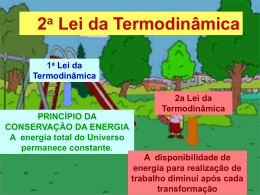
TERMODINÂMICA CARNOT (1796-1832) Físico francês, sadi Carnot nasceu em Paris. Em 1824 publicou sua famosa tese "Réflexions sur la puissance motrice du feu et sur les machines propes à développer cette puissance". Nela estabeleceu as características ideais de uma máquina térmica, que funciona num ciclo térmico, conhecido com ciclo de Carnot. O ciclo de Carnot, que é reversível, desenvolve-se em quatro fases, duas isotérmicas à temperatura constante e duas adiabáticas, sem trocar de calor com o ambiente. álgEBra linEar Editora da Universidade Estadual de Maringá Reitor Prof. Dr. Décio Sperandio Vice-Reitor Prof. Dr. Mário Luiz Neves de Azevedo Diretor da Eduem Prof. Dr. Ivanor Nunes do Prado Editor-Chefe da Eduem Prof. Dr. Alessandro de Lucca e Braccini Conselho Editorial Presidente Prof. Dr. Ivanor Nunes do Prado Editor Associado Prof. Dr. Ulysses Cecato Vice-Editor Associado Prof. Dr. Luiz Antonio de Souza Editores Científicos Prof. Adson C. Bozzi Ramatis Lima Profa. Dra. Ana Lúcia Rodrigues Profa. Dra. Analete Regina Schelbauer Prof. Dr. Antonio Ozai da Silva Prof. Dr. Clóves Cabreira Jobim Profa. Dra. Eliane Aparecida Sanches Tonolli Prof. Dr. Eduardo Augusto Tomanik Prof. Dr. Eliezer Rodrigues de Souto Prof. Dr. Evaristo Atêncio Paredes Profa. Dra. Ismara Eliane Vidal de Souza Tasso Prof. Dr. João Fábio Bertonha Profa. Dra. Larissa Michelle Lara Profa. Dra. Luzia Marta Bellini Profa. Dra. Maria Cristina Gomes Machado Profa. Dra. Maria Suely Pagliarini Prof. Dr. Manoel Messias Alves da Silva Prof. Dr. Oswaldo Curty da Motta Lima Prof. Dr. Raymundo de Lima Prof. Dr. Reginaldo Benedito Dias Prof. Dr. Ronald José Barth Pinto Profa. Dra. Rosilda das Neves Alves Profa. Dra. Terezinha Oliveira Prof. Dr. Valdeni Soliani Franco Profa. Dra. Valéria Soares de Assis Equipe Técnica Projeto Gráfico e Design Fluxo Editorial Artes Gráficas Marketing Comercialização Marcos Kazuyoshi Sassaka Edneire Franciscon Jacob Mônica Tanamati Hundzinski Vania Cristina Scomparin Edilson Damasio Luciano Wilian da Silva Marcos Roberto Andreussi Marcos Cipriano da Silva Norberto Pereira da Silva Paulo Bento da Silva Solange Marly Oshima Formação de Professores EM FÍSICA - EAD Arlindo Antonio Savi Cesar Canesin Colucci TERMODINÂMICA Maringá 2010 10 Coleção Formação de Professores em Física - EAD Apoio técnico: Rosane Gomes Carpanese Normalização e catalogação: Ivani Baptista - CRB 9/331 Revisão Gramatical: Tania Braga Guimarães Projeto Gráfico: Carlos Alexandre Venancio Edição e Diagramação: Renato William Tavares Capas: Arlindo Antonio Savi Kellis Germano de Freitas Dados Internacionais de Catalogação na Publicação (CIP) S267t Savi, Arlindo Antonio Termodinâmica 10 / Arlindo Antonio Savi, César Canesin Colucci.-- Maringá: Eduem, 2010. 131p.: il. (Coleção Formação de Professores em Física - EAD, v. 10) ISBN 978-85-7628-240-2 1. Física - Termodinâmica. I. Savi, Arlindo. Antonio. II. Colucci, César Canesin. CDD 21. ed. 536.7 Copyright © 2010 para o autor Todos os direitos reservados. Proibida a reprodução, mesmo parcial, por qualquer processo mecânico, eletrônico, reprográfico etc., sem a autorização, por escrito, do autor. Todos os direitos reservados desta edição 2010 para Eduem. Endereço para correspondência: Eduem - Editora da Universidade Estadual de Maringá Av. Colombo, 5790 - Bloco 40 - Campus Universitário 87020-900 - Maringá - Paraná Fone: (0xx44) 3011-4103 / Fax: (0xx44) 3011-1392 http://www.eduem.uem.br / [email protected] S umário Sobre os autores ................................................................................... 5 Apresentação da coleção ..................................................................... 7 Apresentação do livro ........................................................................... 9 1 Estrutura da termodinâmica..................................................................11 2 preliminares Matemática .....................................................................21 3 teoria Cinética Elementar ................................................................... 35 4 a 1ª lei da termodinâmica...................................................................51 5 a 2ª lei da termodinâmica ................................................................. 69 6 potenciais termodinâmicas ................................................................ 87 7 a 3ª lei da termodinâmica .................................................................117 8 referências / Bibliografia................................................................... 131 3 S obre os autores Arlindo Antonio Savi Graduação em Física pela Faculdade Filosofia Ciências e Letras de Rio Claro (1972). Mestrado pela Universidade Estadual de Campinas (1978). Atualmente é professor adjunto do Departamento de Física da UEM, onde atua desde 1978. Cesar Canesin Colucci Bacharel em Física pela Universidade Estadual de Campinas. Obteve seu Mestrado (1978) sobre supercondutividade e seu doutorado (1993) trabalhando com materiais magnéticos pela mesma Universidade. Em 1993 foi pesquisador visitante no Max Planck Institut (StuttgartAlemanha). Desde 1983 é professor do Departamento de Física da Universidade Estadual de Maringá e atualmente ocupa o cargo de Professor Associado. 5 A presentação da Coleção EmborarelativamenterecentenoBrasil,aEducaçãoaDistânciafoiimaginadaeimplantada com relativo sucesso, há muito tempo em diversas partes ao redor do mundo. Já em 1833, na Suécia, uma publicação se referia ao ensino por correspondência, e poucos anos depois, na Alemanha, foi fundada a primeira escola por correspondência destinadaaoensinodelínguas.Comoadventodatransmissãoradiofônica,asfacilidadessetornaramreaiseastrocasdeinformaçõesseagilizarame,consequentemente, aEducaçãoaDistânciaexperimentouumcrescimentosignificativo.Fatosemelhante ocorreucomaevoluçãodossetoresdecomunicaçãotelevisiva,edefinitivamente,a Educação a Distância se consolidou incorporando novas formas de comunicação. O Ministério da Educação, através da Secretaria de Educação a Distância (SEED) tem promovido uma ampla difusão de vários cursos a distância, em parceria com diversas Instituições Públicas de Ensino Superior (IPES). O curso de Física em EAD da UniversidadeEstadualdeMaringá(UEM)foiimplantadocomtotalapoiodessesórgãosoficiais.Possuidisciplinasidênticaseomesmoconteúdoprogramáticodocurso presencial. Entretanto, existem pontos entre ambos, que não podem convergir devido ao enfoque: enquanto o curso presencial requer uma metodologia característica, com a relação professor-discente acontecendo quase que exclusivamente dentro de um espaçofísicopróprio,ocursoadistânciadeveabrangereconsiderararelaçãoespaçotemporal para efetivar o aprendizado. A coleção que ora apresentamos reflete essa preocupação. Os volumes foram escritos por professores que possuem experiência suficienteparaelaboraroconteúdoadequadoacadadisciplinae,deformabastante consistente,elegerostópicosexigidosparaaformaçãodeumlicenciadoemFísica.O leitorperceberáque,mesmodentrodeumúnicolivroescritopordiversosautores, a linguagem não é uniforme e os enfoques são diferenciados; enfim, preservamos tantoquantopossívelasparticularidadesrespeitando-seasexperiênciasindividuaise, certamente,issoserefletenaapresentaçãodoconteúdoenoestilodeexposiçãodo 7 tErModinÂMiCa material didático. Adicionalmente uma parcela do corpo docente do Departamento de Física – UEM tem se dedicado à tarefa de produção de textos direcionados a Educação a Distância, os Departamentos de Matemática, de Química, de Fundamentos da Educação e de Informáticatêmcontribuídocomostextospertinentesàsdisciplinasqueusualmente ministramnamodalidadePresencial.Aofinaldoquartoano,acoleçãocontarácom maisdetrintavolumes.Essesforamgeradoscomoobjetivodeproporcionaraodiscente da Educação a Distância um material produzido pelo empenho de um conjunto deprofessoresqueacreditamqueaEducaçãoaDistânciasejaumaalternativapara supriradeficiênciadeprofessoresdeFísicanoensinomédio.Percebe-setambémque nãoéamodalidadedeensinoquedeterminaoaprendizado,maseledepende,acima detudo,doesforçoedadedicaçãodecadaum.Esperamosqueessacoleçãosejauma forma de tornar essa tarefa mais fácil de Física em EAD. Sonia Maria Soares Stivari Organizadora da Coleção 8 A presentação do livro A primeira exposição à Termodinâmica ocorreu na disciplina Física II e o objetivo, naquelemomento,eraodeabordar,adequadaebrevemente,umconteúdoparagraduandosingressantes. Emboratenhasidodadoumtratamentointrodutório,naqueladisciplina,osconceitos apresentadossãodeespecialrelevânciaparaprosseguiroestudodaTermodinâmica.Basicamente,astrêsleisqueanalisamosnoscapítulos6,7e8,dovolumeII,deFísicaGeral,são essenciaisparaumestudocontinuado:osfundamentosdaTermodinâmicaestãocontidos lá.Algunstópicoseresultadosnãoforamabordados,edecertaforma,nempoderiamter sido.Existemalgumasrazõesparaqueumtratamentomaisaprofundadosejafeitosomenteagora:umadelaséqueoembasamentomatemáticodoestudantenoiníciodoquarto semestreébastantesuperioràqueledosegundosemestre.Aoutra,équeacreditamosque ocontatocomdiversosfenômenosfísicosesuasdescriçõesadequadas,aolongodocurso, facilitaoaprendizadomesmodedisciplinasquenãoestão,pelomenosaparentemente, relacionadas. Opropósitodestelivroébastantemodesto.Elefoielaboradoparaauxiliaroestudanteaatingirumestágionoqualsuaconcepçãodeideiasfundamentaisnãosãosomente corretas e precisas, mas também fazê-lo se sentir à vontade com o conteúdo. Desnecessário dizerqueolivrocumpreumapartedesseobjetivo.Aoutradeveficarporcontadoleitor. Oqueseespera,então,éessetextopossaajudá-lodealgumaformanoentendimentoda Termodinâmica. O AUTOR 9 1 Estrutura da Termodinâmica 1.1 introdução 1.2 noções preliminares 1.3 Equlíbrio temodinâmico e temperatura 1.4 a lei Zero da termodinâmica 1.5 temperatura e termometria 11 tErModinÂMiCa 1 ESTRUTURA DA TEMODINÂMICA 1.1 Introdução A Termodinâmica trata das propriedades da matéria sob circunstâncias nas quais a noção de temperatura e calor não podem ser ignoradas. Sua apresentação atual envolve as leis da Termodinâmica e os princípios relacionados, uma forma concisa de resultados empíricos e interpretações teóricas durante um período de mais de 150 anos. É exatamente por isso que a Termodinâmica permanece como um ramo formidável da Física, e a sua sólida base experimental oferece um alto grau de confiabilidade e é nisso que reside sua força. Paradoxalmente, essa é também sua fraqueza: historicamente, a Termodinâmica não se desenvolveu baseada em qualquer modelo atômico ou mecânico da matéria que poderia permitir observar como ela funciona. A ampla generalidade de suas leis pode ser aplicada a sistemas que são descritos por uns poucos parâmetros, independentes de como eles se comportam em nível microscópico. Por exemplo, a primeira lei é baseada no princípio da conservação da energia e nada esclarece ou indica de como esse princípio pode estar ligado à natureza íntima da matéria. A definição das variáveis termodinâmicas, embora realizada de forma operacional (isto é, define-se a quantidade prescrevendo como medi-la) é suficiente para caracterizar adequadamente o sistema. Existem quatro leis na Termodinâmica, que chamamos lei zero, primeira, segunda e terceira. Cronologicamente, somente a terceira está corretamente enumerada. A segunda começou a ser formulada em 1824, com o trabalho de Sadi Carnot; sua forma final foi enunciada por Clausius em torno de 1850. A primeira lei apareceu logo depois com os trabalhos de Mayer, baseando-se nas experiências de Joule. A terceira teve sua origem nos trabalhos de Nernst, no início do século XX, e a lei zero só foi estabelecida 20 anos depois. 1.2 Noções Preliminares Muitas vezes é oferecida ao estudante de Física a seguinte proposta: “considere um sistema”. O sistema é algo sobre o qual devemos focalizar nossas atenções – um pedaço do Universo cujas características e comportamento deveríamos nos esforçar para entender. A vizinhança de um sistema seria a parte remanescente do Universo que afeta diretamente a evolução desse sistema. Existem sistemas que podem ser considerados como favoritos. Em mecânica, tem-se um forte apreço pelos sistemas consistindo em partículas que caem em queda livre, ou um pequeno bloco que desliza (com ou sem atrito) por um plano inclinado. Trata-se, então, de observar como reagem esses sistemas quando submetidos ao agente externo, que identificamos como a gravidade, e como ela atua sobre eles. A Termodinâmica também tem seus favoritos: um deles, provavelmente o primeiro da lista, consiste em certa quantidade de gás encerrada em um volume cilíndrico dotado de um pistão móvel. Os detalhes a serem estudados são os efeitos da troca de energia entre o gás e a vizinhança. Considere inicialmente os sistemas citados na parte de mecânica. Eles (e muito provavelmente, diversos outros) possuem em comum um alto grau de simplicidade. Nos sistemas mecânicos, as leis de Newton podem predizer o comportamento das partículas, e os resultados dessas previsões podem ser verificados pelo experimento; o sucesso obtido com esses experimentos simples faz aumentar a confiabilidade nas leis físicas que foram utilizadas para descrever a evolução do sistema. O sistema gás-pistão serve de forma apropriada para se iniciar o estudo Termodinâmico porque ele ilustra os princípios em um nível quase intuitivo. Em certa extensão, a dificuldade encontrada pelos iniciantes em adquirir os conceitos da Termodinâmica, está na falta de experiências anteriores com os aspectos térmicos, mesmo com os mais simples sistemas como cilindro com pistão. Como primeiro passo para se entender o comportamento de um sistema físico, precisamos escolher as quantidades que o descrevem e os efeitos da vizinhança. Essas quantidades medidas, diretamente ou indiretamente, são chamadas de macroscópicas. Por exemplo, no gás confinado no cilindro, as variáveis macroscópicas podem ser a temperatura, a pressão, a densidade do gás, o volume. As quantidades macroscópicas usadas na descrição do sistema termodinâmico são geralmente chamadas de variáveis termodinâmicas. Poderíamos descrever o sistema em nível microscópico. Para isso, deve-se introduzir um modelo conceitual para o sistema: esse modelo requer uma construção em nível atômico e emprega 12 variáveis microscópicas – quantidades que não são diretamente mensuráveis. Por exemplo, a massa e a energia cinética de um átomo ou de uma molécula são variáveis microscópicas. Entretanto, a massa total do gás pode ser medida, mas não a massa de átomo em particular. A descrição microscópica do sistema é ponto de partida da teoria cinética dos gases, ou da mecânica estatística para sistemas mais gerais. A Termodinâmica é formulada em termos de variáveis macroscópicas e, portanto, não considera a estrutura íntima da matéria. Seu propósito é definir quantidades físicas apropriadas (variáveis de estado), que caracterizem as propriedades macroscópicas da matéria (macroestados), de forma menos ambígua quanto possível e relacioná-las por meio de equações de validade universal (equações de estado). A Termodinâmica não pode e nem dará as razões porque certa equação de estado descreve o sistema. Ela se restringe a fazer algumas asserções em relação às variáveis de estado, desde que seja conhecida a equação de estado. Estrutura da termodinâmica ALGUMAS DEFINIÇÕES As variáveis termodinâmicas podem ser classificadas de acordo com sua dependência em relação ao tamanho do sistema (como medido pela sua massa ou pelo seu volume). (1) – uma variável é chamada de extensiva quando seu valor depende do tamanho do sistema. A massa, a energia, o volume são exemplos de variáveis extensivas. (2) – uma variável é dita intensiva quando seu valor independe do tamanho do sistema. A pressão, a temperatura, a densidade são variáveis intensivas. Uma maneira prática para decidir se uma variável é extensiva ou intensiva consiste em imaginar uma divisão introduzida no sistema de tal forma que ele seja dividido em duas partes (não necessariamente iguais). Denominemos y a variável que desejamos identificar, então, para o subsistema 1 temos y1 correspondente ao parâmetro y e para o subsistema 2, temos y 2 . Se acontecer que y1 + y 2 = y , dizemos que a variável termodinâmica é extensiva (ou aditiva, como é fácil perceber). Se ocorrer que y1 = y 2 = y então a variável é intensiva. Com esse procedimento é imediato concluir que a energia interna do sistema, a massa, o próprio volume são quantidades extensivas. Por outro lado, a pressão, a temperatura, a densidade, são grandezas intensivas. Observe que a razão entre duas quantidades extensivas é uma grandeza intensiva. Os sistemas podem ser caracterizados considerando os tipos de paredes que o delimitam. (a) – Sistemas Isolados: denominam-se sistemas isolados aqueles que estão impedidos de qualquer tipo de interação com suas vizinhanças. Não trocam calor e nem matéria: suas paredes são impermeáveis a qualquer tipo de troca com o ambiente. Em particular, a energia total (mecânica, elétrica, magnética...) é uma grandeza conservada. O impedimento da troca de energia (na forma de calor) é conseguido por paredes adiabáticas. (b) – Sistemas Fechados: nesse caso é permitida somente a troca de energia entre o sistema e a vizinhança, vedada a possibilidade de troca de partículas (massa). (c) – Sistemas Abertos: para esse tipo de sistema é permitida tanto a troca de energia como a troca de massa. As paredes adiabáticas são construídas com material cujo isolamento térmico seja perfeito. Alguns estudantes podem se sentir pouco confortáveis em relação a essa perfeição de uma parede adiabática: como a Termodinâmica do mundo real pode utilizar uma peça de ficção tal como uma parede adiabática? Isso pode levá-los a pensar que a Termodinâmica está baseada em fantasia. A noção de parede adiabática é válida e útil em Termodinâmica da mesma forma que uma superfície sem atrito é usada em Mecânica: o atrito cinético entre duas superfícies pode ser reduzido drasticamente por polimento e lubrificação ou mesmo utilizando um colchão de ar entre os dois corpos. Entretanto, ele não pode ser completamente eliminado. Com isso, as forças de atrito podem ser ignoradas quando comparadas às outras forças que atuam sobre o sistema. As paredes adiabáticas são consideradas sob essa condição: o isolamento térmico deve ser conseguido utilizando-se materiais de alto grau de isolamento térmico e baixa capacidade térmica de tal forma que a energia interna do sistema seja constante durante todo o tempo. A falta do perfeito isolamento térmico pode ser então ignorada. A Termodinâmica de equilíbrio, que ora começamos a estudar, trata de sistemas com um número bastante grande de partículas. Em geral, os sistemas possuem o número (ou da ordem) 23 de Avogrado de partículas, ou seja, N ≈ N Avogrado ≈ 6 × 10 . Para se ter uma ideia do que ele 13 tErModinÂMiCa representa, podemos fazer uma experiência pensada: você retira um litro de água do mar, numera todas as partículas desse litro de água e depois devolve ao mar. Agite, mexa, até que as partículas rotuladas estejam homogeneamente distribuídas em todos os oceanos. Novamente, retire um litro 4 de água: nesse litro existem da ordem de 10 partículas que você rotulou inicialmente! A conclusão é óbvia: existem muito mais partículas em um litro de água do que litros de água em toda superfície líquida que cobre a Terra. A Termodinâmica trata de sistemas (em equilíbrio) que possuem número de partículas dessa ordem. 1.3 Equilíbrio Termodinâmico e Temperatura Quando observamos um sistema fechado percebe-se que ele tende ao estado de equilíbrio termodinâmico se esperarmos um tempo suficiente. Obviamente, esse “tempo suficiente” não pode ser especificado à priori porque ele depende das características do sistema. O intervalo pode variar de frações de segundo até valores que atingem minutos, ou horas ou mesmo tempos maiores. Por exemplo, a expansão livre de um gás contido em certo volume, demanda um tempo de equilíbrio muito curto; entretanto, o equilíbrio envolvendo certa reação química pode levar alguns minutos. Percebe-se, então, que o conceito de “tempo suficiente” é bastante relativo: ele é determinado pelas características de cada sistema e está vinculado ao tempo de medição que efetuamos dos parâmetros termodinâmicos. Para perceber o significado do que se chamamos de tempo medição, imagine a expansão livre: se quisermos determinar a pressão final do gás, bastaria uma espera de alguns segundos para obter um valor que vai se manter constante. No outro extremo, a oxidação natural de um metal exposto à atmosfera ambiente é uma situação de não-equilíbrio, mas o processo é tão lento que, por exemplo, a determinação da massa do metal é comparativamente muito rápida para que exista uma discrepância perceptível entre duas medições consecutivas. Nesse caso, pode-se dizer que existe um equilíbrio termodinâmico porque as quantidades termodinâmicas variam muito lentamente. De qualquer forma, convém se lembrar de que as variáveis termodinâmicas são definidas (e medidas) somente no equilíbrio. Para que um sistema esteja em equilíbrio termodinâmico, três condições devem ser satisfeitas: (1) O sistema deve estar em equilíbrio mecânico e livre de qualquer força ou torque externos; (2) O sistema deve estar em equilíbrio químico. Nenhuma reação pode estar ocorrendo; (3) As propriedades mensuráveis do sistema devem ser espacialmente uniformes e não podem variar com o tempo. Entretanto, a condição (3) deve ser considerada com certo cuidado quando tratamos com sistemas heterogêneos. Por exemplo, certo volume fechado, contendo líquido e seu vapor, está em equilíbrio termodinâmico desde que ambas as fases satisfaçam as condições de equilíbrio. Porém, a uniformidade espacial não se cumpre nesse caso. Em relação às propriedades mensuráveis, deve-se entender, por exemplo, que a temperatura é uniforme em todo e qualquer ponto do sistema. Temperatura é uma variável de estado que é desconhecida em mecânica e em eletrodinâmica. Ela é especialmente introduzida para a Termodinâmica e sua definição está intimamente relacionada com o conceito de equilíbrio térmico. Igualdade de temperatura entre dois sistemas é a condição para que exista equilíbrio térmico entre eles. A palavra temperatura tem algum significado para cada estudante antes de estudar Termodinâmica. Entretanto, o significado apreendido da experiência cotidiana é bastante qualitativo e muitas vezes restrito a intervalos da percepção humana. É necessário dar uma definição operacional da temperatura para que façamos algum progresso e configurá-la como uma variável termodinâmica. Isso é um requisito logicamente necessário para que a ideia de igualdade de temperatura possa ser utilizada. Esse procedimento foi prescrito no sexto capítulo, do volume II, da disciplina Física Geral, e seria proveitoso ao estudante que consultasse esse capítulo novamente. Equilíbrio térmico é sinônimo de igualdade de temperatura entre dois sistemas termodinâmicos. Devemos notar que essa condição de equalização de temperatura não envolve e nem requer uma escala termométrica. Uma situação semelhante ocorre com a primeira lei de Newton para partículas: ela indica a definição operacional de resultante nula. Não precisamos de uma definição operacional de força (dada pela segunda lei) para reconhecer uma condição de força nula porque aceleração nula define força resultante nula. Equilíbrio térmico define a igualdade de temperatura, independente da escala termométrica adotada. 