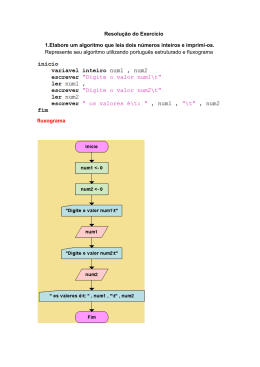
UNIVERSIDADE TECNOLÓGICA FEDERAL DO PARANÁ
PROGRAMA DE PÓS-GRADUAÇÃO EM ENGENHARIA ELÉTRICA E
INFORMÁTICA INDUSTRIAL
DIEGO HUMBERTO KALEGARI
ALGORITMO DE EVOLUÇÃO DIFERENCIAL PARALELO
APLICADO AO PROBLEMA DA PREDIÇÃO DA ESTRUTURA DE
PROTEÍNAS UTILIZANDO O MODELO AB EM 2D E 3D
DISSERTAÇÃO DE MESTRADO
CURITIBA
2010
DIEGO HUMBERTO KALEGARI
ALGORITMO DE EVOLUÇÃO DIFERENCIAL PARALELO
APLICADO AO PROBLEMA DA PREDIÇÃO DA ESTRUTURA DE
PROTEÍNAS UTILIZANDO O MODELO AB EM 2D E 3D
Dissertação apresentada ao Programa de Pósgraduação em Engenharia Elétrica e Informática
Industrial da Universidade Tecnológica Federal do
Paraná como requisito parcial para obtenção do grau
de “Mestre em Ciências” – Área de Concentração:
Informática Industrial.
Orientador:
Prof. Dr. Heitor Silvério Lopes
CURITIBA
2010
Dados Internacionais de Catalogação na Publicação
K14a
Kalegari, Diego Humberto
Algoritmo de evolução diferencial paralelo aplicado ao problema da
predição da estrutura de proteínas utilizando o modelo AB em 2D e 3D /
Diego Humberto Kalegari. — 2010.
126 p. : il. ; 30 cm
Orientador: Heitor Silvério Lopes
Dissertação (Mestrado) – Universidade Tecnológica Federal do Paraná.
Programa de Pós-graduação em Engenharia Elétrica e Informática Industrial.
Área de concentração: Informática industrial, Curitiba, 2010.
Bibliografia: p. 105-109
1. Bioinformática. 2. Controle preditivo. 3. Programação paralela
(Computação). 4. Proteínas. 5. Peptídeos. 6. Computação evolucionária.
7. Engenharia elétrica – Dissertações. I. Lopes, Heitor Silvério, orient.
II. Universidade Tecnológica Federal do Paraná. Programa de Pósgraduação em Engenharia Elétrica e Informática Industrial. III. Título.
CDD (22. ed.) 621.3
Biblioteca Central da UTFPR, Campus Curitiba
AGRADECIMENTOS
À minha esposa Bárbara pelo apoio, dedicação e as palavras de incentivo, que me permitiram a não desistir e perseguir meus objetivos. Agradeço também a ela pela compreensão e
paciência que teve comigo nesses últimos meses, além da ajuda preciosa na revisão do texto.
Aos meus pais, José e Dilma, pelo incentivo e apoio em todos os momentos. Em especial à
minha mãe. Às minhas irmãs, avós e demais familiares que me apoiam.
A meu orientador, Prof. Heitor, por toda a sua dedicação, paciência e orientação durante a
realização deste projeto. Gostaria de, em especial, agradecê-lo por ter acreditado em mim e ter
sido compreensivo nos momentos em que precisei viajar à trabalho.
Aos colegas do laboratório de Bioinformática da UTFPR, em especial ao César, que me
deram suporte.
A todas as pessoas que participaram direta ou indireta nessa minha caminhada acadêmica.
Meu Muito Obrigado.
RESUMO
Kalegari, Diego Humberto. Algoritmo de Evolução Diferencial Paralelo aplicado ao Problema
da Predição da Estrutura de Proteínas utilizando o modelo AB em 2D e 3D . 126 f. Dissertação
– Programa de Pós-graduação em Engenharia Elétrica e Informática Industrial, Universidade
Tecnológica Federal do Paraná. Curitiba, 2010.
A problema da predição da estrutura de proteínas (PPEP) é bastante conhecido na bioinformática. A identificação da conformação nativa de uma proteína permite predizer a sua função
no organismo. Este conhecimento também é útil no desenvolvimento de novos fármacos ou
na compreensão do mecanismo de várias doenças. Várias técnicas tem sido propostas para resolver este problema. Porém, o alto custo envolvido levou ao surgimento de vários modelos
que simplificam, em parte, as estruturas proteicas. No entanto, mesmo com os modelos mais
simplificados, a complexidade do problema traz inúmeros desafios computacionais na busca da
sua conformação nativa. Este trabalho utiliza o algoritmo evolucionário denominado Evolução
Diferencial (ED) para solucionar o PPEP, representando as proteínas com o modelo AB (toy
model), em duas e três dimensões (2D e 3D). O trabalho apresenta a implementação de duas
versões da ED, paralelizadas num ambiente de processamento em cluster, com Message Passing Interface e arquitetura mestre-escravo. Para a configuração dos operadores do algoritmo de
ED, foram realizados vários estudos com diferentes configurações para ambos os modelos, e
análises estatísticas determinaram quais os melhores valores. Além disso, foram criados dois
operadores especiais: dizimação e mutação espelhada. O primeiro pode ser considerado um
operador genérico, que pode ser utilizado em qualquer problema; o segundo é específico para
o problema em questão. Além do algoritmo de ED básico, também foi proposta uma versão
auto-adaptável, em que alguns de seus parâmetros são atualizados no decorrer da evolução. Os
experimentos realizados utilizaram 4 sequências de aminoácidos de benchmark geradas a partir
da sequência de Fibonacci, contendo entre 13 e 55 aminoácidos. Os resultados dos algoritmos
de ED paralelos foram comparados com os resultados obtidos em outros trabalhos. O algoritmo
de ED é capaz de obter resultados excelentes, competitivos com os métodos especializados,
apesar de não atingir o ótimo conhecido em algumas instâncias. Os resultados promissores
obtidos nesse trabalho mostram que o algoritmo de ED é adequado para o problema. Em trabalhos futuros poderão ser estudados novos operadores especiais ou outras técnicas de inspiração
biológica, buscando melhorar os resultados.
Palavras-chave: Bioinformática, Problema da Predição da Estrutura de Proteínas, Dobramento
de Proteína , Modelo AB, Computação Evolucionária, Evolução Diferencial, Computação Paralela, Auto-adaptabilidade
ABSTRACT
Kalegari, Diego Humberto. An improved parallel differential evolution approach for protein
structure prediction using a 2D and 3D off-lattice model. 126 f. Dissertação – Programa de Pósgraduação em Engenharia Elétrica e Informática Industrial, Universidade Tecnológica Federal
do Paraná. Curitiba, 2010.
Protein structure prediction is a well-known problem in bioinformatics. Identifying protein native conformation makes it possible to predict its function within the organism. Knowing this
also helps in the development of new medicines and in comprehending how some illnesses work
and act. During the past year some techniques have been proposed to solve this problem, but
its high cost made it necessary to build models that simplify the protein structures. However,
even with the simplicity of these models identifying the protein native conformation remains a
highly complex, computationally challenging problem. This paper uses an evolutionary algorithm known as Differential Evolution (DE) to solve the protein structure prediction problem.
The model used to represent the protein structure is the Toy Model (also known as the AB
Model) in both 2D and 3D. This work implements two versions of the ED algorithm using a
parallel architecture (master-slave) based on Message Passing Interface in a cluster. A large
number of tests were executed to define the final configuration of the DE operators for both
models. A new set of special operators were developed: explosion and mirror mutation. We
can consider the first one as a generic operator, because it can be used in any problem. The
second one is more specific because it requires previous knowledge of the problem. Of the two
DE algorithm implemented, one is a basic DE algorithm and the second is a self-adaptive DE.
All tests executed in this work used four benchmark amino acid sequences generated from the
Fibonacci sequence. Each sequence has 13 to 55 amino acids. The results for both parallel DE
algorithms using both 2D and 3D models were compared with other works. The DE algorithm
achieved excellent results. It did not achieve the optimal known values for some sequences, but
it was competitive with other specialized methods. Overall results encourage further research
toward the use of knowledge-based operators and biologically inspired techniques to improve
DE algorithm performance.
Keywords: Bioinformatics, Protein Structure Prediction, Protein Folding, Toy Model (AB
Model), Evolutionary Computation, Differential Evolution, Parallel Computation, self-adaptive
LISTA DE FIGURAS
FIGURA 1 – ESTRUTURA α DOS AMINOÁCIDOS ENCONTRADOS NAS ESTRUTURAS PROTEICAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
FIGURA 2 – LIGAÇÃO PEPTÍDICA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
FIGURA 3 – CLASSES DOS AMINOÁCIDOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
FIGURA 4 – DOBRAMENTO BASEADO EM HIDROFOBICIDADE . . . . . . . . . . . . . .
FIGURA 5 – ESTRUTURA PRIMÁRIA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
FIGURA 6 – ESTRUTURA SECUNDÁRIA DE UMA PROTEÍNA . . . . . . . . . . . . . . . . .
FIGURA 7 – ESTRUTURA TERCIARIA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
FIGURA 8 – ESTRUTURA QUATERNÁRIA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
FIGURA 9 – DOBRAMENTOS DO MODELO HP . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
FIGURA 10 – FORMAÇÃO DE ÂNGULOS DIEDRAIS . . . . . . . . . . . . . . . . . . . . . . . . . . . .
FIGURA 11 – REPRESENTAÇÃO GENÉRICA DE UMA PROTEÍNA COMPOSTA DE
9 (NOVE) AMINOÁCIDOS BASEADA NO MODELO AB EM 2D . . . . . .
FIGURA 12 – REPRESENTAÇÃO GENÉRICA DE UMA PROTEÍNA COMPOSTA DE
9 (NOVE) AMINOÁCIDOS BASEADA NO MODELO AB EM 3D . . . . . .
FIGURA 13 – PROCESSO DE MUTAÇÃO DIFERENCIAL PARA GERAÇÃO DE UM
VETOR DOADOR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
FIGURA 14 – REPRESENTAÇÃO DOS INDIVÍDUOS PARA AS SEQUÊNCIAS DE
FIBONACCI NO MODELO AB 2D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
FIGURA 15 – REPRESENTAÇÃO DOS INDIVÍDUOS PARA AS SEQUÊNCIAS DE
FIBONACCI NO MODELO AB 3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
FIGURA 16 – REPRESENTAÇÃO GRÁFICA DO PROCESSO DE CONVERSÃO DE
ÂNGULOS PARA COORDENADAS CARTESIANAS EM 2D . . . . . . . . . .
FIGURA 17 – ROTAÇÃO EM TORNOS DOS EIXOS CARTESIANOS NO ESPAÇO
3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
FIGURA 18 – EXEMPLO DE ROTAÇÃO EM TORNO DO EIXO Y NO ESPAÇO 3D .
FIGURA 19 – REPRESENTAÇÃO GRÁFICA DO PROCESSO DE CONVERSÃO DE
ÂNGULOS PARA COORDENADAS CARTESIANAS EM 3D . . . . . . . . . .
FIGURA 20 – ESTRUTURA DO MODELO MESTRE/ESCRAVO . . . . . . . . . . . . . . . . . . .
FIGURA 21 – REPRESENTAÇÃO GRÁFICA DE UMA PROTEÍNA COM 8 AMINOÁCIDOS NO MODELO 2D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
FIGURA 22 – REPRESENTAÇÃO GRÁFICA DE UMA PROTEÍNA COM 8 AMINOÁCIDOS NO MODELO 2D DEPOIS DA MUTAÇÃO ESPELHADA . . . . . . .
FIGURA 23 – COMPARAÇÃO ENTRE OS RESULTADOS OBTIDOS PARA A ESTRATÉGIA DE EVOLUÇÃO COM A SEQUÊNCIA DE 13 AMINOÁCIDOS NO MODELO 2D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
FIGURA 24 – COMPARAÇÃO ENTRE OS RESULTADOS OBTIDOS PARA A ESTRATÉGIA DE EVOLUÇÃO COM A SEQUÊNCIA DE 13 AMINOÁCIDOS NO MODELO 3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
FIGURA 25 – COMPARAÇÃO ENTRE OS RESULTADOS OBTIDOS PARA A ESTRATÉGIA DE EVOLUÇÃO COM A SEQUÊNCIA DE 21 AMINOÁCIDOS NO MODELO 2D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
19
20
21
22
23
23
24
25
31
34
34
35
40
51
52
54
56
57
60
63
71
72
78
79
80
FIGURA 26 – COMPARAÇÃO ENTRE OS RESULTADOS OBTIDOS PARA A ESTRATÉGIA DE EVOLUÇÃO COM A SEQUÊNCIA DE 21 AMINOÁCIDOS NO MODELO 3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81
FIGURA 27 – COMPARAÇÃO ENTRE OS RESULTADOS OBTIDOS PARA A ESTRATÉGIA DE EVOLUÇÃO COM A SEQUÊNCIA DE 34 AMINOÁCIDOS NO MODELO 2D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81
FIGURA 28 – COMPARAÇÃO ENTRE OS RESULTADOS OBTIDOS PARA A ESTRATÉGIA DE EVOLUÇÃO COM A SEQUÊNCIA DE 34 AMINOÁCIDOS NO MODELO 3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 82
FIGURA 29 – COMPARAÇÃO ENTRE OS RESULTADOS OBTIDOS PARA A ESTRATÉGIA DE EVOLUÇÃO COM A SEQUÊNCIA DE 55 AMINOÁCIDOS NO MODELO 2D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 83
FIGURA 30 – COMPARAÇÃO ENTRE OS RESULTADOS OBTIDOS PARA A ESTRATÉGIA DE EVOLUÇÃO COM A SEQUÊNCIA DE 55 AMINOÁCIDOS NO MODELO 3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 83
FIGURA 31 – COMPARAÇÃO ENTRE AS DISTRIBUIÇÕES DE F COM CR=0,85 NO
MODELO AB 2D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 86
FIGURA 32 – COMPARAÇÃO ENTRE AS DISTRIBUIÇÕES DE F COM CR=0,85 NO
MODELO AB 3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 87
FIGURA 33 – AJUSTE DE F E CR COMBINADOS PARA O MODELO 2D APÓS
100.000 GERAÇÕES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89
FIGURA 34 – AJUSTE DE F E CR COMBINADOS PARA O MODELO 2D APÓS
350.000 GERAÇÕES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89
FIGURA 35 – AJUSTE DE F E CR COMBINADOS PARA O MODELO 3D APÓS
100.000 GERAÇÕES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 90
FIGURA 36 – AJUSTE DE F E CR COMBINADOS PARA O MODELO 3D APÓS
350.000 GERAÇÕES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 91
FIGURA 37 – DISTRIBUIÇÃO DOS RESULTADOS UTILIZANDO DIZIMAÇÃO PARA
O MODELO 2D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92
FIGURA 38 – DISTRIBUIÇÃO DOS RESULTADOS UTILIZANDO A DIZIMAÇÃO
PARA O MODELO 3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 93
FIGURA 39 – DISTRIBUIÇÃO DA MUTAÇÃO ESPELHADA PARA O MODELO 2D 95
FIGURA 40 – MELHOR CONFORMAÇÃO OBTIDA COM O ALGORITMO ED NO
MODELO 2D PARA A SEQUÊNCIA DE 13 AMINOÁCIDOS . . . . . . . . . . 116
FIGURA 41 – MELHOR CONFORMAÇÃO OBTIDA COM O ALGORITMO ED NO
MODELO 2D PARA A SEQUÊNCIA DE 21 AMINOÁCIDOS . . . . . . . . . . 116
FIGURA 42 – MELHOR CONFORMAÇÃO OBTIDA COM O ALGORITMO ED NO
MODELO 2D PARA A SEQUÊNCIA DE 34 AMINOÁCIDOS . . . . . . . . . . 117
FIGURA 43 – MELHOR CONFORMAÇÃO OBTIDA COM O ALGORITMO ED NO
MODELO 2D PARA A SEQUÊNCIA DE 55 AMINOÁCIDOS . . . . . . . . . . 117
FIGURA 44 – MELHOR CONFORMAÇÃO OBTIDA COM O ALGORITMO ED AUTOADAPTÁVEL NO MODELO 2D PARA A SEQUÊNCIA DE 13 AMINOÁCIDOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 118
FIGURA 45 – MELHOR CONFORMAÇÃO OBTIDA COM O ALGORITMO ED AUTOADAPTÁVEL NO MODELO 2D PARA A SEQUÊNCIA DE 21 AMINOÁCIDOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 119
FIGURA 46 – MELHOR CONFORMAÇÃO OBTIDA COM O ALGORITMO ED AUTOADAPTÁVEL NO MODELO 2D PARA A SEQUÊNCIA DE 34 AMINOÁ-
CIDOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 119
FIGURA 47 – MELHOR CONFORMAÇÃO OBTIDA COM O ALGORITMO ED AUTOADAPTÁVEL NO MODELO 2D PARA A SEQUÊNCIA DE 55 AMINOÁCIDOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 120
FIGURA 48 – MELHOR CONFORMAÇÃO OBTIDA COM O ALGORITMO ED NO
MODELO 3D PARA A SEQUÊNCIA DE 13 AMINOÁCIDOS . . . . . . . . . . 122
FIGURA 49 – MELHOR CONFORMAÇÃO OBTIDA COM O ALGORITMO ED NO
MODELO 3D PARA A SEQUÊNCIA DE 21 AMINOÁCIDOS . . . . . . . . . . 122
FIGURA 50 – MELHOR CONFORMAÇÃO OBTIDA COM O ALGORITMO ED NO
MODELO 3D PARA A SEQUÊNCIA DE 34 AMINOÁCIDOS . . . . . . . . . . 123
FIGURA 51 – MELHOR CONFORMAÇÃO OBTIDA COM O ALGORITMO ED NO
MODELO 3D PARA A SEQUÊNCIA DE 55 AMINOÁCIDOS . . . . . . . . . . 123
FIGURA 52 – MELHOR CONFORMAÇÃO OBTIDA COM O ALGORITMO ED AUTOADAPTÁVEL NO MODELO 3D PARA A SEQUÊNCIA DE 13 AMINOÁCIDOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 125
FIGURA 53 – MELHOR CONFORMAÇÃO OBTIDA COM O ALGORITMO ED AUTOADAPTÁVEL NO MODELO 3D PARA A SEQUÊNCIA DE 21 AMINOÁCIDOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 125
FIGURA 54 – MELHOR CONFORMAÇÃO OBTIDA COM O ALGORITMO ED AUTOADAPTÁVEL NO MODELO 3D PARA A SEQUÊNCIA DE 34 AMINOÁCIDOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 126
FIGURA 55 – MELHOR CONFORMAÇÃO OBTIDA COM O ALGORITMO ED AUTOADAPTÁVEL NO MODELO 3D PARA A SEQUÊNCIA DE 55 AMINOÁCIDOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 126
LISTA DE TABELAS
TABELA 1
TABELA 2
TABELA 3
TABELA 4
TABELA 5
TABELA 6
TABELA 7
TABELA 8
TABELA 9
TABELA 10
TABELA 11
TABELA 12
TABELA 13
TABELA 14
TABELA 15
TABELA 16
TABELA 17
TABELA 18
TABELA 19
TABELA 20
– ESTRATÉGIAS DE EVOLUÇÃO ED . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
– SEQUÊNCIAS DE BENCHMARK . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
– RESULTADOS OBTIDOS NOS EXPERIMENTOS REALIZADOS NOS
MODELOS AB 2D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 49
– RESULTADOS OBTIDOS NOS EXPERIMENTOS REALIZADOS NOS
MODELOS AB 3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 49
– CONFIGURAÇÕES PARA AJUSTE DA ESTRATÉGIA . . . . . . . . . . . . . 68
– CONFIGURAÇÕES PARA AJUSTE DE F . . . . . . . . . . . . . . . . . . . . . . . . . 68
– CONFIGURAÇÕES PARA AJUSTE DE CR . . . . . . . . . . . . . . . . . . . . . . . 69
– VALORES DE TESTE PARA DEFINIÇÃO DO MAXCOUNT PARA A
DIZIMAÇÃO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70
– VALORES DE MAXMIRROR PARA O OPERADOR DE MUTAÇÃO
ESPELHADA DO MODELO EM 2D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72
– AJUSTE DE NÚMERO INDIVÍDUOS POR ESCRAVO PARA O ALGORITMO DE ED PARALELO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 76
– RESULTADOS DO AJUSTE DE PARÂMETROS PARA A ESTRATÉGIA DE EVOLUÇÃO COM A SEQUÊNCIA DE 13 AMINOÁCIDOS NO
MODELO 2D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 78
– RESULTADOS DO AJUSTE DE PARÂMETROS PARA A ESTRATÉGIA DE EVOLUÇÃO COM A SEQUÊNCIA DE 13 AMINOÁCIDOS NO
MODELO 3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
– RESULTADOS DO AJUSTE DE PARÂMETROS PARA A ESTRATÉGIA DE EVOLUÇÃO COM A SEQUÊNCIA DE 21 AMINOÁCIDOS NO
MODELO 2D1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 80
– RESULTADOS DO AJUSTE DE PARÂMETROS PARA A ESTRATÉGIA DE EVOLUÇÃO COM A SEQUÊNCIA DE 21 AMINOÁCIDOS NO
MODELO 3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 80
– RESULTADOS DO AJUSTE DE PARÂMETROS PARA A ESTRATÉGIA DE EVOLUÇÃO N=34 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81
– RESULTADOS DO AJUSTE DE PARÂMETROS PARA A ESTRATÉGIA DE EVOLUÇÃO COM A SEQUÊNCIA DE 34 AMINOÁCIDOS NO
MODELO 3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 82
– RESULTADOS DO AJUSTE DE PARÂMETROS PARA A ESTRATÉGIA DE EVOLUÇÃO COM A SEQUÊNCIA DE 55 AMINOÁCIDOS NO
MODELO 2D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 82
– RESULTADOS DO AJUSTE DE PARÂMETROS PARA A ESTRATÉGIA DE EVOLUÇÃO COM A SEQUÊNCIA DE 55 AMINOÁCIDOS NO
MODELO 3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 83
– ANÁLISE DE VARIÂNCIA PARA VALORES DE F DO MODELO AB
2D COM CR=0,85 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85
– RESULTADOS DO TESTE DE TUKEY PARA COMPARAÇÕES EN-
TABELA 21
TABELA 22
TABELA 23
TABELA 24
TABELA 25
TABELA 26
TABELA 27
TABELA 28
TABELA 29
TABELA 30
TABELA 31
TABELA 32
TABELA 33
TABELA 34
TABELA 35
TABELA 36
TABELA 37
TABELA 38
TABELA 39
TABELA 40
TABELA 41
TABELA 42
TABELA 43
TRE MÉDIAS DE F COM CR=0,85 NO MODELO AB 2D . . . . . . . . . . . . . 86
– ANÁLISE DE VARIÂNCIA PARA VALORES F DO MODELO AB 3D
COM CR=0,85 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 87
– RESULTADOS DO TESTE DE TUKEY PARA COMPARAÇÕES ENTRE MÉDIAS F COM CR=0,85 NO MODELO AB 3D . . . . . . . . . . . . . . . . 88
– ANÁLISE DE VARIÂNCIA PARA A DIZIMAÇÃO (MAXCOUNT)
MODELO 2D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 91
– ESTATÍSTICAS DA ENERGIA MÍNIMA GERADA NO PROCESSO
DE DIZIMAÇÃO NO MODELO AB 2D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92
– ANÁLISE DE VARIÂNCIA PARA A DIZIMAÇÃO (MAXCOUNT)
MODELO 3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 93
– ESTATÍSTICAS DA ENERGIA MÍNIMA GERADA NO PROCESSO
DE DIZIMAÇÃO NO MODELO AB 3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
– ANÁLISE DE VARIÂNCIA PARA O MAXMIRROR NO MODELO
2D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
– ESTATÍSTICAS DA ENERGIA MÍNIMA GERADA NO PROCESSO
DE MUTAÇÃO ESPELHADA NO MODELO AB 2D . . . . . . . . . . . . . . . . . . 95
– RESULTADOS ED AUTO-ADAPTÁVEL PARA SEQUÊNCIA DE 34
AMINOÁCIDOS NOS MODELOS 2D E 3D . . . . . . . . . . . . . . . . . . . . . . . . . . 96
– RESULTADOS ED COM PARÂMETROS AJUSTADOS PARA AS SEQUÊNCIAS DE BENCHMARK MODELO 2D . . . . . . . . . . . . . . . . . . . . . . . . . 97
– RESULTADOS ED COM PARÂMETROS AJUSTADOS PARA AS SEQUÊNCIAS DE BENCHMARK MODELO 3D . . . . . . . . . . . . . . . . . . . . . . . . . 97
– RESULTADOS ED AUTO-ADAPTÁVEL PARA AS SEQUÊNCIAS DE
BENCHMARK MODELO 2D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97
– RESULTADOS ED AUTO-ADAPTÁVEL PARA AS SEQUÊNCIAS DE
BENCHMARK MODELO 3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 98
– RESULTADOS OBTIDOS NOS EXPERIMENTOS COM OS BENCHMARKS NO MODELO AB 2D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 98
– COMPARAÇÃO ENTRE O MELHOR BENCHMARK NO MODELO
AB 2D COM OS MELHORES RESULTADOS DO PRESENTE TRABALHO 99
– RESULTADOS OBTIDOS NOS EXPERIMENTOS COM OS BENCHMARKS NO MODELOS AB 3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 99
– COMPARAÇÃO ENTRE O MELHOR BENCHMARK NO MODELO
AB 3D COM OS MELHORES RESULTADOS DO PRESENTE TRABALHO 99
– LISTA DOS AMINOÁCIDOS PROTEINOGÊNICOS . . . . . . . . . . . . . . . 111
– CLASSIFICAÇÃO DOS AMINOÁCIDOS SEGUNDO CRITÉRIOS DE
HIDROFOBICIDADE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 113
– ÂNGULOS DAS MELHORES CONFORMAÇÕES OBTIDOS PELO
ALGORITMO DE ED PARA AS SEQUÊNCIAS DE BENCHMARK MODELO 2D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 115
– ÂNGULOS DAS MELHORES CONFORMAÇÕES OBTIDOS PELO
ALGORITMO DE ED AUTO ADAPTÁVEL PARA AS SEQUÊNCIAS DE
BENCHMARK MODELO 2D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 118
– ÂNGULOS DAS MELHORES CONFORMAÇÕES OBTIDOS ALGORITMO DE ED PARA AS SEQUÊNCIAS DE BENCHMARK MODELO
3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 121
– ÂNGULOS DAS MELHORES CONFORMAÇÕES OBTIDOS PELO
ALGORITMO DE ED AUTO ADAPTÁVEL PARA AS SEQUÊNCIAS DE
BENCHMARK MODELO 3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 124
LISTA DE SIGLAS
HP
Hidrofóbico-Polar
AG
Algoritmo Genético
ED
Evolução Diferencial
PDP
Problema do Dobramento de Proteína
PPEP
Problema Predição da Estrutura de Proteínas
PDB
Protein Data Bank
RMN
Ressonância Magnética Nuclear
CE
Computação Evolucionária
PG
Programação Genética
PSO
Particle Swarm Optimization
ACO
Ant Colony Optimization
VLSI
Very Large-Scale Integration
PVM
Parallel Virtual Machine
MPI
Message Passing Interface
SPMD
Single Program Multiple Data
MIMD
Multiple Instruction Multiple Data
ANOVA
Análise de Variância
SUMÁRIO
1 INTRODUÇÃO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1.1 MOTIVAÇÃO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1.2 OBJETIVOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1.2.1 Objetivo Geral . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1.2.2 Objetivos Específicos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1.3 ESTRUTURA DA DISSERTAÇÃO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2 FUNDAMENTAÇÃO TEÓRICA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.1 PROTEÍNAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.2 ESTRUTURA DE PROTEÍNAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.3 DOBRAMENTO DE PROTEÍNAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.4 O PROBLEMA DO DOBRAMENTO DE PROTEÍNAS . . . . . . . . . . . . . . . . . . . . . . . . .
2.5 MÉTODOS COMPUTACIONAIS QUE ABORDAM O PPEP . . . . . . . . . . . . . . . . . . . .
2.5.1 Modelagem por Métodos Baseados em Padrões . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.5.2 Modelagem por Métodos ab initio . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.6 TOY MODEL - MODELO AB . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.7 COMPUTAÇÃO EVOLUCIONÁRIA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.7.1 Evolução Diferencial . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.8 COMPUTAÇÃO PARALELA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3 METODOLOGIA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.1 BENCHMARKS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.2 MODELAGEM DO PPEP UTILIZANDO ED . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.3 REPRESENTAÇÃO DOS INDIVÍDUOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.3.1 Modelo AB em duas dimensões (2D) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.3.2 Modelo AB em três dimensões (3D) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.4 DEFINIÇÃO E IMPLEMENTAÇÃO DA FUNÇÃO DE FITNESS . . . . . . . . . . . . . . . .
3.4.1 Modelo AB em duas dimensões (2D) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.4.2 Modelo AB em três dimensões (3D) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.5 METODOLOGIA DE PARALELIZAÇÃO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.6 AJUSTE DE PARÂMETROS DA ED . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.7 DIZIMAÇÃO (EXPLOSÃO) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.8 MUTAÇÃO ESPELHADA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.9 ADAPTAÇÃO DINÂMICA DOS PARÂMENTROS DE CONTROLE DA ED . . . . .
