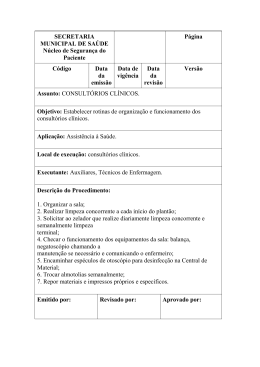
Guia para realização de estudos não clínicos e clínicos para registro de alfainterferona como produto biológico pela via de desenvolvimento por comparabilidade Agência Nacional de Vigilância Sanitária | Anvisa Guia para Realização de Estudos Não Clínicos e Clínicos para Registro de Alfainterferona como Produto Biológico pela Via de Desenvolvimento por Comparabilidade Brasília 2011 Copyright © 2011. Agência Nacional de Vigilância Sanitária. É permitida a reprodução parcial ou total desta obra, desde que citada a fonte.Depósito Legal na Biblioteca Nacional, conforme Decreto n.º 1.825, de 20 de dezembro de 1907. 1ª edição. Diretor-Presidente Adjunto do Diretor-Presidente Dirceu Aparecido Brás Barbano Luiz Roberto da Silva Klassmann Diretores Adjuntos de Diretores Jaime César de Moura Oliveira Luciana Shimizu Takara José Agenor Álvares da Silva Neilton Araujo de Oliveira Maria Cecília Martins Brito Luiz Armando Erthal Chefe de Gabinete Vera Maria Borralho Bacelar Gerência Geral de Medicamentos Norberto Rech Gerência de Avaliação de Segurança e Eficácia Laura Gomes Castanheira Coordenação de Registro de Produtos Biológicos Marcelo Mário Matos Moreira Participação direta: Brenda Gomes Valente Bernardo Luiz Moraes Moreira Fanny Nascimento Moura Flávia Regina Souza Sobral Daniela Marreco Cerqueira Juliana Bertoli da Silva Laura Gomes Castanheira Marcelo Mário Matos Moreira Marcos Fernando Galves da Silva Maria Fernanda Reis e Silva Thees Neemias Silva de Andrade Patrícia Ferrari Andreotti Raimundo Nonato Vieira Júnior Rodrigo Martins Bretas Silmara Crisitane da Silveira Elaboração e edição AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA SIA Trecho 5, Área Especial 57, Lote 200 71205-050, Brasília – DF Tel.: (61) 3462-6000 Home page: www.anvisa.gov.br Sumário 1 Introdução.....................................................................................................................................................4 2 Considerações gerais....................................................................................................................................4 3 Estudos não clínicos......................................................................................................................................5 4 Estudos clínicos............................................................................................................................................6 5 Imunogenicidade...........................................................................................................................................7 6 Extrapolação de Indicações...........................................................................................................................7 7 Plano de Farmacovigilância...........................................................................................................................7 8 Referências..................................................................................................................................................8 Guia para realização de estudos não clínicos e clínicos para registro de alfainterferona como produto biológico pela via de desenvolvimento por comparabilidade 3 1 Introdução O presente guia traça as exigências clínicas e não-clínicas para o registro de alfainterferona humana recombinante na Anvisa, como produto biológico, utilizando a via da desenvolvimento individual . A seção não-clínica trata das exigências fármacotoxicológicas, e a seção clínica aborda as exigências para os estudos de farmacocinética, farmacodinâmica, eficácia e de segurança, assim como os aspectos de farmacovigilância. São apresentados ainda sugestões de protocolos não clínicos e clínicos. Neste guia trazemos orientações gerais para a condução de estudos não-clínicos e clínicos para registro de alfainterferona humana recombinante pela via da comparabilidade. Porém, caso o pesquisador/instituição consiga comprovar a segurança desses fármacos por outros estudos científica e tecnicamente mais viáveis, os dados apresentados serão avaliados pela ANVISA e o desvio das orientações do guia deverá sempre ser justificado. São abordados neste Guia somente os requisitos para os estudos não clínicos e clínicos aplicáveis para o registro de alfainterferona humana recombinante pela via da comparabilidade, sendo que todos os demais itens das normas específicas devem ser cumpridos para o registro sanitário do produto. 