14 1.4 A Lei Zero da Termodinâmica Em matemática, a propriedade transitiva é introduzida como um postulado devido à consequência elementar de outros postulados. A forma apresentada é: Estrutura da termodinâmica A=B e B=C, então A=C . Uma lei análoga existe em Termodinâmica, embora ela não seja nem um postulado, nem uma verdade lógica, mas antes o estabelecimento de um fato experimental, que indica o equilíbrio térmico entre os corpos. Esse fato, muitas vezes conhecido como a lei zero da Termodinâmica, pode ser enunciado da seguinte forma: todos os sistemas que estão em equilíbrio térmico com um dado sistema, estão em equilíbrio térmico entre si. Essa é a propriedade transitiva a que nos referimos no início da seção. Se considerarmos as relações TA =Treferência , TB =Treferência , ,TN =Treferência , então, TA =TB = =TN . Como dissemos, essa igualdade não se refere a qualquer escala termométrica em particular, porém, se fizermos determinada escolha por uma delas, todas as leituras devem ser conduzidas usando essa mesma escala. Portanto, sistemas que estão em equilíbrio térmico entre si possuem uma propriedade intensiva comum, que chamamos de temperatura. Assim, sistemas que não estão em equilíbrio térmico entre si, têm temperaturas diferentes. A Termodinâmica não nos conta nada sobre o tempo requerido para que dois corpos entrem em equilíbrio térmico: as preocupações estão direcionadas para os estados inicial e final do processo. A figura 1 mostra o equilíbrio térmico entre dois corpos, A e B, aferido pelo uso de uma escala termométrica. Figura 1 – Teste esquemático da lei zero da Termodinâmica. 1.5 Temperatura e Termometria Pelo que foi exposto nas seções anteriores, percebe-se que o parâmetro que chamamos de temperatura, tem papel muito importante na Termodinâmica. Portanto, convém abordar alguns fatos relacionados a ela. De certa forma, o que iremos tratar abaixo já foi feito no volume II, de Física Geral: provavelmente, para muitos estudantes, isso serve mais como um tópico de recordação, acrescido de uma discussão mais detalhada. A noção intuitiva e ingênua sobre a sensação de quente ou frio para caracterizar temperatura é insuficiente: precisamos de um conceito mais apropriado e útil para torná-la cientificamente aceitável. O procedimento de se medir a temperatura está ligado a uma equação de estado, isto é, envolve uma relação entre a quantidade observada (por exemplo, o volume, a resistência elétrica) e a temperatura. Isso requer uma definição operacional de temperatura: devemos especificar como a temperatura pode ser medida. Termômetros são caracterizados por uma substância termométrica e uma variável termométrica. O exemplo mais trivial é o termômetro caseiro, com um líquido (álcool colorido ou mercúrio), sendo a substância termométrica e o comprimento da coluna líquida como a variável termométrica. Na escala termodinâmica a temperatura é definida em termos de calor. Assim, a definição de temperatura é independente das propriedades peculiares da substância termométrica. Claramente, essa característica é de extrema importância porque a temperatura registrada depende somente do estado do sistema e não do material usado como substância termométrica. Uma escala empírica é conseguida por uma definição arbitrária em termos de alguma 15 tErModinÂMiCa variável termométrica. Uma relação matemática elementar é geralmente adotada. Por exemplo, um termômetro a gás a volume constante (alguns outros) essa relação é linear: a temperatura é definida em termos da pressão do gás, P, por: T(P) = bP (1). T(X) = bX (2), De forma mais geral, podemos escrever: como uma relação linear definindo a temperatura T(X) em termos do valor de alguma variável termométrica X. A constante de proporcionalidade b é determinada elegendo-se a temperatura para algum ponto fixo padrão. O funcionamento do termômetro a gás de volume constante está descrito à página 99, do volume II, de Física Geral. O ponto fixo escolhido por uma convenção internacional é o ponto triplo da água: a escolha, embora arbitrária, é de extrema conveniência devido à acessibilidade e a fácil reprodução. O funcionamento de um termômetro a gás se baseia no fato experimental que a pressão de um gás, mantido a volume constante, aumenta linearmente com a temperatura e isto é verdade para qualquer gás com baixa densidade de tal forma que podemos considerá-lo ideal. Coloca-se certa quantidade de gás em um recipiente rígido (para manter seu volume constante) que tem um manômetro acoplado. Em seguida, mergulha-se este volume em um banho de água e gelo em equilíbrio, anota-se o valor da pressão. O outro ponto de referência é determinado usando água em ebulição, correspondendo à outra pressão registrada pelo manômetro. Estes dois pontos são colocados em gráfico de temperatura x pressão e traça-se uma reta passando por eles. Qualquer outra temperatura pode ser obtida permitindo que nosso termômetro interaja termicamente com o sistema cuja temperatura se deseja medir. A figura abaixo é um esboço gráfico do comportamento deste termômetro. As três curvas representam diferentes tipos de gases e com densidades diferentes. Figura 2: Gráfico T (oC) versus p (unidade arbitrária). Extrapolando-se estas retas para pressões tendendo a zero, obtém-se o valor de −273.15 oC . Você poderia suspeitar de que este valor seria diferente para gases diferentes, mas o resultado é sempre o mesmo para qualquer tipo de gás, desde que seja considerado ideal (baixa densidade). Outro ponto que poderia ser questionado neste experimento seria o comportamento deste gás a baixas temperaturas: à medida que se abaixa a temperatura, o gás sofre uma transformação de fase e se torna líquido. Isso é verdade desde que a pressão seja suficiente para ocorrer a liquefação. Note, entretanto, que o procedimento envolve a diminuição sistemática da pressão do gás, e isso evita a transformação pra a fase líquida. Em todos os casos, independente da natureza do gás ou da baixa pressão inicial (para considerá-lo ideal), a pressão vai a zero quando a temperatura o é de −273.15 C . Este valor sugere um caso universal porque não depende da natureza da substância usada no termômetro e também deve representar um limite inferior para processos físicos. Por isso, esta temperatura é definida como zero absoluto e serve de base para a escala Kelvin de temperatura. Por um acordo internacional, a temperatura da água em seu ponto triplo tem recebido o valor de 273.15 K , onde K é a abreviação para a unidade de temperatura, chamada Kelvin. Com esse valor estabelecido, podemos voltar às equações (1) e (2). Se X triplo representa o valor da variável termodinâmica no ponto triplo da água, tem-se: 273.16 K=bX triplo ∴ b= 16 273.15 K . X triplo A escala de temperatura empírica é agora definida pela relação: T(X)=273.15 K X X triplo (3). Estrutura da termodinâmica Em particular, quando escolhemos a pressão do gás como variável termodinâmica em um termômetro a volume constante, podemos escrever (3) na forma: T(P) = 273.15 K P Ptriplo . (4). Exemplo 1 Utilizando um termômetro a gás de volume constante de modesta precisão, um estudante encontra um valor de 1.37 para a razão entre a pressão de vapor e a pressão do ponto triplo. Qual é a temperatura do ponto de vapor? Solução O uso direto de relação (4) nos dá: Tvapor = 273.15 K ×1.37 ≅ 374.21K . A relação (3) é completamente geral e depende da escolha da variável termométrica. Por exemplo, se usarmos a resistência elétrica de um resistor ou a tensão de um termopar temos, respectivamente: T(R) = 273.15 K R R triplo (5) e T(ε ) = 273.15 K ε ε triplo (6). A escolha apropriada de um termômetro depende, dentre outras coisas, da faixa de temperatura envolvida e das características do sistema. Para determinar a temperatura de fusão de um metal (em geral, eles possuem alto ponto de fusão), não podemos utilizar um termômetro clínico: mesmo se o bulbo resistisse (fato que não acontece), o intervalo da escala não é apropriada. Da mesma maneira, o uso de um termopar não é recomendável porque ele pode sofrer a fusão antes do material (a exceção aqui é o termopar Platina/ Platina-Ródio). Para altas temperaturas (acima de 1500 K), o termômetro óptico é mais conveniente. Ele compara a emissividade de luz emitida pelo sólido incandescente com uma escala previamente calibrada e a temperatura pode ser determinada diretamente. Para medidas de temperaturas intermediárias (da ambiente até 700 K), o uso de termopares é largamente aceito. Entretanto, algumas vezes as características do sistema restringem sua utilização: para registrar a leitura de forma confiável, o termopar deve ficar tão próximo quanto possível da amostra e, em certos casos, existe a limitação experimental que impede esse arranjo. Medidas em baixas temperaturas (da ambiente até próximas da liquefação do Hélio ≈ 4 K ) alternam o uso de termômetros de resistência com termopares. Obviamente, as escolham são conformadas com as características do sistema e com o grau de precisão que se exige na determinação da temperatura. 17 tErModinÂMiCa Anotações 18 Estrutura da termodinâmica Anotações 19 tErModinÂMiCa Anotações 20 2 Preliminares Matemática 2.1 introdução 2.2 diferenciais 2.3 aplicações de derivadas parciais em termodinâmica 2.4 derivadas parciais de ordem superior 21 2 PRELIMINARES MATEMÁTICA 2.1 Introdução tErModinÂMiCa Esse capítulo tem por objetivo servir como uma revisão matemática necessária para o desenvolvimento da Termodinâmica. Alguns conceitos e definições são apresentados de forma analítica e, sempre que possível, os significados geométricos são utilizados para enfatizar e complementar o desenvolvimento analítico. A experiência tem demonstrado que, na maioria dos casos, alguns conceitos são melhor compreendidos ao se utilizar um diagrama, ou um esboço com apelo geométrico, enfim, algo que possibilite ao estudante associar a parte analítica com aspectos visuais. E esse empreendimento é tanto mais eficaz e mais abrangente se for acompanhado de exemplos envolvendo situações físicas. Muitos dos assuntos tratados aqui já foram abordados nas disciplinas de Cálculo e, portanto, devem servir para como uma revisão e também como uma referência rápida para dar maior segurança ao estudante. 2.2 Diferenciais Sob o ponto de vista termodinâmico, isto é, em escala macroscópica, as propriedades observáveis dos sistemas são geralmente contínuas. Por exemplo, a pressão atmosférica varia suavemente com a altitude. De forma semelhante, sua temperatura exibe uma dependência espacial contínua. A difusão de uma substância em outra pode ser analisada através da concentração, e a relação matemática que descreve esse processo é baseada na hipótese das continuidades espacial e temporal da concentração (2ª lei de Fick). O comportamento gráfico da curva da concentração versus a variável espacial é caracterizado por uma função suave e contínua que decresce à medida que aumenta a distância do centro de difusão. Obviamente, para tempos suficientemente longos, a concentração se torna uniforme e o sistema atinge a homogeneidade: a essa situação é que chamamos de estado estacionário. A continuidade das propriedades físicas permite uma concisa formulação matemática da Termodinâmica. Duas construções matemáticas são especialmente úteis: a diferencial e a derivada parcial. Esses conceitos se fazem presentes em todo o domínio da Termodinâmica (e também em diversos outros campos da Física). A diferencial é útil em relacionar variações de duas ou mais quantidades interdependentes. Vamos considerar inicialmente uma função que depende somente de uma variável: x é nossa variável independente e denotamos por y(x) ou por f(x) a variável dependente. dy é uma quantidade única e não a razão dx entre dy e dx. Entretanto, é comum o uso da expressão dy = y′(x) dx com certa arbitrariedade no Os livros de Cálculo enfatizam que a expressão sentido de “passar o dx multiplicando”, como se ele fora um número. Porém, o uso dessa relação pode ser justificado se pensarmos que ela seja uma diferencial da variável dependente y. A derivada de y(x) é definida como: ∆y ∆x →0 ∆x y ′( x ) = lim (7), onde ∆y = y( x + ∆x ) − y( x ) . Para pequenos valores de ∆x pode-se esperar que y ′( x ) esteja ∆y , ou ∆x ∆y ≈ y ′( x )∆x para pequenos valores de ∆x . próximo ao quociente Usando essa relação, podemos reescrever ∆y = y( x + ∆x ) − y( x ) : ∆y = y( x + ∆x ) − y( x ) ⇒ y( x + ∆x ) = ∆y + y( x ) , portanto, y( x + ∆x ) ≈ y ′( x )∆x + y( x ) 22 (8). Exemplo 2 Vamos utilizar a expressão (8) para avaliar o valor de onde o argumento é dado em radianos. preliminares Matemática Solução Nesse caso, y ( x) = sen x , x = 0 e ∆x = 0.1 . y′( x) = cos x . (8) ⇒ sen(0 + = 0.1) sen(0.1) ≈ cos 0 × (0.1) + 0 ∴ sen(0.1) ≈ 0.1 . O valor real de é 0.0998. O desvio relativo percentual é da ordem de 0.16%. O próximo exemplo mostra como se propaga um desvio quando se utiliza uma quantidade para calcular um novo valor numérico. A propagação de erros é um dos itens tratados em Física I, parte experimental. Exemplo 3 Suponha que desejamos estimar o volume de uma esfera medindo sua circunferência. O ato de medir carrega em si uma incerteza: diversos fatores influenciam na precisão e um deles está vinculado à precisão do instrumento usado. De qualquer forma, considere que a medida da circunferência tenha sido expressa por 162 cm, dentro de uma incerteza de 0.20 cm. Matematicamente, a medida pode ser escrita: = C 162.0 ± 0.2 0 cm . O que se pede aqui é avaliar como essa incerteza afeta o resultado final do cálculo do volume, ou, de forma equivalente, que desvio no volume teremos devido a esse desvio na medida da circunferência. Solução O volume da esfera e sua circunferência são dados, respectivamente, por: V(r ) = 4 3 πr e C(r ) = 2πr . 3 Nesse caso, a identificação é feita da seguinte forma: , e a variável independente e não r, como somos levados a fazer instintivamente. Como não foi medido o raio, mas somente sua circunferência, precisamos trabalhar com essa quantidade. C = 2πr ⇒ r = C C3 ∴ r3 = 3 . 2π 8π Portanto, o volume é expresso em função do comprimento C: V ( C) = C3 6π 2 (9). A incerteza em V pode ser obtida usando-se a relação (8): dV ∆C . Então, dC C2 (162) 2 (0.20)=266 cm3 . ∆V= 2 ∆C= 2π 2π 2 Incerteza em V = ∆V = Para avaliar o desvio relativo, considere o valor de 162 cm para obter: V(162)= Assim, o desvio relativo é (162)3 =71795 cm3 . 2 6π ∆V 266 = ≈ 0.0037 ou cerca de 0.37%. V 71795 Note que inicialmente tínhamos um desvio de 0.2 cm, que corresponde a 0.12% da medida da circunferência. Esse desvio afeta a incerteza do volume em 0.37%, ou seja, um desvio três vezes maior do que o inicial: observe então o expoente da quantidade C na relação (9). Isso não é uma coincidência. 23 tErModinÂMiCa A figura 3 sugere as características geométricas das quantidades usadas no desenvolvimento que ora fizemos. A quantidade ∆= y y(x + ∆x) − y(x) é a variação em y que resulta se continuarmos ao longo da curva e dy é a variação em y se seguirmos ao longo da reta tangente desde x até x + ∆x . Essa variação em y é a diferencial de y e é dada por: dy = y′(x)∆x . Figura 3 – A diferencial dy é uma aproximação linear para a variação ∆y . Na figura é fácil perceber que quanto menor for ∆x , mais próximo se tornam os valores de ∆y e dy. No caso limite onde ∆x → 0 , a igualdade pode ser escrita como: dy dy = y′(x)dy = dx dx (10). A Termodinâmica trata de situações para as quais x e y(x) representam quantidades mensuráveis, isto é, propriedades físicas da matéria. A natureza contínua dessas propriedades permitem que consideremos variações suficientemente pequenas de tal forma que podemos usar a relação (10) para relacionar as variações em x e y. Exercícios 2.1 - Achar ∆y e dy para a função y( x ) = x 2 − x em x = 10 e ∆x = 0.1 . [Lembre-se de que ∆y = y( x + ∆x ) − y( x ) ]. 2.2 - Encontre os quocientes ∆x ∆y dy = 0.01 , qual a diferença e para a parábola y = x 2 . Se x y y ∆y dy e ? y y 2.3 - Encontre ∆y e dy para a função y( x ) = x em x = 4 e ∆x = 0.35 . Qual a diferença numérica entre numérica entre as duas quantidades? Obtenha o desvio absoluto e o relativo entre as duas quantidades. Observe que para esse caso dy > ∆y . DERIVADAS PARCIAIS A necessidade de derivadas parciais aparece quando mais de uma variável independente está presente. Considere uma função z( x , y) de duas variáveis independentes, x e y. A igualdade abaixo é a mais comum para representar uma função de duas variáveis: f ( x , y) = z = z ( x , y) . Geometricamente z( x , y) pode ser pensada como a altura de uma superfície acima do plano xy. Uma superfície tridimensional pode ser representada por uma equação da forma f ( x , y, z) = 0 . A superfície de uma esfera de raio R, centrada na origem, pode ser escrita como: f ( x , y, z ) = x 2 + y 2 + z 2 − R 2 = 0 . 24 A figura 4 representa geometricamente o significado das derivadas parciais para uma função de duas variáveis independentes. Note que as quantidades ∆z e dz , quando somente uma das variáveis independentes é deixada variar, é muito semelhante ao caso de funções com uma única variável. A notação para simbolizar a derivada parcial apresenta certas diferenças com aquela usada para funções de uma variável. Denotamos por: preliminares Matemática ∂z ∂f ou a derivada da função z( x , y) ou f ( x , y) em relação à x. ∂x y ∂x y O subscrito y nos parênteses é para ser entendido que a variável y deve ser mantida constante ao derivarmos em relação à x. Outra notação comum para essa operação é z x ou f x , entretanto, seu uso deve ser evitado quando tratamos simultaneamente com vetores sob o risco de acontecer alguma interpretação errônea entre as derivadas e as componentes vetoriais. Figura 4 – Derivadas parciais para uma função de duas variáveis independentes. A derivada da função em relação à variável y é similar àquela expressa para x: ∂z ∂f ou . ∂y x ∂y x Estabelecidas as notações convenientes para as derivadas parciais, o processo limite que as define são: z( x + ∆x , y) − z( x , y) ∂z desde que esse limite exista ( = ser finito e único). = lim ∆x ∂x y ∆x →0 ∂z z( x , y + ∆y) − z( x , y) = lim desde exista esse limite. ∆y ∂y x ∆y→0 A diferencial da função z( x , y) é definida como: = dz ∂z ∂z ∆x + ∆y ∂x y ∂y x (11). Quando ∆x e ∆y tendem a zero, a expressão (11) pode ser escrita na forma: ∂z ∂z dx + dy ∂x y ∂y x (12). ∆z = z( x + ∆x , y + ∆y) − z( x , y) (13). = dz O incremento ∆z da função é dado por: 25 tErModinÂMiCa Exemplo 4 Considere a função z( x , y) = x 3 y . Encontrar dz e ∆z no ponto (1,2) para ∆x =∆y =0.01 . Solução Usamos (13) para calcular ∆z : = ∆z z (1.01, 2.01) − z (1, 2) ⇒ = ∆z (1.01)3 × (2.01) − (1)3 × (2) ∴ ∆z ≈ 0.071 dz = ( 3x y ) 2 (1,2) × (0.01) + ( x3 ) ∂z ∂z 2 3 = 3x y e = x . ∂ y ∂x (1,2) × (0.01) ⇒ dz = 6 × 0.01 + 1× 0.01 ∴ dz = 0.07 Exemplo 5 Obter a diferencial da pressão para um gás ideal. Solução A equação de estado de um gás ideal é dada por PV = nRT. nRT . Temos, então, a variável P em função de T e V. V PV= nRT ⇒ P= ∂P ∂P = dP dT + dV ∂T V ∂V T = dP nR nRT dT − 2 dV . V V Exercícios 2.4 - Considere a equação de estado de um gás ideal. Estimar a variação da pressão, ∆p , quando a temperatura e o volume variam simultaneamente. A temperatura inicial é 273.15 K e a final é 274 K, enquanto o volume inicial é 10 litros e o final é 9.9 litros. Após calcular ∆p , use a equação do gás ideal para obter p1 ( V1 , T1 ) e p 2 ( V2 , T2 ) . Calcular ∆p = p 2 − p1 e compare com o primeiro resultado que você obteve. 2.5 - Encontrar a expressão para dS, sabendo-se que a entropia de um mol de gás ideal é dada em termos da pressão e da temperatura pela relação: S(T,V) = C p ln T − R ln P + So . Temos considerado z( x , y) como uma função das variáveis independentes x e y e sua diferencial é dada por (12): ∂z ∂z = dz dx + dy ∂x y ∂y x (12). Do ponto de vista formal, podemos considerar igualmente válida a escolha de x como variável dependente e y e z como sendo independentes, ou seja, x = x ( y, z) . Então, a diferencial dx é dada por: ∂x ∂x = dx dy + dz ∂z y ∂y z (14). Ambas as relações, (12) e (14) são válidas simultaneamente, isto é, elas são formas alternativas de relacionar variações na tripla interdependente x, y e z. Podemos, então, substituir (14) em (12): ∂z ∂z ∂x ∂x = dz dy + dz + dy . ∂x y ∂y z ∂z y ∂y x 26 Podemos rearranjar os termos da expressão: ∂z ∂x ∂z ∂x ∂z = dz + + dy dz ∂x y ∂z y ∂x y ∂y z ∂y x (15). preliminares Matemática Essa relação é válida para toda e qualquer variação em y e z. Se, por exemplo, mantivermos y constante e deixarmos somente z variar temos dy = 0 e dz ≠ 0 . Assim, o primeiro termo entre colchetes à direita de (15) deve ser unitário, enquanto o segundo se anula pelo fato de dy ser nulo. Então, temos: 1 ∂z ∂x ∂z =1 ⇒ = ∂x y ∂z y ∂x y ∂x ∂z y (16). Esse resultado é conhecido, algumas vezes, como teorema da inversão. Dissemos que a relação (15) é válida para quaisquer variações de y e z. Podemos, então, fazer dz = 0 e dy ≠ 0 para obter: ∂z ∂x ∂z = 0 + dy ∂x y ∂y z ∂y x E como dy ≠ 0 , o termo entre colchetes deve se anular: ∂z ∂z ∂x = − , ou usando a forma apropriada de (16), podemos escrever: ∂x y ∂y z ∂y x ∂x ∂y ∂z = −1 ∂y z ∂z x ∂x y (17). Note que cada derivada em (17) envolve as três coordenadas em “ordem cíclica”. Exemplo 6 Verifique a relação (17) para a função z = x 3 y . Solução ∂x ∂y e também a derivada , precisamos obter x ( y, z) e Como usaremos a derivada ∂z x ∂y z y( x , z ) : z x = y 13 = z 1 3 y −1 3 y= z . x3 Então, as três derivadas que constituem a (17) são: ∂z 2 = 3x y (I) ∂x y ∂x z1 3 = − 3y 4 3 ∂y z 1 ∂y = 3 ∂z x x (II) (III). A multiplicação desses três resultados dá: z1 3 3x 2 y × − 43 3y 1 z1 3 × = − . x3 y1 3 x Observe a igualdade para compará-la com a função original z = x 3 y ou 3 z1 3 3 z z x3y = 1 . Dessa −1 3 = −1 e, portanto, (17) se comparação é imediato concluir que ( −1) 1 3 = yx y x cumpre. Exercícios 2.6 - Verifique se a igualdade (17) é satisfeita para a relação x 2 + y2 + z2 − R 2 = 0 (I). 27 tErModinÂMiCa Sugestão: evite calcular a derivada de, por exemplo, x = − y 2 − z 2 + R 2 . Ao invés, derive diretamente a expressão (I): x 2 = − y 2 − z 2 + R 2 para obter cíclica para obter as outras derivadas. Ou faça cada uma delas. ∂x y = − e use a permutação x ∂y z Antes de prosseguirmos na análise de sistemas físicos, usando os resultados matemáticos obtidos, é conveniente ressaltar alguns pontos referentes a sistemas termodinâmicos reais. Todo processo infinitesimal em Termodinâmica deve satisfazer o requisito que ele represente uma mudança em alguma quantidade que é pequena em relação a si mesmo e grande em comparação com o efeito produzido no comportamento de umas poucas moléculas. A razão disso é que as variáveis termodinâmicas tais como o volume, a pressão, a temperatura não possuem significado quando aplicadas a sistema compostos de poucas partículas, ou, de forma equivalente, as variáveis termodinâmicas são grandezas macroscópicas. 