4 RESULTADOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4.1 AJUSTE DA PARALELIZAÇÃO DO ALGORITMO DE ED . . . . . . . . . . . . . . . . . . . .
4.2 RESULTADOS PARA AJUSTE DE PARÂMETROS . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4.2.1 Ajuste da Estratégia de Evolução . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4.2.2 Ajuste dos parâmetros da ED (constante de diferenciação (F) e crossover (CR)) . . .
4.2.3 Ajustes Dizimação . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4.2.4 Ajuste de parâmetros da mutação espelhada . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4.2.5 Resultados do algoritmo de ED auto-adaptável . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4.2.6 Resultados do algoritmo final de ED com os parâmetros ajustados . . . . . . . . . . . . . . . .
15
16
17
17
17
17
19
19
22
25
26
28
28
30
33
36
38
43
47
47
50
51
51
51
52
52
55
62
67
69
70
72
75
75
77
78
84
90
94
96
96
4.2.7 Resultados do algoritmo final de ED auto-adaptável . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97
4.2.8 Comparação com outras abordagens . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 98
5 DISCUSSÃO DOS RESULTADOS E CONCLUSÃO . . . . . . . . . . . . . . . . . . . . . . . . . . . 101
5.1 DISCUSSÃO DOS RESULTADOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101
5.2 CONCLUSÃO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 102
5.3 TRABALHOS FUTUROS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 103
REFERÊNCIAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 105
ANEXO A -- AMINOÁCIDOS PROTEINOGÊNICOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 111
ANEXO B -- CLASSIFICAÇÃO DOS AMINOÁCIDOS SEGUNDO O MODELO HP 113
ANEXO C -- ÂNGULOS E CONFORMAÇÕES DOS DOBRAMENTOS OBTIDOS
COM OS ALGORITMOS DE ED . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 115
15
1
INTRODUÇÃO
As proteínas são os compostos orgânicos mais abundantes nas células vivas e existem aos
milhares, desde peptídeos de tamanho relativamente pequeno a polímeros gigantescos. Além de
estarem nas células, as proteínas exibem uma grande diversidade de funções biológicas (função
estrutural: dando rigidez, consistência e elasticidade aos tecidos; função hormonal: realizam
função específica em estruturas do corpo; função de defesa do organismo; função energética;
função enzimática; função de conduzir gases, além de várias outras), e são instrumentos moleculares por meio dos quais a informação genética é expressa (LEHNINGER; NELSON; COX,
2001).Visando mapear essas estruturas, existe hoje uma grande quantidade de dados sobre as
proteínas, mas pouco se sabe sobre elas, pois o conhecimento extraído é infinitamente pequeno
quando comparado à sua complexidade estrutural.
As proteínas são formadas por estruturas menores denominadas aminoácidos, sendo que
todas são constituídas partindo-se de um conjunto ubíquo de 20 (vinte) aminoácidos, combinados sequencialmente através de ligações peptídicas, um tipo especial de ligação covalente, para
a formação dos peptídios/polipeptídios formadores da molécula proteica. Os aminoácidos presentes nas proteínas são diferentes entre si devido a vários fatores, tais como as cadeias laterais
e a afinidade com a água, além de tantas outras características.
O processo pelo qual ocorre a formação desses polipeptídios é denominado de Dobramento
de Proteína. É através do dobramento que uma proteína atinge sua conformação nativa, ou seja,
a conformação tridimensional final. A função que uma determinada proteína exerce está diretamente ligada à sequência de aminoácidos que a compõe, assim como à estrutura tridimensional
formada por eles (LEHNINGER; NELSON; COX, 2001). Uma proteína mal-dobrada pode
perder suas funções normais, adquirindo funções diferentes ou nenhuma função. Quando essas
proteínas mal-dobradas são combinadas entre si podem prejudicar a formação celular ou causar
doenças sérias (como fibrose cística, a doença da vaca louca ou alguns tipos de câncer). Além
disso, determinar a função proteica auxilia no desenvolvimento de novos fármacos, pois, conhecendo a sua estrutura tridimensional, pode-se identificar quais compostos podem ser ligados
ao sítio ativo da proteína de forma mais adequada.
16
Devido à grande importância de se entender como as proteínas adquirem sua conformação
nativa, pesquisadores ligados a diversas áreas têm dedicado inúmeros esforços para entender
esse processo de formação, o qual é estudado pela Bioinformática, que tem como um de seus
objetivos adquirir conhecimento sobre as funções das proteínas.
Para lidar com a complexidade das estruturas proteicas, vários modelos surgiram ao longo
dos anos visando a simplificação das informações contidas nas sequências. Dentre os modelos
simplificados mais estudados na Bioinformática estão o Modelo Hidrofóbico-Polar HP (DILL,
1985) e o Toy Model ou Modelo AB (STILLINGER; HEAD-GORDON; HIRSHFELD, 1993)
em duas e três dimensões. Ambos simplificam os aminoácidos da estrutura proteica em dois
tipos, diferenciando-os por meio da característica de afinidade com a água, além de delimitarem
o comprimento e os ângulos entre as ligações. O objetivo dos modelos é encontrar a energia
livre em função das conformações estruturais permitidas para ela, sendo que o ponto mínimo
global corresponde à conformação estrutural nativa de uma proteína. Isto é, determinar a energia
mínima significa encontrar a estrutura regular da proteína associada a essa função, ou seja, a
sua conformação nativa.
Diversas técnicas computacionais foram aplicadas a estes modelos visando encontrar a energia mínima, tais como: a simulação de Monte Carlo (CHIKENJI; KIKUCHI; IBA, 1999);
Algoritmo Genético ( AG) (LOPES; SCAPIN, 2005); colônias de formigas (SHMYGELSKA;
HOOS, 2003; ZHARG; LI, 2007); Evolução Diferencial ( ED) (BITELLO; LOPES, 2007),
entre outros.
1.1
MOTIVAÇÃO
O Problema do Dobramento de proteína s( PDP) ou Problema Predição da Estrutura de Pro-
teínas ( PPEP) é um dos problemas mais desafiadores da área da Bioinformática, e vêm sendo
explorado a fundo nos últimos anos por meio da aplicação de várias técnicas computacionais
nos diferentes modelos proteicos, visando encontrar uma técnica que permita a predição da
estrutura com mais eficiência e rapidez.
A motivação principal do trabalho é o estudo do algoritmo de evolução diferencial, uma técnica de computação evolucionária recente que tem se mostrado muito promissora nas áreas em
que ela tem sido aplicada (PLAGIANAKOS; TASOULIS; VRAHATIS, 2008), e a sua aplicação
ao problema da predição da estrutura de proteínas tanto bidimensional quanto tridimensional.
Outra motivação é a possibilidade de aplicar técnicas de computação paralelas ao algoritmo
de evolução diferencial, reduzindo o tempo de processamento, e fazendo com que essa técnica
17
possa ser aplicada com eficiência a sequências de proteínas relativamente longas.
1.2
OBJETIVOS
1.2.1
Objetivo Geral
O objetivo geral do trabalho é aplicar o algoritmo de Evolução Diferencial (STORN; PRICE,
1997) ao problema da predição da estrutura de proteínas (ou dobramento de proteínas), utilizando como base o Modelo AB (Toy Model) (STILLINGER; HEAD-GORDON; HIRSHFELD, 1993) em duas e três dimensões.
1.2.2
Objetivos Específicos
Os objetivos específicos são:
• Propor a melhor configuração para os parâmetros de controle do algoritmo de ED;
• Utilizar recursos de computação paralela para reduzir o tempo de processamento das
sequências de aminoácidos analisadas.
• Implementar um algoritmo de ED auto-adaptável e comparar o seu desempenho com o
desempenho do ED básico.
• Aplicar os algoritmos ED básico e ED auto-adaptável a um benchmark de sequências de
proteínas com o modelo AB, tanto em duas quanto em três dimensões.
1.3
ESTRUTURA DA DISSERTAÇÃO
A presente dissertação está organizada em cinco capítulos, sendo que o primeiro, do qual faz
parte essa seção, é composto pela Introdução, Motivação e Objetivos que levaram à elaboração
desse trabalho.
O Capítulo 2 traz uma abordagem sobre o Problema Predição da Estrutura de Proteínas,
baseada na revisão da literatura com relação aos conceitos teóricos por trás das proteínas, e
expõe as pesquisas existentes que visam solucionar esse problema. Ainda nesse capítulo, é
apresentada uma revisão literária sobre computação evolucionária, a qual levou à definição do
algoritmo de Evolução Diferencial, bem como os conceitos de computação paralela utilizada
para melhorar a velocidade do algoritmo de ED.
18
O Capítulo 3, por sua vez, descreve detalhes das abordagens utilizadas para a implementação dos modelos AB em 2D (duas dimensões) e 3D (três dimensões), assim como do processo
utilizado para definir e ajustar os parâmetros do algoritmo de ED para a solução do problema.
Além disso, referido capítulo também apresenta a proposta de dois novos operadores, quais
sejam, a dizimação e a mutação espelhada, que foram criados na tentativa de melhorar o desempenho do algoritmo. Ademais, apresenta a proposta de um algoritmo de ED auto-adaptável,
assim como o processo de paralelização do algoritmo.
Já no Capítulo 4, são apresentados os resultados obtidos para o ajuste de parâmetros, assim
como os melhores resultados obtidos para os modelos AB em 2D e 3D utilizando os algoritmos
propostos.
Por fim, o Capítulo 5 apresenta a discussão dos resultados obtidos, a conclusão sobre o
trabalho e os trabalhos futuros.
Ao final da dissertação foram incluídos anexos que mostram os aminoácidos presentes em
uma proteína, sua nomenclatura, abreviações e fórmulas, além da classificação por hidrofobicidade. Também são apresentados os resultados finais obtidos, com os valores dos ângulos das
conformações e a representação gráfica para ambos os modelos (2D e 3D).
19
2
FUNDAMENTAÇÃO TEÓRICA
2.1
PROTEÍNAS
Os aminoácidos são unidades monoméricas (monômeros 1 ) que podem ser unidas por meio
de um tipo de ligação covalente específica, denominada ligação peptídica. As ligações entre
aminoácidos formam os peptídios, moléculas pequenas contendo poucos aminoácidos. Os peptídios se ligam e formam os polipeptídios que, por sua vez, quando ligados entre si, formam os
polímeros, os quais são compostos por uma infinidade de subunidades semelhantes ou idênticas
e são chamados de proteínas.
Os aminoácidos presentes nas proteínas são α-aminoácidos, ou seja, eles apresentam um
grupo carboxila e um grupo amino, ligados ao mesmo átomo de carbono (o carbono α). Esses
aminoácidos são distintos dos aminoácidos menos comuns, pois são frequentemente referidos
como aminoácidos primários, padrões ou normais (LEHNINGER; NELSON; COX, 2001). Existem inúmeros tipos de aminoácidos presentes nos seres vivos, mas apenas 20 (vinte) podem
ser encontrados na formação das estruturas proteicas. A listagem destes, assim como a suas
abreviaturas, podem ser encontradas no Anexo A.
A diferença entre os aminoácidos é dada pela cadeia lateral, denominada de grupo ou radical (R), que está ligada diretamente ao carbono-α. A Figura 1 representa a estrutura comum de
todos os aminoácidos α das proteínas, exceto da prolina, que é a exceção, por ser um aminoácido cíclico.
Figura 1: Estrutura α dos aminoácidos encontrados nas estruturas proteicas
Fonte: Adaptado de (LEHNINGER; NELSON; COX, 2001)
1 Molécula,
de massa molecular geralmente pequena, capaz de ligar-se a outras moléculas da mesma espécie,
constituindo longas cadeias (LEHNINGER; NELSON; COX, 2001)
20
A ligação peptídica, que une os aminoácidos (Figura 2) ocorre por meio da desidratação,
da seguinte maneira: o grupo carboxila de um aminoácido perde uma hidroxila (OH), enquanto
o grupo amino do outro aminoácido perde um hidrogênio (H). Esses grupos OH e H liberados
formam uma molécula de água ((H2 0)), e o carbono do grupo α-carboxila se liga ao nitrogênio
do grupo α-aminoácido formando um dipeptídeo (LEHNINGER; NELSON; COX, 2001).
Figura 2: Ligação peptídica entre dois aminoácidos
Fonte: Adaptado de (ALBERTS, 2002)
Os 20 (vinte) aminoácidos podem ser classificados em cinco classes principais, diferenciadas pelas propriedades de sua cadeia lateral. As classes são (LEHNINGER; NELSON; COX,
2001):
• Não-polares e alifáticos: nessa classe os aminoácidos são hidrofóbicos e não-polares.
• Aromáticos: são relativamente apolares (hidrofóbicos).
• Não carregados, mas polares: esses aminoácidos são mais solúveis em água, ou hidrofílicos, se comparados com os aminoácidos não polares, pois contêm grupos funcionais que
formam pontes de hidrogênio com a água.
• Carregados positivamente (básicos): os radicais mais hidrofílicos são aqueles que são
positiva ou negativamente carregados.
• Carregados negativamente (ácidos): os dois aminoácidos, com radicais com uma carga
líquida negativa em pH 7, são o aspartato e o glutamato, sendo que cada um deles possui,
na molécula, um segundo grupo carboxila.
A Figura 3 mostra a classificação dos aminoácidos nas cinco classes.
21
Figura 3: Classes dos Aminoácidos encontrados em proteínas
Fonte: (LEHNINGER; NELSON; COX, 2001)
Analisando as classes principais, pode-se perceber que todas são baseadas, de certa forma,
no grau de hidrofobicidade dos aminoácidos. Sendo assim, pode-se simplificar ainda mais as
classes, dividindo os aminoácidos em apenas duas, quais sejam: hidrofóbicos e hidrofílicos.
Os aminoácidos hidrofóbicos, também chamados de não polares, possuem cadeias laterais
compostas por hidrocarbonetos que tendem a se agrupar mais ao centro da estrutura quando na
presença de água, minimizando, assim, o contato com ela.
Os aminoácidos hidrofílicos ou polares, por sua vez, apresentam cadeias laterais com elementos como oxigênio e nitrogênio, por exemplo, que tendem a ficar em contato com a água
normalmente, podendo fazer ligações de hidrogênio com outras moléculas polares. A hidrofobicidade é uma característica importante para a formação de uma proteína, pois a tendência é que
a estrutura nativa apresente os aminoácidos hidrofóbicos no interior da estrutura, se protegendo
das moléculas de água, e os polares no seu exterior, facilitando as ligações com as moléculas de
hidrogênio da água.
Com essa classificação de hidrofobicidade percebe-se que o processo de formação das pro-
22
teínas (o seu dobramento), influencia a distribuição das cadeias laterais no decorrer das suas
estruturas, fazendo com que os aminoácidos hidrofóbicos se concentrem no interior da estrutura e os polares no seu exterior (Figura 4).
Figura 4: Dobramento baseado em hidrofobicidade
Fonte: Adaptado de (ALBERTS, 2002)
A compreensão do dobramento, assim como da sua estrutura final (nativa), é muito importante para a identificação e o estudo da funcionalidade de uma proteína, pois a sua função
depende, quase que invariavelmente, das interações com outras moléculas. Essas interações são
importantes, tendo em vista que podem ocasionar mudanças na conformação da proteína.
2.2
ESTRUTURA DE PROTEÍNAS
Uma proteína contém diversas ligações peptídicas elementares entre os aminoácidos. Sendo
possível a rotação livre em torno de muitas dessas ligações, a proteína pode assumir um número
ilimitado de conformações. A conformação final, que é a estrutura tridimensional singular
assumida por uma proteína, determinará a função química ou estrutural dessa.
A estrutura de uma proteína é subdividida em quatro níveis de organização, crescentes em
complexidade: estrutura primária, secundária, terciária e quaternária.
A estrutura primária (Figura 5) apresenta a menor complexidade. Esse nível corresponde à
sequência de aminoácidos que constituem uma determinada proteína, sendo que as suas quantidades, seus tipos e a sequência em que estão diferenciam uma proteína de outra. As forças
23
existentes entre os aminoácidos, assim como as ligações entre eles, serão as grandes responsáveis por determinar a conformação nativa (arranjo tridimensional) da proteína. Os outros
níveis derivam desse primeiro (LEHNINGER; NELSON; COX, 2001).
Figura 5: Estrutura primária, representada pela sua cadeia de aminoácidos
A estrutura secundária (Figura 6) refere-se à conformação local de uma parte da sequência
de aminoácidos. Essa conformação local apresenta, muitas vezes, padrões regulares de enovelamento. Tais padrões ocorrem devido à possibilidade das rotações livres, em que as ligações
do carbono-α com os grupamentos amina e carboxila sofrem rotações resultantes das forças
de atração e repulsão entre as moléculas. Os tipos mais comuns de enovelamento podem ser
classificados em dois grupos: as α-hélices e as folhas-β (β -sheet, estruturas representadas a
esquerda e a direita da Figura 6, sendo que as folhas-β são vistas de cima e lateralmente). Além
desses também existem as estruturas em grampo.
Figura 6: Estrutura secundária de uma proteína
Fonte: Adaptado (ALBERTS, 2002)
A estrutura terciária, por sua vez, corresponde ao arranjo tridimensional de todos os aminoácidos em uma proteína (Figura 7). A diferença básica entre a estrutura secundária e a terciária
está na distância de interação das forças de ligação entre os aminoácidos dentro das sequências.
Em uma estrutura terciária, a interação das forças ocorre a longa distância, ocasionando a formação de novos enovelamentos. Por outro lado, em uma estrutura secundária, essa interação
24
ocorre entre aminoácidos próximos entre si. A interação da estrutura terciária faz com que as
cadeias hidrofóbicas longas perturbem as estruturas locais (secundárias), fazendo com que essas
se curvem para o interior da proteína, para ficarem o mais distante possível das extremidades
e, assim, se protegerem quando em meio aquoso, deixando as partes hidrofílicas no exterior da
estrutura.
Figura 7: Estrutura terciária de uma proteína
Fonte: Adaptado (LEHNINGER; NELSON; COX, 2001)
Já a estrutura quaternária (Figura 8) é formada por duas ou mais cadeias polipeptídicas
separadas (também chamadas de subunidades), idênticas ou não, que formam enovelamentos
denominados molécula de proteína.
25
Figura 8: Estrutura quaternária de uma proteína.
Fonte: Adaptado (LEHNINGER; NELSON; COX, 2001)
2.3
DOBRAMENTO DE PROTEÍNAS
As proteínas são compostas de diferentes segmentos de uma ou mais cadeias polipeptídi-
cas que tendem a se enovelarem umas sobre as outras (Figura 8). Esse enovelamento tem
uma forma compacta em relação aos polipeptídeos, gerando também a diversidade estrutural
necessária para que as proteínas executem e adquiram um conjunto de funções biológicas específico (LEHNINGER; NELSON; COX, 2001).
O processo de enovelamento se inicia a partir do momento em que as cadeias de polipeptídios, ao serem transcritas no ribossomo, encontram-se em um estado em que a sua energia livre
2
é alta e, dessa maneira, estão altamente suscetíveis às forças existentes entre as moléculas da
própria cadeia. É através dessas forças que os enovelamentos se formam, surgindo, assim, o que
se chama de processo de dobramento ou simplesmente dobramento de proteína. O dobramento
é o processo pelo qual a sequência linear de aminoácidos contidos em uma cadeia polipeptídica dá origem à conformação tridimensional de uma proteína. Esta conformação também é
conhecida como final, nativa ou natural (HARTL, 1996).
Hoje, o número de estruturas proteicas tridimensionais conhecido é de 66.961 (sessenta e
seis mil novecentos e sessenta e um), segundo o Protein Data Bank ( PDB3 ), em 03 de agosto de
2 Energia
livre é a diferença entre a energia interna de um sistema e o produto da sua temperatura absoluta e
entropia. Mede o nível de desordem de uma molécula, quanto menor a energia, menor a desordem.
3 O PDB é considerado uma das principais bases de dados de proteínas. Nele estão armazenadas as estruturas
terciárias de proteínas obtidas através de vários métodos experimentais.
26
2010. A informação estrutural das proteínas tem revolucionado a compreensão das suas funções
e até mesmo os caminhos evolutivos pelos quais as proteínas atingiram o presente estado. A
predição da estrutura das proteínas tem sido estudada para que seja possível concluir sobre
como o dobramento atinge sua conformação nativa.
A conformação nativa da proteína é o estado em que ela apresenta a sua organização estrutural máxima, e em que o grau de eficiência de utilização de energia é o maior possível, ou seja,
a energia livre será a menor, adquirindo, assim, sua função como transporte e armazenamento
de nutrientes, catálise, defesa do organismo, regulação, entre outros.
O processo de dobramento tende a ocorrer de forma natural e espontânea, obedecendo às
leis da termodinâmica, de tal maneira que a energia é conservada. Isso significa que a energia
das moléculas de proteína tende a permanecer em sua conformação final (estado de menor
energia livre) durante a maior parte do tempo.
2.4
O PROBLEMA DO DOBRAMENTO DE PROTEÍNAS
Uma vez determinado como o dobramento de uma proteína ocorre a fim de formar sua
conformação nativa, é possível alcançar inúmeros benefícios, tais como: predizer a função da
proteína no organismo, aplicar esse conhecimento no desenvolvimento de novos fármacos e
descobrir a causa de doenças.
Diversos pesquisadores têm buscado determinar como o processo de dobramento ocorre
(KARPLUS; SHAKHNOVICH, 1992), porém ainda não existe uma teoria capaz de explicar
adequadamente esse processo.
Atualmente, existem alguns métodos experimentais que são capazes de determinar a estrutura tridimensional das proteínas, como a Cristalografia por Raios X, a Espectroscopia por
Ressonância Magnética Nuclear ( RMN) e a Microscopia Crio-eletrônica.
A Cristalografia por Raios X, também denominada de método estático de identificação, é
uma técnica que permite identificar a estrutura de proteínas através da utilização de feixes de
raios X, por meio da proteína cristalizada. Essa contém vários canais e poros preenchidos com
solventes por onde os feixes se difundem em várias direções, devido à simetria do agrupamento
de átomos que, por difração, dá lugar a um padrão de intensidade que pode ser interpretado
segundo a distribuição dos átomos cristalizados. Isso se dá por meio de métodos físicos e
complexas análises dos dados experimentais coletados. A qualidade do modelo tridimensional
apresentado está diretamente ligado à resolução dos dados coletados e do grau de refinamento
(LEHNINGER; NELSON; COX, 2001; ALBERTS, 2002; BRANDEN; TOOZE, 1999).
27
A Microscopia Crio-eletrônica, que, assim como a Cristalografia por Raio X, é considerada um método estático, identifica a estrutura por meio do congelamento rápido de amostras,
as quais posteriormente serão analisadas em um nível microscópico para a identificação das
estruturas.
Já a Espectroscopia por RMN, considerada um método dinâmico, é um processo em que
ocorre a absorção ressonante de energia eletromagnética. Essa absorção ocorre por meio das energias rotacionadas nos átomos (spin nucleares), causadas pela transição gerada pelos elétrons
envolvidos nas ligações químicas entre os níveis de energia (níveis desdobrados em função do
campo magnético). A referida transição é gerada pelos elétrons envolvidos nas ligações químicas. Na RMN, os aminoácidos são diferenciados através da cadeia lateral de cada aminoácido.
Os métodos experimentais para a determinação da estrutura de proteínas, como citado anteriormente, já mapearam milhares de proteínas; porém, essa quantidade ainda é muito pequena
quando comparada ao número de proteínas existentes na natureza. Entretanto, esses métodos
apresentam alguns problemas, pois exigem sofisticados métodos físicos, além de um conjunto
de dados experimentais contendo informações relevantes. Ademais, por meio da Espectroscopia
por RMN não é possível identificar estruturas proteicas muito grandes, enquanto a Cristalografia, além de depender muito dos dados coletados, é um método muito dispendioso tanto em
tempo quanto em custo.
Devido às dificuldades em se compreender o processo de dobramento, assim como às dificuldades encontradas nos métodos experimentais, tem sido utilizada uma alternativa baseada
em abordagens teóricas e práticas para a predição da estrutura das proteínas, criando assim o
PPEP.
O PPEP e o PDP se confundem na literatura, pois o PPEP foca em determinar a estrutura final tridimensional (estrutura terciária) baseado em informações da estrutura primária, enquanto
o PDP foca em descobrir os caminhos pelos quais as proteínas são dobradas de maneira natural
durante a sua síntese (LOPES, 2008). Sendo assim, para determinar a conformação nativa, o
PPEP utiliza-se de uma função matemática baseada no modelo para determinar a energia livre
final de uma conformação, visando encontrar o menor valor de energia, valor esse que representa a conformação nativa de uma proteína.
O problema do dobramento pode ser visto como um problema computacional de otimização. Para isso, é necessário que sejam desenvolvidos modelos que abstraiam as informações
das proteínas necessárias para o dobramento. Essas informações devem levar em conta o custo
computacional e ao mesmo tempo as caraterísticas físico-químicas das proteínas e do dobramento.
28
A distinção entre os modelos para a determinação da estrutura de uma proteína pode ser
feita com base no método utilizado para a identificação desta. O método pode ser por padrões
ou ab initio. O primeiro é um método que se baseia em estruturas proteicas previamente conhecidas, visando encontrar a estrutura da proteína em estudo por meio de comparações. Já os
métodos ab initio não utilizam conhecimento prévio das estruturas, porém exploram a superfície de energia livre, identificando a conformação que apresenta a menor energia livre. Os
métodos computacionais serão descritos em detalhes na seção 2.5.
Uma vez que a determinação da estrutura de uma proteína está ligada à busca da conformação em que esta apresenta menor energia livre, os modelos existentes devem levar em conta
os princípios da termodinâmica, na busca da minimização da energia livre.
A minimização é definida por meio do modelo que descreve a função de ajuste, potencial ou
fitness. Essa função potencial leva em consideração os graus de liberdade presentes na molécula
proteica. O grau de liberdade é relativo aos ângulos formados entre os aminoácidos no espaço,
quais sejam, um ângulo de rotação e um de torção, que estão presentes em cada aminoácido,
sendo que o seus valores determinarão a estrutura tridimensional da proteína.
2.5
MÉTODOS COMPUTACIONAIS QUE ABORDAM O PPEP
2.5.1
Modelagem por Métodos Baseados em Padrões
A modelagem por métodos baseados em padrões tem por base a tentativa de identificar a
conformação nativa de uma proteína por meio da comparação desta com as estruturas de outras
proteínas previamente determinadas. Esses modelos são computacionalmente mais simples do
que os modelos ab inito descritos na seção 2.5.2, pois eles utilizam informações previamente
conhecidas. Por outro lado são muito dependentes dessas informações, o que prejudica os
resultados caso as sequências analisadas não apresentem estruturas tridimensionais previamente
mapeadas. Dentre as modelagens por métodos baseados em padrões, destacam-se a abordagem
por homologia e por threading.
A modelagem por homologia baseia-se no princípio de que uma proteína tenha semelhança
a uma ou mais proteínas conhecidas. Isto é, a homologia tenta predizer a estrutura de uma
proteína até então desconhecida por meio de outras proteínas homólogas. Sendo assim, surge
a necessidade de avaliar a similaridade da sequência estudada com a conhecida, para prever
aproximadamente a estrutura desta (GINALSKI; GRISHIN, 2001). Existem abordagens que
se baseiam no fato de que um grupo de sequências proteicas possuem apenas uma sequência
ancestral comum (DOOLITTLE, 1986). De outro lado, há outras abordagens que acreditam
29
que existe mais de um ancestral comum para uma determinada proteína, já que é difícil a comprovação da existência ou não de um ancestral em proteínas que apresentam menos de 30% de
resíduos idênticos na mesma posição.
O processo de análise de um modelo homólogo segue os seguintes passos:
• realizar o alinhamento das sequências de aminoácidos da proteína que se quer determinar
a estrutura e da proteína homóloga;
• analisado o alinhamento, é gerado um modelo da cadeia principal da proteína;
• uma vez abordagem a cadeia principal, as cadeias laterais devem ser colocadas no modelo.
A maior dificuldade encontrada nessa modelagem ocorre quando as cadeias laterais são
adicionadas ao modelo, pois os aminoácidos podem assumir várias conformações, e é essa
informação que tem grande importância na determinação do modelo tridimensional da proteína,
ou seja, sua conformação nativa.
A modelagem por threading, assim como a homóloga, utiliza o conhecimento de estruturas
de proteínas previamente mapeadas para determinar a estrutura desconhecida de uma proteína.
Geralmente esse modelo é utilizado quando não são encontradas proteínas homólogas. Essa
abordagem utiliza as proteínas cadastradas no PDB, em que uma proteína é escolhida aleatoriamente do banco e sua sequência é comparada com a da proteína analisada, verificando se esta
apresenta um alinhamento que se adeque à estrutura comparada.
O alinhamento das proteínas pode ser realizado por dois métodos: i) o alinhamento sequênciasequência, em que se busca encontrar o melhor alinhamento entre os aminoácidos da sequência
da proteína analisada e os da sequência da proteína escolhida do PDB, por meio de inserções e
remoções; e ii) o alinhamento sequência-estrutura, em que a sequência de busca é sobreposta à
estrutura tridimensional, geralmente na estrutura secundária, que é conhecida, mantendo as propriedades físico-químicas e verificando se elas se encaixam, determinando, assim, a estrutura
ou parte da estrutura final da proteína analisada.
Uma vez alinhadas as estruturas seguindo um dos métodos citados anteriormente, as interações de pareamento hidrofóbico são utilizadas para determinar se o alinhamento pode ou não
ser utilizado para definir a estrutura da proteína (BAXEVANIS; OUELLETTE, 2004).
