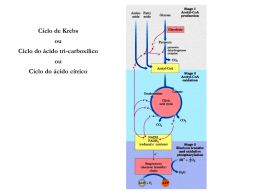
SIMULAÇÃO COMPUTACIONAL DOS COMPOSTOS DE COORDENAÇÃO FORMADOS DURANTE O PROCESSO PECHINI: CITRATOS DE ZINCO E COBALTO Regineide de Oliveira Lima, Ótom Anselmo de Oliveira, Robson Fernandes de Farias* Departamento de Química, Universidade Federal do Rio Grande do Norte [email protected] INTRODUÇÃO O método Pechini, também chamado método dos precursores poliméricos para a preparação de cerâmicas, envolve a capacidade que alguns ácidos orgânicos (e.g. ácido cítrico) α-hidroxicarboxílicos possuem para formação de quelatos com a maioria dos cátions. Quando um álcool polihídrico é adicionado aos quelatos, sob aquecimento e agitação, ocorre a formação de um éster, devido à condensação entre o álcool e o quelato ácido. O polímero formado apresenta grande homogeneidade na dispersão dos íons metálicos e um tratamento térmico adequado é realizado para a eliminação da parte orgânica e obtenção da fase cerâmica desejada. Largamente difundido em química de materiais, o método Pechini é empregado para a síntese de uma grande variedade de compostos, desde pigmentos cerâmicos nanométricos e materiais ferroelétricos, a pós cerâmicos para fins eletrocatalíticos [1,2]. Contudo verifica-se, analisando-se as centenas de trabalhos publicados todos os anos na literatura, que não existe, efetivamente, um estudo sistemático, capaz de correlacionar os conhecimentos básicos sobre a química de coordenação dos diferentes metais de transição empregados, com as propriedades finais dos pós cerâmicos obtidos. Tendo-se em vista os múltiplos usos e aplicações da química teórica e da modelagem molecular [3], no presente trabalho utiliza-se o programa Spartan [4] para a modelagem de compostos (citratos, mais especificamente), de zinco e cobalto, a fim de estabelecer-se quais as estruturas dos compostos e quais os sítios de coordenação empregados pelo ligante (ácido cítrico). PROCEDIMENTOS Tendo-se em vista o maior tempo computacional requeridos pelos métodos Hartree-Fock e os métodos baseados na Teoria do Funcional da Densidade (esses últimos, usualmente mais eficazes na modelagem de compostos de coordenação), e tendo-se ainda em vista que o objetivo da parte inicial desse estudo não é obter valores acurados para as entalpias de formação dos compostos mas, em uma dada série de compostos possíveis, estabelecer, por comparação, qual o termodinamicamente mais estável (ou seja, qual o que exibe entalpia de formação mais exotérmica), foi empregado, primeiramente, um método semi-empírico (AM1) em função do menor tempo computacional requerido. Foram modelados, ao todo, 4 compostos, sendo 2 de zinco (II) e 2 de cobalto (II), considerando-se, em cada caso, diferentes estereoquímicas de coordenação do íon citrato ou do ácido cítrico (comportando-se como ligante bi e/ou monodentado). Para os compostos de zinco assumiu-se uma geometria tetraédrica, e para os compostos de cobalto uma geometria octaédrica, tendo-se em vista que são essas as geometrias mais comuns para compostos de coordenação envolvendo esses cátions [5]. Para os dois compostos de Zn(II), foi assumida uma carga neutra, enquanto para os compostos de Co(II) assumiu-se carga neutra ou -1. Os sítios de coordenação considerados foram a carbonila (carbonilas ligadas aos carbonos 1 ou 2) e o grupo O- a ela ligado (após desprotonação de um dos grupos OH). RESULTADOS E DISCUSSÕES Os resultados