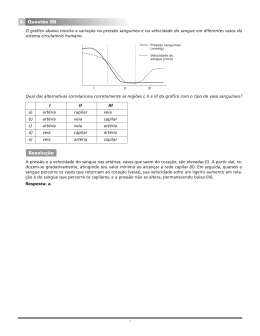
Electroforese Capilar A electroforese capilar é uma técnica separativa muito sensível, que foi desenvolvida com base nos conhecimentos adquiridos com a cromatogafia líquida de alta eficiência (HPLC). Embora esteja relacionada com a electroforese clássica em gel, difere desta de muitas formas. A electroforese capilar permite a separação de biomoléculas com uma eficiência muito superior à que é possível em HPLC, e permite também a quantificação de pequenas moléculas que não podem ser analisadas por electroforese em gel. Electroforese de zona A electroforese de zona,que inspirou o desenvolvimento da electroforese capilar, é uma técnica semi-manual largamente usada em bioquímica. Baseia-se na migração de espécies electricamente carregadas numa solução, sob o efeito de um campo eléctrico. Os catiões são atraídos para o cátodo (terminal negativo) e os aniões para o ânodo (terminal positivo). As espécies são suportadas num meio adequado, geralmente um material plástico coberto de uma substância porosa (gel) impregnada com um electrólito tampão. As extremidades deste sistema gel são colocadas em reservatórios independentes contendo o mesmo electrólito e ligadas por um circuito eléctrico a um gerador de corrente contínua (figura 1). Figura 1 - Princípio da electroforese de zona. A amostra é colocada na forma de uma banda transversal entre duas placas isoladoras. Os solutos migram de um extremo para o outro sob o efeito de vários factores: potencial aplicado (500 V ou mais, no caso de moléculas pequenas), carga e dimensões das moléculas, temperatura, forma e viscosidade do meio, num tempo que pode variar entre minutos ou horas. Cada composto na mistura é caracterisado pela sua mobilidade relativa, Rf. Ao contrário da cromatografia em camada fina (TLC) não é possível identificar uma frente do eluente, por isso é necessário um marcador interno para medir as distâncias relativas de migração. 1 Princípios da electroforese capilar A figura seguinte representa uma montagem típica em electroforese capilar. Neste exemplo, aplicase uma voltagem de ~30kV entre os extremos de uma solução contida num capilar de sílica fundida, de 50 cm de comprimento e diâmetro interno 25 a 75 µm. Figura 2 - Montagem para electroforese capilar. A amostra pode ser injectada colocando o capilar num tubo contendo a amostra e aplicando pressão (ou campo eléctrico) neste ou sucção à saída do capilar. Diferentes solutos terão mobilidades diferentes, e por isso migram através do capilar a velocidades diferentes. A técnica serve para analisar iões, mas pode ser modificada de modo a analisar também moléculas neutras. Este capilar pode ser introduzido dentro de uma célula viva, e analisar directamente o seu conteúdo. Ou então, se a célula for pequena, pode ser introduzida, inteira, dentro do capilar. (Figura 3) Figura 3 - Microfotografias mostrando (a) imagem fluorescente de uma única célula, intacta, imediatamente após a sua injecção no capilar; (b) resíduos celulares começando a separar-se, 10s após a aplicação da voltagem. (S. N. Krylov et al, Anal. Chem. 2000, 72, 872) 2 Vimos que, em cromatografia, há três causa para o alargamento dos picos cromatográficos: A - percursos múltiplos; B - difusão longitudinal; C - transferência de massa. A eficiência de uma coluna cromatográfica, medida pela altura equivalente de um prato teórico, H, varia com a velocidade do eluente, v, de acordo com a equação de van Deemter: H = A+ B +C ⋅v v (eq. 1) Usando uma coluna capilar, desaparece a contribuição dos percursos múltiplos (A=0), e reduzimos assim a altura equivalente de um prato teórico, H, melhorando a resolução Rs. Em electroforese capilar, desaparece também o termo C, pois não há transferência de massa entre fases. A única causa de alargamento dos picos é a difusão longitudinal. Isto faz com que a electroforese capilar seja uma técnica muito mais eficiente do que as técnicas cromatográficas. Enquanto a eficiência em HPLC varia tipicamente entre 5000 e 50 000 pratos teóricos, em electroforese capilar varia entre 50 000 a 500 000 pratos. (Figura 4) Figura 4 - Comparação entre as larguras dos picos na análise do álcool benzílico (C6H5CH2OH) em electroforese capilar