30
2.5.2
Modelagem por Métodos ab initio
A modelagem por métodos ab initio não depende do conhecimento prévio de estruturas
de proteínas, sejam essas homólogas ou cadastradas no PDB, mas, sim, tenta determinar a
conformação nativa tridimensional por meio de uma busca no espaço de possíveis conformações
(VULLO, 2002). Esses métodos fazem a busca explorando o espaço de valores da energia livre
das conformações, pois sabe-se que a proteína apresenta a sua energia mínima no momento em
que ela atinge a sua conformação nativa (KHIMASIA; COVENEY, 1997).
Para determinar a energia mínima de uma conformação durante o dobramento, utiliza-se
uma função de minimização, também conhecida com função de fitness ou de adequabilidade.
A função de minimização é baseada nas leis da física de movimentação em campos potenciais
(dinâmica molecular), ou seja, nas interações entre os átomos presentes na sequência (VULLO,
2002). Sendo assim, a função deve conter parâmetros que reproduzam as propriedades energéticas, dinâmicas e estruturais das proteínas.
Esses métodos ab inito utilizam modelos que representam a estrutura de uma proteína de
forma simplificada (KOLINSKI; SKOLNICKB, 2004), ou seja, existem modelos em que o
resultado final do dobramento pode representar fielmente uma proteína dobrada, mas outros
podem apresentar resultados que não lembrem a proteína real.
A representação simplificada de uma proteína pode ser baseada em dois modelos: i) modelos discretos, também conhecidos como modelos baseados em treliças (lattice); e ii) modelos
livres ou contínuos não treliçados (off-lattice).
Os modelos baseados em treliças (lattice) para a modelagem do PPEP foram, inicialmente,
propostos por Unger e Moult (UNGER; MOULT, 1993), posteriormente seguido por diversos outros grupos de pesquisa (KOLINSKI; SKOLNICKB, 2004; BRANDEN; TOOZE, 1999;
KHIMASIA; COVENEY, 1997).
Nos modelos treliça, os aminoácidos são posicionados em um ponto de uma grade (lattice).
A grade utilizada é geralmente quadrada ou cúbica, sendo que um ponto dela pode ser ocupado somente por um único aminoácido. Como esses aminoácidos estão ligados a outros, os
seus aminoácidos adjacentes estarão distribuídos na grade, por meio de um comprimento fixo
(geralmente de valor unitário para cada eixo da grade).
Apesar de simplificados, os modelos discretos ainda preservam algumas características de
uma proteína real, como, por exemplo, as interações entre os resíduos, estejam esses conectados
ou não (KHIMASIA; COVENEY, 1997).
31
Nesses modelos, a determinação da posição do aminoácido atual na grade depende da
posição do seu antecessor, e assim por diante.
O dobramento dos aminoácidos nesse tipo de modelo ocorre por meio do tipo de deslocamento que um aminoácido sofreu em relação ao seu antecessor. O número de deslocamentos
de aminoácidos possíveis em uma grade é n-1, em que n é o número de aminoácidos. Os
movimentos possíveis para o deslocamento de um aminoácido dependerão do tipo de grade utilizada. Em uma grade quadrada, a proteína será simplificada em uma estrutura bidimensional,
e os possíveis deslocamentos serão: esquerda (E), direita (D), cima (C), baixo (B). Já em uma
grade cúbica, que representa o dobramento de forma tridimensional, os deslocamentos poderão
ser os mesmos da quadrática, mais frente (F) e trás (T) (KRASNOGOR et al., 1999). O modelo lattice mais estudado atualmente é denominado de Modelo Hidrofóbico-Polar (HP) (LAU;
DILL, 1989; LOPES; SCAPIN, 2005).
A Figura 9 mostra dobramentos HP em duas dimensões à esquerda e em três dimensões à
direita.
Figura 9: Dobramentos no Modelo HP
Fonte: Adaptado (SHMYGELSKA; HOOS, 2005)
Como citado, o dobramento ocorre por meio de deslocamentos, e a sua representação é
comumente denominada de coordenadas internas. Já o sistema de coordenadas Cartesianas,
que mapeia os aminoácidos na grade, é relativo à geometria espacial absoluta, ou seja, a posição
dos aminoácidos é determinada de forma absoluta, independentemente de cálculos em relação
à posição do resíduo anterior (KRASNOGOR et al., 1999).
O modelo de energia livre HP é baseado em duas características importantes das proteínas
(RICHARDS, 1977; LAU; DILL, 1989), quais sejam:
32
• Hidrofobicidade: é a principal força de interação dos aminoácidos para o desenvolvimento da conformação nativa de pequenas proteínas.
• Estruturas nativas compactas: muitas proteínas possuem estruturas nativas compactas e
têm núcleos compactos com grande concentração de resíduos hidrofóbicos, assim como
uma área mínima de superfície não-polar exposta ao solvente com uma área polar altamente exposta.
O modelo HP simplifica os 20 (vinte) aminoácidos da estrutura proteica em duas categorias:
monômeros Hidrofóbicos/Não polares (H) e resíduos Hidrofílicos/Polares (P). A classificação
dos aminoácidos nesse modelo pode ser encontrada no anexo B. Devido a essa distinção, a
estrutura de uma proteína pode ser simplificada e representada por meio de uma sequência de
letras H e P H,P.
A conformação que uma determinada sequência atinge baseada no modelo HP está diretamente associada ao nível de energia livre, que, nesse caso, é proporcional ao número de contatos
não locais entre os monômeros hidrofóbicos não vizinhos.
Os modelos HP, apesar de simplificarem as estruturas proteicas, são computacionalmente
complexos. Esse modelo de PPEP, tanto para 2D quanto para 3D, pode ser classificado como um
problema computacional NP-completo4 (CRESCENZI et al., 1998; BERGER; LEIGHTON,
1998).
No modelos não treliçados (off-lattice), os aminoácidos não ficam restritos à grade (lattice).
Sendo assim, seus parâmetros podem assumir quaisquer valores contínuos. Nesses modelos,
os aminoácidos estão dispostos no espaço, de modo que apenas um aminoácido ocupe uma
determinada posição.
O dobramento do aminoácido atual também ocorre levando em conta o posicionamento
do aminoácido anterior, mas a sua posição dependerá do par de ângulos diedrais (ϕ,ψ )5 , que
ocorre com a cadeia principal de proteína (BRANDEN; TOOZE, 1999).
Existem modelos off-lattice que levam em consideração os ângulos da cadeia lateral (MERKLE
et al., 1996; SCHULZE-KREMER, 1993), mas a grande maioria dos modelos estudados atualmente utilizam apenas os dois ângulos diedrais (modelo 3D). Já para o modelo 2D é necessário
apenas um ângulo diedral.
4A
classe de problemas que possui algoritmos não determinísticos, cujo passo de reconhecimento pode ser
realizado por um algoritmo polinomial do tamanho de entrada, é chamada de NP. Sendo assim, um problema X é
denominado de NP-completo se não existe um algoritmo polinomial capaz de solucionar o problema.
5 Ângulo diedral é o ângulo formado por dois planos concorrentes. Para obter esse ângulo, basta tomar o ângulo
formado por duas retas quaisquer perpendiculares aos planos concorrentes.
33
Dentro os modelos off-lattice, o Toy Model, conhecido também como Modelo AB, é o
mais estudado. Esse modelo será utilizado no presente trabalho, tanto em duas como em três
dimensões. A descrição do modelo está na próxima seção.
2.6 TOY MODEL - MODELO AB
O Modelo AB é baseado nos mesmos fundamentos do modelo HP. A diferença básica está
na distribuição dos monômeros no decorrer do dobramento, pois o modelo AB não limita a
posição dos monômeros em uma matriz, mas pode posicionar os monômeros em qualquer lugar
do espaço ou do plano. O dobramento ocorre por meio dos ângulos diedrais dos monômeros
interconectados por ligações.
Esse modelo também é conhecido como Toy Model, e foi apresentando primeiramente por
Stillinger e Head-Gordon (STILLINGER; HEAD-GORDON; HIRSHFELD, 1993). Utilizando
o princípio da hidrofobicidade, assim como o modelo HP, os aminoácidos são convertidos em
dois grupos: monômeros A (hidrofóbico) e monômeros B (hidrofílico/polar).
O cálculo da energia livre do modelo AB é diferente do modelo HP, tornando aquele um
pouco mais complexo, tanto em termos estruturais, para a definição da conformação das proteínas, como computacionalmente. O valor das forças ou energia entre os aminoácidos é representado por números reais. Esse valor positivo indica que existe atração entre os aminoácidos
ligados da cadeias. Por outro lado, um valor negativo indica que há repulsão entre eles. No
modelo HP, as ligações entre os monômeros H-H (hidrofóbico-hidrofóbico) apresentam energia
igual a 1, e as outras ligações H-P (hidrofóbico-polar) e P-P (polar-polar) possuem energia 0.
No modelo AB em 2D, as ligações entre A-A (hidrofóbico-hidrofóbico) continuam apresentando valor de energia igual a 1; já as ligações B-B (polar-polar) apresentam energia igual
a +1/2; e as ligações A-B (hidrofóbico-polar) apresentam energia igual a -1/2. Analisando
os valores de energia predefinidos, percebe-se, no presente modelo, que as ligações entre os
aminoácidos AA apresentam uma energia de atração grande, enquanto entre os BB apresentam uma energia de atração média e entre os AB apresentam características de repulsão média
(STILLINGER; HEAD-GORDON; HIRSHFELD, 1993). Já no modelo em 3D, tanto as ligações B-B quanto as ligações entre monômeros diferentes (A-B, B-A) apresentam valor de
energia igual a +1/2, enquanto as ligações A-A mantêm o valor de energia igual a 1 (HSU;
MEHRA; GRASSBERGER, 2003; BACHMANN; ARKM; JANKE, 2005).
A força das ligações entre os aminoácidos será considerada na avaliação da energia do
dobramento (fitness do dobramento). As ligações entre os monômeros estão agrupadas através
34
de ângulos diedrais formados entre elas, sendo que esses ângulos são relativos ao monômero
predecessor. A Figura 10 mostra a formação de um ângulo diedral ϕ, formado entre os planos
que referenciam os monômeros A e B, para uma sequência formada por 4 (quatro) monômeros.
Figura 10: Formação de ângulos diedrais
Os ângulos diedrais formados são conhecidos também como ângulos de torção e apresentam
valores restritos entre -180 e 180 graus ou −π e π radianos.
A Figura 11 apresenta a representação hipotética de uma proteína no modelo AB em 2D. A
proteína é composta por 9 (nove) aminoácidos, sendo que cada um está conectado ao próximo
da cadeia por meio do ângulo de torção, que é responsável pelo seu dobramento na cadeia.
No caso do dobramento 2D, o modelo é composto de n-2 ângulos necessários para gerar o
dobramento, pois a ligação entre os dois primeiros aminoácidos é fixa com ângulo 0 ◦ .
Figura 11: Representação Genérica de uma proteína composta de 9 (nove) aminoácidos baseada
no Modelo AB em 2D
A Figura 12 apresenta a representação hipotética de uma proteína no modelo AB em 3D. A
35
proteína é composta por 9 (nove) aminoácidos, sendo que cada um está conectado ao próximo
da cadeia por meio de dois ângulos, um de rotação e um de torção, formados entre os planos xy
e zy, traçados em cada um dos aminoácidos da cadeia,
Figura 12: Representação Genérica de uma proteína composta de 9 (nove) aminoácidos baseada
no Modelo AB em 3D
A energia livre do dobramento do modelo 2D (EI ) é composta, basicamente, por duas
partes: i) a energia intermolecular; e ii) a energia potencial. A energia potencial é a energia
formada entre os monômeros não conectados, ou seja, é aquela em que as forças dos aminoácidos não interconectados vão exercer sobre o aminoácido atual em um dobramento. Essa energia
é conhecida como potencial de Lennard-Jones. Por sua vez, a energia intermolecular é a energia que depende apenas dos ângulos entre os monômeros e representa os backbones potenciais
(HSU; MEHRA; GRASSBERGER, 2003; BACHMANN; ARKM; JANKE, 2005).
O modelo matemático que descreve a energia livre do dobramento AB (EI ) para uma proteína com N monômeros em duas dimensões é definido pela Equação 1:
EI =
N−2 N
CI (σi , σ j )
1
1 N−2
(1
−
cos
θ
)
+
4
( 12 −
)
k
∑
∑
∑
4 k=1
ri6j
i=1 j=i+2 ri j
(1)
O primeiro somatório representa o custo para dobrar os monômeros interconectados na
sequência por meio do ângulo θk . O segundo termo, denominado potencial de Lennard-Jones,
depende das distâncias entre os aminoácidos não adjacentes no decorrer da estrutura e é influenciado pelas forças das ligações entre aminoácidos da sequência analisada, sendo que σi =A para
monômeros hidrofóbicos, σ j =B para monômeros polares e ri j é a distância entre os aminoácidos
i e j, tal que:
36
CI (σi , σ j ) =
+1
seσi , σ j =A
+1/2
−1/2
(2)
seσi , σ j =B
seσi , 6= σ j
A Equação 3 descreve a energia livre (EII ) do modelo AB para três dimensões (IRBäCK et
al., 1997; BACHMANN; ARKM; JANKE, 2005):
N−2
EII = −k1
N−3
N−2
∑ (bk .bk+1) − k2
∑ (bk .bk+2) + 4
k=1
k=1
N
∑ ∑
(
i=1 j=i+2
CII (σi , σ j ) CII (σi , σ j )
−
)
ri12j
ri6j
(3)
Na Equação 3, bk corresponde ao vetor unitário que representa as ligações entre os monômeros
k e k+1. O produto vetorial bk . bk+1 pode ser escrito como cos ωk . Os valores de k1 e k2 foram
determinados através de uma série de testes realizados (IRBäCK et al., 1997; BACHMANN;
ARKM; JANKE, 2005), sendo que k1 foi definido como -1 e k2 como +1/2. O segundo termo
da equação calcula a energia baseada nas interações causadas pela torção dos monômeros. Já o
terceiro termo é o cálculo do potencial de Lennard-Jones, no qual
C (σ ,σ )
CII (σi ,σ j )
− II r6i j
ri12j
ij
representa
as interações de atração independentemente dos tipos de monômeros que estão interagindo.
(
CII (σi , σ j ) =
2.7
+1
seσi , σ j =A
+1/2
seσi , σ j =B ou σi 6= σ j
(4)
COMPUTAÇÃO EVOLUCIONÁRIA
A Computação Evolucionária ( CE) surgiu nos anos 60 e é uma área que abrange métodos
computacionais inspirados na Teoria da Evolução de Darwin. A CE é inspirada no mecanismo de seleção natural das espécies, em que os indivíduos mais aptos e melhor adaptados têm
mais chances de sobreviver e se reproduzir. Tais métodos são aplicados não só nos estudos da
vida artificial, mas também na solução de vários problemas de ciência de computação, engenharia, biologia, entre outros (MICHALEWICZ; FOGEL, 2004), especialmente os relacionados
à otimização complexa de problemas não-lineares, estocásticos e com componentes temporais,
nos quais as técnicas de otimização mais tradicionais não se mostraram eficientes.
Para resolver problemas utilizando esses paradigmas, as possíveis soluções são representadas sob a forma de indivíduos em uma população. Estes indivíduos evoluem através de procedimentos genéricos e adaptáveis de acordo com regras de seleção e operadores genéticos.
37
A CE engloba uma família de algoritmos inspirados na Teoria Evolutiva de Darwin que
foram desenvolvidos independentemente, e que já demonstraram ser capazes de resolver problemas complexos, apesar das limitações de hardware existentes na época em que foram propostos.
Esses algoritmos são: Algoritmos Genéticos (AG) (FRASE, 1957; HOLLAND, 1962; BREMERMANN, 1962), Programação Evolutiva (FOGEL, 1962) e Estratégias Evolutivas (RECHENBERG, 1965). Além desses, alguns outros algoritmos utilizam técnicas evolutivas relacionadas
como a Programação Genética ( PG) (KOZA, 1992).
Existem também algoritmos baseados em técnicas bio inspiradas, quais sejam: i) inteligência por enxame de partículas (traduzido do inglês Particle Swarm Optimization ( PSO) (KENNEDY;
EBERHART; SHI, 2001); ii) otimização por Colônia de Formigas (do inglês (Ant Colony Optimization) ( ACO)); iii) sistemas imunológicos artificiais; e iv) Evolução Diferencial (ED)
(STORN; PRICE, 1997). O último algoritmo citado será objeto de estudo do presente trabalho
e é considerado por muitos como um algoritmo evolucionário.
Os algoritmos evolutivos possuem algumas peculiaridades e não podem ser prontamente
utilizados, já que necessitam ser adaptados a cada problema ao qual será aplicado. Basicamente,
esses algoritmos utilizam o conceito de uma população composta por indivíduos, sendo que
cada um é representado de acordo com a característica do algoritmo utilizado. Os indivíduos são
as soluções candidatas para o problema, sendo que, para avaliar a adequação deles, é utilizada
a função de adequação, adaptabilidade ou fitness.
Apesar das abordagens acima citadas terem sido desenvolvidas de forma independente, seus
algoritmos possuem uma estrutura comum (exceto PSO, ACO e imunológicamente inspirados).
A estrutura básica de um algoritmo evolutivo é apresentada no Algoritmo 1 (MICHALEWICZ;
SCHOENAUER, 1996).
t ← 0;
inicialize P(t);
avalie P(t);
while condição de parada não encontrada do
t ← t + 1;
selecione P(t) a partir de P(t − 1);
altere P(t) utilizando operadores genéticos;
avalie P(t) utilizando a função de fitness;
end
Algoritmo 1: Algoritmo Evolutivo Padrão
Os algoritmos evolutivos apresentam uma população P na geração t contendo n indivíduos,
38
solução candidata para o problema. Cada um dos indivíduos é avaliado através da função de
adaptação (fitness), conforme já exposto. Sendo assim, uma nova população é formada na
próxima interação t+1 a partir da seleção dos indivíduos mais adaptados. Durante o processo,
alguns indivíduos são submetidos à alteração por meio dos operadores, com o intuito de formar
novos indivíduos. O algoritmo é executado até chegar à condição de parada (n gerações máximo, tempo de execução, valor do fitness dentre outras), o que indica que a solução ótima ou
aceitável para o problema foi encontrada.
2.7.1
Evolução Diferencial
A Evolução Diferencial (ED) (STORN; PRICE, 1997, 2005) é um dos mais recentes métodos heurísticos de computação evolucionária. Inicialmente, esse método foi proposto para resolver os problemas de otimização matemática (zeros de polinômios), e rapidamente provou ser
prático e eficiente na solução de outros problemas de otimização.
A ideia básica da ED é utilizar a diferença entre dois indivíduos, a qual é calculada de forma
simples e rápida por meio de um operador linear, denominado de diferenciação, o que torna a
ED um método evolucionário único (FEOKTISTOV, 2006).
O algoritmo da ED é de implementação fácil e seu conceito é realmente simples. Além
disso, a ED tem se mostrado altamente flexível podendo ser utilizada em diversos problemas
de otimização, trazendo resultados muito satisfatórios (PLAGIANAKOS; TASOULIS; VRAHATIS, 2008).
O conceito de ED surgiu a partir do algoritmo de Genetic Annealing (Anelamento Genético)
desenvolvido por K. Price (PRICE, 1994), um algoritmo combinatorial baseado em populações
de indivíduos que serão avaliados por meio de critérios de anelamento via limiares derivados da
média de desempenho da população.
Posteriormente, Storn contatou Price para utilizar o genetic annealing, visando resolver o
problema polinomial de Chebyshev. Para adequar o algoritmo ao problema, substituiu-se a utilização de vetores binários (bit-array) por ponto flutuante (floating-point) e a utilização de operações entre vetores (aritméticas) em vez de operações lógicas. Essas mudanças transformaram
o algoritmo, sendo que esse passou da aplicação a problemas combinatoriais para a aplicação
a problemas contínuos, surgindo, assim, o conceito de Evolução Diferencial (STORN; PRICE,
1997). O algoritmo foi finalizado com a remoção total do fator de anelamento (annealing), pois
detectou-se que a mutação diferencial, introduzida ao algoritmo, combinada com a recombinação discreta e a seleção pareada dispensam esse fator. Existem várias estratégias de mutação
39
(STORN; PRICE, 1997) que foram classificadas em quatro grupos: aleatório, direto, local e
híbrido (FEOKTISTOV; JANAQI, 2004).
O elemento principal que diferencia a ED dos outros métodos de computação evolucionária
é o conceito da mutação diferencial, ou simplesmente mutação ou diferenciação. A mutação é
o processo pelo qual dois indivíduos (vetores) que foram selecionados aleatoriamente por meio
de adição da diferença ponderada entre eles gera um terceiro indivíduo.
Esse processo ocorre da seguinte maneira: as variáveis do indivíduo selecionado aleatoriamente são misturadas às variáveis de um segundo indivíduo, também escolhido aleatoriamente,
para resultar o chamado vetor tentativa (trial). O vetor tentativa é então submetido à avaliação realizada pela função de fitness. Caso o valor da avaliação seja melhor (menor no caso
de um problema da minimização, ou maior caso o problema seja de maximização) do que o
gerado pelo vetor candidato, substituirá esse na população; caso contrário, o vetor candidato
será descartado e uma nova iteração será iniciada.
A ED apresenta parâmetros de controle importantes: i) tamanho da população (NP); ii)
dimensão do problema (nDim); iii) estratégia de evolução; iv) constante de diferenciação, ou
fator de peso (F) v) constante de crossover, ou cruzamento (CR); e vi) critério de parada.
A Figura 13 mostra o processo de mutação na evolução diferencial para a geração de um
indivíduo doador, em uma população de NP indivíduos. A mutação ocorre no plano bidimensional X1 e X2, no qual foram distribuídos todos os NP indivíduos. Dentre esses, denominados
aqui de vetores, são selecionados aleatoriamente três: X α , X β e X γ . Em uma determinada
geração q é realizada a diferença entre os vetores X β e X γ , que é multiplica por um fator F,
sendo denominada de diferença ponderada. O resultado da diferença ponderada é utilizado para
perturbar o vetor X α (candidato), gerando um vetor doador V (q+1) .
O valor de F é conhecido como fator de peso, e é um número real que, na evolução diferencial de Storn e Price (STORN; PRICE, 1997), é constante para todas as gerações e pertence ao
intervalo [0,1]. O seu valor controlará a amplitude da diferença ponderada entre os vetores X β
e X γ . Deve-se ressaltar que a diferença ponderada pode ser feita entre dois ou mais vetores, dependendo do tamanho da população. Estudos provaram que, para populações maiores, o uso de
quatro vetores para calcular a diferença ponderada se mostrou mais eficiente (STORN; PRICE,
2005).
O crossover na ED utiliza o mesmo princípio da computação evolucionária, ou seja, é
introduzido visando aumentar a diversidade da população. Nesse caso, ele será aplicado nos
indivíduos que sofrem a mutação e no caso da Figura 13 é gerado um vetor tentaiva denominado
40
Figura 13: Processo de mutação diferencial para geração de um vetor doador
Fonte: Adaptado (BITELLO; LOPES, 2007)
de V (q+1) . A crossover ocorre baseada na regra da Equação 5.
(
(q+1)
Ui
=
(q+1)
se ri menor ou igual a CR
(q)
se ri maior que CR, para i=1
Vi
Xd,i
(5)
Na regra CR, a constante de crossover compreendida no intervalo [0,1] representa a probabilidade do vetor tentativa herdar as variáveis do vetor doador V (q+1) ou do vetor candidato
X α . Para isso, um valor real ri entre [0,1] é aleatoriamente gerado. Se o valor de ri for maior
do que CR, o vetor tentativa herda mais características do vetor candidato; caso contrário, ele
herda mais características do vetor doador. Se CR=1, isto indica que todas as variáveis herdadas
pelo vetor U (q+1) vêm do vetor doador. Caso CR seja igual a 0, U (q+1) herda todas as variáveis
provenientes do vetor candidato.
O cruzamento na ED pode ser de dois tipos: i) binomial (bin) (STORN; PRICE, 1997), que
indica que a operação de cruzamento é controlada por uma série de experimentos binomiais,
em que o cruzamento é executado em cada variável sempre que ri for menor que CR ; e ii)
exponencial (exp) (STORN; PRICE, 1997), que indica que a operação de cruzamento é controlada por uma série de experimentos exponenciais, ou seja, o cruzamento é executado em uma
determinada porção de variáveis enquanto o valor de ri for menor do que CR. Caso seja maior
o cruzamento, não é mais executado, preservando os valores das variáveis.
41
Na literatura existem alguns valores utilizados frequentemente para F e CR (STORN; PRICE,
1997; FEOKTISTOV, 2006). Porém, o ajuste desses valores depende diretamente da natureza
do problema. Assim, são necessários testes para o refinamento e ajuste dos valores. O valor
de F mais utilizado é 0,7, já para CR o valor é de 0,85. Além disso, a literatura aponta que a
ED não é tão sensível ao CR, ou seja, o valor escolhido para F tende a influenciar muito mais a
evolução do que o CR.
Uma vez calculado o vetor tentativa, é realizada a seleção. O fitness do vetor tentativa é
calculado e comparado com o do vetor candidato. Como citado anteriormente, a substituição
do vetor tentativa pelo vetor candidato dependerá da natureza do problema (minimização ou
maximização). Se o vetor tentativa for considerado melhor que o vetor candidato, substitui esse
na próxima geração. Caso contrário, o vetor candidato permanecerá na próxima geração.
A geração de novos indivíduos a partir da população é conhecida, em ED, como a estratégia
de evolução. Essa, por sua vez, varia o número de indivíduos que são selecionados para o
cálculo da diferença ponderada, o tipo de seleção do vetor candidato que será perturbado e o
tipo de cruzamento.
A representação das estratégia em ED se dá por ED/α/β /γ, onde:
• α identifica o método de seleção do vetor candidato que será perturbado para a geração
de vetor tentativa. O vetor candidato pode ser selecionado por duas formas: a) aleatória
(rand), ou seja, qualquer vetor da população poder ser selecionado; b) melhor indivíduo
(best), ou seja, o indivíduo com o melhor custo será perturbado.
• β identifica o número de diferenças ponderadas que será necessário para a perturbação
do vetor cantidato. Geralmente, apresenta o valor 1 ou 2, identificando que dois ou quatro
vetores serão utilizados para calcular as diferenças.
• γ identifica o tipo do cruzamento que, conforme já exposto, pode ser bin (binário) ou exp
(exponencial).
Os tipos de estratégias, assim como a sua formulação, estão representados na Tabela 1. As
estratégias nem sempre funcionam bem para todos os problemas. Sendo assim, a determinação
de qual utilizar deve ser realizada através de testes de tentativa e erro.
Uma vez selecionada a estratégia e aplicada ao problema, surge a necessidade de verificar
se os novos indivíduos são ou não melhores do que o indivíduo candidato. Para essa seleção,
a ED utiliza a função do fitness. O algoritmo avalia o custo do vetor tentativa U(q+1) e o do
(q)
vetor candidato Xd . Caso o custo do vetor candidato seja menor que o custo do vetor tentativa,
42
Tabela 1: Estratégias de Evolução ED mais utilizadas na literatura
Notação
Fórmula Mutação
q
q
q
(q+1)
ED/rand/1/bin
V
= Xα + F (Xβ - Xγ )
q
q
q
ED/best/1/bin
V(q+1) = Xbest + F (Xβ - Xγ )
q
q
q
q
q
ED/rand/2/bin
V(q+1) = Xα + F (Xλ - Xβ + Xγ - Xσ )
q
q
q
q
q
ED/best/2/bin
V(q+1) = Xbest + F (Xα - Xβ + Xγ - Xσ )
q
q
q
q
q
ED/rand-to-best/2/bin V(q+1) = Xold + F (Xbest - Xold + Xγ - Xσ )
q
q
q
ED/rand/1/exp
V(q+1) = Xα + F (Xβ - Xγ )
q
q
q
ED/best/1/exp
V(q+1) = Xbest + F (Xβ - Xγ )
q
q
q
q
q
ED/rand/2/exp
V(q+1) = Xα + F (Xλ - Xβ + Xγ - Xσ )
q
q
q
q
q
ED/best/2/exp
V(q+1) = Xbest + F (Xα - Xβ + Xγ - Xσ )
q
q
q
q
q
ED/rand-to-best/2/exp V(q+1) = Xold + F (Xbest - Xold + Xγ - Xσ )
o vetor candidato continua na população na próxima geração, caso contrário, o vetor tentativa
substitui o vetor candidato na geração seguinte, nos termos já explanados. A formalização deste
procedimento é a seguinte:
le f t{
(q+1)
será substituído por X(q+1)
(q+1)
será substituído por Xd
q
, então Xα
q
, então Xα
Se f (U (q+1) ) < f (Xα )
Se f (U (q+1) ) < f (Xα )
(q+1)
(6)
O algoritmo de ED pode ser escrito como a seguir, onde: NP é o tamanho da população,
NDIM é tamanho de cada indivíduo, F é o fator de peso, CR é a constante de cruzamento e GEN
é o número de gerações.