2 Considerações gerais A alfainterferona (2a e 2b) humana recombinante é uma proteína conhecida e bem caracterizada formada por 165 aminoácidos. A proteína não glicosilada possui um peso molecular de aproximadamente 19,240 Da. A proteína contem duas pontes dissulfeto, uma entre os resíduos de cisteína 1 e 98 e outro entre os resíduos de cisteína 29 e 138. A seqüência contém sítios de O-glicosilação potenciais. Métodos físico químicos e biológicos estão disponíveis para caracterização das proteínas. A alfainterferona alfa recombinante é aprovada para diferentes indicações como hepatite viral B e C, leucemia, linfoma, carcinoma de células renais e mieloma múltiplo e pode ser usada isoladamente ou em combinação com outros medicamentos. A alfainterferona pode apresentar diferentes efeitos farmacodinâmicos. A importância destes efeitos farmacodinâmicos em cada uma das aplicações clínicas ainda é desconhecida. De maneira geral o uso de alfainterferona em oncologia tem diminuído consideravelmente e vem sendo substituído por outros tratamentos. A dose e o esquema terapêutico necessários para atingir a resposta desejada variam muito de acordo com as diferentes indicações. Geralmente a alfainterferona é utilizada por via subcutânea, apesar de também poder ser usada por via intramuscular ou intravenosa. O tratamento com alfainterferona 2a e 2b está associado com diferentes reações adversas como febre, mal estar, fadiga, mialgia e pode estar associado também a eventos adversos psiquiátricos, hematológicos e renais. A terapia com alfainterferona pode levar ao desenvolvimento de auto-anticorpos. Uma variedade de distúrbios auto-imunes como problemas na tireóide, artrite reumatóide, lupus eritematoso sistêmico, neuropatias e vasculite tem sido observados com o uso de alfainterferona. Tanto anticorpos neutralizantes quanto não neutralizantes foram observados durante o uso de alfainterferona. Este guia não trata das exigências de qualidade. Para os aspectos relativos à qualidade, aplicam-se os princípios conforme dispostos no Guia para realização do exercício de comparabilidade para registro de produtos biológicos pela via da comparabilidade e a RDC55/10 e suas atualizações. De acordo com a RDC 55/10 o produto biológico comparador (PBC) citado no decorrer deste guia é a alfainterferona humana recombinante registrada na Anvisa mediante apresentação de um dossiê completo. 4 Guia para realização de estudos não clínicos e clínicos para registro de alfainterferona como produto biológico pela via de desenvolvimento por comparabilidade 3 Estudos não clínicos Antes de iniciar o desenvolvimento clínico, devem-se realizar estudos não-clínicos. Os estudos não-clíncos devem ser de natureza comparativa e projetados para detectar diferenças na resposta fármaco-toxicológica entre a alfainterferona que se pretende registrar e o PBC, e não apenas para avaliar a resposta propriamente dita. A abordagem tomada precisará ser completamente justificada no resumo não-clínico. Estudos Farmacodinâmicos Estudos in vitro: A fim de comparar as alterações na atividade entre a alfainterferona que se pretende registrar e o PBC, devem-se fornecer dados de uma série de bioensaios comparativos (ligação com receptor, efeito antiviral em cultura celular, efeitos anti-proliferativos em linhagens de células tumorais) sendo que vários destes ensaios já farão parte do dossiê de qualidade no exercício de comparabilidade. Estudos in vivo: Deve-se comparar a atividade farmacodinâmica in vivo da alfainterferona que se pretende registrar e do PBC em: • um modelo farmacodinâmico animal adequado (por exemplo avaliação dos efeitos em marcadores farmacodinâmicos, como atividade 2’,5’oligoadenilato sintetase no soro). Se possível, essas avaliações devem ser realizadas como parte de um estudo de toxicidade. e/ou • modelo animal tumoral adequado e/ou • modelo animal antiviral adequado Estudos de Toxicologia Devem-se fornecer dados de, pelo menos, um estudo de toxicidade de dose repetida em espécies relevantes. O estudo deve durar pelo menos 4 semanas. Estudos toxicocinéticos devem ser realizados como parte do estudo de toxicidade de dose repetida e deverá incluir a avaliação quanto o potencial de formação de anticorpos. Devem-se fornecer os dados sobre a tolerância local em pelo menos uma espécie. Se possível, esses testes de tolerância local podem ser realizados como parte de um estudo de toxicidade de dose repetida. Estudos de farmacologia de