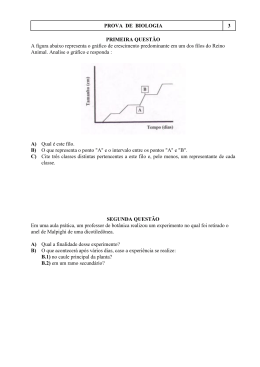
UNIVERSIDADE FEDERAL DO RIO GRANDE DO NORTE CENTRO DE BIOCIÊNCIAS PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIAS BIOLÓGICAS ABINADABE JACKSON DE MELO METAGENÔMICA: BUSCA DE NOVOS GENES ENVOLVIDOS COM A BIODEGRADAÇÃO DE HIDROCARBONETOS E SÍNTESE DE BIOSSURFACTANTES NATAL – RN 2012 ABINADABE JACKSON DE MELO METAGENÔMICA: BUSCA DE NOVOS GENES ENVOLVIDOS COM A BIODEGRADAÇÃO DE HIDROCARBONETOS E SÍNTESE DE BIOSSURFACTANTES Dissertação de mestrado apresentada ao Programa de Pós-Graduação em Ciências Biológicas da Universidade Federal do Rio Grande do Norte, como requisito para obtenção do título de Mestre em Ciências Biológicas. Orientador: Prof.ª Dra Lucymara Fassarella Agnez NATAL – RN 2012 Catalogação da Publicação na Fonte / Bibliotecário Raimundo Muniz de Oliveira CRB15-429 Melo, Abinadabe Jackson de. Metagenômica: busca de novos genes envolvidos com a biodegradação de hidrocarbonetos / Abinadabe Jackson de Melo. – Natal, RN, 2012. 66 f. : il. Orientadora Lucymara Fassarella Agnez. . Dissertação (Mestrado em Ciências Biológicas) – Universidade Federal do Rio Grande do Norte. Centro de Biociências. Programa de Pós Graduação em Ciências Biológicas. 1. Genômica microbiana – Dissertação 2. Metagenômica – Dissertação. 3. Microrganismo – Dissertação. 4. Biodegradação. 5. Hidrocarboneto. I. Agnez, Lucymara Fassarella. II. Título. RN CDU 575.111:579 A minha família, a pedra angular da minha vida, que sempre está presente em todos os momentos me ensinando a lutar com muita determinação pelos meus sonhos. AGRADECIMENTOS A minha família, especialmente, a meu pai José Cícero de Melo e a minha mãe Maria Selma dos Santos de Melo, pelas lições e por investirem tudo na minha formação. A minha orientadora professora Dra Lucymara Fassarela por tudo, pela confiança, pela brilhante orientação, pelos sábios conselhos que levarei para vida toda. Aos “metagenômincos”, Rita, Del, Uaska, Fernanda, Larissa “briba “ André Fonseca, Fabíola Marques, Sinara Carla, Jéssica Paiva, Edson, Tatiana Pereira,Bia e Ralfo, pelo espírito de equipe e de união em todos os momentos. Sem eles eu não conseguiria! A família LBMG/LAMA, amigos inenarráveis ! Especialmente a Thayse, Daniele Maria, Juliana, Julianeee, Fafa, Kellya, Lelinha, Iza, Aninha Sales Mariènne,Amanda, Isabella (a galega), Déborah, Joana, Jana Dara, Tatiana Bressel, Ana Paula, Marcos Felipe, Felipe, Gabriel Gralha, Adilson, Naldo,Leonam Cris, Cecília, Karol Melo, Sara, Paulinho, Jessica Fernanda, Fanny, Djair, Luiz Sodré, Rai, Daniel Chaves, Fabão, Vivi, Rayssa,Geka e as professoras Silvia Regina e Katia Scortecci ! As Dras Viviane Amaral e Keronninn Bessa pelas críticas e sugestões na banca de qualificação. A Dra Magnólia Fernandes Florêncio de Araújo pela participação na banca de defesa. Ao Dr Demetrius A.M Araujo Araújo pela participação na banca de defesa. Ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPQ) pelo apoio financeiro. A Deus, sobretudo, a quem devo toda honra e toda glória para todo sempre ! RESUMO Atividades industriais, derramamentos de petróleo e seus derivados, bem como a combustão incompleta de combustíveis fósseis têm causado um grande acúmulo de hidrocarbonetos no meio ambiente. O número de microrganismos no planeta é estimado em 1030, sendo os procariotos os mais abundantes. Eles colonizaram diversos ambientes durante milhares de anos, incluindo aqueles considerados extremos e representam uma fonte inexplorada de diversidade genética e metabólica com um grande potencial biotecnológico. Sabe-se que muitos microrganismos possuem vias metabólicas complexas atuando na biodegradação de hidrocarbonetos derivados de petróleo e, em muitos ecossistemas, existe uma comunidade autóctone capaz de realizar essa função. A metagenômica tem revolucionado a Microbiologia permitindo o acesso às comunidades microbianas não cultiváveis, sendo uma potente ferramenta para elucidação de suas funções ecológicas, dos perfis metabólicos, bem como para identificação de novas biomoléculas. Assim, o presente estudo aplicou abordagens metagenômicas não apenas para seleção funcional de genes envolvidos nos processos de biodegradação e biossurfactação de hidrocarbonetos derivados do petróleo, mas também para descrição da composição taxonômica e metabólica de dois metagenomas de microbiota aquática. Foram analisadas 123.116 (365 ± 118 pb) e 127.563 sequências (352 ± 120 pb) dos metagenomas marinho e estuarino, respectivamente. Oito clones foram encontrados, sendo quatro envolvidos na biodegradação de petróleo e quatro capazes de emulsificar querosene, indicando a habilidade de sintetizar biossurfactantes. Portanto, as análises metagenômicas realizadas foram eficientes não apenas na busca de bioprodutos de interesse biotecnológico como também na análise do perfil funcional e taxonômico dos metagenomas estudados. Palavras chave: Genômica microbiana. Metagenômica. Microrganismo. Biodegradação. Hidrocarboneto. ABSTRACT Industrial activities, oil spills and its derivatives, as well as the incomplete combustion of fossil fuels have caused a great accumulation of hydrocarbons in the environment. The number of microorganisms on the planet is estimated at 10 30 and prokaryotes the most abundant. They colonized diverse environments for thousands of years, including those considered extreme and represent an untapped source of metabolic and genetic diversity with a large biotechnological potential. It is also known that certain microorganisms have the enzymatic capacity to degrade petroleum hydrocarbons and, in many ecosystems, there is an indigenous community capable of performing this function. The metagenomic has revolutionized the microbiology allowing access uncultured microbial communities, being a powerful tool for elucidation of their ecological functions and metabolic profiles, as well as for identification of new biomolecules. Thus, this study applied metagenomic approaches not only for functional selection of genes involved in biodegradation and emulsification processes of the petroleum-derived hydrocarbons, but also to describe the taxonomic and metabolic composition of two metagenomes from aquatic microbiome. We analyzed 123.116 (365 ± 118 bp) and 127.563 sequences (352 ± 120 bp) of marine and estuarine metagenomes, respectively. Eight clones were found, four involved in the petroleum biodegradation and four were able to emulsify kerosene indicating their abilities in biosurfactants synthesis. Therefore, the metagenomic analyses performed were efficient not only in the search of bioproducts of biotechnological interest and in the analysis of the functional and taxonomic profile of the metagenomes studied as well. Key words: Microbial genomic. Metagenomic. Microorganism.Biodegradation.Hydrocarbon. SUMÁRIO 1 INTRODUÇÃO........................................................................................................ 14 1.1 PETRÓLEO: BENEFÍCIOS, IMPACTOS E BIORREMEDIAÇÃO........................... 14 1.2 Biodegradação De Hidrocarbonetos........................................................................ 15 1.2.1 GENES E ENZIMAS ENVOLVIDOS COM A BIODEGRADAÇÃO DE HIDROCARBONETOS POR BACTÉRIAS.............................................................. 16 1.3 18 BIOSSURFACTANTES............................................................................................ 1.3.1 GENES ENVOLVIDOS COM A SÍNTESE DE BIOSSURFACTANTES BACTERIANOS....................................................................................................... 20 1.4 METAGENÔMICA: DESCOBERTA DO DESCONHECIDO................................... 21 1.4.1 METAGENÔMICA: ABORDAGEM FUNCIONAL E BASEADA EM SEQUÊNCIA.. 23 2 JUSTIFICATIVA....................................................................................................... 26 3 OBJETIVOS............................................................................................................. 27 3.1 GERAIS.................................................................................................................... 27 3.2 ESPECÍFICOS.......................................................................................................... 28 4 MATERIAL E MÉTODOS......................................................................................... 28 4.1 COLETA DAS AMOSTRAS AMBIENTAIS............................................................... 28 4.2 ANÁLISES FÍSICO-QUÍMICAS............................................................................... 29 4.3 EXTRAÇÃO DO eDNA............................................................................................. 29 4.4 CONSTRUÇÃO DAS BIBLIOTECAS METAGENÔMICAS...................................... 29 4.4.1 TRATAMENTO DAS EXTREMIDADES DOS FRAGMENTOS GERADOS POR SONICAÇÃO............................................................................................................ 30 4.4.2 PURIFICAÇÃO DOS INSERTOS TRATADOS........................................................ 30 4.4.3 PREPARAÇÃO DO VETOR PBC SK................................................................ 30 4.4.4 CLONAGEM DO EDNA..................................................................................... 31 4.5 SEÇÃO FUNCIONAL......................................................................................... 33 4.5.1 TESTE QUALITATIVO DE BIODEGRADAÇÃO DE HIDROCARBONETOS.... 33 4.5.2 HABILIDADE DE SINTETIZAR BIOSSURFACTANTES................................... 33 4.5.3 CONFIRMAÇÃO DA PRESENÇA DOS INSERTOS AMBIENTAIS.................. 34 4.6 SEQUENCIAMENTOS....................................................................................... 35 4.7 ANÁLISES DE BIOINFORMÁTICA.................................................................... 36 5 RESULTADOS................................................................................................... 38 5.1 BIBLIOTECAS METAGENÔMICAS................................................................... 38 5.2 ANÁLISES FÍSICO-QUÍMICAS DAS AMOSTRAS AMBIENTAIS..................... 38 5.3 HABILIDADE DE SINTETIZAR BIOSSURFACTANTES................................... 39 5.4 BIODEGRADAÇÃO DE HIDROCARBONETOS................................................ 42 5.5 DADOS GERAIS, COMPOSIÇÃO TAXONÔMICA E METABÓLICA DOS METAGENOMAS............................................................................................... 44 6 DISCUSSÃO...................................................................................................... 50 7 CONCLUSÕES.................................................................................................. 56 8 REFERÊNCIAS BLIOGRÁFICAS..................................................................... 57 LISTA DE FIGURAS Figura 1. Esquema mostrando as abordagens metagenômicas: análise funcional e análise baseada em sequência................................ 24 Figura 2. Esquema resumindo a estratégia usada nesse estudo.............. 37 Figura 3. Teste de emulsificação............................................................... 41 Figura 4. Teste de confirmação da atividade de biodegradação............... 42 Figura 5. Curvas de rarefação dos metagenomas EST (azul) e MAR (vermelho)................................................................................... Figura 6. Composição taxonômica relativa (domínio) dos metagenomas EST e MAR............................................................................ Figura 7. 45 Composição taxonômica (classe) dos metagenomas EST e MAR..................................................................................... Figura 9. 45 Composição taxonômica (filo) dos metagenomas EST e MAR..................................................................................... Figura 8. 44 Composição metabólica (nível 1 do MG-RAST) 46 dos metagenomas............................................................................. 47 Figura 10. Perfil metabólico representado pelo mapa de vias do KEGG..... 48 Figura 11. Perfil de distribuição de algumas proteínas relacionada à atividade de biodegradação de hidrocarbonetos (a) e de alguns genes envolvidos na síntese de biossurfactantes conhecidos (b)............................................................................ 49 LISTA DE TABELAS Tabela 1. Análise físico-química de amostras de água do estuário jundiaí/potengí (Natal/RN) e da praia de pirangí do norte (Parnamirim/RN)......................................................................... Tabela 2. Clones positivos para o teste de emulsificação de querosene.................................................................................. Tabela 3. 39 Melhores resultados do blastx dos clones positivos para síntese de biossurfactantes........................................................ Tabela 4. 38 40 Melhores resultados do blastx dos clones positivos para atividade de biodegradação........................................................ 