43
Inicialize população Pop ← rand();
Calcule f itness para cada indivíduo gerado;
best ← null;
for i = 0 to GEN do
for j = 0 to NP do
escolha aleatoriamente os indivíduos Vetores r1 ,r2 ,r3 ;
vetorcandidato ← indívíduo aleatório;
crie vetor tentativa X ← S(r, F,CR, Pop);
verifique se os indivíduos do vetor criado estão dentro dos valores do problema;
calcule f itness do vetortentativa;
if vetortentativa for melhor que vetorcandidato then
candidato ← vetortentativa;
if f itnessvetortentativa for melhor que best then
best ← vetortentativa;
end
end
end
end
Algoritmo 2: Algoritmo de Evolução Diferencial (ED)
2.8
COMPUTAÇÃO PARALELA
A computação paralela e distribuída surgiu devido à necessidade de processar grandes
quantidades de dados em pouco tempo, e também para possibilitar a solução de problemas
complexos dividindo-os em pequenas tarefas. A computação evoluiu na última década para
um modelo integrador de componentes de custo reduzido e de elevada capacidade, através do
surgimento dos computadores com processadores multi-core.
O início da computação paralela se deu na década de 70, com o processador vetorial Illiac IV. Posteriormente, surgiu o primeiro Cray I, um processador vetorial com pipeline 6 . Já
os multiprocessadores de baixo custo se tornaram disponíveis após 1984. Em 1987, surgiu o
conceito da VLSI (Very Large-Scale Integration), integração em larga escala que possibilitou
o surgimento dos computadores pessoais e o desenvolvimento de dispositivos computacionais
compostos por vários processadores que trabalham em conjunto para realizar uma tarefa especí6 Forma
de execução de tarefas como uma "cascata", em que execuções diferentes ocorrem com sobreposição e
defasagem no tempo.
44
fica (QUINN, 1994).
A definição dada por Hwang e Zhiwei (HWANG; ZHIWEI, 1998) define o processamento
paralelo como uma forma eficiente de processamento de informações, em que se deve dar ênfase
à exploração dos eventos concorrentes de um processo computacional. O conceito básico por
trás do processamento paralelo é o de dividir uma tarefa em várias partes, sendo que essas
serão distribuídas entre os vários processos. Esses, por sua vez, cooperam entre si por meio
de comunicação e sincronismo, para realizar a tarefa melhorando o desempenho e reduzindo o
tempo de processamento na obtenção do resultado final.
O paralelismo se caracteriza pela execução concorrente de dois ou mais processos, a qual
ocorre quando esses processos estão em andamento (iniciados mas não terminados). Essa execução concorrente dos processos pode ser classificada em dois tipos: i) paralelismo real; e
ii) pseudo-paralelismo (QUINN, 1994). O que os diferencia é que no paralelismo real existe
a sobreposição de processos, tal que todos os processos em paralelo estão em andamento simultaneamente, utilizando os múltiplos recursos existentes. Já no pseudo-paralelismo existe a
ocorrência intercalada de partes de processos concorrentes, em que será possível, em um determinado tempo, apenas um processo ser executado no único recurso disponível.
A execução do paralelismo pode ocorrer de forma síncrona, ou seja, a tarefa é dividida de
forma idêntica entre os vários processos que são sincronizados através de uma única unidade
de controle. O paralelismo pode ser também assíncrono, em que os processos executam tarefas
diferentes interagindo entre si.
Como já mencionado, o principal objetivo da paralelização de algoritmos ou tarefas é a
redução do tempo de execução envolvido. Existe uma infinidade de métricas que podem ser
utilizadas para determinar se a paralelização é eficiente ou não. A métrica mais utilizada é conhecida como speedup, definida como o aumento da velocidade observada em um computador
paralelo (processo paralelo) com p elementos de processamento em relação a um computador
sequencial (HWANG; ZHIWEI, 1998). O speedup é definido na Equação 7:
Sp =
Tr
Tp
(7)
Onde:
• S p = speedup observado em um computador com p elementos de processamento;
• Ts = tempo de execução do programa em um único elemento de processamento;
• T p = tempo de execução do mesmo programa em um computador paralelo, com p ele-
45
mentos de processamento.
Nos casos em que se atinge uma distribuição perfeita dos processos a serem executados em
paralelo, obtém-se o valor máximo de speedup, Sp = p (nesse caso, Tp = Ts/p). Existem vários
fatores que podem afetar o tempo de processamento de um algoritmo paralelizado. São eles:
sobrecarga de comunicação e de sincronismo, balanceamento inadequado de carga e algoritmos
ineficientes para explorar o paralelismo do problema a ser solucionado. Existem casos em
que speedup obtido pode ser maior do que o previsto. Esse fator é conhecido como anomalia
de speedup ou speedup superlinear. Isto pode ser causado pelas características do algoritmo
paralelo ou pelas características da plataforma de hardware (utilização da memória cache dos
elementos de processamento) (FOSTER, 1993; QUINN, 1994).
Visando à paralelização dos processos e fazendo com que esses cooperem entre si por meio
de máquinas distribuídas, conectadas e interligadas por meio de uma rede de computadores, é
necessário que estes comuniquem-se entre si. Para viabilizar essa cooperação, surgiram alguns
modelos de comunicação baseados em um protocolo de troca de mensagens ou Message Passing
Models. Nos modelos Message Passing, os processos possuem acesso à memória local, a partir
da qual as informações são enviadas para a memória local do processo remoto. Nesse modelo,
o programador é responsável pela sincronização das tarefas. Dentre os modelos de troca de
mensagens mais utilizados estão o PVM (Parallel Virtual Machine) (GEIST et al., 1994) e o
MPI (Message Passing Interface) (PACHECO, 1996).
O modelo PVM surgiu em 1989, nos laboratórios da Emory University e Oak Ridge National Laboratory, com o objetivo de criar e executar aplicações paralelas em hardware já existentes. Referido modelo foi amplamente definido, sendo aceito facilmente por vários pesquisadores
e usuários, pois proporcionava que vários computadores heterogêneos interligados em rede se
comportassem como um único recurso computacional. Por meio desse modelo, vários problemas computacionais puderam ser resolvidos com maior rapidez e menor custo.
Já o MPI teve sua primeira versão publicada em 1994, sendo resultado de um Fórum aberto
formado por pesquisadores, empresas e usuários que, reunidos, definiram a sua sintaxe e o
conjunto de suas rotinas, padronizando o conceito de Message Passing. Esse modelo também
foi criado para ser executado em máquinas heterogêneas. A sua implementação fica a cargo do
desenvolvedor, pois nele está somente o funcionamento lógico das operações. Devido às suas
extensões, o MPI é mais fácil de ser utilizado, pois gera uma flexibilidade maior em relação ao
PVM, embora suas interfaces sejam bastante semelhantes.
Tanto no MPI quanto no PVM existem componentes em comum para a realização e ex-
46
ecução da paralelização. São eles: rotinas de gerência de processos, com as funções de inicializar, finalizar, determinar o número e identificar os processos; rotinas de comunicação de
grupos, com as funções de broadcast, sincronização de processos entre outras; e as rotinas de
comunicação ponto a ponto, em que a comunicação é feita entre dois processos.
Para o presente trabalho foi escolhido trabalhar com o MPI devido à segurança, pois provê
uma interface de comunicação confiável. O usuário não precisa se preocupar com possíveis
falhas na comunicação. Além disso, a utilização do MPI permite a escalabilidade entre processos, de tal maneira que uma aplicação pode criar subgrupos que permitem operações de
comunicação coletiva para melhorar a execução.
47
3
METODOLOGIA
Este trabalho apresenta uma proposta para a solução do Problema do Dobramento de Proteínas baseada no Toy Model (Modelo AB) por meio da utilização do algoritmo de Evolução
Diferencial paralelizado em um cluster Beowulf. Um cluster, também conhecido como um
aglomerado de computadores, é formado por um conjunto de computadores que utiliza-se de
um tipo especial de sistema operacional classificado como sistema distribuído. É construído
muitas vezes a partir de computadores convencionais, sendo que estes vários computadores são
ligados em rede e comunicam-se através do sistema de forma que trabalham como se fosse uma
única máquina de grande porte. Há diversos tipos de cluster. Um tipo famoso é o cluster da
classe Beowulf, constituído por diversos nós escravos gerenciados por um só computador
A primeira abordagem do dobramento foi a da utilização do algoritmo de ED em modelo
AB bidimensional. Já a segunda abordagem é um pouco mais complexa, já que aproxima a
estrutura das proteínas ainda mais da realidade, por utilizar a ED em um modelo AB tridimensional.
Para ambas as abordagens os testes foram realizados com algoritmo de ED sequencial e
com o mesmo algoritmo paralelizado (seção 3.5), visando, além da melhora de desempenho, a
melhoria dos resultados.
Os resultados obtidos foram comparados com vários trabalhos apresentados na literatura,
objetivando identificar se o algoritmo utilizado pode ser considerado como uma alternativa
promissora para a solução do Problema Predição da Estrutura de Proteínas (PPEP). Os resultados da literatura para os modelagem em 2D e 3D são apresentados na seção 3.1.
3.1 BENCHMARKS
O Modelo AB surgiu a partir dos modelos lattice, mais especificamente do modelo HP,
visando à melhoria dos dobramentos e, assim, dando mais liberdade aos aminoácidos. Desde
que referido modelo foi proposto por Stillinger e Head-Gordon (STILLINGER; HEAD-GORDON;
48
HIRSHFELD, 1993), ele tem sido objeto de estudo de vários pesquisadores, tanto em sua modelagem 2D quanto em 3D, sendo que os métodos mais utilizados para a solução do problema
foram: i) redes neurais (STILLINGER; HEAD-GORDON; HIRSHFELD, 1993); ii) Conventional Metropolis Monte Carlo (STILLINGER; HEAD-GORDON, 1995); iii) Pruned-Enriched
Rosenbluth method (HSU; MEHRA; GRASSBERGER, 2003); iv) Multicanonical Monte Carlo
(BACHMANN; ARKM; JANKE, 2005); v) Annealing Contour Monte Carlo (ACMC) (LIANG,
2004); vi) Simulated Tempering (IRBäCK et al., 1997); vii) métodos motivados biologicamente
(GORSE, 2001, 2002); viii) dinâmica molecular (TORCINI; LIVI; POLIT, 2001); ix) colônias
de formigas (ZHARG; LI, 2007); x) Conformational Space Annealing (KIM; LEE; LEE, 2005),
dentre outros.
O método da evolução diferencial para a solução do PPEP, objeto do presente trabalho,
tanto no modelo AB quanto em outros modelos, tem sido pouco utilizado. Apenas um trabalho
e um artigo foram encontrados: i) A Differential Evolution Approach for Protein Folding Using
a Lattice Model (BITELLO; LOPES, 2007); e ii) A differential evolution approach for protein
structure optimisation using a 2D off-lattice model (artigo publicado originado deste trabalho e
dos resultados aqui descritos) (KALEGARI; LOPES, 2010).
Todos os estudos realizados utilizam as mesmas sequências de aminoácidos para os testes.
Essas sequências foram geradas a partir da sequência de Fibonacci e foram as mesmas com
as quais Stillinger e Head-Gordon (STILLINGER; HEAD-GORDON; HIRSHFELD, 1993)
realizaram seus experimentos. Sendo assim, foram utilizadas essas mesmas sequências para
que seja possível a comparação dos resultados.
A sequência de Fibonacci para o modelo AB é gerada por meio da Equação 8
?
S0 = A, S1 = B, Si+1 = Si−1
Si
(8)
Na Equação 8, o * é chamado de operador de concatenação, ou seja, as primeiras sequências geradas pela fórmula são S2 =AB, S3 =BAB, S4 =ABBAB, etc. O tamanho das sequências
é dado por Ni+1 =Ni−1 +Ni . As sequências de Fibonacci mais utilizadas nos trabalhos estão
representadas na Tabela 2.
Tabela 2: Sequências de BenchMark
N
13
21
34
55
Sequência
ABBABBABABBAB
BABABBABABBABBABABBAB
ABBABBABABBABBABABBABABBABBABABBAB
BABABBABABBABBABABBABABBABBABABBABBABABBABABBABBABABBAB
49
Os resultados dos experimentos realizados em diversos trabalhos com as sequências de
Fibonacci para o Modelo AB em duas dimensões (2D) estão representados na Tabela 3 e para o
mesmo modelo em três dimensões (3D) estão representados na Tabela 4.
Na Tabela 3, E representa a energia mínima; Emin representa a energia obtida através do
método da conjugação de gradientes, com configuração inicial através do PERM (Pruned Enriched Rosenbluth Method) (HSU; MEHRA; GRASSBERGER, 2003); E∗min representa a energia mínima obtida por Stillinger e Head-Gordon (STILLINGER; HEAD-GORDON, 1995);
E perm representa a menor energia obtida por meio de uma rodada completa do PERM (HSU;
MEHRA; GRASSBERGER, 2003); EACMC representa a menor energia utilizando o Annealing
Contour Monte Carlo Method (LIANG, 2004); ECSA representa a energia por meio do Conformational Space Annealing(CSA) (KIM; LEE; LEE, 2005); e, por fim, EPSO representa a energia
utilizando o PSO (ZHARG; LI, 2007).
Tabela 3: Resultados obtidos nos experimentos realizados nos modelos AB 2D
N
13
21
34
55
Emin
-3,2939
-6,1976
-10,7001
-18,5154
E∗min
-3,2235
-5,2881
-8,9749
-14,4089
Modelo AB 2D
E perm
EACMC
-3,2167 -3,2941
-5,7501 -6,1979
-9,2195 -10,8060
-14,9050 -18,7407
ECSA
-3,2941
-6,1980
-10,8060
-18,9110
EPSO
-3,2941
-6,1977
-10,7036
-18,4236
Já a Tabela 4 apresenta os melhores resultados obtidos na literatura para o modelo AB em
três dimensões (3D) em que E representa a energia mínima; EMUCA representa a energia obtida
através do multicanônico; E∗ELP representa a energia mínima obtida por meio da minimização
ELP (BACHMANN; ARKM; JANKE, 2005); EACMC representa a menor energia obtida por
meio do Annealing Contour Monte Carlo Method; EACMC+ representa a menor energia utilizando o Annealing Contour Monte Carlo Method melhorado por meio ajustes de metropolis
(LIANG, 2004); e ECSA , por fim, representa a energia por meio do Conformational Space Annealing(CSA) (KIM; LEE; LEE, 2005).
Tabela 4: Resultados obtidos nos experimentos realizados nos modelos AB 3D
N
13
21
34
55
EMUCA
-26,496
-52,915
-97,272
-169,654
Modelo AB 3D
EELP
EACMC
EACMC+
-26,498 -26,363 -26,507
-52,917 -50,860 -51,718
-97,261 -92,746 -94,043
-172,696 -149,481 -154,505
ECSA
-26,4714
-52,7865
-97,7321
-173,9803
50
3.2
MODELAGEM DO PPEP UTILIZANDO ED
A aplicação do algoritmo de ED no problema do dobramento de proteínas baseado no mod-
elo AB, tanto em 2D quanto em 3D, passa pela codificação dos indivíduos, pela definição
e implementação da função de fitness e pela definição dos seguintes parâmetros da evolução
diferencial: i) estratégia de evolução; ii) fator de peso (F); iii) constante de cruzamento (CR);
iv) tamanho ou número de indivíduos da população inicial (NP); e v) número de gerações (MaxGen).
O processo de codificação dos indivíduos de uma população é muito importante, pois eles,
ao representarem a conformação de uma determinada sequência de aminoácidos, tornam-se
possíveis soluções para o PPEP.
Como citado na seção 2.6, o Modelo AB simplifica os aminoácidos presentes em uma
proteína em dois grupos: i) monômeros A (hidrofóbico); e ii) monômeros B (hidrofílico/polar).
Ambos são conectados entre si por meio de ângulos de torção, que apresentam valores restritos
entre -180 e 180 graus ou π e -π radianos, para formar a conformação nativa de uma proteína.
São esses ângulos que compõem cada individuo da população em ambos os modelos (2D e
3D). Para validar os ângulos que compõem um indivíduo gerado aleatoriamente ou por meio
do processo de recombinação do algoritmo de ED, utilizou-se um processo de verificação e
correção dos valores, com a finalidade de verificar se algum indivíduo possui ângulos fora do
intervalo. Desta maneira, garante-se que o indivíduo possa continuar na próxima geração. Com
a utilização da ED, os ângulos dos indivíduos podem ser codificados utilizando variáveis reais
(representação em ponto flutuante), o que pode ser apontado como uma vantagem de referido
modelo. Sendo assim, os ângulos em radianos poderão ser codificados sem a necessidade de
realizar uma normalização.
A codificação dos indivíduos é baseada em ângulos, e a quantidade deles presentes no
indivíduo depende do modelo, 2D ou 3D. Assim como a codificação, a definição dos parâmetros
de ED e a função de fitness também dependem do modelo, 2D ou 3D, que será apresentado em
detalhes nas próximas seções. A população inicial para os dois modelos é gerada de maneira
aleatória, por meio da utilização de um algoritmo que gera valores aleatórios para cada um dos
ângulos, dentro da restrição apresentada, que compõe os indivíduo da população inicial.
51
3.3
REPRESENTAÇÃO DOS INDIVÍDUOS
3.3.1
Modelo AB em duas dimensões (2D)
A sequência proteica representada pela Figura 11 na seção 2.6 indica como os indivíduos,
para o PPEP, devem ser compostos. Para representar o dobramento de uma sequência composta
de N aminoácidos no modelo AB em 2D, são necessários N-2 ângulos, pois, por definição do
modelo, o ângulo entre a ligação dos primeiros dois aminoácidos da sequência é 0 ◦ , e o último
aminoácido da sequência apresenta apenas ligação com o aminoácido anterior, não necessitando
da representação angular para o próximo aminoácido.
A Figura 14 traz um exemplo de representação de um indivíduo para a sequência Fibonacci
composta por 13 (treze) aminoácidos, para o modelo AB em 2D. A figura apresenta um vetor
de 11 (onze) posições de P0 até P10 , ou seja, N-2, em que N é o número de aminoácidos e os 11
(P0 a P10 ) representam os ângulos de rotação. Todos os ângulos são representados em radianos
e devem estar dentro da restrição entre -π e π.
Figura 14: Representação dos indivíduos para as sequências de Fibonacci no modelo AB 2D
3.3.2
Modelo AB em três dimensões (3D)
A Figura 12, na seção 2.6, mostra a representação de uma sequência de 9 (nove) aminoácidos no modelo AB em 3D, evidenciando que a posição de cada aminoácido no espaço é composta por um conjunto de dois ângulos, denominados rotação e torção, sendo ambos ângulos
diedrais nos planos xy e zy, respectivamente. Para dobrar a sequência no espaço são necessários
N-2 ângulos de rotação, os quais são formados entre as ligações dos aminoácidos. Essa modelagem é a mesma apresentada no problema em duas dimensões. Além dos ângulos de rotação,
são necessários N-3 ângulos de torção, os quais são responsáveis por rotacionar o aminoácido
no espaço. Frisa-se que a quantidade de ângulos de torção é menor do que a de rotação, pois
as ligações entre o primeiro e o segundo aminoácido, assim como a do segundo com o terceiro,
são fixas, ou seja, não apresentam variação no eixo Cartesiano Z.
Uma vez identificada a quantidade de ângulos necessária para representar o dobramento,
52
pode-se definir a composição dos indivíduos que serão utilizados na população inicial do problema PPEP. Para representar o dobramento de uma sequência composta de N aminoácidos no
modelo AB em 3D, é necessário de N-2 ângulos de rotação e N-3 ângulos de torção. Esses
ângulos são utilizados na função de fitness, que exige a conversão das coordenadas polares para
Cartesianas.
A Figura 15 apresenta um exemplo de representação de um indivíduo para a sequência
de Fibonacci composta por 13 (treze) aminoácidos, para o modelo AB em 3D, mostrando um
vetor de 21 (vinte e uma) posições de P0 até P21 , ou seja, N-5, em que: N é o número de
aminoácidos; as primeiras 11 (onze) posições (P0 a P10 ) representam os ângulos de rotação; e
as próximas 10 (dez) posições (P11 a P20 ) representam os ângulos de torção. Todos os ângulos
são representados em radianos e devem estar dentro da restrição entre -π e π.
Figura 15: Representação dos indivíduos para as sequências de Fibonacci no modelo AB 3D
3.4
DEFINIÇÃO E IMPLEMENTAÇÃO DA FUNÇÃO DE FITNESS
3.4.1
Modelo AB em duas dimensões (2D)
Com os indivíduos devidamente representados, pode-se implementar a função de fitness
definida pela Equação 1, já apresentada na seção 2.6, para o problema do PPEP em duas dimensões. Essa função é responsável pelo cálculo da energia mínima de um indivíduo. Esse
indivíduo, denominado de vetor tentativa, será a entrada da função, que, além disso, tem a sequência de proteínas como um valor de entrada que será carregado de um arquivo texto. Para
o cálculo da energia, os aminoácidos A e B da sequência de entrada serão substituídos por 1 e
-1, respectivamente, valor esse que será utilizado para calcular a energia entre as ligações A-A,
A-B, B-A e B-B no cálculo do potencial de Lennard-Jones.
A implementação da função de fitness para o modelo AB em 2D é dividida em duas partes:
53
i) conversão dos valores dos ângulos que compõem um determinado indivíduo em coordenadas
Cartesianas x e y; e ii) implementação da formulação matemática responsável pela avaliação da
conformação.
A conversão dos ângulos em coordenadas Cartesianas é necessária uma vez que o cálculo da
energia mínima apresentada na equação 1 utiliza a posição Cartesiana de cada um dos aminoácidos para o cálculo da distância de Lennard-Jonnes, que compõe o segundo termo da equação.
A posição de cada aminoácido no plano xy é representada por P(x,y), em que P é o ponto no
plano. No presente trabalho, o ponto (aminoácido) é representado pela letra A.
Por definição, os dois primeiros aminoácidos apresentam entre si uma ligação com ângulo igual a 0◦, sendo o aminoácido A1 colocado na origem do sistema Cartesiano A1 =(0,0) e
A2 =(0,1). Uma vez definidos e plotados os aminoácidos iniciais, os próximos serão convertidos
em pontos x e y a partir desses pontos iniciais.
Para calcular a posição do próximo aminoácido, no caso A3 , é necessária a utilização de
conceitos de transformações geométricas que, utilizadas na rotação e translação, são baseadas
na origem. Dessa maneira, para o posicionamento de A3 é necessário que o vetor formado pelos
pontos antecessores (A1 e A2 ) seja projetado em relação à origem. Essa projeção é realizada
através da translação de A2 para a origem, subtraindo as coordenadas x e y dos pontos A2 e
A1 , como apresentado na Equação 9, em que Pr(a,b) é o ponto que representa o vetor projetado
entre a origem (0,0) (KINDLE, 1968).
"
Pr(a, b) =
a
#
"
=
b
x1
#
"
−
y1
x2
#
(9)
y2
O conceito de rotação é utilizado para rotacionar um determinado ponto, neste caso A3 , de
um certo ângulo (primeiro ângulo do indivíduo a ser avaliado) ω em relação à origem. A matriz
de rotação R para um sistema de 2D é definida pela Equação 10.
R=
"
#
cos(θ ) −sen(θ )
sen(θ )
cos(θ )
(10)
A rotação aplicada ao ponto Pr(a,b) resulta na seguintes equações:
xr = a ∗ cos(θ ) − b ∗ sen(θ )
y = a ∗ sen(θ ) + b ∗ cos(θ )
r
(11)
54
Uma vez rotacionado o ponto, deve-se utilizar o conceito de translação para projetar esse
ponto em relação ao antecessor, uma vez que o ponto já é baseado em relação a origem do
sistema. Para isso, as coordenadas inteiras x e y serão adicionadas às coordenadas inteiras do
ponto antecessor, no caso A2 (x2 ,y2 ), gerando as coordenadas finais do ponto que representa o
aminoácido A3 . A translação funciona através da Equação 12, em que Pt(xt ,yt ) representam o
ponto final transladado.
"
Pt(xt , yt ) =
xt
yt
#
"
=
xr
yr
#
"
+
x2
#
(12)
y2
A Figura 16 mostra o processo de conversão do ângulo de um aminoácido A3 igual a − π2
em um ponto cartesiano. O primeiro gráfico apresenta a fixação dos aminoácidos A1 e a A2
representados pelos pontos em vermelho; o segundo, mostra o ângulo que será utilizada para
rotacionar o ponto A3 ; e. por fim, o terceiro apresenta a fixação do ponto A3 após a rotação
em relação à origem. Os 3 (três) gráficos inferiores apresentam a translação do ponto A3 em
relação ao ponto antecessor A2 .
Figura 16: Representação gráfica do processo de conversão de ângulos para coordenadas Cartesianas em 2D
O processo de transformação dos ângulos pode ser utilizado para qualquer aminoácido
predecessor de uma sequência composta por 3 (três) aminoácidos ou mais. Uma vez definido
o processo de conversão dos ângulos em pontos, a segunda parte da função de fitness pode
55
ser implementada conforme a Equação 12. O algoritmo da função de fitness é mostrado no
Algoritmo 3.
∗trial Vetor que representa o indivíduo de entrada da função;
a, b variáveis auxiliares v1, v2 componentes da Energia do Modelo AB 2D;
Energia Energia mínima c = força entre as ligações, d = distância de Leonard-Jones;
QuantidadeAnimoacidos recebe tamanho da sequência da proteína do arquivo de
entrada;
Posiciona a primeira ligação entre os aminoácidos 0 e 1;
Aminoacido[0] ← [0, 0] Aminoacido[1] ← [0, 1]
for i = 0 to QuantidadeAnimoacidos − 1 do
CorrigeValorTentativa() //corrige vetor tentativa se ângulo fora do intervalo -π até
π;
ConverteCatesianos(Angulos) //converte ângulos em coordenadas Cartesianas
end
v1 ← 0 v2 ← 0;
for i = 1 to QuantidadeAminoacidos − 1 do
v1 ← v1 +CalculoEnergiaPotencial(Aminoacido[i]);
end
for i = 1 to QuantidadeAminoacidos − 2 do
for j = i + 2 to QuantidadeAminoacidos do
c ← CalculodaEnergiadasLigacoes(Aminoacido[i], Aminoacido[ j]);
d ← CalculodaDistanciaLennardJones(c, Aminoacido[i]);
v2 ← v2 +CalculoPotencialdoLennardJones(d);
end
end
Energia ← v1 + v2;
Algoritmo 3: Algoritmo para o cálculo da energia mínima para o modelo 2D
3.4.2
Modelo AB em três dimensões (3D)
Os indivíduos da população são representados por vetores com 2N-5 posições, sendo N
igual ao número de aminoácidos da sequência, e cada posição representada por um ângulo.
As primeiras N-2 posições representam os ângulos de rotação, e de N-1 até 2N-5 representam
os ângulos de torção. A função de fitness definida pela Equação 3, apresentada na seção 2.6,
modela o problema do PPEP em três dimensões.
56
Como os indivíduos são representados por vetores de ângulos, é necessária a conversão
destes ângulos em coordenadas Cartesianas utilizadas para o cálculo da energia mínima para o
modelo. A função de fitness desse modelo também é dividida em duas partes: i) conversão dos
ângulos em coordenadas Cartesianas x,y,z; e ii) implementação do cálculo da energia mínima
para o indivíduo da Equação 3.
Para converter os ângulos de rotação e torção, responsáveis pelo posicionamento de um determinado aminoácido no espaço, é necessário utilizar novamente conceitos de transformações
geométricas. As transformações geométricas são espaciais (3D) e podem ser obtidas por meio
da generalização das transformações em 2D. Para realizar tais transformações, são utilizadas as
matrizes de translação e rotação no espaço.
A matriz de translação de um ponto no espaço é uma simples extensão das transformações
em um plano. No R3 é dada pela Equação 13. O componente homogêneo de um vetor da
coordenada (chamado normalmente w) nunca será alterado, geralmente pode se supor esse valor
é sempre 1 e ignorá-lo. E tx , ty , tz são constantes que são somadas às coordenadas para transladar
os pontos (KINDLE, 1968; LIMA, 2002).
1
0
y 0
0 =
z 0
w
0
x
0
0 0 tx
x
1 0 ty
y
0 1 tz
z
0 0 1
1
(13)
As matrizes de rotação no espaço, entretanto, são mais complexas do que as matrizes de
rotação no plano. Sabe-se que, no espaço, um determinado corpo pode ser rotacionado em torno
dos três eixos cartesianos (x,y,z), como ilustra a Figura 17.
Figura 17: Rotação em tornos dos eixos cartesianos no espaço 3D
As colunas da matriz são coordenadas dos vetores da base da transformada. Sendo assim,
pode-se facilmente derivar as matrizes de cada uma dessas rotações. Para exemplificar o fator
da rotação, esta será analisada em torno do eixo y. A Figura 18 demonstra a posição dos vetores
57
unitários da base canônica, depois desses terem sofrido uma rotação de θy .
Figura 18: Exemplo de rotação em torno do eixo y no espaço 3D
Baseando-se na Figura 18, as coordenadas Cartesianas dos vetores da base canônica, depois
da rotação, são dadas pelas seguintes componentes da Equação 14.
cos(θy )
î =
0
−sen(θ
)
y
0
jˆ =
1
0
sen(θy )
k̂
=
1
cos(θy )
(14)
A rotação pode ser escrita na forma de matriz de rotação de acordo com a Equação 15.
0 sen(θy ) 0
0
y
0
1
0
0
Ry = 0 =
z −sen(θy ) 0 cos(θy ) 0
1
0
0
0
1
x
0
cos(θy )
x
y
z
1
(15)
De maneira análoga, pode-se chegar às matrizes de rotação em torno dos eixos x e z, quais
sejam a Equação 16 e a Equação 17, respectivamente.
