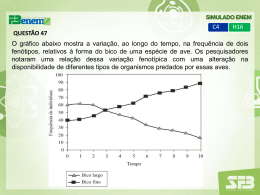
LIPOPROTEÍNAS E DISLIPIDÉMIAS Por não serem solúveis na água, os lípidos circulam no plasma sob a forma de lipoproteínas. Os ácidos gordos livres circulam ligados à albumina. Transporte dos lípidos Classificação e função das lipoproteínas Quilomicra: Transporte de triglicéridos exógenos VLDL: Transporte de triglicéridos endógenos LDL: Transporte de colesterol do fígado para outros tecidos HDL: Transporte do colesterol dos tecidos periféricos e outras lipoproteínas para o fígado. Fig. 1- Estrutura de uma lipoproteína. Tabela I - Caracterização das lipoproteínas pela sua composição (% em peso) Lipoproteína Densidade Proteínas PL COL EC TG <1,006 2 9 1 3 85 VLDL 0,95-1,006 10 18 7 12 50 LDL 1,006-1,063 23 20 8 37 10 HDL 1,063-1,210 55 24 2 15 4 (g/ ml) Qilomicron PL = Fosfolípidos EC = Ésteres de colesterol COL = Colesterol TG = Triglicéridos Quilomicra e VLDL Na tabela I pode-se ver a caracterização das lipoproteínas. Os quilomicra são responsáveis pelo transporte de lípidos da dieta para a circulação sanguínea. A sua formação aumenta com a quantidade de triglicéridos absorvidos. A maior parte das VLDL plasmáticas são de origem hepática. Elas transportam os triglicéridos do fígado para os tecidos extrahepáticos. A apolipoproteína Apo-B (Tabela II) é essencial para a formação dos quilomicra e VLDL. Na abetalipoproteinémia, uma doença rara, a Apo-B não funciona normalmente devido a um defeito na proteína de transferência dos triacilgliceróis, o que previne a formação de lipoproteínas contendo esta apolipoproteína – nestas condições ocorre a acumulação de lípidos no intestino e fígado. Tabela II - Classificação das proteínas presentes nas lipoproteínas: Função Apolipoproteína (Apo-) AI Cofactor da LCAT (activa a LCAT); estrutura das HDL AII Estrutura das HDL B100 Estrutura das LDL e VLDL; liga-se a receptores das LDL B48 Estrutura dos quilomicra CI Estrutura das VLDL e HDL CII Activador de LPL; estrutura dos estrutura dos quilomicra; VLDL e HDL CIII Inibidor de LPL; quilimicrons, VLDL e HDL E Ligação/ receptor; estrutura quilimicrons, VLDL e HDL LCAT = Lecitina-colesterol-acil-transferase LPL = Lipoproteína lipase 1 dos Os quilomicra e as VLDL são rapidamente catabolisados. Os triglicéridos dos quilomicra e das VLDL são hidrolisados pela lipoproteína lipase (LPL), presente nas paredes dos capilares, coração, tecido adiposo, baço, pulmões, medula renal, aorta, diafragma, glândulas mamárias lactantes e no fígado dos recém nascidos. Os fosfolípidos e a apolipoproteína C-II (presentes nos quilomicra e VLDL) são cofactores da LPL, pelo que os quilomicra e as VLDL possuem o substrato e os cofactores da enzima. Pela acção da LPL formam-se quilomicra remanescentes e VLDL remanescentes ou IDL (lipoproteína de densidade intermédia) extremamente ricos em colesterol e ésteres de colesterol devido à perda de triacilgliceróis. Assim, as VLDL são precursoras das IDL e estas das LDL. LDL e HDL As LDL possuem a apolipoproteína Apo B-100, reconhecida por receptores específicos (receptores para as LDL). Aproximadamente 30% da LDL é degradada em tecidos extrahepáticos, enquanto 70% da LDL é degradada no fígado. Existe uma correlação positiva entre a incidência de aterosclerose e a concentração plasmática de LDL, com altos níveis de colesterol. As HDL são sintetizadas e secretadas a partir do fígado e intestino. As HDL secretadas do intestino (de novo) não contêm Apo-C ou Apo-E, apenas contêm Apo-A. Desta forma, a ApoC e Apo-E são sintetizadas no fígado e transferidas do fígado para o intestino, passando depois para o plasma. Uma das funções principais das HDL é o facto de servirem como armazenadoras da Apo-C e Apo-E, requeridas para o metabolismo das VLDL e quilomicra. As HDL nascentes têm uma forma de disco, sendo constituídas por uma bicamada de