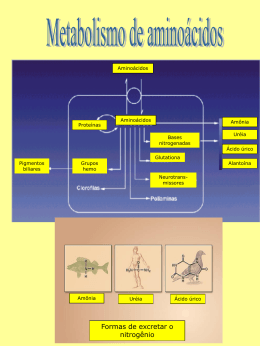
DEFICIÊNCIA TARDIA DE ORNITINA TRANSCARBAMILASE: RELATO DE UMA HISTÓRIA FAMILIAL 1 Autores 1 1 Maria Luiza Wey Vieira , Gabriela Biava de Carvalho Silva , Júlio Boschini Filho , Sandro Blasi Esposito 1 1 1 , Debora Rodrigueiro , Marta Wey Vieira 1 Instituição PUC SP - Pontifícia Universidade Católica de São Paulo (Praça Dr José Ermírio de Moraes, 290) Resumo OBJETIVOS Relatar uma história familial de deficiência tardia de ornitina transcarbamilase (OTC), a dificuldade de diagnóstico até a ocorrência de um caso fatal. Relatamos aqui uma família portadora da mutação c.158T>C, no éxon 2 do gene OTC. MÉTODOS Descrição clínica e análise retrospectiva de prontuário, com revisão de literatura a respeito da patologia, e discussão de terapia proposta. RESULTADOS A paciente LACS, 54 anos, foi encaminhada para aconselhamento genético-clínico por apresentar história familial de três óbitos em meninos com idade entre 11 e 19 anos, de causa não esclarecida. A paciente referia que seu filho, com 19 anos de idade e dois sobrinhos, de 11 anos e 12 anos de idade, filhos de sua irmã, haviam falecido em decorrência de um quadro agudo, com vômitos e declínio do nível de consciência, evoluindo para edema cerebral de causa não-esclarecida. Após revisão de prontuário dos pacientes falecidos observamos que todos haviam evoluído com insuficiência hepática aguda e causa mortis relacionada ao edema cerebral. Durante a investigação genéticoclínica identificamos outro sobrinho, filho de outra irmã da paciente, que evoluiu com descompensação metabólica. Neste caso foi possível realizar a dosagem de uréia que resultou extremamente elevada, 400mg/dL, levantando-se a hipótese de deficiência tardia de OTC. Este último paciente, apesar do tratamento, evoluiu de forma grave indo a óbito cerca de 7 dias após o início dos sintomas. Foi solicitado para a paciente LACS o estudo molecular para o gene OTC e identificada a mutação c.158T>C, no éxon 2 desse gene. Através da análise do heredograma levantamos os casos suspeitos de possíveis portadores da mutação e iniciamos a investigação molecular dos mesmos e encaminhamento para tratamento dietético e acompanhamento clínico. CONCLUSÃO A deficiência de ornitina transcarbamilase é uma rara deficiência enzimática envolvida no metabolismo da uréia, com herança ligada ao cromossomo X. Essa enzima tem expressão no fígado e na mucosa do intestino delgado. Sua falta acarreta aumento do nível da uréia plasmática, podendo provocar danos irreversíveis ao indivíduo, se não diagnosticada e tratada a tempo. A crise de hiperamoninemia decorrente da deficiência dessa enzima está associada a edema cerebral agudo, resultando em perda neurológica e morte. A partir da análise genético-clínica e estudo do heredograma foi possível realizar o diagnóstico e iniciar intervenção terapêutica para os casos suspeitos, bem como discutir a possibilidade de transplante hepático. Palavras-chaves: ciclo da uréia, ornitina transcarbamilase, deficiência metabólica tardia, herança ligada ao X, herança familial Agência de Fomento:
Baixar