ionização negativo e com detecção por MRM (m/z, 247/217). O volume de solução injetada foi sempre 20 µL. Exceto quando mencionado no texto, a fase móvel empregada foi sempre MeOH (100%), em vazão de 1,0 mL/min. As demais condições utilizadas foram aquelas descritas nas tabelas 8 e 9. As massas de material certificado empregadas inicialmente foram de cerca de 20 mg e posteriormente as condições foram otimizadas para a extração de cerca de 2 mg, visando atender às massas de amostras de fuligem disponíveis. Foram pesados cerca de 20 mg de SRM 2975 em tubo de centrífuga e foram realizadas quatro etapas de extração em banho de ultra-som (4 mL de DCM, 3 min cada). Ao término de cada etapa de sonicação, o extrato parcial foi submetido à centrifugação (5 min, 3500 RPM) e após cada etapa de centrifugação o sobrenadante foi retirado e transferido para um balão volumétrico de 50 mL. Ao término das quatro etapas de extração, o extrato bruto foi avolumado para 50 mL com MeOH e a concentração de 1-NPi resultante era de cerca de 15 µg/L em composição final de DCM/MeOH (30:70). A Figura 15 representa o procedimento empregado. 83 20 mg SRM 2975 4 mL DCM U.S.; 3 min. Centrifugar (5min; 3500 RPM) 1) Repetir 4 vezes; 2) Adicionar MeOH qsp...50 mL Retirar sobrenadante e transferir Extrato bruto (DCM/MeOH; 30:70) (50 mL; [1-NPi]~15 µg/L) 1) Alíquota 1mL CLAE-EM-EM 2)Filtração Figura 15: Procedimento de extração de cerca de 20 mg SRM 2975 em condições otimizadas Em seguida, foram avaliados dois procedimentos de extração de cerca de 2 mg de material certificado SRM 2975: Com o primeiro procedimento (Figura 16), foram realizados diversos experimentos nos quais foram pesados cerca de 2 mg de SRM 2975 em tubo de centrífuga, e foram realizadas quatro etapas de extração em banho de ultrasom (1 mL de DCM; 5 min cada). Ao término de cada etapa de sonicação, o extrato parcial foi submetido à centrifugação (5 min, 3500 RPM) e após cada etapa de centrifugação o sobrenadante foi retirado e transferido para uma proveta. Ao término das quatro etapas de extração, o extrato bruto foi avolumado para 13 mL com MeOH e a concentração de 1-NPi resultante era de cerca de 6 µg/L em composição final de DCM/MeOH (30:70). No segundo experimento (Figura 17), foram realizados diversos experimentos pesados cerca de 2 mg de SRM 2975 em tubo de centrífuga, o qual foi submetido a seis etapas de extração em banho de ultra-som (1 mL de DCM; 5 min cada). Ao término de cada etapa de sonicação, o extrato parcial foi 84 submetido à centrifugação (5 min; 3500 RPM) e após cada etapa de centrifugação o sobrenadante foi retirado e transferido para uma proveta. Ao término das seis etapas de extração, o extrato bruto foi avolumado para 20 mL com MeOH e a concentração de 1-NPi resultante era de cerca de 4 µg/L em composição final de DCM/MeOH (30:70). As Figuras 16 e 17 representam os dois procedimentos avaliados. 2 mg SRM 2975 1 mL DCM U.S.; 5 min. Centrifugar (5min; 3500 RPM) 1)Repetir 4 vezes 2) Adicionar MeOH qsp...13 mL Retirar sobrenadante e transferir Extrato bruto (DCM/MeOH; 30:70) (13 mL; [1-NPi]~ 6 µg/L) 1) Alíquota 1mL CLAE-EM-EM 2)Filtração Figura 16: Procedimento de extração de cerca de 2 mg SRM 2975 em condições otimizadas, após 4 etapas de extração 85 2 mg SRM 2975 1 mL DCM; U.S.; 5 min. Centrifugar (5min; 3500 RPM) Repetir 6 vezes; adicionar MeOH qsp...20 mL Retirar sobrenadante e transferir Extrato bruto 1) Alíquota 1mL (DCM/MeOH; 30:70) (20 mL; [1-NPi]~4 µg/L) CLAE-EM-EM 2)Filtração Figura 17: Procedimento de extração de cerca de 2 mg SRM 2975 em condições otimizadas, após 6 etapas de extração Em todas as condições avaliadas (Figuras 15, 16 e 17) eram adicionados ao extrato volumes suficientes de metanol para se obter uma composição semelhante a dos padrões. A recuperação de 1-NPi calculada em ambos os casos é apresentada na Tabela 11. Tabela 11: Avaliação da recuperação de 1-NPi em SRM 2975 Massa de SRM 2975 Etapas de extração Recuperação (%) 20 mg 4 102% 2 mg 4 80 a 100%. 2 mg 6 76 a 91%. Como se pode verificar, a recuperação do método usando 2 ou 20 mg do material certificado apresentou resultados comparáveis. O mesmo foi observado com relação ao número de etapas para a massa de 2 mg. A importância da otimização do método para massas muito pequenas estava relacionada às massas de fuligem de diesel disponíveis para a análise, que encontravam-se na faixa de 2 mg. 86 5.11) Avaliação da recuperação de 1-NPi em papel de filtro Além dos estudos de extração em condições otimizadas, também foram realizados outros estudos para avaliar a recuperação da metodologia desenvolvida. Nos experimentos que envolveram avaliação da recuperação em papel de filtro, foram utilizados papéis de filtro fornecidos pelo CENPES. Estes experimentos foram realizados nas mesmas condições analíticas de CLAE-EM descritas no ítem 5.10. Foram realizados dois experimentos individuais para avaliação da recuperação de 1-NPi em solução. No primeiro deles foram adicionados 50 µL de solução de 1-NPi 150 µg/L (7,5 ng de 1-NPi) e no segundo foram adicionados 250 uL de solução de 1-NPi 150 µg/L (37,5 ng de 1-NPi) a ¼ de papel de filtro previamente recortado e condicionado com 1mL de MeOH. Em ambos os casos, o papel foi processado segundo o procedimento