fosfolípidos, Apo-A e colesterol. Estas lipoproteínas são semelhantes às partículas encontradas nos pacientes com uma deficiência na enzima plasmática lecitina:colesterol aciltransferase (LCAT). De facto a LCAT e a Apo-A-I, activador da LCAT, ligam-se ao 2 disco. A catálise da LCAT coverte os fosfolípidos de superfície e o colesterol livre em ésteres de colesterol e lisolecitina. Os ésteres de colesterol não polares migram para o interior hidrofóbico da bicamada, enquanto a lisolecitina é transferida para a albumina plasmática. A reacção continua conduzindo à formação de um centro apolar que empurra a bicamada e forma uma pseudomicela. A LCAT é assim responsável pela remoção de colesterol nãoesterificado de outras lipoproteínas e dos tecidos. O fígado é o local final para a degradação dos ésteres de colesterol das HDL, pelo que as HDL têm sido implicadas no transporte de colesterol dos tecidos para o fígado por um processo conhecido como transporte reverso do colesterol. Este transporte envolve a tomada e esterificação do colesterol pela HDL3, que se torna maior e menos densa, formando a HDL2. A lipase hepática hidrolisa fosfolípidos e triglicéridos da HDL, permitindo que os ésteres de colesterol sejam libertados para o fígado, conduzindo novamente à formação de HDL3. As concentrações plasmáticas de HDL variam inversamente com as concentrações de triglicéridos e directamente com a actividade da lipoproteína lipase. As concentrações plasmáticas de HDL2 são inversamente proporcionais à incidência de aterosclerose coronária, uma vez que reflectem a eficiência de captação de colesterol dos tecidos. A presença de Apo- 3 AI protege contra a aterosclerose. A Apo-AI associa-se aos fosfolípidos e colesterol formando a preβ-HDL, a forma mais potente da HDL na indução do efluxo de colesterol dos tecidos extrahepáticos. DISLIPIDÉMIAS As dislipidémias ou hiperlipidémias são doenças resultantes de alterações do metabolismo das lipoproteínas em que se observam níveis plasmáticos anormais dessas lipoproteínas. Podem ser devidas a mutações nos genes que codificam para os componentes das lipoproteínas ou para os seus receptores (Tabela III), ou podem ser uma manifestação secundária de outras doenças (Tabela IV). As dislipidémias podem ainda ser classificadas com base no aspecto do plasma sanguíneo após permanecer 12 horas a 4ºC e no seu conteúdo em colesterol e triglicéridos (Classificação de Fredrickson, Tabela V). A classificação de Fredrickson define 6 tipos de hiperlipoproteinemia, sendo os Tipos IIa, IIb e IV os mais comuns, enquanto o Tipo III tem uma frequência intermédia (1: 5000 indivíduos) e os Tipos I e V são raros. Tabela III - Causas genéticas das dislipidémias: Doença Defeito genético Fredrickson Hipercolesterolémia familiar Redução do número de receptores LDL funcionais. Hipertrigliceridémia familiar Possível defeito genético. Hiperlipidémia familiar combinada Possível defeito genético. IIa ou IIb Risco CHD IV ou V IIa, IIb, IV ou V CHD Redução da síntese de apo CII (cofactor da LPL). I Pancreatite Incapacidade na síntese de apo B. I Pancreatite Normal Deficiências em vitaminas lipossolúveis; defeitos neurológicos Defeitos neurológicos; armazenamento dos ésteres de colesterol em locais anormais Redução nos níveis de LPL Deficiência da lipoproteína lipase funcional. Deficiência da Apo C-II Abetalipoproteinémia Incapacidade na síntese de apo A. Analfalipoproteinémia (doença de Tangier) Normal 4 Tabela IV – Causas de hiperlipidémia secundária: Doença Diabetes Mellitus Excesso de álcool Disfunção renal crónica Drogas (diuréticos, tiazida, bloqueadores-β não selectivos) Hipotiroidismo Síndrome nefrótico Disfunção lipídica Aumento Aumento Aumento Aumento triglicéridos triglicéridos triglicéridos triglicéridos Aumento colesterol Aumento colesterol Tabela V – Classificação das Dislipidémias