58
1
0
0
0
y 0 cos(θ ) −sen(θ )
x
x
Rx = 0 =
z 0 sen(θy ) cos(θx )
1
0
0
0
x
0
0
x
0
y
Rz = 0
z
1
cos(θz ) −sen(θz )
0
1
= sen(θz ) cos(θz )
0
0
0
0
0
0
0
1
0 0
0 0
0 0
1 0
0 1
x
y
z
1
x
(16)
y
z
1
(17)
Com as matrizes de rotação para os eixos x,y,z pode-se gerar a matriz de rotação genérica
MGR, conforme a Equação 18 para qualquer uns dos eixos. A rotação pode ser definida por um
eixo de rotação dado pelo vetor unitário u=(x,y,z)T e por um ângulo de rotação θ .
tx2 + c txy − sz txz + sy
2 + c tyz − sx
MGR =
txy
+
sz
ty
txz − sy tyz + sx
(18)
tz2 + c
Em que (x,y,z) são as coordenadas do vetor u e:
t = 1 − cos(θ )
s = sen(θ )
c = cos(θ )
(19)
Uma vez conhecidas as matrizes de translação e rotação necessárias para rotacionar os pontos em relação à origem, aplica-se esses conceitos ao problema com a finalidade de posicionar
os aminoácidos. Para isso, contudo, é necessário decompor o problema em sub-problemas mais
simples para os quais se conhecem as matrizes de transformação.
• O primeiro passo é converter os pontos que representam os aminoácidos A1 e A2 para a
origem do sistema. Para isso, é necessário realizar uma translação nesses pontos, representada na Equação 20.
• O próximo passo é aplicar uma rotação em torno do eixo z, de modo a levar os pontos A1
e A2 ao plano xy. A rotação é aplicada para um ângulo de rotação θ definido no indivíduo
59
que está sendo analisado, utilizando a matriz MGR (equação 18), o que resulta no ponto
00
A2 .
• O terceiro passo é aplicar a torção. Para isso é necessária uma nova rotação, sendo que a
mesma deve ser feita em torno do eixo y, de modo a levar ao plano xz. Essa rotação utiliza
00
o ângulo de torção θ do indivíduo e é aplicada ao ponto resultante da rotação anterior A2 ,
00
utilizando novamente a matriz MGR, o que resulta no ponto A3 .
• Como ambas as rotações foram realizadas em relação à origem, é necessário transladar o
00
ponto A3 , adicionando o valor do ponto A2 , uma vez que esse é o ponto do aminoácido
antecessor a A3 , tomado como referência.
1
0
T (−x1 , −y1 , −z1 ) =
0
0
0 0 −x1
1 0 −y1
0 1 −z1
0 0 1
A Figura 19 mostra o processo de conversão dos ângulos de rotação e torção iguais a
(20)
−π
2 ,
de um aminoácido A3 , em pontos Cartesianos x,y,z.
Como já descrito, a função de fitness para o modelo AB 3D também é dividida em duas
partes, quais sejam: i) conversão das coordenadas polares para coordenadas Cartesianas; e ii)
cálculo da energia mínima. A energia mínima entre as ligações dos aminoácidos adjacentes
é utilizada para o cálculo do potencial de Leonnard-Jones. Para que o valor de energia seja
utilizado corretamente, de acordo com o termo CII da equação 3, é necessário diferenciar o tipo
do aminoácido anterior, seja ele A ou B.
O Algoritmo 4, que será invocado na chamada ConverteAngulosemPontos() da função de
fitness, calcula a conversão dos ângulos.
60
Figura 19: Representação gráfica do processo de conversão de ângulos para coordenadas Cartesianas em 3D
dim ← (QuantidadeAminoacidos − 2);
Aminoacido[0] ← [0, 0, 0] Aminoacido[1] ← [0, 1, 0] for i = 1 to
QuantidadeAminoacidos − 1 do
CorrigeValorTentativa();
ConverteOrigem() //Converte em relação a origem ;
RotacaoGenerica(Aminoacido[i]) //Rotaciona eixo Z;
RotacaoGenerica(Aminoacido[i]) //Rotaciona eixo Y;
Tranlacao()
end
Algoritmo 4: Algoritmo de Conversão de ângulos para coordenas Cartesianas para o modelo
AB 3D
A conversão do fitness invoca uma função denominada RotacaoGenerica, a qual apresenta
a implementação de uma matriz genérica de rotação para qualquer um dois eixos, dependendo
dos seu valores de entrada. Ademais, essa função é utilizada para realizar a rotação dos vetores,
61
através do ângulo θ .
De outro lado, o Algoritmo 5 implementa o cálculo da energia mínima representado na
Equação 3.
∗trial vetor que representa o indivíduo de entrada da função;
Energia energia mínima;
QuantidadeAnimoacidos ← sequencia ;
//Posiciona a primeira ligação entre os aminoácidos 0 e 1;
Aminoacido[0] ← [0, 0, 0] Aminoacido[1] ← [0, 1, 0]
ConverteAngulosemPontos();
Ev1 = 0;
for i = 1 to QuantidadeAminoacidos − 1 do
Ev1 = Ev1 +CalculoEnegiaPotencialRotacao(Aminoacido[i], Aminoacido[i − 1]);
end
Ev2 = 0;
for i = 1 to QuantidadeAminoacidos − 2 do
Ev2 = Ev2 −CalculoEnegiaPotencialTorcao(Aminoacido[i], Aminoacido[i − 1]);
end
Ev3 = 0;
for i = 0 to QuantidadeAminoacidos − 2) do
for j = (i + 2) to QuantidadeAminoacidos do
if Aminoacido[i].Tipo ==0 A0 and
(Aminoacido[i].Tipo == Aminoacido[ j].Tipo) then
c = 1;
end
else
c = 0.5;
end
d = CalculoDistanciaLennardJones(Aminoacido[i], Aminoacido[ j]);
Ev3 = Ev3 +CalculoPotencialLennardJones(d);
end
end
Energia = Ev1 + Ev2 + Ev3;
Algoritmo 5: Algoritmo para cálculo da energia mínima do modelo AB 3D
62
3.5
METODOLOGIA DE PARALELIZAÇÃO
Como citado anteriormente, o PPEP é um problema computacional complexo, classificado
como NP-completo, ou seja, é um problema em que não é possível obter a solução em tempo
menor do que exponencial. Além disso, é muito provável que, para chegar à melhor solução,
seja necessário passar por várias outras equivalentes para o problema, e, muitas vezes, essa
solução não é a solução ótima, mas aceitável.
À medida em que um aminoácido é acrescentado à sequência de entrada, o espaço de busca
cresce exponencialmente aumentando assim a quantidade de cálculos e comparações necessária
para tentar se chegar à solução. Dessa maneira, frisa-se que não será diferente com o algoritmo objeto desse trabalho. Visando diminuir o tempo de processamento do algoritmo para
se solucionar o problema de maneira eficiente e rápida, optou-se por empregar a paralelização
utilizando um cluster Beowulf.
Para que o paralelismo apresente a eficiência desejada, ou seja, para que se possa implementar e executar um algoritmo ou um programa de forma paralela, em um sistema com múltiplos
processadores (cluster) (SARKAR; HENNESSY, 1986), é necessário que três problemas fundamentais sejam resolvidos:
• Identificação do paralelismo potencial;
• Particionamento do programa em tarefas sequenciais;
• Escalonamento das tarefas na execução concorrente.
De acordo com Grit (GRIT, 1990), além dos problemas acima, são também críticos para o
desempenho dos sistemas paralelos através da utilização de passagem de mensagens:
• A proporção entre a velocidade de comunicação e a capacidade de computação;
• O custo da sobrecarga na criação de tarefas.
Olhando para o problema do PPEP, quando aplicado o algoritmo de ED para a sua solução,
identifica-se o cálculo da função de fitness (definição da energia mínima de cada indivíduo
do população) como objeto de paralelismo, ou seja, o cálculo do fitness dos indivíduos será
distribuído entre vários processos.
Para que a distribuição do fitness ocorra, utiliza-se o modelo de implementação paralela
conhecido como Single Program Multiple Data ( SPMD) (ALMASI; GOTTLIEB, 1994; PACHECO,
63
1995), que é um caso especial da Multiple Instruction Multiple Data ( MIMD)1 , em que todos os
processos executam o mesmo programa. Esse modelo pode ser utilizado tanto em sistemas de
memória compartilhada como em sistemas de memória distribuída (passagem de mensagens),
sendo que no presente trabalho é utilizado o último.
Para que um programa seja executado no sistema de memória distribuída, é necessário
definir, no início da execução, qual a quantidade de processos que esse programa executa ao
mesmo tempo. A distinção entre as tarefas que são executadas por cada um dos processos se
dá pelo rank, que é um identificador que cada processo recebe na inicialização. As tarefas são
distribuídas nos processos seguindo o modelo SPMD mestre/escravo (master/slave), em que o
mestre é o processo responsável por distribuir os dados e controlar a execução das tarefas e
os outros, os escravos, são responsáveis por executar as tarefas retornando seus resultados ao
processo mestre. A Figura 20 representa o modelo SPMD mestre/escravo composto de 6 (seis)
processos, em que M é o processo mestre e S1 até S5 , os escravos.
Figura 20: Estrutura do modelo Mestre/Escravo
Na arquitetura mestre/escravo, implementada para o PPEP com a utilização do algoritmo
de ED, o processo mestre é responsável pela inicialização da população e distribuição dos indivíduos entre os processos escravos, que os utilizam para executar a tarefa a eles designada, qual
seja o cálculo do fitness. O processo mestre, por sua vez, espera, por meio de um semáforo de
sincronização, até que todos os escravos enviem os resultados para ele.
Os processos escravos inicializados ficam aguardando os indivíduos que serão executados.
Nesta implementação, um escravo pode receber um ou mais indivíduos para o cálculo da energia mínima. Concluído o cálculo dos indivíduos recebidos, o escravo envia os resultados ao
processo mestre.
Após receber todos os resultados, o mestre continua executando a análise dos indivíduos e
1É
um tipo de arquitetura de computação conjugada. Consiste em processos diferentes que executam programas iguais compartilhando memória comum e cálculos coincidentes. Cada processador tem acesso à memória
compartilhada através do barramento lógico.
64
seus resultados propostos pelo algoritmo de ED, de modo a verificar se existe algum indivíduo
melhor do que o melhor encontrado até agora.
A quantidade de indivíduos que fazem parte da população inicial afeta a paralelização do
algoritmo de ED, pois a distribuição dos indivíduos ou dos pacotes de indivíduos (denominação dada quando mais de um indivíduo é enviado para um escravo) que o mestre envia para
os escravos deverá ser a mesma. Isto cria uma relação de proporcionalidade entre o número
de indivíduos da população, a quantidade de escravos e a quantidade de indivíduos que cada
escravo deverá processar. Essa relação é representada pela Equação 21, em que o número de
indivíduos processados por cada escravo Indslaves será resultante da divisão de T pop (tamanho
da população) por Slaves (número de escravos). Deve-se lembrar que a divisão deve ser exata,
ou seja, todos os escravos devem receber a mesma quantidade de indivíduos.
Indslaves =
Tpop
Slaves
(21)
A definição da melhor proporção de indivíduos por escravo é determinada empiricamente
através de testes com diversas configurações, visando avaliar o tempo de processamento do
algoritmo paralelizado, pois de nada adianta paralelizar o algoritmo e esse apresentar um tempo
de execução maior do que o algoritmo sequencial.
Na arquitetura paralela, a troca de mensagens entre os processos é realizada através de
uma rede ethernet, ou seja, o desempenho do sistema paralelizado pode ser prejudicado pelas
redes físicas em que os computadores estão conectados, devido ao tempo de latência da rede2 ,
consequência da duração da troca de mensagens.
Como citado na seção 2.8, o método de paralelização utilizado para a implementação do
algoritmo no SPMD mestre/escravo foi a passagem de mensagens (MPI). Isto é feito através
da comunicação ponto-a-ponto (SNIR et al., 1996), em que um processo envia uma mensagem
e outro a recebe. A comunicação ponto-a-ponto no MPI pode ser de dois tipos: bloqueante e
não-bloqueante.
A função de envio bloqueante retorna somente quando o buffer da mensagem (fornecido
como parâmetro da função) pode ser reutilizado pelo processo. Quando uma mensagem é enviada de forma não bloqueante, a função de envio pode retornar antes que o buffer possa ser
alterado novamente. Para isso, deve-se garantir que qualquer alteração no buffer seja realizada
somente quando há certeza de que os dados da mensagem não serão mais alterados.
2 Latência
perceptíveis.
é a diferença de tempo entre o início de um evento e o momento em que seus efeitos tornam-se
65
Na recepção bloqueante de mensagens, o processo destinatário é bloqueado até que a mensagem desejada seja armazenada no buffer fornecido pelo processo. A função de recepção
não-bloqueante, no entanto, pode retornar antes que a mensagem tenha sido recebida. Neste
caso, uma função de verificação deve ser executada para indicar quando a mensagem realmente
está disponível ao receptor.
No presente trabalho, foi utilizada a comunicação bloqueante entre os processos. Todos
os processos escravos, quando inicializados, executam uma função de recepção bloqueante à
espera dos dados do processo mestre. Além disso, dependendo da quantidade de indivíduos
da população e da quantidade de escravos, um escravo poderá processar mais de um indivíduo,
como definido na Equação 21. Para que os indivíduos sejam enviados corretamente pelo mestre
aos escravos, é necessário que eles sejam empacotados antes de serem enviados, e, quando recebidos pelos processos escravos, é necessário que estes desempacotem os dados transformandoos em indivíduos, para a execução da tarefa.
O algoritmo de ED paralelizado (Algoritmo 6) apresenta alguns conceitos de MPI, como a
inicialização e a identificação de cada processo através de um identificador único denominado
rank, além de mostrar as chamadas de comunicação através das funções MPISend,MPIRecv,
bem como das funções de empacotamento e desempacotamento (MPIPack,MPIUnPack). No
algoritmo, NP representa o tamanho da população; NDIM, o tamanho dos indivíduos da população; F, o fator de peso; CR, a constante de cruzamento; GEN, o número de gerações; e, por
fim, NSLAVES, o número de processos escravos.
66
MPIInit() Inicialização do MPI rank ← MPICommRank() ;
if rank == 0 then
Inicialize população Pop ← rand(); f itnessperslave ← NP/NSLAV ES;
for i = 0 to GEN do
for v = 0 to NSLAV ES do
torank ←= rank + (v + 1);
for f = 0 to f itnessperslave do
crie vetor tentativa X ← S(r, F,CR, Pop);
MPIPack(energiaIndTentativa);
MPIPack(IndTentativa);
end
MPISend(MPIPackIn f o,torank);
end
while 1 do
MPIReceive(rankreceived);
if rankreceiced! = 0 then
countreceived + +;
end
if countreceived == NSLAV ES then
break;
end
end
for f = 0 to f itnessperslave do
MPIUnPack(energiaIndTentativa);
MPIUnPack(IndTentativa);
if energiaIndTentativa melhor que vetor candidato then
candidato ← IndTentativa;
if f itness trial melhor best then
best ← IndTentativa;
end
end
end
MPISend(MPIPackIn f o,torank);
end
if rank! = 0 then
waittoCalFitness(NSLAV ES);
end
end
Algoritmo 6: Algoritmo de Evolução Diferencial Paralelizado
67
O algoritmo de ED paralelo utiliza uma função denominada waittoCalFitness (Algoritmo
7), a qual é utilizada para sincronizar os processos escravos com o processo mestre. A função
waittoCalFitness é executada pelos processos escravos, ou seja, pelos processos que possuem
um rank diferente de 0 (zero), quando eles estiverem aguardando o recebimento de indivíduos
do processo mestre.
if rank! = 0 then
rank ← 0;
while 1 do
MPIRecv(MPIPackIn f o,torank);
for f = 0 to f itnessperslave do
MPIUnPack(energiaIndTentativa);
MPIUnPack(IndTentativa);
calcule f itness do vetor Tentativa (IndTentativa);
energiaIndTentaiva ← EnergyFunction; recebe o valor de energia
MPIPack(energiaIndTentativa);
MPIPack(IndTentativa);
MPISend(MPIPackIn f o, rank);
end
end
end
Algoritmo 7: Algoritmo de espera para sincronização dos processos escravos
3.6
AJUSTE DE PARÂMETROS DA ED
Para que o algoritmo de ED apresente soluções ótimas para o PPEP, quando comparado
aos resultados obtidos por outros algoritmos, é necessário ajustar seus parâmetros de controle.
O ajuste é muito importante, apesar de vários estudos apresentarem sugestões de valores para
os parâmetros (STORN; PRICE, 1997; MEZURA-MONTES; VELáZQUES-REYES.J; Coello
Coello, 2006; VESTERSTROEM; THOMASEN, 2004). De fato, nem sempre os valores utilizados em um determinado problema levarão ao mesmo desempenho quando aplicados a um
outro problema.
Os seguintes parâmetros da ED foram ajustados para o problema do PPEP, e são considerados os três principais da ED, conforme outros trabalhos já apresentados: i) estratégia de
evolução; ii) constante de diferenciação, ou fator de peso F; e iii) crossover, ou cruzamento CR.
Para isso, vários experimentos foram realizados com os parâmetros da ED.
68
Quanto à constante de crossvoer (CR), Storn, Price e Feoktistov (STORN; PRICE, 1997;
FEOKTISTOV, 2006) concluíram que o algoritmo de ED não apresenta grande sensibilidade
a alterações nela. Além disso, sugeriram o valor 0,85 para este parâmetro, para a maioria dos
problemas, o qual foi definido inicialmente como constante em todos os experimentos. De outro
lado, os valores de F e a estratégia de evolução variaram juntos.
Após a obtenção dos primeiros resultados que geraram o artigo (KALEGARI; LOPES,
2010), decidiu-se realizar uma análise de combinação entre os valores de F e CR, para verificar
se o CR=0,85 é realmente o melhor valor para esse parâmetro.
Os primeiros testes foram realizados para definir a estratégia de evolução que seria utilizada
em cada modelo. Para isso, foram criadas configurações utilizando as 3 (três) estratégias de
evolução mais utilizadas na literatura: rand/1/exp, randtobest/1/exp e best/1/exp. Para cada
estratégia foram utilizados 4 (quatro) valores diferentes da constante de diferenciação F, visando
identificar a melhor estratégia, independentemente do valor de F. O valor de CR utilizado nesse
primeiro momento foi CR=0,85, conforme mencionado. As configurações utilizadas para os
testes estão na Tabela 5.
Tabela 5: Configurações para ajuste da Estratégia
Estratégia
CR F1 F2 F3
F4
rand/1/exp
0,85 0,4 0,6 0,8 0,95
randtobest/1/exp 0,85 0,4 0,6 0,8 0,95
best/1/exp
0,85 0,4 0,6 0,8 0,95
Uma vez definida a estratégia de evolução (vide capítulo 4, seção 4.2, para verificar qual
será a estratégia utilizada para os próximos testes nos modelos em 2D e em 3D, respectivamente), será necessário ajustar a constante de diferenciação F e o valor de CR.
Em um primeiro momento, o valor de CR continuou constante em 0,85. Já o parâmetro
F, considerado o parâmetro de maior impacto durante o processo de evolução, sofreu variação
de valores apresentados na Tabela 6. Esses testes foram realizados na sequência de 34 (trinta e
quatro) aminoácidos, pois ela é considerada a sequência intermediária, devido ao seu tamanho
e complexidade de dobramento.
Tabela 6: Configurações para ajuste de F
F 0,4
0,5 0,6 0,7 0,8 0,9 0,95
Posteriormente, objetivando verificar qual a melhor combinação dos valores de F e CR para
os dois modelos, bem como aferir se CR=0,85 realmente é o melhor valor a ser utilizado pelo algoritmo de ED para solucionar o problema do dobramento de proteínas, foi criada uma variação
69
de combinações, em que os valores de F utilizados nos testes foram os mesmos apresentados na
Tabela 6 e os valores de CR os representados na Tabela 7.
Tabela 7: Configurações para ajuste de CR
CR 0,4 0,5 0,6 0,7
0,8 0,85 0,9
0,95
A combinação dos valores de F e CR apresentados nas duas tabelas (Tabela 6 e Tabela 7,
respectivamente) gera um conjunto de 56 (cinquenta e seis) testes. Para cada teste foram executadas 50 (cinquenta) rodadas com uma população de 100 (cem) indivíduos, durante 100.000
(cem mil) e 350.000 (trezentos e cinquenta mil) iterações, o que significa que foram realizados 112 (cento e doze) testes. Frisa-se que as rodadas foram realizadas para dois valores de
iterações a fim de identificar se o número delas influencia no resultado final.
3.7
DIZIMAÇÃO (EXPLOSÃO)
O algoritmo de ED pode levar a população a convergir rapidamente para um mínimo ou
máximo local fazendo com que a diversidade genética da população, fator fundamental para o
bom desempenho do algoritmo, seja perdida rapidamente. Isto faz com que todos os indivíduos
da população fiquem muito parecidos entre si. Esse fenômeno é conhecido como convergência
prematura e impede que ocorram melhorias futuras nos indivíduos da população.
Caso o processo evolutivo da ED por meio da mutação diferencial e do crossover consiga
gerar um indivíduo que explore uma outra parte do espaço de busca, isso seria considerado um
evento casual, já que a população não apresenta diversidade. Dessa maneira, pode-se dizer que
é inútil deixar a população evoluindo até que atinja o critério de parada, pois a evolução que
acontecerá será mínima. Então, foi utilizada uma estratégia, chamada de dizimação (SCAPIN,
2005).
A dizimação consiste em reinicializar a população inicial a partir de uma regra que constata
que a evolução não está ocorrendo ou que ela, mesmo ocorrendo, não apresenta resultados satisfatórios, ou seja, os indivíduos gerados apresentam valores de energia mínima muito próximos,
com a evolução apenas nas casas decimais, quando comparados com o valor de energia mínima
do melhor indivíduo da população.
O algoritmo de ED armazena o melhor indivíduo e o seu fitness, que, no caso do PPEP, é a
energia mínima. Essa energia representa um valor utilizado para comparar o valor de energia do
indivíduo candidato. Caso esse indivíduo apresente um valor melhor de energia mínima, será
armazenado.
70
Analisando os resultados da energia mínima obtidos durante a evolução, percebe-se que
existem momentos em que há uma estagnação dos valores, o que indica que a diversidade da
população é muito pequena.
Para definir a estagnação do valor da energia durante a evolução, foi definido um contador,
que é incrementado quando não ocorre evolução ou quando ela ocorre de maneira não significativa, ou seja, quando a evolução ocorre apenas nas casas decimais menos significativas da
energia resultante.
É necessário criar algumas regras para incrementar e zerar o contador da dizimação, que
será chamado de nonevolution. Esse contador será iniciado em 0 e é zerado toda vez que o processo de evolução apresentar um indivíduo melhor do que o melhor indivíduo até o momento,
o que pode ser constatado pela diferença maior ou igual a 0,5 entre o melhor atual e o melhor
obtido. Caso contrário, o contador nonevolution será incrementado em 1. Se a evolução continuar não ocorrendo e o contador continuar sendo incrementado, é necessário definir um critério
de parada, isto é, um valor máximo que o contador poderá atingir. Quando o contador atingir
esse critério de parada a população será dizimada, ou seja, todos os indivíduos da população
serão reinicializados, seguindo o mesmo princípio da população inicial. Uma vez reinicializada a população, o primeiro indivíduo da população recebe o melhor indivíduo encontrado
até então, e a evolução continua.
O valor máximo que o contador nonevolution pode atingir até que a população seja dizimada é definido como MAXCOUNT e é determinado por meio de testes em que os valores
testados estão apresentados na Tabela 8.
Tabela 8: Valores de teste para definição do MAXCOUNT para a dizimação
1000
3.8
2000 3000
4000 5000
10000 15000 20000 30000 40000 50000
MUTAÇÃO ESPELHADA
Tentando melhorar ainda mais o desempenho do algoritmo de ED aplicado ao problema do
dobramento de proteína, foi implementado um operador especial denominado de operador de
mutação espelhada. Esse operador é um operador específico, que poderá ser aplicado apenas ao
problema tratado neste trabalho, o PPEP, pois utiliza informações e conhecimentos prévios do
problema.
A ideia do operador de mutação espelhada é realizar uma busca local na sequência de
aminoácidos quando for detectado que a evolução não está ocorrendo de maneira satisfatória.
71
A utilização desse operador segue o mesmo princípio da dizimação, em que um contador é
definido e incrementado cada vez que o candidato analisado apresenta uma evolução inexpressiva, da quarta casa decimal para baixo. Caso a evolução seja superior, esse contador será zerado. Quando o valor do contador atingir o valor máximo (MAXMIRROR), o que será definido
por meio de testes, é realizada a busca local.
O operador de mutação, como citado, nada mais é do que uma busca local realizada em um
indivíduo de uma sequência, invertendo um de seus ângulos.
A Figura 21 mostra uma sequência composta por 8 (oito) monômeros (ABABABAB), em
que os monômeros A são representados pela circunferências pretas e os B, por brancas, sendo
que a circunferência preta maior representa o monômero inicial da sequência. O indivíduo
que representa essa sequência, seguindo o modelo AB 2D, apresenta N-2 ângulos e 6 ângulos,
sendo que esses últimos estão definidos no vetor (45; -45; -90; 45; -45; -90) em graus ou
(0,785398163; -0,785398163; -1,57079633; 0,785398163; -0,785398163; -1,57079633;) em
radianos, representação utilizada pelo algoritmo.
Figura 21: Representação gráfica de uma proteína com 8 aminoácidos no modelo 2D
A inversão dos ângulos na busca local ocorre a partir do terceiro aminoácido, já que os dois
primeiros são ligados entre si por uma ângulo fixo de 0◦ , e são utilizados como referência para
as próximas ligações. O ângulo que representa o dobramento do aminoácido será espelhado, o
que significa, na prática, que, se esse ângulo for de -90◦ , ele passará a ser de 90◦ .
Importante salientar que, sendo um ângulo espelhado, esse espelhamento afeta os ângulos
entre os outros aminoácidos conectados a ele. Essa mudança é feita para manter os aminoácidos
adjacentes nas mesmas posições anteriores à mutação espelhada.
A Figura 22 apresenta a mesma sequência de 8 (oito) aminoácidos da Figura 21 depois de
passar por um ciclo de mutação espelhada.
72
Figura 22: Representação gráfica de uma proteína com 8 aminoácidos no modelo 2D depois da
mutação espelhada
Cada vez que um ângulo é espelhado, uma nova avaliação do fitness desse dobramento
é realizada. Caso o valor da energia resultante seja melhor do que o resultado da sequência
sem a mutação, o indivíduo é substituído na população por essa nova conformação. Caso seja
pior, a mutação espelhada passa a ser realizada no próximo aminoácido da sequência, até que
seja detectada alguma melhoria no fitness. Se todos os ângulos possíveis forem espelhados e
não for encontrada nenhuma solução melhor do que a original, esse dobramento continuará na
população. De qualquer maneira, encontrando ou não uma solução melhor, o contador será
reiniciado, dando chance a outro indivíduo de sofrer esse processo na busca da indivíduos mais
próximos da conformação nativa da sequência.
Para definir qual será o valor de MAXMIRROR, ou seja, qual o valor máximo que o contador atinge para realizar a mutação espelhada, foram realizados alguns testes com valores pré
estipulados, apresentados na Tabela 9. A ideia desses testes, além de definir esse valor, também
é de verificar se a mutação espelhada trouxe benefícios nos resultados obtidos até então para o
modelo AB em 2D.
Tabela 9: Valores de MAXMIRROR para o operador de mutação espelhada do modelo em 2D
5
3.9
10 20 30
40 50
ADAPTAÇÃO DINÂMICA DOS PARÂMENTROS DE CONTROLE DA ED
A Evolução Diferencial, conforme já citado na seção 3.6, vem se mostrando um método
evolucionário eficiente quando seus parâmetros de controle são ajustados corretamente para
o problema proposto. Sabe-se que o ajuste de parâmetros não é um processo trivial, sendo
necessária a realização de vários testes arbitrando diversos valores para os parâmetros.
73
Buscando solucionar o problema do ajuste de parâmetros, foram propostos recentemente
mecanismos de adaptação e de auto-adaptação para controlar e atualizar os parâmetros de ED
de forma dinâmica, sem a necessidade do conhecimento prévio do problema em que o algoritmo
será utilizado (ABBASS, 2002; TEO, 2006; LIU; LAMPINEN, 2002; LIU; LAMPINE, 2005).
A ideia de criar métodos de adaptação e de auto-adaptação visa tornar o algoritmo de ED
mais robusto, por meio da atualização automática dos parâmetros de controle, independente da
função de fitness do problema, e sem a necessidade de ajuste de parâmetros utilizando o método
da tentativa e erro. Além disso, a taxa de convergência do algoritmo pode ser melhorada quando
os parâmetros de controle são atualizados para valores apropriados durante os vários estágios
da evolução.
Aproveitando que a literatura vem utilizando frequentemente o valor de CR=0,85 (STORN;
PRICE, 1997; FEOKTISTOV, 2006), esse valor será mantido, assim como a estratégia de
evolução DE/rand/1/exp, que foi definida através dos testes na seção 4.2 que trata dos resultados. Ademais, a adaptação ocorrerá apenas na constante de diferenciação F utilizada no método
de adaptação desenvolvido no algoritmo de ED conhecido como JADE (Jingqiao’s Adaptive
Differential Evolution) (ZHANG; SANDERSON, 2009). A cada geração, esse fator de diferenciação, definido com F j , é gerado automaticamente para cada indivíduo da população, por meio
de uma distribuição normal conforme a Equação 22, em que µF é atualizado com base na regra
apresentada na Equação 23. Caso o valor de F seja maior que 1 (um) ou menor que 0 (zero),
deverá ser gerado novamente até que F j fique dentro dos limites.