ilustrado na Figura 16. A concentração de 1-NPi nos extratos resultantes era de cerca de 0,6 µg/L e de 3 µg/L, respectivamente e ambos os extratos foram obtidos em composição final de (DCM/MeOH; 30:70). Também foram realizados dois experimentos individuais para avaliação da recuperação de 1-NPi proveniente do material certificado SRM 2975 (36 mg/Kg). Em ambos, foram adicionados cerca de 2 mg de SRM 2975 (72 ng de 1NPi) a ¼ de papel de filtro previamente recortado e condicionado com 1mL de MeOH. No primeiro experimento, o papel foi processado e analisado conforme procedimento ilustrado na Figura 16. A concentração de 1-NPi no extrato resultante era de cerca de 6 µg/L em composição final de DCM/MeOH (30/70). No segundo experimento realizado, o papel foi processado e analisado conforme procedimento ilustrado na Figura 17. A concentração de 1-NPi no extrato resultante era de 4 µg/L em composição final de DCM/MeOH (30/70). A recuperação da extração de 1-NPi foi avaliada também fortificando-se filtro em branco com solução deste NHPA em dois níveis: 7,50 ng e 37,50 ng. Os resultados obtidos encontram-se na Tabela 12. 87 Tabela 12: Avaliação da recuperação de 1-NPi em solução Nível de fortificação Recuperação (%) 7,5 ng 102 37,5 ng 114 A recuperação obtida nos dois experimentos indicou que havia recuperação adequada para a determinação de 1-NPi, independente da presença do filtro. Na avaliação da recuperação de 1-NPi de 2 mg da amostra certificada (SRM 2975) na presença do filtro, recuperações um pouco menores (73 a 74%) foram obtidas indicando que neste caso pode haver alguma interferência da matriz na extração do NHPA de interesse, embora estes valores sejam ainda aceitáveis nos níveis de concentração deste estudo. Outros experimentos de controle foram realizados. Uma solução contendo 1-NPi foi submetida ao ultra som durante 1 hora e não foi observada degradação desta substância. Brancos de solventes e de filtro também demonstraram não haver contaminação detectável por 1-NPi. 5.12) Determinação de 1-NPi em amostras reais de FD 5.12.1) Análise de FD depositada nos filtros BR1 e BR2 Foram fornecidos pelo CENPES BR as seguintes amostras de fuligem de diesel: • Filtro BR1 (4,56 + 0,12,00 mg): Euro III: ETC, European transient cycle; oscila rotação e carga; coleta é feita durante todo o ensaio (cerca de 30 minutos). • Filtro BR2 (2,09 + 0,08 mg): Euro II – R49 (ensaio “13 pontos”); Euro III – ESC, European steady cycle; estabiliza rotação e a carga e faz a coleta 88 Foi realizada a extração de cerca de ½ do filtro BR1 (2,35 mg de fuligem) e de cerca de ½ do filtro BR2 (1,08 mg de fuligem), segundo o procedimento apresentado na Figura 18. A análise dos extratos foi realizada nas condições otimizadas das Tabelas 7 e 9. A quantificação dos experimentos foi feita através de curva de calibração com figuras de mérito semelhantes às da Tabela 10. Após a pesagem, os filtros foram recortados em pequenos pedaços e processados separadamente em tubos de centrífuga, que foram submetidos a seis etapas de extração em banho de ultra-som. Entre cada etapa de sonicação, os extratos parciais eram submetidos à centrifugação (5 min; 3500 RPM) e após a retirada do sobrenadante eram transferidos para uma proveta de 25 mL. Ao término de todo o processo, os extratos brutos totais foram avolumados para 20 mL com MeOH e foram analisados (Figura 18). Em cada caso, uma alíquota de 5 mL do extrato bruto original (20 mL; DCM/MeOH, 30:70) foi concentrada (N2) até o volume final de 0,5 mL. Após filtração, foram realizadas análises nas mesmas condições de CLAE-EM apresentadas no item 5.10 e também com volume de injeção igual a 100 µL. A Figura 18 representa o procedimento descrito para ambas as amostras: 89 Fuligem de Diesel BR 1 mL DCM; U.S.; 5 min. Centrifugar (5min; 3500 RPM) Repetir 6 vezes; adicionar MeOH qsp...20 mL Retirar sobrenadante e transferir 1) Alíquota 5mL 2) N2 (g); Vf=0,5mL 3) Filtração Vinj = 20 µL Extrato bruto original Vf = 20 mL (DCM/MeOH; 30:70) 1) Alíquota 1mL 2)Filtração CLAE-EM-EM Vinj = 100 µL Figura 18: Procedimento de extração e tratamento das amostras de Fuligem de Diesel da BR A quantificação dos experimentos foi feita através de curva de calibração com figuras de mérito semelhantes às da Tabela 10. O tratamento das amostras seguiu o procedimento otimizado com material certificado, apesar da pequena massa de fuligem de diesel que apresentavam (cerca de 2 a 4 mg). O filtro BR1 foi o primeiro a ser analisado por ser aquele que apresentava a maior massa de fuligem. A concentração de 1NPi calculada tanto para o filtro BR1, quanto para o filtro BR2, no extrato original e nas alíquotas de 20 e de 100 µL (Figura 18) injetadas após concentração por um fator de dez vezes, estava abaixo dos limites de detecção e quantificação determinados. 