43 14 1 INTRODUÇÃO 1.1 PETRÓLEO: BENEFÍCIOS, IMPACTOS E BIORREMEDIAÇÃO Atividades industriais, derramamentos de petróleo e seus derivados (tais como gasolina, diesel e querosene), bem como a combustão incompleta de combustíveis fósseis têm causado um grande acúmulo de hidrocarbonetos no meio ambiente (SANTOS et al., 2011a). O petróleo é uma mistura heterogênea de compostos orgânicos, incluindo hidrocarbonetos alifáticos (n-alcanos), alicíclicos e aromáticos, com pequenas quantidades de compostos como oxigênio, nitrogênio e enxofre, que variam em propriedades físicas e composição de acordo com o reservatório de origem (HAMME et al., 2003). Em virtude de ainda ser a principal fonte energética do mundo, este recurso natural é responsável por grandes movimentações financeiras na economia mundial, gerando emprego e renda para famílias do mundo inteiro. Neste cenário, o Brasil certamente deverá alcançar uma posição de destaque no ranking dos grandes produtores a partir da exploração da recentemente descoberta camada pré-sal. Todavia, como se sabe, a utilização em larga escala desse recurso gera impactos ambientais decorrentes da poluição dos solos, aquíferos, mares e da atmosfera proveniente da alta produção, distribuição e consumo dos derivados do petróleo. Aproximadamente 0,08 – 0,4% da produção mundial de petróleo acabam chegando aos oceanos (BARTHA et al., 1986) gerando, sobretudo, impactos ecológicos a ecossistemas marinhos e terrestres (SANTOS et al., 2011b). Para minimizar os danos, estratégias visando à contenção e remoção do contaminante livre ou dissolvido vêm sendo aplicadas na remediação de áreas contaminadas. Vários métodos podem ser empregados para remover hidrocarbonetos do solo e de água subterrânea, tais como extração a vapor do solo, bombeamento e biorremediação (MARIANO, 2006). Tratamentos físicos separam os contaminantes do solo sem destruí-los ou modificá-los quimicamente, mas apresentam muitas limitações, destacando-se o alto custo e eficiência relativamente baixa. Processos biológicos, por outro lado, são alternativas promissoras para 15 remover esses contaminantes, principalmente devido à simplicidade e eficiência de custo quando comparados a outros métodos tradicionais (ALEXANDER, 1994 apud MARIANO, 2006). Microrganismos podem degradar ou produzir hidrocarbonetos dependendo da presença de vias metabólicas específicas para cada função em certas condições ambientais (PEIXOTO et al., 2011). Biorremediação pode ser considerada como uma nova tecnologia para tratar locais contaminados mediante o uso de agentes biológicos capazes de modificar ou decompor poluentes alvos. Estratégias de biorremediação incluem: a utilização de microrganismos autóctones, ou seja, do próprio local, sem qualquer interferência de tecnologias ativas de remediação (biorremediação intrínseca ou natural); a adição de agentes estimulantes como nutrientes, oxigênio e biossurfactantes (bioestimulação); e a inoculação de consórcios microbianos enriquecidos (bioaumento) (BENTO et al., 2003). O benefício desses processos é a mineralização do poluente e a transformação em gás carbônico, água e biomassa. Assim, a biorremediação é bastante pertinente, já que é natural e seus produtos finais não apresentam riscos ao meio ambiente. 1.2 BIODEGRADAÇÃO DE HIDROCARBONETOS Muitos microrganismos possuem a capacidade de degradar hidrocarbonetos derivados de petróleo e, em muitos ecossistemas, existe uma comunidade autóctone de microrganismos capazes de realizar essa função, os quais são denominados de hidrocarbonoclásticos (KATAOKA, 2001 apud MARIANO, 2006). Alguns degradam alcanos, compostos aromáticos, e outros degradam tanto hidrocarbonetos parafínicos quanto os aromáticos (ATLAS, 1995). Alcanos na faixa de C10 e C26 são vistos como os mais facilmente degradados, porém aromáticos de baixo peso molecular tais como benzeno, tolueno e xileno, que estão entre os compostos tóxicos encontrados no petróleo, são também muito facilmente biodegradados por diversos microrganismos marinhos (ATLAS, 1995). Os primeiros estudos da utilização de hidrocarbonetos por microrganismos foram realizados por Sohnger e Kaserer em 1906. Em 1913, Sohnger relatou que 16 gasolina, querosene, parafina e óleo de parafina poderiam ser oxidados a gás carbônico, água e traços de ácidos orgânicos por microrganismos, esses pertenciam principalmente aos gêneros Mycobacterium e Pseudomonas (BUSHNELL e HAAS, 1941). Posteriormente, as seguintes espécies de bactérias estudadas pelos métodos tradicionais da microbiologia como Acinetobacter sp., Aeromonas sp., Bacillus sp., Escherichia coli, Flavobacterium sp., Klebsiella cepacia, Micrococcus luteus, Moraxella phenylpiruvica, Nocardia sp., Ochrobactrum anthropi, Pseudomonas aeruginosa, Pseudomonas sp., Proteus mirabilis, Vibrio sp., Rhodococcus sp., Streptomyces sp., Vibrio fisheri e Xanthomonas maltophilia, foram identificadas como hicrocarbonoclásticas (SORKHOH et al., 1990; AL-HADHRAMI et al., 1995). De modo geral, vazamentos de petróleo provocam um aumento na atividade e abundância de microrganismos degradadores de hidrocarbonetos no local, proporcionando uma alteração na estrutura das comunidades, além de afetar a função e a integridade do ecossistema (EL-TARABILY, 2002; BRADDOCK et al., 1996; ATLAS, 1995). A proporção de microrganismos degradadores aumenta de menos de 1%, em ambientes marinhos não contaminados, para mais de 10%, em ambientes marinhos contaminados (ATLAS, 1981). Assim, mudanças na riqueza, diversidade e padrões estruturais das comunidades microbianas refletem alterações ambientais, que podem ser utilizadas estrategicamente tanto no monitoramento de impactos quanto como bioindicadores para biorremediação. 1.2.1 GENES E ENZIMAS ENVOLVIDOS COM A BIODEGRADAÇÃO DE HIDROCARBONETOS POR BACTÉRIAS A biodegradação dos inúmeros componentes de petróleo ocorre sob diferentes condições ambientais (aeróbica ou anaeróbica em variados pHs e salinidades), graças a uma miríade de enzimas e vias metabólicas (PEIXOTO et al., 2011). A degradação anaeróbica é catalisada por bactérias anaeróbicas tais como as bactérias redutoras de sulfato, usando diferentes aceptores finais de elétrons como nitrato, sulfato e ferro (III) (HAMME et al., 2003). No metabolismo anaeróbico, geralmente os compostos são convertidos a benzoil-CoA, que é alvo da enzima 17 benzoil-CoA redutase (BCR). Vale ressaltar, que esse tipo de degradação é fundamental em processos de biorremediação onde a disponibilidade de oxigênio é bastante limitada como em mangues e aquíferos, por exemplo. (SANTOS et al., 2011a). Os genes envolvidos nos processos de degradação de compostos orgânicos estão não apenas localizados em cromossomos, mas em diferentes plasmídeos, transposons e elementos genéticos integrativos, cujo embaralhamento e fenômenos de transferência horizontal têm levado a evolução de novas vias degradativas (PHALE et al., 2007). A base genética e bioquímica da biodegradação de alifáticos saturados (alcanos), hidrocarbonetos aromáticos e policíclicos aromáticos são as mais estudadas. Os aspectos fundamentais do metabolismo de n-alcanos e os genes envolvidos são conhecidos há muito. A via de degradação desse composto extensivamente caracterizada é codificada pelo plasmídeo OCT de Pseudomonas oleovorans (VAN BEILEN et al.,1994). Os principais genes, as vias metabólicas envolvidos na degradação aeróbica de hidrocarbonetos policíclicos aromáticos, bem como um modelo para o metabolismo de alcano, incluindo a localização das proteínas ALK e a regulação dos genes alk foram revisados por Van Beilen et al (2001). De acordo com esse trabalho, o operon alkBFGHJKL codifica as enzimas necessárias para conversão de alcanos a acetil-coenzima A (CoA), ao passo que alkST codifica uma rubredoxin redutase (ALKT) e um regulador positivo do operon alkBFGHJKL (ALKS). O catabolismo aeróbico de hidrocarbonetos é mais rápido em virtude da vantagem metabólica de ter oxigênio (O2) como aceptor de elétron (CAO et al., 2009). Hidrocarbonetos aromáticos tais como benzeno, tolueno, xileno e naftaleno podem ser degradados nessas condições. A degradação desses compostos normalmente serve como etapa inicial na formação de catecol, que uma vez formado, pode ser consumido gerando compostos que entram no ciclo do ácido cítrico sendo completamente degradados a CO2 (PEIXOTO et al., 2011). A hidroxilação de um grupo alquil catalisada por oxigenases é normalmente a primeira etapa na degradação de compostos orgânicos. Há diversos tipos de hidroxilases de grupo alquil, incluindo CYP153, uma enzima da superfamília 18 citocromo p450 (CYP), e alcano hidroxilase (codificada por alkB) (YERGEAU et al., 2012). Embora os anéis aromáticos também precisem ser hidroxilados para serem degradados, a etapa crucial na degradação de hidrocarbonetos aromáticos é a abertura do anel aromático hidroxilado, que é catalisada por dioxigenases de clivagem do anel aromático. Existem três tipos dessas dioxigenases: intradiol, extradiol e gentisato/homogentisato, que podem ser diferenciadas com base em seu substrato e na posição onde ocorre a fissão do anel em relação aos grupos hidroxilas (HARAYAMA et al., 1992). Como aromáticos são componentes reduzidos, aumentando o estado de oxidação do núcleo aromático, esses compostos ficam mais susceptíveis a degradação permitindo aos microrganismos usá-los como única fonte de carbono e energia. Isso só é possível pela incorporação de oxigênio molecular mediante a participação das oxigenases, visto que o oxigênio, devido a sua estrutura química peculiar, é de baixa reatividade (HAYAISHI et al., 1955). Mono-oxigenases (tal como metano mono-oxigenase) introduzem apenas um átomo de oxigênio a um substrato, ao passo que dioxigenases introduzem dois átomos ao mesmo substrato (PEIXOTO et al., 2011). Portanto, enzimas como catecol dioxigenase, benzeno dioxigenase, tolueno dioxigenase, bifenil dioxigenase, clorobenzeno dioxigenase, naftaleno dioxigenase e outras, que estão envolvidas na clivagem do anel aromático, são responsáveis pela capacidade de degradação de compostos aromáticos em um grande número de espécies bacterianas (BRODERICK, 1999). 1.3 BIOSSURFACTANTES Durante degradadores a biodegradação geralmente de produzem hidrocarbonetos, moléculas os adjuvantes microrganismos chamadas de biossurfactantes. Entretanto, é comum o uso industrial de moléculas sintéticas com propriedades semelhantes denominadas surfactantes (MULLIGAN, 2005). Em geral, surfactantes são moléculas anfipáticas que, em solução aquosa, podem reduzir 19 significativamente a tensão superficial por acumular-se na interface e facilitar a formação de emulsões entre líquidos de diferentes polaridades (MILLER, 1995). Biossurfactantes são surfactantes naturais (biológicos) sintetizados por microrganismos, fungos, plantas e animais, incluindo os humanos (CHRISTOFI; IVSHINA, 2002). Assim como os surfactantes sintéticos, os biológicos também apresentam uma porção hidrofílica e outra hidrofóbica (apolar). A parte hidrofóbica constitui-se tipicamente de ácidos graxos de cadeia longa, ao passo que a outra porção pode ser um carboidrato, aminoácido, peptídeos cíclicos, fosfato, ácido carboxílico ou um álcool (MULLIGAN, 2005). Eles estão agrupados em glicolipídeos, lipopeptídeos, fosfolipídeos, ácidos graxos, lipídeos neutros e componentes poliméricos (BIERMANN et al., 1987). Estas moléculas têm sido exploradas para uma variedade de aplicações industrial e de biorremediação (BODOUR et al., 2003). De uma perspectiva clínica, é sabido que alguns têm atividade antibiótica (VOLLENBROICH et al., 1997; BECHARD et al., 1998) e que pelo menos um biossurfactante rhamnolipídio produzido por Pseudomonas aeruginosa tem um papel na sua patogênese (SINGH et al., 2000), além de estudos demonstrarem dados que sugerem importante envolvimento no crescimento e sobrevivência microbiana no ambiente. Foi demonstrado que a estrutura do biossurfactante tem um efeito na solubilização e degradação dos n-alcanos (ZANG; MILLER, 1992) e sua presença pode levar a um aumento na concentração de compostos hidrofóbicos na fase aquosa, o que é conseguido através da formação de emulsões óleo/água e consequente solubilização (CHRISTOFI; IVSHINA, 2002). A formação de emulsões estáveis de petróleo e água por microrganismos pode aumentar a degradação desse poluente (BREDHOLT et al., 1998). Nos últimos anos, muitos estudos têm mostrado que biossurfactantes podem também solubilizar e mobilizar componentes orgânicos sorvidos no solo (ZANG; MILLER 1992; SCHEIBENBOGEN et al., 1994; LAFRANCE; LAPOINTE, 1998; PAGE et al., 1999). Além disso, os biossurfactantes também têm potenciais aplicações na agricultura, em cosméticos, indústrias farmacêuticas, detergentes, produtos de cuidado pessoal, processamento de alimentos, indústria têxtil, processamento e 20 tratamento de metais, processamento de papel e indústrias de tintas (BANAT et al., 2000). Desse modo, esses compostos biológicos apresentam um enorme potencial biotecnológico como adjuvantes na biorremediação de áreas afetadas por petróleo e hidrocarbonetos derivados, uma vez que são menos tóxicos e menos persistentes do que os surfactantes sintéticos (MULLIGAN, 2005). 