☰
Explorar
Assinar em
Inscrever-se
Envio
×
Baixar
Sem categoria
Ferreira (2014) - observatorio.faculdadeguanambi.edu.br.
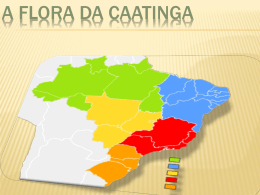
A FLORA DA CAATINGA – Luis Henrique
Caatinga - Colégio Rio Branco
Jair da Silva Mello
Caatinga - Unicamp
análise sensorial de alimentos - Universidade Castelo Branco
O DOMÍNIO DAS CAATINGAS
A seca no NORDESTE. - Ensinando a Viver | Pirlimpimpim Baby
"Laboratório de Triagem de Amostras" (Dra. Vera Gonçalves, CDA-SP)
Slide 1 - Biodados
Webquest Phabricia (1778131) - Educar e Crescer