2.3 Aplicações de Derivadas Parciais em Termodinâmica O cálculo de derivadas parciais de uma função matemática é operação de rotina: a manipulação de derivadas parciais no Cálculo é um treinamento pelo qual se atinge prática e confiança para aplicações. O uso de derivadas parciais em Termodinâmica é fundamental, pois diversas quantidades podem ser representadas por elas. Diversas substâncias sofrem variações no volume quando são aquecidas a pressão constante. O volume de um fluido (gás ou líquido) ou de sólido pode ser considerado como uma variável dependente de duas outras variáveis independentes, a pressão e a temperatura: V = V ( p, T ) . Se a temperatura é variada ligeiramente de T para T + ∆T , enquanto a pressão é mantida constante, o volume da amostra varia de uma quantidade que é proporcional a ∆T . Ainda mais, essa variação no volume é proporcional também ao próprio volume. Essas afirmações, baseadas em fatos experimentais, você já encontrou no capítulo 6, do volume II, de Física Geral. ∆V = Vγ∆T . O fator de proporcionalidade γ é chamado de coeficiente de expansão volumétrica e sua determinação experimental permite escrever 1 ∆V V ∆T γ= O quociente medido (18). ∆V ∂V corresponde à derivada parcial . Mais precisamente, ∆T ∂T p ∆V ∂V = lim ∂T p ∆T →0 ∆T (19). ∂V é adequadamente representada pelo ∂T p Para pequenas variações em T e V, a derivada parcial quociente (mensurável) ∆V , e vice versa. Com isso, o coeficiente de expansão volumétrica é ∆T identificado como a quantidade γ= 1 ∂V V ∂T p (20). Outra quantidade relacionada com derivadas parciais é a compressibilidade isotérmica, κ. A definição analítica dessa grandeza é dada por: κ=− 1 ∂V V ∂p T (21). ∂V é negativo para a maioria das substâncias ∂p T O sinal negativo é devido ao fato de que o termo 28 (elas se contraem quando pressionadas) e, portanto, o sinal negativo é para assegurar que κ seja uma quantidade positiva. Fisicamente, ele mede com que extensão o volume de um sistema é diminuído quando a pressão aumenta e o nome compressibilidade é bastante apropriado. O valor de κ para sólidos e líquidos varia muito pouco com a temperatura e pressão, e, portanto, pode ser considerado constante para intervalos razoáveis de p e T. Uma das dificuldades em se aplicarem os resultados da parte II para analisar o comportamento de sistemas físicos, está na identificação das variáveis matemáticas, x, y e z com as variáveis termodinâmicas. O exemplo seguinte mostra como se procede. preliminares Matemática Exemplo 7 Queremos determinar a relação entre as quantidades γ e κ. Solução As definições de γ e κ envolvem as derivadas do volume em relação a T e a V, respectivamente. ∂x ∂y ∂z = −1 , não parece imediato o que se deve Observando a relação (17), ∂y z ∂z x ∂x y identificar com as variáveis termodinâmicas V, p e T. Mas (17) foi obtida a partir de ∂z ∂z ∂x = − ∂x y ∂y z ∂y x (I). Agora aparecem duas derivadas da variável z e, como dissemos, tínhamos duas derivadas de V: parece conveniente identificarmos z com o volume do sistema. As outras duas variáveis podem ser escolhidas por qualquer critério pessoal. Façamos, então, as correspondências: z↔V x↔T y↔p. Com essa escolha, as derivadas parciais na relação (I), ficam: ∂V ∂V ∂T = − ∂T p ∂p V ∂p T (II). As definições de γ e κ possibilitam as mudanças necessárias em (II): γ= 1 ∂V ∂V ⇒ = γV V ∂T p ∂T p κ=− ∂V 1 ∂V ⇒ = − κV . V ∂p T ∂p T Então, (II) pode ser escrita na forma: ∂T κ ∂T γV = κV ∴ = . γ ∂p V ∂p V Esse é o resultado final. Tivéssemos escolhido (critério pessoal) x ↔ p e y ↔ T , a relação que obteríamos seria o recíproco: 1 1 γ ∂p = ⇒ = . κ γ ∂T κ ∂T V ∂p V Verifique essa afirmação, refazendo os cálculos. Exercícios 2.7 - Usando a equação de estado para o gás ideal, mostre que: (a) O coeficiente de expansão volumétrica é dado por γ = 1 T . (b) A compressibilidade isotérmica é κ = 1 p . 2.8 - Uma equação aproximada para um gás real à temperatura e pressão moderadas, considerando o tamanho das moléculas, é dada por: p(V − b) = RT , onde R e b são constantes. 29 Mostre que: tErModinÂMiCa (a) γ = 1T . 1 + bp RT (b) κ = 1p . 1 + bp RT 2.9 - A densidade volumétrica é definida por ρ = m , onde m é a massa do corpo e V seu volume. V Quando aquecido (a pressão constante), sua densidade e seu volume variam. Mostre que a variação relativa da densidade é dada por: dρ ρ = −γ dT . Sugestão: sabendo-se que ρ = ρ(T ) e V = V (T ) , derive a expressão da densidade em relação à temperatura. Lembre-se de que γ = − γ= − 1 dV . V dT p 2.4 1 ∂V e, como V é somente função da temperatura, V ∂T p Derivadas Parciais de Ordem Superior Você deve ter notado que as derivadas parciais de uma função z = z( x , y) , ∂z ∂z 2 x ou , podem ser funções de x e de y. Por exemplo, a função z( x , y) = y e ∂x y ∂y x ∂z ∂z 2 x x tem derivadas = y e e = 2ye . Portanto, podemos derivar novamente essas ∂ ∂ x y y x funções como usualmente se fez com a z( x , y) . As segundas derivadas parciais são dadas pelas relações: ∂2z ∂ ∂z = 2 ∂x ∂x y ∂x y (22) ∂2z ∂ ∂z = 2 ∂y ∂y x ∂y x (23) As derivadas (22) e (23) são semelhantes às segundas derivadas para funções de uma variável, mas funções de duas ou mais variáveis também admitem derivadas mistas: ∂ ∂z ∂2z = ∂y ∂x y ∂y∂x ∂ ∂z ∂2z = ∂x ∂y x ∂x∂y (24). Muitas vezes, dependendo da função z( x , y) , a ordem de derivação é essencial. No primeiro caso de (24), deriva-se a função em relação à x, mantendo-se y constante e, em seguida, deriva-se o resultado com respeito à y, mantendo-se x constante. No segundo caso, inverte-se a ordem. Exemplo 8 2 Encontrar as derivadas parciais da função z ( x, = y ) xy 2 + e x y , usando as relações (22), (23) e (24). Solução (22) ⇒ 2 2 ∂2z ∂ ∂z ∂ 2 z ∂ 2 x2y e x y 2xy(2xy) + 2ye x y 2 = (y + e 2x y) ∴ 2 = = ∂x ∂x y ∂x y ∂x ∂x y ∂ 2z 2 ⇒ 2 = (4 x 2 y 2 + 2 y)e x y . ∂x y 30 (23) ⇒ 2 ∂2z ∂ ∂z ∂ 2 z ∂ 2 x2y = = + ∴ (2yx x e ) 2x + x 4 e x y . 2 = 2 ∂y ∂y x ∂y x ∂y ∂y x preliminares Matemática As relações (24) dão: 2 2 2 2 ∂2z ∂ = (y 2 + 2xye x y ) =2y + 2xe x y + 2xyx 2 e x y =2y + 2x(1 + x 2 ye x y ) . ∂y∂x ∂y 2 2 2 2 ∂2z ∂ = (2xy + x 2 e x y ) =2y + 2xe x y + 2x 3 ye x y =2y + 2x(1 + yx 2 e x y ) . ∂x∂y ∂x Para essa função, ambas as derivadas mistas são iguais. No exemplo 8, as derivadas mistas apresentaram os mesmos resultados, mas nem sempre isso ocorre. Quando a função original z( x , y) tiver primeira e segunda derivadas contínuas para todo e qualquer ponto em algum domínio, então a ordem de derivação nas derivadas mistas é irrelevante: ∂ ∂z ∂ ∂z = ∂y ∂x y ∂x ∂y x (25). Felizmente, a maioria das funções que aparecem nas aplicações físicas é continua e tem derivada contínua. Em particular, na Termodinâmica relações do tipo (25) aparecem frequentemente. Exemplo 9 Na termodinâmica, a entropia S e a pressão p de uma substância podem ser expressas como derivadas parciais de uma função chamada energia livre de Helmholtz, F = F( V, T ) : ∂F S = − ∂T V ∂F p = − . ∂V T ∂p ∂S = considerando a continuidade das derivadas da função F. ∂V T ∂T V Mostrar que Solução ∂ 2 F ∂ ∂F ∂ ∂ = (−S) = − (S) ⇒ ∂V∂T ∂V ∂T V T ∂V T ∂V T 2 ∂ F ∂ ∂F ∂ ∂ = ⇒ (− p ) = − (p ) ∂T∂V ∂T ∂V T V ∂T V ∂T V (I). (II). Como as derivadas mistas são contínuas, (I) e (II) são iguais e podemos escrever: ∂p ∂S ∂V = ∂T . T V A igualdade acima é conhecida como uma das relações de Maxwell da Termodinâmica. Ela se mostra bastante útil porque permite calcular a entropia de um sistema (que não é uma quantidade diretamente mensurável experimentalmente) em termos da dependência entre p, V e T, que são razoavelmente fáceis de obter. 31 tErModinÂMiCa Anotações 32 preliminares Matemática Anotações 33 tErModinÂMiCa Anotações 34 3 Teoria Cinética Elementar 3.1 introdução 3.2 o gás ideal 3.3 Modelo para o gás ideal 3.4 Colisões e distribuição de velocidades 35 tErModinÂMiCa 3 TEORIA CINÉTICA ELEMENTAR 3.1 Introdução No volume Física Geral II, capítulo 7, usamos o fato que a energia cinética das partículas que compõem um gás ideal é função somente da temperatura, isto é, E = E (T ) . A soma das energias cinéticas de cada uma das partículas é o que se chamou de energia interna do gás. Queremos, agora, mostrar que essa relação é verdadeira. O modelo que usaremos consiste em considerar o gás como uma coleção enorme de partículas – átomos e moléculas – que se move incessantemente e de forma caótica. Esse modelo leva à equação de estado para o gás ideal e fornece uma interpretação mecânica da temperatura. 3.2 O Gás Ideal Temos notado previamente que todos os gases podem ser descritos satisfatoriamente pela mesma equação de estado, desde que a pressão não seja demasiadamente alta e nem a temperatura seja muito baixa. Por exemplo, qualquer gás à temperatura ambiente e pressão atmosférica pode ser considerado como ideal. A relação entre as variáveis termodinâmicas é dada pela equação de estado: PV = nRT ou PV = NkT (26) R onde N é número de partículas e k= . Utilizaremos N o ou N Av. para denotar o número de No Avogrado. Usaremos P, ao invés de p, como temos feito para evitar confundi-la com o momento linear, p, da partícula. O valor constante R depende das unidades utilizadas: R = 8.318×107 erg/K ; R = 8.318 Joule / K ; R = 1.987 cal / K ou R = 0.082 litro.atm / K . TAMANHO DA PARTÍCULA E O TAMANHO DO RECIPIENTE. Não podemos ver as moléculas que compõem o ar que aspiramos. Presumimos, então, que átomos e moléculas devem ser pequenos. Mas de que tamanho? Qual o volume ocupado por uma dessas partículas típicas? A resposta a essa questão não demanda um microscópio eletrônico ou um conhecimento de Mecânica Quântica. Uma estimativa confiável do tamanho atômico pode ser baseada na observação de que sólidos e líquidos são praticamente incompressíveis. Somente com altíssimas pressões se consegue alterar o volume de um sólido de alguns percentuais. Contrastando com esse comportamento, os gases são facilmente compressíveis e esse fato sugere que suas partículas se encontram distantes umas das outras, enquanto que para sólidos e líquidos elas estão mais fortemente próximas. Para estimar o tamanho atômico vamos considerar que o volume do sólido (ou o líquido) seja igual ao volume total de seus constituintes: isso significa ignorar o espaço entre os átomos. Parece uma aproximação? Não pode ser ruim porque se houvesse grandes espaços entre eles, seria fácil comprimi-los e tornar seus átomos mais próximos quando sob pressão. E, experimentalmente, sabe-se que para pequenas variações de volume, altíssimas pressões são requeridas. Considere um mol de uma substância, como por exemplo, o cobre. Seja V seu volume e M sua massa molecular dada em gramas. O número de átomos desta amostra é igual ao número 23 de Avogadro N o ≈ 6 × 10 . Em seguida, considere que o volume V seja composto por esses N o átomos: isso corresponde a dividir o volume V em sub-volumes de mesmos tamanhos – um para cada partícula. Assim, cada uma delas ocupa um minúsculo cubo de aresta d e volume d 3 . Com essa configuração, d é uma medida do tamanho atômico e coletivamente essas caixas cúbicas fornecem o volume V da substância: V = Nod3 . A densidade é expressa por: = ρ 36 massa de um mol M M . = = volume de um mol V N o d 3 Resolvendo para d, temos: M d = ρN o 13 (27). Substituindo N o pelo seu valor numérico, escrevemos: 13 13 1.66 M 1.66 M −8 d≈ ×10 cm = Å ρ ρ (28), onde Å é a unidade de comprimento chamada angstrom, cujo valor numérico é 10 1.6 M A tabela seguinte mostra que o fator ρ teoria Cinética Elementar −8 cm. 13 é essencialmente o mesmo para diversos sólidos e líquidos elementares. Isso indica que a unidade angstrom caracteriza o tamanho atômico. 13 (1.66M ρ ) SÓLIDOS M (g) ρ ( g / cm3 ) Li Al Zn Ag Au Pb 6.9 27.0 65.4 107.9 197.0 207.2 0.53 2.7 7.1 10.5 19.3 11.3 2.8 2.6 2.5 2.6 2.6 3.1 200.6 4.0 20.2 13.6 0.12 1.21 2.9 3.8 2.9 (Å) LÍQUIDOS Hg He Ne TABELA 1. Dimensões atômicas características. Os valores indicam que o volume de cada célula, d 3 , contendo uma partícula é da ordem de 10−23 cm3 , ou seja, o volume atômico é dessa ordem. Vamos analisar como esse volume atômico se compara com uma amostra de gás ocupando certo volume: queremos estimar o volume ocupado pelo gás quando comparado com o volume do recipiente que o contém. Um mol de átomos de um gás ocupa um volume de 22400 cm3 sob condições normais 23 de temperatura e pressão, ou seja, N o = 6 × 10 partículas ocupam esse volume. Se cada átomo possui volume estimado da ordem de 10−23 cm3 , então se comprimíssemos esses átomos de tal forma que obtivéssemos um sólido em certa região do recipiente, o volume ocupado por eles seria de apenas 1 4000 do volume total, ou, dito de outra forma, os átomos na fase gasosa estão dispersos em um volume cerca de 4000 vezes sua própria dimensão. Todos os cálculos efetuados são estimativos, e não resultados rigorosos, mas a ordem de grandeza dos parâmetros encontrados está correta. O próximo objetivo é estabelecer um modelo para o gás ideal. A equação de estado para o modelo possibilitará uma importante interpretação: a temperatura de um gás revelará como ela está relacionada com a energia cinética média das partículas. 3.3 Modelo Para O Gás Ideal Para iniciar a elaboração de um modelo é necessário estabelecer algumas hipóteses que permitam esboçar a linha de desenvolvimento e, se possível, antecipar as limitações impostas ao modelo, baseadas em observações experimentais. O conjunto de quatro hipóteses define um modelo particular de um gás. Nosso propósito de deduzir a equação de estado supõe implicitamente que o gás está em equilíbrio térmico com as paredes do recipiente. (a) O gás consiste de um grande número de partículas (átomos para um gás monoatômico) que obedecem às leis de Newton. (b) O movimento das partículas é caótico. 37 tErModinÂMiCa (c) O volume ocupado pelos próprios átomos é desprezível comparado com o volume do recipiente e também essas partículas são consideradas como pontos materiais no sentido da Mecânica Newtoniana. (d) As colisões entre as partículas e as paredes são perfeitamente elásticas e o único tipo de energia a ser considerada é a cinética translacional. Cada item expresso para a formulação do modelo permite críticas e considerações. Por exemplo, supor que os átomos obedeçam às leis de Newton parece ser um contra-senso depois do desenvolvimento da Mecânica Quântica. Entretanto, a fundamentação da Teoria Cinética dos Gases foi baseada em termos da Mecânica Newtoniana, e seu sucesso posterior por si poderia justificar apropriadamente o uso da Física Clássica. Reconhecidamente, o modelo apresenta dificuldades porque os átomos são descritos de maneira mais conveniente pelo uso da Teoria Quântica, e certos paradoxos surgem na Teoria Cinética se insistirmos na utilização do contexto clássico. As anomalias no calor específico são historicamente significantes porque indicaram que algo estava errado com os fundamentos da Física do século XIX. Por ora, vamos nos concentrar no sucesso da Teoria Cinética como ela foi originalmente usada, isto é, dentro da descrição clássica. A hipótese de que os choques com as paredes são perfeitamente elásticos é, na realidade, bastante simplista, mas justificável em última análise: as partículas ao se chocarem com as paredes sofrem o que se denomina de adsorção por um curto intervalo de tempo e se afastam com velocidades correspondentes à temperatura da parede. Se existe equilíbrio térmico entre o gás e as paredes, não há transferência de energia entre ambos e o módulo das velocidades das partículas permanece constante. Esse fato permite validar a hipótese de choques totalmente elásticos. EQUAÇÃO DE ESTADO Escolhidas as hipóteses para o modelo, deve-se, então, estabelecer um plano para efetuar os cálculos. A tarefa requer a determinação da pressão do gás, que nada mais é do que a força normal média, por unidade de área, devido às colisões entre as partículas e as paredes do recipiente. O gás está contido em um volume de dimensões L x , L y e L z . A componente da velocidade paralela à parede não varia nem em módulo e nem em sentido. Somente a componente perpendicular à parede tem seu sentido alterado, mas o módulo permanece constante porque o choque é perfeitamente elástico. Inicialmente, vamos supor que todas as partículas possuem a mesma componente da velocidade, v x . Esta restrição é bastante forte, porém, mais tarde podemos relaxá-la pelo uso de um valor médio (quadrático). Por ora, com a intenção de simplificar o desenvolvimento, essa hipótese é conveniente. Figura 5 – Representação de uma colisão entre uma partícula e a parede. 38 Pela figura 5, com o eixo x na direção perpendicular à parede, podemos ver que a componente x do momento linear, p x , varia de − p x (antes da colisão) até + p x (após a colisão). Assim, a variação do momento linear da partícula é dada por: ∆p x ≡ (p x )depois − (p x )antes = p x − (− p x ) = 2p x teoria Cinética Elementar (29). Em termos de velocidade a relação (29) pode ser escrita como ∆p x = 2mv x (30). A terceira de lei de Newton afirma que a parede do recipiente sofreu a mesma variação de momento linear, porém, em sentido oposto: momento transferido para a parede = 2mvx . Aqui utilizamos essa quantidade em módulo porque vamos tratar de pressão, uma grandeza não negativa. O momento transferido à parede por uma partícula do gás não é observável: instrumentos de medida respondem ao efeito combinado de um número muito grande de tais colisões. Na figura 6 está esboçado um cilindro que contém diversas partículas na iminência de se chocarem com a parede. Se a molécula está prestes a colidir com uma dada área A da parede em um intervalo de tempo dt, então sua posição inicial deve ser vx dt (somente partículas dentro do cilindro mostrado na figura 6 alcançam a área A no intervalo dt). Figura 6 – Moléculas na iminência de colidirem com certa área A. O volume desse cilindro é A vx dt , e vamos supor que a densidade de partículas seja uniforme em qualquer região dentro do recipiente, isto é, N / V = constante. Assim, o número de partículas, n ′ , dentro desse pequeno cilindro imaginário, é: n′ = N A v x dt V (31). Como estamos supondo que as partículas executam um movimento caótico, o número delas que possui componente v x em direção à parede é somente a metade do valor expresso por (31): a outra metade está se afastando da parede. Portanto, o número de colisões que ocorrem no intervalo de tempo é dado por: = n n′ 1 N = A v x dt 2 2V (32). A variação do momento linear sofrida pela parede no intervalo dt é dada pelo produto 2m v x × n : = dp x 1N A v x (dt) × 2m v x 2V ∴ = dp x NAmv 2x dt V (33). 39 tErModinÂMiCa A taxa de variação temporal do momento linear é a força resultante que atua sobre a área A, na direção x: dp x dt = Fx ⇒ Fx = NAmv 2x V (34). A pressão P é definida como o módulo da força exercida pela área considerada: Fx N ∴ P = mv 2x V A P= (35). A pressão do gás é a mesma para qualquer área A e, portanto, poderíamos escolher essa área como sendo toda a parede perpendicular à direção x, isto é, A = L y L z , ou, de forma equivalente, construir cilindros cuja soma das áreas cobririam toda a parede: o resultado final desse procedimento é o mesmo dado por (35). Mas não existe nada de preferencial na direção x: refazendo-se os mesmos cálculos para as duas outras direções, y e z, podemos concluir que: P= N mv 2y V N mv 2z V P= e O módulo da velocidade da partícula está relacionado com as componentes por: v 2 = v 2x + v 2y + v 2z (36). O valor médio de (36) é dado por: (v ) 2 média ( ) = v 2x ( ) + v 2y média ( ) + v 2z média média (37). Considerando ainda o fato de não haver direção preferencial para o movimento das partículas do gás, (v ) 2 x média ( ) = v 2y média ( ) = v 2z média . Então, a relação (37) pode ser rescrita como: v 2x = ( ) 1 2 v 3 média . Substituindo essa expressão em (35), temos finalmente a igualdade: P= ( ) N 1 m v2 V 3 média 1 2 ∴ PV = 3 Nm v ( ) média (38). Temos estabelecido uma relação entre a pressão do gás é a velocidade quadrática média das partículas. Mas a equação (38) é pouco reveladora porque o valor das velocidades das partículas não é acessível de imediato. Poderíamos elaborar um pouco mais a relação (38) para expressá-la em termos da energia cinética total: = ( E c )TOTAL 1 1 . = m ( v2 ) Nm ( v 2 ) ∑ média média 2 todas as 2 particulas Substituindo em (38), temos: PV = 2 ( E c )TOTAL 3 (39). Podemos comparar (39) com a equação de estado para o gás ideal: PV = nRT ∴ nRT = 2 (E c )TOTAL . 3 Finalmente, podemos estabelecer a relação entre a energia cinética e a temperatura: ( E c )TOTAL = 40 3 nRT 2 (40). Esse resultado extraordinariamente simples mostra que a energia cinética total, composta pela soma das energias cinéticas de cada uma das partículas, é diretamente proporcional à temperatura absoluta T. De forma alguma isso representa uma definição de temperatura porque as medidas de energia cinética de uma partícula são bastante difíceis de serem obtidas e, portanto, torna inviável uma definição operacional de temperatura. O resultado (40) foi utilizado no capítulo 7, do volume Física Geral II, sem que fosse dada uma justificativa baseada em um modelo mecânico-molecular. Na equação (40), n significa o número de mols da amostra gasosa, e o número de partículas, N, está relacionado com n pela relação: N = nN Avogadro teoria Cinética Elementar (41). A energia cinética de cada partícula é: ( E c= )partícula O quociente ( E c )TOTAL 3 nRT 3 R = = T . N 2 nN Av. 2 N Av. R é identificado como a constante de Boltzmann, k, cujo valor numérico, em N Av. unidades do SI, é 1.38 × 10 dada por: −23 joules / molécula. K . Então, a energia cinética por partícula é 3 kT 2 ( E c )partícula = (42). Exemplo 10 Avalie as velocidades quadráticas médias de um átomo de hélio, e de uma molécula de nitrogênio e de uma molécula de hidrogênio, todas à temperatura ambiente. Dados: m= 6.64 ×10−27 kg , m = 4.65 ×10−26 kg e m= 3.32 ×10−27 kg . hélio N2 H2 Solução Inicialmente precisamos definir velocidade quadrática média para uma partícula: Definição: v q − média = ( v 2 ) média . Para calcular os valores pedidos, usamos a relação (42): (E c )partícula = 1 m(v 2 )média ∴ 2 = v q-média Portanto, = ( v2 ) média 1 3 m ( v2 ) = kT . média 2 2 3kT . m Então, com os valores das massas das partículas e com T = 300 K , temos: (v = ) q − média hëlio 3 ×1.38 ×10−23 × 300 ∴ 6.64 ×10−27 ( v= ) 3 ×1.38 ×10−23 × 300 ∴ 4.65 ×10−26 v (= ) 3 ×1.38 ×10−23 × 300 3.32 × 10−27 q − média N 2 q − média H 2 ∴ (v ) q − média hëlio (v ) q − média N 2 (v ) ≈ 1370 m / s . ≈ 516 m / s . q − média H 2 ≈ 1950 m / s . As moléculas de hidrogênio são as que possuem as maiores velocidades quadráticas média. Uma fração significativa dessas partículas se move com velocidade muito maior do que essa e, portanto, conseguem escapar do campo gravitacional terrestre ( v escape ≈ 11000 m / s ). Esse fato é responsável pela baixíssima concentração de hidrogênio na atmosfera, embora ele seja o elemento mais abundante no Universo. 41 Exercícios tErModinÂMiCa 3.1 - Verifica-se que quatro partículas, escolhidas ao acaso, possuem velocidades de 500, 550, 600 e 700m/s. Encontrar a velocidade quadrática média e a velocidade média desse conjunto. 3.2 - (a) Achar a energia cinética média de uma partícula de um gás ideal a uma temperatura de 300 K. (b) Encontre a energia cinética de um mol desse gás. Compare seu resultado com a energia cinética de algo macroscópico, como por exemplo, um atleta correndo. (c) Suponha que o gás seja o oxigênio. Calcule a velocidade quadrática média das moléculas. −8 3.3 - Usando-se uma bomba de vácuo do tipo difusora, é possível atingir pressões da ordem de 10 Torr. Qual o número de moléculas em 1 cm3 ? Encontre a energia cinética média das partículas contidas nesse volume. Considere T = 300K . É comum a interpretação da pressão como sendo a razão do módulo da força por unidade de área. Entretanto, se insistirmos em entendê-la assim, pode-se chegar a conclusões errôneas dos tipos “existe uma pressão nas paredes do recipiente, mas não existe pressão em nenhum ponto no interior da massa gasosa” ou ainda “um gás não confinado por paredes materiais, não exibe pressão”. Ambas as afirmações são incorretas, e uma interpretação mais adequada será apresentada agora. Usando a relação (39), suprimindo-se os subscritos, podemos definir uma densidade de energia interna (que se resume em energia cinética): PV= onde u ≡ 2 2 E ⇒ P= u, 3 3 E é a densidade de energia interna. Esse resultado é significante, pois ele relaciona, de V forma muito simples, a pressão com a densidade de energia e permite uma nova interpretação da quantidade P: a pressão é a medida da densidade de energia. Esse ponto de vista é bastante útil quando se consideram diferentes tipos de energia envolvida: cada uma delas contribui para a pressão total através de sua densidade de energia. Um exemplo dessa situação é uma nuvem estrelar de hidrogênio que pode se contrair sob ação devido à sua própria interação gravitacional. O sistema possui três distintos tipos de energia que contribuem para sua pressão interna: a energia cinética de translação de seus átomos, a energia potencial gravitacional responsável por sua contração e devido a essa contração, os átomos de hidrogênio emitem energia radiante na forma de fótons. A ideia de a pressão estar relacionada com a densidade de energia pode ser estendida a situações fora do contexto de gases formados por ponto materiais. Na eletrodinâmica é definida uma densidade de energia do campo elétrico que está relacionada com a pressão de radiação. Os detalhes da situação serão tratados na disciplina específica. 