1.3.1 GENES ENVOLVIDOS COM A SÍNTESE DE BIOSSURFACTANTES BACTERIANOS Estudos de genética molecular e bioquímica da síntese de biossurfactantes bacterianos são melhor compreendidos para as espécies de Bacillus e Pseudomonas e têm revelado operons, enzimas e as vias metabólicas necessárias para sua produção. Surfactina, um lipopeptídeo produzido por Bacillus subtilis e rhamnolipideo, um glicolipídeo produzido por Pseudomonas aeruginosa foram os primeiros a serem decifrados e são os mais estudados. Além desses, os genes envolvidos na síntese de outros surfactantes microbiológicos menos conhecidos tais como viscosina, amphisina, putisolvina, arthrofactina, alasan, serrawentina, lichenisina e iturina também foram elucidados, embora mais estudos sejam necessários para uma compreensão mais acurada dos processos bioquímicos e de regulação da síntese dessas biomoléculas (PALASHPRIYA et al., 2008). A biossíntese de surfactina, o primeiro e melhor conhecido biossurfactante de B.subtilis, é catalisada não-ribossomicamente por um complexo multienzimático peptídeo sintetase mais conhecido como surfactina sintetase e consiste de três subunidades protéicas: SRFA, SRFB (ou COMA) e SRFC (PALASHPRIYA et al., 2008). Essa peptídeosintetase é codificada por três ORFs dispostas no operon srfA (FABRET et al., 1995). A ORF srfaa contem o sítio ativo para adição de Glu, Leu e D-Leu; srfab tem o sítio ativo para adição de Val, Asp, e D-Leu; ao passo que srfac para Leu (GALLI et al., 1994), mas codifica também uma tioesterase de motivo tipo 1 responsável pela terminação do peptídeo. Há ainda a ORF srfad que não está envolvida na sínese de surfactina. Alguns kilobases downstream do operon srfA está 21 o gene sfp, que também é fundamental para síntese desse biossurfactante. SFP é uma fosfopantenil transferase necessária para ativação da surfactina sintetase através de modificação pós traducional (LAMBALOT et al., 1996). Espécies de Pseudomonas são o segundo maior grupo de bactérias produtoras de biossurfactantes, especificamente rhamnolipídios. Durante o crescimento de P. aeruginosa as principais moléculas produzidas são monorhamolipídeos e di-rhamnolipídeos através de duas reações sucessivas de transferência de glicosil, de modo que cada uma é realizada por uma rhamnosiltransferase diferente (BURGER et al., 1963). Quatro genes denominados rhla, b, r e i estão dispostos em um operon, transcritos no sentido 5´-rhlabri-3´ e são suficientes para produção de rhamnolipideo (OCHSNER et al., 1995). A síntese de mono-ramnolipidios é catalisada pela enzima rhamnosiltransferase 1, codificada pelos genes rhla e rhlb. A proteína RHLA, que reside no periplasma, presumivelmente está envolvida na síntese e/ou transporte de substratos precursores de rhamnosiltransferase ou na estabilização de RHLB, uma proteína citoplasmática estrutural (OCHSNER et al., 1994). Uma rhamnosiltransferase 2 é codificada pelo gene rhlc, cuja expressão parece ser coordenadamente regulada com rhla e rhlb pelo mesmo sistema de quorum sensing que é codificado pelos genes rhlr e rhli (RAHIM et al. 2001). 1.4 METAGENÔMICA: DESCOBERTA DO DESCONHECIDO O número de microrganismos no planeta é estimado em 1030, sendo os procariotos os mais abundantes (TURNBAUGH et al., 2008). Eles colonizaram diversos ambientes durante milhares de anos, incluindo aqueles que, para a maioria dos organismos, são considerados extremos. Além de colonizar o ambiente, os microrganismos habitam outros organismos e são imprescindíveis para manutenção da vida na Terra, visto que são os principais componentes das cadeias alimentares, participam dos ciclos biogeoquímicos, mantêm diversas relações simbióticas com organismos superiores (PEIXOTO et al., 2011) e representam uma fonte inexplorada de diversidade genética e metabólica com potencial biotecnológico (DANIEL et al., 2011). Todavia, 22 apenas 1% deles pode ser cultivado em laboratório pelos métodos tradicionais (AMANN et al., 1990). Nesse contexto, a metagenômica tem revolucionado a Microbiologia abrindo caminho para o acesso pleno a comunidades microbiológicas de ecossistemas complexos, tornando-se uma potente ferramenta para exploração da ecologia (BIDDLE et al., 2008), do perfil metabólico dessas comunidades (DELONG et al., 2006; TRINGE et al., 2005), bem como para identificação de novas biomoléculas a partir da construção de bibliotecas contendo DNA de diversas amostras ambientais tais como solo (HÅRDEMAN et al., 2007; VOGET et al., 2006; WASCHKOWITZ et al., 2009), sedimentos de superfícies contaminadas (ABULENCIA et al., 2006), águas subterrâneas (UCHIYAMA et al., 2005), águas superficiais de rios (WU et al., 2009), geleiras (SIMON et al., 2009), solo do deserto antártico (HEATH et al., 2009), rúmen de búfalo (DUAN et al., 2009), dentre outros. O termo metagenoma é derivado do conceito estatístico de meta-análise, o processo de combinar estatisticamente análises separadas, e genômica, a análise ampla do material genético de um organismo (SCHLOSS; HANDELSMAN, 2003). Embora atribua-se a Handelsman et al (1998) a elaboração do termo, este pode ser adequadamente aplicado para alguns estudos anteriores como o trabalho de Torsvik et al (1990) que purificaram DNA de uma fração bacteriana isolada do solo; a pesquisa de Schmidt et al (1991) que, com o intuito de minimizar a seleção de sequências particulares, caracterizaram sequências de rRNA 16S de uma população de picoplancton a partir da clonagem direta em fago tendo E. coli como cepa hospedeira; e o trabalho de Healy et al (1995), no qual obtiveram clones expressando celulase de um ambiente termofílico a partir da montagem das então denominadas zoobibliotecas. Sendo assim, a metagenômica consiste na análise, independente de cultivo, de genomas presentes em dado hábitat (RIESENFELD et al., 2004). Esse excitante campo de pesquisa vem obtendo significativo progresso nos últimos anos graças aos avanços não apenas no desenvolvimento de tecnologias de sequenciamento de última geração, cuja aplicação resultou na criação de grandes bancos de dados derivados de vários ambientes, tais como solo e mar (SIMON e DANIEL, 2009), mas também na geração de novos algoritmos, softwares e ferramentas de bioinformática, 23 permitindo análises mais robustas da enorme quantidade de dados gerados, o que vem abrindo caminho para novas aplicações tais como metagenômica comparativa, metatranscriptômica e metaproteômica (CHISTOSERDOVA, 2010; SJÖLING et al., 2008). Além disso, abordagens metagenômicas são ferramentas plausíveis para o acesso ao perfil taxonômico e metabólico dos mais variados ambientes, como também para descoberta de produtos de interesse biotecnológico provenientes da inexplorada diversidade genética de 99% das espécies de microrganismos, embora esses não possam ser cultivados pelos métodos padrões da microbiologia (AMANN et al., 1990). 1.4.1 METAGENÔMICA: ABORDAGEM FUNCIONAL E BASEADA EM SEQUÊNCIA Estudo de metagenomas podem ser divididos em análise funcional e análise baseada na sequência (GABOR et al., 2007) (Figura 1). A análise funcional corresponde à seleção de clones de uma biblioteca metagenômica para um fenótipo particular como tolerância a elevadas concentrações de sal, produção de antibióticos e inúmeras atividades enzimáticas específicas, e envolve ainda a identificação da origem filogenética do DNA clonado (DINSDALE et al., 2008). A construção de bibliotecas metagenômicas é sempre uma tarefa desafiadora e laboriosa, pois começa com o isolamento de eDNA (DNA ambiental) de alta qualidade apropriado para clonagem. A qualidade e quantidade da extração de eDNA depende das características bióticas e abióticas (propriedades físicoquímicas) da amostra. Por exemplo, um problema relacionado ao isolamento de DNA diretamente do solo, tipo de amostra mais comum em projetos de metagenoma funcional, é a possibilidade de co-extração de ácidos húmicos e outros contaminantes, tornando crucial, passos de purificação mais eficientes (RAJENDHRAN; GUNASEKARAN, 2008). Já a extração de DNA de amostras de águas marinhas normalmente tem baixo rendimento, visto que os microrganismos estão mais dispersos na coluna d’água (KENNEDY et al., 2010). Dependendo do tamanho do inserto desejado, bibliotecas metagenômicas são montadas usando plasmídeos (até 15 Kb), fosmídeos, cosmídeos (ambos até 40 24 Kb), ou cromossomos artificiais de bactérias (>40 Kb) como vetores (SIMON; DANIEL, 2011). A escolha do vetor depende da qualidade do DNA, dos genes alvos, e da estratégia de varredura. Bibliotecas contendo pequenos insertos podem ser aplicadas para identificação de novos biocatalisadores codificados por um só gene ou por pequenos operons, enquanto bibliotecas de grandes insertos são necessárias para buscar clusters gênicos maiores, que codificam vias complexas (DANIEL, 2005). Amostra Ambiental Extração de eDNA Construção de bibliotecas metagenômicas Sequenciamento de genes marcadores filogenéticos Bioprospecção Acesso à diversidade taxonômica Sequenciamento do eDNA Acesso ao potencial metabólico Figura 1. Esquema mostrando as abordagens metagenômicas: análise funcional e análise baseada em sequência. Na construção de uma biblioteca metagenômica, o eDNA é extraído da amostra, fragmentado por processos físicos ou enzimáticos, clonado no vetor escolhido e por fim este é transformado num hospedeiro (normalmente Escherichia coli). A partir da biblioteca metagenômica faz-se a bioprospecção por novos genes e biomoléculas. O mesmo DNA metagenômico ainda pode ser sequenciado para estudos de biodiversidade e do potencial metabólico da microbiota escolhida. Adaptada de Sleator et al (2008) e Daniel et al (2011). O passo subsequente à clonagem nos vetores é a transformação num hospedeiro desejado, sendo as cepas de Escherichia coli as mais usadas em estudos de metagenoma funcional. Como existem diferenças significativas no modo de expressão entre diferentes grupos taxonômicos de procariotos e apenas 40% de atividades enzimáticas podem ser detectados pela clonagem aleatória em E. coli 25 (GABOR et al., 2004) outros hospedeiros, tais como Streptomyces spp (WANG et al., 2000), Thermus thermophilus (ANGELOV et al., 2009), Sulfolobus solfataricus (ALBERS et al., 2009) e outras Proteobacterias (CRAIG et al., 2010) têm sido aplicados para aumentar a faixa de atividades detectáveis nos ensaios funcionais. Três diferentes tipos de seleção funcional foram descritos na literatura: a) detecção direta de fenótipos específicos de clones individuais; b) complementação heteróloga de linhagens hospedeiras ou mutantes; c) expressão gênica induzida (SIMON; DANIEL, 2009). No primeiro, uma função enzimática pode ser identificada em clones individuais pela adição do substrato da enzima ao meio de cultura (proteases, lipases); no segundo linhagens hospedeiras exigem complementação por genes exógenos para crescimento em condições seletivas, de modo que apenas clones recombinantes levando o gene alvo e produzindo o correspondente produto gênico funcional são capazes de crescer (SIMON et al., 2009). A terceira consiste num sistema de expressão gênica induzida pelo substrato para identificação de novos genes catabólicos (UCHIYAMA et al., 2005). A análise baseada na sequência consiste tanto no sequenciamento completo de clones contendo âncoras filogenéticas como o gene 16S rRNA e o gene de reparo de DNA radA , que indicam o grupo taxonômico e informação funcional dos organismos dos quais derivaram tais clones (SINGH et al., 2009), quanto no sequenciamento aleatório de todo metagenoma para buscar âncoras filogenéticas em genomas reconstruídos (RIESENFELD et al., 2004; HOFF et al., 2008). A principal desvantagem em relação à abordagem funcional é a limitação quanto à identificação de apenas novos membros de famílias gênicas conhecidas. Em geral, genes alvos são identificados por primers aplicados em abordagens envolvendo hibridização e PCR, ou por sondas derivadas de regiões conservadas de genes ou produtos gênicos conhecidos (DANIEL, 2005; HANDELSMAN, 2004). Assim, embora a seleção baseada na sequência não seja a mais indicada para prospecção de novos genes ou produtos gênicos inteiros, é possível encontrar novos genes ao sequenciar todo o metagenoma e, a partir de análises de bioinformática, inferir a função de novos genes a partir daqueles conhecidos (DANIEL, 2005; HANDELSMAN, 2004), como foi feito para a descoberta de novas enzimas funcionais tais como quitinase, álcool oxirredutases, diol desidratases, e 26 outras que conferem resistência a antibióticos (SIMON; DANIEL, 2009). A vantagem dessa estratégia de seleção é a independência da expressão de genes exógenos no hospedeiro utilizado para construção de uma biblioteca metagenômica (LORENZ et al., 2002). Genes novos são um grande desafio a ser vencido, pois se não é observada similaridade com sequências já descritas, em geral não tem sido possível inferir funções. Apesar da análise funcional ser a mais indicada para identificação de clones que expressem um fenótipo particular, seguida da caracterização genética e bioquímica, a análise do perfil taxonômico e metabólico do metagenoma como todo permite uma melhor compreensão de fenômenos. Desta forma, análises baseadas em função e em sequência se complementam, tornando a pesquisa mais robusta. 2 JUSTIFICATIVA Diversos estudos têm identificado novos produtos gênicos a partir da abordagem de seleção funcional, dentre eles citam-se: esterase/lipase (HENNE et al., 2000), protease (GUPTA et al., 2002), álcool oxidoredutase (KNIETSCH et al., 2003), celulase (REES et al, 2003), dehidratase (KNIETSCH et al., 2003b), amidase (GABOR et al., 2004), amilase e β-lactamase (GABOR, 2004), poliphenol oxidase (BELOQUI et al., 2006) endoglucanase (PANG et al., 2009), lipase termoestável alcalina (MEILLEUR, 2009), e metaloproteases (WASCHKOWITZ et al., 2009), novas DNA polimerases (SIMON et al., 2009), genes que conferem resistência ao estresse salino (KAPARDAR et al., 2010), dentre outros. Apesar do potencial biotecnológico associado à metagenômica, trabalhos relativos à identificação de novos genes envolvidos na biodegradação de hidrocarbonetos e na síntese de biossurfactantes por abordagem metagenômica ainda são escassos. Desta forma a exploração da diversidade microbiana ambiental poderá contribuir não somente quanto ao desenvolvimento de estratégias de biorremediação dos impactos ambientais, mas também quanto à exploração e ao refinamento de petróleo, recurso natural que ainda é a principal fonte energética mundial. 