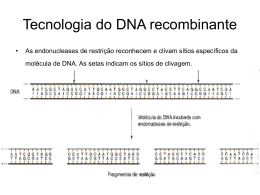
WISE2 Algoritmos inteligentes para busca em DNA http://www.sanger.ac.uk/Software/Wise2/ O que é o WISE2 ? Wise2 é um pacote orientado à comparação de seqüências de DNA ao nível de tradução, desprezando erros de seqüenciamento e Introns. Wise2 é um pacote orientado à comparação de biopolímeros, comumente seqüências de DNA e proteínas. Tradução Processo pelo qual a informação genética ( que é estocada na seqüência de nucleotídeos numa molécula de mRNA) é traduzida em uma seqüência de aminoácidos Intron Exon-GU..............................................AG-Exon DNA Exon 1 Intron A Exon 2 Intron B Exon 3 transcrição Transcrito Primário: 5’ Exon 1 Intron A Exon 2 Códon n Intron B Códon n+1 Exon 3 3’ --------------------- Processamento do RNA mRNA Exon 1 Exon 2 Exon 3 tradução Polipeptídeo -- Concorrentes: •BLAST package ( NCBI) Sequence Searching •Fasta package (Bill Pearson) Sequence Searching •SAM package (UC Santa Cruz) HMM •HMMER package (Sean Eddy) HMM Os HMMER e Pfam podem ser classificados como parceiros !! Quais as vantagens do WISE2? •O ponto forte do Wise2 é a comparação de seqüência de DNA a nível de sua tradução protéica . •Implementação dos algoritmos mais robusta: Manuseio de grandes pedaços de DNA sem estouro de memória; •Integração tecnológica faz do WISE2 o parceiro ideal para o HMMER e Pfam; •Design permite reutilização e alteração do código. Desvantagens • O algoritmo GENEWISE não tenta predizer um gene inteiro, mas regiões que apresentam homologia com a proteína. - porém é confiável! •Velocidade (Maior preocupação com a algoritmos corretos que com velocidade) Opções -u -v ( início e final de cadeia de DNA a ser comparada) Scripts em Pearl - Blastwise e Halfwise • Até o momento dispõe somente de arquivos de freqüencia de genes em Humanos e Vermes. • Uso de memória Linear com perda de desempenho quando uso de matrizes ultrapassam 20 MB Modos 4 programas principais executáveis: • Genewise Cadeia proteica vs Seqüência simples de DNA • Genewisedb Banco de dados de proteínas vs banco de dados de seqüências de DNA. • Estwise Cadeia proteica vs Seqüência simples de cDNA/EST • Estwisedb Banco de dados de proteínas vs banco de dados de seqüências de cDNA/EST. OPÇÕES -u Posição inicial no DNA -v Posição final no DNA -trev Comparação do reverso -tfor (default) Comparação standard -both Comparação nos dois sentidos -s Posição inicial na proteína - não aplicável a HMM -t Posição final na proteína - não