90 Figura 19: Cromatogramas de 1NPi obtidos por CLAE-EM-APCI, com detecção por MRM em modo de ionização negativo. a) extrato de fuligem de diesel depositada no filtro BR1 após reconcentração (10x) e com injeção de 100 µL; b) padrão 1µg/L (20 µL) O sinal do 1-NPi esteve abaixo do LD (1,5 mg/Kg) na injeção de 20 µL de ambos os extratos. A reconcentração de ambos os extratos (10x), embora sujeita a perdas, também não permitiu a detecção de 1-NPi, indicando que as concentrações de 1-NPi estavam abaixo de 1,5 mg/Kg. Para avaliar de modo aproximado a concentração de 1-NPi nas amostras de FD fornecidas pela Petrobrás, o extrato foi reconcentrado 10 vezes com nitrogênio gasoso e foram injetados 100 µL desta solução nas condições habituais. Os resultados obtidos permitem estimar uma concentração de 0,080 mg de 1NPi/kg de fuligem de diesel no extrato BR1 e 0,2 mg de 1NPi/kg de fuligem de diesel no extrato BR2. Do ponto de vista toxicológico e de Saúde Pública, este resultado é de interesse, pois indica que há pequena contaminação do ar pela emissão desta fuligem. Este resultado é conseqüência das características também das amostras de óleo diesel estudadas (Euro II e Euro III), que têm processos de fabricação muito controlados e restritivos. 91 5.12.2) Extração de Fuligem de Diesel de automóvel Foi realizada a análise de uma amostra de fuligem de diesel de um automóvel Jeep Land Rover, movido à diesel, coletada no estacionamento do Campus do Valonguinho, da Universidade Federal Fluminense, na cidade de Niterói, RJ. A coleta foi realizada durante o dia, por volta de 13hs, com o auxílio de uma espátula de metal. A parte superior do interior do cano de escapamento do veículo foi raspada de forma a soltar a camada de fuligem aderida às paredes internas sobre um pedaço de papel alumínio, inserido na parte inferior do cano. A raspagem da fuligem foi realizada na posição mais distante da abertura do cano de descarga para o exterior quanto possível, considerando–se as dimensões do cano e da espátula usada. A fuligem coletada foi imediatamente transportada ao laboratório e armazenada em geladeira, em frasco recoberto com alumínio, ao abrigo da luz. Foram realizados dois procedimentos de extração de cerca de 2 mg desta fuligem. O primeiro deles foi realizado segundo o procedimento descrito na Figura 17, no qual foram extraídos 2,90 mg da fuligem coletada. A concentração de 1NPi calculada estava abaixo do limite de detecção do método. Um segundo experimento de extração foi realizado pela extração de 2,20 mg de fuligem, pelo procedimento envolvendo reconcentração sob fluxo de nitrogênio (Figura 20). Foram pesados cerca de 2 mg da fuligem do automóvel em tubo de centrífuga, o qual foi submetido a seis etapas de extração em banho de ultra-som (1 mL de DCM; 5 min cada). Ao término de cada etapa de sonicação, o extrato parcial foi submetido à centrifugação (5 min; 3500 RPM) e após cada etapa de centrifugação, o sobrenadante foi retirado e transferido para uma proveta de 10 mL. Ao término das seis etapas de extração, foram adicionados 1,5 mL de MeOH ao extrato bruto e a solução resultante foi concentrada sob fluxo de N2 até o volume final de 1 (um) mL, que foi filtrado e analisado. Na injeção de 20 µL deste extrato, obteve-se área de 1-NPi comparável à área desta substância no padrão de concentração 1 µg/L. A partir destes 92 resultados, foi estimado que a concentração de 1-NPi nesta amostra era da ordem de 0,5 mg/Kg. Fuligem de automóvel 1 mL DCM; U.S.; 5 min. Centrifugar (5min; 3500 RPM) 1) Repetir 6 vezes; 2) adicionar 1,5 mL de MeOH Retirar sobrenadante e transferir 1) N2 (g); Vf= 1 mL 2) Filtração Extrato Final (Vf= 1 mL) CLAE-EM-EM Figura 20: Procedimento de extração alternativo empregado no tratamento da amostra de fuligem de cano de descarga de automóvel movido a diesel. Estes resultados devem ser tomados com cautela já que se trata de apenas uma amostra e, portanto, sem representatividade estatística. Ademais, a temperatura do cano de descarga é alta, o que deve levar a perda considerável de substâncias relativamente voláteis com HPAs, hidrocarbonetos e o 1-NPi. Entretanto nossos resultados indicam que a metodologia tem potencial para ser aplicada a amostras de campo, já que neste caso a restrição para a quantidade de amostra é menor, pois podem ser coletadas maiores massas e/ou avaliadas amostras combinadas de fuligem de diesel. 93 5.13) Avaliação de outros NHPAs em FD 5.13.1) Otimização da separação de 1-nitropireno e outros NHPAs Foram estudados parâmetros de separação cromatográfica para alguns NHPAs selecionados (1-NPi, 9-NA, 2-NFl), em coluna Vydac 201TP54 (ODS; 4.6mm x 5um x 250mm). Os tempos de retenção das substâncias estudadas foram determinados em diferentes composições de fase móvel do sistema MeOH:H2O, em diferentes vazões. A aquisição de dados foi feita segundo as condições de CLAE-EM descritas no item 5.10. A separação de NHPAs selecionados (1-NPi, 9-NA e 2-NFl) foi otimizada na coluna Vydac 201TP54 (ODS; 4,6mm x 5mm x 250mm), usada na determinação de 1-NPi. O tempo de retenção de cada substância foi determinado em vazão de 1,0, 0,8 e 0,6 mL/min e foram testadas diferentes composições de fase móvel do sistema MeOH:H2O. A Tabela 13 apresenta os tempos de retenção obtidos. Os resultados obtidos demonstram que há aumento na retenção dos NHPAs conforme a proporção de água é aumentada na fase móvel. Este fato provocou aumento na largura dos picos cromatográficos, redução da relação sinal/ruído e, conseqüentemente, piora do limite de detecção e do limite de quantificação. Entretanto, foi verificado que até uma proporção MeOH/H2O (80:20), ainda era possível obter uma boa quantificação dos NHPAs. Na fase móvel MeOH/H2O (70:30), os picos cromatográficos foram largos, com perda de resolução e redução de sinal significativa, o que indica ser apropriado trabalhar em sistema que contenha no máximo 20% de H2O. Além disso, na proporção MeOH/H2O (70:30), o tempo de total de análise seria alto e maior do que o desejado, uma vez que o 1-NPi apresentou tr = 21,5 min. Em vazão de 0,8 mL/min também não houve melhora na resolução de 9NA e 2-NFl em relação à vazão de 1,0 mL/min. A composição MeOH/H2O (70:30) não foi testada com esta vazão pois já havia apresentado resultados insatisfatórios na vazão anteriormente testada. 