3.4 Colisões E Distribuição De Velocidades No modelo proposto na seção anterior para um gás ideal, não consideramos a possibilidade de que ocorressem choques entre as partículas. Para analisar a situação em que se permitem as colisões, usaremos uma argumentação bastante simples e tentaremos responder, por exemplo, qual é o tempo médio entre as colisões, ou, que distância a partícula percorre entre duas sucessivas colisões. Na segunda parte desta seção, introduziremos a distribuição de velocidades para as partículas que compõem o gás. Considere N partículas (esféricas) ocupando certo volume V. Para simplificar a situação, suponha que apenas uma partícula esteja se movendo e todas as outras restantes estejam em repouso (adiante daremos o fator de correção que admite o movimento simultâneo de todas as moléculas). Cada partícula tem raio r e ela vai colidir somente com partículas cujas distâncias estejam em um cilindro de raio 2r. A figura 7 mostra a situação descrita. A molécula que se move irá colidir com qualquer outra cujo centro esteja no interior desse cilindro de comprimento vdt . O volume desse cilindro é π( 2r ) 2 vdt = 4πr 2 vdt . Uma vez sendo constante a densidade do 42 gás, existem N V partículas por unidade de volume; portanto, dentro desse cilindro existem dN partículas cujos centros estejam dentro desse cilindro: teoria Cinética Elementar N dN = 4π r 2 vdt . V O número de colisões por unidade de tempo é dado por: dN 4π r 2 vN = dt V (43). dN 4 2 π r 2 vN = dt V (44). Uma análise mais detalhada, considerando o movimento simultâneo das partículas, indica que o valor de (43) deve ser corrigido por um fator multiplicativo 2 (isso é razoavelmente intuitivo porque o número de colisões deve aumentar): Esse resultado traduz o número de colisões por unidade de tempo que uma partícula sofre ao se deslocar dentro do recipiente. Se (44) é o número de colisões, o tempo médio entre as colisões é o inverso dessa relação: t médio = V 4 2 π r 2 vN (45). Queremos estimar qual seria o caminho livre médio entre duas colisões sucessivas. Essa grandeza é denotada por λ e sua definição é dada por: λ ≡ vt médio ∴ λ = V 4 2 π r2N (46). Observe que o livre caminho médio independe da velocidade da partícula. Entretanto, a relação (46) indica que ele é inversamente proporcional à densidade do gás, N V , e também é inversamente proporcional à seção de choque da partícula. Quaisquer das três relações, (44), (45) ou (46) pode ser modificada usando a equação de estado do gás ideal. Por exemplo, (46) pode ser escrita como: λ= kT 4 2 π r 2P (47). Quando se aumenta a temperatura do gás, mantendo-se a pressão constante, há uma expansão, e, portanto o livre caminho médio deve crescer. Por outro lado, aumentando-se a pressão à temperatura constante, o volume diminui e λ sofre uma redução. Exemplo 11 Estime o caminho livre médio e o tempo médio de colisão considerando a velocidade média como sendo v q − média , para as moléculas de oxigênio a 300 K e pressão de 1 atm. O raio da molécula é de 2 × 10 −10 m. Solução O caminho livre médio é calculado diretamente através de (47). A única parte que demanda atenção é a escolha adequada para a constante de Boltzmann, pois a pressão é dada em atm. O valor de k pode ser obtido, por exemplo, pela última relação da constante R dada na seção II: k= R N Av. ⇒ k= = λ 0.082 litro.atm / K ∴ k = 1.36 ×10-25 litro ⋅ atm / molécula ⋅ K . 23 6.02 ×10 (47) ⇒ 1.36 ×10-25 × 300 4.08 ×10-23 litro . = ∴ λ 17.77 × 4 ×10−20 m 2 4 2π (2 ×10−10 ) 2 ×1 Transformando litro para m 3 , temos: λ ≈ 5.7 ×10−8 m . 43 tErModinÂMiCa O valor encontrado para o caminho livre médio indica que a partícula percorre distâncias muito grandes, comparadas ao seu tamanho, antes de se chocar com outra partícula. Para se obter o tempo médio de colisão precisamos conhecer a velocidade quadrática média, v q − média para a molécula de oxigênio. Do exemplo anterior podemos calculá-la simplesmente trocando a massa, por exemplo, da molécula de hidrogênio pela do oxigênio: v (= ) q − média O 2 3 ×1.38 ×10−23 × 300 5.31×10−26 ∴ (v ) q − média O 2 ≈ 483 m / s . Com a definição de λ em (46) envolve duas grandezas já conhecidas, o tempo médio é dado por: t= médio λ vq − média ⇒ t= médio 5.7 ×10−8 ∴ t médio ≈ 1.2 ×10−10 s . 483 10 Esse valor é realmente notável: a molécula sofre, em média, 10 colisões por segundo. Os resultados encontrados permitem compreender porque ao se abrir um vidro de perfume em um ambiente fechado, o olfato percebe as moléculas aromáticas depois de certo tempo. Se elas percorressem trajetórias retilíneas do frasco até o observador, com v q = média da ordem da velocidade do som no ar, o tempo de percepção para registrar o odor seria extremamente pequeno. O seu movimento errático através do ar faz esse tempo aumentar consideravelmente. Como é a distribuição das velocidades das partículas constituintes de um gás? Primeiramente, temos que nos convencer de que realmente existe uma distribuição de velocidades, isto é, as partículas possuem velocidades diferentes, umas se movendo mais rapidamente do que outras. Mas isso acontece realmente? Por que elas não possuem a mesma velocidade? É uma pergunta pertinente, porque se você imaginar certa quantidade de gás encerrada em um volume V, após um tempo suficiente onde se permitiram todos os tipos de colisões, o sistema entrando em equilíbrio térmico, poderíamos pensar que todas as partículas apresentassem a mesma velocidade. Estamos usando o termo velocidade de forma pouco correta: o termo rapidez é mais indicado por se tratar de uma quantidade escalar, e não vetorial como pressupõe o termo velocidade. Ou, simplesmente utilizar a denominação “módulo da velocidade”. Que fique entendido, então, que por velocidade queremos dizer rapidez ou módulo da velocidade, e, portanto, uma quantidade escalar. Para responder à pergunta sobre a existência de uma distribuição de velocidades das partículas, podemos supor que as partículas tenham a mesma massa e se comportem como esferas rígidas. A colisão pode ser imaginada como uma semelhante àquela envolvendo duas bolas de bilhar rígidas e que durante o processo existem a conservação do momento linear e a energia mecânica (choque perfeitamente elástico). Se a colisão não for frontal, as velocidades finais de ambas as partículas terão componentes em direções diversas daquelas iniciais. Assim, é possível acontecer que a partícula mais rápida ganhe mais velocidade após o choque e aquela de menor velocidade, tenha sua velocidade final diminuída. As partículas que formam o gás, por pequenas que sejam, possuem dimensões e choques não frontais acontecem com extrema frequência. Existe uma evidência muito forte e definitiva de que as partículas de um gás obedecem a uma distribuição de velocidades: as medidas experimentais. A figura 8 é um esquema de um aparato utilizado para se determinar a distribuição de velocidades. Para descrever os resultados obtidos pelo experimento definimos uma função f(v) chamada função distribuição. Sua forma analítica será fornecida a seguir, juntamente com a definição de alguns valores médios. Figura 7 – Colisão não- frontal entre duas partículas com dimensões não nulas. 44 Suponha que um gás ideal tenha N partículas e ocupa um volume V, e, portanto, a densidade de partículas é dada por n = N V . A função distribuição de velocidades para essas partículas é expressa pela relação: m f(v) = 4π n 2π kT teoria Cinética Elementar 32 v 2 e-mv 2 2kT (48). Figura 8 – A partícula com velocidade v está passando pela fenda do primeiro disco giratório. Quando ela atinge o segundo disco giratório, ambos giraram de um ângulo θ = ωt . Se a velocidade da partícula é tal que v = ωx θ , ela passa pela segunda fenda e atinge o detector. Variando-se a velocidade angular é possível registrar as diversas velocidades das partículas que compõem o feixe. Esse resultado é um dos mais importantes da Teoria Cinética dos Gases e foi obtida primeiramente por Maxwell por considerações de Mecânica Estatística. Pelo fato de ele ter utilizado a distribuição de Boltzmann, a equação (48) é mais conhecida como distribuição de Maxwell – Boltzmann para velocidades de partículas. A figura 9 mostra o comportamento gráfico de (48) para três temperaturas diferentes. Comentaremos mais acerca dessas curvas logo mais adiante. Conhecendo-se, então, a forma analítica de f(v) é possível obter diversas quantidades: a velocidade mais provável vmp , a velocidade média v m e a velocidade quadrática média v q − média . Esta última grandeza já foi definida no exemplo 10 em termos da temperatura e da massa da partícula. Esperamos que uma vez conhecida f(v), ambos os resultados coincidam. A velocidade média é definida como: vm ≡ ∞ 1 v f(v)dv n ∫0 (49). A velocidade quadrática média é a raiz quadrada do valor dado por: ∞ 1 2 v f(v)dv n ∫0 (50). df(v) =0 permite calcular vmp dv (51). (v 2 ) média ≡ A velocidade mais provável é obtida igualando a zero a derivada da função f(v): Figura 9 – A distribuição de velocidades dada por (48). 45 tErModinÂMiCa Observando a relação (48), vemos que a dependência com a velocidade (a uma dada temperatura fixa) é composta pelo produto de duas funções: v 2 exp − αv 2 . Para valores pequenos de velocidade, a função cresce aproximadamente como uma parábola, e à medida que v aumenta a exponencial suprime o crescimento parabólico levando a função a valores que tendem a zero. O resultado dessa combinação faz com que a função apresente um ponto de máximo para algum valor da velocidade: esse ponto é que se define como a velocidade mais provável. O que a curva está indicando é o comportamento das velocidades das partículas: é relativamente baixa a probabilidade de se encontrar partículas com baixas e com altas velocidades. Ou, dito de forma diferente, existem relativamente poucas partículas com baixas e com altas velocidades. Outra característica que pode ser observada na figura 9 é que o pico da curva se desloca para a direita quando a temperatura cresce: isso indica que a velocidade mais provável aumenta quando o gás é aquecido, e, simultaneamente a curva se torna mais “achatada”. Entretanto, não só a velocidade mais provável aumenta com a temperatura. A velocidade média e a velocidade quadrática média também crescem com a temperatura. No exemplo seguinte serão calculados alguns valores médios para essa distribuição, e essas afirmações serão comprovadas. Os crescimentos, entretanto, não são diretamente proporcionais a T, mas dependem de T . ( ) Exemplo 12 Dada a distribuição de Maxwell para as velocidades das partículas, encontre (a) a velocidade mais provável; (b) a velocidade média e (c) a velocidade quadrática média. Solução m É dada a função f(v) = 4π n 2π kT 32 v 2 e-mv 2 2kT (48). (a) Para calcular a velocidade mais provável derivamos f(v) e igualamos a zero: 32 m df(v) d 2 -mv 2 2kT 0 ⇒ 0. = = 4π n v e dv dv 2 kT π v=v mp m 2π kT 32 O fator numérico 4π n é irrelevante nos cálculos e, portanto, será suprimido. Ficamos, então, com a expressão após efetuar a derivada: 2 − mv2 kT 2kT 2mv 2 m . + v 2 e-mv 2kT − = 0 ⇒ 2 − v mp = 0 ∴ v mp = 2ve 2kT kT m vmp Esta é a expressão que determina a velocidade mais provável. Seu significado físico é que se ao se escolher aleatoriamente uma partícula do gás, é mais provável que a partícula tenha essa velocidade. Ou, dito de outra forma, a probabilidade é máxima quando v = vmp . (b) A definição de v média (49) nos dá: 32 32 ∞ m m ∞ 3 -mv2 2kT 1 2 -mv 2 2kT vm v 4π n = v e dv = dv ⇒ v m 4π v e n ∫0 2π kT 2π kT ∫0 A integral envolve a função gama e pode ser encontrada em tabelas especializadas (veja, por exemplo, Spiegel- tabela de integrais, coleção Schaum). O resultado é: 32 2 m 1 2kT v m 4π = = ∴ vm 2π kT 2 m 8kT . πm Observe que a velocidade média é maior do que a velocidade mais provável. (c) A velocidade quadrática média é dada pela raiz quadrada da integral: 12 v q-média = 46 1 ∞ (v ) média ∫ v 2 f(v) dv . = n 0 2 Substituindo a função f(v) na expressão acima, temos: 12 v q-média m 3 2 ∞ 4 -mv2 2kT dv . = 4π ∫v e 2π kT 0 teoria Cinética Elementar Novamente, a integral pode ser encontrada em tabelas de integrais. O resultado: 12 v q-média 52 m 3 2 3 2kT π = 4π . m 2π kT 8 12 v q-média 52 m 3 2 3 2kT π = 4π , que após as simplificações se torna: 2π kT 8 m v q-média = 3kT . m Esse resultado já havia sido obtido anteriormente (exemplo 10). O valor de v q = média é maior do que os outros dois determinados: v q-média > v m > v mp . Exercícios 3.4 - A distribuição de Maxwell é dada, algumas vezes, em função da energia cinética da partícula, ε. Mostre que (48) pode ser escrita como: 32 8π m −ε kT . f (ε ) = εe m 2π kT 3.5 - Usando a distribuição do problema anterior, mostre que a função atinge seu máximo para ε = kT . 3.6 - O dióxido de carbono, ( CO2 ), tem massa molecular de 44.0 g/mol. Se a temperatura desse gás é 300 K, encontre: (a) velocidade mais provável; (b) a velocidade média; (c) a velocidade quadrática média. Observe o uso correto do sistema de unidades. Recomenda-se utilizar o SI. (3.7) Usando a função distribuição de velocidades de Maxwell – Boltzmann (48) e o resultado para v média do exemplo 12, verifique se é falsa ou verdadeira a igualdade: 1 1 . = v média v média 1 1 1 = é dada por: v média v média n A definição de ∞ 1 ∫ v f(v) dv . 0 47 tErModinÂMiCa Anotações 48 teoria Cinética Elementar Anotações 49 tErModinÂMiCa Anotações 50 4 A 1ª Lei da Termodinâmica 4.1 trabalho 4.2 processos quase-Estáticos e processos reversíveis 4.3 alguns tipos de trabalho 4.4 Energia interna e Calor 4.5 a primeira lei da termodinâmica 51 tErModinÂMiCa 4 A 1ª LEI DA TERMODINÂMICA 4.1 Trabalho Os conceitos de trabalho e calor estão intimamente relacionados. No volume Física Geral II já abordamos ambas as grandezas, entretanto, é conveniente se fazer uma breve revisão dos pontos mais importantes envolvendo esses dois conceitos. A forma mais intuitiva e prática de se introduzir o trabalho é através da mecânica utilizandose uma força F, agindo sobre uma partícula quando ela se desloca de uma quantidade infinitesimal ds: o trabalho infinitesimal, dW, realizado por essa força F é dado pelo produto escalar entre a força e o deslocamento: dW = F . ds (52). Alguns autores usam um “d cortado” [đ ] para representar o trabalho infinitesimal. Essa notação será explicada no decorrer desse capítulo. Adiantamos que essa simbologia é meramente utilizada para lembrar que o trabalho, assim como o calor, não são variáveis de estado. Para se determinar o trabalho total realizado por essa força quando a partícula se desloca de uma quantidade não infinitesimal ao longo de uma curva especificada, os trabalhos infinitesimais devem ser somados, isto é, devemos realizar uma integração (que deve ser feita ao longo de um caminho previamente escolhido, ligando o ponto inicial e o final). Podemos, então, escrever para o trabalho total: F A →B W= B O simbolismo B B dW ∫ F.ds ∫= A (53). A ∫ dW significa “adicione todos os elementos infinitesimais de trabalho realizado A entre os pontos A e B”. Como dizemos acima, é necessário especificar o caminho (ou trajetória) F que une os pontos A e B. O resultado WA → B , em geral, depende desse caminho e, por isso, é dito ter “dependência de trajetória’”. O exemplo a seguir mostra a dependência do resultado para duas trajetórias distintas. Exemplo 13 Uma partícula se move no plano xy sob influência de uma força que possui somente a componente x: Fx = Cxy . Calcular o trabalho realizado por essa força quando a partícula sai da origem e se desloca até o ponto (1,1). Considere, primeiramente, o percurso ao longo de uma reta unindo os pontos. Em seguida, escolha o caminho ao longo de uma parábola, passando pelos pontos dados. Solução A constante C que aparece na expressão da força é para dar as unidades apropriadas. Suas dimensões podem ser determinadas da seguinte forma no SI: kg.m kg [Newton] = [C][m][m] ⇒ 2 = [C].[m 2 ]∴[C]= . 2 s m.s Observe que, usualmente, as constantes possuem unidades; somente algumas são números puros. A força F de que precisamos para calcular o trabalho pode ser identificada a partir da componente dada: F = Fx ˆi + Fy ˆj + Fz kˆ = Cxyˆi + 0ˆj + 0kˆ . 52 O deslocamento infinitesimal ds (ou muitas escrito como dr) é sempre dado, em coordenadas cartesianas, por: ds = dxˆi + dyˆj + dzkˆ . a 1ª lei da termodinâmica (I) Cálculo do trabalho ao longo do segmento de reta que une os pontos A = (0,0) e B = (1,1) . A equação da reta nesse caso é y = x e, portanto, a expressão da força é: F= Cxyˆi + 0ˆj + 0kˆ= Cx 2 ˆi + 0ˆj + 0kˆ . WAF= →B 1 ∫ (Cx ˆi + 0ˆj + 0kˆ ).(dxˆi + dyˆj + dzkˆ ) . 2 0 Note que temos agora uma integração simples em apenas na variável x. Como as componentes da força nas direções ˆj e kˆ são nulas, a integração se restringe somente à variável x. Portanto, o trabalho realizado é dado por: F A →B W = 1 ∫ Cx dx 2 F A →B ⇒W 0 x3 = C 3 1 F A →B ∴ W 0 C = 3 . (II) Podemos utilizar o mesmo procedimento para obter o trabalho da força F ao longo da 2 parábola y = x . Agora a forma analítica de F é dada por: F= Cxyˆi + 0ˆj + 0kˆ= Cx 3ˆi + 0ˆj + 0kˆ . Então, o cálculo do trabalho se resume em resolver a integral: F A →B W = 1 ∫ Cx dx 0 3 F A →B ⇒W x4 = C 4 1 ∴ WAF→B = 0 C. 4 Observe que os valores encontrados em (I) e em (II) não são os mesmos, indicando que o trabalho depende do caminho escolhido. Entretanto, para alguns tipos de forças a integral fornece sempre o mesmo valor, independentemente do caminho escolhido: são as forças chamadas conservativas. O trabalho mecânico de uma força é a forma mais familiar de trabalho. Entretanto, em termodinâmica o trabalho realizado pelo sistema (ou sobre ele), pode aparecer sob variadas formas: nesse contexto incluem-se todas as formas de energia transferida com exceção do calor. Exercícios 4.1 - Dada a força F= (x,y) Ax 2 yˆi + Bxy 2 ˆj [N], (a) Quais são as unidades das constantes A e B? (b) Calcular o trabalho realizado por essa força quando a partícula evolui desde a origem até o ponto (1,1) ao longo do percurso formado pelo eixo x e a seguir ao longo do eixo vertical. (c) Refaça o item (b), considerando um novo percurso formado pelo eixo y e por um eixo horizontal. 4.2 - Achar o trabalho realizado pela força F (x,y,z) = Axˆi + Byˆj + Czkˆ [N], ao longo da reta que une a origem ao ponto (1, 1, 1) . 4.2 Processos Quase-Estáticos e Processos Reversíveis No exemplo 13, e também nos exercícios propostos, as forças consideradas apresentavam uma propriedade que permitia calcular as integrais de forma inequívoca: tais forças eram definidas para todos e quaisquer valores das variáveis independentes. Isto significa que para qualquer ponto especificado, o valor da função F fica bem caracterizado e univocamente determinado. Do ponto de vista matemático, essa situação é bastante desejável para o cálculo do trabalho. Entretanto, 53 tErModinÂMiCa quando analisamos a evolução de um sistema físico, nem sempre é possível se conhecer o valor da variável termodinâmica de interesse para todos os pontos (ou e forma equivalente, a todo instante). Por exemplo, durante uma expansão livre arbitrária de certa quantidade de gás encerrada em um volume inicial, a variação da pressão não fica univocamente determinada, e a falta de conhecimento desta função ponto a ponto é um severo impedimento para se calcular o trabalho realizado pelo gás. Na verdade, quando consideramos a representação do processo em um diagrama pV, a ausência da curva ligando os pontos inicial e final inviabiliza a avaliação do trabalho realizado. Em outras palavras, a expressão WA →B = VB ∫ p dV VA não pode ser integrada até que p seja especificada como função de V. Os fatos expostos acima para o caso onde se considera o trabalho mecânico realizado pelo sistema, pode ser estendido a situações semelhantes nas quais as variáveis termodinâmicas de interesse, não são caracterizadas univocamente. Portanto, necessitamos impor algumas restrições que irão permitir o cálculo dos variados tipos de trabalho. O procedimento é idealizar uma evolução cujo processo ocorra de maneira quase-estática. Durante um processo quase-estático, o sistema está em todos os instantes infinitesimalmente próximo a um estado de equilíbrio termodinâmico. Uma equação de estado é, portanto, válida para todos esses estados intermediários. Essas condições quase-estáticas não podem ser rigorosamente alcançadas no laboratório, mas podem ser aproximadas com alto grau de precisão. A importância do processo quase-estático é perfeitamente entendível. Queremos descrever processos em termos das variáveis termodinâmicas do sistema, e elas são bem definidas somente para estados de equilíbrio. Mas, qual o critério para se configurar um comportamento quase-estático? Um dos critérios mais funcionais para caracterizar esses processos pode ser estabelecido da seguinte forma: seja τ processo o tempo característico do processo; por exemplo, o tempo requerido para a variável termodinâmica sofrer uma variação mensurável. Associada à taxa temporal na qual o sistema se aproxima do equilíbrio, também existe um tempo característico que denotamos por τ equilíbrio . Para que o processo possa ser considerado quase-estático, nós impomos a condição: τ equilíbrio < τ processo (54). O que essa desigualdade está indicando é que o tempo necessário para que o sistema atinja o equilíbrio termodinâmico deve ser muito menor comparado com o tempo de medida de qualquer mudança nas variáveis termodinâmicas. A condição (54) significa que os processos irreversíveis ocorrem muito rápidos, sempre mantendo o sistema em estados próximos ao de equilíbrio. Para ilustrar o que estamos discutindo, considere um processo no qual uma barra metálica é aquecida lentamente. A condução de calor é um processo irreversível que permite atingir uma temperatura homogênea do metal. Se o calor é adicionado muito lentamente, mantendo a fonte de calor a uma temperatura ligeiramente superior àquela do metal, então a temperatura da barra aumentará uniformemente. Em outras palavras, termômetros posicionados em diferentes pontos da barra indicarão a mesma leitura se a condutividade térmica do material for suficientemente efetiva em comparação com a taxa de aquecimento. Para a condução de calor, τ equilíbrio é o tempo para o qual uma variação da temperatura seja percebida ao longo de a barra metálica. O tempo requerido para adicionar uma quantidade suficiente de calor para produzir uma variação detectável na temperatura é τ processo . Uma rápida condução de calor e um aquecimento lento calor cooperam para tornar válida a igualdade (54), caracterizando um processo quase-estático. Percebe-se, então, que o conceito de processos quase-estáticos é relativo e está ligado ao tempo de relaxação τ do sistema. Por tempo de relaxação, entende-se o tempo necessário para que um sistema termodinâmico, uma vez deslocado da situação de equilíbrio, retorne a um novo estado de equilíbrio. Esse tempo requerido depende da natureza detalhada das interações que ocorrem entre as partículas que constituem o sistema; o cálculo da taxa temporal com que o sistema se aproxima desse novo estado de equilíbrio é uma tarefa bastante difícil, mesmo para sistemas simples. Durante esse intervalo de tempo não se pode utilizar, obviamente, a descrição da termodinâmica de equilíbrio. Uma situação bastante comum pode servir para exemplificar o conceito de processo quase-estático, considerando o tempo de relaxação. Suponha um sistema constituído por um gás 54 encerrado em um cilindro que possua um pistão móvel. A questão que se coloca é essa: com que taxa deve-se puxar (ou empurrar) esse pistão para que ocorra um processo quase-estático? Ou, quão lentamente devemos variar o volume para caracterizar um processo quase-estático? A resposta mais ingênua a essas perguntas poderia ser: devemos proceder de tal forma que o tempo entre dois deslocamentos sucessivos infinitesimais seja maior do que o tempo de relaxação do sistema. Uma resposta mais quantitativa, entretanto, pode ser obtida: a perturbação criada pelo deslocamento se propaga, aproximadamente, com a velocidade do som no gás. Se o volume do cilindro for V, então o tempo de relaxação pode ser escrito, com boa aproximação, como τ = V 1 3 v som . Para sistemas macroscópicos esse valor é da ordem de alguns milissegundos. Portanto, se a expansão (ou compressão) entre dois estados de equilíbrio acontecer a uma taxa de, digamos 0.1 segundo, o processo pode ser considerado quase-estático: a todo instante o sistema está em equilíbrio. a 1ª lei da termodinâmica O conceito de processo reversível está intimamente relacionado com processos quaseestáticos. O uso da reversibilidade de uma evolução será usado de forma extensiva quando abordarmos a segunda lei da termodinâmica. Antes de definir o que seja um processo reversível, vamos transcrever o que alguns reconhecidos autores dizem sobre ele: ► “Transições quase-estáticas são, de fato, reversíveis, mas de forma alguma é ó b v i o que todas as transições reversíveis são quase-estáticas.” [H. A. Buchdahl] ►“Todo processo reversível coincide com um quase-estático.” [H. B. Callen] Kestin] ►“Todo processo reversível é quase-estático, mas a inversa não é verdadeira.” [ J . É fácil perceber que, para cada autor, a definição de processo reversível é entendida de forma diferente. O que parece ser um problema é, na verdade, devido ao fato de que os termos quaseestático e reversível não são definidos de maneira única. O exemplo de um cilindro munido de êmbolo serve admiravelmente bem para introduzir a noção de processo reversível. A figura 10 sugere como uma compressão pode ser conduzida quase estaticamente. Figura 10 – Um exemplo de uma compressão quase-estática de um gás. Considere, inicialmente, o comportamento do sistema quando não existe atrito entre o pistão e as paredes do cilindro. A adição ou a remoção de um grão de areia devem ser entendidas como produzindo uma variação infinitesimal nas vizinhanças. Em qualquer estágio podemos parar a compressão e iniciar a expansão pela retirada de um grão de areia, desde que não exista atrito. Se a areia é removida grão a grão, revertendo a ordem da adição, o gás percorrerá o caminho termodinâmico inverso daquele seguido pela compressão. Um ponto importante a ser notado é que a inversão pode ser iniciada por uma variação infinitesimal nas vizinhanças. A compressão quase-estática sem atrito, assim descrita, é um exemplo de um processo reversível. Uma definição mais geral pode ser estabelecida: “Um processo reversível é um processo quase-estático que pode ser revertido por uma variação infinitesimal nas vizinhanças.” 