aplicável a HMM -gap [no] default [12] gap penalty -ext [no] default [2] extension penalty -matrix default [blosum62.bla] Matriz de Comparação. Estima a probabilidade de comparações de aminoácidos -hmmer especifica que o modelo proteico é do tipo HMM -hname Nomeia o HMM -init DEFAULT GENEWISE genewise protein.pep cosmid.dna compara uma seqüência proteica a uma de DNA genewise -hmmer pkinase.hmm cosmid.dna compara uma seqüência proteica ( HMM) a uma de DNA . GENEWISEdb genewisedb protein.pep human.fa compara uma seqüência proteica a um banco de DNA genewisedb -hmmer pkinase.hmm human.fa compara uma seqüência proteica (HMM) a um banco de DNA genewisedb -prodb protein.pep -dnas cosmid.dna compara um banco de seqüências proteicas a uma seqüência de DNA genewisedb -pfam Pfam -dnas cosmid.dna compara um banco de seqüências proteicas (HMM) a uma seqüência de DNA genewisedb -prodb protein.pep human.fa compara um banco de seqüências proteicas a um de seqüências de DNA genewisedb -pfam Pfam human.fa compara um banco de seqüências proteicas (HMM) a uma seqüência proteica ESTWISE estwise protein.pep singleest.fa compara uma seqüência proteica a uma de DNA estwise -hmmer pkinase.hmm singleest.fa compara uma seqüência proteica (HMM) a uma de DNA ESTWISEdb estwisedb protein.pep est.fa compara uma seqüência proteica a um banco de DNA estwisedb -hmmer pkinase.hmm est.fa compara uma seqüência proteica (HMM) a um banco de DNA estwisedb -prodb protein.pep -dnas singleest.fa compara um banco de seqüências proteicas a uma de DNA estwisedb -pfam Pfam -dnas singleest.fa compara um banco de seqüências proteicas (HMM) a uma de DNA estwisedb -prodb protein.pep est.fa compara um banco de seqüências proteicas a um banco de DNA Genewise example •Usage •genewise <protein-file> <dna-file> in fasta format Options. In any order, '-' as filename (for any input) means stdin Dna [-u,-v,-trev,-tabs,-fembl,-both] Protein [-s,-t,-g,-e,-m] HMM [-hmmer,-hname] Model [-codon,-gene,-cfreq,-splice,-subs,-indel,-intron,-null] Alg [-kbyte,-alg] Output [-pretty,-genes,-para,-sum,-cdna,-trans,-ace,-embl,-diana] ..cont [-gff,-gener,-alb,-pal,-block,-divide] Standard [-help,-version,-silent,-quiet,-errorlog] Query protein: roa1_drome Comp Matrix: blosum62.bla Gap open: 12 Gap extension: 2 Start/End local Target Sequence HSHNRNPA Strand: forward Gene Paras: human.gf Codon Table: codon.table Subs error: 1e-05 Indel error: 1e-05 Model splice? model Model codon bias? flat Model intron bias? tied Null model