94 A vazão de 0,6 mL/min, empregada apenas com fase móvel de 100% MeOH, também não melhorou a resolução do par 9-NA e 2-NFl (Tabela 13). Vazões maiores que 1,0 mL/min não foram avaliadas sistematicamente pois impediam a resolução do DCM e do 1-NPi. A partir destes resultados a composição de MeOH/H2O (80:20) foi utilizada para a separação dos NHPAs de interesse. No caso em que o objetivo da análise foi apenas a determinação de 1-NPi, MeOH 100% foi empregado na fase, reduzindo assim o tempo total de análise dos extratos. Tabela 13: Tempos de retenção (tr) de 1-NPi, 9-NA e 2-NFl em coluna Vydac tr (min.) NHPA MeOH V=1,0 mL/min MeOH/H2O MeOH/H2O MeOH/H2O (90/10) (80/20) (70/30) 1NPi 3,5 5,0 9,0 21,5 9NA 3,0 3,5 5,0 9,5 2NFl 3,2 3,7 5,3 9,2 NHPA MeOH MeOH/H2O MeOH/H2O MeOH/H2O (90/10) (80/20) (70/30) V=0,8 mL/min 1NPi 4,4 6,2 12,2 - 9NA 3,8 4,5 6,7 - 2NFl 3,9 4,6 7,0 - NHPA Tr (min) - - - V=0,6 mL/min MeOH 1NPi 5,9 - - - 9NA 5,0 - - - 2NFl 5,2 - - - 95 5.13.2) Avaliação preliminar de outros NHPAs no material certificado SRM 2975 Os extratos obtidos em condições otimizadas (Figuras 16 e 17) foram guardados em geladeira (8°C) após troca das tampas usadas dos vials por tampas novas e alguns deles foram analisados posteriormente visando a identificação de outros NHPAs além do 1-NPi. Para a análise de outros NHPAs, foi empregada detecção em modo de varredura. As demais condições de CLAE-EM descritas no ítem 5.10 foram empregadas, exceto a composição e a vazão da fase móvel, tendo sido empregada fase composta por MeOH:H2O (80/20) em vazão de 0,8 mL/min. Os extratos de SRM 2975 estudados anteriormente foram analisados em modo de varredura para avaliação de outros NHPAs. A fase móvel MeOH/H2O (80:20) foi utilizada uma vazão de 0,8 ml/min, nas condições instrumentais (Tabelas 7 e 9 ) descritas para a determinação de 1-NPi. Além do 1-NPi (tr = 12,2 min), o único NHPA identificado foi o 9-NA (tr = 6,7 min) (Figura 21). 1-NPi 9-NA Figura 21: Cromatogramas em CLAE-EM-APCI-MRM de 9-NA e 1-NPi em extrato de SRM 2975 A quantificação do 9-NA foi realizada com uma curva de calibração na faixa de 5 a 25 µg/L deste NHPA (70%MeOH:30%DCM). A aquisição de dados foi feita a partir do sistema CLAE (20 µL; FM= 80%MeOH:20% H2O; 1,0 mL/min.) e a detecção feita por MRM (m/z 193) em modo de ionização APCI negativo. 96 As áreas em cada ponto tiveram desvios relativos menores que 5%. Bom coeficiente de correlação foi obtido (0,9955). O limite de detecção estimado a partir desta curva para o 9NA foi de 1,92 µg/L. A concentração deste NHPA não foi estimada no padrão, pois foi considerado que deve haver pré-concentração do extrato, o que não foi estudado com mais detalhes neste trabalho. Ademais não há valor certificado ou valor de referência para o 9-NA no material SRM 2975. A comparação das áreas de 1-NPi e 9-NA calculadas nestes extratos indicou que a concentração deste NHPA era cerca de 10 vezes menor do que a do 1-NPi no SRM 2975 e comparável ao limite de detecção para o 9-NA. Estimativas preliminares, usando a área do menor padrão, indicam que este valor situa-se numa faixa inferior a 1 mg/kg. 97 CAPÍTULO 6 Conclusões Os padrões de fragmentação de NHPAs foram estudados neste trabalho. A ionização negativa e a formação dos íons [M-30]- parecem ser dois processos comuns a todos os NHPAs estudados. O uso de MRM considerando o íon de peso molecular [M-] e o [M-30]- possibilitaram a detecção de NHPAs, em particular o 1-NPi em níveis muito baixos (LD = 1,5 mg/kg) e compatível com a determinação deste NHPA em fuligem de diesel. As amostras avaliadas apresentaram concentrações de 1-NPi abaixo do limite de detecção, o que do ponto de vista de toxicologia e da saúde pública tem alta importância devido às características mutagênicas desta substância. As características de tratamento do óleo diesel empregado nos testes para a obtenção de fuligem parecem influenciar este baixo teor de 1-NPi já que o tratamento deste combustível, neste caso, é bastante restritivo. Outros NHPAs não foram quantificados no material certificado e nas amostras de diesel estudadas. Na amostra certificada 9-nitroantraceno foi detectado com concentrações (estimadas) de 1 ordem de grandeza menores que as do 1-nitropireno. O método desenvolvido apresenta potencial para aplicação em amostras de fuligem de diesel e em amostras de outras origens e características, que 98 contenham NHPAs e onde a massa de amostra não seja a principal limitação, como foi o caso das amostras estudadas neste trabalho. O prosseguimento deste trabalho será focado na determinação de NHPAs em fuligem de diesel coletada em veículos automotores onde a limitação de massa de amostra não é um impeditivo para o trabalho analítico. A avaliação das figuras de mérito na determinação de outros NHPAs pela metodologia desenvolvida também está em curso neste momento. Outro aspecto de interesse a ser mais avaliado é a perda de NHPAs na etapa de reconcentração com nitrogênio, o que permitirá a análise de massas de fuligens menores. 