27 O Brasil está entre os países autossuficientes em petróleo e a exploração plena da camada pré-sal recentemente descoberta deverá impulsioná-lo cada vez mais para as primeiras posições do ranking mundial. No âmbito estadual, no ano de 2010, o Rio Grande do Norte (RN), o maior produtor do petróleo nacional, em terra, produziu aproximadamente 333,9 milhões de barris (terra) e 185,7 milhões de barris (mar), tendo 3.569 poços produtores em terra enquanto no mar possui apenas 100 poços (ANP, 2011), consequentemente o estado é também um potente poluidor dos seus ecossistemas. Neste contexto, projetos metagenomas que busquem novos genes e biocatalisadores de interesse biotecnológico voltados para biorremediação são urgentes, uma vez que a utilização do petróleo com seus impactos ambientais ainda continuarão por décadas. 3 OBJETIVO 3.1 GERAIS O presente trabalho tem como objetivos gerais identificar novos genes envolvidos na biodegradação de hidrocarbonetos e na síntese de biossurfactantes por seleção funcional em duas bibliotecas metagenômicas de microbiota aquática e, analisar a composição taxonômica e metabólica de ambos metagenomas. 3.2 ESPECÍFICOS Construir duas bibliotecas metagenômicas utilizando DNA extraído de amostras de água de estuário e mar; Analisar fisico-quimicamente as amostras (pH, temperatura, oxigênio dissolvido, condutividade / salinidade e metais ); Selecionar clones envolvidos na biodegradação de hidrocarbonetos e/ou síntese de biosurfactantes; Sequenciar e analisar in silico os clones selecionados; 28 Analisar comparativamente os perfis taxonômico e metabólico dos metagenomas; Identificar nos metagenomas o perfil de distribuição de genes e proteínas envolvidos em biodegradação de hidrocarbonetos e síntese de biossurfactantes. 4 MATERIAL E MÉTODOS 4.1 COLETA DAS AMOSTRAS AMBIENTAIS Os pontos de coleta foram o estuário do rio Jundiaí/Potengí – Natal/RN e a praia de Pirangi do Norte – Parnamirim/RN. O estuário localiza-se no litoral oriental do RN, entre a praia de Santa Rita (ao norte) e o Parque das Dunas (ao sul) situando-se na Bacia Potengi (Secretaria de Meio Ambiente e Recursos Hídricos/RN – SEMARH, 2009). São diversos os tipos de efluentes que o estuário recebe das mais diversas atividades: indústrias e distritos industriais, imunizadoras, estações de tratamento de efluentes, carcinicultura e um hospital. Assim, é um ambiente cujos microrganismos podem apresentar capacidades metabólicas interessantes do ponto de vista industrial. A praia de Pirangí possui águas tranquilas e transparentes e situa-se no litoral sul do estado, à aproximadamente 20 km da capital Natal. Com o auxílio de uma rede de fitoplâncton com malha de 22 µm de porosidade, foram coletados, em maré alta, 4 L de água superficial filtrada de um volume total de aproximadamente 200 L nos dois pontos de coleta: No estuário (S 05º 46.921’, W 035º 14.844’) e outro no mar (S 05º 58.856’, W 035º 07, 225’). As amostras foram mantidas no gelo em frascos âmbar, previamente esterilizados com uma solução contendo ácido sulfúrico 100% e ácido nítrico 50% (6:1) e depois autoclavados, até o momento da extração do eDNA no laboratório em um período de até 24 h. 4.2 ANÁLISES FÍSICO-QUÍMICAS Os dados de condutividade/salinidade, pH, temperatura e oxigênio dissolvido foram obtidos em in situ usando um condutivímetro e um pH-metro. As 29 concentrações de nitrito, nitrato, chumbo, cromo, cobre, ferro, níquel e zinco nas amostras de água foram obtidas por espectrofotometria (Espectrofotômetro Micronal B572) utilizando o kit Vacu-vials® (CHEMetrics) segundo especificações do fabricante. 4.3 EXTRAÇÃO DO eDNA Para extração do eDNA foi utilizado o kit Power Water® DNA Isolation (MoBio Laboratories), com adaptações. Antes da extração, um gradiente de 0,1 – 1 L das amostras de água foram filtradas em membranas com poro de 0,2 µm (Millipore) com auxílio de uma bomba a vácuo (Millipore, WP6111560) para padronização do volume mínimo necessário para extração do eDNA. Logo após, as membranas contendo toda microbiota aderida foram submetidas aos procedimentos especificados pelo fabricante do kit. O sucesso da extração foi verificado através de eletroforese em gel de agarose 0,7%, 80 V, 80 mA e as concentrações dosadas em espectrofotômetro nanovue 18V DC (GE Healthcare Bio-Sciences AB). No final do processo, as amostras de eDNA resultante de cada extração foram precipitadas com 1/10 V de glicogênio (20 mg/mL), 1/10 V de NaOAc 3M (pH 5,2) e 2 V de etanol absoluto, e mantidas por 1 h em freezer - 80 ºC, seguida por centrifugação a 12.100 rpm por 20 min, lavagem dos precipitados com etanol 70%, nova centrifugação a 12.100 rpm por 20 min, secadas em speed vacuum (Eppendorf AG) por 10 min a temperatura ambiente, eluidas em TE (Tris-HCL pH 8; EDTA pH 8) e estocadas a - 20 ºC. 4.4 CONSTRUÇÃO DAS BIBLIOTECAS METAGENÔMICAS Para construção das bibliotecas metagenômicas foram utilizados ~20 µg de eDNA de microbiota estuarina e ~15 µg de microbiota marinha. Ambas as amostras de eDNA foram sonicadas 4 seg, potência 10 (Vibra cell /Sonics) em volume final de 300 µl em TE, para fragmentação. 30 4.4.1 TRATAMENTO DAS EXTREMIDADES DOS FRAGMENTOS GERADOS POR SONICAÇÃO Visto que a sonicação é um processo de fragmentação aleatória, antes da etapa de clonagem, foi necessário tratar as extremidades dos fragmentos de eDNA com as enzimas T4 DNA Polimerase (Biolabs) e T4 Polinucleotídeo Quinase (Biolabs), para gerar pontas cegas e fosforilar a extremidade 5’ dos fragmentos, respectivamente. Para tanto, na reação de polimerização foi utilizado: 1,5 µg de DNA sonicado, 1X tampão NEB2 (Biolabs), 0,5 mM de dNTPs, 2 U de enzima T4 DNA Polimerase (Biolabs) e água ultrapura autoclavada para o volume final de 80 µL. A reação foi mantida a 12 ºC por 30 min e logo após a 75 ºC por 20 min, em termociclador, para inativação da enzima. Na reação de fosforilação, foi adicionado ao mesmo tubo da reação anterior: 1x tampão da quinase (Biolabs), 2 mM de ATP, 10 U de T4 Polinucleotide quinase (Biolabs) e água ultrapura autoclavada para o volume final de 90 µL. A reação foi incubada por 10 min a 37 ºC e, para inativação da enzima, foi mantida a 65 ºC por mais 10 min. 4.4.2 PURIFICAÇÃO DOS INSERTOS TRATADOS Após o tratamento mencionado, foi realizada uma eletroforese em gel de agarose 0,7%, 27 V, 27 mA, por 24 h, para purificação dos fragmentos na faixa de 1,5 a 2 kb utilizando o kit GFX PCR DNA and Gel Band (Ge Healthcare) de acordo com as instruções do fabricante. Um nova eletroforese em gel de agarose 0,7%, 80 V, 80 mA, foi feita para confirmação da purificação. 4.4.3 PREPARAÇÃO DO VETOR PBC SK O vetor de clonagem utilizado neste trabalho foi o plasmídeo pBC SK (Stratagene) o qual foi digerido utilizando a enzima EcoRV (Amersham), seguido por desfosforilação das extremidades do vetor linearizado a fim de diminuir a probabilidade de recircularização. 31 A reação de digestão consistiu em 20 µg de pBC, 20 U de EcoRV (Amersham), 1x tampão H (Amersham) e água ultrapura autoclavada para o volume final de 70 µL, sendo mantida por 1 h a 37 ºC. Confirmada a digestão através de eletroforese em gel de agarose 0,7%, 80 V, 80 mA, foi realizada a reação de desfosforilação das extremidades do vetor. Para esta reação, 6,5 µg de pBC digerido foi desfosforilado com 5 U da enzima fosfatase alcalina CIP (Biolabs), 1X tampão NEB2 (Biolabs) e água ultrapura autoclavada para o volume final de 60 µL, sendo a reação mantida por 1 h a 37 ºC. Posteriormente, foi realizada eletroforese em gel de agarose 0,7%, (20 V, 20 mA, por 24 h) do vetor desfosforilado com o fim de isolar do gel apenas o pBC em sua forma linear, o que foi feito com o Kit GFX PCR DNA and Gel Band (Ge Healthcare). Após purificação, uma nova eletroforese em gel de agarose 0,7% permitiu estimar a concentração do vetor desfosforilado e linearizado. 4.4.4 CLONAGEM DO eDNA Partindo das concentrações conhecidas de cada amostra de eDNA (65 ng/µL MAR e 70 ng/µL EST ) e do vetor desfosforilado, foi estabelecido o volume necessário de eDNA para cada reação de ligação seguindo a razão 3:1 (eDNA : vetor). Assim, foi utilizado 1 µL de vetor, 4U de T4 DNA Ligase (Biolabs), 1X tampão da Ligase (Biolabs) e água ultrapura autoclavada completando o volume final para 25 µL. As reações foram mantidas a 16 ºC por 16-18 h e, em seguida, precipitadas para dessalinização e/ou retirada de impurezas com 1/10 V de glicogênio (20 mg/mL), 1/10 V de NaOAc 3M (pH 5,2) e 2 V de etanol absoluto, e mantidas por 1 h em freezer - 80 ºC, seguida por centrifugação a 12.100 rpm por 20 min, lavagem dos precipitados com etanol 70%, nova centrifugação a 12.100 rpm por 20 min e, então, permitidas secar a temperatura ambiente antes da adição de 15 µL de água ultrapura autoclavada para ressuspender a ligação precipitada e purificada. Para inserir o DNA transformante na cepa bacteriana DH10B [F- endA1 recA1 galE15 galK16 nupG rpsL ΔlacX74 Φ80lacZΔM15 araD139 Δ(ara,leu) 7697 32 mcrA Δ(mrr-hsdRMS-mcrBC) λ-] (GRANT et al.,1990), foi necessário torná-la eletrocompetente. Para tanto, ela foi submetida a sucessivas lavagens com água ultrapura e glicerol 10% gelados. Assim, uma colônia isolada da cepa foi crescida sob agitação a 37 ºC por 16-18 h em 2 mL de meio de cultura LB (Luria-Bertani) líquido. No dia seguinte, a cultura foi transferida para erlenmeyer de 2 L contendo 250 mL de meio LB líquido e mantida a 37 ºC sob agitação de 180 rpm. Obtida a densidade bacteriana entre 0,6-0,8, a cultura foi então distribuída em tubos de 50 mL, os quais foram centrifugados a 4 ºC por 15 min a 3.200 rpm. O precipitado obtido foi ressuspendido em 10 mL de água ultrapura autoclavada gelada e em seguida, o volume de água foi completado para 45 mL e novamente centrifugado a 4 ºC por 15 min a 3.200 rpm, sendo o processo repetido completando, entretanto, o volume para 25 mL antes de nova centrifugação. O precipitado foi ressuspendido em 5 mL de glicerol 10% gelado e mais uma vez centrifugado a 4 ºC por 15 min a 3.200 rpm. Uma alíquota única de 750 μL de glicerol 10% gelado foi utilizada para ressuspender o precipitado de um dos tubos, sendo a mesma transferida para o tubo seguinte, ressuspendendo o seu precipitado. O procedimento foi repetido até que os precipitados de todos os tubos estivessem unidos em um só tubo. Concluído esse passo, alíquotas de 55 μL foram distribuídas em microtubos previamente resfriados, e mantidos em freezer - 80 ºC até o momento do uso. Para a transformação bacteriana, 5 µL de cada reação de ligação foi adicionado a 55 μL de bactéria competente. A eletroporação foi realizada a 1800 V (Multiporator, Eppendorf) sendo o volume total ressuspendido em meio líquido LB previamente resfriado e incubado por 1 h a 37 ºC sob agitação constante de 180 rpm. A seleção dos transformantes foi realizada através de plaqueamento em meio LB agar suplementado com 25 µg/mL do antibiótico cloranfenicol e 40 µg/mL de Xgal e incubação a 37 ºC por 22 h. As colônias brancas obtidas foram inoculadas em placas de 96 poços contendo meio LB suplementado com 25 µg/mL de cloranfenicol por 22 h a 37 ºC sob agitação a 200 rpm para ser feito estoque, em microplacas de 96 poços, com glicerol 50% e mantidas em freezer - 80 ºC. 33 4.5 SELEÇÃO FUNCIONAL Montadas as bibliotecas metagenômicas, deram-se início aos ensaios funcionais em busca dos genes de interesse para este estudo, sendo analisada inicialmente a biblioteca contendo eDNA de microbiota estuarina (EST). Para a realização dos testes de seleção funcional, as colônias das placas-estoque foram inoculadas, com o auxílio de um replicador, em placas de 96 poços. 4.5.1 TESTE QUALITATIVO DE BIODEGRADAÇÃO DE HIDROCARBONETOS Para detecção da atividade de biodegradação de hidrocarbonetos do petróleo, foi realizado teste qualitativo onde 10 µL de cada poço da cultura crescida em placa de 96 poços foram transferidos para seu poço correspondente em placa de 96 poços (com tampa) contendo em cada um 180 µL de meio LB líquido suplementado com 25 µg/mL de cloranfenicol, mais uma gota de óleo árabe leve (OAL), adicionado com auxílio de uma seringa (para aplicação de insulina), correspondendo a uma alíquota pipetada de ~6 µL de água destilada. Para confirmação da atividade de biodegradação citada, placas de poliestireno com 24 poços (com tampa), contendo 100 µL de cultura de cada clone, 1,8 mL de meio LB líquido suplementado com 25 µg/mL de cloranfenicol e uma gota de óleo árabe leve (OAL), foram utilizadas. Nesse ensaio, DH10B com plasmídeo vazio (DH10B +pBC Ø) e LB não inoculado foram os controles. Em ambos os ensaios, a placa-teste foi incubada em estufa a 30 ºC por 15 dias, fotografada no primeiro e no décimo quinto dia de incubação. 