55 tErModinÂMiCa Note que um processo reversível não é necessariamente um que evolui em uma direção e então retorna ao ponto inicial. Reversível implica que ele pode ser revertido. A presença de atrito é fatal para a reversibilidade. Se existe atrito, podemos ainda comprimir o gás quase-estaticamente pela lenta adição de grãos de areia, mas, e é aqui que a reversibilidade é perdida, a compressão não pode ser revertida por uma variação infinitesimal nas vizinhanças: a remoção de um grão (variação infinitesimal) não reverterá o processo. Necessitamos variações finitas (não-infinitesimais) nas vizinhanças para inverter a compressão tornando-a uma expansão. O conceito de reversibilidade é importante, não somente porque nos permite, quando utilizada de maneira apropriada, fornecer os cálculos pertinentes de um dado processo que de outra forma seriam impossíveis de se obter, mas porque a reversibilidade é essencial para estabelecer critérios para a ocorrência de processos espontâneos na Natureza. Diga-se aqui que processos espontâneos são irreversíveis. A figura 11 mostra a diferença fundamental entre um processo quase-estático e um processo arbitrário (rápido) para a compressão e expansão de um gás. Os diagramas pV para ambos os processos são também mostrados. Figura 11 – Processos quase-estáticos e não quase-estáticos. Para enfatizar esses pontos discutidos, gostaríamos de comentar mais alguns aspectos acerca da reversibilidade (ou da falta dela). Considere dois blocos metálicos a temperaturas diferentes e isolados um do outro. Inicialmente, ambos estão separadamente em equilíbrio; é, então, permitida a troca de calor entre eles através de uma barra, de baixíssima condutividade térmica, ligando os dois corpos. Certamente, a situação pode ser estabelecida como sendo um processo quase-estático: a troca de calor acontece de maneira extremamente lenta e após um tempo suficientemente longo (que depende, por exemplo, da diferença de temperatura entre ambos), o equilíbrio térmico é estabelecido. Entretanto, esse não é um processo reversível. O fluxo de calor não pode ser revertido em qualquer instante e uma vez estabelecido o equilíbrio térmico, é impossível restaurar as condições iniciais dos blocos e da vizinhança. Observe que poderíamos tentar reverter a evolução enquanto não ocorresse o equilíbrio, mas isso demanda que o corpo de menor temperatura seja aquecido acima da temperatura do bloco que inicialmente estava a uma temperatura mais elevada. Mas, ao fazer isso a variação nas vizinhanças não se configura como sendo infinitesimal porque existe uma diferença finita (não-infinitesimais) de temperatura entre os dois corpos. Somente nos casos onde se tem uma diferença de temperatura infinitesimal, pode-se considerar tal processo como reversível. 56 Uma importante consequência da reversibilidade é que o trabalho realizado pelo sistema pode ser expresso em termos das coordenadas (ou variáveis) termodinâmicas e suas variações. Na próxima seção discutiremos alguns tipos de trabalho que aparecem mais frequentemente em termodinâmica. 4.3 a 1ª lei da termodinâmica Alguns Tipos de Trabalho Podemos agora considerar os diversos tipos de trabalho e a evolução dos sistemas é suposta acontecer de forma quase-estática. Por sistema hidrostático entende-se um sistema constituído por um gás encerrado em um recipiente de volume V e munido de um pistão móvel com aquele mostrado na figura 10. Esse sistema foi considerado no livro Física Geral II, no capítulo 7, e o desenvolvimento que iremos fazer é muito semelhante àquele. Quando o êmbolo se movimenta, podemos construir um diagrama escolhendo o eixo x para representar as mudanças do volume e o eixo y indica as variações da pressão. Assim, as variações do volume e das pressões do sistema durante a expansão ou da compressão são indicadas simultaneamente no mesmo diagrama. Esse tipo de construção é chamado indicador ou diagrama pV. A convenção utilizada no curso introdutório de termodinâmica será mantida aqui: o trabalho realizado pelo sistema é considerado positivo e o trabalho realizado sobre o sistema, negativo. A figura 12 enfatiza esses aspectos: em (a) o trabalho é positivo (o sistema realiza trabalho sobre as vizinhanças); em (b) o trabalho realizado pelo sistema é negativo (o meio realiza trabalho sobre o sistema); na parte (c) as curvas dos dois processos anteriores são adicionadas e, juntas, elas formam uma figura fechada a que chamamos ciclo. O conceito de ciclo é bastante importante e será usado no capítulo sobre a segunda lei da termodinâmica quando analisarmos um dos mais importantes ciclos, o ciclo de Carnot. No volume Física Geral II, foi mostrado que o trabalho realizado pelo sistema em um processo quase-estático é dado pela integral: B WA →B = ∫ p dV (55). A Figura 12 – O diagrama pV. A simplicidade do integrando, pdV , dessa expressão (e pelo fato de ser encontrado inúmeras vezes) pode fazer com que sua mais importante característica seja esquecida. O trabalho realizado pelo gás (ou sobre ele) é dado somente em termos das variáveis termodinâmicas do sistema. A natureza da força externa e outras propriedades da vizinhança não aparecem na expressão. Esse resultado é possível somente pelo fato de se considerar uma evolução quase-estática e sem atrito (processo reversível). Se houver atrito, um termo extra deve aparecer em (55) representando o trabalho realizado pela força dissipativa. O trabalho infinitesimal, dW = pdV , é muitas vezes chamado de trabalho termodinâmico porque sua expressão contém unicamente variáveis termodinâmicas do sistema. Em contraste, o trabalho realizado por forças de atrito não são classificadas de trabalho termodinâmico porque tais forças não são variáveis termodinâmicas. Simplesmente usa-se a expressão trabalho, sem adjetivos, para se referir a grandeza definida por (55). O valor dessa integral, como já dissemos, depende do caminho escolhido. Isso significa que, fixados dois pontos (A e B, ou i e f) no diagrama pV, o trabalho vai depender da forma da curva que liga esses dois pontos. Considere a figura 13, na qual temos marcado os pontos i e f para representarem os estados inicial e final, respectivamente. Obviamente, existem infinitas possibilidades de se ligarem esses dois pontos e o diagrama mostra algumas curvas ligando esses dois pontos. 57 tErModinÂMiCa Figura 13 – O trabalho depende da trajetória representada no diagrama pV. Por exemplo, uma isobárica a partir de i até o ponto a e a seguir através de uma isocórica (ou isovolumétrica) terminando em f. O trabalho realizado pelo sistema é numericamente igual a área sob a curva (numericamente porque devemos acrescentar as unidades consistentes para que o trabalho seja expresso em Joules). Nesse caso a trabalho é dada por 2P0 V0 . Uma segunda possibilidade seria primeiramente uma evolução isocórica até o ponto b, e a seguir uma isobárica até f. Nesse caso, o valor do trabalho é o dobro daquele calculado anteriormente, ou seja, W = P0 V0 . A reta inclinada conectando os dois pontos representa um processo no qual tanto a pressão como o volume variam simultaneamente. O cálculo do trabalho nesse caso pode ainda ser obtido geometricamente pela área do trapézio sob a curva (ou adicionar as áreas do retângulo e do triângulo que determinam o trapézio). O resultado, para esse caso é W = 3 P0 V0 2 (verifique). É interessante observar que esses resultados, obtidos de forma geométrica através da área, independem da natureza da substância: basta obter a área sob a curva para se determinar o trabalho. Note que para cada percurso (ou caminho) escolhido temos um resultado, indicando uma vez mais que o trabalho depende não só dos pontos inicial e final, mas da curva que descreve o processo. O uso da relação (55) para o cálculo do trabalho, digamos, mecânico, pode não ser uma tarefa fácil. O exemplo seguinte ilustra esse ponto. Exemplo 14 −2 Suponha uma amostra de cobre de massa 10 kg. De forma quase-estática e isotérmica (300 K) aplica-se uma pressão que cresce desde 0 atm até 1000 atm. Encontre o trabalho realizado 3 pela amostra nesse processo. É dada a densidade ρ cobre = 8.93 g / cm a 300 K. Solução O trabalho é dado por (55): Wi →f = f ∫ pdV . i Mas o que é dV nesse processo? Sabemos que o volume depende da temperatura e também da pressão. Assim, ∂V ∂V = dV dp + dT . ∂T p ∂p T A compressibilidade isotérmica é dada definida por: κ=− 1 ∂V . V ∂p T Como o processo acontece a temperatura constante, o segundo termo na diferencial dV se anula. ∂V : ∂p T Então, podemos escrever, substituindo ∂V −κ Vdp . dV = dp ⇒ dV = ∂p T 58 Com isso podemos calcular o trabalho realizado pelo sólido: a 1ª lei da termodinâmica pf f ∫ ∫ Wi →f = pdV ⇒ Wi →f = p(− κVdp) . i pi Temos ainda um pequeno problema para realizar a integração: o integrando apresenta duas variáveis, κ e V que dependem da pressão. Nesse ponto convém refletir antes de fazer contas: para um sólido, o volume é praticamente insensível à variação de pressão; da mesma forma, o módulo isotérmico de compressibilidade também se mantém constante para uma ampla faixa de pressão. Com essas aproximações a integral pode ser resolvida imediatamente: Wi→= f pf pf pi pi ∫ p(−κ Vdp) ⇒ Wi→f ≈ −κ V ∫ p dp ∴ Wi →f ≈ − κV 2 (p f − p i2 ) . 2 Os dados fornecidos para o problema permitem avançar um pouco mais. O volume não foi dado, mas conhecemos a densidade e a massa da amostra. Portanto, pela relação V = m , pode ρ 10−2 kg ∴ V ≈ 1.12 × 10-6 m3 . Devemos ter atenção especial para 8.93 × 103 kg/m3 −11 −1 as unidades: é recomendável utilizar o SI. A constante κ vale 0.73 × 10 Pa , e, sabendo-se mos obter V = 5 que 1 atm vale 1.013 × 10 Pa , o valor do trabalho realizado pelo sistema é: Wi →f ≈ −0.042 J . O sinal negativo significa que foi feito trabalho sobre a massa de cobre. O valor apresentado indica que quando um gás é comprimido (mesmo a uma pressão extremamente alta) dentro de um recipiente metálico, o trabalho realizado sobre as paredes é desprezível comparado àquele feito sobre o gás. Embora o último exemplo tenha tratado do cálculo do trabalho realizado sobre uma amostra metálica, é comum o estudante associar trabalho com uma expansão ou compressão de um gás (ideal ou não). Entretanto, na termodinâmica o conceito de trabalho envolve diversas situações para as quais a relação dW = pdV requer alterações específicas considerando as características do sistema. Se um fio de comprimento inicial L é submetido a uma tração (ou tensão) F e seu novo comprimento é L + dL, o trabalho infinitesimal realizado pelo fio é dado por: dW = −F dL (56). O sinal negativo é usado porque um acréscimo no comprimento L significa uma expansão do fio, e isso deve ser feito por um trabalho sobre ele, portanto, um trabalho negativo. O cálculo do trabalho f total W = ∫ dW somente pode ser obtido se o processo for reversível, isto é, o valor da tração F i deve ser conhecido em cada etapa do processo. Como outro exemplo, considere uma película de face dupla preenchida por um líquido. A película é mantida dentro de uma moldura rígida com um dos lados móvel, como mostra a figura 14. Sobre o fio existe uma força exercida pelo filme. Ela é proporcional ao comprimento L que resulta de uma força de tensão por unidade de área do líquido: isso é conhecido como a tensão superficial σ cujas unidades são N/m. Por exemplo, para a interface água/ar a 35 oC, σ = 0.072 N / m . Uma força de tensão é o oposto de uma força de compressão: ela puxa ao invés de empurrar. 59 tErModinÂMiCa Figura 14 – Uma película dupla esticada em uma moldura. A origem física da tensão superficial está no fato das moléculas em um líquido exercem forças atrativas entre si que mantém o líquido coeso. Uma molécula no interior do líquido terá uma energia potencial menor, pois ela está cercada em todas as direções por outras moléculas, enquanto uma molécula na superfície sente uma força atrativa somente em uma direção. Portanto, uma molécula na superfície tentará se mover para o interior do líquido. No equilíbrio, as partículas mais internas tenderão a puxar as outras mais externas resultando uma tensão superficial. Desde que a película tem duas superfícies, a força requerida para manter estacionário o lado móvel da armação é: F = 2σL . Essa é a força que um agente externo deve aplicar sobre o lado móvel para que ele não se desloque em uma contração. Portanto, esse lado aplica o mesmo módulo, porém em sentido contrário sobre o fio (via película): F = −2σL Se agora esse lado é puxado reversivelmente (ou seja, a força externa aplicada é ligeiramente maior do que aquela exercida pelo fio) de uma distância dx, o trabalho infinitesimal realizado pelo sistema é dado por: dW = −2σ Ldx (57). Nesse processo a área total (parte superior e a inferior) do filme tem sido aumentada de dA = 2 Ldx e, portanto o trabalho infinitesimal realizado pelo sistema pode ser escrito como: dW = −σ dA (58). O trabalho total, após a película ter sua área variada reversivelmente de uma quantidade finita (não infinitesimal), é dado pela integral: Af Wi→f = − ∫ σ dA (59). Ai Embora tenhamos considerado uma geometria muito particular para calcular o trabalho, o resultado se aplica para qualquer arranjo que contenha uma película. De forma semelhante, a expressão para f o trabalho realizado por um gás, Wi →f = do recipiente. ∫ pdV , também é válido independentemente da forma i A seguir é fornecida uma tabela que lista os principais tipos de trabalho encontrados na termodinâmica. Deve ficar claro que todos os processos considerados são reversíveis e, caso contrário as relações não são válidas. 60 SISTEMA GRANDEZA INTENSIVA GRANDEZA EXTENSIVA TRABALHO INFINITESIMAL Gás-pistão Pressão (Pascal) Volume (m3) PdV (Joules) Fio F (Newtons) L (m) −FdL (Joules) Película σ (N/m) A (m2) −σ dA (Joules) Pilha reversível ε (Volt) Q (Coulomb) −ε dQ (Joules) Sólido magnético H (Oe) M (pólo.cm) − HdM (ergs) a 1ª lei da termodinâmica Tabela 4.1 – Trabalho realizado por sistemas simples. Para finalizar a análise de trabalho que desenvolvemos até agora, queremos analisar o trabalho realizado em um ciclo termodinâmico. Um processo cíclico pode resultar no desempenho do trabalho líquido de um sistema. Uma situação bastante apropriada para isso é considerar um diagrama pV. A sequência da figura 15 mostra as diversas etapas para obter o trabalho líquido em um processo reversível. Em (a), o trabalho de expansão é positivo e corresponde à área hachurada. A parte (b) indica uma compressão e o trabalho é negativo e representado pela área abaixo da curva. Em (c) é mostrado o trabalho líquido positivo que corresponde à área determinada pela curva traçada no sentido horário. Finalmente, em (d) tem-se outro ciclo percorrido no sentido anti-horário, cujo trabalho líquido é agora negativo. De forma geral, o trabalho realizado de forma quase-estática (mas não reversível) pela pressão ao considerar um ciclo completo (o sistema retornando ao ponto inicial), pode ser escrito como: Wciclo = ∫ pdV (60). C Figura 15 – Um dos possíveis ciclos representado no diagrama pV. 61 tErModinÂMiCa O símbolo ∫ C significa que a integração é feita sobre um ciclo completo ao longo de uma curva C. Geometricamente ∫ pdV representa a área delimitada pela curva C descrevendo C o ciclo no diagrama pV. Uma trajetória horária sempre representa um trabalho positivo, enquanto que uma trajetória anti-horária se refere a um trabalho negativo. Esses conceitos serão essenciais no capítulo sobre a segunda lei da termodinâmica, especialmente ao se estudar o ciclo de Carnot. Exercícios 4.3 - (a) Mostre que o trabalho realizado isotermicamente por um gás ideal é dado por: Wi→f = nRTln Vf . Vi (b) Calcule o trabalho realizado por 2 mols desse gás quando seu volume de 4 litros é diminuído até 1 litro e sua temperatura é mantida a 273 K. O que significa o sinal negativo? 4.4 - Calcular o trabalho realizado por 1 mol de gás durante a expansão quase-estática e isotérmica desde um volume Vi até um volume Vf , sabendo-se que a equação de estado é: (a) p(V-b)=RT com R e b constantes . B (b) pV=RT 1 − onde R é constante e B uma função de T . V 4.5 - Durante a expansão reversível e adiabática de um gás ideal, a pressão e o volume estão relacionados em qualquer instante por: γ pV= cte = K , onde γ = c p c v > 1 . Mostre que o trabalho realizado na expansão desde um estado inicial i até o estado final f, pode ser escrito como: Wi→f = p i Vi − p f Vf . γ −1 4.6 - Um mol de gás ideal é submetido ao ciclo mostrado. Se o sistema evolui de forma quaseestática, mostre que o trabalho líquido realizado pelo gás é dado por: Wciclo = p1 (V1 − V2 ) + RT ln V1 V2 Figura para o problema 4.6. (4.7) A pressão aplicada sobre 100 gramas de um metal é aumentada isotermicamente e quase – estaticamente desde 0 até 1000 atm. Supondo que sua densidade de 10 g/cm3 e sua compressibilidade −7 −1 isotérmica de 6.75 × 10 atm se mantenham constantes durante o processo, calcular o trabalho, em Joules, realizado pelo sólido. 62 4.4 Energia Interna e Calor A termodinâmica utiliza frequentemente os conceitos de temperatura, de energia e de calor, todos eles se relacionando de maneira muito próxima. É comum acontecer que as dificuldades que aparecem aos estudantes provêm de certa confusão entre esses três conceitos. A temperatura é, fundamentalmente, uma medida da tendência que certo corpo de ceder energia. Assim, em geral, quando sua energia cresce, também aumenta sua temperatura. Entenda que isso não é uma definição de temperatura: é meramente uma afirmação sobre temperatura que ocorre ser verdadeira. Para tornar mais claro os conceitos, gostaríamos muito de dar a você uma definição precisa de energia. Infelizmente, não podemos fazer isso. Energia é o conceito dinâmico mais fundamental da Física, e, portanto, não se pode explicá-la em termos mais fundamentais. Porém, pode-se elaborar uma lista das várias formas com que ela se apresenta: cinética, eletrostática, gravitacional, nuclear – e acrescentar que, enquanto uma forma pode se converter em outra, a quantidade total de energia no Universo não varia. Isso revela uma das leis mais fundamentais já descobertas na Física: a conservação da energia. Suponha um sistema, que esteja termicamente isolado por efetivas paredes adiabáticas. Nessas condições, o sistema pode realizar trabalho somente à custa de sua energia. Mas o que significa a expressão “a energia do sistema”? Se a vizinhança (ou o meio, ou ambiente) realiza trabalho sobre esse sistema, percebemos que a energia do sistema sofreu um acréscimo. Essa percepção, até certo ponto intuitiva, pode ser usada para uma definição operacional da energia interna do sistema. Seja, então, W o trabalho realizado nesse processo pelo sistema sobre suas vizinhanças, que é executado desde um estado inicial até outro estado final de equilíbrio. Denotamos por E a energia interna do sistema (alguns autores utilizam a letra U para simbolizar a energia interna; portanto, é muito provável que você encontre essa notação para definir a mesma quantidade que chamamos E). Essa é a quantidade que desejamos definir operacionalmente. Pela conservação da energia, escrevemos: a 1ª lei da termodinâmica Trabalho realizado Energia interna de um Energia interna de um = + pelo sistema no ambiente sistema em seu estado final sistema em seu estado inicial Em termos simbólicos essa igualdade fica: E i = E f + Wi →f . Já temos uma definição operacional do trabalho e, portanto, essa relação é apropriada para se definir operacionalmente a variação da energia interna do sistema. Rearranjando os termos podemos reescrever a igualdade: E f − E i = − Wi →f (61). Essa é variação da energia interna do sistema; se W era o trabalho realizado pelo sistema, então − W é o trabalho realizado sobre o sistema pela vizinhança (o subscrito na grandeza é agora desnecessário). A relação (61) é chamada de forma adiabática da primeira lei da termodinâmica. O estudante pode ficar confuso sobre o sinal negativo que aparece do lado esquerdo de (61), mas ele é necessário para que a igualdade seja consistente com a conservação da energia. Note que, mesmo de forma operacional, ainda não definimos a energia interna: somente sua variação em um processo no qual o sistema evolui desde um estado inicial até um estado de equilíbrio final. Isso é permissível porque podemos selecionar um valor particular de certo estado e atribuir o ele o valor zero de sua energia interna. O procedimento é muito semelhante àquele de se eleger o “zero” da energia potencial para analisar situações mecânicas e eletromagnéticas: na verdade, em todos os campos da Física trabalha-se com variações de energia e não com seu valor absoluto (mesmo porque a determinação desse valor absoluto seria tão difícil quanto desnecessário). Outro comentário muito pertinente se refere à natureza da energia interna. Um valor definitivo de E está associado a cada estado de equilíbrio de um sistema. Por essa razão, a energia E caracteriza uma função (ou variável) de estado: o valor de E para um dado estado independe da história prévia do sistema. Dito de outra forma, o caminho termodinâmico que o sistema percorre para levá-lo até um estado final de equilíbrio, não afeta o valor de E para esse estado. Se você se lembra de alguns conceitos tratados no curso de Cálculo, existe uma relação muito estreita aqui: a diferencial dE é uma diferencial exata. Isto significa que sua integração depende somente dos pontos inicial e final, mas independe da trajetória escolhida. Situação contraria àquela observada por dW: o trabalho depende tanto dos pontos inicial e final como também do caminho escolhido para a integração; isso significa que o trabalho não é uma variável de estado (como foi dito no início da parte I desse capítulo, alguns autores utilizam đW para representar uma diferencial não exata). Por essa razão que não se diz que existe certa quantidade de trabalho em um sistema. Entretanto, é perfeitamente válido dizer o sistema possui certa energia interna. 