segundo Fredrickson: As dislipidemias são factores de risco para a ocorrência de doença cardíaca coronária, nomeadamente o aumento do colesterol-LDL, assim como a baixa dos níveis de colesterolHDL e o aumento dos triglicéridos. A determinação do colesterol total e do perfil lipídico em jejum, dá-nos informação sobre o risco de doença cardiovascular (Tabela VI). A razão colesterol total/ colesterol HDL, é um parâmetro útil para avaliar o transporte de colesterol dos e para os tecidos periféricos e uma razão de colesterol total/ colesterol HDL acima de 5 é um indicativo de risco cardiovascular aumentado. 5 Tabela VI- Factores de risco para as doenças cardiovasculares: * Idade * História familiar de doença de coração prematura * Tabagismo * Hipertensão * Obesidade * Sedentarismo * Diabetes Mellitus * Altos níveis de colesterol LDL * Baixos níveis de colesterol HDL A decisão de tratar uma dislipidémia baseia-se nos dados laboratoriais obtidos, nomeadamente nos níveis de colesterol, triglicéridos e colesterol-HDL. O tratamento destas doenças envolve medidas dietéticas em que é reduzido o consumo carne e recomendado o consumo de peixe, vegetais e frutas (Fig. 4). A redução de peso corporal e o exercício físico, são medidas importantes uma vez que levam a uma melhor tolerancia à glicose e à redução da tensão arterial. Por vezes é necessário associar a estas medidas uma terapêutica medicamentosa que diminua os níveis de colesterol (Tabela VII). Fig. 4 - Redução de lípidos na dieta. 6 Tabela VII - Fármacos utilizados no controlo dos níveis séricos de lípidos: Grupo farmacológico Resinas que sequestram ácidos biliares Principal acção Bloqueiam a reabsorção dos ácidos biliares e reduzem o colesterol total e LDL Inibidores da HMG-CoA redutase Inibem a síntese do colesterol e reduzem o colesterol total e LDL Fibráceos Activam a lipoproteína lipase (LPL) e reduzem os níveis de TG, colesterol total e LDL. Podem ainda aumentar o colesterol HDL. Ácido nicotínico e derivados Inibem a lipólise ao nível dos adipócitos e reduzem os níveis de TG. Aumentam o colesterol HDL 7 Caso Clínico J.M.D. é um indivíduo do sexo masculino, com 15 anos de idade, que vem ao médico por lhe terem surgido na palma das mãos vários nódulos de cor amarela clara e consistência elástica (xantomas). Apresenta ainda um nódulo semelhante no calcanhar do pé direito. O estudo dos lípidos plasmáticos mostra: colesterol total 20,2 mmol/L (N: 3.9±0.5 mmol/L); triglicéridos 1.9 mmol/L (N: 1.35±0.5 mmol/L); colesterol-HDL 1.4 mmol/L (N: 0.35±0.5 mmol/L). A electroforese das lipoproteínas plasmáticas mostra uma concentração elevada das LDL e HDL. Perante o quadro clínico é posta a hipótese de diagnóstico de hipercolesterolémia familiar. Com base nesta hipótese foi feito o estudo lipídico dos seus progenitores, que mostrou: Pai: Colesterol total 9.0 mmol/L; triglicéridos 1.3 mmol/L. Mãe: “ “ 8.7 mmol/L; “ 1.0 mmol/L. Questões: 1. Explique porque razão os níveis de colesterol são mais baixos nos progenitores do que no filho. 2. Que tipo de transmissão genética lhe é sugerida? 3. A hipercolesterolémia familiar é devida a mutações dos genes que codificam a síntese do receptor das LDL. Identifique, no esquema da Fig. 5, as várias etapas onde a síntese de receptor pode ser afectada. 4. Porque é que as concentrações séricas de LDL e HDL estão aumentadas? 5. Acha que o doente corre o risco de desenvolver doença cardíaca coronária? Porquê? 6. Como medida terapêutica poderia prescrever resinas sequestradoras dos ácidos biliares? Explique a razão desta medida terapêutica. 8 Síntese do Receptor das LDL: N Estrutura do Receptor das LDL: Domínio ligação LDL 1 2 NOligossacarídeo RER OOligossacarídeo Golgi 3 Vesícula secretora Membrana plasmática 4 Receptor das LDL Fig. 5 – Síntese e estrutura do receptor das LDL. 9 Domínio transmembranar
Download