4.5.2 HABILIDADE DE SINTETIZAR BIOSSURFACTANTES Os clones candidatos que apresentaram indício de atividade de biodegradação em placas de 96 poços foram avaliados quanto à capacidade de sintetizar biossurfactantes usando o teste de emulsificação (IQBAL et al., 1995) com adaptações. Então, para uma varredura preliminar, eles foram inoculados em placas de 96 poços contendo em cada poço 1 mL de meio YPG (yeast peptone glucose) pH 34 6,5, composto por extrato de levedura 10 g/L, peptona 10 g/L e glicose 10 g/L (ZEINALI et al., 2007), suplementado com 25 µg/mL de cloranfenicol, e crescidos por 22 horas a 37 ºC a 200 rpm. No dia seguinte, foi realizado o teste tanto com cultura quanto com o sobrenadante do seguinte modo: em microtubos foi adicionado 0,5 mL de querosene, 1 mL da cultura ou do sobrenadante livre de células, e para melhor visualização 20 µL de azul de metileno 0,05%; os tubos foram agitados em vortex por 30 seg e imediatamente após, fez-se a análise da capacidade de emulsificação do querosene. SDS 0,1% foi usado como controle positivo e DH10B + pBC Ø foi o controle negativo. Os clones candidatos tidos como positivos foram retransformados em DH10B e submetidos ao mesmo teste de emulsificação a fim de confirmar a atividade observada. Nesse ensaio, três colônias isoladas dos clones selecionados foram inoculadas em 2 mL de meio LB suplementado com 25 µg/mL de cloranfenicol e crescidas por 16-18 h a 37 ºC em 180 rpm. No dia seguinte, 200 µL da cultura foi transferida para um tubo de ensaio de 50 mL contendo 9,8 mL de YPG, mais 25 µg/mL de cloranfenicol, e permitidos crescer a 37 ºC a 180 rpm até atingirem a densidade óptica (D.O 600nm) de 0,4 e 0,8. Na medida em que atingiam as referidas D.O, 2 mL da cultura ou do sobrenadante foram adicionados nos seus respectivos tubos de ensaio de 15 mL contendo 2 mL de querosene e 200 µL de azul de metileno e logo submetidos a agitação em vortex por 1 min. O índice de emulsificação (E24%) foi aferido pela razão entre a altura da camada de emulsão e a altura total do sistema (medidas com paquímetro) multiplicada por 100. SDS 0,1% e DH10B + pBC Ø foram os controles, como supracitado. E24% = altura da camada emulsificada x 100 altura total do sistema 4.5.3 CONFIRMAÇÃO DA PRESENÇA DOS INSERTOS AMBIENTAIS O isolamento do DNA plasmideal dos clones positivos foi feito através de miniprep (SAMBROOK; RUSSELL, 2001). A fim de verificar a presença dos insertos clonados em pBC SK foi feita dupla digestão como segue: 0,5 µg de DNA 35 plasmideal, 3 U das enzimas XhoI e PstI (Amersham), 1X tampão NEB 2 e água ultrapura autoclavada para o volume final de 25 µL. A reação foi mantida a 37 ºC por 2 h. Foi realizada eletroforese em gel de agarose 0,7%, 80 V, 80 mA usando o pBC linear vazio e o marcador de peso molecular 1kb DNA ladder (Invitrogen) como controles para visualizar a presença dos insertos e seus tamanhos relativos. 4.6 SEQUENCIAMENTOS Os insertos ambientais dos clones positivos para os testes funcionais foram sequenciados em ambas as extremidades com os primers T3 e T7, separadamente, utilizando o sequenciador automático Mega-Bace 1000 e o kit DYEnamic ET dye terminator (MegabaceTM) (Ge Healthcare). Para a reação, foram utilizados 250 ng de DNA, 4 µL de ET Kit, 3,2 pMol de primer e água ultrapura autoclavada para o volume final de 10 µL. A reação constituiu de uma etapa inicial de desnaturação por 2 seg a 95 ºC, seguida por 35 ciclos de 20 seg a 95 ºC para desnaturação, 15 seg para anelamento a 45 ºC e 1 seg a 60 ºC para extensão. Em seguida, para retirada de sais e/ou resquícios da reação anterior, foi realizada reação de precipitação utilizando 1/10 do volume de acetato de amônio 7,5 M, 2,5 V de etanol absoluto. A placa foi agitada em vortex, mantida por 15 min a temperatura ambiente e centrifugada a 4 ºC, 2.430 rpm por 30 min O sobrenadante foi descartado por inversão em papel absorvente e o precipitado lavado com 150 µL de etanol a 70% e a placa centrifugada a 4 ºC por 10 min a 2.430 rpm. A placa invertida sobre papel absorvente foi brevemente centrifugada até 1.215 rpm e o DNA no poço foi ressuspendido em 10 µL de tampão de amostra. A placa protegida da luz, utilizando papel alumínio, foi mantida a 4 ºC por 16 horas, quando, ao final desse período foi injetada no sequenciador automático Mega-Bace 1000. Para o pirosequenciamento (Sequenciador 454/Roche) dos metagenomas desse estudo, alíquotas de eDNA EST (354,1 ng/µL; razão 260/280 de 1,79) e MAR (240,2 ng/µL; razão 260/280 de 1,60) foram enviadas para o Laboratório Nacional de Computação Científica – Rio de Janeiro/RJ (LNCC). 36 4.7 ANÁLISES DE BIOINFORMÁTICA Após montagem dos contigs usando o programa codoncode aligner 3.7.1, estes foram comparados com sequências de proteínas na base de dados GenBank não redundante utilizando o BlastX do pacote BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). As ORFs (preditas no ORF Finder) foram submetidas ao CLUSTALW para alinhamento com o melhor resultado do blastx de cada uma. A composição taxonômica e o perfil metabólico dos metagenomas foram obtidos com o programa MG-RAST 3.0 (MEYER F. et al., 2008) utilizando a configuração padrão e análises estatísticas feitas com STAMP v.2.0.0 (PARKS e BEIKO, 2010) desconsiderando as sequências não classificadas e usando “Fisher’s exact test, two side, DP: Asymptotic - 0,95“. Para verificar a abundância nos metagenomas tanto de proteínas envolvidas nos processos de biodegradação quanto de genes conhecidos relacionados à síntese de biossurfactantes, foi feita uma busca no banco de dados UniProtKB/Swiss-Prot (http://web.expasy.org/docs/swiss-prot_guideline.htmL) por sequências de referência curadas; fez-se tblastn contra os metagenomas EST e MAR usando um e-value de 10-5 como ponto de corte e os resultados foram submetidos ao programa HMMER 3.0 (FINN R.D. et al., 2011) para verificação da presença de domínios funcionais. Por fim, os hits redundantes foram excluídos e os demais contabilizados. A figura 2 apresenta um resumo da metodologia empregada nesse estudo. 37 Figura 2. Esquema resumindo a estratégia usada nesse estudo. Parte do mesmo eDNA usado para construção das bibliotecas metagenômicas (abordagem funcional) também foi pirosequenciado (454) para análises comparativas do perfil taxonômico e metabólico dos metagenomas. 38 5 RESULTADOS 5.1 BIBLIOTECAS METAGENÔMICAS Foram construídas duas bibliotecas metagenômicas contendo eDNA de microbiota estuarina (EST) e marinha (MAR). A biblioteca EST é constituída por 1152 clones e a biblioteca MAR com 1344 clones. Os ensaios de seleção funcional foram realizados apenas com os clones da biblioteca EST. 5.2 ANÁLISES FÍSICO-QUÍMICAS DAS AMOSTRAS AMBIENTAIS A tabela 1 mostra que os metais cobre, nitrito e níquel apresentaram-se em quantidade superior aos níveis permitidos pelo CONAMA em ambas amostras considerando águas salobras de classe 2 (para EST) e águas salinas de classe 1 (para MAR). Tabela 1. Análise Físico-química de Amostras de Água do Estuário Jundiaí/Potengí (Natal/RN) e da Praia de Pirangí do Norte (Parnamirim/RN) PARÂMETROS FISICO - QUÍMICOS UNIDADE ÁGUAS SALINAS CLASSE 1* MAR ÁGUAS SALINAS CLASSE 2* EST Nitrito Nitrato Chumbo Cromo Cobre Ferro Níquel Zinco Oxigênio Dissolvido pH Salinidade/Condutividade Temperatura mg/L mg/L mg/L mg/L mg/L mg/L mg/L mg/L mg/L O2 0-14 MiliS/cm ºC 0.070 0.400 0.010 0.050 0.005 0.300 0.025 0.090 ≥ 6.0 6,5 – 8,5 - 0.900** 0.130 0.000 0.003 0.540** 0.240 0.200** 0.000 6.31 9.0 0π*** 28,5 0.200 0.700 0.210 1.100 0.008 0.074 0.120 ≥4 6,5 – 9.0 - 0.240** 0.420 0.000 0.000 0.750** 0.300 0.100** 0.000 6,5 9.0 0π*** 28 * Valores máximos permitidos pela resolução 357/2005 do CONAMA ** Níveis acima dos permitidos pelo CONAMA. *** Valor acima da capacidade de leitura do condutivímetro usado. 39 5.3 HABILIDADE DE SINTETIZAR BIOSSURFACTANTES Os clones candidatos que já haviam apresentado indício de degradação de OAL também foram submetidos à varredura inicial para identificar uma possível habilidade de emulsificação, já que os biossurfactantes são moléculas adjuvantes no processo de biodegradação (CHRISTOFI; IVSHINA, 2002). Do montante testado quatro clones confirmaram a atividade de emulsificação de querosene em DO 600 nm 0,4 e/ou 0,8. Tanto a cultura quanto o sobrenadante livre de células dos clones positivos para atividade de síntese de biossurfactantes foram capazes de formar emulsão com o querosene, mas com diferenças na fase de crescimento bacteriano. Enquanto os clones 7A4 e 1G12 produziram biossurfactante na fase lag (DO 600 0,4) e na fase exponencial (DO600 0,8), o clone 3C2 apresentou atividade apenas na fase exponencial e o clone 6E4 só na fase lag (Tabela 2). Tabela 2. Clones Positivos para o Teste de Emulsificação de Querosene TESTE DE EMULSIFICAÇÃO Cultura Sobrenadante (E24 % ± dp) Clones DO600 0.4 (E24 % ± dp) DO600 0.8 DO600 0.4 7A4 __ 91,08 % ± 9,4 85,33 % ± 2,8 1G12 86,33 % ± 11,0 3C2 __ 6E4 DH10B + pBC Ø DO600 0.8 91,33 % ± 4,0 89 % ± 7,9 87 % ± 5 __ 85,66 % ± 11,0 70,66 % ± 7,5 __ 88,66 % ± 2,8 __ 74 % ± 11,5 __ 53,33 % ± 3,51 52,33 % ± 2,5 55,66 % ± 3,7 58,66 % ± 2,8 SDS 0,1 % __ = sem atividade. (E24 %= 99 %) 40 A análise BlastX das sequências dos clones positivos para síntese de biossurfactantes indicou similaridade a uma proteína hipotética de Magnetococcus sp. para a sequência do clone 7A4; as sequências codificadas pelos clones 1G12, 3C2 e 6E4 foram similares a mesma proteína hipotética de Candidatus Koribacter versatilis (Tabela 3). Para inferir uma provável função dessas proteínas hipotéticas foram feitas buscas por domínios conservados no CDD. Tabela 3. Melhores Resultados do BLASTX dos Clones Positivos para Síntese de Biossurfactantes CLONES ORF / TAMANHO BLASTX E-VALUE (Ident.) CLUSTALW Identidade Forte similaridade Frame = -3 / 201aa Proteína hipotética [Magnetococcus sp. MC-1] 0.022 (38 %) 5.15 % 3.49 % 7A4 Frame = -3 / 240aa Proteína hipotética Acid345_4742 [Candidatus Koribacter versatilis Ellin345] 2e-22 (41 %) 24.48 % 12.86 % 1G12 Frame = -2 / 172aa Proteína hipotética Acid345_4742 [Candidatus Koribacter versatilis Ellin345] 1e-24 (42 %) 12.86 % 16.49 % 3C2 Frame = -1 / 236aa Proteína hipotética Acid345_4742 [Candidatus Koribacter versatilis Ellin345] 7e-24 (42 %) 25.59 % 11.42 % 6E4 Os resultados mostraram que a sequência do clone 7A4 tem um domínio NADB (Rossmann-fold NAD(P)H/NAD(P)(+) binding) encontrado geralmente em dehidrogenases de vias metabólicas tais como glicólise e em outras enzimas redox. De modo geral, a família de proteínas que compartilham esse domínio possui também um segundo domínio que é responsável por especificamente ligar-se a um 41 substrato e catalisar uma reação enzimática particular. As proteínas hipotéticas codificadas pelas sequências dos clones 1G12, 3C2 e 6E4 apresentaram um domínio de função desconhecida (DUF898), uma grande família de proteínas do banco de dados Pfam que não foram caracterizadas. Assim, futuros ensaios funcionais específicos permitirão a caracterização dessas novas proteínas de origem metagenômica. A figura 3 mostra o ensaio para o teste de emulsificação usado. SDS 0,1 % 1G12 DHA10B+pBC Ø A SDS 0,1 % DHA10B+pBC Ø 7A4 B Figura 3. Teste de emulsificação. A) Teste com a cultura do clone 1G12 (DO 600 0.8); B) Teste com o sobrenadante do clone 7A4 (DO600 0.8). SDS 0,1 % e DH10B são os controles positivo e negativo, respectivamente. 42 5.4 BIODEGRADAÇÃO DE HIDROCARBONETOS Nos ensaios funcionais para atividade de biodegradação de hidrocarbonetos de OAL, do total de 1152 clones testados até o momento 14% apresentaram indício de atividade em placas de 96 poços após 15 dias de incubação a 30 °C, dos quais 4 confirmaram a atividade em placas de 24 poços no mesmo período de incubação. Como pode ser visto na figura 4. A) B) SDS 0,1 % 1G12 DHA10B+pBC Ø DH10B +pBC Ø LB não inoculado DH10B+pBCØ 5G9 7B9 6B7 5H9 7D6 DH10B+pBCØ 5G9 7B9 6B7 5H9 7D6 LB 3A9 5H10 6C8 6E1 LB LB 3A9 5H10 6C8 6E1 LB Clones em duplicata As setas indicam atividade após 15 dias/30 ºc. Figura 4. Teste de confirmação da atividade de biodegradação. a) placa no 1º dia do ensaio; b) placa no 15º dia de incubação e mapa de localização dos clones na placa. 43 A análise por BlastX das sequências dos clones positivos para o teste de biodegradação de OAL indicou similaridade a um receptor dependente de TonB de Pseudomonas sp. tanto para o clone 6E1 quanto para o 6B7; já a sequência do clone 5H9 apresentou similaridade a uma proteína hipotética de Xanthobacter autotrophicus Py2, e a do 6C8 foi similar ao fator sigma 24 da RNA polimerase de Roseiflexus castenholzii DSM 13941 (Tabela 4). Não foi encontrada a presença de domínio na proteína hipotética do clone 5H9. Porém, é possível que novos domínios que ainda não foram descritos no banco de dados estejam presentes nessa proteína. Tabela 4. Melhores BLASTX dos Clones Positivos para Atividade CLONES ORF /Resultados doBLASTX E-VALUE CLUSTALW de Biodegradação TAMANHO (Ident.) Identidade Forte similaridade Receptor dependente de TonB [Pseudomonas fluorescens PfO -1] 7e-15 (38 %) 5.88% 2.60% Frame = 2/203aa Receptor dependente de TonB [Pseudoalteromonas sp. BSi20311] 5e-04 (68 %) 6.02% 4.86% 5H9 Frame = 3/201aa Proteína hipotética [Xanthobacter autotrophicus Py2] 5e-19 (75 %) 5.15% 3.49% 6C8 Frame = -2 /237aa Fator sigma 24 da RNA polimerase [Roseiflexus castenholzii DSM 13941] 14.88% 12.81% 6E1 Frame -1 /136aa 6B7 3e-04 (55 %) 44 5.5 DADOS GERAIS,COMPOSIÇÃO TAXONÔMICA E METABÓLICA DOS METAGENOMAS. Após passarem pelo controle de qualidade do MG-RAST, o metagenoma de microbiota aquática marinha (MAR) ficou com 123.116 sequências (45.000.469 pb) com uma média de 365 ± 118 pb e média percentual de conteúdo GC de 47 ± 11%, ao passo que o metagenoma de microbiota aquática estuarina (EST) com 127.563 sequências (44.976.324 pb) com uma média de 352 ± 120 pb e média percentual de conteúdo GC de 45 ± 10%. Os metagenomas apresentaram uma riqueza de espécies praticamente idênticas com as curvas tendendo a um plateau como pode ser obervado na figura 5. Figura 5. Curvas de rarefação dos metagenomas EST (em azul) e MAR (em vermelho). Conforme a figura 6, nos dois metagenomas observa-se uma predominância do domínio Bacteria, seguido por Eukaryota, mais abundante no metagenoma MAR, Viruses, e Archaea. Porém, nota-se uma elevada proporção de sequências não assinadas pelo MG-RAST, as quais representam o enorme potencial genético ainda desconhecido desses ambientes, mas que precisa ser explorado. 