99 REFERÊNCIAS BIBLIOGRÁFICAS Alfheim, I.; Lofroth, G.; Moller, M.; Bioassay of extracts of ambient particulate matter, Environmental Health Perspectives 47: 227-238 (1983) Amorisco, A.; Losito, I.; Carbonara, T. et al.; Photocatalytic degradation of phenyl-urea herbicides chlortoluron and chloroxuron: characterization of the byproducts by liquid chromatography coupled to electrospray ionization tandem mass spectrometry, Rapid Communications in Mass Spectrometry 20 (10): 1569-1576 (2006) Anacleto, J.F.; Ramaley, L.; Benoit, F.M.; Boyd, R.K.; Quilliam, M.; Comparison of liquid-chromatography mass-spectrometry interfaces for the analysis of polycyclic aromatic-compounds, Analytical Chemistry 67:4145-4154 (1995) Arey, J.; Zielinska, B.; Atkinson, R.; Winter, A.M.; Ramdahl, T.; Pitts, J.N.Jr.; The formation of nitro-PAH from the gas-phase reactions of fluoranthene and pyrene with the OH radical in the presence of NOx, Atmospheric Environment 20, 2339-2345, (1986) Arey, J.; Zielinska, B.; Warger, W.P.; Atkinson, R.; Winter, A.M.; The contribution of nitrofluoranthenes and nitropyrenes to the mutagenic activity of 100 ambient particulate organic-matter collected in southern-california, Mutation Research 207: 45-51, (1988) Atkinson, R.; Atmospheric transformations of automotive emissions. In: Air Pollution, the Automobile, and Public Health (Watson, A.Y.; Bates, R.R.; Kennedy, D.; eds.) Washington: National Academy Press, 99-132 (1988) Atkinson, R.; Arey, J.; Zielinska, B.; Aschmann, S.M.; Kinetics and nitroproducts of the gas-phase OH and NO3 radical-initiated reactions of naphthalene-D8, fluoranthene-D10, and pyrene, International Journal of Chemical Kinetics. 22, 999-1014, (1990) Badger, G.M., Lewis, G.E.; Napier, I.M.; The formation of aromatic hydrocarbons at high temperatures .8. The pyrolysis of acetylene, Journal of the Chemical Society: 2825-2827 (1960) Baek, S.O.; Field, R. A.; Goldstone, M.E.; Kirk, P.W.; Lester, J.N.; Perry, R.; A review of atmospheric polycyclic aromatic-hydrocarbons - sources, fate and behavior, Water, Air and Soil Pollution 60 (3-4): 279-300 (1991) Barranco, A.; Alonso-Salces, R.M.; Bakkali, A.; Berrueta, L.A.; Gallo, B.; Vicente, F.; Sarobe, M.; Solid-phase clean-up in the liquid chromatographic determination of polycyclic aromatic hydrocarbons in edible oils, Journal of Chromatography A 988: 33-40 (2003) Bayona, J.M.; Casellas, M.; Fernández, P.; Solanas, A.M.; Albaigés, J.; Sources and seasonal variability of mutagenic-agents in the Barcelona city aerosol, Chemosphere 29 (3): 441-450 (1994) Beije, B.; Moller, L.; 2-nitrofluorene and related-compounds - prevalence and biological effects, Mutation Research 196: 177-209 (1988) Bezabeh, D.Z.; Bamford, H.A.; Schantz, M.M.; Wise, S.A.; Determination of nitrated polycyclic aromatic hydrocarbons in diesel particulate-related standard 101 reference materials by using gas chromatography/mass spectrometry with negative ion chemical ionization, Analytical and Bioanalytical Chemistry 375 (5): 381-388 (2003) Blumer, M.; Polycyclic aromatic-compounds in nature, Scientific American, 234 (3), 34-45 (1976) Boffetta, P.; Jourenkova, N.; Gustavsson, P.; Cancer risk from occupational and environmental exposure to polycyclic aromatic hydrocarbons, Cancer Causes & Control 8 (3): 444-472 (1997) Bonfanti, L.; Careri, M.; Mangia, A.; Manini, P.; Maspero, M.; Simultaneous identification of different classes of hydrocarbons and determination of nitropolycyclic aromatic hydrocarbons by means of particle beam liquid chromatography mass spectrometry, Journal of Chromatography A, 728: 359369 (1996) Butler, J.D.; Crossley, P.; Reactivity of polycyclic aromatic-hydrocarbons adsorbed on soot particles, Atmospheric Environment 15 (1): 91-94 (1981) Castello, G.; Gerbino, T.C.; Analysis of polycyclic aromatic-hydrocarbons with an ion-trap mass detector and comparison with other gas-chromatographic and high-performance liquid-chromatographic techniques, Journal of Chromatography, 642 (1-2): 351-357 (1993) Ciccioli, P.; Cecinato, A. Brancaleone, E.; Frattoni, M.; Zacchei, P.; Miguel, A.H.; Vasconcellos, P.D.; Formation and transport of 2-nitrofluoranthene and 2nitropyrene of photochemical origin in the troposphere, Journal of Geophysical Research-Atmospheres, 101(D14): 19567-19581 (1996) Cvacka, J.; Barek, J.; Fogg, A.G.; Moreira, J.C.; Zima, J.; High-performance liquid chromatography of nitrated polycyclic aromatic hydrocarbons, Analyst 123 (2): 9R-18R (1998) 102 Dole, M.; Hines, R.L.; Mack, L.L.; Mobley, R.C.; Ferguson, L.D.; Alice, M.B.; Molecular beams of macro ions, Journal of Chemical Physics 49: 2240 (1968). Finlayson-Pitts, B.J.; Pitts, J.N.Jr.; Atmospheric Chemistry: Fundamental and Experimental Techniques. Wiley, New York. (1986) Furton, K.G.; Jolly, E.; Pentzke, G.; Recent advances in the analysis of polycyclic aromatic-hydrocarbons and fullerenes, Journal of Chromatography 