63 tErModinÂMiCa Existem diversos tipos de mecanismos pelos quais energia pode ser adicionada ao sistema ou retirada dele. Entretanto, na termodinâmica esses mecanismos são usualmente classificados em duas categorias: calor e trabalho. Calor é definido como qualquer fluxo espontâneo de energia de um corpo a outro devido a uma diferença de temperatura entre eles. O mecanismo pode ser diferente para cada situação, mas em cada um desses processos a energia transferida é chamada calor. Por exemplo, o calor transferido por condução se dá através de contato molecular: partículas que se movem rapidamente colidem com aquelas de baixas velocidades, dando-lhes certa quantidade de energia no processo. Através da convecção, o movimento de certa quantidade de massa de um gás ou líquido com temperatura maior, expande e ascende em um campo gravitacional. E, finalmente, o processo chamado radiação que consiste na emissão de ondas eletromagnéticas, principalmente na região do infravermelho para objetos a temperatura ambiente, mas inclui também a luz visível para corpos mais quentes como o filamento de uma lâmpada ou a superfície solar. 4.5 A Primeira Lei da Termodinâmica As definições e os comentários das seções precedentes permitem agora estabelecer a primeira lei da termodinâmica. Os créditos pela formulação da primeira lei são divididos por Robert Mayer e James Prescott Joule, que, trabalhando independentemente e por diferentes motivações, ambos chegaram às mesmas conclusões: ► Calor é uma das muitas formas de energia. ► Energia é conservada – ela pode ser transformada, mas não pode ser criada ou destruída. Em 1842, Mayer apresentou a primeira determinação experimental do equivalente mecânico do calão, uma unidade denominada por razões históricas de caloria. O valor proposto por Mayer estava com um desvio de 20% do valor aceito presentemente de: 1 cal =4.186 Joules =4.186 ×107 ergs =0.0040 Btu . James Prescott Joule foi um fabricante inglês de cerveja que, apoiado em sua fé, acreditava que a lei da conservação da energia era algo místico baseado na religião. Em uma série de experimentos que durou 40 anos, ele mediu o equivalente mecânico do calor. Seus dados confirmaram que a dissipação de certa quantidade de energia mecânica ou elétrica, sempre resultava ser proporcional a uma quantidade de calor. A forma geral da primeira lei da termodinâmica reconhece que uma variação na energia interna de um sistema pode ser acompanhada por uma troca de calor, uma realização de trabalho, ou uma combinação de ambos os processos. calor adicionado aparece como ao sistema uma variação na energia interna e/ou é usado para . realizar trabalho A formulação matemática dessa afirmação expressa a primeira lei da termodinâmica em sua forma mais geral: Q = ∆E + W (62). Convém notar que Q é o nome dado para a quantidade líquida de calor adicionada ao sistema. Se Q acontece ser negativo, isso simplesmente significa que o sistema rejeitou certa quantidade de calor. Em um processo adiabático Q = 0 e a relação (62) reverte à forma adiabática (61). O trabalho W que consta da relação (62) é o trabalho realizado pelo sistema. Se ele for precedido de um sinal negativo, significa que a vizinhança (ou o meio) realizou trabalho sobre o sistema. A convenção de sinais que estamos usando é aceita pela maioria dos autores, mas ela não é consensual. Por isso, convém manter certa atenção sobre os sinais dessas grandezas quando consultar outros livrostextos. A relação (62) pode ser lida da seguinte forma: quando adicionamos calor ao sistema, parte desse calor é usada para aumentar a energia interna, e outra parte é utilizada para que o sistema realize trabalho. A figura 16 apresenta os dois processos que fazem variar a energia interna de um sistema, composto por um gás, encerrado em um cilindro de volume variável. 64 a 1ª lei da termodinâmica Figura 16 – Fornecimento de calor a um sistema. A primeira lei da termodinâmica nada mais é do que a expressão para a conservação da energia. Assim, calor e trabalho são manifestações diferentes de uma única quantidade que chamamos energia. Exemplo 15 6 Um sistema absorve 1.2 × 10 cal de calor e realiza um trabalho de sobre o meio. Determine a variação da energia interna desse sistema. Solução Primeiramente vamos escolher o SI como o sistema de unidades. Assim, W = 1.7 ×107 J. Aplicando a primeira lei dada por (62), temos: 1.7 ×1014 ergs Q ≈ 5 ×106 J e Q = ∆E + W ∴ ∆E = Q − W . Os valores tanto de W como de Q são positivos e, portanto, ∆E =5 ×106 − 1.7 ×107 ⇒ ∆E =−1.2 ×106 J. O sinal negativo indica que a energia interna decresceu no processo: podemos dizer que a 7 quantidade de calor fornecida não foi suficiente para a realização de 1.7 × 10 J de trabalho. O sistema utilizou parte de sua própria energia para suprir esse trabalho porque a quantidade de calor fornecida foi insuficiente. Em situações onde o processo termodinâmico ocorre lentamente, podemos imaginá-lo como uma sequência contínua de pequenas variações. Matematicamente podem-se representar as quantidades que constituem a primeira lei por infinitesimais. Essa forma “ miniaturizada” da primeira lei é: dQ = dE + dW (63). Certamente, o conteúdo físico da lei permanece inalterado ao escrevê-la nessa forma. Entretanto, essa forma apresenta algumas inconveniências e sutilezas para as quais devemos estar atentos. Isto diz respeito à dependência do caminho, pois tanto o trabalho como o calor dependem da escolha do caminho. O fato de escrevermos dQ e dW não implica que a existência das funções Q e W que podem nos levar, erroneamente, a supor que seja verdadeiro existirem quantidades de calor o trabalho contidos no sistema. Portanto, as diferenciais dQ e dWsão matematicamente infinitesimais, mas não são diferenciais exatas, elas simplesmente denotam pequenas quantidades de calor e trabalho. O que é relevante na relação (63) pode ser percebido escrevendo: dE = dQ - dW. A diferença entre duas diferenciais inexatas nos fornece uma diferencial exata. E somente a relação dQ - dW origina uma diferencial exata: qualquer outra combinação, como por exemplo, 2dQ - 3dW, não é capaz de resultar em uma diferencial exata. 65 tErModinÂMiCa Exercícios 14 4.8 - O meio realiza 1.7 × 10 ergs de trabalho sobre um sistema que simultaneamente absorve 1.2 ×106 cal de calor. Encontre a variação da energia interna do sistema. 4.9 - Uma bateria é conectada aos terminais de um resistor que está imerso em água para se preparar um bom café. Você classificaria o fluxo de energia da bateria para resistor como “calor” ou “trabalho”? E sobre o fluxo de energia do resistor para a água? 4.10 - Dê exemplo de um processo no qual não se adiciona calor, mas sua temperatura cresce. Dê também um exemplo no qual ocorre o oposto: adiciona-se calor, mas sua temperatura se mantém constante. 4.11 - Em 1842, Mayer estava concluindo suas observações sobre o equivalente mecânico do calor. o Nessa ocasião ele estabeleceu que a quantidade de calor para elevar um grama de água de 0 C o a 1 C equivale à energia adquirida por essa massa de água solta de uma altura de 365 m. Qual o 2 valor que Mayer havia determinado para o equivalente mecânico do calor? (Usar g = 9.8m / s ). 4.12 - Um gás ideal é submetido a um processo cíclico mostrado abaixo. Para cada uma das etapas A, B e C determine os sinais (positivo, negativo ou nulo), (a) do trabalho feito sobre o gás. (b) da variação da energia interna do gás. (c) do calor fornecido ao gás. (d) Considerando agora o ciclo completo, determine o sinal de cada uma das grandezas acima. Figura para o problema 4.12 −2 4.13 - Considere 4 × 10 mols de hélio dentro de um cilindro de volume inicial 1 litro e a uma atmosfera de pressão. De alguma maneira esse gás sofre uma expansão até atingir um volume de 2 litros de tal forma que sua pressão cresça proporcionalmente à raiz quadrada do volume. (a) Esboce um diagrama pV para esse processo. (b) Calcule o trabalho realizado pelo gás supondo que não aconteceu nenhum outro tipo de trabalho, exceto o trabalho mecânico. (c) Qual a variação da energia interna do gás? (d) Encontre a quantidade de calor adicionada, ou retirada, durante esse processo. 66 a 1ª lei da termodinâmica Anotações 67 tErModinÂMiCa Anotações 68 5 A 2ª Lei da Termodinâmica 5.1 introdução 5.2 Máquinas térmicas 5.3 Ciclo de Carnot 5.4 Formulações da segunda lei 5.5 teorema de Carnot 5.6 a Entropia e o Ciclo de Carnot 69 tErModinÂMiCa 5 A 2ª LEI DA TERMODINÂMICA 5.1 Introdução A primeira lei da termodinâmica coloca uma restrição quantitativa no processo de conversão de energia. As transformações podem alterar a forma, mas não a quantidade de energia. A segunda lei impõe limites sobre os processos de conversão de calor em trabalho. O resultado da segunda lei é que a completa conversão de calor em trabalho é impossível. Deve-se, entretanto, observar que não existe nenhuma objeção em converter qualquer quantidade de trabalho em calor, mas note que o inverso não é possível. Historicamente, o conteúdo da termodinâmica começou com o estudo das propriedades básicas de máquinas térmicas. Entende-se por máquina térmica qualquer dispositivo que, operando em ciclos, absorva calor, converta parte dele em trabalho, e rejeite o restante. Colocada em termos da limitação das máquinas térmicas, a segunda lei pode ser estabelecida dizendo-se que nenhuma delas pode absorver calor e convertê-lo integralmente em trabalho. Essa é a orientação prática da segunda lei. Indubitavelmente, a ciência da termodinâmica começou com a análise do problema de como se construir a melhor e mais eficiente máquina térmica. Essa tarefa foi realizada de forma brilhante pelo engenheiro francês Sadi Carnot, e esse é um dos poucos casos onde a engenharia tem contribuído fundamentalmente para a teoria física. No período em que Carnot viveu, a primeira lei da termodinâmica (conservação da energia) não era conhecida. Entretanto, os argumentos de Carnot foram tão cuidadosamente desenvolvidos, que eles são válidos mesmo embora a primeira lei não tivesse sido estabelecida. Algum tempo depois, Clausius elaborou uma maneira mais simples que seria compreendida mais facilmente do que a o raciocínio sutil de Carnot. Mas descobriu-se que Clausius supôs, não a conservação da energia, mas que o calor era conservado de acordo com a teoria do calórico, que logo depois mostrou ser falsa. Tem sido dito muitas vezes que a lógica de Carnot estava errada. Mas ela estava bastante correta, e de fato, somente a versão simplificada de Clausius (que todos leem) estava incorreta. A famosa segunda lei da termodinâmica foi, então, descoberta antes da primeira lei e seria interessante desenvolver os argumentos de Carnot, mas isso não será feito: usaremos a primeira desde o início, embora se possa obter resultados consistentes sem sua utilização. Nas seções subsequentes, a análise de máquinas térmicas será feita de modo frequente, não só pela importância tecnológica (que foi suficiente para ter ocorrido a revolução industrial), mas também pelo interesse intrínseco baseado no desenvolvimento de um dos principais ramos da Física. Associado às máquinas térmicas, o conceito de reservatório térmico é de importância fundamental. Um reservatório térmico é um corpo com temperatura uniforme cuja massa (ou capacidade térmica) é suficientemente grande para que sua temperatura permaneça inalterada quando ocorrer uma absorção ou rejeição de calor. O conceito é relativo: se um cubo de gelo é colocado em um balde de água, a grande massa de água do balde funciona com um reservatório térmico para o cubo de gelo. Entretanto, se essa massa de água tiver sua temperatura aumentada e for misturada à água de uma piscina, então, a piscina serve de reservatório térmico para o volume que estava no balde. Se o mesmo cubo de gelo fosse colocado em uma xícara de chá quente, seria possível registrar uma variação da temperatura da mistura e, nesse caso, o chá não pode ser considerado reservatório térmico. Para encerrar essa introdução, gostaríamos de tratar duas situações: a primeira envolvendo a expansão de um gás ideal; a segunda se refere a um processo reversível de troca de calor. Suponha que certa quantidade de um gás ideal esteja encerrada em um volume inicial, cujo recipiente seja munido de um êmbolo que pode deslizar sem atrito. O gás é, então, submetido a um processo isotérmico no qual seu volume é duplicado. Como a energia interna de um gás ideal é função somente da temperatura e ela se manteve constante, ∆E = 0 . Então, pela primeira lei, Q= W= nR ln 2 e todo o calor foi transformado em trabalho (o fator 2 é devido à absorvido pelo gás duplicação do volume). Esse resultado contraria a segunda lei? Ela afirma que a conversão total de calor em trabalho é impossível, e, no entanto, pelo exposto acima, esse fato ocorreu. Não existe contradição aqui: a segunda lei diz também que a máquina térmica deve operar em ciclo, e a situação final é bastante diferente da inicial, portanto, não fica caracterizado um ciclo. Uma vez alcançado o estágio final, o processo cessa e o sistema não se encontra no mesmo estado inicial, a não ser que um agente externo comprima novamente o gás, realizando trabalho sobre ele. Embora tenhamos considerado processos reversíveis na seção 2, do capítulo IV, queremos comentar sobre a troca de calor entre dois corpos. A situação será usada extensivamente na análise de máquinas térmicas e, portanto, sua conceituação é bastante importante. Sem dúvida, perdemos alguma coisa se a máquina apresentar partes nas quais ocorre atrito: a melhor máquina é aquela livre de atrito. A idealização pode ser comparada aos casos mecânicos, nos quais foi analisada a conservação da energia mecânica: a inexistência de atrito entre quaisquer superfícies. Nesse sentido, nossas máquinas térmicas são aparatos perfeitamente livres de atrito. Adicionalmente a isso, precisamos considerar o análogo do movimento sem atrito, a transferência de calor “sem atrito”. Em última instância, queremos transferir 70 calor em certo sentido tal que o fluxo possa ser revertido por apenas uma variação infinitesimal. Se a diferença de temperatura é finita, digamos de alguns graus, esse processo é impossível. Entretanto, se temos certeza de que o calor flui de um corpo a outro, cujas temperaturas são essencialmente iguais (mas não exatamente iguais) e diferem infinitesimalmente para que o fluxo de calor aconteça no sentido desejado, esse fluxo é dito ser reversível. A figura 17 mostra os processos reversíveis de troca de calor. Se fornecermos uma pequena quantidade de calor na parte à esquerda do corpo, então, o fluxo de calor se propaga para a direita; se retiramos pequena quantidade de calor da parte à esquerda do corpo, o calor fluirá da direita para a esquerda. Essas quantidades de calor, em ambos os casos, são tais que determinam variações infinitesimais de temperaturas. a 2ª lei da termodinâmica Figura 17 – Transferência reversível de calor. 5.2 Máquinas Térmicas Máquinas Térmicas são dispositivos que operando em ciclos termodinâmicos, (1) – realizam algum trabalho líquido à custa da transferência de calor de um corpo a uma temperatura elevada para outro a uma temperatura mais baixa; ou, (2) – transferem calor de algum corpo que está a uma temperatura baixa para outro a uma temperatura mais elevada à custa de um trabalho externo (Figura 18). A figura 1-a esboça o funcionamento de um motor. Combustível é transformado em energia, na forma de calor, parte dessa energia faz com que um móvel se locomova e outra parte é transferida para o ar atmosférico. Na figura 1b, o esboço é o de um refrigerador. Utilizamos energia elétrica, na forma de trabalho, para transferirmos calor de um lugar que está frio, dentro do refrigerador, para um local que está mais quente, o ambiente da cozinha. De acordo com a Primeira Lei da Termodinâmica ΔE = Q – W, tendo em vista que tanto um motor como um refrigerador funcionam em ciclos, ∆E = 0 , pois E é coordenada termodinâmica do sistema, consequentemente, a integral cíclica do calor é igual à integral cíclica do trabalho, ou seja, Q = W . Figura 18 - Digramas esquemáticos de um motor e um refrigerador. Para um sistema que executa um ciclo, existem somente duas hipóteses para as grandezas Q e W. Hipótese 1 Q = W > 0 (positivos). Quando Q = W > 0, convencionalmente, esta expressão indica que calor está sendo fornecido para o sistema e que trabalho é realizado pelo sistema. Mostraremos através de um exemplo como estas formas de energia atuam em um sistema que executa um ciclo. Cabe ressaltar que esta interpretação vale para qualquer ciclo. 71 tErModinÂMiCa A figura 19 apresenta um ciclo, idealizado, de um motor composto por duas transformações adiabáticas e duas transformações isovolumétricas. Em um motor, certa quantidade calor QE é transferida de um reservatório quente, a alta temperatura, para um sistema capaz de realizar trabalho útil, e certa quantidade calor QS é transferida para um reservatório frio. No desenvolvimento a seguir estaremos considerando ambos, Q E e Q S , como quantidades positivas. Do estado 1 até o estado 2 (adiabática), o sistema realiza trabalho sobre o meio exterior (ou simplesmente, o trabalho é realizado pelo sistema); do estado 3 até o 4 (outra adiabática), o meio exterior realiza trabalho sobre o sistema. O trabalho total W realizado pelo sistema para completar um ciclo é dado pela diferença entre estas duas quantidades, ou seja, pela área hachurada formada pelo ciclo. Observe que nos processos isovolumétricos 2 → 3 e 4 → 1 , o trabalho mecânico é nulo (dV = 0). Figura 19 - Ciclo idealizado de um motor que opera com um gás ideal. Considerando que as transformações 1 → 2 e 3 → 4 são adiabáticas, não existe troca de calor entre o meio exterior e o sistema. Quando o sistema evolui do estado 2 para o 3, calor é transferido dele para o meio exterior, e vamos denominar esta quantidade de QS (negativo). Por outro lado, para evoluir do estado 4 até o estado 1, o calor entra no sistema, e nós vamos chamar esta quantidade de QE (positivo). De acordo com esta nomenclatura, a quantidade de calor Q para o sistema realizar este ciclo é igual à Q E − Q s , com QS sendo diferente de zero, pois senão o sistema não completa o ciclo. Voltando à lei de conservação de energia aplicada no ciclo, escrevemos Q = Q E − QS = W ∴ Q E = W + QS (64). Esta expressão mostra que a energia QE, entrando no sistema na forma de calor, apenas uma parte dela pode ser utilizada para realizar trabalho, uma vez que uma fração desta energia QS tem que, necessariamente, ser transferida para o meio exterior. Agora, é conveniente introduzir o conceito de eficiência térmica para uma máquina que opera em ciclos. A eficiência é definida como a razão entre o que é produzido (trabalho útil, W) e o calor que é transferido do reservatório quente (energia QE): η térmica = benefício W = custo QE (65). Usando a relação (64), a eficiência térmica pode ser escrita como ηtérmica = Q W Q E − QS = = 1− S QE QE QE (66). A eficiência é máxima quando Q S = 0 . Essa seria a máquina dos sonhos: todo calor fornecido é convertido em trabalho, ciclo após ciclo. Infelizmente, esse aparato não pode ser construído. A limitação não é devido ao desenho, à substância utilizada ou qualquer outro fator envolvendo tecnologia ou escolha criteriosa de novos materiais. Note que, mesmo sendo uma máquina idealizada e reversível, o rendimento é sempre menor que a unidade. Esse fato reflete uma propriedade da Natureza, não uma característica particular da máquina térmica. O exemplo seguinte envolve a aplicação de uma máquina térmica bastante familiar – o motor de combustão interna de um automóvel. Ele realmente não absorve calor, mas podemos simular que a energia térmica vem de fora e não de dentro (queima do combustível) e tratá-lo, também, como uma máquina térmica. 72 Exemplo 16 A potência do motor de um automóvel é de 100 HP e sua eficiência é igual a 30%. Sabendo que a queima do combustível fornece 35 000 kJ/kg ao motor, determinar a taxa de transferência de calor para o meio e a vazão em massa de combustível consumido em kg/s. a 2ª lei da termodinâmica Solução A potência de um motor é dada por: W(trabalho útil) = W tempo kW = W 100Hp × 0,7355 = 73,55kW Hp P = A energia do combustível que entra é QE = Ou, a potência que entra é: W eficiência 73,55 = W Q = = 245,16kW E e 0,3 Consequentemente, a taxa de transferência de calor para o meio é = Q −W = 171,61kW Q S E Obs.- Esses resultados mostram que para este motor funcionar, 171,61 kJ de energia são necessariamente transferidas para o meio exterior, e como veremos adiante, essa quantidade não é devido tão somente a perdas por irreversibilidade. A quantidade em massa por unidade de tempo do combustível é calculada por: Q ∆m 245.16 E = = m = = 7.0×10−3 kg/s ∆t → 0 ∆t queima 35000 lim Isto significa que um motor a gasolina queima 7,00 gramas de combustível por segundo. Fica como exercício mostrar que um motor, com o mesmo rendimento que este e com uma potência maior, consome maior quantidade de combustível. Exemplo 17 Calcular a eficiência termodinâmica de uma máquina térmica cujo ciclo de operação está esboçado na figura abaixo. Suponha que a substância de trabalho seja um gás ideal. Figura 20 – Ciclo termodinâmico para o exemplo 17 Solução A expansão isobárica 1 → 2 determina uma absorção de calor e, consequentemente, a um aumento de temperatura do gás: Q E = C p ∆T = C p (T2 − T1 ) . A descompressão 2 → 3 resulta em uma diminuição da temperatura e uma rejeição de calor pelo sistema. 73 Q S = C v ∆T = C v T3 − T2 . tErModinÂMiCa Como T2 > T3 , podemos retirar o módulo de Q S simplesmente escrevendo T2 − T3 porque essa quantidade é positiva. Portanto, Q S = C v (T2 − T3 ) . O ciclo é completado pela evolução adiabática 3 → 1 e, portanto nenhuma troca de calor está envolvida. A eficiência termodinâmica (66) nos dá η =1− A razão C p C v é escrita como: Qs Qe =1− C v (T2 − T3 ) . C p (T2 − T1 ) Cp Cv = γ∴ Cv 1 = . Cp γ Substituindo na expressão da eficiência, temos a relação final: η =1− 1 (T2 − T3 ) 1 (1 − T3 T2 ) ∴ η =1− γ (T2 − T1 ) γ (1 − T1 T2 ) Exercícios 5.1 - Usando o resultado obtido para eficiência térmica no último exemplo e a equação de estado dos gases ideais, mostre que se pode escrever η =1− 1 (1 − P3 P2 ) . γ (1 − V1 V2 ) 5.2 - Determine a eficiência do ciclo do exemplo 17, usando os dados da figura. Considere os casos de um gás ideal monoatômico ( γ = 5 / 3 ) e diatômico ( γ = 7 / 5 ). 5.3 - Uma máquina térmica opera com um mol de gás ideal segundo o diagrama pV. Mostre que a eficiência é dada por η= γ −1 . V1 γP2 + P2 − P1 V2 − V1 Figura para o problema (5.3). 