e HPLC. (S. Fazio et al, Am. Biotech. Lab., Jan. 1990, p. 10) Mobilidade Electroforética Quando um ião de carga q colocado num campo eléctrico E (V/m) sofre uma força exercida: F=q.E (eq. 2) Em solução, a força de fricção do meio opõe-se ao movimento da carga: F = f . ve (eq. 3) onde ve é a velocidade do ião e f é o coeficiente de fricção. O ião atinge uma velocidade constante quando a força de aceleração iguala a força de fricção: q . E = f . ve (eq. 4) 3 Então, ve = ( q / f ). E = µe . E (eq. 5) µe é a mobilidade electroforética do ião. É a constante de proporcionalidade entre a velocidade do ião e a força do campo eléctrico. A mobilidade é directamente proporcional à carga do ião e inversamente proporcional ao coeficiente de fricção. Para moléculas de tamanho semelhante, a mobilidade aumenta com a carga. Para partículas esféricas de raio r movendo-se num fluido de viscosidade η, o coeficiente de fricção (f) é dado pela equação de Stokes: f=6πηr (eq. 6) A mobilidade é q/f, logo diminui quando o raio da partícula aumenta. A maior parte das moléculas não são esféricas, mas a eq. de Stokes define um raio hidrodinâmico da molécula com base na sua mobilidade observada, como se este fosse esférica. Electroosmose Acima de pH 2, o interior do capilar de sílica fundida está revestido de grupos silanol carregados - negativamente (Si-O ). Forma-se uma dupla camada eléctrica na superfície do capilar (figura 5). Figura 5 - (a) Dupla camada eléctrica formada pelas cargas negativas à superfície da sílica e por catiões adsorvidos. (b) A predominância de catiões na zona difusa da dupla camada produz um fluxo electroosmótico global no sentido do cátodo, quando se aplica um campo eléctrico. Uma camada de catiões imóveis adsorvidos à superfície neutraliza parcialmente a carga eléctrica negativa. A restante carga negativa é neutralizada por um excesso de catiões solvatados, móveis, na 4 proximidade da parede do capilar. Estes formam a zona difusa da dupla camada, que pode estender-se até uma distância de cerca de 10 nm da superfície do capilar. Na presença de um campo eléctrico, os catiões são atraídos para o cátodo e os aniões para o ânodo. O excesso de catiões na zona difusa da dupla camada faz com que esta se mova no sentido do cátodo, arrastando consigo toda a solução. A este efeito chama-se electroosmose, é causado pelos catiões adsorvidos a cerca de 10 nm de distância das paredes do capilar, e causa um fluxo uniforme de toda a solução no sentido do cátodo. Este fluxo difere do fluxo hidrodinâmico (característico da cromatografia), que é causado pela diferença de pressão entre os extremos de um tubo (Figura 6). Figura 6 - (a) A electroosmose causa um movimento uniforme ao longo de mais de 99,9% da secção recta do capilar. A velocidade cai bruscamente junto às paredes do capilar. (b) O fluxo hidrodinâmico é caracterizado por um perfil parabólico de velocidades (fluxo laminar), em que a velocidade máxima ocorre no centro do tubo, e a velocidade junto às paredes é nula. A constante de proporcionalidade entre a velocidade electroosmótica (veo) e o campo aplicado é a mobilidade electrooosmótica (µeo) veo = µeo . E (eq. 7) O facto de o fluxo electroosmótico ser uniforme contribui para a alta resolução da electroforese capilar. Qualquer efeito que diminua a uniformidade causa o alargamento das bandas e diminui a resolução. A mobilidade aparente de um ião resulta da soma da sua mobilidade electroforética e da mobilidade electroosmótica da solução: µap = µe + µeo (eq. 8) Para um catião, que se move na mesma direcção que o fluxo electroosmótico, µe e µeo têm o mesmo sinal, por isso µap > µe. Os aniões são transportados por electroforese na direcção oposta da electroosmose, por isso para aniões os dois termos da equação têm sinais opostos. A pH neutro ou alto, a electroosmose transporta 5 aniões para o cátodo, porque é mais rápida do que a electroforese. A pH baixo, a electroosmose é mais fraca e os aniões podem nunca atingir o detector. Se quisermos separar aniões a pH baixo, é necessário inverter a polaridade do instrumento de modo a que o extremo onde está o detector seja positivo, e onde se coloca a amostra seja negativo (ao inverso do que está representado na figura 