syn Algorithm 623 Find start end points: [25,1387][346,3962] Score 87719 Recovering alignment: Alignment recoveredExplicit read offone 94% genewise output Score 253.10 bits over entire alignment Scores as bits over a synchronous coding model Genewisedb genewisedb road.pep hngen.fa Ex 1 Warning: The bits scores is not probablistically correct for single seqs See WWW help for more info roa1_drome HSHNRNPA 187 LNGKMVDVKKALPKQNDQQGGGGGR +NG +V+KAL KQ R VNGHNCEVRKALSKQEMASASSSQR G:G[ggt] 2405 gagcatggaagctacgagagttacaGGTATGCT Intron 4 tagaagatgactcaaatcgcccgag <1-----[2481 : 2793] gtccctataacgagaggtttaccaa genewise fly.pep human.genomic > genewise.out fly.pep >ROA1_DROME P07909 HETEROGENEOUS NUCLEAR RIBONUCLEOPROTEIN A1 (HNRNP CORE PROTEIN A1-A) (PEN REPEAT CLONE P9). MVNSNQNQNGNSNGHDDDFPQDSITEPEHMRKLFIGGLDYRTTDENLKAHFEKWGNIVDVVV MKDPRTKRSRGFGFITYSHSSMIDEAQKSRPHKIDGRVVEPKRAVPRQDIDSPNAGATVKKLFV GALKDDHDEQSIRDYFQHFGNIVDINIVIDKETGKKRGFAFVEFDDYDPVDKVVLQKQHQLNG KMVDVKKALPKQNDQQGGGGGRGGPGGRAGGNRGNMGGGNYGNQNGGGNWNNGGNNW GNNRGGNDNWGNNSFGGGGGGGGGYGGGNNSWGNNNPWDNGNGGGNFGGGGNNWNNGG NDFGGYQQNYGGGPQRGGGNFNNNRMQPYQGGGGFKAGGGNQGNYGGNNQGFNNGGNNR RY genewise fly.pep human.genomic > genewise.out >HSHNRNP human.genomic ACGCAAAGCTAGGACAAACTCCCGCCAACACGCAGGCGCCGTAGGTTCACTGCCTACTCCTGCCCGCCATTTCACGTGTTCTCAGAGGCAGGTGGAACTTCTTAATGC GCCTGCGCAAAACTCGCCATTTTACTACACGTGCGGTCAACAAGAGTTCATTGCAAAAAAATTGTTACCTCCTAGCTGCTTGTCTAATACATAGTGTTAATCATGCTTT GCCAAGCGACTTGACTGTAATATTTGCGCGTGGAAGATTAAAAAGATGTTAAACACCCAAGGTAGATTCAAATGTGAATGATTGGTCGGTTGGCCAATCAGACTGGTT AACAATAACATTACTCGGGAACCAATGGACTCCAAGGGGTGGAGACGGCGTAGAACGACCGAAGGAATGACGTTACACAGCAATGTGGCACCACAGGCCAATAGCAG GGGGAAGCGATTTCAAGTATCCAATCAGAGCTGTTCTAGGGCGGAGTCTACCAATGCCGAAAGCGAGGAGGCGGGGTAAAAAAGAGAGGGCGAAGGTAGGCTGGCA GATACGTTCGTCAGCTTGCTCCTTTCTGCCCGTGGACGCCGCCGAAGAAGCATCGTTAAAGTCTCTCTTCACCCTGCCGTCATGTCTAAGTCAGAGGTGAGTTAGGCG CGCTTTCCCACTTGAATTTTTTCCTCTCCCTTTCCTGAATCGGTAAGATGCTGCTGGGTTTCGTTCCTTGCACCAGCCCATTCTACAGTTCCTTCGGTCGCTGCCACGG CCTACCCCTCCCAAAGTTCAAGTCGCCATTTTGTCCTCTTGATCGCCATGAGGCCGCTCTCCGCCAACCATGTGTTATCATGCGGGACTCGTTACTCGTAGCAAAATTC TTAGGCACACAGGATCTTTGTCTTTTTTTAAACCTTGCCTTGGTGAGCGAGTTTTCTAAAGAGCGATTAGTCCCATTGTGGAGATGCACCCCTACCGCCCAAGCCTTTG TTGCGCGTGCGTCGGAAGGCGACTAGGGACGCATGCGCTTGCGATTTCCTAGCACTCCCAACTCCAGCATACGGCCTCCCTTGATAGGCAGAAGCACGTGTCTTGTTG CGACCTGAACGAACAATAAGTGCTAGGTACACAGTTGGTGTCTAGTTTTTCTTTTCCTCGATGGAAATTGTTTCGTGTTGTAGCCCATTTAACACTTCCCCCTCCCCCC ACTCTAGTCTCCTAAAGAGCCCGAACAGCTGAGGAAGCTCTTCATTGGAGGGTTGAGCTTTGAAACAACTGATGAGAGCCTGAGGAGCCATTTTGAGCAATGGGGAAC