5.4 - A figura mostra um ciclo imaginário de uma máquina térmica trabalhando com um gás ideal. Mostre que a eficiência é dada por η = 1− γ 74 (V1 V2 ) − 1 (P3 P2 ) − 1 a 2ª lei da termodinâmica Figura para o problema 5.4 Um refrigerador é uma máquina térmica operando no sentido inverso, aproximadamente. Na prática, ele pode trabalhar de maneira completamente diferente. Porém, se estivermos preocupados somente o que ele realiza, não como ele o faz, podemos inverter as setas da figura 19, o ciclo é percorrido no sentido contrário e acontece que as quantidades de calor, Q E e Q S , são trocadas de posições e seus sentidos invertidos. Nesse caso, temos que em (1) ocorre a transferência de calor da fonte fria para a fonte quente e (2) é realizado trabalho sobre o sistema. A hipótese 2 trata dessas características e a figura 20 mostra um diagrama pV para o funcionamento de um refrigerador. Hipótese 2 Q = W < 0 (negativos) [REFRIGERADORES] Essas condições se verificam quando o calor que sai do sistema é maior do que o calor que entra: Q = Q E − Q S < 0 . Além disso, o ciclo no diagrama pV é percorrido no sentido antihorário: isso significa que o meio realiza trabalho sobre o sistema. Em refrigeradores domésticos e em aparelhos de ar condicionado, esse trabalho é realizado por um motor elétrico. Ambos resfriam um volume específico e rejeitam calor para o exterior. O ar condicionado expele esse calor para regiões extra-ambiente, enquanto para um refrigerador doméstico a troca ocorre em suas proximidades. Portanto, se você estiver pensando em resfriar o ambiente mantendo aberta a porta de geladeira, isso é um péssimo procedimento: o resultado líquido é a elevação da temperatura ambiente, e não seu decréscimo. Figura 20 – Ciclo idealizado de um refrigerador que opera com um gás perfeito. Do estado 4 até o 3, o sistema realiza trabalho sobre o meio exterior, e do estado 2 para o 1, o trabalho é realizado sobre o sistema. Note que W2 →1 > W4 → 3 e, consequentemente, o trabalho líquido é negativo. Nos processos que envolvem troca de calor: do estado 1 para o 4, o calor é transferido para o meio exterior, e do estado 3 para o 2, calor é absorvido pelo sistema. Como podemos definir a eficiência de um refrigerador? O parâmetro relevante ainda é a razão “benefício/custo”, e nesse caso o benefício é o calor retirado do ambiente pelo sistema e o custo é caracterizado pelo trabalho realizado sobre ele. Como a função de um refrigerador é retirar calor de uma região fria e rejeitar calor em uma região quente, define-se o coeficiente de 75 desempenho (COD) como sendo tErModinÂMiCa = COD benefício Q E (calor extraído do reservatório frio) = custo W(energia gasta) (67). Como esse coeficiente é positivo, o trabalho W deve ser considerado como uma grandeza positiva, já que Q E > 0 . Basta, então, escrever: W = QS − Q E > 0 . Substituindo na expressão (67), temos COD = QE 1 = QS QS − Q E QE (68) −1 Da definição (D), vemos que o COD é inversamente proporcional à variável básica de economia – o custo por Joule extraído. Altos coeficientes de eficiência revelam otimização de refrigeração: paga-se menos para resfriar determinado volume. Exemplo 18 Um refrigerador consome 150 Watts [J/s] de potência e transfere para o ambiente 400 Watts [J/s]. (a) determinar a taxa de transferência de calor (energia por unidade de tempo) para o interior do refrigerador; (b) o coeficiente de desempenho. Solução = 150 J/s ; Q = 400 J/s . Dados: W S (a) de acordo com a lei de conservação de energia, a taxa de transferência de calor para o interior do refrigerador é: = Q −W = 250 W = 400 W − 150 W∴ Q Q E S E (b) o coeficiente de desempenho pode ser calculado, em regime estacionário, pela relação (67): = COD Q 250W E = ≈ 1.67 . W 150W Note que ao contrário de uma máquina térmica o coeficiente de desempenho de um refrigerador é sempre superior a 100%. 5.3 Ciclo De Carnot Existem diversos ciclos termodinâmicos representativos de máquinas térmicas e alguns deles se encontram nos problemas propostos na seção anterior. Outro exemplo é o ciclo de Stirling, desenvolvido antes da termodinâmica se firmar como ciência, e que foi analisado no exemplo 8.3, do volume II, de Física Geral. Entretanto, existe um ciclo que requer um estudo mais completo por sua relevância histórica e científica – o ciclo de Carnot. O engenheiro francês Sadi Carnot se colocou a seguinte questão: dentre todos os ciclos termodinâmicos possíveis, trabalhando entre duas temperaturas fixas, qual seria o de maior rendimento? O ciclo de Carnot é de interesse especial por, pelo menos, duas razões. A primeira diz respeito ao Teorema de Carnot, que trata da eficiência de máquinas térmicas. A segunda é o fato que a eficiência do ciclo de Carnot é uma propriedade universal, independente da substância de trabalho utilizada na máquina térmica. Veremos adiante que essa característica leva à definição de uma escala termodinâmica de temperatura. Por ora, queremos discutir o ciclo propriamente dito. O ciclo de Carnot é uma série de processos reversíveis, consistindo de dois ramos adiabáticos e de dois ramos isotérmicos. A figura 21 ilustra um ciclo de Carnot no qual se utiliza um gás (não necessariamente ideal) como substância de trabalho (na verdade, a substância de trabalho nem mesmo precisa ser um gás; entretanto, para ser específico, vamos considerar que ela seja um gás ideal). 76 a 2ª lei da termodinâmica Figura 21 – Uma máquina térmica simples efetuando um ciclo de Carnot. O diagrama pV está esboçado na figura 22. As setas indicam a sequência dos processos. Durante a expansão isotérmica 1 → 2 calor Q E é absorvido de um reservatório a alta temperatura: o gás expande, realizando trabalho sobre a vizinhança (meio). A expansão continua sob condição adiabática no ramo 2 → 3 , isto é, o gás é impedido de trocar calor com o ambiente. Na compressão isotérmica 3 → 4 , o meio realiza trabalho sobre o gás e certa quantidade de calor Q s é rejeitada pelo sistema para um reservatório a baixa temperatura. O ciclo é completado pela compressão adiabática 4 → 1 , na qual o meio realiza trabalho sobre o sistema, fazendo com que sua energia interna aumente. A área hachurada nos dá o trabalho líquido realizado pelo sistema. Convém notar que todas as transformações envolvidas no ciclo são reversíveis. Em particular, as isotermas ocorrem com troca de calor devido a uma diferença infinitesimal entre os reservatórios e o sistema. Esse ponto foi discutido extensivamente no final da Introdução desse capítulo. Embora o ciclo de Carnot seja bastante eficiente, ele é horrivelmente impraticável. O fluxo de calor é tão lento durante os processos isotérmicos que para se retirar uma quantidade apreciável de trabalho, precisaríamos esperar uma “eternidade”. Portanto, não se preocupe em instalar uma máquina de Carnot em seu carro, pois ao mesmo tempo em que você veria aumentar a razão km/litro, na estrada você seria ultrapassado até por pedestres. Figura 22 – Ciclo de Carnot representado no diagrama pV. 5.3.1 O Rendimento do Ciclo de Carnot Usaremos um gás ideal para obter o rendimento de uma máquina operando segundo um ciclo de Carnot, mas o resultado é válido de forma geral. Na expansão isotérmica 1 → 2 , a energia interna se mantém constante e, portanto, o calor é igual ao trabalho realizado pelo gás: = QE W = nRT1 ln 1→ 2 V2 >0 V1 V V4 = = = QS W nRT2 ln nRTln 3 > 0 3→ 4 V3 V4 (69). (70). O quociente entre os valores (69) e (70) fornece, QS QE = T2 ln(V3 / V4 ) × T1 ln(V2 / V1 ) (71). 77 tErModinÂMiCa Para finalizar a análise precisamos somente encontrar uma relação entre V3 V4 e V2 V1 . Isso pode ser conseguido observando os ramos adiabáticos, nos quais pVγ é uma constante. Independentemente do processo considerado, o gás continua sendo ideal, isto é, a equação de estado pV = nRT permanece verdadeira. Para qualquer adiabática (gás ideal) podemos escrever as equações γ pV= K= constante e pV = nRT 1 Da primeira, a pressão pode ser escrita como p = K 1 V γ . Usamos essa relação na segunda: K1 nR 1 V = nRT ⇒ V1−γ = T = K 2 T ∴ Vγ −1 = γ V K1 K 2T ou, finalmente, temos TVγ −1 = K 3 (72). Essa relação é válida para qualquer ponto de qualquer adiabática. Em particular, ela se cumpre para os pontos de interesse (1), (2), (3) e (4). Os pontos (1) e (2) são caracterizados pelas variáveis ( p1 , V1 , T1 ) e ( p 2 , V2 , T1 ), respectivamente. Os pontos (3) e (4) são caracterizados por ( p 3 , V3 , T2 ) e ( p 4 , V4 , T2 ) , respectivamente. Agora, os pontos (2) e (3) estão sobre a adiabática, portanto, a relação (72) fornece T1 V2γ −1 = K 3 = T2 V3γ −1 (73). Da mesma forma, os pontos (1) e (4) estão sobre a outra adiabática, e, portanto, temos T1 V1γ −1 = K 3 = T2 V4γ −1 (74). Dividindo (J) por (K), temos T1 V2γ −1 T2 V3γ −1 V2γ −1 V3γ −1 V2 = ∴ γ −1 = γ −1 ⇒ T1 V1γ −1 T2 V4γ −1 V1 V4 V1 γ −1 V = 3 V4 γ −1 ∴ V2 V3 = . V1 V4 Substituindo esse resultado na relação (71), temos QS QE = T2 T1 (75). porque a razão dos logaritmos é unitária. Essa é uma das expressões que estávamos buscando. Embora ela tenha sido obtida através de um gás ideal obedecendo ao ciclo de Carnot, sabemos que ela é verdadeira para qualquer máquina térmica reversível. A outra expressão é a eficiência do ciclo de Carnot. η Carnot = 1 − QS QE . Usando (75), a eficiência do ciclo de Carnot fica: η Carnot = 1 − T2 T1 (76). Esta é a expressão da eficiência para uma máquina térmica operando segundo o ciclo de Carnot. Nenhuma outra, trabalhando entre as temperaturas T1 e T2 , dá um rendimento superior a este. Isso é fácil perceber porque a máquina térmica de Carnot opera em ciclos reversíveis. Observe que as temperaturas devem sempre ser expressas em Kelvin. 78 Exemplo 19 O diagrama PV de um ciclo de Carnot, mostrado na figura 22, apresenta duas isotérmicas e duas adiabáticas. Mostrar que essas curvas têm tangentes negativas em qualquer ponto. Isso significa que na construção desse tipo de diagrama as curvas devem apresentar declividade negativa. a 2ª lei da termodinâmica Solução A substância de trabalho é um gás ideal e, portanto, a equação de estado é dada por pV = nRT e γ constante a equação para as adiabáticas é pV= K= 1 Para as isotermas, temos, ∂ nRT nRT pV p ∂p ∂p = ∴ = − 2 = − 2 = − < 0. V V V ∂V isotrema ∂V V isoterma ∂V isoterma Para as adiabáticas escrevemos K1 ∂ K1 pVγ ∂p ∂p = ⇒ = − γ = − γ ∴ γ Vγ +1 Vγ +1 ∂V adiabática ∂V V adabática ∂V adiabática p ∂p = −γ < 0 . V ∂V adiabática Esses resultados mostram que as declividades das curvas só permitem derivadas negativas. Exemplo 20 Um inventor alega que ter construído um motor que, em certo intervalo de tempo, absorve 110 MJ de calor a 415 K e rejeita 50 MJ a 212 K; simultaneamente esse motor realiza um trabalho de 16.7 kW × hora. Você investiria dinheiro nesse projeto? Solução As unidades não estão padronizadas: uma escolha recomendada é trabalhar no SI. kJ kW × hora = × 3600s ∴ kW × hora = 3600 kJ = 3.6 MJ s Pelos dados que o inventor nos forneceu, podemos calcular a eficiência (ou rendimento) de sua máquina: ηinventor = W 16.7(3.6MJ) = ≈ 0.55 ou 55% . QE 110 MJ A eficiência para o ciclo de Carnot para esse dispositivo é: ηCarnot = 1− Tfrio 212 = 1− ∴ηCarnot ≈ 0.49 ou 49% . Tquente 415 Como o rendimento alegado é maior do que o máximo teórico previsto para o ciclo de Carnot, a melhor decisão é não investir. 5.4 Formulações da Segunda Lei Existem dois aspectos marcantes sobre as pesquisas de Carnot. Primeiro, ele encontrou que a eficiência com que o calor pode ser convertido em outras formas de energia depende da natureza dos processos cíclicos empregados, mas não dependia da substância de trabalho – o material que sofre a transformação cíclica. Segundo, Carnot descobriu o mais eficiente processo cíclico para converter calor em outras formas de energia. As conclusões de Carnot são verdadeiramente monumentais: sem o benefício de uma teoria correta do calor, ele estabeleceu o padrão último de eficiência de uma máquina térmica. As análises subsequentes de Clausius e de Lord Kelvin levaram ao estabelecimento da segunda lei da termodinâmica. O enunciado de Kelvin da segunda lei é esse “É impossível dispor de processo cujo único resultado seja o de converter calor, extraído de um único reservatório, totalmente em trabalho”. 79 tErModinÂMiCa Evidentemente, a palavra “único” no enunciado é uma qualificação fundamental. Muitos processos podem ser realizados nos quais um sistema converte calor integralmente em trabalho. Um exemplo fiel dessa situação foi considerado na Introdução desse capítulo em uma expansão isotérmica de um gás ideal. Em tais processos o resultado é que o estado final difere do estado inicial. Portanto, não aconteceu um único resultado. O enunciado de Clausius para a segunda lei afirma que “É impossível obter um processo cujo único resultado seja extrair calor de um reservatório a transferi-lo para outro reservatório com temperatura maior”. A equivalência entre os dois enunciados pode ser estabelecida na forma de Se Kelvin é falso, então Clausius é falso e Se Clausius é falso então Kelvin é falso. Certamente a demonstração de equivalência lógica não comprova a segunda lei. Como todas as outras leis da Natureza, a segunda lei da termodinâmica sobrevive somente porque ela está de acordo com os experimentos. Nenhuma quantidade de evidências experimentais poderia estabelecêla como verdadeira. A evidência que suporta a segunda lei é o fracasso de todas as tentativas de se construir máquinas de movimento perpétuo de segunda espécie. Fama, fortuna, imortalidade científica instantaneamente seriam conseguidas por alguém que violasse a segunda lei. Exercícios 5.5 - Uma máquina de Carnot opera com um reservatório quente a 620 K e absorve 550 J de calor a esta temperatura por ciclo, e fornece 335 J para o reservatório frio. a) – Qual o trabalho produzido por ciclo? b) – Encontre a temperatura da fonte fria. c) – Qual é a eficiência desta máquina? 5.6 - Suponha que 0.20 mol de um gás ideal diatômico ( γ = 1.4 ) passe por um ciclo de Carnot. O 6 reservatório quente está a 227 ºC e o reservatório está a 27 ºC. A pressão inicial é 10 Pa e, durante a expansão isotérmica a 227 ºC, seu volume dobra. O diagrama pV para o ciclo está mostrado na figura. Figura para o problema (5.5). (a) Achar a pressão e o volume para cada um dos pontos a, b, c e d. (b) Calcule Q , W e ∆E no ciclo todo e em cada etapa. (c) Determine a eficiência a partir dos resultados obtidos no item (b) e compare com aquele calculado diretamente pela relação (76). 5.7 - Como o ciclo de Carnot é reversível, podemos inverter os sentidos das transformações para obter um refrigerador de Carnot (reversível, obviamente). A definição do coeficiente de desempenho (COD) é dada pela equação (68): COD = 80 QE . QS − Q E Podemos alterar a notação dos parâmetros da seguinte forma: Q E → Q frio e Q S → Q quente . É exatamente isso que faz um refrigerador: ele retira certa quantidade de calor do reservatório frio e rejeita calor para um reservatório quente. Mostre que para um refrigerador de Carnot temos (COD) Carnot = onde a 2ª lei da termodinâmica Tfria Tquente − Tfria Tfria corresponde a Q fria e Tquente corresponde a Q quente . A expressão de (COD) Carnot mostra um fato curioso: quando Tquente → Tfria o coeficiente de desempenho aumenta. Isto significa que quando menos precisamos do refrigerador, melhor ele funciona! 5.5 O Teorema de Carnot A eficiência de uma máquina de Carnot apresenta um limite que não pode ser excedido nem mesmo pelo mais inteligente projeto. O Teorema de Carnot estabelece que “Nenhuma máquina operando entre dois reservatórios térmicos pode ser mais eficiente que uma máquina de Carnot operando entre os mesmos reservatórios. A demonstração desse fato pode ser feita supondo que alguém lhe diga que desenvolveu uma máquina X cuja eficiência é maior do que a de Carnot. Isto é, η X > η Carnot (77). Podemos acoplar a máquina X a um refrigerador de Carnot escolhido de tal forma que todo trabalho realizado pela máquina X seja utilizado no refrigerador (figura 23). O aparato X executa um número inteiro de ciclos no processo de retirar certa quantidade de calor Q ´′quente da fonte quente ( Tquente ), realizar trabalho W, e rejeitar a quantidade Q ′fria = Q ′quente − W para a fonte fria ( Tfria ). O refrigerador pode ser ajustado para trabalhar com todo trabalho liberado pela máquina X, e também perfazer o mesmo número de ciclos de X. Usando a desigualdade (77) e a definição de eficiência térmica, temos W Q′quente > W Q quente (78). Figura 23 – Acoplamento de uma máquina térmica X com refrigerador de Carnot. O lado direito da desigualdade é a eficiência da máquina de Carnot. A desigualdade (78) permite escrever Q quente > Q′quente (79). O trabalho da máquina X é totalmente utilizado pelo refrigerador. Entretanto, após cada ciclo já sabemos que o trabalho é dado pela diferença entre as quantidades de calor absorvida e rejeitada: W = Q quente − Q fria (80) 81 e essa quantidade deve ser igual ao trabalho realizado pela máquina X. Portanto, tErModinÂMiCa Q quente − Q fria = Q′quente − Q′fria (81). Podemos reescrever (81) como Q quente − Q′quente = Q fria − Q′fria = Q > 0 (82). O calor Q é positivo devido à desigualdade (79). Isto significa que conseguimos retirar calor de uma fonte fria, transferi-lo para outra fonte quente, sem a necessidade de realizar trabalho. Assim, a combinação da máquina X com um refrigerador de Carnot, age como um refrigerador perfeito, violando a segunda lei da termodinâmica. Então algo deve estar errado e a suspeita (fundamentada) recai sobre a desigualdade inicial η X > η Carnot . Ela não pode ser verdadeira: nenhuma máquina térmica pode ter eficiência maior do que uma máquina de Carnot. O estudante pode estranhar o uso de valores absolutos no desenvolvimento. Algumas vezes eles são necessários, entretanto, em outras são redundantes e seu uso pode ser suprimido. Por exemplo, na figura 22, vemos que Q ′quente é positivo (calor entra no sistema) e, nesse caso, o uso do módulo é desnecessário; porém, Q ′fria é uma quantidade negativa (sai do sistema) e na diferença entre eles é necessário o uso de módulo em Q ′fria . O módulo usado para o trabalho em (80) é desnecessário porque W está saindo do sistema, mas para o refrigerador o valor absoluto deve ser considerado para igualarmos ambos na equação (81). Alguns autores não utilizam valores absolutos, dizendo que as grandezas, Q e W, são positivas. Na próxima seção aboliremos o uso do módulo. 5.6 A Entropia e o Ciclo De Carnot A segunda lei da termodinâmica foi enunciada na forma de uma impossibilidade de se conseguir uma máquina perfeita. Nesse aspecto, ela difere bastante das outras leis da Física: ela não foi formulada em termos de uma equação ou de relação quantitativa. Entretanto, ela pode adquirir essa característica quantitativa através do conceito de entropia. A lei zero se refere a um parâmetro denominado temperatura; a primeira lei define uma variável de estado – a energia interna do sistema. A segunda lei introduz uma nova variável de estado, que Clausius chamou de entropia. Uma troca de calor reversível, como acontece no ciclo de Carnot, só é possível se a temperatura do sistema diferir infinitesimalmente das temperaturas dos reservatórios. As quantidades de calor absorvida e rejeitada nas transformações isotérmicas no ciclo de Carnot estão relacionadas com as temperaturas correspondentes pela relação (75), QS QE = T2 T1 (75) onde T1 é a temperatura da fonte quente e T2 é a temperatura do reservatório frio. O calor Q S tem valor negativo (sai do sistema) e podemos escrever a igualdade na forma seguinte, com T1 → Tquente e T2 → Tfria , − QS QE = ∴ Tfria Tquente Q QE + S = 0 , com Q S < 0 Tquente Tfria (83). A equação (83) pode ser interpretada da seguinte maneira: como Q E e Q S são as únicas transferências de calor no ciclo fechado, a relação (83) nos diz que a soma algébrica da quantidade Q T , calculada ao longo do ciclo, é zero. Analisando teoricamente esse simples resultado, Clausius concluiu que a quantidade Q T apresentava um significado especial. Em 1865, ele denominou essa quantidade de entropia, uma palavra de origem grega significando transformação (e talvez, também, por ela lembrar energia). Porém, o que se medem são variações de entropia. Para um processo que receba uma quantidade Q de calor à temperatura T, ∆S = 82 Q T (84) Entretanto, Clausius não explicou o que realmente seja a entropia. Ludwig Boltzmann analisou a questão nos anos subsequentes, e, em 1887, conseguiu dar uma interpretação microscópica dessa variável de estado. Analogamente à energia, a entropia não pode ser destruída; diferentemente da energia, a entropia pode ser criada. Comumente, a letra S é utilizada para nomear essa variável. Porém, o que se define são variações de entropia, e usa-se o símbolo ∆S . Assim, a relação (83) pode ser escrita como ∆S quente + ∆S fria = 0 [processo isotérmico reversível] a 2ª lei da termodinâmica (85). Podemos generalizar a definição da variação de entropia para incluir qualquer processo reversível, independentemente de ele ser ou não isotérmico. Vamos imaginar o processo como uma série de etapas infinitesimais reversíveis e, em cada uma delas, uma quantidade infinitesimal de calor dQ é absorvida a uma temperatura T. Em seguida, somamos (na verdade, realizamos uma integração) sobre todos os quocientes dQ/T envolvidos no processo: 2 dQ rev. [variação da entropia em um processo reversível] T 1 ∆S ≡ S2 − S1 =∫ (86). A notação dQ rev. é para enfatizar que a troca de calor é realizada reversivelmente. No próximo capítulo abordaremos novamente o conceito de entropia. A relação (83) é válida para processos que ocorrem um ciclo de Carnot. Queremos, agora, generalizar o resultado para um ciclo reversível qualquer. A figura 24 mostra o diagrama pV desse ciclo arbitrário juntamente com algumas isotermas. Figura 24 – Ciclo reversível arbitrário e uma família de isotermas. Esse ciclo arbitrário pode ser aproximado com bastante precisão, ligando-se as isotermas adjacentes (com diferença dT de temperatura entre elas) com pequenos segmentos de linhas adiabáticas convenientemente escolhidos. Com isso, formamos inúmeros ciclos de Carnot ao longo da curva representativa do ciclo arbitrário. O resultado dessa aproximação está mostrado na figura 25. Ao percorrer a sequência dos inúmeros micro ciclos de Carnot é equivalente – em termos de trabalho realizado e calor transferido – a percorrer a série de isotermas e adiabáticas que aproximam o ciclo real. E essa aproximação é tanto mais confiável à medida que aumentamos o números de micro ciclos. A equivalência ocorre porque ao percorrer dois micro ciclos adjacentes existe uma única isoterma comum e percorrida em sentidos opostos, e isso dá um resultado nulo em termos de transferência de calor e de trabalho realizado ao longo desse ramo. A situação está mostrada na figura 26. A generalização da equação (83) para a sequência dos diversos micro ciclos de Carnot pode ser escrita como N Qi i i ∑T =0 (87). No limite de diferenças de temperaturas infinitesimais ( N → ∞ ) entre pares de isotermas, a sequência de ziguezagues converge para o ciclo arbitrário e temos: ∫ dQ rev. = 0 [ciclo reversível] T (88). 83 tErModinÂMiCa A circunferência sobre a integral indica que o processo de integração deve ser efetuado em um ciclo, começando e terminando no mesmo ponto. Figura 25 – O ciclo arbitrário aproximado por uma sequência de micro ciclos de Carnot. Figura 26 – Dois micro ciclos adjacentes com uma única isoterma. Um teste que permite decidir se uma quantidade é ou não uma variável de estado consiste em integrar sua diferencial ao longo de uma curva fechada arbitrária: se o resultado dessa integração se anular, pode-se concluir que a quantidade é uma variável de estado. Observando a expressão (88), a diferencial dQ/T nada mais é do que a diferencial dS da função entropia S. ∫ dQ rev. = T dS ∫= 0 (89). Podemos, portanto, concluir que a entropia S é uma variável de estado. Essa conclusão é equivalente a dizer que para um processo reversível, a diferença de entropia ∆S entre dois estados de equilíbrio A e B não depende da trajetória escolhida ligando esses dois pontos. B dQ rev. T qualquer A ∆S = ∫ (90). caminho A relação (90) nos diz que a variação de entropia quando o sistema evolui de um estado A até outro estado B (ambos de equilíbrio), pode ser obtida por qualquer caminho que liga esses pontos. Entretanto, convém ressaltar que todo o processo deve acontecer de forma reversível. Na seção 2 do próximo capítulo voltaremos a considerar a função de estado chamada entropia. 84 a 2ª lei da termodinâmica Anotações 85 tErModinÂMiCa Anotações 86 6 Potenciais Termodinâmicas 6.1 introdução 6.2 Mais sobre Entropia 6.3 potenciais termodinâmicos 6.4 relações de Maxwell 87 tErModinÂMiCa 6 POTENCIAIS TERMODINÂMICOS 6.1 Introdução As aplicações das leis da termodinâmica, que temos feito nos capítulos anteriores, envolviam processos cíclicos. Entretanto, existem muitos processos termodinâmicos que não são cíclicos. Por exemplo, reações químicas são governadas pelas leis da termodinâmica e cujo estado final não apresenta as mesmas características do estado inicial. O objetivo desse capítulo é aplicar as leis da termodinâmica a processos que envolvam transformações da matéria. Uma complicação que surge imediatamente é que essas transformações ocorrem muito frequentemente em sistemas que não são isolados e, portanto, podem interagir com a vizinhança, trocando calor e também na forma mecânica, realizando trabalho. A energia do sistema não pode apresentar um valor fixo, ao invés, sua temperatura é mantida fixa, através da interação com as vizinhanças. De forma semelhante, em muitos casos não é o volume que se mantém constante, mas a pressão do sistema que permanece invariável. Assim, nossa tarefa é desenvolver ferramentas conceituais necessárias para compreender os processos que se desenvolvem a temperatura constante e a pressão constante. 