2). A mobilidade aparente de uma espécie (µap) é a razão da velocidade (v) pelo campo eléctrico (E): Ld / t V / Lt µap = v / E = (eq. 9) Onde Ld é o comprimento da coluna desde a injecção ao detector; t é o tempo de migração do soluto desde a injecção até ao detector; V é a diferença de potencial aplicada entre os dois extremos do capilar e Lt é o comprimento total da coluna. A mobilidade electroosmótica (µeo) é a velocidade de uma espécie neutra (vn) dividida pelo campo eléctrico: µeo = vn / E = Ld / t n V / Lt (eq. 10) Onde tn é o tempo de migração de uma espécie neutra. A mobilidade eléctroforética (µe) de um analito é calculada pela diferença µe = µap - µeo (eq. 11) Para ter uma boa precisão, as mobilidades são medidas relativamente a um padrão interno. As mobilidades relativas não devem alterar-se de ensaio para ensaio. Resolução e número de pratos teóricos Tal como em cromatografia, a eficiência em electroforese capilar é medida pelo número de pratos 2 2 teóricos, N = Ld / σ . Quando a única causa para o alargamento dos picos é a difusão logitudinal, pode deduzir-se que N= µ ap ⋅ v Ld 2D ⋅ (eq. 12) Lt onde D é o coeficiente de difusão do soluto. + Para iões pequenos, como K , N pode ser da ordem de 100 000. Para proteínas, de baixo coeficiente de difusão, N pode ser da ordem dos 3 000 000... O factor de resolução, tal como em cromatografia, é a razão entre a diferença dos tempos de migração e a largura na base média dos picos de dois componentes A e B. Em electroforese capilar, relaciona-se com a eficiência da coluna e com a razão de velocidades (γ=vA/vB) como: Rs = N (γ − 1) 4 (eq. 13) 6 Técnicas em Electroforese Capilar Electroforese capilar de zona Este é o tipo de electroforese mais largamente usado, e em que o soluto migra através do capilar. O electrólito pode ser um tampão ácido (fosfato, citrato, etc), um tampão básico (borato), ou uma substãnica anfotérica. O fluxo electroosmótico aumenta com o pH do suporte. Electroforese capilar micelar electrocinética Esta técnica é uma variante da electroforese capilar de zona que permite separar moléculas neutras e iões. Adiciona-se à fase móvel um agente tensioactivo aniónico ou catiónico, de modo a formar micelas. Estas têm a forma esférica, sendo o seu interior imiscível com a solução aquosa, e conseguem enclausurar eficientemente compostos neutros por interacções hidrofílicas/hidrofóbicas. Figura 7 - Micelas de dodecilsulfato de sódio têm cargas negativas à superfície, e conseguem transportar moléculas neutras contra o fluxo electroosmótico. As moléculas neutras (a cheio) estão em equilíbrio entre a solução exterior e o interior das micelas. Quanto mais tempo está dentro das micelas, mais uma molécula se atrasa relativamente ao fluxo electroosmótico. Electroforese capilar em gel Esta técnica é a transposição da electroforese em gel clássica para um capilar. Nestas condições, o fluxo electroosmótico é relativamente fraco. O capilar é cheio com um electrólito impregnado num gel que minimiza os fenómenos de difusão e convecção. Este tipo de electroforese é ideal para separar oligonucleótidos, mas não é adequado a moléculas frágeis e termicamente instáveis, como proteínas. 7 Focagem isoeléctrica capilar Esta técnica consiste em criar um gradiente de pH estável e linear num capilar. Cada componente migra e fixa-se no pH que corresponde ao seu ponto isoeléctrico (quando a sua carga global é nula). Então, mantendo o campo eléctrico e aplicando pressão hidroestática, as espécies separadas são empurradas para o detector. Este processo tem uma resolução muito boa, e permite a separação de peptídeos que difiram de apenas 0,02 unidades de pH no ponto isoeléctrico. Electrocromatografia capilar Nesta técnica de separação a migração eléctrica de iões é combinada aos efeitos de adsorção que caracterisam a cromatografia usando uma coluna capilar (diâmetro 75 µm) contendo uma fase estacionária de partículas muito finas (1-3 µm) . Isto é possível devido à inexistência de queda de pressão ao longo da coluna. Bibliografia: D. C. Harris, "Quantitative Chemical Analysis", 6th. ed., Freeman, 2002. F. Rouessac, A. Rouessac, "Chemical Analysis - Modern Instrumentation and Techniques", John Wiley & Sons, 2001. 8
Baixar