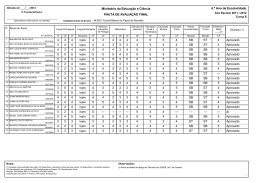
FICHA PARA VERIFICAÇÃO DA ROTULAGEM DE DISPOSITIVOS MÉDICOS NÃO ACTIVOS Dispositivo Médico, Classe: Nome comercial: Fabricante/Mandatário: Distribuidor: Requisitos essenciais de acordo com o estabelecido no ponto 13 do Anexo I do Decreto-Lei nº 30/2003, de 14 Fevereiro (esta informação não dispensa a consulta da legislação aplicável). Sim 1. Identificação do dispositivo e/ou conteúdo da embalagem. 2. Nome e endereço do fabricante, precedido da menção “fabricado por” ou o correspondente símbolo harmonizado. 3. Nome e endereço do mandatário, se aplicável. 4. Marcação CE1, e se aplicável código do ON2, que realizou a avaliação da conformidade. 5. Informações redigidas em língua portuguesa3 de forma legível, para uma correcta e segura utilização do dispositivo. 6. Informações apresentadas sob a forma de símbolos harmonizados (EN 980, 1996), ou outros4 cuja descrição deve estar presente na documentação que acompanha o Dispositivo Médico. 7. Condições especiais de armazenamento ou manuseamento. 8. Código de lote, precedido da menção “Lote” ou do símbolo harmonizado, sempre que o processo de fabrico seja em série. Não 1 A “Marcação CE deve ser aposta pelo fabricante de modo visível, legível e indelével no dispositivo ou na sua embalagem esterilizada, se praticável e adequado, bem como nas instruções de utilização e na embalagem comercial”, como disposto no ponto 2 do artigo 7º do mesmo diploma. A isenção da obrigatoriedade desta marcação verifica-se no caso do dispositivo destinado a investigação clínica, ou nos dispositivos médicos feitos por medida, ou no âmbito de feiras ou exposições e outras demonstrações (pontos 4 e 5 do Artigo 5º do Decreto-Lei nº 30/2003, de 14 de Fevereiro de 2003). 2 Organismo Notificado 3 De acordo com o disposto no ponto 6 do artigo 5º do Decreto-Lei 30/2003, de 14 de Fevereiro. 4 Que não estejam previstos na norma NP EN 980 Pág 1 / 2 Sim Não SE APLICÁVEL: 9. Menção “Estéril” e respectivo Método de Esterilização ou os correspondentes símbolos harmonizados. 10. Menção “Dispositivo feito por medida”, quando é fabricado de acordo com uma prescrição para um determinado doente. 11. Menção “Exclusivamente para investigação clínica”. 12. Data limite de utilização5 : (ano/mês), ou uso símbolo harmonizado. 13. Indicação que o dispositivo é “Descartável” (uso único). 14. Instruções particulares de utilização. 15. Advertências ou Precauções a tomar . 16. Finalidade prevista, sempre que esta não seja evidente. 17. Menção de que “o dispositivo inclui como parte integrante uma substância derivada do sangue humano” . Embora não esteja previsto na legislação, poderá ser mencionada na rotulagem e/ou no folheto de instruções do dispositivo a identificação do distribuidor. Neste caso, o fabricante deverá autorizar o distribuidor a apor a sua identificação e/ou logotipo e se o ON estiver envolvido na avaliação de conformidade do dispositivo, este deverá avaliar todas as menções propostas. Os dados relativos ao distribuidor não poderão substituir ou minimizar a identificação do fabricante e da sua sede social, uma vez que de acordo com a legislação, é o fabricante que é responsável pela colocação do dispositivo no mercado, garantindo a sua conformidade com os requisitos essenciais aplicáveis desde a concepção à utilização prevista, incluindo a responsabilidade pela vigilância de eventuais incidentes ou quase incidentes que possam ocorrer. 5 Apenas aplicável se estiver em causa a segurança, i.e., se o dispositivo se alterar ao longo do tempo e não cumprir com o desempenho para o qual foi concebido. Pág 2 / 2
Baixar