642 (1-2): 33-45 (1993) Gallagher, J.; Heinrich, U.; George, M.; Hendee, L.; Phillips, D.H.; Lewtas, J.; Formation of DNA-adducts in rat lung following chronic inhalation of diesel emissions, carbon-black and titanium-dioxide particles, Carcinogenesis, 15 (7): 1291-1299 (1994) Glusker, J.P. X-Ray analyses of polycyclic hydrocarbon metabolite structures, Chapter 7 in: Polycyclic Hydrocarbons and Carcinogenesis, Harvey, R.G.; American Chemical Society, Washington, DC, USA, p.125-185, (1985) Grosjean, D.; Fung, K.; Harrison, J.; Interactions of polycyclic aromatichydrocarbons with atmospheric pollutants, Environmental Science & Technology 17: 673-679, (1983) Guillén, M.D.; Polycyclic aromatic-compounds - extraction and determination in food, Food Additives and Contaminants 11 (6): 669-684 (1994) Henderson, T.R.; Sun, J.D.; Royer, R.E.; Clark, C.R.; Li, A.P.; Harvey, T.M.; Hunt, D.H.; Fulford, J.E.; Lovette, A.M.; Davidson, W.R.; Triple-quadrupole mass-spectrometry studies of nitroaromatic emissions from different dieselengines, Environmental Science & Technology 17: 443-449 (1983) Horning, E.C. et al., Atmospheric-pressure ionization (API) mass-spectrometry solvent-mediated ionization of samples introduced in solution and in a liquid 103 chromatograph effluent stream, Journal of Chromatographic Science 12: 725 (1974) IARC: Diesel and Gasoline Engine Exhausts and Some Nitroarenes, Lyon, International Agency for Research on Cancer, 322 pp; IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Humans, Volume 46, (1989) Ingelse, B.A. Van Dam, RCJ, Vreeken, RJ.; Determination of polar organophosphorus pesticides in aqueous samples by direct injection using liquid chromatography-tandem mass spectrometry, Journal of Chromatography A, 918 (1): 67–78 (2001) International Programme on Chemical safety (IPCS). Environmental Health Criteria 202: Selected non-heterocyclic polycyclic aromatic hydrocarbons, W.H.O.; Genebra, (1998) International Programme on Chemical safety (IPCS), Environmental Health Criteria 229: Selected Nitro- and Nitro-Oxy-Polycyclic Aromatic Hydrocarbons, W.H.O.;Genebra (2003) Iribarne, J.V.; Thomson, B.A.; Evaporation of small ions from charged droplets, Journal of Chemical Physics 64 (6): 2287-2294 (1976) Jager, J.; Detection and characterization of nitro-derivatives of some polycyclic aromatic-hydrocarbons chromatography - by fluorescence application to quenching air-pollution after analysis, thin-layer Journal of Chromatography 152 (2): 575-578 (1978) Josephy, P.D.; Molecular Toxicology, Chapter 18 in: Polycyclic Aromatic Hydrocarbons Carcinogenesis, Oxford University Press, NY, USA, p.315-351. 1997. 104 Kamens, R.M.; Fan, Z.; Yao, Y.; Chen, D.; Chen, S.; Vartiainen, M.; A methodology for modeling the formation and decay of nitro-PAH in the atmosphere, Chemosphere 28 (9), 1623-1632 (1994) Karancsi, T.; Slegel, ,P.J.; Reliable molecular mass determination of aromatic nitro compounds: elimination of gas-phase reduction occurring during atmospheric pressure chemical ionization, Journal of Mass Spectrometry 34 (9): 975 (1999) Knewstubb, P.F.; Sugden, S.B.; Mass-spectrometric studies of ionization in flames .1. The spectrometer and its application to ionization in hydrogen flames, Proceedings of the Royal Society of London series a-mathematical and physical sciences A, 255: 520 (1966) Kootstra, P.R.; Straub, M.H.C.; Stil, G.H.; van der Velde, E.G.; Hesselink, W.; Land, C.C.J., Solid-phase extraction of polycyclic aromatic-hydrocarbons from soil samples, Journal of Chromatography A 697 (1-2): 123-129 (1995) Korfmacher W.A, Rushing L.G., Arey J., Zielinska B., Pitts Jr J.N.; Identification of mononitropyrenes and mononitrofluoranthenes in air particulate matter via fused-silica gas-chromatography combined with negative-ion atmosphericpressure ionization mass-spectrometry, Journal of High Resolution Chromatography & Chromatography Communications 10 (12): 641-646 (1987) Lemière, F.; Guide to LC-MS, Dezembro, (2001) Lewtas, J.; Chuang, J.; Nishioka, M.; Petersen, B.; Bioassay-directed fractionation of the organic extract of SRM-1649 urban air particulate matter, International Journal of Environmental Analytical Chemistry 39 (3): 245-256 (1990) Levsen, K., The analysis of diesel particulate, Fresenius Zeitschrift fur Analytische Chemie 331 (5): 467-478 (1988) 105 Librando, V.; Fazzino, S.D.; Quantification of polycyclic aromatic-hydrocarbons and their nitro-derivatives in atmospheric particulate matter of Augusta city, Chemosphere, 27 (9): 1649-1656 (1993) Madl, T.; Sterk, H.; Mittelbach, M.; Tandem mass spectrometric analysis of a complex triterpene saponin mixture of chenopodium quinoa, Journal of The American Society for Mass Spectrometry 17 (6): 795-806 (2006) Menzie, C.A.; Potoki B.B.; Santodonato, J.; Exposure to carcinogenic PAHs in the environment, Environmental Science & Technology 26: 1278-1284 (1992) Moller, L.; Lax, I.; Lennart, C.; Nitrated polycyclic aromatic-hydrocarbons - a risk assessment for the urban citizen, Environmental Health Perspectives 101: 309313 (1993) Moreira, J.C.; Barek, J.; Analysis of carcinogenic nitrated polycyclic aromatichydrocarbons - a review Química Nova, 18: 362-367 (1995) Moriwaki, H.; Funakasa, K.; Uebori, M.; Direct determination