45 Valor de p corrigido Intervalo de confiança 95% Não assinado Proporção (%) Diferença entre as proporções (%) Figura 6. Composição taxonômica (Domínio) dos metagenomas EST e MAR. Proteobacteria foi o filo mais abundante em ambos metagenomas, mas com uma proporção maior no estuário (EST), seguido de Bacteriodetes (mais abundante em MAR), Actinobacteria (mais frequente em EST), Cyanobacteria (maior em MAR) e Firmicutes, para o qual não se observa diferença entre as proporções de sequências correlatas (Figura 7). Valor de p corrigido Intervalo de confiança 95% Não assinado Proporção (%) Diferença entre as proporções (%) Figura 7. Composição taxonômica (Filo) dos metagenomas EST e MAR. 46 Essa predominância de Proteobacteria é refletida também na composição taxonômica em nível de classes, pois as mais abundantes pertencem a esse filo. Como mostra a figura 8, Alphaproteobacteria foi a mais frequente, com perfis praticamente semelhantes em ambos metagenomas, seguido de Gammaproteobacteria, mais abundante em EST, Flavobacteria, mais frequente em MAR, Actinobacteria, Betaproteobacteria, essas também mais abundantes em EST, e Deltaproteobacteria com uma proporção ligeiramente maior em MAR. Valor de p corrigido Intervalo de confiança 95% Não assinado Não assinado Proporção (%) Diferença entre as proporções (%) Figura 8. Composição taxonômica (Classe) dos metagenomas EST e MAR. Os perfis metabólicos dos metagenomas EST e MAR em nível 1 do MGRAST são bastante similares com pequenas diferenças em poucas categorias. Por exemplo, a presença de sequências relacionadas ao metabolismo de compostos aromáticos, função de interesse nesse estudo, possui praticamente a mesma proporção nos dois metagenomas como indicado pela figura 9. 47 Intervalo de confiança 95% Phagos, Prophagos,ElementosTransponíveis e Plasmideos Carboidratos Aminoácidos e Derivados Metabolismo de DNA Metabolismo de Fósforo Valor de p corrigido Metabolismo de RNA Metabolismo de Compostos Aromáticos Parede Celular e Cápsula Metabolismo Secundário Fotossíntese Transporte de Membrana Aquisição e Metabolismo de Ferro Resposta ao Estresse Cofatores, Vitaminas, Grupos Prostéticos, Pigmentos Nucleosídeos e Nucleotídeos Ácidos Graxos, Lipídeos e Isoprenoides Proporção (%) Diferença entre as proporções (%) Figura 9. Composição metabólica (Nível 1 do MG-RAST) dos metagenomas. Do mesmo modo, nos perfis representados pelo mapa de vias do KEGG observa-se que a maioria das vias, destacadas pelas zonas de sobreposição em rosa, é comum a ambos metagenomas, (Figura 10). A figura 11 indica que os perfis de distribuição de alguns genes que codificam proteínas envolvidas na síntese de biossurfactantes conhecidos são praticamente idênticos para os dois metagenomas. Apenas o gene rhlb do operon rhlABRI de Pseudomonas sp. esteve presente no metagenoma EST (1,98%). Para os demais genes desse mesmo operon não houve sequências similares em nenhum dos metagenomas. Da mesma forma, verifica-se um padrão na distribuição das proteínas envolvidas nos processos de biodegradação de hidrocarbonetos. Porém, metano mono-oxigenase (7,21%) e ALKS (0,96%) estão presentes apenas. 48 Figura 10. Perfil metabólico representado pelo mapa de vias do KEGG. Em rosa a sobreposição entre as amostras, demonstrando a ocorrência de hits similares às proteínas das vias. As áreas realçadas em azul simbolizam resultados únicos em MAR e em vermelho as vias únicas em EST. (Figura em paisagem). 49 HITS A) HITS B) Figura 11. Perfis de distribuição de proteínas relacionada à atividade de biodegradação de hidrocarbonetos (a) e de genes envolvidos na síntese de biossurfactantes conhecidos (b). 50 6 DISCUSSÃO O presente trabalho aplicou abordagens metagenômicas de seleção funcional para buscar novos genes de interesse biotecnológico envolvidos nos processos de biodegradação de hidrocarbonetos e de síntese de biossurfactantes, e abordagens baseadas em sequência para analisar os perfis taxonômico e metabólico de dois metagenomas de microbiota aquática marinha e estuarina. Os receptores dependentes de TonB consistem em uma família de proteínas de membrana externa de bactérias Gram-negativas que atuam no reconhecimento e internalização de complexos Fe3+-sideróforos, bem como de grandes moléculas (>800 Da), tais como a vitamina B12, por transporte ativo (BRAUN et al., 1998). A biodegradação de hidrocarbonetos realizada por bactérias, um fenômeno importante para remediação ambiental, requer a captação desses compostos através da membrana celular sendo a primeira etapa da biodegradação (BERG et al., 2008). Além de genes codificantes de enzimas, as vias de degradação de hidrocarbonetos monoaromáticos tais como benzeno, tolueno, etilbenzeno e xileno (BTEX) incluem genes codificantes de proteínas de membrana externa. Visto que a membrana externa de bactérias Gram-negativas é envolta por uma camada lipopolissacarídica hidrofílica (NIKAIDO, 2003), essas proteínas de membrana são necessárias para facilitar o transporte de compostos hidrofóbicos através da membrana externa (BERG et al., 2008). Assim, as proteínas metagenômicas dos clones 6E1 e 6B7 similares a tais receptores (Tabela 4) podem estar atuando na captação dos hidrocarbonetos tornando-os substratos acessíveis às vias metabólicas da cepa de E. coli DH10B. O reconhecimento dos promotores em Eubacteria é realizado pelo fator de iniciação sigma (σ), uma subunidade que se liga a RNA polimerase e inicia a transcrição. As células têm diversos fatores sigmas alternativos que possuem propriedades de reconhecimento de promotores diferentes. A célula pode fazer escolhas de seu repertório de fatores sigmas para alterar seu programa transcricional em resposta a condições específicas de estresse e alterações morfológicas (GRUBER et al., 2003). 51 A bactéria Gram-negativa E. coli, cujos mecanismos de resposta ao estresse estão bem elucidados, tem dois compartimentos, o citoplasma e o envelope que inclui a membrana interna, periplasma e a membrana externa. Sabe-se que sistemas separados de resposta ao estresse celular atuam em cada compartimento (ALBA et al., 2003). A regulação da resposta ao estresse de envelope, também chamada de resposta ao estresse extracitoplasmático, difere da resposta ao estresse citoplasmático porque os sinais de indução são gerados no envelope. Existem duas vias de resposta ao estresse de envelope bem estudadas, uma dirigida pelo fator σE ECF (do inglês extracytoplasmic function) ou fator sigma 24, e a outra pelo sistema CpxAR (RAIVIO et al., 2001). O fator σE, codificado pelo gene rpoe de E. coli, é ativado por proteínas desenoveladas no envelope, bem como por estresse de parede celular, para transcrever diversos genes envolvidos no restabelecimento da integridade do envelope, enzimas que atuam na biossíntese de lipA (um componente de lipopolisacarídeo ou LPS) e genes codificantes de proteases (ALBA et al., 2003; LONETTO et al., 1994). Foi mostrado que outro fator sigma 54 (σ54) é crucial para a expressão de várias vias catabólícas codificadas por plasmídeos ou cromossomicamente tais como aquela para o metabolismo de m-xileno codificada pelo plasmídeo pWW0 TOL (RAMOS et al., 1997). Visto que o petróleo é uma mistura complexa de hidrocarbonetos tóxicos, a presença do fator σE no clone 6C8 (Tabela 4) pode contribuir para a resposta ao estresse através da ativação transcricional de genes relacionados não apenas ao restabelecimento da integridade do envelope, mas também daqueles possivelmente envolvidos nos processos de biodegradação de hidrocarbonetos. A capacidade dos sobrenadantes livres de células de emulsificar o querosene apresentada pelos clones obtidos nesse estudo (Tabela 2) é interessante do ponto de vista biotecnológico, uma vez que produtos liberados no meio extracelular simplificam passos de purificação e, portanto, diminui os custos de produção (ABOUSEOUD et al., 2008). Algumas bactérias e leveduras excretam biossurfactantes iônicos que emulsificam o substrato de carbono no meio de crescimento como os ramnolipídios produzidos por diferentes Pseudomonas sp. e os soforolipídios, produzidos por várias Torulopsis sp (KARANTH, 1999). 52 No entanto, outros microrganismos são capazes de modificar a estrutura da sua parede celular através da síntese de lipopolissacarídios ou biossurfactantes nãoaniônicos, como por exemplo, Candida lipolytica e C. tropicalis, que produzem lipopolissacarídios ligados à parede celular quando crescem em n-alcanos, e Rhodococcus erythropolis, Mycobacterium sp. e Arthrobacter sp., que sintetizam o biossurfactante não-aniônico trealose corinomicolatos (KARANTH, 1999). Do mesmo modo, a formação de emulsão entre a cultura dos clones encontrados nesse trabalho e o querosene (Tabela 2) indica também uma produção de biossurfactantes destinados à parede celular. Embora a habilidade emulsificante seja uma boa avaliação da produção de biossurfactante ela não é correlacionada com a redução da tensão superficial e viceversa (YOUSSEF et al., 2004). Os biossurfactantes de baixo peso molecular como glicolipídeos, lipopeptideos e fosfolipídios diminuem eficientemente a tensão superficial enquanto os polímeros de alto peso molecular são melhores em estabilizar emulsões (ROSENBERG, 1999). Emulsan, um biossurfactante polimérico produzido por Acinetobacter calcoaceticus, é um bom bioemulsificante, porém reduz a tensão superficial para 52 mN/m (ROSENBERG et al., 1979), quando é sabido que um bom surfactante pode possibilitar a diminuição da tensão superficial da água destilada de 72 mN/m para 30mN/m (MULLIGAN, 2005). Assim, a avaliação da tensão superficial trará uma conclusão mais precisa a respeito dos biossurfactantes produzidos pelos clones metagenômicos. Nos testes para verificar a habilidade de emulsificação dos clones, os índices obtidos nesse estudo (Tabela 3) foram superiores ao obtido pela cepa produtora de biossurfactante Halomonas salaria M27, isolada de sedimento marinho com histórico de exposição a hidrocarbonetos, a qual apresentou índice de emulsificação (E24%) de 42.3% em óleo diesel (OLIVERA et al., 2008). Em outro estudo que visava uma maior produtividade de biossurfactante por Pseudomonas fluorescens Migula 1895-DSMZ, foram obtidos valores de E24% de 55% para diesel e querosene, 50% para heptano e 45% para óleo de girassol utilizando diferentes condições de cultura (ABOUSEOUD et al., 2008). Foram demonstrados ainda, não apenas o efeito da fonte carbono na produção de ramnolipidio por Pseudomonas aeruginosa EM1 refletida no índice de 53 emulsificação do querosene que alcançou valores de 9%, quando cultivado em hexano, a 71%, quando em óleo de soja, glicose ou glicerol (WU et al., 2008), mas também valores de E24% de 46 a 99% para querosene; 23 a 100% para gasolina e de 40 a 95% para OAL, sendo esses valores alcançados a partir de um consórcio bacteriano obtido a partir de amostras de sedimento com histórico de derramamento de petróleo (KREPSKY et al., 2007). De modo geral, têm sido relatados valores de E24% similares, bem como inferiores e superiores aos obtidos nesse trabalho, entretanto, a maioria dos surfactantes de origem microbiana são específicos, solubilizando ou emulsionando hidrocarbonetos diferentes de forma distinta (KREPSKY et al., 2007; ABOUSEOUD et al., 2008), o que, por conseguinte, direciona a sua aplicação, sendo sugerido, portanto, testar os biossurfactantes obtidos pelos clones metagenômicos em outros substratos além do querosene. Desse modo, posteriores ensaios de caracterização funcional permitirá um maior esclarecimento de todos os processos propostos tanto para atividade de biodegradação de hidrocarbonetos quanto para síntese de novos biossurfactantes. A análise da composição taxonômica dos metagenomas estudados revelou a predominância de Proteobacteria (Figura 7), o principal filo do domínio Bacteria que inclui uma variedade de patógenos, tais como Escherichia, Salmonella, Vibrio, Helicobacter dentre outros gêneros. Outros gêneros são de vida-livre e responsáveis pela fixação de nitrogênio. Possuem diversos tipos de metabolismos incluindo bactérias aeróbias, anaeróbicas facultativas ou obrigatórias, quimioautotróficas e heterotróficas. Dentre as classes desse filo, Alphaproteobacteria e Gammaproteobacteria foram as mais abundantes nos metagenomas analisados, mas a segunda foi significativamente mais frequente em EST (Figura 8). Alguns Gammaproteobacteria participam do processo de oxidação de metano,o que pode explicar sua abundancia no estuário, visto que esse é um ambiente com intensa produção metano. Hailian et al (2006) estudando a diversidade e distribuição de bactérias heterotróficas pigmentadas, em águas do oceano pacífico ao sul da china e em um estuário, usando métodos tradicionais de cultivo e analise de sequencias do gene 16S rRNA, observou uma predominância em águas marinhas de gêneros 54 pertencentes à Alphaproteobacteria e Gammaproteobacteria, enquanto no estuário foram