obtidos encontram-se resumidos na Tabela 1. Tabela 1. Dados resultantes da modelagem para compostos de Zn(II) e Co(II) com ácido cítrico. Citrat. = íon citrato (-1). Acitr. = ácido cítrico. Ângulo de ligação O-Zn-O Momento dipolar (D) ∆Hf (kJ mol-1) Tetraedro distorcido 60,3 e 58,5 3,02 -2.397,6 0,75 Tetraedro distorcido 59,7 e 59,7 3,64 -2.368,8 0 - Piramidal 42,1 e 69,5 - -370,3 -1 - Piramidal* 42,4; 65,1 e 65,8 - 242,4 Carga Geometria de Millikan para o cátion metálico Composto Carga Zn(Citrat.)2 0 0.76 Zn(Citrat.)2# 0 Co(Citrat.)2(Acitr.)2 Co(Citrat.)3 # Para esse composto, as ligações são feitas via grupo COO- do carbono 2, e não do carbono 1. *Geometria em tono do Co(II). Comprando-se os dados resumidos na Tabela 1, podemos constatar que o composto de Zn com dois íons citrato com entalpia de formação mais exotérmica é aquele que envolve ligaçõess via grupo COO- do carbono 1, em comparação com aquele que forma ligações via grupo COO- do carbono 2. Contudo, o momento de dipolo para o primeiro composto é cerca de 21% maior. Assim, podemos inferir que no caso da formação de composto neutro, com relação cátion:citrato igual a 1:2, a ligação dar-se-á, preferencialmente, via grupo COO- do carbono 1, sendo que o composto formado por ligação via carbonila do carbono 2, sendo mais polar, exibirá, possivelmente, forças atrativas intermoleculares mais altas. . Para os compostos de cobalto, verifica-se que o composto formado com três íons citrato exibe uma entalpia de formação endotérmica, enquanto o composto formado com dois íons citrato e duas moléculas de ácido cítrico, exibe uma entalpia de formação exotérmica. Logo, devemos esperar a formação do segundo, e a não formação do primeiro, ou sua formação em quantidade apreciavelmente menor. Figura 1. Estrutura para o composto Zn(Citrat.)2. Uma vez que, no método Pechini, adiciona-se primeiramente o precursor (nitrato) de um dos metais (geralmente, a proporção cátions metálicos:ácido cítrico é de 1:3), para em seguida adicionar-se o precursor do outro metal, podemos inferir que, no sistema destinado à obtenção de um pó cerâmico misto Zn-Co, os compostos termodinamicamente mais estáveis serão os citratos de Zn(II). Caso a adição do precursor de cobalto seja efetuada antes da adição do precursor de zinco, é possível supor-se que, após a adição do nitrato de Zn, os íons citratos já eventualmente coordenador ao cobalto, irão romper suas ligações, a fim de coordenar-se ao zinco, possivelmente com a formação de ligações em ponte (citratos mistos, Zn-Co, portanto). Contudo, caso a adição do precursor de cobalto seja efetuada após a adição do nitrato de zinco, não deve-se esperar um tal fenômeno, de forma que, em vez de citratos mistos, tenhamos, isso sim, uma mistura de citratos de Zn(II) e Co(II). Pode-se ainda supor que, esses dois possíveis “cenários”, tenham influência sobre as propriedades finais do pó cerâmico obtido. REFERÊNCIAS [1] J.T. Son, K.S. Park, H.G. Kim, H.T. Chung, J. Mater. Sic., 39 (2004) 3635. [2] Chr. Arginusis, T. Damjanovic, O. Schneider, J. Mater Chem., 41 (2006) 8059. [3] C.H. Morgon, K. Coutinho (Eds.), Métodos de química teórica e modelagem molecular, Livraria da Física, São Paulo, 2007. [4] SPARTAN, Wavefunction Inc., 18401 Von Karman Avenue, Suite 370, Irvine, CA 92612 USA. [5] N.N. Greenwood, A. Earnshaw, Chemistry of the Elements, Butterworth-Heinemann, Cambridge, 1984.
Baixar