GCTCACGGACTGTGTGGTAAGATTTGGAAGGGACAAAGCAGTAAAACAGCCGATTTCCTTGGCTTATCTTGGTGCAGTCTTCTCCGAATGCTTATGAAAGTAGTTAAT AGCATTATAGTTAGAGCTTTGTTGGCAAAGGAACGTCCTGCTTTGATTTTAAAAGCTAACCTCTTAAATCTAAGGGTAGTGGGAAACTGGACGAACTTTTTATAAAAGG CTGGTGTAAAGTTTCCTATTGCCCTATTCAAAGTTAAAATAACAAAAGCTTTTGCGGTCAGACTTTGTGTTACATAAATTAACACTGTTCTCAGGTAATGAGAGATCCA AACACCAAGCGCTCTAGGGGCTTTGGGTTTGTCACATATGCCACTGTGGAGGAGGTGGATGCAGCTATGAATGCAAGGCCACACAAGGTGGATGGAAGAGTTGTGGA ACCAAAGAGAGCTGTCTCCAGAGAAGTGAGTGGGTTTTTTTTCTTCTTCTTCTTAAACTTACTTGGATATGTGCTGCTATGAACTTAAGATTCGGGAGTTTTCTAAACTT ACCAAAATTTTTTATTCGAGTATAGGCTTTGCTAATCTAAACCTATGGTTTTTCTCCTATTAGGATTCTCAAAGACCAGGTGCCCACTTAACTGTGAAAAAGATATTTGT TGGTGGCATTAAAGAAGACACTGAAGAACATCACCTAAGAGATTATTTTGAACAGTATGGAAAAATTGAAGTGATTGAAATCATGACTGACCGAGGCAGTGGCAAGAA AAGGGGCTTTGCCTTTGTAACCTTTGACGACCATGACTCCGTGGATAAGATTGTCAGTAAGTATCAGATAGTGGCATTTAGTAAGGGTTCCACAATCTGTATGGCATTC TAAACCCTGATACCATGTTGTATCTATGTTTTTTTTTTAGTTCAGAAATACCATACTGTGAATGGCCACAACTGTGAAGTTAGAAAAGCCCTGTCAAAGCAAGAGATGG CTAGTGCTTCATCCAGCCAAAGAGGTATGCTTGTTGCTTAATTAAACCTTAAAGGTAACTTTGAGTTACTCCAGTATGAATGATTTAATGCTTAAACTTCATGTCTTAAG GTCGAAGTGGTTCTGGAAACTTTGGTGGTGGTCGTGGAGGTGGTTTCGGTGGGAATGACAACTTCGGTCGTGGAGGAAACTTCAGTGGTCGTGGTATGTATGGTTTAT CTACATGTAGTTCTGACTTCTCACCATCTTTGCTATGAAGATTTTACAGTACGGGAACTGCATTCAGAATGTCACTTTAAGTCCAAGTCATACTTAAAACTTGAAACTTT TTCTTACAGGTGGCTTTGGTGGCAGCCGTGGTGGTGGTGGATATGGTGGCAGTGGGGATGGCTATAATGGATTTGGCAATGATGGTAAGTTTTTTAGGAATAAGTAGA GAAAAATTCCTGGCAACCTGGATCTTTAGAATAGGTTAGTAGAGACTAAAATTCTGGTGCATGTCAAACTCAACTTTGCCCATAACACGCATGCTGTGAGCAGGCCTTC AGCCGTTACACTTGCACAAGTTTTCATTGTCAAATACTTTTGTCTTATTGAGAAGAATTGTATTCTTGTAGGTGGTTATGGAGGAGGCGGCCCTGGTTACTCTGGAGGA AGCAGAGGCTATGGAAGTGGTGGACAGGGTTATGGAAACCAGGGCAGTGGCTATGGCGGGAGTGGCAGCTATGACAGCTATAACAACGGAGGCGGAGGCGGCTTTG GCGGTGGTAGTGGTAGGTATCCAGTGATCCAAGTACTTGGTGTGACAGCTAGATTAGCCTTTTAGAGCTTGGGTTCTGGTGCTGTTGAAGCATTGTGTGGTACACTGC ATGGTATATTAAAAACAAATGGGCTTGCTATGCTACCTCCTCCTAGCTTTAAGCTGGGGCCGCCTCACTCCCAAATAGTAGAGATAAGTGGATAGTGTTGTCTTTGAGT TAGATTAGTATCATAGAAGGATTTAGTATTTTAACTCCTTTGGGACCTTAGGCGCTTAGTTGATGTATCCAAGATACTTCTGCTTGCTGTGGCCCTGGATCCGTGAAGG CCTTCAAGGCTGAAGGGTATGCTTGTGCCACTCTGAAAATCTCTTTATTTTATGTCATGGTGAGTTAGGCCAGTTTTCTTTGTATTACTGGATTATTCAACTGAATGCCT TTCCCAGAGAATGAAATGCAAAGATTGGAGTCACCATAGTTTGGGAGAAAGGAAGGCTGATAACTCAACCTTATTTTATTCTGACTGCTAAACAGAATTGGAAACTAA CATCATCCTCAGGTAACAGATAAAGGCCCTCTTTCCCATTCATAGGAAGCAATTTTGGAGGTGGTGGAAGCTACAATGATTTTGGGAATTACAACAATCAGTCTTCAAA