of NO2-substituted pyrenes by liquid chromatography-atmospheric pressure chemical ionizationmass spectrometry, Analytical Sciences 16: 1247-1249 (2000) Nielsen, T.; Seitz, B.; Ramdahl, T.; Occurrence of nitro-PAH in the atmosphere in a rural area, Atmospheric Environment 18, 2159-2165 (1984) Nielsen, T.; Ramdahl, T.; Determination of 2-nitrofluoranthene and 2nitropyrene in ambient particulate matter - evidence for atmospheric reactions, Atmospheric Environment 20, 1507-1509 (1986) Niessen W.M.A.; Tinke, ,A.P., Liquid-chromatography mass-spectrometry general-principles and instrumentation, Journal of Chromatography A, 703 (12): 37-57 (1995) 106 Niessen, W.M.A.; Lc-MS in quantitative analysis, Reviews in Analytical Chemistry, 19 (3-4): 289-301 (2000) Nishioka, M.G.; Howard, C.C.; Contos, D.A.; Ball, L.M.; Lewtas, J.; Detection of hydroxylated nitro aromatic and hydroxylated nitro polycyclic aromaticcompounds in an ambient air particulate extract using bioassay-directed fractionation, Environmental Science & Technology 22: 908-915 (1988) Ohgaki, H.; Negishi, C.; Wakabayashi, K.; kusama, K.; Sato, S.; Sugimura, T.; Induction of sarcomas in rats by subcutaneous injection of dinitropyrenes, Carcinogenisis 5: 583-585 (1984) Paputa-Peck, M.C.; Marano, R.S.; Schuetzle, D.; Riley, T.L.; Hampton, C.V.; Prater, T.I.; Skewes, L.M.; Jensen, T.E.; Ruehle, P.H.; Bosch, L.C.; Duncan, W.P.; Determination of nitrated polynuclear aromatic-hydrocarbons in particulate extracts by capillary column gas-chromatography with nitrogen selective detection, Analytical Chemistry 55: 1946-1954 (1983) Pereira Netto, A.D.; Tese de Doutorado; Depto de Físico Química, IQ, UFRJ, Rio de Janeiro, RJ, 1999 Pereira Netto, A.D.; Moreira, J.C. Arbilla, G.; Ferreira, L.F.V.; Oliveira, A.S.; Barek, J.; Evaluation of human contamination with polycyclic aromatic hydrocarbons (PAHs) and their nitrated derivatives (NHPAs): a review of methodology, Química Nova 23: 765-773 (2000) Pereira Netto, A.D.; Barreto, R.P.; Moreira, J.C.; Arbilla, G.; Preliminary comparison of PAH in total suspended particulate samples taken at Niteroi and Rio de Janeiro cities, Brazil, Bulletin of Environmental Contamination and Toxicology, 66:36 (2001) Pereira Netto, A.D.; Muniz , F.C.; Laurentino, Rego, E.C.P.; Identification of polycyclic aromatic hydrocarbons in street dust of niteroi city, RJ, Brazil, Bulletin of Environmental Contamination and Toxicology, 68 (6): 831-838 (2002a) 107 Pereira Netto, A.D.; Barreto, R.P.; Moreira, J.C.; Arbilla, G.; Polycyclic aromatic hydrocarbons in total suspended particulate of Niteroi, RJ, Brazil: a comparison of summer and winter samples, Bulletin of Environmental Contamination and Toxicology, 69 (2):173-180 (2002b) Perez, S.; Barcelo, D.; Determination of polycyclic aromatic hydrocarbons in sewage reference sludge by liquid chromatography-atmospheric-pressure chemical-ionization mass spectrometry, Chromatographia, 53 (9-10): 475–480 (2001) Pitts, J.N. Jr.; Van Cauwenberghe, K.; Grosjean, D.; Schimid, J.P.; Fitz, D.R.; Belser, W.L.; Knudson, G.B.; Hynds, P.M.; Atmospheric reactions of polycyclic aromatic-hydrocarbons - facile formation of mutagenic nitro-derivatives Science 202 :515 (1978) Pitts, J.N. Jr.; Photo-chemical and biological implications of the atmospheric reactions of amines and benzo(a)pyrene. Phil. Trans. R. Soc. Lond A290 (1376): 551-576 (1979) Pitts, J.N. Jr.; Atkinson R.; Sweetman, J. A.; Zielinska, B.; Determination of 2nitrofluoranthene and 2-nitropyrene in ambient particulate organic-matter evidence for atmospheric reactions, Atmospheric Environment 19 (10), 16011608, (1985a) Pitts, J.N.Jr.; Zielinska, B.; Sweetman, J.A.; Atkinson, R.; Winter, A.M.; Reactions of adsorbed pyrene and perylene with gaseous N2O5 under simulated atmospheric conditions, Atmospheric Environment 19 (6): 911-915, (1985b) Pitts, J.N. Jr.; Nitration of gaseous polycyclic aromatic-hydrocarbons in simulated and ambient urban atmospheres - a source of mutagenic nitroarenes, Atmospheric Environment 21 (12): 2531-2547, (1987) 108 Pryor, A.W. (Ed.): Free Radicals in Biology, vol. 5, p.161. Academic Press, New York. 1982 Raat, W.K. de.; Boers, J.P.; Bakker, G.L.; Meijere, F.A. de.; Hooimeijer, A.; Lohman, P.H.M.; Mohn, G.R.; Contribution of PAH and some of their nitrated derivatives to the mutagenicity of ambient airborne particles and coal fly-ash, Science of the Total Environment, 153 (1-2): 7-28 (1994) Ramdahl, T.;. Zielinska, B.; Arey, A.; Atkinson, R.; Winter, A.M.; Pitts, J.N.Jr.; Ubiquitous occurrence of 2-nitrofluoranthene and 2-nitropyrene in air, Nature 321: 425-427 (1986) Rappaport, S.M.; Wang, Y.Y.; Wei, E.T.; Sawyer, R.; Watkins, B.E.; Rappaport, H.; Isolation and identification of a direct-acting mutagen in diesel-exhaust particulates Environmental Science & Technology 14 (12): 1505-1509 (1980) Rosenkranz, H.S.; Mutagenicity of chloroalkene epoxides in bacterial systems Mutation Research 101(2): 115-125 (1982) Rosenkranz, H.S.; Mermelstein, R., Mutagenicity and genotoxicity of nitroarenes - all nitro-containing chemicals were not created equal, Mutation Research 114 (3): 217-267 (1983) Rosenkranz, H.S.; Mutagenic and carcinogenic nitroarenes in diesel emissions risk identification, Mutation