segurança, toxicologia de reprodução, mutagenicidade e carcinogenicidade não são exigências de rotina para os testes não-clínicos de um produto medicinal biológico que contenha alfainterferona humana recombinante. Para o caso do desenvolvimento não clínico, deverão ser realizados estudos comparativos utilizando a alfainterferona eleita como produto comparador (alfainterferona registrada na Anvisa através da apresentação de um dossiê completo) e a alfainterferona que se pretende registrar. Devem ser utilizadas no mínimo duas espécies relevantes se disponíveis e devem ser conduzidos estudos de tolerância local, toxicidade aguda e toxicidade de dose repetida. Avaliação comparativa de parâmetros de farmacocinética e farmacodinâmica e de imunogenicidade (sempre que possível) também são necessários. Guia para realização de estudos não clínicos e clínicos para registro de alfainterferona como produto biológico pela via de desenvolvimento por comparabilidade 5 4 Estudos clínicos Estudos de Farmacocinética/ Farmacodinâmica As propriedades farmacocinéticas / farmacodinâmicas da alfainterferona que se pretende registrar e do PBC devem ser comparadas em um estudo randomizado de dose única em voluntários saudáveis. Os parâmetros farmacocinéticos primários recomendados é a área sobre a curva (AUC) e os parâmetros secundários são Cmax, T1/2 e CL/F. As margens de equivalência devem ser pré-especificadas e apropriadamente justificadas. Estudos Farmacodinâmicos: Estão disponíveis diferentes marcadores farmacodinâmicos como β2 microglobulina, neopterin e atividade de 2’,5’oligoadenilato sintetase no soro que são relevantes para mensurar a interação entre a alfainterferona e o sistema imune. As doses selecionadas devem estar na região linear ascendente da curva dose resposta. Mesmo que a importância destes efeitos farmacodinâmicos ainda seja desconhecida nas diferentes indicações, uma avaliação completa comparativa após administração da alfaepoetina que se pretende registrar e o PBC podem fornecer dados úteis. Após a realização dos estudos não clínicos necessários com resultados satisfatórios recomenda-se a realização de um estudo Fase I, comparativo, duplo cego, com voluntários saudáveis e um período de wash out de 4 semanas. O objetivo deste estudo é a comparação dos parâmetros de farmacocinética e farmacodinâmica e coleta de dados iniciais de segurança. Eficácia Clínica População do estudo O mecanismo de ação da alfainterferona compreende uma série de diferentes efeitos não relacionados. A demonstração da similaridade de efeito entre o produto que se pretende registrar e o PBC é necessária. Isto pode ser feito em uma população virgem de tratamento com hepatite C (HCV) conforme delineado pela indicação do PBC. Outras populações devem ser estudadas de acordo com as indicações desejadas. Delineamento e duração do estudo Um grupo paralelo, randomizado para comparação entre os produtos por pelo menos 48 semanas é recomendado. O estudo deverá ser duplo cego no mínimo até o ponto em que os dados das análises primárias forem gerados. Se não for possível, uma justificativa deve ser apresentada e todas as ações para eliminar possíveis vieses devem ser claramente identificadas no protocolo. A posologia (dose, via de administração e metodologia de administração) deve ser a mesma do PBC. A alfainterferona deverá ser administrada juntamente com o tratamento padrão para hepatite C utilizado em conjunto com o PBC. O estudo deve ser delineado de modo a permitir que a análise de eficácia primária seja realizada na décima segunda semana para todos os pacientes recrutados. De preferência uma população homogênea é recomendada, por exemplo, um genótipo viral único. Caso uma população heterogênea seja selecionada deverá ser realizada a estratificação de acordo com o genótipo viral. Desfechos Primários: resposta virológica avaliada pelo número de pacientes com ausência de detecção de RNA de HCV por PCR quantitativo na décima segunda semana. A metodologia utilizada e a linha de corte aplicada deverão ser justificadas. Um decréscimo de 2 log no crescimento viral deverá ser um desfecho co-primário Secundário: resposta virológica na quarta semana e ao final do tratamento; resposta virológica sustentada (24 semanas após o fim do tratamento); alterações nos parâmetros bioquímicos hepáticos incluindo níveis de transaminase e morbidade. 