TTTTGGACCCATGAAGGGAGGAAATTTTGGAGGCAGAAGCTCTGGCCCCTATGGCGGTGGAGGCCAATACTTTGCAAAACCACGAAACCAAGGTATGGTATCTATGTA ATTTTGGATAATGTCAAAAGAGTGTCTGTAGCTACTGCTGGGAAGAAAGCCCTTTAACTGCTATGTCTGGGCAGCAAAACGTTTATAGTTTAGAACCTTCAGAAAGTGA TAATTTGATCACAAATTAGAAAAATCATGGGACCTCTTTACCACCTCCCTTGTAGTAGGGCCATTTTTAAATGGCCAGACACTTGAATTTAACTTTTATTATCCCAAATA TGAAAACATTACTGTTGGCACTTTGAAACTTTAAAAGAAAAATTGTACTTTTCAGGTGGCTATGGCGGTTCCAGCAGCAGCAGTAGCTATGGCAGTGGCAGAAGATTT TAATTAGGTAAGTAAGCACCTTTTTGTGTGTTGACATAATTTTTTAAATTGCTGATGAACCCAATAACCCTAATGTAGCTGAGCAGTGCAACATAGTTAACATTATAATT GCAGTAATTGTGGATATAAAGTTAATATTCAGATCAGCAAAATTTGTGGGAAACAAACTTGATATTGGATTGTAGCCTTGAGTCTTAATATGTTTAGATTAACAACTCT ATTCCATATTGTTCAACAGGAAACAAAGCTTAGCAGGAGAGGAGAGCCAGAGAAGTGACAGGGAAGCTACAGGTTACAACAGATTTGTGAACTCAGC Query protein: Comp Matrix: Gap open: Gap extension: Start/End Target Sequence Strand: Start/End (protein) Gene Paras: Codon Table: Subs error: Indel error: Model splice? Model codon bias? Model intron bias? Null model Algorithm ROA1_DROME blosum62.bla 12 2 default HSHNRNPA forward default human.gf codon.table 1e-05 1e-05 model flat tied syn 623 genewise.out genewise output Score 253.10 bits over entire alignment Scores as bits over a synchronous coding model Warning: The bits scores is not probablistically correct for single seqs ROA1_DROME HSHNRNPA ROA1_DROME HSHNRNPA ROA1_DROME HSHNRNPA ROA1_DROME HSHNRNPA 26 EPEHMRKLFIGGLDYRTTDENLKAHFEKWGNIVDVV EPE +RKLFIGGL + TTDE+L++HFE+WG + D V EPEQLRKLFIGGLSFETTDESLRSHFEQWGTLTDCV 1206 gcgccaactaggtatgaaggacaactgctgacagtg acaatgatttggtgtaccaagtggataaggctcagt gcaggggcctaggctaattgcggcttgagagcgctg 62 VMKDPRTKRSRGFGFITYSHSSMIDE VM+DP TKRSRGFGF+TY+ +D VMRDPNTKRSRGFGFVTYATVEEVDA 1314 GTAAGAT Intron 1 CAGgaagcaaactagtgtgatgagggggg <0-----[1314 : 1608]-0>ttgacacagcggtgttcacctaatac agataccgctgctgtcatctggggta 88 AQKSRPHKIDGRVVEPKRAVPRQ DID A +RPHK+DGRVVEPKRAV R+ D AMNARPHKVDGRVVEPKRAVSRE DSQ 1687 gaagaccagggagggcaaggtagGTGAGTG Intron 2 TAGgtc ctacgcaataggttacagctcga<0-----[1756 : 1903]-0>aca tgtagacggtaatgaagatccaa tta 114 SPNAGATVKKLFVGALKDDHDEQSIRDYFQHFGNIVDINIVIDKETGKK P A TVKK+FVG +K+D +E +RDYF+ +G I I I+ D+ +GKK RPGAHLTVKKIFVGGIKEDTEEHHLRDYFEQYGKIEVIEIMTDRGSGKK 1913 acggctagaaatgggaaggaggcccagttgctgaaggagaaagcgagaa gcgcatctaatttggtaaacaaaatgaataaagatattattcaggggaa aatccatgagatttctaactaatcaatttagtaatagtacgtcactcga Alignment 1 Score 35.31 (Bits) Ex 2 SEED EM:HS453C12 1 CAPNN-PCSNGGTCVNTPGGSSDNFGGYTCECPPGDYYLSYTGKRC CA++ C++ +CVN + +++C+C PG Y L+ + K C CAEGGHGCQH--QCVNAWA-------MFHCTCNPG-YKLAADNKSC 132851 tggggcgtcc