encontrados isolados bacterianos afiliados principalmente a Actinobacteria e Firmicutes. Embora não tenha sido mais abundante em relação aos outros filos, no presente estudo Actinobacteria também foi mais frequente em EST do que em MAR (Figura 8). As bactérias desse filo são fundamentalmente saprófitas, e as mais conhecidas são de solo, onde contribuem significativamente para ciclagem de polímeros complexos como lignocelulose, hemicelulose, pectin, queratina e quitina, e produtoras de metabólitos secundários, portanto com elevado valor do ponto de vista farmacológico e comercial. Porém, há evidencias de que Actinomycetes, um grupo importante desse filo, pode ser encontrado na água e em sedimento do mar (MORAN et al.,1995). Um estudo sobre a diversidade e abundancia de comunidades microbiológicas do mar argentino e do estuário do Rio La Plata usando a técnica de pirosequenciamento foi realizado por Peressuti et al (2010). Seus resultados indicaram que Bacteriodetes (Flavobacteria) foi dominante no mar, seguido de Proteobacteria (Gammaproteobacteria), enquanto no estuário o segundo foi mais abundante, como foi observado no metagenoma do estuário do Rio Potengí (EST). Andreote et al (2012), também usou a técnica de pirosequenciamento para analisar o potencial metabólico e taxonômico de microbiomas de sedimento de mangues brasileiros e encontrou uma predominância de Deltaproteobacteria e Gammaproteobacteria, o que corrobora com os dados de Dos Santos et al (2011), que também usou pirosequenciamento e detectou a predominância desses grupos em mangues sob condições naturais e após a simulação de um derramamento de petróleo. Em sedimentos de mangues os organismos relacionados a redução de sulfato são Deltaproteobacteria, os quais são abundantes indicando a importância de tal metabolismo nesse ambiente (ANDREOTE et al., 2012) Abordagens baseadas em sequência permitiu inferir uma semelhança no perfil de distribuição nos dois metagenomas das proteínas e dos genes elencados. Isso indica que esses ambientes possuem um potencial metabólico natural parecido para síntese de biossurfactantes tais como surfactina, para biodegradação de hidrocarbonetos aromáticos como indicado pela quantidade de sequências similares 55 a benzeno dioxigenase, naftaleno dioxigenase, bifenil dioxigenase, tolueno dioxigenase, catecol 1,2 dioxigenase, catecol 2,3 dioxigenase e para o metabolismo de alcanos intermediado pelas alcano-hidroxilases ALKB e CYP153 (Figura 11). Esses enzimas atuam na clivagem do anel aromático e são responsáveis pela capacidade de degradação de compostos aromáticos em um grande número de espécies bacterianas (BRODERICK, 1999). Como aromáticos são componentes reduzidos, aumentando o estado de oxidação do núcleo aromático, esses compostos ficam mais susceptíveis a degradação permitindo aos microrganismos usá-los como única fonte de carbono e energia. Isso só é possível pela incorporação de oxigênio molecular mediante a participação das oxigenases (mono e dioxigenases), visto que o oxigênio, devido a sua estrutura química peculiar, é de baixa reatividade (HAYAISHI et al., 1955). Um estudo de análise metagenômica de amostras de solos, contaminados e não contaminados com diesel, do alto ártico canadense também verificou a abundancia de sequências proteicas de referências de alcanos-hidroxilases (como CYP153 e ALKB) e dioxigenases tais como catecol 2,3 dioxigenase e protocatechuato 4,5-dioxigenase (extradiol), além de catecol 1,2 dioxigenase e protocatechuato 3,4-dioxigenase (intradiol) como um indicativo da atividade de biodegradação de sua comunidade microbiológica antes desse ambiente ter sido submetido à tratamento de biorremediação, após um mês do início do tratamento e após um ano em comparação com a amostra de solo não contaminado usado como controle (YERGEAU et al., 2012). Apesar dos perfis metabólicos serem praticamente idênticos, como mostrado pela composição metabólica de nível 1 do MG-RAST e pelo mapa de vias do KEGG, (Figuras 9 e 10) foi verificada a presença de sequências similares a metanooxigenase apenas no metagenoma de microbiota estuarina (Figura 11). Isso pode ser explicado pela produção desse gás decorrente da atividade de decomposição da matéria orgânica característica do ecossistema, revelando um potencial biotecnológico peculiar para novos bioprodutos relacionados ao metabolismo de metano. A presença em ambos metagenomas de sequências similares aos genes e ás proteínas conhecidas,mostra também que esses ambientes são uma rica fonte de 56 produtos potencialmente aplicáveis a biorremediação e refino de hidrocarbonetos derivados do petróleo. A biodiversidades da microbiota desses ambientes representam o quanto há de inexplorado em relação aos aspectos ecológicos e biotecnológicos. O que ressalta a necessidade de mais estudos dessa natureza que poderão permitir novos insights sobra a dinâmica das comunidades microbianas, cuja variabilidade genética as torna uma fonte inestimável de bioprodutos biotecnologicamente aplicáveis nas mais diversas áreas, incluindo-se a biorremediação de ambientes impactados por hidrocarbonetos derivados do petróleo. 7 CONCLUSÕES A aplicação de abordagens metagenômicas de seleção funcional foi eficiente na busca de clones portando sequências de novos genes envolvidos na biodegradação de hidrocarbonetos e na síntese de biossurfactantes, visto que oito clones biotecnologicamente promissores foram encontrados a partir de uma biblioteca de eDNA de microbiota estuarina. As análises das sequências metagenômicas geradas por pirosequenciamento indicaram perfis taxonômicos dos metagenomas estudados com predominância do domínio Bacteria, representado principalmente por Proteobacteria, cujas classes Gammmaproteobacteria e Alphaproteobacteria foram as mais frequentes. Os perfis metabólicos dos metagenoma estudados foram praticamente idênticos com uma distribuição parecida de genes e proteínas envolvidos com a biodegradação de hidrocarbonetos e síntese de biossurfactantes. 57 8 REFERENCIAS BIBLIOGRÁFICAS ABULENCIA, C. B., et al. Environmental whole genome amplification to access microbial populations in contaminated sediments. Appl. Environ. Microbiol. v.72, p. 3291–3301, 2006. ABOUSEOUD, M., YATAGHENE, A., AMRANE, A., MAACHI, R. Biosurfactant production by free and alginate entrapped cells of Pseudomonas fluorescens. J Ind Microbiol Biotechnol, v. 35, p. 1303 – 1308, 2008. ALBA, B.M., GROSS, C.A. Regulation of the Escherichia coli σE - dependent envelope stress response. Mol. Microbiol, Inpress, 2003. ALBERS, S.-V., et al. Production of recombinant and tagged proteins in the hyperthermophilic archaeon Sulfolobus solfataricus. Appl. Environ. Microbiol. v. 72, p. 102–111, 2006. AL-HADHRAMI , M. N.; LAPPIN-SCOTT, H. M.; FISHER, P. J. Bacterial survival and n-alkane degradation within Omani crude oil and a mousse. Marine Pollution Bull., v.30, p. 403-408, 1995. ALEXANDER, M. Biodegradation and bioremediation. 302 p, Academic Press, 1994. AMANN, R.J., et al. Combination of 16S rRNA targeted oligonucleotide probes with flow-cemetry for analyzing mixed microbial populations. Appl Environ Microbiol. v.56, p. 1919–1925, 1990. ANGELOV, A., M. MIENTUS, S. LIEBL, and W. LIEBL. A two-host fosmid system for functional screening of (meta)genomic libraries from extreme thermophiles. Syst. Appl. Microbiol. v.32, p.177–185, 2009. ANP - AGÊNCIA NACIONAL DO PETRÓLEO, GÁS NATURAL E BIOCOMBUSTÍVEIS, Anuário estatístico 2011. Disponível em: <http://www.anp.gov.br>. Acesso em: 23 mar 2012. ANDREOTE, F.D et al. The microbiome of Brazilian mangrove sediments as revealed by metagenomics. Plos One, v.7, p. 1-14, 2012. ATLAS, R.M.Petroleum biodegradation and oil spill bioremediation. Marine Pollution Bulletin, v. 31. p. 178-182, 1995. ATLAS, R.M. Microbial Degradation of Petroleum Hydrocarbons: an Environmental Perspective. Microbiol Rev, v. 45, p. 180-209, 1981. 58 BANAT, I.M., MAKKAR, R.S., CAMEOTRA, S.S. Potential commercial applications of microbial surfactantes. Appl Microbial Biotechnol, v. 53. p. 495-508,2000. BERG, V.D.B., HEARM, E.M., PATEL, D.R. Outer – membrane transport of aromatic hydrocarbons as a first step in biodegradation. PNAS, v. 105, p. 8601- 8606, 2008. BARTHA, R. Biotechnology of petroleum pollutant biodegradation. Microbiol Ecol v. 12. p. 155-172. BECHARD, J., et al. Isolation and partial chemical characterization of an antimicrobial peptide produced by a strain of Bacillus subtilis. J. Agric. Food Chem, v.46, p. 5355–5361, 1998. BELOQUI, A., et al, Novel polyphenol oxidase mined from a metagenome expression library of bovine rumen: biochemical properties, structural analysis, and phylogenetic relationships. The Journal of Biological Chemistry, v.281, p. 22933–22942, 2006. BENTO, F. M.., CAMARGO, F. A. O., OKEKE, B. Bioremediation of soil contaminated by diesel oil, Brazilian Journal of Microbiology, v.34, p. 65-68, 2003. BIERMANN, M. et al. Surfactants in consumer products, theory, technology and application. Springer-Verlag, Heidelberg, 1987. BIDDLE, J. F., S. FITZ-GIBBON, S. C. SCHUSTER, J. E. BRENCHLEY, and C. H. HOUSE. Metagenomic signatures of the Peru Margin sub sea floor biosphere show a genetically distinct environment. Proc. Natl. Acad. Sci. U. S. A. v. 105, p. 10583– 10588, 2008. BODOUR, A.A., et al. Distribution of biosurfactant-producing bacteria in undisturbed and contaminated arid southwestern soils. Appl Environ Microbiol, v. 69, p. 3280– 3287, 2003. BRADDOCK, J. F., LINDSTROM, J. E.,YEAGER, T. R., RASLEY, B. T., BROWN, E. J. Patterns of microbial activity in oiled and unoiled sediments in Prince William Sound EXXON VALDEZ. Proceedings of the Exxon Valdez oil spill symposium. Bethesda: Am Fish Soc, v.18, p. 94-108, 1996. BREDHOLT, H., et al. Emulsification of crude oil by an alkane-oxidizing Rhodococcus species isolated from seawater. Canadian Journal of Microbiology, v. 44, p. 330–340, 1998 BRODERICK, J.B. Catechol dioxigenases. Essays in Biochemistry, v. 34, p. 173189, 1999. BURGER, M.M., GLASER, L., BURTON, R.M. The enzymatic synthesis of a rhamnose-containing glycolipid by extracts of Pseudomonas aeruginosa. J Bio/ Chem, v. 238, p. 2595-2602, 1963. 59 BUSHNELL, L. D., HAAS, H. F. The utilization of certain hydrocarbons by microorganisms. J. Bacteriol., v. 41, p. 653-673, 1941. CAO, B., NAGARAJAN, K., LOH, K.C. Biodegradation of aromatic compounds: current status and opportunities for biomolecular approaches. Applied Microbiology and Biotechnology, v. 85, p. 207–228, 2009. CRAIG, J. W., et al. Expanding small-molecule functional metagenomics through parallel screening of broad-host-range cosmid environmental DNA libraries in diverse Proteobacteria. Appl. Environ. Microbiol. v.76, p. 1633–1641, 2010. CHISTOSERDOVA, L. Recent progress and new challenges in meta- genomics for biotechnology. Biotechnol Lett. v. 32, p. 1351–1359, 2010. COOPER, D.G., GOLDENBERG, B.G. Surface-active agents from two Bacilllus species. Applied and Environmental Microbiology, v.53 p. 224-229, 1987. CHRISTOFI, N., IVSHINA, I.B. Microbial surfactants and their use in field studies of soil remediation. Journal of Applied Microbiology, v. 93, p. 914-929, 2002. DANIEL, R. The metagenomics of soil. Nat. Rev. Microbiol. v. 3, p. 470–478, 2005. DELONG, E. F., et al. Community genomics among stratified microbial assemblages in the ocean’s interior. Science, v. 311, p. 496–503, 2006. DOS SANTOS, H et al. Mangrove bacterial diversity and the impact of oil contamination revealed by pyrosequencing: bacterial proxies for oil pollution. Plos One, v. 6, p. 1 – 8, 2011. DUAN, C. J., et al. Isolation and partial characterization of novel genes encoding acidic cellulases from metagenomes of buffalo rumens. J. Appl. Microbiol. v. 107, p. 245–25, 2009. DINSDALE, E. A. et al. Functional metagenomic profiling of nine biomes. Nature v.452, p. 629–632, 2008. El-TARABILY, K.A. Total microbial activity composition of a mangrove sediment are reduced by oil pollution at a site in the Arabian Gulf. Can. J. Microbiol, v. 48, p. 176– 182, 2002. FABRET C, QUENTIN Y, GUISEPPI A, BUSUTTIl J, HAIECH J, DENIZOT F: Analysis of errors in finished DNA sequences: the surfactin operon of Bacillus subtilis as an example. Microbiology. v.141, p. 345-350, 1995. GABOR, EM., et al. Construction, characterization, and use of small-insert gene banks of DNA isolated from soil and enrichment cultures for the recovery of novel amidases. Environ. Microbiol, v.6, p. 948–958, 2004. 60 GABOR, E., et al. Updating the metagenomics toolbox. Biotechnol J, v.2, p. 201– 206, 2007. GEORGIOU, G., LIN, S.C., SHARMA, M.M. Surface-active compounds from microorganisms. Bio ⁄ Technology, v.10, p. 60–65, 1992. GRANT, S.G.N. et al. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation restriction mutants. Proc. Natl. Acad, v.87, p.46454649, 1990. GRUBER, T.M., GROSS, C.A. Multiple sigma subunits and the partitioning of bacterial transcription space. Annu. Rev. Microbiol, v. 57, p. 441- 466, 2003. GUPTA, R., et al. Bacterial alkaline proteases: molecular approaches and industrial applications. Appl. Microbiol. Biotechnol. v. 59, p. 15–32, 2002. GALLI G, RODRIGUEZ F, COSMINA P, PRATESI C, NOGAROTTO R, de FERRA E GRANDI G: Characterization of the suffactin synthetase multi-enzyme complex. Biochim Biophys Acta. v. 1205, p. 19-28, 1994. GABOR, E. M., W. B. ALKEMA, D. B. JANSSEN. Quantifying the accessibility of the metagenome by random expression cloning techniques. Environ. Microbiol. v. 6, p. 879 –886. 