Research, 140 (1): 1-6 (1984) Ryno, M.; Rantanen, L.; Papaioannou, E.; et al.; Comparison of pressurized fluid extraction, soxhlet extraction and sonication for the determination of polycyclic aromatic hydrocarbons in urban air and diesel exhaust particulate matter, Journal of Environmental Monitoring 8 (4): 488-493 (2006) Salmeen, I.T.; Durisin, A.M.; Prater, T.J.; Riley, T.; Shuetzle, D.; Contribution of 1-nitropyrene to direct-acting ames assay mutagenicities of diesel particulate extracts 109 Mutation Research 104 (1-3): 17-23 (1982) Schauer, C.; Niessner, R.; Poschl, U.; Analysis of nitrated polycyclic aromatic hydrocarbons by liquid chromatography with fluorescence and mass spectrometry detection: air particulate matter, soot, and reaction product studies, Analytical and Bioanalytical Chemistry, 378 (3): 725-736 (2004) Scheepers, P.T.J.; Bos, R.P.; Combustion of diesel fuel from a toxicological perspective .1. Origin of incomplete combustion products, International Archives of Occupational and Environmental Health 64 (3): 149-161 (1992) Schuetzle, D.; S-C Lee, F.; Prater, T.J.; The identification of polynuclear aromatic hydrocarbon (PAH) derivatives in mutagenic fractions of diesel particulate extracts, International Journal of Environmental Analytical Chemistry 9 (2) 93-144 (1981) Schuetzle, D.; Riley, T.L.; Prater, T.J.; Harvey, T.M.; Hunt, D.F., Analysis of nitrated polycyclic aromatic hydrocarbons in diesel particulates, Analytical Chemistry 54 (2): 265-271 (1982) Schuetzle, D.; Sampling of vehicle emissions for chemical-analysis and biological testing, Environmental Health Perspectives 47: 65-80 (1983) Schuetzle, D.; Lewtas, J.; Bioassay-directed chemical-analysis in environmental-research, Analytical Chemistry 58 (11): 1060 (1986) Shahin, M.M.; Mass-spectrometric studies of corona discharges in air at atmospheric pressures, Journal of Chemical Physics 45 (7): 2600 (1966) Smyth, W.F.; Leslie, J.C.; McClean, S. Hannigan, B.; McKenna, H.P.; Doherty, B.; Joyce, C.; O’Kane, E.; The characterisation of selected antidepressant drugs using electrospray ionisation with ion trap mass spectrometry and with quadrupole time-of-flight mass spectrometry and their determination by highperformance liquid chromatography/electrospray ionisation tandem mass 110 spectrometry, Rapid Communications In Mass Spectrometry 20 (11): 16371642 (2006) Soriano, S.; Pereira-Netto, A.D.; Cassela, R.J.; Determination of Cu, Fe, Mn and Zn by flame atomic absorption spectrometry in multivitamin/multimineral dosage forms or tablets after an acidic extraction, Journal of Pharmaceutical and Biomedical Analysis (2006), in press Straube, E.A.; Dekant, W.; Volkel, W.; Comparison of electrospray ionization, atmospheric pressure chemical ionization, and atmospheric pressure photoionization for the analysis of dinitropyrene and aminonitropyrene LCMS/MS, Journal of the American Society for Mass Spectrometry, 15 (12): 18531862 (2004) Sweetman, J. A.; Karasek, F.W.; Schuetzle, D.; Decomposition of nitropyrene during gas-chromatographic mass-spectrometric analysis of air particulate and fly-ash samples, Journal of Chromatography 247 (2): 245-254 (1982) Sweetman, J. A.; Zielinska, B.; Atkinson, R.; Ramdahl, T.; Winter,A .M.; Pitts, J.N.Jr.; A possible formation pathway for the 2-nitrofluoranthene observed in ambient particulate organic-matter, Atmospheric Environment 20 (1): 235-238, (1986) Tákats, Z.; Vékey, K.; Electrospray and atmospheric pressure chemical ionisation of aromatic compounds in dichloromethane solvent, European Mass Spectrometry 4 (5): 365-370 (1998) Thomson, B.A.; Iribarne, J.V.; Field-induced ion evaporation from liquid surfaces at atmospheric-pressure, Journal of Chemical Physics 71 (11): 4451– 4463 (1979) Tokiwa, H.; Morita, H.; Ohnish, Y.; Mutagenicity and carcinogenicity of nitroarenes and their sources in the environment, Crc Critical Reviews in Toxicology 17 (1): 23-60 (1986) 111 Varghese, A.J.; Withmore, G.T.; Binding of nitroreduction products of misonidazole to nucleic-acids and protein, Cancer Clinical Trials 3 (1): 43-46 (1980) Vasconcellos, P.C.; et al.; Chemical composition of aerosol collected in the amazon forest, Química Nova 21 (4): 385-393 (1998) Williams, T.T.J.; Perreault, H.; Selective detection of nitrated polycyclic aromatic hydrocarbons by electrospray ionization mass spectrometry and constant neutral loss scanning, Rapid Communications in Mass Spectrometry 14 (16): 1474-1481 (2000) Wise, S.A.; Sander, L.C.; May, W.E.; Determination of polycyclic aromatichydrocarbons by liquid-chromatography, Journal of Chromatography, 642 (1-2): 329-349 (1993) Whitehouse, C.N.; Dreyer, R.N.; Yamashita, M.; Fenn, J.B.; Electrospray interface for liquid chromatographs and mass spectrometers, Analytical Chemistry 57 (3): 675-679 (1985) Yamashita, M.; Fenn, J.B.; Negative-ion production with the electrospray ionsource, Journal of Physical Chemistry 88 (20): 4671–4675 (1984) Zeleny, J.; Industry application of electrospray technology, Physical Review., 10: 1–6 (1917) Zwirner-Baier, I.; Neumann, H.G.; Polycyclic nitroarenes (nitro-pahs) as biomarkers of exposure to diesel exhaust, Mutation Research-Genetic Toxicology and Environmental Mutagenesis 441 (1): 135-144 (1999) 112
Baixar