ctgagtg atctatacg tacgggaaat gcaggaggaa agtacgc ttagcgacg aatccaaagg ttggattcgc atctcgc gccccccac CGAAATCGCT Alignment 2 Score 45.92 (Bits) SEED EM:HS453C12 1 CAPNN-PCSNGGTCVNTPGGSSDNFGGYTCECPPGDYYLSYTGKRC CA+++ C + CVN+PG +Y+C+C++G +L+ + + C CAEGTHGCEH--HCVNSPG-------SYFCHCQVG-FVLQQDQRSC 134919 tgggacgtgc ctgatcg ttttctcgg tgcccgcaat gcagcaggaa agtaccg catgagatg tttaaaaggg ttagctatgc ccctcac ctctccatc tacggcggcc tggcatggggcgcaggttctctatacaccccccgcccccggctgccaggctctgcggcctcaccttggaacta cagggcaagagcttcttccagggggtgaggttttcggtgcagaccacctcccgcggcagcacagcatagcgca gaaagtagtggtcagtgtctgagggagacagaggtctgtctggggtgggccttgggctctgacccctcgggat ccacattccagagatgggaatgaccctcctgctccccacaccacctctagcaccacagtctggacagtcccaa ctgggagtaggactcccttctctccttgggaaaaggcatgcagagatggcacagtattgggggcctgcacaca caggggacttaggatctagcccaggctgaggaagcaggaaactgagggaaaaggaggcaaaggtttgggcagg aggtaagaggaagaaggaaagggctgtaggggttatctcaccattggccagacccaggggtttgaaggaggca gtgggagtgactgtgttggtggagtcgatgaagttgagagaggcgcagaagatccctgagaggacattactga gctccttccaagatttatccacactggatagagacacaaatccactcactgtcctggggctacctctgctccc tctttcaaagtccacagctggctgctaaacctatgataggaggaggctgtattcttaactattagacgggcca gttgatggagctggaacattgctgcccccagccagcccacttgctgggtctcatcctactcagccccttcttc ctcactctcctctggacatctctgcatccccatgggtctctgctcaggtgattcttccttccttgcaagcctt tgctaacttctttctgcctaccttcatgatccggctccagtgcttacctcccctccacgaagcctttcctgcc ctccttaagcacagtctcctctgtgcagtcacggttctgaccatccaagcatcttactaggtccctcctggga gatggctaggtggcagcagcatcgtgtcctgaccaccttttctccctaactaggctgtaagcaacttgaggac aaggaccagtctgggtcatctatgtacttcccctgacaccatggaaagcgcctcatgtatcagagctgaaatg agctcactgatcttccttgaatgtgctgggctgggcaaaacaatgcatactaccctgtgtatactctgggaat aaaggtaagtcctgattctactatcatggtgagaagtcttatatccaaaaaagctcactgaacatgggaaaaa caactgttctaggatttcataaaaacatcaaattaaattaatgttcttttcttggagaaatatcaaaagagat ttgctctcagtaatagagaaagcataaaacttaataagcactagaaagaattctaagcatttgctccacattt caggcaattacgggctgagggaagacagtgacagcagagtagacaggaaagggtaggggagccagagttgagg caagagagaaagtcttggcaagctggggagttactgcttattccttattccttagtgttgtccaggagctttt gataattctatgttcagagcttttcaactgctccaatccttaagcctcaaataaaaatggcaaacttgaagcc ggaaagctctactcaaaccataaacatgcttcatttggtatgcacaacattgacccgcacagcactcaaaaaa tttttaaattacttgctgatatttgaatttgccaattttcacattaaattccagatttctggtatctcttgaa aaatgaggccaggtgtggtggctcttgcctgtaatcccaacactttgggaggctgaggcaggaggatCGCTtg aacccaggagttcgagaccagcctgggcaatatagtgagaccttgtttctacaaaaaatttttagaaacattt gactctgaccacattaggcctctattcccacatggcaacaatccatagaagctgagtggcagagctgtcctcg
Download