Fj = rand(µ, 0, 1)
(22)
Na Equação 23 o valor de µF deve ser inicializado em 0,5 e atualizado a cada geração
baseados na constante c (um valor positivo entre 0 e 1 geralmente definido como 0,5) e na
média SF.
µF = 1 − c ∗ µF + c ∗ media(SF)
(23)
A Equação 24 apresenta o cálculo da média Lehmer (ZHANG; SANDERSON, 2009), media(SF) dos F j utilizados até o momento.
∑ Fj2
media(SF) =
∑ Fj
(24)
74
O Algoritmo 8 mostra como funciona a adaptabilidade do valor de F durante o processo
de evolução, em que NP é o tamanho da população, NDIM é tamanho dos indivíduos, F é a
constante de diferenciação, CR constante de cruzamento e GEN é número de gerações.
Inicialize população Pop ← rand();
µF ← 0.5;
c ← 0.5;
for i = 0 to GEN do
SF ← 0;
for j = 0 to NP do
Gere valor aleatório para F j =rand(µ,1);
escolha aleatoriamente os indivíduos Vetores r1 ,r2 ,r3 ;
crie vetor tentativa X ← S(r, Fj ,CR, Pop);
verifique se o vetor criado pertence aos valores de restrição do problema;
calcule f itness do vetor tentativa (trial);
if trial melhor que vetor candidato then
candidato ← trial;
if f itness trial melhor best then
best ← trial;
end
SF ← Fj ;
end
end
µ ← (1 − c)µ + c ∗ media(SF);
end
Algoritmo 8: Algoritmo de ED-adaptável
75
4
RESULTADOS
Este capítulo é dividido em duas partes. A primeira apresenta os resultados obtidos durante
o processo de ajuste de parâmetros do algoritmo de ED, além dos resultados dos testes de auto
adaptação desses parâmetros, visando a melhoria dos resultados finais. A segunda parte apresenta os melhores resultados obtidos com o algoritmo de ED, já com os parâmetros ajustados,
bem como os resultados obtidos utilizando o algoritmo auto-adaptável para os dois modelos
apresentados nesse trabalho.
4.1
AJUSTE DA PARALELIZAÇÃO DO ALGORITMO DE ED
A paralelização do algoritmo ED para a solução do PPEP utilizando a arquitetura master-
slave (em português, mestre-escravo) visa reduzir o tempo de processamento do algoritmo sequencial de ED implementado. Para isso foram realizados testes variando o tamanho do população inicial (T pop ) e o número de processos escravos (Slaves), valores esses que, quando
aplicados à Equação 21, determinam o número de indivíduos que cada um dos processos escravos processa durante cada uma das gerações do algoritmo. Todos os testes executados foram
realizados utilizando as máquinas do cluster Beowulf e o modelo AB 2D.
O algoritmo de ED foi configurado utilizando valores conhecidos e definidos na literatura
para a estratégia de evolução, F e CR, sendo que os valores dos parâmetros são estratégias de
evolução rand/1/exp, F=0,7 e CR=0,85 (STORN; PRICE, 1997; FEOKTISTOV, 2006). Além
disso, o número de gerações foi definido como sendo de 100.000.
Para determinar os tempos de processamento, foram executadas 50 (cinquenta) rodadas para
cada uma das configurações mostradas na Tabela 10. Para as rodadas do algoritmo sequencial de
ED, utilizou-se a premissa apresentada por (STORN; PRICE, 1997), que define que o tamanho
da população ideal para o ED é de duas a três vezes maior do que a dimensão do problema. Para
esse caso, a dimensão do problema é baseada no número de ângulos que serão necessários para
dobrar a sequência de aminoácidos composta de 34 (trinta e quatro) monômeros para o modelo
AB 2D. Como o número de ângulos é N-2, onde N é o tamanho da sequência, a dimensão do
76
problema será 32 (trita e dois), ou seja, cada indivíduo será composto de 32 (trita e dois) ângulos
que representarão o dobramento.
Os testes estão apresentados na Tabela 10. Vale ressaltar que, nesses testes, não foram
levados em consideração os resultados da energia mínima do modelo, uma vez que os valores
dos parâmetros do algoritmo foram testados posteriormente para cada um dos modelos apresentados. Contudo, foi considerado o tamanho da população, tendo em vista a necessidade de
encontrar um tamanho de população adequada para o paralelismo. A relação do número de
indivíduos da população com o número de processos deve ser gerada na proporção exata, sem
que sobrem indivíduos avulsos, pois, caso isso aconteça, esses nunca serão processados, pelo
fato de que o processo mestre distribui os indivíduos de maneira igual a todos os escravos.
Tabela 10: Ajuste de número indivíduos por escravo para o algoritmo de ED paralelo
T pop
10
20
40
100
10
20
50
100
50
100
200
100
200
400
150
300
900
200
400
100
Slaves IndSlave
2
5
2
10
2
20
2
50
5
2
5
4
5
10
5
20
10
5
10
10
10
20
20
5
20
10
20
20
30
5
30
10
30
30
40
5
40
10
1
1
Tmedio (s)
122,467
70,733
122,9
240,456
135,5
75,733
151,167
270,4
180,567
297,5
480,733
342,3
503,767
821,667
649,233
1028,967
2099,233
822,533
1286,167
586,45
Tmedio/ind (µs)
122,467
70,733
122,900
240,456
135,500
75,733
151,167
270,400
180,567
297,500
480,733
342,300
503,767
821,667
649,233
1028,967
2099,233
822,533
1286,167
586,45
A Tabela 10 apresenta cinco colunas. A primeira traz o tamanho da população (T pop ) que
está sendo avaliada. Na segunda está o número de processos escravos (Slaves) em que serão
distribuídos os indivíduos. A terceira apresenta a relação T pop /Slaves, isto é, o número de
indivíduos que cada escravo processa (IndSlave ). A quarta coluna, Tmedio (s), representa o tempo
médio (em segundos) de processamento total do algoritmo durante todas as 50 rodadas. Por
fim, a quinta coluna apresenta o tempo médio (em µs) de processamento de um único indivíduo
para a configuração apresentada (Tmedio/ind (µs)). A última linha da tabela mostra os valores
77
correspondentes à execução sequencial do algoritmo.
Analisando-se a tabela, infere-se que a ED, na forma sequencial, necessita da 586,45 segundos para processar 100 (cem) indivíduos, por 100.000 (cem mil) gerações, sendo que o tempo
médio de processamento de um único indivíduo é de 586,45 µs. Ademais, é possível concluir
que o tempo de processamento menor (Tmedio/ind (s)) para um único indivíduo acontece quando
o tamanho da população é de 20 (vinte) indivíduos e o número de escravos é de 2 e 5 (70,733 e
75,733 µs, respectivamente).
Como essa relação entre tamanho da população e número de escravos foi utilizada para as
quatro sequências de benchmark, é necessário encontrar uma relação de tempo e um tamanho
de população na Tabela 10 que se adapte da melhor maneira ao problema. Considerando a
proporcionalidade tida como ideal para o tamanho da população (o número de indivíduos da
população deve ser de 2 a 3 vezes maior que o número de variáveis do problema), a relação
que mais se aproxima para satisfazer as quatro sequências é a de 100 (cem) indivíduos para 2
(dois) processadores escravos. Os resultados mostram que houve um speedup de 41%, com o
tempo médio de 240,456 µs, comparado com o tempo médio do algoritmo sequencial através
da Equação 7.
Sendo assim, para todos os testes realizados nas seções seguintes, o número de indivíduos
da população foi fixado em 100 (cem) para ambos os modelos, e o número de processos escravos
foi de 2 (dois) processos.
4.2
RESULTADOS PARA AJUSTE DE PARÂMETROS
Os resultados obtidos nesses experimentos definiram os parâmetros de controle que foram
utilizados na evolução diferencial para os testes nas sequências de Fibonacci, na busca da melhor conformação para as sequências proteicas apresentadas.
Para a realização dos testes, utilizou-se uma máquina com processador Intel Quad-Core
com 1 GB de RAM, com 4 núcleos, ou seja, em uma única máquina podem ser criados 4
processos MPI. Definiu-se por utilizar a configuração mestre-escravo, com três processos MPI,
sendo 1 (um) processo mestre e 2 (dois) processos escravos.
A população inicial, composta de 100 (cem) indivíduos, é representada por ângulos entre
-π e π e gerada com a utilização do algoritmo gerador de números aleatórios Mersenne Twister
(MATSUMOTO; NISHIMURA, 1998).
78
4.2.1
Ajuste da Estratégia de Evolução
O ajuste de parâmetros visa ajustar a estratégia de evolução que será utilizada posteriormente para solucionar o PPEP em duas dimensões. Para esses testes, foram selecionadas
as estratégias de evolução rand/1/exp, randtobest/exp, best/1/exp e o valor de CR foi fixado
em CR=0,85 seguindo as conclusões obtidas em trabalhos anteriores (STORN; PRICE, 1997;
FEOKTISTOV, 2006). A população de 100 indivíduos foi submetida a evolução durante 100.000
gerações a cada rodada. Para cada um dos conjuntos de testes apresentados na Tabela 5 foram
realizadas 50 rodadas, sendo que os valores médios de cada conjunto são apresentados em
forma de tabela e de gráfico de colunas, para facilitar a visualização do melhor resultado para
cada conjunto, em cada uma das sequências de Fibonacci nos dois modelos (2D e 3D).
Inicia-se a análise dos resultados obtidos para a identificação da melhor estratégia de evolução
utilizando a sequência de Fibonacci composta de 13 (treze) aminoácidos (ABABABABAB),
para o modelo 2D, que estão representados na Tabela 11 e na Figura 23.
Tabela 11: Resultados do ajuste de parâmetros para a Estratégia de Evolução com a sequência de
13 aminoácidos no modelo 2D
Estratégia
F=0,4
F=0,6
F=0,8
rand/1/exp
-2,3698 -3,0846 -2,818175
randtobest/1/exp -2,8966 -2,40744 -2,4482
best/1/exp
-1,7874 -2,03556 -2,029895
F=0,95
-3,199
-2,4488
-2,43509
Figura 23: Comparação entre os resultados obtidos para a Estratégia de Evolução com a sequência
de 13 aminoácidos no modelo 2D
A Figura 23, para o modelo 2D, mostra que a estratégia rand/1/exp, foi a melhor para os
valores de F diferentes de 0,4, resultando em médias de energias mínimas menores.
A Tabela 12 e a Figura 24 apresentam os resultados dos testes para definição das estratégias de evolução para a sequência de Fibonacci de 13 (treze) aminoácidos quando aplicadas ao
79
modelo 3D.
Tabela 12: Resultados do ajuste de parâmetros para a Estratégia de Evolução com a sequência de
13 aminoácidos no modelo 3D
Estratégia
F=0,4
F=0,6
F=0,8 F=0,95
rand/1/exp
-25,282 -24,876 -24,177 -25,634
randtobest/1/exp -24,21 -24,45 -24,315 -24,254
best/1/exp
-25,282 -24,876 -24,177 -24,254
Figura 24: Comparação entre os resultados obtidos para a Estratégia de Evolução com a sequência
de 13 aminoácidos no modelo 3D
Na Figura 24, modelo 3D, mostra também que a estratégia rand/1/exp foi a melhor para
valores de F (0,4, 0,6 e 0,95), resultado os melhores valores de energia mínima.
Para a sequência de Fibonacci de 13 (treze) aminoácidos em ambos os modelos, F=0,95
combinado com rand/1/exp, apresentaram os melhores resultados médios de energia mínima.
Frise-se que esse fator será levado em conta para concluir que a estratégia rand/1/exp é a melhor.
Concluída a análise para a sequência de Fibonacci de 13 (treze) aminoácidos, passou-se à
análise para identificar a melhor estratégia de evolução para a sequência composta de 21 (vinte
e um) aminoácidos (BABABBABABBABBABABBAB).
Os resultados obtidos para referida sequência no modelo 2D estão representados na Tabela
13 e na Figura 25.
80
Tabela 13: Resultados do ajuste de parâmetros para a Estratégia de Evolução com a sequência de
21 aminoácidos no modelo 2D
Estratégia
F=0,4
F=0,6
F=0,8
rand/1/exp
-4,16997 -4,71337 -5,85342
randtobest/1/exp -4,14425 -4,14425 -4,11337
best/1/exp
-4,00382 -4,99087 -4,24525
F=0,95
-6,1698
-6,10168
-5,5076
Figura 25: Comparação entre os resultados obtidos para a Estratégia de Evolução com a sequência
de 21 aminoácidos no modelo 2D
Quanto ao modelo 3D, os resultados do ajuste da estratégia de evolução para a sequência
de 21 (vinte e um) aminoácidos estão na Tabela 14 e na Figura 26.
Tabela 14: Resultados do ajuste de parâmetros para a Estratégia de Evolução com a sequência de
21 aminoácidos no modelo 3D
Estratégia
F=0,4
F=0,6
F=0,8
F=0,95
rand/1/exp
-47,287814 -41,081998 -39,069098 -40,230462
randtobest/1/exp -37,664098 -38,160066 -37,717346 -38,826142
best/1/exp
-41,337796 -39,625116 -38,587728 -39,658664
Os resultados das combinações de estratégia e F obtidos tanto para o modelo 2D quanto
para o modelo 3D, mostram que a melhor estratégia para ambos os modelos é a rand/1/exp.
Cabe ressaltar que a combinação de estratégia com F=0,6, no modelo em 2D, demonstrou que a
melhor estratégia seria a best/1/exp, porém, a média obtida com as combinações entre a estratégia rand/1/exp e F=0,8 e F=0,95, são respectivamente 17% e 23% melhores quando comparados
a best/1/exp.
Em seguida fez-se a análise da sequência de Fibonacci composta de 34 (trinta e quatro)
aminoácidos (ABBABBABABBABBABABBABABBABBABABBAB). Os resultados obtidos
81
Figura 26: Comparação entre os resultados obtidos para a Estratégia de Evolução com a sequência
de 21 aminoácidos no modelo 3D
para a identificação da melhor estratégia de evolução para essa sequência no modelo 2D está
representado na Tabela 15 e na Figura 27.
Tabela 15: Resultados do ajuste de parâmetros para a Estratégia de Evolução N=34
Estratégia
F=0,4
F=0,6
F=0,8
rand/1/exp
-8,01358 -8,48379 -8,26192
randtobest/1/exp -5,5069 -6,51339 -8,52439
best/1/exp
-7,0763 -7,44486 -6,99854
F=0,95
-9,38861
-8,507254
-8,59036
Figura 27: Comparação entre os resultados obtidos para a Estratégia de Evolução com a sequência
de 34 aminoácidos no modelo 2D
Para o modelo 3D, a Tabela 16 e a Figura 28 trazem os resultados obtidos para a sequência
de 34 (trinta e quatro) aminoácidos quando testada com diferentes estratégias e valores de F.
Após os testes, os resultados obtidos das combinações de estratégia e F, tanto para o modelo
2D quanto para o 3D, mostram que, para ambos os modelos, a estratégia rand/1/exp apresentou
as melhores médias quando combinada com valores de F=0,4, F=0,6 e F=0,95.
82
Tabela 16: Resultados do ajuste de parâmetros para a Estratégia de Evolução com a sequência de
34 aminoácidos no modelo 3D
Estratégia
F=0,4
F=0,6
F=0,8
F=0,95
rand/1/exp
-56,44271 -55,986174 -53,500054 -54,73163
randtobest/1/exp -51,51487 -52,037726 -53,023246 -54,1803
best/1/exp
-54,4726 -53,405436 -53,629136 -53,763082
Figura 28: Comparação entre os resultados obtidos para a Estratégia de Evolução com a sequência
de 34 aminoácidos no modelo 3D
Para a combinação com F=0,8, a estratégia que se mostrou mais promissora foi a best/1/exp.
Ainda, quando comparados os valores das médias obtidas para a combinação de F=0,8 e best/1/exp
com as outras combinações, percebe-se que essa não apresenta os melhores resultados, levando
à conclusão de que a melhor estratégia para a sequência de 34 (trinta e quatro) aminoácidos
para ambos modelos é a estratégia rand/1/exp.
Por fim, analisou-se a sequência de Fibonacci composta de 55 (cinquenta e cinco) aminoácidos (BABABBABABBABBABABBABABBABBABABBABBABABBABABBABBABABBAB).
Os resultados obtidos para a identificação da melhor estratégia de evolução para essa sequência
no modelo 2D está representado na Tabela 17 e na Figura 29.
Tabela 17: Resultados do ajuste de parâmetros para a Estratégia de Evolução com a sequência de
55 aminoácidos no modelo 2D
Estratégia
F=0,4
F=0,6
F=0,8
F=0,95
rand/1/exp
-13,5088 -13,3473 -13,2191 -12,0495
randtobest/1/exp -13,9396 -13,4649 -13,3248 -13,2387
best/1/exp
-11,35 -12,6688 -13,6584 -13,9483
No modelo 3D, o ajuste da estratégia com estratégia de evolução combinados com os val-
83
Figura 29: Comparação entre os resultados obtidos para a Estratégia de Evolução com a sequência
de 55 aminoácidos no modelo 2D
ores de F para a sequência de 55 (cinquenta e cinco) aminoácidos está apresentado na Tabela
18 e na Figura 30.
Tabela 18: Resultados do ajuste de parâmetros para a Estratégia de Evolução com a sequência de
55 aminoácidos no modelo 3D
Estratégia
F=0,4
F=0,6
F=0,8
F=0,95
rand/1/exp
-83,82171333 -74,48780435 -73,55394848 -74,83023478
randtobest/1/exp -70,11471515 -70,65593939 -70,41704839 -72,95410968
best/1/exp
-72,95410968 -70,49590645 -71,80576957 -73,942588
Figura 30: Comparação entre os resultados obtidos para a Estratégia de Evolução com a sequência
de 55 aminoácidos no modelo 3D
Avaliando-se os resultados para identificar qual é a melhor estratégia a ser utilizada, percebese que, para a sequência de 55 aminoácidos, tanto a estratégia rand/1/exp quanto a randtobest/1/exp apresentaram resultados muito próximos para o modelo 2D, quando combinadas
84
com os valores de F=0,6 e F=0,8. Contudo, para um F=0,95, essas estratégias apresentaram
o pior resultado, apesar disso não significar que a estratégia seja ruim para ser utilizada na
resolução do problema, tendo em vista que, quando combinada com outros valores de F, os
resultados foram melhores.
Por outro lado, para o modelo em 3D, as combinações entre estratégias e F=0,6, F=0,8 e
F=0,95 apresentaram resultados muito semelhantes. Entretanto, na combinação com F=0,4, a
estratégia que divergiu e apresentou os melhores resultados foi a rand/1/exp, apresentando um
resultado em média 15% superior às combinações do mesmo F=0,4 com outras estratégias, e
também quando essa é comparada com outras combinações entre estratégia e F. Desse modo,
pode-se afirmar que a estratégia rand/1/exp é a melhor estratégia para esse modelo.
Uma vez combinados todos os resultados dos testes com as quatro sequências de Fibonacci,
pode-se afirmar que a estratégia que apresentou os melhores resultados para o PPEP foi a
rand/1/exp. Essa estratégia se apresentou como a melhor para as sequências de 13, 21 e 34
aminoácidos em ambos modelos, além de ter se mostrado viável para a sequência de 55 (cinquenta e cinco) aminoácidos no modelo 2D e a ideal para a mesma sequência no modelo 3D.
Dessa maneira, todos os experimentos seguintes utilizaram esta estratégia.
4.2.2
Ajuste dos parâmetros da ED (constante de diferenciação (F) e crossover (CR))
Depois da definição da estratégia de evolução rand/1/exp por meio dos testes apresentados
na seção 4.2.1 para os ambos modelos, o passo seguinte é o ajuste dos parâmetros: constante de
diferenciação (F) e crossover (CR). É com essa combinação de va lores que todas as sequências
foram testadas posteriormente, visando encontrar a melhor conformação nativa (conformação
que apresenta a menor energia mínima).
ETAPA 1: CR=0.85, F variável, para os modelos AB 2D e 3D
Em um primeiro momento, decidiu-se utilizar o valor de CR fixo igual a 0,85, definido por
uma série de experimentos apresentados na literatura (STORN; PRICE, 1997; FEOKTISTOV,
2006) que combinados os valores de F, leva às melhores soluções (menor energia livre) para os
dois modelos.
Para cada um dos valores de F que estão apresentados na Tabela 6, foram executadas 50
rodadas do algoritmo de ED com uma população inicial de 100 (cem) indivíduos, evoluindo
durante 350.000 (trezentos e cinquenta mil) gerações. Os resultados das energias mínimas
obtidas durante essas 50 gerações foram analisados por meio de métodos estatísticos.
O método estatístico utilizado para comparar os diferentes valores de F para um CR fixo em
85
0,85 foi a análise da variância com um único fator ( ANOVA one way). O principio da ANOVA
é o de estudar a variabilidade dos dados de forma a identificar que parcela desta variabilidade
é devida aos efeitos dos diferentes tratamentos e que parcela dela é devida aos erros aleatórios
não controláveis (ao acaso) (TRIOLA, 1998).
Experimentos com um único fator são aqueles onde existe uma única variável de interesse
no estudo, e a análise permite comparar três ou mais tratamentos (no caso valores de F) simultaneamente que estão sendo investigados. A hipótese inicial H0, é de que todos os tratamentos
são iguais, isto é, no cada de não rejeição dessa hipótese conclui-se pela igualdade entre os tratamentos envolvidos (valores de F). No caso de rejeição de H0, conclui-se que pelo menos dois
tratamentos são diferentes, esta é a hipótese H1, e nesse caso, novos testes são realizados para
identificar quais tratamentos diferem. Nesse trabalho foi utilizado o teste de Tukey (TRIOLA,
1998) para localizar as diferenças.
Formulação das hipóteses:
• H0: Não há diferença significativa de energia livre mínima entre os níveis de F com CR
fixo 0,85 para o modelo AB em 2D.
• H1: Há diferença significativa de energia livre mínima entre os níveis de F com CR fixo
0,85 para o modelo AB em 2D.
Tabela 19: Análise de variância para valores de F do modelo AB 2D com CR=0,85
Fonte
Grau
de variação
de liberdade
Tratamentos (valores de F)
6
Erro (ao acaso)
686
Soma
de Quadrados
126,4437
128,7895
Quadrado
F
Médio
de Snedecor
21,07394
112,2508
0,187740
Valor de p
0,00012
Os resultados da ANOVA na Tabela 19 indicam o valor de p < 0,05 (5% de significância),
portanto rejeita-e a hipótese H0, ou seja, há diferenças significativas de energia mínima gerada
quando combinados os valores de F com CR fixo em 0,85. Para ilustrar foi gerado o gráfico
box-plot (Figura 31) que mostra o comportamento de cada conjunto de valores de F, sua média
e o intervalo de 68% e 95% das observações.
Analisando o box-plot observa-se que não há diferença entre as médias de F=0,7 e F=0,8,
além de serem as melhores médias. A localização das diferenças foi verificada por testes de
comparações entre F‘s utilizando teste de Tukey com nível de significância de 0,05. A Tabela
20 apresenta os resultados das comparações.
A Tabela 20 mostra que F=0,7 e F=0,8 são grupos estatisticamente iguais, pois as médias
seguidas de mesma letra indicam que não diferem entre si pelo teste de Tukey para nível de
86
Figura 31: Comparação entre as distribuições de F com CR=0,85 no modelo AB 2D
Tabela 20: Resultados do teste de Tukey para comparações entre médias de F com CR=0,85 no
modelo AB 2D
F
Energia Média
Desvio padrão
0,4
0,5
0,6
0,7
0,8
0,9
0,95
-8,21938 a
-8,41861 b
-8,76108 c
-9,36953 d
-9,42368 d
-9,14434 e
-8,99303 f
0,470828
0,436740
0,488510
0,437201
0,477237
0,350945
0,347927
Limite confiança
-95%
-8,31328
-8,50571
-8,85851
-9,45672
-9,51886
-9,21433
-9,06243
Limite confiança
+95%
-8,12547
-8,33150
-8,66365
-9,28233
-9,32850
-9,07434
-8,92364
significância de 5%. Observa-se que F=0,8 apresentou a melhor média e o intervalo mostra a
probabilidade de que 95% das observações deste grupo estão entre -9,51886 e -9,32850. Com
isso, conclui-se que a combinação F=0,8 e CR=0,85, em um primeiro momento, é a melhor
para o modelo 2D.
Para o modelo AB em 3D foi realizada a mesma análise estatística.
Formulação das hipóteses:
• H0: Não há diferença significativa de energia livre mínima entre os níveis de F com CR
fixo 0,85 para o modelo AB em 3D.
• H1: Há diferença significativa de energia livre mínima entre os níveis de F com CR fixo
0,85 para o modelo AB em 3D.
87
Os resultados dessa análise estão na Tabela 21.
Tabela 21: Análise de variância para valores F do modelo AB 3D com CR=0,85
Fonte
Grau
de variação
de liberdade
Tratamentos (valores de F)
6
Erro (ao acaso)
343
Soma
de Quadrados
1791,836
1818,667
Quadrado
F
Médio
de Snedecor
298,6393
56,32328
5,302237
Valor de p
0,0003
Na Tabela 21, o valor de p < 0,05 indica rejeição da hipótese H0. Logo, há diferença
de energia gerada entre os níveis de F com CR=0,85 para o modelo 3D. O box-plot ilustra as
distribuições de valores de F com CR=0,85 (Figura 32).
Figura 32: Comparação entre as distribuições de F com CR=0,85 no modelo AB 3D
Na Figura 32 observa-se que F=0,4 gerou a distribuição com menor média (o melhor resultado) e grau de variabilidade semelhante aos demais. A localização das diferenças por meio
de teste estatístico, pode ser verificada na Tabela 22, que apresenta os resultados dos testes de
comparações entre F’s utilizando teste de Tukey com nível de significância de 0,05.
O grupo F=0,4 apresentou a melhor média e o intervalo mostra a probabilidade de que
95% das observações deste grupo estão entre -56,3419 e 55,0834, ou seja, para o modelo 3D a
configuração inicial é F=0,4 e CR=0,85.
ETAPA 2: CR variável, F variável, para modelos AB 2D e 3D.
Em uma segunda etapa, decidiu-se verificar se realmente o valor de CR=0,85 seria o valor
ideal para os modelos. Assim testou-se todas as 56 (cinquenta e seis) combinações dos valores
88
Tabela 22: Resultados do teste de Tukey para comparações entre médias F com CR=0,85 no modelo
AB 3D
F
Energia Média
Desvio padrão
0,4
0,5
0,6
0,7
0,8
0,9
0,95
-55,7126
-48,0057
-53,7662
-52,4879
-52,7924
-54,0959
-54,2008
2,214000 a
2,250893 b
2,295673 c
2,404471 cd
2,169830 cde
2,372933 cef
2,399328 cf
Limite confiança
-95%
-56,3419
-48,6453
-54,4186
-53„1713
-53,4090
-54,7703
-54,8827
Limite confiança
+95%
-55,0834
-47,3660
-53,1138
-51,8046
-52,1757
-53,4215
-53,5189
de F (Tabela 6) com valores de CR (Tabela 7) para cada um dos modelos. Além da combinação
desses parâmetros, optou-se por realizar duas sequências de testes com dois valores de número
de gerações (100.0000 e 350.000).
A variação no número de gerações que o algoritmo de ED evolui tem como finalidade principal verificar se esse número pode influenciar na definição da melhor combinação dos parâmetros, já que uma quantidade maior de evoluções pode levar a melhores resultados (no caso do
presente trabalho, valores menores de energia mínima). Nesses experimentos, o tamanho da
população foi mantido constante em 100 (cem) indivíduos, sendo que, para cada um dos experimentos, o algoritmo foi executado por 50 (cinquenta) rodadas.
Os resultados, para ambos os modelos e número de gerações, podem ser divididos em
dois momentos. Em um primeiro momento, foram analisados os valores de F e CR separadamente, em que foi levado em conta a média efetiva das energias obtidas para cada parâmetro
isoladamente. Em um segundo momento, a análise avalia os valores de F e CR em conjunto,
verificando qual a melhor combinação. Vale lembrar que os melhores valores serão sempre os
que apresentam a menor energia livre.
MODELO 2D
A Figura 33 mostra os resultados de F e CR combinados pelo algoritmo de ED durante 50
rodadas para um número de gerações igual a 100.000.
Observa-se na Figura 33 que a melhor combinação dos valores de F e CR está em F=0,6
com CR=0,9, que apresentou valor de energia mínima melhor do que nas outras combinações.
A Figura 34 traz os resultados de F e CR combinados pelo algoritmo de ED durante 50
rodadas para um número de gerações igual a 350.000.
O melhor valor de energia é obtido com a combinação de F=0,8 com CR=0,85, sendo
F=0,8 com CR=0,9 também pode ser utilizado. Comparando os dois gráficos para o modelo
89
Figura 33: Ajuste de F e CR combinados para o modelo 2D após 100.000 gerações
Figura 34: Ajuste de F e CR combinados para o modelo 2D após 350.000 gerações
2D, percebe-se que o aumento de gerações em 3,5 vezes apresenta uma melhora significativa
nos valores das energias médias obtidas.
MODELO 3D
A Figura 35 mostra os resultados de F e CR combinados pelo algoritmo de ED durante 50
rodadas para um número de gerações igual a 100.000.
Observa-se na Figura 35 que os melhores valores de energia mínima resultaram das combinações entre F=0,4 os valores de CR, exceto quando CR=0,6. Ressalte-se que o melhor resul-
90
Figura 35: Ajuste de F e CR combinados para o modelo 3D após 100.000 gerações
tado é obtido para a combinação F=0,4 com CR=0,7.