2004. HAILIAN, DU., et al. Diversity and distribution of pigmented heterotrophic bacteria in marine environments. FEMS Microbiol Ecol, . v. 57, p. 92–105, 2006. HAMME, J.D., SINGH. A., WARD, O.P. Recent advances In petroleum microbiology. Microbiology and Molecular Biology Reviews, v. 67, p. 503–549, 2003. HANDELSMAN, J., RONDON, M. R., Brady, S. F., Clardy, J., Goodman, R. M., Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products, Chem Biol. v. 5, p. 245-249, 1998. HARAYAMA, S., KOK, M., NEIDLE, E.L. functional and evolutionary relationships among diverse oxygenases. Annual Review of Microbiology, v. 46, p. 565–601, 1992. HÅRDEMAN, F., S. SJO¨LING. Metagenomic approach for the isolation of a novel low-temperature-active lipase from uncultured bacteria of marine sediment. FEMS Microbiol. Ecol. v. 59, p. 524–534, 2007. HEALY, F. G., et al. Direct isolation of functional genes encoding cellulases from the microbial consortia in a thermophilic, anaerobic digester maintained on lignocellulose, Appl Microbiol Biotechnol. v. 43, p. 667-74. 1995. 61 HEATH, C., et al. Identification of a novel alkaliphilic esterase active at low temperatures by screening a meta- genomic library from Antarctic desert soil. Appl. Environ. Microbiol. v. 75, p. 4657–4659, 2009. HENNE, A., et al. Screening of environmental DNA libraries for the presence of genes conferring lipolytic activity on Escherichia coli. Appl. Environ. Microbiol, v. 66, p. 3113–3116, 2000. HOFF, K.J., et al. Gene prediction in metagenomic fragments: a large scale machine learning approach. BMC Bioinformatics, v.9, p. 217, 2008. IQBAL, S., KHALID, Z. M., MALIK, K. A. Enhanced biodegradation and emulsification of crude oil and hyperproduction of biosurfactants by a gamma ray induced mutant of Pseudomonas aeruginosa, Lett Appl Microbiol. v. 21, p. 176-9, 1995. KAPARDAR, R.K. et al., Identification and characterization of genes conferring salt tolerance to Escherichia coli from pond water metagenoma. BioresourceTechnology, v.101, p. 3917–3924, 2010. KARANTH, S., LYSON, K., MCCANN, S. M. Effects of cholinergic agonists and antagonists on interleukin-2-induced corticotropin-releasing hormone release from the mediobasal hypothalamus. Neuroimmunomodulation, v. 6, p. 168 – 174, 1999. KATAOKA, A. P. A. G. Biodegradação de resíduo oleoso de refinaria de petróleo por microrganismos isolados de “landfarming”. Tese (Doutorado) – Instituto de Biociências, Unesp – Rio Claro, 2001. KENNEDY, JONATHAN et al. Marine metagenomics: new tools for the study and exploitation of marine microbial metabolism. Marine drugs. v. 8, p. 608-628, 2010. KNIETSCH, A., et al. Metagenomes of complex microbial consortia derived from different soils as sources for novel genes conferring formation of carbonyls from short-chain polyols on Escherichia coli. J. Mol. Microbiol. Biotechnol, v. 5, p. 46– 56, 2003. KNIETSCH, A., et al. Construction and screening of metagenomic libraries derived from enrichment cultures: generation of a gene bank for genes conferring alcohol oxidoreductase activity on Escherichia coli. Appl. Environ. Microbiol, v. 69, p. 1408–1416, 2003b. KREPSKY, N., DA SILVA, F. S., FONTANA, L. F., CRAPEZ, M. A. Alternative methodology for isolation of biosurfactant-producing bacteria, Braz J Biol, v. 67, p. 117-124, 2007. LAFRANCE, P., LAPOINTE, M. Mobilisation and co-transport of pyrene in the presence of Pseudomonas aeruginosa UG2 biosurfactants in sandy soil columns. Ground Water Monitoring and Remediation, v. 18, p.139–147, 1998. 62 LAMBALOT RH, GEHRING AM, FLUGEL RS, ZUBER P, LACELLE M, MARAHIEL MA, REID R, KHOSLA C, WALSH CT: A new enzyme superfamily the phosphopantetheinyl transferases. Chem Bio. v. 3, p. 923-936, 1996. LORENZ, P., ECK, J. Metagenomics and industrial applications. Nature, v.3, p. 510516, 2005. LONETTO, M. A., et al. Analysis of the Streptomyces coelicolor sigE gene reveals the existence of a subfamily of eubacterial RNA polymerase σ factors involved in the regulation of extracytoplasmic functions. Proc. Natl. Acad. Sci, v. 91, p. 7573 7577, 1994. MARIANO, A. P. Avaliação do potencial de biorremediação de solos e de águas subterrâneas contaminados com óleo diesel. 2006. 162f. Tese (doutorado) – Universidade Estadual Paulista, Instituto de Geociências e Ciências Exatas, Rio Claro, São Paulo. 2006. MEILLEUR, C., et al. Isolation and characterization of a new alkali-thermostable lipasecloned from a metagenomic library. J Ind Microbiol Biotechnol, v. 36, p. 853– 861, 2009. MILLER, Raina M. Biosurfactant-facilitated Remediation of Metal-contaminated Soils. Environ Health Perspect, v. 103, p. 59-62,1995. MORAN, M.A et al. Evidence for indigenous Streptomyces populations in a marine environment determined with a 16S rRNA probe. Appl Environ Microbiol, v.61, p. 3695-3700. MORIKAWA, M., et al. A new lipopeptide biosurfactant produced by Arthrobacter sp. strain MIS38, J Bacteriol, v. 175, p. 6459-66, 1993. MULLIGAN, C. N. Environmental applications for biosurfactants. Environmental Pollution, v. 113, p. 183 –198, 2005. NIKAIDO, H. Molecular basis of outer membrane permeability revisited. Microbiol Mol Biol Rev, v. 67, p. 593 – 656, 2003. OCHSNER UA, REISER J, FIECHTER A, WITHOIT B: Production of Pseudomonas aeruginosa rhamnolipid biosurfactants in heterologous hosts. App/Environ Microbiol, v. 61, p. 3503- 3506, 1995. OCHSNER UA, FIECHTER A, REISER J: Isolation, characterization, and expression in Escherichia coil of the Pseudomonas aeruginosa rhlAB genes encoding a rhamnosyltransferase involved in rhamnolipid biosurfactant synthesis. J B/o/Chern, v. 269, p. 19787-19795, 1994. 63 OLIVERA, N. L., et al. Isolation and characterization of biosurfactant-producing Alcanivorax strains: hydrocarbon accession strategies and alkane hydroxylase gene analysis, Res Microbiol. v.160, p. 19-26, 2008. PAGE, C.A., et al. Biosurfactant solubilization of PAHs. Environmental Engineering Science, v.16, p. 465–474, 1999. PALASHPRIYA,D., SOUMEN,M.,RAMKRISHNA.,S. Genetic regulation of the biosynthesis of microbial surfactants: an overview. Biotechnology and Genetic Engineering Reviews, v. 25, p. 165-186, 2008. PANG, H., et al, Identification of cellulase genes from the metagenomes of compost soils and functional characterization of one novel endoglucanase. Curr Microbiol, v. 58, p.404– 408, 2009. PARK, A.J., et al. Enhancing solubilization of sparingly soluble organic compounds by biosurfactants produced by Nocardia erythropolis. Water Environment Research, v.70, p. 351–355, 1998. PARKS, D.H. BEIKO, R.G. Identifying biologically relevant differences between metagenomic communities. Bioinformatics, v.26, p.715-721, 2010. PEIXOTO, R S; VERMELHO, A.; ROSADO, A. S. Petroleum-degrading enzymes: bioremediation and new prospects. Enzyme research, v. 2011, p. 1-7, 2011. PHALE, P., et al. Metabolic diversity in bacterial degradation of aromatic compounds. A Jounal of Integrative Biology, v.11, p. 252-279, 2007. PRESSUTTI,S.R., et al. Estudio comparativo de La estructura del bacterioplancton em águas del mar Argentino mediante el método de pirosecuenciacíon 454 tag. Revista Argentina de Microbiología, v. 42, p. 288-297, 2010. RAHIM,R., et al. Cloning and functional characterization of Pseudomonas aeruginosa rhlC gene tha encodes rhamnosiltransferase 2, an enzyme responsible for dirhamnolipid biosynthesis. Molecular Microbiology, v. 40, p. 708-718, 2001. RAIVIO, T., SILHAVY, T. Periplasmic stress and ECF sigma factors. Annu. Rev. Microbiol, v. 55, p. 591–624, 2001. RAJENDHRAN, J., GUNASEKARAN, P., Strategies for accessing soil metagenome for desired applications, Biotechnol Adv, v. 26, p. 576-90, 2008. RAMOS, J.L., MARQUÉS, S., TIMMIS, K.N. Transcriptional control of the Pseudomonas TOL plasmic catabolic operons is achieved through an interplay of host factors and plasmid - encoded regulators. Annu. Rev. Microbiol, v. 51, p. 341– 373, 1997. 64 REES, H.C., et al. Detecting cellulase and esterase enzyme activities encoded by novel genes present in environmental DNA libraries. Extremophiles, v.7, p. 415– 421, 2003. RIESENFELD, C.S., SCHOSS, P.D., HANDELSMAN, J. Metagenomics: genomic analysis of microbial communities. Annu Rev Genet, v.38, p. 525–552, 2004. ROSENBERG, E., RON, E. Z. High - and low - molecular - mass microbial surfactants. Appl Microbiol Biotechnol, v. 52, p. 154 – 162, 1999. ROSENBERG, E., ZUCKERBERG, A., RUBINOVITZ, C., GUTNICK, D. L. Emulsifier of Arthrobacter RAG-1: isolation and emulsifying properties. Appl Environ Microbiol, v. 37, p. 402 – 408, 1979. SANTOS, H.F., et al. Bioremediation of mangroves impacted by petroleum. Water, Air, and Soil Pollution. v. 216, p. 329–350, 2011a. SANTOS, H. F., et al. Mangrove bacterial diversity and the impact of petroleum contamination revealed by pyrosequencing: bacterial proxies for petroleum pollution. PLoS One, v. 6, no. 3, Article ID e16943, 2011b. SAMBROOK, J., RUSSELL, D. W. Molecular cloning: a laboratory manual. 3rd. New York: Cold Spring Harbor Laboratory Press, 2001. SCHMIDT, T. M., DELONG, E. F., PACE, N. R. Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing, J Bacteriol v.173, p. 43714378, 1991. SECRETARIA DE MEIO AMBIENTE E RECURSOS HÍDRICOS (SEMARH/RN). Sistema de Informações. Disponível em: HTTP://WWW.semarh.rn.gov.br/consulta/cBaciaDetalhe.asp?CodigoEstadual=08. Acesso em 09 out 2011. SCHEIBENBOGEN, K., et al. Enhanced removal of selected hydrocarbons from soil by Pseudomonas aeruginosa UG2 biosurfactants and some chemical surfactants. Journal of Chemical Technology and Biotechnology, v. 59, p. 53–59, 1994. SILVA, R.C.B. Prospecção de genes de interesse biotecnológico: uma abordagem metagenômica. 2009. 70f. Dissertação (mestrado) – Universidade Federal do Rio Grande do Norte, Centro de Ciências Biológicas, Natal, Rio Grande do Norte. 2009. SIMON, C., HERATH, J., ROCKSTROH, S., DANIEL, R. Rapid identification of genes encoding DNA polymerases by function-based screening of metagenomic libraries derived from glacial ice. Appl Environ Microbiol. v. 75, p. 2964–2968, 2009. 65 SINGH, P. K., et al. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature, v. 407, p. 762–764, 2000. SINGH, J., et al . Metagenomics: concept, methodology, ecological inference and recent advances. Biotechnol. J., v.4, p. 480–494, 2009. SORKHOH, N. A., GHANNOUM, M. A., IBRAIM, A. S., STRETTON, R. J., RADWAN, S. S. Crude oil and hydrocarbon-degrading strains of Rhodococcus rhodochous isolated from soil and marine environment in Kuwait. Environ. Pollution, v. 65, p.117, 1990. TORSVIK, V., GOKSOYR, J., DAAE, F. L High diversity in DNA of soil bacteria. Appl Environ Microbiol. v. 56, p. 782-787, 1990, TRINGE, S. G., et al. Comparative metagenomics of microbial communities. Science, v. 308, p. 554–557, 2005. TURNBAUGH, P. J.,GORDON, J.I. An invitation to the marriage of metagenomics and metabolomics. Cell. v. 134, p. 708–713, 2008. UCHIYAMA, T., T. ABE, T. IKEMURA, K. WATANABE. Substrate- induced geneexpression screening of environmental metagenome libraries for isolation of catabolic genes. Nat. Biotechnol. v.23, p. 88–93, 2005. VAN BEILEN, J.B., et al. Analysis of Pseudomonas putida alkane degradation gene cluster sand flanking insertion sequences: evolution and regulation of the alk genes. Microbiology, v. 147, p. 1621–1630, 2001. VAN BEILEN, J.B., WUBBOLTS, M.G., WITHOLT., B. Genetics of alkane oxidation by Pseudomonas oleovorans. Biodegradation, v. 5, p. 161–174, 1994. VOLLENBROICH, D., et al. Antimycoplasma properties and application in cell culture of surfactin, a lipopeptide antibiotic from Bacillus subtilis. Appl. Environ. Microbiol, v.63, p. 44–49, 1997. VOGET, S., H. L. STEELE, W., STREIT, R. Characterization of a metagenomederived halotolerant cellulase. J. Biotechnol. v. 126, p. 26–36, 2006. YERGEAU, E., et al. Metagenomic Analysis of the bioremediation of dieselcontaminated canadian high arctic soils. PLoSONE, v. 7, p. 1-10, 2012. YOUSSEF, N. H. Comparison of methods to detect biosurfactant production by diverse microorganisms, J Microbiol Methods, v. 56, p. 339-347. 2004. WASCHKOWITZ, T., ROCKSTROH, S., DANIEL, R. Isolation and characterization of metalloproteases with a novel domain structureby construction and screening of metagenomic libraries. Applied and Environmental Microbiology, v. 75, p. 2506– 2516, 2009. 66 WASCHKOWITZ, T., S. ROCKSTROH, R. DANIEL. Isolation and characterization of metalloproteases with a novel domain structure by construction and screening of metagenomic libraries. Appl. Environ. Microbiol. v.75:2506–2516, 2009. WANG, G.-Y.-S., et al. Novel natural products from soil DNA libraries in a streptomycete host. Org. Lett. v.2, p. 2401–2404, 2000. WU, C., B. SUN. Identification of novel esterase from metagenomic library of Yangtze River. J. Microbiol. Biotechnol. v.19, p. 187–193, 2009. WU, J. Y., YEH, K. L., LU, W. B., LIN, C. L., CHANG, J. S. Rhamnolipid production with indigenous Pseudomonas aeruginosa EM1 isolated from oil- contaminated site. Bioresour Technol, v. 99, p. 1157-1164. 2008. ZANG, Y., MILLER, R.M. Enhanced octadecane dispersion and biodegradation by Pseudomonas rhamnolipid surfactant (biosurfactant). Applied and Environmental Microbiology, v. 58, p. 3276–3282, 1992. ZEINALI, M., VOSSOUGHI, M., ARDESTANI, S. K. Characterization of a moderate thermophilic Nocardia species able to grow on polycyclic aromatic hydrocarbons, Lett Appl Microbiol. v. 45,p . 622-628, 2007.
Download