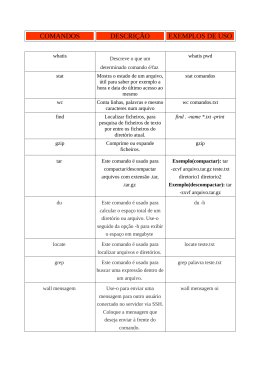
Análise de Composição Bacteriana com o QIIME Artur Trancoso Lopo de Queiroz – CPqGM/FIOCRUZ-BA RESUMO: A comunidade bacteriana que habita determinado nicho e, consequentemente, é capaz de exercer alguma influência sobre o mesmo é determinada Microbiota ou microbioma. No corpo humano estima-se que existam 10 vezes mais células de microrganismos do que células humanas. Esses microrganismos apresentam um grande impacto na modulação e formação do sistema imunológico. As bactérias comensais auxiliam na proteção contra as patogênicas através da competição direta pelo mesmo nicho. As plantas também apresentam uma ampla gama de relações com microrganismos simbiontes, onde a troca de nutrientes entre os parceiros simbióticos é uma parte importante da relação. A variação da composição de microrganismos pode afetar o vegetal de forma deletéria, trazendo-lhe deficiências nutricionais, por exemplo. A variação na composição das comunidades de microrganismos está associada com diversas patologias, como por exemplo o autismo, doença de Lyme, úlceras diabéticas, doenças intestinais e, já fora do escopo da saúde humana, associa-se a alterações na eficiência agrícola e no branqueamento de corais. Devido a sua importância, a determinação da comunidade microbiana tem sido alvo de estudos. A primeira abordagem foi utilizando metodologia de cultivo. Entretanto, o crescimento em meios nos permite identificar apenas 1% da comunidade microbiana, devido à seleção que ocorre durante o crescimento nas placas de Petri. Para evitar o viés de seleção que ocorre no cultivo, técnicas moleculares foram empregadas, como a PCR e sequenciamento do gene 16S. O custo de identificação bacteriana através do sequenciamento de Sanger é alto, devido a necessidade de realizar diversas clonagens para garantir uma boa cobertura dos organismos. As novas tecnologias de sequenciamento de alta vazão permitem uma ampla cobertura dos microrganismos, alcançando uma adequada descrição da comunidade. Para avaliar o microbioma de algumas amostras serão utilizados dados de sequenciamento pelo 454 que serão avaliados utilizando o programa qiime. Primeiro passo: Certifique-se que seus dados estão na mesma pasta, juntamente com o arquivo com os barcodings. Na nossa aula serão os arquivos: Fasting_Map.fna, com as sequencias em formato fasta, o Fasting_Map.quals com os valores de qualidade e Fasting_Map.txt com as informações de amostras e barcodings O conteúdo do Fasting_Map.txt: para abrir o programa abra um terminal, navegue até a sua pasta (/home/$user/qiime_tutorial) e execute o comando qiime (macqiime, pois executei em mac) $qiime PS: o $ na frente quer dizer que é uma linha de comando, não escrevam no terminal Iremos separar os dados das sequencias pelos barcodes do arquivo .txt. para isso iremos utilizar o comando do qiime “split_libraries.py", porém antes disso veremos os parâmetros que cada comando possui em seu manual. Para isso basta colocar a flag “-h” após o comando: $split_libraries.py -h Os parâmetros mais importantes são os abaixo: Assim, iremos separar as seqs por biblioteca com o comando: $split_libraries.py -m Fasting_Map.txt -f Fasting_Example.qual -o split_library_output/ Fasting_Example.fna -q Após isso, os arquivos de saída estão na pasta split_library_output. Para como a análise ocorreu execute o comando: $less split_library_output/split_library_log.txt O programa recebeu 1339 sequencias cruas, escreveu 1337 nos 9 PC.s e a tabela mostra quantas sequencias tem em cada amostra. Agora iremos determinar a que OTU (Unidades taxonômicas operacionais), que são os clusters das sequencias de cada grupo taxonômico das amostras que estão sendo analisadas. O QIIME analisa os OTUS em vez de todo o conjunto de sequências. O qiime vai atribuir a cada OTU dados, como a taxonomia, abundância, e história evolutiva de cada OTU em comparação com os outros. Para isso execute o comando: $pick_de_novo_otus.py qiime_parameters.txt -i split_library_output/seqs.fna -o otus -p Este comando irá realizar a clusterização dos OTUS baseado na similaridade das sequencias entre os reads. Após montar os clusters o programa gera a sequencia representativa de cada cluster. Os OTUS são alinhados de novo utilizando o MUSCLE ou comparando com um alinhamento já existente com o PyNAST. Em dados com mais de 10k sequencias, o alinhamento de novo é muito lento, sendo recomendado o PyNAST. Após isso o alinhamento final é filtrado e a arvore filogenética representativa é gerada. Finalmente o qiime gera a tabela de OTUs. Entretanto, o output ainda não está formatado adequadamente, caso esteja curioso use o comando less para verificar o otu_table.biom. Bem legal, não é? Caso queira ver os resultados finais use o comando: $ summarize_taxa_through_plots.py -i otus/otu_table.biom -o taxa_summary -m Fasting_Map.txt Esse script irá gerar novas tabelas para cada nível taxomômico. Por exemplo, as classificações de classe estão no “taxa_summary/otu_table_L3.txt”. O resultado mais formatado estão nos arquivos HTML na “taxa_summary/taxa_summary_plots”: Com isso você já pode verificar as comunidades bacterianas de cada amostra. Verifique se existem diferenças entre o grupo FAST e CONTROL. Alpha rarefraction: para gerar os gráficos de rarefação, é necessário determinar os parâmetros antes no arquivo. Para isso execute o comando: $echo alpha_diversity:metrics shannon,PD_whole_tree,chao1,observed_species > alpha_parameters.txt isso irá gerar um arquivo txt com os parâmetros que vc irá precisar. Para gerar os gráficos execute o comando: $ alpha_rarefaction.py -i otus/otu_table.biom -m Fasting_Map.txt -o arare p alpha_parameters.txt -t otus/rep_set.tre Este comando gera as tabelas rarefied, calcula as métricas da alpha diversity para cada tabela, agrupa os resultados e gera os gráficos. Para verificar os gráficos vá em ”arare/alpha_rarefaction_plots/rarefaction_plots.html": Assim você definiu as comunidades bacterianas de cada amostra e a sua variação entre amostras e grupos analisados.
Baixar