A Figura 36 traz os resultados de F e CR combinados pelo algoritmo de ED durante 50
rodadas para um número de gerações igual a 350.000.
Observa-se na Figura 36 que o F=0,4 gera melhores energias mínimas quando combinado
com os valores de CR. Sendo F=0,4 com CR=0,85 a melhor média.
4.2.3
Ajustes Dizimação
Para verificar a significância ou não da dizimação durante o processo evolutivo do algoritmo
de ED optou-se por utilizar a sequência de 34 aminoácidos, por ser a sequência média dentre
as sequências de Fibonacci. Os testes utilizaram 100.000 gerações e 100 rodadas. A variável
do estudo foi MAXCOUNT que identifica número de gerações máximas sem evolução antes
que ocorra a dizimação da população. Com os resultados das médias de energia mínima obtidas
para os diferentes valores de MAXCOUNT (Tabela 8) foram realizados dois testes de ANOVA:
I) entre MAXCOUNT 1000, 2000, 3000, 4000, 5000, 10000, 15000, 20000, 25000 e 30000; II)
entre MAXCOUNT 35000, 40000, 45000 e 50000.
Formulação das hipóteses para o modelo 2D:
91
Figura 36: Ajuste de F e CR combinados para o modelo 3D após 350.000 gerações
• H0: Não há diferença significativa de energia entre os níveis de MAXCOUNT.
• H1: Há diferença significativa de energia entre os níveis de MAXCOUNT.
A Tabela 23 apresenta os resultados dos testes estatísticos para a dizimação no modelo 2D,
em que os valores de p < 0,05 indicam rejeição da H0. Assim há diferenças significativas
entre os valores de MAXCOUNT no modelo AB 2D, ou seja, a dizimação pode influenciar o
resultado final do algoritmo, melhorando a energia mínima. O box-plot (Figura 37) foi gerado
para ilustrar as distribuições dos valores de MAXCOUNT e a Tabela 24 apresenta as médias e
os resultados do teste de Tukey para comparações.
Tabela 23: Análise de variância para a Dizimação (MAXCOUNT) modelo 2D
Grau de
liberdade
Tratamento
I
9
II
3
Soma de
Quadrados
Tratamento
214,1076
3,037696
Quadrado
Médio
Tratamento
23,78973
1,012565
Grau de
liberdade
Erro
543
190
Soma de Quadrado
F
Quadrados
Médio
de
Erro
Erro
Snedecor
108,4014 0,199634 119,1665
31,49532 0,165765 6,108444
Observa-se na Figura 37 que MAXCOUNT=30000 gerou a distribuição com valor médio
menor (o melhor resultado).
Na Tabela 24 são apresentados para cada valor de MAXCOUNT, estatística da média,
desvio padrão e o intervalo de confiança da média, de 95% de confiança para a energia mínima,
Valor
de p
0,0002
0,000547
92
Figura 37: Distribuição dos resultados utilizando dizimação para o modelo 2D
sendo que médias seguidas de mesma letra não diferem entre si pelo teste de Tukey para nível
de significância de 5%.
Tabela 24: Estatísticas da energia mínima gerada no processo de Dizimação no modelo AB 2D
MAXCOUNT
Energia Média
Desvio padrão
1000
2000
3000
4000
5000
10000
15000
20000
25000
30000
35000
40000
45000
50000
-7,64040 a
-7,93238 b
-8,28247 c
-8,64264 d
-8,73070 d
-9,19448 e
-9,43640 f
-9,37592 fg
-9,23608 egh
-9,50384 fgi
-9,16811 ehj
-9,44631 fgik
-9,17693 ehj
-9,39401 fghik
0,615067
0,464807
0,423265
0,411403
0,353397
0,430770
0,471767
0,432688
0,392137
0,435663
0,365954
0,426500
0,370542
0,462457
Limite confiança
-95%
-7,80512
-8,05042
-8,38996
-8,74712
-8,82044
-9,30388
-9,58159
-9,50297
-9,34752
-9,63176
-9,27211
-9,57153
-9,28223
-9,52979
Limite confiança
+95%
-7,47569
-7,81434
-8,17498
-8,53817
-8,64095
-9,08509
-9,29122
-9,24888
-9,12464
-9,37593
-9,06410
-9,32108
-9,07162
-9,25823
A dizimação MAXCOUNT=30000 apresentou a melhor média e o melhor intervalo, tendo
em vista que 95% das observações deste grupo apresentaram energias livres entre -9,63176
e -9,37593. Uma vez que MAXCOUNT=30000 apresentou redução dos valores de energia
livre mínima, quando comparados aos valores do algoritmo sem este operador, a dizimação foi
aplicada ao algorimto final de ED para o modelo AB 2D.
Formulação das hipóteses para o modelo 3D:
93
• H0: Não há diferença significativa de energia entre os níveis de MAXCOUNT.
• H1: Há diferença significativa de energia entre os níveis de MAXCOUNT.
Tabela 25: Análise de variância para a Dizimação (MAXCOUNT) modelo 3D
Grau de
liberdade
Tratamento
I
9
II
3
Soma de
Quadrados
Tratamento
3909,868
16,53983
Quadrado
Médio
Tratamento
434,4297
5,513276
Grau de
liberdade
Erro
490
196
Soma de Quadrado
F
Quadrados
Médio
de
Erro
Erro
Snedecor
5187,537 10,58681 41,03500
1188,589 6,064229 0,909147
Os resultados na tabela 25, mostram que para a análise I o valor de p < 0,05 indica rejeição
da H0, há diferença de energia mínima gerada entre os níveis de MAXCOUNT. Na análise II
como p > 0,05 não se rejeita H0, não há diferença entre os níveis de MAXCOUNT.
O box-plot (Figura 38) mostra as distribuições dos valores de MAXCOUNT e a Tabela 26
apresenta as médias e os resultados do teste de Tukey para comparações, sendo que as médias
seguidas de mesma letra não diferem entre si para nível de significância de 5%.
Figura 38: Distribuição dos resultados utilizando a dizimação para o modelo 3D
Observa-se que o grupo MAXCOUNT=30000 apresentou a melhor média e o intervalo
mostra a probabilidade de que 95% das observações deste grupo estão entre -67,4458 e 66,2868.
Para os dois modelos AB, tanto em 2D quanto em 3D, a dizimação apresenta significância,
podendo ocasionar a melhoria dos resultados de energia mínima. O valor de MAXCOUNT para
os dois modelos foi fixado em 30000 no algoritmo final.
Valor
de p
0,00032
0,437595
94
Tabela 26: Estatísticas da energia mínima gerada no processo de Dizimação no modelo AB 3D
MAXCOUNT
Energia Média
Desvio padrão
1000
2000
3000
4000
5000
10000
15000
20000
25000
30000
35000
40000
45000
50000
-56,9068 a
-60,7432 bc
-59,9908 bc
-61,1384 cd
-62,4586 d
-63,9156 ef
-64,8899 eg
-65,2869 g
-63,5910 df
-66,8663 h
-66,2256 h
-66,2664 h
-66,9278 h
-66,6370 h
5,026298
3,216264
3,462069
2,297793
4,314620
2,687521
3,579007
1,923404
2,547305
2,039130
2,390997
2,265593
2,798119
2,361709
4.2.4
Limite confiança
-95%
-58,3353
-61,6572
-60,9747
-61,7914
-63,6848
-64,6794
-65,9071
-65,8335
-64,3150
-67,4458
-66,9052
-66,9103
-67,7230
-67,3082
Limite confiança
+95%
-55,4784
-59,8291
-59,0068
-60,4853
-61,2324
-63,1519
-63,8728
-64,7402
-62,8671
-66,2868
-65,5461
-65,6225
-66,1326
-65,9658
Ajuste de parâmetros da mutação espelhada
A mutação espelhada, um operador especial criado para tentar melhorar os resultados do
dobramento para as sequências, foi analisada inicialmente para o modelo AB em 2D, em que
alguns valores de MAXMIRROR foram testados. MAXMIRROR é o valor máximo que o
contador pode atingir para realizar a mutação espelhada. Os testes para MAXMIRROR foram
executados para 50 rodadas com o modelo AB 2D, usando os seguintes valores dos parâmetros
do algoritmo ED: estratégia rand/1/exp e F=0,8 CR=0,85. Os testes visam verificar se esse
novo operador é significantivo ou não para ser utilizado no algoritmo final. Para verificar a
significância do operador após os testes, foi utilizada para a análise dos resultados a ANOVA.
Formulação das hipóteses no modelo 2D:
• H0: Não há diferença significativa de energia entre os níveis de MAXMIRROR.
• H1: Há diferença significativa de energia entre os níveis de MAXMIRROR.
Tabela 27: Análise de variância para o MAXMIRROR no modelo 2D
Fonte de
variação
Tratamentos (MAXMIRROR)
Erro (ao acaso)
Grau de
liberdade
5
536
Soma de Quadrado
Quadrados
Médio
1,539999 0,308000
86,65908 0,161677
F
de Snedecor
Valor
de p
1,905027
0,091797
Na Tabela 27, os resultados da análise de variância mostra que p > 0,05 indica não rejeição
95
da H0, portanto não há diferença significativa de energia entre os níveis de MAXMIRROR, ou
seja, são estatisticamente iguais.
Figura 39: Distribuição da mutação espelhada para o modelo 2D
O box-plot (Figura 39) apresenta as distribuições dos valores de energia mínima para os
diferentes valores de MAXMIRROR. Observa-se visualmente que são semelhantes.
A Tabela 28 mostra a média, o devio padrão e os limites de confiança de 95% para a média.
Tabela 28: Estatísticas da energia mínima gerada no processo de mutação espelhada no modelo
AB 2D
MAXMIRROR
Energia Média
Desvio padrão
5
10
20
30
40
50
-9,39130
-9,48385
-9,47538
-9,45525
-9,50337
-9,36287
0,426284
0,441796
0,391152
0,371901
0,380572
0,380896
Limite confiança
-95%
-9,47767
-9,57151
-9,55299
-9,56569
-9,57888
-9,43845
Limite confiança
+95%
-9,30493
-9,39618
-9,39776
-9,34481
-9,42786
-9,28730
É possível perceber, da análise conjunta da tabela e do gráfico, que os valores médios de
MAXMIRROR são muito próximos, isto é, estatisticamente iguais. A melhor média de energia
mínima de MAXMIRROR (-9,50337) se comparada à média da melhor energia mínima de
MAXCOUNT (-9,50384) não apresenta um ganho significativo.
Uma vez que o operador dizimação apresentou um resultado semelhante ao operador de
mutação espelhada optou-se por desconsidera-lo nos modelos, pois a complexidade de implementação e o custo computacional elevado, não compensam a sua utilização.
96
4.2.5
Resultados do algoritmo de ED auto-adaptável
Utilizando os conceitos de auto-adaptabilidade propostos no capítulo 3, seção 3.9, com o
parâmetro F auto-adaptável, testes foram realizados para ambos modelos (2D e 3D), visando
identificar se os resultados são satisfatórios.
Para ambos os modelos utilizou-se o algoritmo de ED com os parâmetros: estratégia rand/1/exp
e CR=0,85, determinados através de testes de desempenho. O valor de F será adaptado automaticamente a cada rodada, seguindo as regras definidas nas equações 23 e 24. O número de
gerações foi definido em 350.000, uma vez que os resultados obtidos para ambos os modelos
foi melhor quando o número de gerações foi maior. Além disto, os testes foram executados para
a sequência de 34 aminoácidos durante 50 rodadas e os resultados médios das energias mínimas
obtidas estão representados na Tabela 29.
Tabela 29: Resultados ED auto-adaptável para sequência de 34 aminoácidos nos modelos 2D e 3D
Sequência
34
4.2.6
Médiaenergia2D
-9,55754
Médiaenergia3D
-85,2752
Resultados do algoritmo final de ED com os parâmetros ajustados
Após o ajuste de parâmetros e a validação dos operadores especiais criados para tentar
melhorar os resultados do algoritmo de ED, chegou-se à configuração final dos parâmetros.
O algoritmo de ED paralelo final será executado para as quatro sequências de bechmark apresentadas. Deve-se ressaltar que o algoritmo utilizado será o mesmo para os dois modelos,
diferenciando apenas o cálculo do fitness, o tamanho do indivíduo de entrada e o valor de F,
uma vez que os resultados apresentaram variações no valor de F para os dois modelos.
O algoritmo final apresenta a seguinte configuração para ambos os modelos:
• População Inicial de 100 indivíduos
• Estratégia de evolução rand/1/exp
• Constante de crossover CR=0,85
• Dizimação MAXCOUNT=30000
• Número de gerações 350.000
O valor de F para cada um dos modelos está definido abaixo:
97
• Modelo 2D: F=0,80
• Modelo 3D: F=0,40
Os resultados obtidos utilizando as configurações descritas acima estão representados nas
Tabelas 30 e 31, que, ademais, apresentam as médias das energias obtidas para cada uma das
sequências em ambos os modelos, assim como o melhor resultado. Esses resultados serão
utilizados na comparação com outros trabalhos. Nas tabelas, a coluna Energiamedia representa
a energia média nas 50 rodadas e a coluna Melhor apresenta o melhor resultado encontrado nas
50 rodadas.
Tabela 30: Resultados ED com parâmetros ajustados para as sequências de benchmark modelo 2D
Sequencia
13
21
34
55
Energiamedia
-2,8999
-5,9880
-9,4237
-11,5240
Melhor
-3,1999
-6,1980
-10,4699
-13,5205
Tabela 31: Resultados ED com parâmetros ajustados para as sequências de benchmark modelo 3D
Sequencia
13
21
34
55
4.2.7
Energiamedia
-24,5465
-46,762225
-83,3553
-138,3495
Melhor
-26,5066
-48,9873
-89,7957
-149,5675
Resultados do algoritmo final de ED auto-adaptável
As Tabelas 32 e 33 apresentam os resultados obtidos a partir da utilização das configurações
descritas na seção 4.2.6, utilizando a auto-adaptação do parâmetro F. Além disso, elas apresentam as médias das energias para cada uma das sequências em ambos os modelos, assim como o
melhor resultado. Esses resultados serão comparados com os resultados de outros trabalhos.
Tabela 32: Resultados ED auto-adaptável para as sequências de benchmark modelo 2D
Sequencia
13
21
34
55
Energiamedia
-2,78459
-5,7880
-9,8577
-15,6743
Melhor
-3,1999
-6,1980
-10,5565
-17,3133
98
Tabela 33: Resultados ED auto-adaptável para as sequências de benchmark modelo 3D
Sequencia Energiamedia
13
-24,77597391
21
-47,1454
34
-85,61326579
55
-147,927
4.2.8
Melhor
-26,507
-50,3613
-92,0962
-157,112
Comparação com outras abordagens
A Tabela 34 apresenta os resultados das energias mínimas obtidas para o modelo AB 2D,
com os resultados de benchmarks, em que: o símbolo E representa a energia mínima para as diversas abordagens; Emin , a energia obtida através do método da conjugação de gradientes, com
configuração inicial através do PERM (Pruned Enriched Rosenbluth method) (HSU; MEHRA;
GRASSBERGER, 2003); E∗min , a energia mínima obtida por Stillinger e Head-Gordon (STILLINGER; HEAD-GORDON, 1995); EPERM , a menor energia obtida por meio de uma rodada
completa do PERM (HSU; MEHRA; GRASSBERGER, 2003); EACMC , a menor energia utilizando o Annealing Contour Monte Carlo Method (LIANG, 2004); ECSA , a energia por meio
do Conformational Space Annealing(CSA) (KIM; LEE; LEE, 2005); EPSO , a energia utilizando
o PSO (ZHARG; LI, 2007); EED , a energia por meio do algoritmo de ED com os parâmetros
ajustados obtidos neste trabalho; e, por fim, EEDad pt , que representa a energia por meio do algoritmo de ED com F auto-adaptável obtidos neste trabalho. Os valores em negrito expressam
as melhores energias mínimas da literatura.
Tabela 34: Resultados obtidos nos experimentos com os benchmarks no modelo AB 2D
N
Emin
13 -3,2939
21 -6,1976
34 -10,7001
55 -18,5154
E∗min
-3,2235
-5,2881
-8,9749
-14,4089
Modelo AB 2D
EPERM
EACMC
ECSA
EPSO
-3,2167 -3,2941
-3,2941 -3,2941
-5,7501 -6,1979
-6,1980 -6,1977
-9,2195 -10,8060 -10,8060 -10,7036
-14,9050 -18,7407 -18,9110 -18,4236
EED
-3,1999
-6,1980
-10,4699
-13,5205
EEDad pt
-3,1999
-6,1980
-10,5565
-17,3133
A Tabela 35 mostra a comparação entre os melhores resultados da literatura com os melhores obtidos nesse trabalho para as 4 (quatro) sequências de Fibonacci no modelo AB 2D.
Na Tabela 35 analisando a coluna (DifEEDxMelhor ) que indica em percentual o diferencial entre
o melhor resultado da literatura e o algoritmo de ED com parâmetros ajustados observa-se que a
para as sequências menores (13, 21, 34) os resultados menos diferem. A coluna DifEEDad pt xMelhor
indica a diferença entre o melhor resultado da literatura e o algoritmo de ED auto-adaptável.
Observa-se que o algoritmo auto-adaptável apresenta menores diferenciais em relação a literatura e ao ED com parâmetros ajustados. Cabe ressaltar que para a sequência de 21 aminoácidos
99
Tabela 35: Comparação entre o melhor benchmark no modelo AB 2D com os melhores resultados
do presente trabalho
N
13
21
34
55
Modelo AB 2D
Melhor literatura
EEED
DifEEDxMelhor
-3,2941
-3,1999
-2,859%
-6,1980
-6,1980
0%
-10,8060
-10,4699
-3,11%
-18,9110
-13,5205
-28%
EEED EDad pt
-3,1999
-6,1980
-10,5565
-17,3133
DifEEDad pt xMelhor
-2,859%
0%
-2,31%
-8,44%
ambos algoritmos propostos chegou-se ao melhor resultado apresentado na literatura.
A Tabela 36 apresenta os resultados das energias mínimas obtidas para o modelo AB 3D,
com os resultados de benchmarks, em que: o símbolo E representa a energia mínima para as
diversas abordagens; EMUCA , a energia obtida através do Multicanonical; E∗ELP , a energia mínima obtida por meio da minimização ELP (BACHMANN; ARKM; JANKE, 2005); EACMC ,
a menor energia obtida por meio do Annealing Contour Monte Carlo Method; EACMC+ , a
menor energia utilizando o Annealing Contour Monte Carlo Method melhorado por meio de
ajustes de Metropolis (LIANG, 2004); ECSA , a energia por meio do Conformational Space Annealing(CSA) (KIM; LEE; LEE, 2005); EDE , a energia por meio do algoritmo de ED com os
parâmetros ajustados obtidos neste trabalho; e, enfim, EDEad pt representa a energia por meio do
algoritmo de ED com F auto-adaptável obtidos neste trabalho. Os valores em negrito expressam
as melhores energias mínimas da literatura.
Tabela 36: Resultados obtidos nos experimentos com os benchmarks no modelos AB 3D
N
EMUCA
13 -26,496
21 -52,915
34 -97,272
55 -169,654
Modelo AB 3D
EELP
EACMC
EACMC+
ECSA
-26,498 -26,363 -26,507 -26,4714
-52,917 -50,860 -51,718 -52,7865
-97,261 -92,746 -94,043 -97,7321
-172,696 -149,481 -154,505 -173,9803
EDE
EDEad pt
-26,4714 -26,507
-48,56454 -50,3613
-89,7957 -92,0962
-149,5675 -157,112
A Tabela 37 mostra a comparação entre os melhores resultados da literatura com os melhores obtidos nesse trabalho para as 4 (quatro) sequências de Fibonacci no modelo AB 3D.
Tabela 37: Comparação entre o melhor benchmark no modelo AB 3D com os melhores resultados
do presente trabalho
N
13
21
34
55
Modelo AB 3D
Melhor literatura
EEED
DifEEDxMelhor
-26,507
-26,4714
-0,134%
-52,917
-48,56454
-8,225%
-97,7321
-89,7957
-8,12%
-173,9803
-149,5675
-14,03%
EEED EDad pt
-26,507
-50,3613
-92.0962
-157,112
DifEEDad pt xMelhor
0%
-4,829%
-6,119%
-9,69%
100
Na Tabela 37 analisando a coluna (DifEEDxMelhor ) que indica em percentual o diferencial entre
o melhor resultado da literatura e o algoritmo de ED com parâmetros ajustados observa-se que
a para a sequência de 13 aminoácidos a diferença em relação ao melhor da literatura foi muito
pequena, quase nula. Já para as demais sequências as diferenças foram maiores, aumentando
de acordo com o tamanho da sequência. A coluna DifEEDad pt xMelhor indica a diferença entre o
melhor resultado da literatura e o algoritmo de ED auto-adaptável. Observa-se que o algoritmo
auto-adaptável apresenta menores diferenciais em relação a literatura e ao ED com parâmetros
ajustados. Para a sequência de 13 aminoácidos chegou-se ao melhor resultado conhecido.
101
5
5.1
DISCUSSÃO DOS RESULTADOS E CONCLUSÃO
DISCUSSÃO DOS RESULTADOS
Os testes iniciais visaram validar os conceitos apresentados na literatura para o algoritmo
de ED no decorrer dos anos, em que vários experimentos foram realizados (STORN; PRICE,
1997; FEOKTISTOV, 2006). Nesses experimentos, conclui-se que o valor de CR não influencia
tanto o processo de evolução e que esse valor inicialmente deve ser de 0,85. Além disso, os
experimentos indicaram que o valor de F e a estratégia de evolução seriam os valores que teriam
maior peso no resultado final do algoritmo.
Dessa maneira, realizou-se uma série de testes para as quatro sequências de benchmark
visando definir primeiramente qual a melhor estratégia para cada modelo. Os testes para ambos os modelos indicaram que a estratégia rand/1/exp seria a estratégia ideal. Deve-se ressaltar
que essa é uma das estratégias mais utilizadas na literatura para a solução de outros problemas. Posteriormente, para cada um dos modelos, utilizando esta estratégia e a sequência de 34
aminoácidos, foram realizados testes que visaram definir qual seria o melhor valor de F. Não
satisfeito da fixação de CR em 0,85 decidiu-se avaliar qual a combinação de CR e F que melhor
poderia satisfazer os resultados. Os testes comprovaram que realmente o valor de CR definido
na literatura em 0,85 é o melhor valor para o problema proposto, sendo que a combinação desse
com F=0,8 e F=0,4 deve ser utilizada nos modelos 2D e 3D, respectivamente.
Para tentar melhorar ainda mais os resultados obtidos, foram desenvolvidos dois novos operadores, conforme apresentado: a dizimação e a mutação espelhada. Os resultados apresentados da dizimação para ambos os modelos foram suficientemente satisfatórios para incorpora-lá
ao algoritmo de ED final, ao contrário da mutação espelhada, pois não apresentou um resultado
satisfatório quando considerados complexidade da implementação e gasto computacional para
executá-la.
Os resultados obtidos utilizando o algoritmo ED final com a dizimação (EED ) estão apresentados nas Tabelas 34 e 36, para os modelos 2D e 3D, respectivamente. Esses resultados,
quando comparados a outros métodos, apresentaram resultados promissores, pois, para algu-
102
mas sequências, conseguiu-se chegar ao melhor resultado obtido até então (Tabelas 35 e 37).
No modelo 2D foi possível atingir o melhor resultado para a sequência de 21 aminoácidos e no
modelo 3D para a sequência de 13 aminoácidos. Já nas outras sequências, os resultados ficaram
um pouco aquém dos melhores já encontrados. Por por outro lado, ficaram melhores do que os
obtidos por outros métodos.
Para tentar melhorar ainda mais os resultados obtidos, optou-se por implementar o algoritmo de evolução diferencial adaptável, em que o valor de CR e a estratégia de evolução e
dizimação são fixados nos valores definidos através dos experimentos da seção 4.2. Para ambos
os modelos o valor de F é variável no decorrer das gerações.
Após a execução dos testes para o ED adaptável (EEDad pt ), os resultados apresentados
foram melhores do que os apresentados pelo algoritmo de ED com dizimação. Em média, a
melhora dos resultados foi percebida para as sequências maiores, de 34 e 55 aminoácidos, pois,
para as outras sequências, o algoritmo anterior já havia apresentado bons resultados. No entanto,
a melhora mais significativa dos resultados foi identificada para a sequência de 55 aminoácidos,
em ambos os modelos. Os resultados melhoraram em aproximadamente 20% para a sequência
em 2D e de 5% para a sequência em 3D, tudo em relação ao algoritmo inicial, quando esse foi
comparado com os melhores resultados para cada uma das sequências de bechmark.
5.2
CONCLUSÃO
A principal contribuição deste trabalho é a apresentação de uma nova metodologia de
otimização para a solução do Problema da Predição da Estrutura de Proteína quando aplicada
ao modelo AB em 2D e 3D: a Evolução Diferencial (ED).
A ED foi estudada a fundo, verificando e ajustando cada um dos parâmetros do algoritmo
que melhor se adequam ao problema, gerando resultados excelentes. Além disso, foi proposta
a evolução diferencial auto-adaptável, em que o valor de F se ajusta a cada geração na busca de
melhores resultados.
O trabalho propôs, ainda, dois novos parâmetros (operadores) para o algoritmo de ED. Um
deles é considerado um operador global, qual seja, a dizimação, que pode ser utilizada em qualquer problema e evita a estagnação da evolução em alguns dos vários mínimos locais existentes
nos problemas. Ele visa, ainda, a dar condições para o algoritmo se recuperar da convergência
prematura, permitindo, assim, uma exploração mais eficiente do espaço de busca. O segundo
parâmetro proposto foi a mutação espelhada, um operador específico para o problema, mas que
apresentou um custo elevado para uma melhora pequena do resultado.
103
É importante ressaltar que os melhores resultados obtidos para ambos os modelos AB, em
2D e 3D, utilizam métodos de otimização especializados com a adição de técnicas de busca
local. Já a ED, mesmo adicionando a dizimação ou utilizando-a auto-adaptável, continua sendo
considerado um método generalista, uma vez que o único operador especializado criado, a
mutação espelhada, foi descartado do algoritmo final.
Comparando todos os resultados obtidos até então na literatura com os resultados obtidos
pelas duas metodologias de ED propostas nesse trabalho, sugere-se que a ED é um método
promissor para a solução de problemas de otimização de estrutura das proteínas.
Contudo, observou-se uma perda de eficiência à medida em que o tamanho das sequências
aumentaram. Isso ocorreu devido a uma série de fatores, tais como: o aumento exponencial do
espaço de busca cada vez que um aminoácido foi adicionado à sequência; a natureza complexa
do problema; a convergência prematura para um mínimo local e a dificuldade para quebrá-lo.
É importante a continuidade de trabalhos de exploração de algoritmos que visem a solucionar o PPEP, com o intuito de gerar uma literatura específica. Espera-se que, com isso, o
conhecimento da biologia, trabalhado pela especifidade e ferramentas de computação, possam
contribuir na compreensão dos fenômenos biológicos, e consequentemente em descobertas para
melhoria das condições de saúde do homem.
5.3
TRABALHOS FUTUROS
Os algoritmos propostos neste trabalho apresentaram resultados excelentes quando com-
parados a outros algoritmos da literatura, mesmo não atingindo as melhores conformações (dobramentos com menor energia mínima) para todas as sequências. Para melhorar ainda mais
esses resultados, podem ser criados outros operadores especiais, tanto genéricos como bioinspirados (dizimação e mutação espelhada, respectivamente). Além disso, pode ser estudada
uma outra forma de aplicar o operador de mutação espelhada, para que esse possa trazer benefícios ao resultado final.
Ademais, é interessante realizar novos estudos com o algoritmo de ED auto-adaptável, propondo outros métodos de auto-adaptação dos parâmetros do algoritmo de ED. Esse conceito de
auto-adaptação também pode ser aplicado nos operadores especiais válidos para o problema.
Ainda, novos testes de ajuste de parâmetros podem ser executados, visando explorar o
espaço de busca dos problemas propostos e, consequentemente, identificar outras configurações
que solucionem o problema.
104
Por fim, outra proposta de paralelização, utilizando o conceito de ilhas, pode ser implementada para melhorar os resultados dos algoritmos de ED. A ideia é inicializar várias populações
de indivíduos utilizando diferentes configurações de parâmetros, até mesmo uma ilha rodando o
algoritmo de ED auto-adaptável. Durante o decorrer das gerações, indivíduos de uma determinada ilha migram para outra e assim por diante, visando aumentar a diversidade da população
nas diferentes ilhas e, por conseguinte, gerar indivíduos melhores.
105
REFERÊNCIAS
ABBASS, H. A. The self adaptive Pareto differential evolution algorithm. In: Proceedings of
IEEE Congress on Evolutionary Computation. Piscataway, NJ, USA: IEEE Press., 2002. p.
831–836.
ALBERTS, B. Molecular Biology of the Cell. 4. ed. New York: Garland Publishing, 2002.
ALMASI, G. S.; GOTTLIEB, A. Highly Parallel Computing. 2. ed. Redwood City, CA,
USA: Benjamin-Cummings Publishing Co., Inc, 1994. 670 p.
BACHMANN, M.; ARKM, H.; JANKE, W. Multicanonical study of coarse-grained off-lattice
models for folding heteropolymers. Physical Review E, v. 71, p. 1–11, 2005.
BAXEVANIS, A. D.; OUELLETTE, B. F. F. Bioinformatics: A Practical Guide to the
Analysis of Genes and Proteins. 3. ed. New York: John Wiley & Sons, 2004. 560 p.