6 Guia para realização de estudos não clínicos e clínicos para registro de alfainterferona como produto biológico pela via de desenvolvimento por comparabilidade Segurança clínica Os dados de segurança devem ser coletados dos pacientes após doses repetidas em um estudo comparativo durante o período de tratamento e mais um período de 24 semanas de seguimento. O número de pacientes deve ser suficiente para a comparação do perfil de eventos adversos. Avaliação de alterações laboratoriais de distúrbios imuno-mediados deve ser incluída. O perfil de segurança entre a alfainterferona que se pretende registrar e o PBC deve ser similar para os eventos adversos mais comuns (febre, mal estar, alopecia, mialgia, leucopenia, anemia e trombocitopenia) Caso os resultados do Estudo Fase I sejam satisfatórios, poderá ser realizado um estudo Fase II/III para avaliação de eficácia e segurança da alfainterferona que se pretende registrar em relação ao produto biológico comparador. Deverá ser um estudo duplo cego comparativo de não inferioridade ou equivalência com pacientes portadores de Hepatite C e duração mínima de 48 semanas. O desfecho primário do estudo deverá ser a remissão viral (avaliada através de PCR quantitativo) na semana 24 e os desfechos secundários serão resposta virológica nas semanas 4, 12 e 48 (avaliada através de PCR quantitativo); deverão ser avaliados também parâmetros bioquímicos (incluindo transaminases) e níveis de morbidade. Avaliação de segurança (tipo, gravidade e incidência de eventos adversos) e de imunogenicidade deverão ser realizadas pelo período de duração do estudo (48 semanas). Caso ocorra algum achado significativo em relação aos parâmetros de segurança e imunogenicidade estudos adicionais poderão ser solicitados. 5 Imunogenicidade Dados comparativos de imunogenicidade (níveis de anticorpos) devem ser coletados durante o período do estudo e adicionalmente durante o período de 24 semanas de seguimento. Os anticorpos quando presentes devem ser avaliados para capacidade neutralizante e o potencial impacto na eficácia do produto. Adicionalmente qualquer potencial de neutralização do efeito endógeno da alfainterferona (desenvolvimento de autoimunidade) deve ser avaliado. Qualquer impacto de imunogenicidade deverá ser avaliado em pacientes que não respondem ao tratamento, pacientes que apresentam perda de resposta no tratamento primário e paciente que apresentam reações adversas inesperadas ou eventos imuno-mediados. 6 Extrapolação de Indicações A princípio a extrapolação de indicações terapêuticas é possível quando o mecanismo de ação e/ou os receptores são os mesmos para os quais a similaridade clínica foi estabelecida através do estudo clínico comparativo. 7 Plano de Farmacovigilância Dentro do procedimento de autorização, o solicitante deve apresentar sempre um plano de farmacovigilância e um plano de minimização de risco sempre que necessário de acordo com a legislação vigente e as diretrizes de farmacovigilância. Deverá ser dada atenção especial as questões relacionadas à imunogenicidade e eventos adversos sérios tardios em pacientes sob administração crônica do produto. Dados de segurança devem ser coletados de pacientes cobrindo todas as indicações aprovadas. Deverá constar no Plano de Farmacovigilância o acompanhamento dos pacientes incluídos no Estudo Fase II/III para avaliação de recidivas. Guia para realização de estudos não clínicos e clínicos para registro de alfainterferona como produto biológico pela via de desenvolvimento por comparabilidade 7 8 Referências Guideline on similar biological medicinal products (CHMP/437/04/draft). Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues (EMEA/CPMP/42832/05/draft). Note for guidance on repeated dose toxicity (CPMP/SWP/1042/99). Note for guidance on toxicokinetics: A Guidance for assessing systemic exposure in toxicological studies (CPMP/ICH/384/95). Note for guidance on non-clinical locale tolerance testing of medicinal products (CPMP/SWP/2145/00). Guideline on risk management systems for medicinal products for human use (EMEA/CHMP 96286/2005). Note for Guidance on Good Clinical Safety Data Management: Definitions and Standards for Expedited Reporting (CPMP/ ICH/377/95). ICH Note for Guidance on Planning Pharmacovigilance Activities (CPMP/ICH/5716/03 - Final approval by CHMP on PHV). Guidance on Planning Pharmacovigilance Activities (CPMP/ 8 Guia para realização de estudos não clínicos e clínicos para registro de alfainterferona como produto biológico pela via de desenvolvimento por comparabilidade Ministério da Saúde
Baixar