BERGER, B.; LEIGHTON, T. Protein folding in the hydrophobic-hydrophilic (HP)
model is NP-complete. In: Proceedings of Second annual International Conference on
Computational Molecular Biology. New York: ACM Press, 1998. v. 5, p. 30–39.
BITELLO, R.; LOPES, H. S. A differential evolution approach for protein folding using a
lattice model. International Journal of Computer Science and Technology, v. 22, n. 6, p.
904–908, 2007.
BRANDEN, C.; TOOZE, J. Introduction to Protein Structure. 2. ed. New York: Garland
Publishing, 1999. 410 p.
BREMERMANN, H. J. Optimization through evolution and recombination. In:
Self-Organizing Systems. Washington, DC: Spartan Books, 1962. p. 93–106.
.
CHIKENJI, G.; KIKUCHI, M.; IBA, Y. Multi-self-overlap ensemble for protein folding:
ground state search and thermodynamics. Physical Review Letters, v. 83, n. 9, p. 1886–1889,
1999.
CRESCENZI, P. et al. On the complexity of protein folding. Journal of Computational
Biology, v. 50, n. 3, p. 423–466, 1998.
DILL, K. A. Theory for the folding and stability of globular proteins. Biochemistry,
Biochemistry, v. 24, p. 1501–1509, 1985.
DOOLITTLE, R. F. Of URFs and ORFs: A Primer on How to Analyze Derived Amino
Acid Sequences. 1. ed. Mill Valley, CA, USA: University Science Books, 1986. 103 p.
FEOKTISTOV, V. Differential Evolution: In Search of Solutions. 1. ed. New York:
Springer-Verlag, 2006. 196 p.
106
FEOKTISTOV, V.; JANAQI, S. Classical identification problem solved by differential
evolution. In: MATOUSEK, R.; OSMERA, P. (Ed.). Proceedings of 10th - International
Conference on Soft Computing. Brno, Czech Republic: Mendel, 2004. p. 64–67.
FOGEL, L. J. Autonomous automata. Industrial Research, v. 4, p. 14–19, 1962.
FOSTER, I. Designing and Building Parallel Programs: Concepts and Tools for Parallel
Software Engineering. 3. ed. Boston: Addison-Wesley Longman Publishing Co., 1993. 430 p.
FRASE, A. S. Simulation of genetic systems by automatic digital computers: I. Introduction.
Australian Journal of Biological Sciences, v. 10, p. 484–491, 1957.
GEIST, A. et al. PVM: Parallel Virtual Machine A Users’ Guide and Tutorial for
Networked Parallel Computing. Cambridge, Massachusetts: MIT Press, 1994. 299 p.
GINALSKI, K.; GRISHIN, N. V. Pratical lessons from protein structure prediction. Nucleic
Acids Research, v. 33, n. 6, p. 3326–3340, 2001.
GORSE, D. Global minimisation of an off-lattice potential energy function using a
chaperone-based refolding method. Biopolymers, v. 59, n. 4, p. 411–426, 2001.
GORSE, D. Application of a chaperone-based refolding method to two- and three-dimensional
off-lattice protein models. Biopolymers, v. 64, n. 3, p. 146–160, 2002.
GRIT, D. H. Sisal on a Message Passing Architecture. In: Proceedings of Joint International
Conference on Vector and Parallel Processing. Berlin: Springer-Verlag, 1990. p. 721–721.
HARTL, F. U. Molecular chaperones in cellular protein folding. Nature, v. 381, p. 571–580,
1996.
HOLLAND, J. H. Outline for a logical theory of adaptive systems. Journal of ACM (JACM),
v. 9, n. 3, p. 297–314, 1962.
HSU, H.-p.; MEHRA, V.; GRASSBERGER, P. Structure optimization in an off-lattice protein
model. Physical Review E, v. 68, n. 037703, p. 20–23, 2003.
HWANG, K.; ZHIWEI, X. Scalable Parallel Computing: Technology, Architecture,
Programming. 1. ed. New York: McGraw-Hill, 1998. 832 p.
IRBäCK, A. et al. Local interactions and protein folding: A 3D off-lattice approach. Chemical
Physics, v. 107, n. 1, p. 273–282, 1997.
KALEGARI, D. H.; LOPES, H. S. A differential evolution approach for protein structure
optimisation using a 2D off-lattice model. International Journal of Bio-Inspired
Computation, v. 2, n. 3/4, p. 242–250, 2010.
KARPLUS, M.; SHAKHNOVICH, E. Protein folding - theoretical studies of thermodynamics
. Protein Folding. 1. ed. New York: W.H Freeman and Company,
and dymanics. In:
1992. cap. 4, p. 127–196.
KENNEDY, J.; EBERHART, R. C.; SHI, Y. Swarm Intelligence. 1. ed. San Francisco, CA,
USA: Morgan Kaufmann Publishers, 2001. 512 p.
107
KHIMASIA, M. M.; COVENEY, P. V. Protein structure prediction as a hard optimization
problem: the genetic algorithm approach. Molecular Simulation, v. 19, n. 4, p. 205–226,
1997.
KIM, S. Y.; LEE, S. B.; LEE, J. Structure optimization by conformational space annealing in
an off-lattice protein model. Physical Review E, v. 72, p. 1–6, 2005.
KINDLE, J. H. Geometria analítica plana e analítica plana e no espaço : resumo da teoria,
problemas resolvidos, problemas propostos. 2. ed. Rio de Janeiro: Ao Livro Técnico, 1968.
244 p.
KOLINSKI, A.; SKOLNICKB, J. Reduced models of proteins an their applications. Polymer,
v. 45, p. 511–524, 2004.
KOZA, J. R. Genetic Programming: On the Programming of Computers by Means of
Natural Selection. 1. ed. Cambridge, Massachusetts: The MIT Press, 1992. 840 p.
KRASNOGOR, N. et al. Protein structure prediction with evolutionary algorithms. In: Genetic
and Evolutionary Computation Conference. New York: Morgan Kaufmann Publishers,
1999. p. 1569–1601.
LAU, K.; DILL, K. A. A Lattice statistical mechanics model of the conformation and sequence
space of proteins. Macromolecules, v. 22, p. 3986–3997, 1989.
LEHNINGER, A. L.; NELSON, L. D.; COX, M. M. Lehninger, Princípios de Bioquímica.
4. ed. São Paulo: Editora Sarvier, 2001.
LIANG, F. Annealing contour Monte Carlo algorithm for structure optimization in an
off-lattice protein model. Chemical Physics, v. 120, n. 14, p. 6756–6763, 2004.
LIMA, E. L. Coordenadas no plano com as soluções dos exercícios: geometria analítica,
vetores e transformações geométricas. 4. ed. Rio de Janeiro: INEP, 2002. 329 p.
LIU, J.; LAMPINE, N. J. A fuzzy adaptive differential evolution algorithm. Soft Computing,
IEEE Press, v. 9, n. 6, p. 448–62, 2005.
LIU, J.; LAMPINEN, J. Adaptative parameter control of differential evolution. In: Proceedings
of 8th International Mendel Conference on Soft Computing. Brno, Czech Republic:
Mendel, 2002. p. 19–26.
LOPES, H. S. Evolutionary algorithms for the protein folding problem: a review and current
. Applications of Computational Intelligence in Bioinformatics and
trends. In:
Biomedicine: Current Trends and Open Problems. 1. ed. Heidelberg: Springer-Verlag,
2008. p. 297–315.
LOPES, H. S.; SCAPIN, M. P. An enhanced genetic algorithm for protein structure prediction
using the 2D hydrophobic polar model. Lecture Notes in Computer Science, v. 3871, p.
238–246, 2005.
MATSUMOTO, M.; NISHIMURA, T. Mersenne twister: a 623-dimensionally equidistributed
uniform pseudo-random number generator. ACM Transactions on Modeling and. Computer
Simulation, v. 8, n. 1, p. 3–30, 1998.
108
MERKLE, L. et al. Hybrid genetic algorithms for polypeptide energy minimization. In:
Proceedings of the 1996 ACM Symposium on Applied Computing. New York: ACM Press,
1996. p. 305–311.
MEZURA-MONTES, E.; VELáZQUES-REYES.J; Coello Coello, C. A. A comparative study
of differential evolution variants for global optimization. In: Proceedings of Genetic and
Evolutionary Computation Conference. New York: ACM Press, 2006. p. 485–492.
MICHALEWICZ, Z.; FOGEL, D. B. How to Solve it: Modern Heuristics. 2. ed. New York:
Springer-Verlag, 2004. 580 p.
MICHALEWICZ, Z.; SCHOENAUER, M. Evolutionary algorithms for constrained parameter
optimization problems. Evolutionary Computation, v. 4, n. 1, p. 1–32, 1996.
PACHECO, P. A User’s Guide do MPI. 1995. Disponível em:
<htts://math.usfca.edu/pub/MPI/mpi-guide.ps>.
PACHECO, P. Parallel Programming with MPI. 1. ed. San Francisco, CA, USA: Morgan
Kaufmann Publishers, 1996. 500 p.
PLAGIANAKOS, V.; TASOULIS, D.; VRAHATIS, M. A review of major application areas
. "A Review of Major Application Areas of Differential
of differential evolution. In:
Evolution. Berlin: Springer-Verlag, 2008. v. 143, p. 197–238.
PRICE, K. Genetic annealing. Dr. Dobb‘s Journal, v. 220, p. 127–132, 1994.
QUINN, M. J. Parallel Computing: Theory and Practice. 2. ed. New York: McGraw-Hill,
1994. 446 p.
RECHENBERG, I. Cybernetic solution path of an experimental problem. Royal Aircraft
Establishment, v. 1122, p. 178–187, 1965.
RICHARDS, F. M. Areas, volumes, packing and protein structures. Annual Review of
Biophysics and Bioengineering, v. 6, p. 151–176, 1977.
SARKAR, V.; HENNESSY, J. Compile time partitioning and scheduling of parallel programs.
In: Proceedings of SIGPLAN - Symposium on Compiler Construction. New York: ACM
Press, 1986. p. 17–26.
SCAPIN, M. P. Um Algoritmo Genético Híbrido Aplicado à Predição da Estrutura de
Proteínas utilizando o modelo Hidrofóbico-Polar dibimensional. Tese (Doutorado) —
Universiodade Tecnologida do Paraná, 2005.
SCHULZE-KREMER, S. Genetic algorithm for protein tertiary structure prediction. In:
Proceedings of the European Conference on Machine Learning. Berlin: Springer-Verllag,
1993. p. 262–279.
SHMYGELSKA, A.; HOOS, H. H. An improved ant colony optimisation algorithm for 2D HP
protein folding problem. Lecture Notes in Computer Science, v. 2671, p. 400–417, 2003.
SHMYGELSKA, A.; HOOS, H. H. An ant colony optimisation algorithm for the 2D and 3D
hidrophobic polar protein folding problem. BMC Bioinformatics, v. 6, p. 1–22, 2005.
109
SNIR, M. et al. MPI: The Complete Reference. 1. ed. Cambridge, Massachusetts: MIT Press,
1996. 350 p.
STILLINGER, F. H.; HEAD-GORDON, T. Collective aspects of protein folding illustrated by
a toy model. Physical Review E, v. 52, p. 2872–2877, 1995.
STILLINGER, F. H.; HEAD-GORDON, T.; HIRSHFELD, C. Toy model for protein folding.
Physical Review E, v. 48, n. 2, p. 1468–1477, 1993.
STORN, R.; PRICE, K. Differential Evolution: A Simple and Efficient Heuristic for Global
Optimization over Continuous Spaces. Journal of Global Optimization, v. 11, p. 341–359,
1997.
STORN, R.; PRICE, K. Differetial Evolution - A Practical Aprroach to Global
Optimization. 1. ed. Berlin: Springer-Verlag, 2005. 538 p.
TEO, J. Exploring dynamic self-adaptive populations in differential evolution. Soft
Computing, v. 10, n. 8, p. 673–686, 2006.
TORCINI, A.; LIVI, R.; POLIT, A. A dynamical approach to protein folding. Biological
Physics, v. 27, p. 181, 2001.
TRIOLA, F. M. Introdução a Estatística. 7. ed. Rio de Janeiro: LTC - Livros Técnicos e
Científicos, 1998. 384 p.
UNGER, R.; MOULT, J. Genetic algorithm for protein folding simulations. Journal of
Molecular Biology, v. 81, p. 231–275, 1993.
VESTERSTROEM, J.; THOMASEN, R. A comparative study of differential evoltion, particle
swarm optimization, and evolutionary algorithm on numerical benchmark problems. In: IEEE
Congress on Evolutionary Computation. [S.l.]: IEEE Press, 2004. p. 1980–1987.
VULLO, A. On the role of machine learning in protein structure determination. Journal of the
Italian Association for Artificial Intelligence, v. 15, n. 2, p. 22–30, 2002.
ZHANG, J.; SANDERSON, A. C. Adaptative Differential Evolution, A Robust Approach
to Multimodal Problem Optimization. 1. ed. Berlin: Springer-Verlag, 2009.
ZHARG, X.; LI, T. Improved particle swarm optimization algorithm for 2D protein folding
prediction. Science And Technology, p. 53–56, 2007.
110
111
ANEXO A -- AMINOÁCIDOS PROTEINOGÊNICOS
Tabela 38: Lista dos Aminoácidos Proteinogênicos
Nome
Alanina
Arginina
Asparagina
Aspartato (Ácido Aspártico)
Cisteína
Glutamina
Glutamato (Ácido Glutânico )
Glicina
Histidina
Isoleucina
Leucina
Lisina
Metionina
Fenilalanina
Prolina
Serina
Treonina
Triptofano
Tirosina
Valina
Abreviações
Fórmula Química
Ala
A
CH3 -CH(NH2 )-COOH
Arg
R
HN=C(NH2 )-NH-CH2 -CH2 -CH2 -CH(NH2 )-COOH
Asn
N
NH2 -CO-CH2 -CH(NH2 )-COOH
Asp
D
HCOO-CH2 -CH(NH2 )-COOH
Cys
C
SH-CH2 -CH(NH2 )-COOH
Gln
Q
NH2 -CO-CH2 -CH2 -CH(NH2 )-COOH
Glu
E
HCOO-CH2 -CH2 -CH(NH2 )-COOH
Gly
G
H-CH(NH2 )-COOH
Hys
H
H-(C3 H2 N2 )-CH2 -CH(NH2 )-COOH
IIe
I
CH3 -CH2 -CH(CH3 )-CH(NH2 )-COOH
Leu
L
CH3 -CH2 -CH2 -CH(NH2 )-COOH
Lys
K
NH2 -CH2 -CH2 -CH2 -CH2 -CH(NH2 )-COOH
Met
M
CH3 -S-CH2 -CH2 -CH(NH2 )-COOH
Phe
F
C6 H5 -CH2 -CH(NH2 )-COOH
Pro
P
CH2 -CH2 -CH2 Ser
S
OH-CH2 -CH(NH2 )-COOH
Thr
T
OH-CH(CH3 )-CH(NH2 )-COOH
Trp
W
Ph-NH-CH=C-CH2 -CH(NH2 )-COOH
Tyr
Y
OH-C6 H2 -CH2 -CH(NH2 )-COOH
Val
V
CH3 -CH(CH3 )-CH(NH2 )-COOH
112
113
ANEXO B -- CLASSIFICAÇÃO DOS AMINOÁCIDOS SEGUNDO O MODELO HP
Os aminoácidos classificados segundo o modelo HP se diferem em dois grupos: i) hidrofóbicos (não-polares) ii) hidrofílicos (polares). Os hidrofílicos podem ser carregados positivamente,
negativamente ou eletricamente neutros. Na tabela 39 está a classificação dos aminoácidos para
o modelo HP, baseada na hidrofobicidade. Essa tabela apresenta os nomes dos aminoácidos
com seus respectivos símbolos e tipos de cadeia lateral. Essa classificação é a mesma utilizada
no modelo AB estudado nesse trabalho.
Tabela 39: Classificação dos Aminoácidos segundo critérios de hidrofobicidade
Hidrofílicos ou Polares
Hidrofóbicos ou não-Polares
Aminoácidos
Cadeia Lateral
Aminoácidos
Cadeia Lateral
Ácido Aspártico Asp D negativa
Alanina
Ala A não-polar
Ácido Glutânico Glu E negativa
Glicina
Gly G não-polar
Arginina
Arg R
positiva
Valina
Val V não-polar
Leucina
Leu L não-polar
Lisina
Lys K
positiva
Histidina
Hys H
positiva
Isoleucina
IIe
I não-polar
Asparagina
Asn N
neutra
Prolina
Pro P não-polar
Fenilalanina Phe F não-polar
Glutamina
Gln Q
neutra
Serina
Ser S
neutra
Metionina Met M não-polar
Treonina
Thr T
neutra
Triptofano Trp W não-polar
Cisteína
Cys C não-polar
Tirosina
Tyr Y
neutra
114
115
ANEXO C -- ÂNGULOS E CONFORMAÇÕES DOS DOBRAMENTOS OBTIDOS
COM OS ALGORITMOS DE ED
Abaixo estão representadas as melhores conformações obtidas com os algoritmos de ED e
de ED auto-adaptável para as quatro sequências de bechmarks, para ambos modelos 2D e 3D.
As tabelas mostram os valores dos ângulos obtidos, enquanto as figuras mostram a conformação
final de cada uma das sequências. Nas figuras, os pontos escuros representam aminoácidos A
(hidrofóbicos) e os pontos claros, aminoácidos B (polares).
Tabela 40: Ângulos das melhores conformações obtidos pelo algoritmo de ED para as sequências
de benchmark modelo 2D
N
13
21
34
55
Ângulos Rotação
0,122017 -0,914766 -1,81045 -0,353436 -0,344119 0,48482 -1,94469 1,43178
-1,50568 -1,49304 1,74276
-0,513719 1,93782 -0,470109 0,364129 -0,188574 -1,65951 1,94368
-0,447273 0,355614 0,312402 1,80768 0,949635 -0,114519 -1,66464 1,94321
-0,437428 0,34786 -0,22524 -1,27756
0,38143 0,198268 1,87574 0,93681 -0,0844027 -0,608405 1,9266 -1,61565
-0,124181 0,933801 1,80756 0,316939 0,381818 -1,52466 1,9248 -0,528318
0,434005 -0,0602672 -0,408558 1,91518 -1,82404 -0,351419 0,357028 -0,991077
1,49238 1,49833 -0,373208 1,94005 -0,561751 -0,193915 0,429822 -1,74624
0,507596 -1,93559 0,495796 -0,195393 0,00919731 -0,460302 -1,93595 -0,605413
-0,13802 0,0350926 1,21499 -0,218141 -0,136593 -0,795126 1,93269 -0,37845
-0,189439 0,425147 -0,369901 1,98102 -1,77117 0,285025 0,480342 1,87415
0,722447 -0,0883586 -0,406094 1,9296 -1,72084 -0,687359 -0,564666 -1,65983
0,207056 -0,225586 -0,432022 1,93581 -1,43796 1,51219 1,48044 -0,346011
1,95054 -1,64149 0,375751 -0,141113 -1,32367 -0,1537 0,323599 0,54637
1,93108 0,474092 -0,0747476 0,23514 -1,72481
116
Figura 40: Melhor conformação obtida com o algoritmo ED no modelo 2D para a sequência de 13
aminoácidos, E=-3,1999
Figura 41: Melhor conformação obtida com o algoritmo ED no modelo 2D para a sequência de 21
aminoácidos, E=-6,1980
117
Figura 42: Melhor conformação obtida com o algoritmo ED no modelo 2D para a sequência de 34
aminoácidos, E=-10,4699
Figura 43: Melhor conformação obtida com o algoritmo ED no modelo 2D para a sequência de 55
aminoácidos, E=-13,5205
118
Tabela 41: Ângulos das melhores conformações obtidos pelo algoritmo de ED auto adaptável para
as sequências de benchmark modelo 2D
N
13
21
34
55
Ângulos Rotação
0,122017 -0,914766 -1,81045 -0,353436 -0,344119 0,48482 -1,94469 1,43178
-1,50568 -1,49304 1,74276
-0,513719 1,93782 -0,470109 0,364129 -0,188574 -1,65951 1,94368 -0,447273
0,355614 0,312402 1,80768 0,949635 -0,114519 -1,66464 1,94321
-0,437428 0,34786 -0,22524 -1,27756
0,32797 -1,0496 -1,78711 -0,29727 -0,454621 0,519674 -1,94551 1,57323
-0,400051 -0,31462 -1,80743 -0,933713 0,120407 1,64561 -1,94604 0,58293
0,603734 0,91993 0,56594 -1,94278 1,66127 0,115137 -0,950209 -1,80733
-0,312423 -0,35536 0,446404 -1,94426 1,58443 -0,522261 0,982266 1,80636
0,539419 -1,93898 0,477557 -0,332417 0,188609 -0,493116 -1,93431 -0,554134
-0,367632 0,212594 1,19716 -0,423748 0,308328 0,566289 -1,93495 0,418303
-0,307985 0,282036 0,468932 -1,94279 1,60381 -0,393552 -0,352106 -1,81005
-0,908282 0,154715 0,503552 -1,93604 1,63186 -0,435861 0,129278 1,61347
0,140351 0,384945 -0,366427 1,9222 -0,473356 -0,107455 0,275248 -1,60604
1,93916 -0,427302 0,371343 0,279359 1,80303 0,978709 -0,106878 -1,71316
1,95144 -0,455306 -0,171136 0,289578 -1,44159
Figura 44: Melhor conformação obtida com o algoritmo ED auto-adaptável no modelo 2D para a
sequência de 13 aminoácidos, E=-3,1999
119
Figura 45: Melhor conformação obtida com o algoritmo ED auto-adaptável no modelo 2D para a
sequência de 21 aminoácidos, E=-6,1980
Figura 46: Melhor conformação obtida com o algoritmo ED auto-adaptável no modelo 2D para a
sequência de 34 aminoácidos, E=-10,5565
120
Figura 47: Melhor conformação obtida com o algoritmo ED auto-adaptável no modelo 2D para a
sequência de 55 aminoácidos, E=-17,3133
121
Tabela 42: Ângulos das melhores conformações obtidos pelo algoritmo de ED para as sequências
de benchmark modelo 3D
N
13
Ângulos Rotação
-2,00123 2,00765 -1,98636-1,99378
2,01299 -1,97374 1,98693 1,98266
2,00189 2,01195 -1,97379
21
1,98575 1,97845 -2,00002 1,99323
-1,99637 -1,9803 -1,97587 1,94608
-2,0171 -1,99616 -1,98306 -1,97794
-2,03801 -1,96848 -2,01559 -2,00059
2,01336 2,00491 -1,99626
34 -1,99448 -1,98524 -1,9854 -2,01118
-2,01936 1,99573 -2,01141 1,9435
-2,03489 -1,96376 -1,97012 -1,85652
-1,43552 1,85437 1,99602 1,97912
-2,01816 2,01679 -1,90312 1,98988
-1,66606 -1,98329 -1,93786 -1,97519
-1,98847 2,03079 1,93586 -1,99663
1,95305 -1,99301 2,03305 1,97195
55 -1,96772 -1,99782 -2,00809 -1,80802
1,89257 -2,00261 -1,27132 -2,0067
2,02023 -2,00177 -0,100961 2,02545
2,00735 -2,0424 -2,0272 -1,97513
-2,02461 1,97497 -1,99129 -1,90771
1,30344 -2,04587 -2,04237 -1,99199
1,6869 -1,93945 -2,01598 1,96446
-1,94537 -1,31763 0,962717 2,00075
-2,01768 1,46133 2,03557 -1,97099
1,99542 1,23559 1,96906 1,73982
-1,93011 -1,94589 2,04905 -0,983467
-1,92158 -1,97696 -1,82371 1,94291
-1,97653 -1,95804 -1,9875 1,26241
1,98634
Ângulos Torção
1,36669 -0,282944 -0,773825 -1,3366
0.310387 -2,50039 2,44781 -2,30847
1,34701 -0,351089
2,34812 1,90117 2,78461 -1,78231
-1,91027 0,725324 2,38625 -0,311267
-1,74888 0,805067 0,287563 1,87578
0,505217 -0,487918 0,847716 2,3632
-1,40963 -0,698326
0,659667 0,366816 0,757538 -1,77884
-2,41058 -0,800736 1,27411 0,70949
1,36973 -0,139073 0,965118 -2,46838
2,02433 2,52307 2,5358 0,283245
1,36113 0,128902 1,7737 -0,717571
-1,16045 -1,90947 -0,31724 -0,789957
-1,30806 -1,61932 0,649807 2,42592
0,853239 1,33558 1,73229
-0,965375 -0,391889 -0,70876 1,07572
0,91171 -2,43633 -0,762454 -2,14886
-0,95519 0,829047 1,24451 -1,26873
1,04151 1,08789 0,476364 0,341006
-1,24064 -0,700732 0,800597 -0,529443
-0,380349 1,53729 0,409255 0,986876
-0,303404 0,211757 -1,36141 -0,848497
-0,166333 1,8681 0,0555848 1,11191
0,945761 0,888932 0,128948 -1,553
-0,594271 1,78909 1,11493 -0,083178
-1,87874 -2,33818 0,0661203 0,42853
0,875875 1,95914 -1,09932 0,315737
0,816258 -0,694396 -0,698457 -0,592489
122
Figura 48: Melhor conformação obtida com o algoritmo ED no modelo 3D para a sequência de 13
aminoácidos, E=-26,4714
Figura 49: Melhor conformação obtida com o algoritmo ED no modelo 3D para a sequência de 21
aminoácidos, E=-48,56454
123
Figura 50: Melhor conformação obtida com o algoritmo ED no modelo 3D para a sequência de 34
aminoácidos, E=-89,7957
Figura 51: Melhor conformação obtida com o algoritmo ED no modelo 3D para a sequência de 55
aminoácidos, E=-149,5675
124
Tabela 43: Ângulos das melhores conformações obtidos pelo algoritmo de ED auto adaptável para
as sequências de benchmark modelo 3D
N
13
21
34
55
Ângulos Rotação
Ângulos Torção
-1,99143 -2,005 -1,97603 -1,9807
1,78119 0,298257 0,776777 1,30329
2,00377 -1,97429 -1,9888 -1,9702
-0,301256 -0,624863 0,684238 -0,839701
-1,98978 2,00129 -1,9735
-1,31482 0,33129
1,99892 -1,94986 -1,99889 1,94
-0,166749 -0,770594 -2,47219 1,2118
1,97249 -1,92599 1,99815 -1,97419
-0,785949 -2,41279 -0,724433 1,87664
-1,99946 1,971 -2,0049 -1,99216
-1,27958 0,36364 0,758153 -1,83949
-2,03131 -1,97847 1,9984 -1,99716
-0,326818 1,3182 -0,762685 0,779714
-1,9953 -2,01789 1,95972
-1,79978 2,80055
-1,99452 -1,94774 -2,03742 -1,97885 0,616526 -0,718699 1,89793 -0,805534
-1,96961 -2,01747 2,00311 -1,99638
-0,402477 -3,03226 0,852622 -1,25059
-2,0382 -1,98622 2,00311 -2,01286
-1,86174 3,14159 -1,35784 -1,95263
1,98287 -1,97822 -1,99242 -2,00419
-0,116714 -1,11322 -1,08583 0,230638
-2,01933 -2,02346 2,01035 2,00994
-1,84929 3,14159 2,28096 -0,616166
-1,99206 2,0176 2,0026 1,99292
2,9025 1,39871 -2,38599 0,250757
-1,98218 -2,03364 -1,99623 -2,00677
1,84932 -0,750339 0,688876 -2,38119
1,95164 -2,00821 -1,96693 1,96671
0,285461 1,77725 3,14159
-1,9353 -2,01484 -1,93479 -1,76554
-0,426657 1,01283 -2,01934 -2,31972
-1,6719 -2,00534 2,02485 2,00181
0,155924 2,46195 2,44422 -0,838596
-2,03391 -1,99634 -2,00954 1,97425
1,80121 -0,726186 2,72816 -0,698718
-1,94583 -1,86281 -1,98874 -1,99484
-1,99189 0,45417 -1,02039 0,270971
-1,96887 -1,79232 -2,01006 -1,8935
-2,01503 0,628091 -0,836068 2,88962
1,99889 -1,98243 2,01795 1,97939
1,12262 1,75339 2,5499 -1,10489
-2,01299 -1,99801 -2,03232 -1,98219
-1,81161 0,121992 -0,158619 2,91486
1,98135 -1,57221 1,86087 1,81848
-2,22855 0,895319 -1,85259 2,08569
-1,9266 2,0292 -1,98253 2,01181
2,28768 0,449114 2,28098 -0,319789
-1,95861 -1,85786 1,53446 1,97218
-1,96337 0,854306 0,930333 -2,18817
2,00677 -1,95164 2,04466 1,91078
-0,379786 -2,16533 -2,26489 -1,76559
-2,02412 -1,95651 -2,00744 -2,02484
-0,7921 0,770109 -1,76985 -0,283059
-1,96201 2,00087 -1,90065 2,0408
1,27518 0,209587 -3,10004 -0,146001
-2,00268
125
Figura 52: Melhor conformação obtida com o algoritmo ED auto-adaptável no modelo 3D para a
sequência de 13 aminoácidos, E=-26,507
Figura 53: Melhor conformação obtida com o algoritmo ED auto-adaptável no modelo 3D para a
sequência de 21 aminoácidos, E=-50,3613
126
Figura 54: Melhor conformação obtida com o algoritmo ED auto-adaptável no modelo 3D para a
sequência de 34 aminoácidos, E=-92,0962
Figura 55: Melhor conformação obtida com o algoritmo ED auto-adaptável no modelo 3D para a
sequência de 55 aminoácidos, E=-157,112
Download