6.2 Mais Sobre a Entropia A primeira lei da termodinâmica expressa a conservação da energia e que a eficiência de qualquer aparato não pode ser maior que um: não se pode realizar mais trabalho do que a quantidade de calor que o sistema absorve. A situação é muitas vezes parafraseada “Você não pode ganhar”. A segunda lei torna as coisas piores – a eficiência de qualquer máquina térmica só atinge o valor unitário se Tfrio = 0 ou Tquente = ∞ , ambas impossíveis de se conseguir na prática. Nesse contexto, essa situação também pode ser parafraseada como “Você nem mesmo pode empatar”. Foi visto que a eficiência, capítulo que tratou da segunda lei da termodinâmica e analisou diversos processos cíclicos envolvendo o que se denomina máquinas térmicas, foi introduzida uma nova variável de estado, a entropia. Gostaríamos de comentar algumas propriedades e características dessa nova função antes de introduzir os potenciais termodinâmicos. Isso é plenamente justificável por uma razão muito simples: ela é menos familiar do que os conceitos de energia interna, de trabalho, e de calor. A não familiaridade, em geral, induz no estudante certa sensação de insegurança; além disso, muitas vezes ela exibe situações para as quais seu entendimento se revela bastante precário. O que propomos a seguir não é garantia de que ao final o estudante seja um “expert” em entropia, mas tão somente que o desenvolvimento poderá ajudá-lo na compreensão da entropia. A variação infinitesimal da entropia para um processo termodinâmico é dada por: dS = dQ R T (91). Essa é a variação da entropia resultante de uma troca reversível de calor dQ com uma fonte de calor a temperatura absoluta T. O subscrito R em dQ é para enfatizar que o processo acontece reversivelmente. Entretanto, ele será suprimido, mas o estudante deve entender que ao usar (91) ou aplicá-la em outras relações derivadas dela, está implícito que o calor foi fornecido (ou retirado) de forma reversível. A entropia S é uma função termodinâmica de estado – uma propriedade do sistema – implicitamente definida para todos os estados de equilíbrio. Da relação (91), vemos que a absorção de calor (dQ > 0) produz um aumento de entropia (dS > 0). É importante ressaltar que (64) é válida somente para processos reversíveis. Exemplo 21 A capacidade térmica C(T ) de um sistema é dada por: C(T) = AT 3 , onde A é uma constante positiva. Se a entropia do sistema é considerada nula a temperatura de 0 K , encontre a entropia em função da temperatura. 88 Solução A quantidade de calor dQ para essa substância pode ser calculada facilmente porque a capacidade térmica foi fornecida: potenciais termodinâmicas dQ = C(T)dT . Usamos agora a relação (64) supondo que se trata de um processo reversível: S T C(T′) ∫0 dS′= ∫0 T′ dT′ ∴S(T)= T AT′3 1 1 3 ∫0 T′ dT′ ⇒ S(T)= 3 AT = 3 C(T) . As unidades (dimensões) de entropia são energia por temperatura: no SI a unidade é Joule/ K. Para processos reversíveis podemos usar (91) para expressar dQ: dQ = TdS (92). Esse é um resultado notável. Ele expressa o calor absorvido (de forma reversível) somente em termos das variáveis termodinâmicas do sistema: nenhuma referência é feita ao meio (ou vizinhança). Situação similar ocorre em conexão com o trabalho dW realizado por um sistema: somente para processos reversíveis é legítimo expressar o trabalho mecânico como dW = pdV . Podemos usar a primeira lei substituindo dQ pela expressão (92): TdS = dE + pdV (93). Uma relação bastante apropriada para descrever um processo reversível. O conceito de entropia permanecerá vago até que possamos responder a pergunta “O que a entropia mede?”. Em 1877, Ludwig Boltzmann visualizou uma maneira probabilística para medir a entropia de um gás ideal: ele definiu a entropia como sendo proporcional ao logaritmo do número de microestados acessíveis ao sistema. Ele foi o primeiro a enfatizar que a entropia de um sistema é uma medida de sua aleatoriedade ou desordem. Desordem em nível molecular é registrada macroscopicamente como um aumento de entropia e essas ideias evoluíram do domínio microscópico da teoria cinética dos gases e da mecânica estatística. A seguir, discutimos um longo exemplo que ilustra a ideia de que entropia é uma medida da desordem do sistema. Exemplo 22 Considere um grama de gelo que está em sua temperatura de fusão. Se adicionarmos uma quantidade de calor (calor latente de fusão) igual a 80 cal, essa massa de gelo sofrerá fusão. A mudança de fase acontece à temperatura constante. Supondo que o processo seja reversível, podemos integrar a equação (91) para obter: ∆Ss→l = L fusão , T onde ∆S s →l ≡ S líquido − S sólido é a variação da entropia e L fusão é o calor latente de fusão, o calor requerido para fundir um grama de gelo: seu valor é de 80 cal e a temperatura é de 273 K. Portanto, temos: = ∆Ss→l 80 cal cal ≈ 0.29 . 273 Kelvin K ≡ S líquido − Ssólido é positivo, indicando que a fusão resulta Observe, primeiramente, que ∆S s →l em um acréscimo de entropia. Em segundo lugar, pode-se concluir que a estrutura molecular do estado final (líquido) é altamente caótica ( = mais desordenada ) em comparação com o estado inicial (sólido). No estado cristalino, as moléculas formam uma estrutura periódica ordenada – no processo de fusão essa ordem é perdida e as moléculas podem se movimentar mais livremente. O valor calculado, ∆Ss →l = 0.29 cal K , sozinho parece ser pouco revelador. Necessitamos outros valores para efeito de comparação e, portanto, vamos obter as variações de entropia para outros dois processos, utilizando a mesma massa de um grama. Inicialmente, ela é aquecida até a sua temperatura de ebulição mantendo-se a pressão constante; em seguida, fornecemos mais calor até sua completa evaporação. 89 tErModinÂMiCa Se adicionarmos calor ao líquido de forma isobárica, sua temperatura aumenta até atingir o ponto de ebulição. Para um acréscimo infinitesimal da temperatura, escrevemos: = dQ mc = Cp dT . p dT A variação de entropia correspondente a esse processo é: dS = Cp dT . T Para muitos líquidos, e em particular a água, C p é praticamente constante no intervalo de temperatura compreendido entre os pontos de fusão e ebulição; assim, tratando C p como constante, a variação de entropia para esse processo é dada por: ∆S= Cp f →e Para um grama de água, 373 dT 373 . ∴ ∆S= Cp ln f →e T 273 273 ∫ C p = 1 cal K e o resultado é: ∆Sf →e ≈ 0.31cal/K . Esse valor é somente ligeiramente maior do aquele obtido devido à fusão do gelo, mas indica que, aumentando-se a temperatura a entropia cresce devido ao crescimento da desordem molecular. Esse fato está fortemente relacionado ao aumento das velocidades médias das partículas com a temperatura. Tendo elevado a temperatura da massa de água até seu ponto de ebulição, ela pode ser vaporizada fornecendo-se mais calor. A variação de entropia no processo de ir do estado líquido ao estado gasoso pode ser calculada de forma análoga ao item inicial, exceto que agora devemos usar o calor latente de vaporização L v , cujo valor a 1 atm é 539 cal / grama : S vapor − S líquido ≡ ∆S l→ v = L vaporização T ⇒ ∆S l→ v = 539 cal ≈ 1.4 cal / K . 373 K Esse valor é grosseiramente cinco vezes maior do que aqueles calculados previamente. O que pode ter acontecido em nível molecular que causou esse aumento substancial da variação da entropia? Uma resposta parcial para isso seria que um grama de água a temperatura de ebulição (373 K) ocupa um volume próximo de 1cm3. À mesma temperatura e pressão o vapor ocuparia um volume de aproximadamente 1700cm3 e, portanto, as moléculas estão enormemente desorganizadas devido ao fato que cada uma delas tem a sua disposição um volume muito maior para se locomover. Dissemos que, parcialmente, a variação da entropia era devido à mudança de fase da massa de água. Mas isso não é tudo. Existe também a contribuição pela quebra das ligações moleculares entre as moléculas: quando uma delas escapa da fase líquida para fazer parte da fase gasosa, ela deixa para trás uma estrutura mais desordenada no líquido: pode-se dizer que sua saída criou um “buraco” no líquido. Tal efeito representa aumento da desordem e, portanto, contribui para a variação total da entropia. Exercícios 6.1 - O hélio líquido, a pressão de 1 atm, evapora a 4.2 K. O calor latente de evaporação é 4.9 cal/grama. Determinar a variação de entropia (por grama) resultante de sua evaporação. 6.2 - O calor específico do cobre (a pressão constante e temperatura próxima a ambiente) é 3.8 ×10−4 J / kg .K . Considere a massa de 20 gramas. (a) – Se a temperatura é aumentada de 273 para 293 K, determine a variação da entropia nesse processo. (b) – Qual a quantidade de calor absorvido? (c) – Se o valor calculado no item (b) fosse absorvido a temperatura média de 283 K, qual deveria ser a variação de entropia? 6.3 - Dez gramas de água a 20 ºC são convertida em gelo a -10 ºC sob pressão constante (ambiente). Supondo que a capacidade térmica por grama (calor específico) da água permanece constante igual a 4.2 J / grama. ºC e que para o gelo a capacidade térmica seja a metade desse valor, e que o calor de fusão de gelo seja 335 J / grama, calcular a variação total de entropia no processo. 90 Temos afirmado que a entropia é uma variável de estado, assim como a energia interna de um sistema. Uma consequência desse fato é que em um ciclo, a variação de entropia (e também da energia interna) é nula. A quantidade infinitesimal dS permite o cálculo para uma transformação finita (não infinitesimal) do estado inicial i até o estado final f: f Sf − Si = Γ∫ i dQ T potenciais termodinâmicas (94). “A entropia de um sistema é uma função das variáveis termodinâmicas cuja variação é igual à integral de dQR T entre os estados inicial e final, integrada ao longo de qualquer caminho reversível, Γ , que liga esses dois estados. Outra relação, conhecida como teorema de Clausius, pode ser obtida integrando (91) ao longo de um ciclo reversível, de modo a se ter uma variação nula da entropia: Γ ∫ dQ =0 T (94'). O conceito de entropia foi primeiro introduzido na Física, por Clausius, na metade do século XIX. Nessa época havia muita confusão com respeito à relação entre calor e trabalho e seus respectivos papeis no funcionamento de máquinas térmicas. Os famosos engenheiros franceses Carnot, Petit, Clément e Désormes tinham pouco conhecimento da primeira lei da termodinâmica. Carnot acreditava que o trabalho líquido de uma máquina térmica era o resultado de uma quantidade de calor deixando um reservatório quente e a mesma quantidade de calor entrando em outro reservatório frio. Petit e Clément calculavam a eficiência de uma máquina térmica usando o trabalho realizado somente em um estágio sem considerar, como Carnot insistia que deveria ser feito, um ciclo completo. Podemos dizer que nas mãos de Clapeyron, Kelvin e Clausius a termodinâmica começou a se consolidar como ciência somente quando ela se divorciou dos desenhos de máquinas térmicas. No final capítulo IV, estabelecemos a expressão “miniaturizada” da primeira lei, equação (63): dQ = dE + dW (63). Notamos que a diferença entre dQ e dW, duas diferenciais não exatas, resulta em uma diferencial exata, ou seja, define uma quantidade infinitesimal dE proveniente de uma função de estado E. Observe que se o processo de adição de calor é efetuado de tal forma que o sistema esteja impedido de realizar trabalho (ou que o meio não possa realizar trabalho sobre o sistema), então a diferencial inexata dQ se transforma em uma diferencial exata: dQ = dE. No caso de ocorrer um processo adiabático, dQ = 0, então a diferencial inexata dW se torna uma diferencial exata: dW = -dE. Em relação à variação infinitesimal de entropia, existe uma diferencial inexata, dQ, dividida pela temperatura absoluta T e esse quociente nos fornece uma diferencial exata. A razão 1/T é conhecida como fator integrante. Entretanto, a relação dS = dQ não nos informa como S depende das variáveis T termodinâmicas T, p, V,... A forma funcional de S pode ser determinada usando-se a primeira lei juntamente com a equação de estado e com a energia interna dada em termos de algumas variáveis termodinâmicas. No exemplo seguinte, analisaremos a variação de entropia de um mol de gás ideal. Exemplo 23 Analisar a variação de entropia para um gás ideal. Solução Para um gás ideal a energia interna só depende da temperatura absoluta, e como foi visto no volume Física Geral II, seção 7.4, a energia interna pode ser escrita como: dE = C v dT (I). C V é o calor específico molar a volume constante, que pode ser considerada como constante. Usando a primeira lei da termodinâmica escrevemos: = dS dQ dE+pdV = T T (II). A equação de estado para um mol de gás ideal é dada por: pV = RT (III). 91 tErModinÂMiCa Da relação (III) temos: p= RT . V Essa relação, juntamente com a (I), são usadas em (II): = dS C v dT dV +R T V (IV). Esse resultado pode ser integrado para dar: S = C v lnT + RlnV + S0 . S 0 é uma constante de integração cujo valor não tem nenhum interesse porque estamos considerando somente variações de entropia. Por exemplo, em um processo que no qual um mol de gás ideal é levado de (T1 , V! ) a outro estado com (T2 , V2 ) a variação de entropia é: S2 −= S1 C v ln T2 V + Rln 2 T1 V1 (V). A constante desconhecida S 0 tem sido cancelada e esse é o resultado final. Entretanto, o procedimento merece alguns comentários. Observe que para se utilizar a relação (II) precisamos conhecer como a energia interna infinitesimal, dE, depende das variáveis termodinâmicas: isso é dado pela equação (I). De forma semelhante, a equação de estado (III) permite substituir a pressão na expressão do trabalho mecânico infinitesimal dW em (II). O estudante pode estar se perguntando porque se utilizou o calor específico molar a volume constante, C v , na expressão (I). Isso está, aparentemente, em desacordo com o fato de aparecer uma diferencial dV em (IV), e que se reflete no resultado final (V), mostrando claramente que houve uma variação do volume no processo. A pergunta a ser respondida para justificar esse procedimento é: porque é legítimo utilizar C v em (I) mesmo quando sabemos que o volume pode variar? A resposta se encontra no volume Física Geral II, à página 124, e damos aqui uma explicação complementar ao que foi discutido. A energia interna de um gás ideal é função somente da temperatura. Portanto, a variação da energia interna em qualquer tipo de processo para um gás ideal depende somente da variação da temperatura; para a mesma variação de temperatura dT, a variação da energia interna pode ser escrita como dE = Cv dT . Desnecessário dizer, novamente, que estamos considerando um processo reversível. A seguir, complementaremos a discussão. COMPLEMENTO VI.1 Para avançar um pouco mais sobre essa questão, podemos reanalisar o desenvolvimento realizado no volume II. A figura 27 mostra o diagrama pV para os dois processos que queremos analisar: o isovolumétrico e o isobárico. A temperatura inicial T1 determina a energia interna E 1 e a temperatura T2 determina a energia interna E 2 . Para generalizar, suponha que n mols de um gás ideal tenha recebido certa quantidade de calor dQ a volume constante: nessas condições, = dE = nC v dT : todo calor o trabalho mecânico dW é nulo. Então, a primeira lei se reduz a dQ absorvido é destinado a aumentar a energia interna do gás. Figura 27 – Dois processos distintos envolvendo um gás ideal. Por outro lado, se deixarmos variar o volume e mantivermos a pressão constante, o trabalho mecânico dW é diferente de zero e temos dQ = dE + pdV para a primeira lei. Se nesse processo você fixar a quantidade de calor, digamos igual àquela do primeiro processo (isovolumétrico), é certo que a temperatura final T2 não seja atingida, porque parte desse calor é 92 convertido em trabalho. Portanto, para se obter o mesmo dT, devemos aumentar a quantidade de calor: é precisamente isso que se faz para que a temperatura T2 seja atingida em um processo isobárico e, consequentemente, a energia interna final seja E 2 . Os dois processos analisados acima podem ser configurados como casos limites: no primeiro tem-se dV = 0 e, no segundo, dp = 0. Se você escolher uma curva diferente das retas vertical (isovolumétrica) ou horizontal (isobárica), ligando os estados inicial e final, ambos, o volume e a pressão devem variar, e a equação do gás ideal (que deve permanecer válida) fornecerá: d(pV) = nRdT ⇒ Vdp + pdV = nRdT ∴ pdV = nRdT − Vdp . potenciais termodinâmicas Então, a variação de entropia pode ser escrita como: dQ dE pdV dT dT V dS = = + =nC v + nR − dp ⇒ T T T T T T dT V dT V dS = n(C v + R) − dp = nCp − dp . T T T T Na última passagem usamos a relação C p = C v + R . A razão V T pode ser substituída usando a equação de estado de um gás ideal: V nR . Assim, temos: = T p dS nCp = dT dp . − nR T p Se considerarmos que C p independente da temperatura, a integração é imediata: S(T,p) = nCp lnT − nRlnp + S0 . O resultado obtido no exemplo 23 é bastante instrutivo para analisar uma situação que se apresenta como um pseudo paradoxo. O estudante está sujeito a encontrar vez por outra tais tipos de paradoxos, que denominamos de pseudo paradoxos. Eles são pedagogicamente importantes e motivadores porque, uma vez resolvidos, o estudante adquire maior compreensão do tema. E isso é parte integrante de sua educação. O segundo tipo, chamados simplesmente de paradoxo, possui certa legitimidade: em certo ponto da história da humanidade, ele forma um paradoxo genuíno – a natureza mostrava um aspecto que não podia ser explicado com o conhecimento teórico disponível. Muitas vezes, a resolução desses tipos de paradoxos tem determinado a modificação das leis físicas existentes. Por ora, queremos discutir o pseudo paradoxo ilustrado pela figura 28. Inicialmente, o gás ideal está confinado na parte esquerda do recipiente enquanto à direita tem-se um volume evacuado. Os dois compartimentos são separados por uma membrana e o volume total é rígido e isolado termicamente do meio ambiente (vizinhança). Considere o que acontece quando, por algum motivo, a membrana se rompe, permitindo que o gás expanda e ocupe todo o volume. Figura 28 – A expansão livre de um gás ideal. As paredes adiabáticas impedem a troca de calor, portanto, dQ = 0 em todo estágio do processo. Além disso, elas sendo rígidas garantem que nenhum trabalho é realizado sobre o sistema e, portanto, o trabalho mecânico pdV também é nulo em cada etapa do processo. A primeira lei da termodinâmica garante que nessas circunstâncias dE = 0, isto é, a variação da energia interna do gás ideal é nula. Como E só depende de T para um gás ideal, então a temperatura se mantém constante ( T1 = T2 ) . Com exceção do volume, todas as variáveis termodinâmicas se mantiveram constantes. Para um processo adiabático (dQ = 0) temos: = dS dQ = 0. T Podemos concluir, então, que é nula a variação de entropia no caso de expansão livre? Mas vamos analisar com um pouco mais cuidado: mesmo a temperatura se mantendo constante, ocorreu uma variação do volume. O fato das partículas terem um volume maior à disposição não implica uma 93 tErModinÂMiCa maior desordem e consequentemente um aumento na entropia? Essa variação pode ser facilmente calculada usando-se o resultado (V) do exemplo 23: S2 −= S1 Rln V2 > 0 , porque o volume final é maior do que o inicial. V1 Temos dois resultados contraditórios (o pseudo paradoxo): um deles fornece uma variação nula de entropia e o outro um valor positivo. Alguém suficientemente insano poderia propor que façamos R → 0 na última relação e os resultados se tornam compatíveis. Péssima escolha! Devemos evitar o non-sense físico. A contradição está em usar a relação dS = dQ/T para processos irreversíveis: ela não se aplica a processos tais como uma expansão livre. Somente para processos reversíveis sua aplicação é legítima. Para uma expansão livre em um volume rígido e adiabático, a variação de entropia do gás ideal é corretamente expressa por S2 − S1 = Rln V2 . V1 Mas pode ainda persistir um ponto de dúvida: sendo o processo irreversível, no desenvolvimento da solução do exemplo 23, foi usada a relação dS = dQ/T na primeira lei da termodinâmica (equação II). A partir daí, obtivemos a expressão (V) que determina a variação de entropia. O procedimento para se calcular a variação de entropia em qualquer processo (reversível ou irreversível) envolve uma integração que deve ser feita ao longo de um caminho que liga os estados inicial e final. Esse caminho deve ser escolhido de tal forma que caracterize um processo reversível. Em síntese, o processo pode ser irreversível, mas a integração para se calcular a variação de entropia deve seguir um processo reversível. Obviamente, a escolha do caminho não pode afetar o resultado final porque a entropia é uma função de estado, isto é, ela depende somente dos estados inicial e final e não da trajetória escolhida. Para finalizar essa seção, vamos discutir um novo diagrama, semelhante àquele pV que se mostrou conveniente para interpretar a trabalho realizado por um fluido. Um diagrama temperaturaentropia (T-S) serve para avaliar o calor absorvido ou rejeitado em um processo. Suponha que um sistema sofre uma transformação reversível na qual a temperatura e a entropia variam como mostradas na figura 29. Figura 29 – Um processo reversível leva o sistema do estado i até ao estado f. A figura mostra que o calor absorvido pelo sistema pode ser interpretado geometricamente. Para uma variação infinitesimal de entropia dS, o calor absorvido é dQ = TdS. Como mostra a figura, TdS corresponde à área hachurada mais forte da faixa com altura T e com largura dS. A área total sob a curva corresponde ao calor total absorvido durante o processo: f ∫ Q = TdS (95) (processo reversível). i Existem casos particulares importantes e úteis. Um processo isotérmico (reversível) é representado no diagrama T-S por uma reta horizontal e o calor absorvido é dado pela área sob ela. No caso de um processo adiabático (reversível), temos: dS = dQ , porém, agora dQ = 0 e podemos concluir que dS = 0 . T Como S é constante, esse processo é denominado de isentrópico e uma reta vertical representa-o no diagrama T-S. 94 Se dois estados estão infinitesimalmente próximos, então dQ dS = T dT dT dQ = TdS ⇒ (96). potenciais termodinâmicas Se o processo acontecer a volume constante (isovolumétrico), dQ dS T v = C= dT v dT v (97). Ocorrendo a pressão constante (isobárico), temos dQ dS T p = C= dT p dT p (98). Se conhecermos a dependência de C v com a temperatura, isto é, C v (T ) , a variação de entropia durante um processo isovolumétrico pode ser calculada por f C (T) Sf − Si = ∫i vT dT (99) (isovolumétrico reversível). Similarmente, para um processo isobárico, conhecendo-se a dependência funcional de C p com a temperatura, a variação de entropia pode ser obtida pela integração f Cp (T) Sf − Si = ∫i T dT (100) (isobárico reversível). Na figura 30 estão representados os processos discutidos acima. Figura 30 – Conjunto de curvas no diagrama T-S. Exemplo 24 Uma importante aplicação do diagrama T-S é dada pelo ciclo de Carnot. No capítulo anterior, o ciclo de Carnot foi extensivamente analisado e agora pretendemos avançar um pouco mais utilizando o diagrama T-S. A figura 31 mostra o ciclo de Carnot representado no diagrama T-S e no diagrama pV. Dois ramos do ciclo são verticais (isentrópicas) indicando as duas adiabáticas (reversíveis); os dois ramos horizontais e representam as duas isotermas (reversíveis). Figura 31 – Diagramas T-S para o ciclo de Carnot. 95 tErModinÂMiCa Durante a transição isotérmica a→b uma quantidade de calor Q1 = T∆S é absorvida pelo sistema. Geometricamente, Q1 é representado pela área sob a reta horizontal superior: um retângulo de altura T1 e base ∆S . Ao longo da isoterma c→d o sistema rejeita calor dado por Q 2 = T2 ∆S que corresponde à área retangular de altura T2 e base ∆S . O calor líquido absorvido no ciclo é Q1 − Q 2 (observe que o sinal negativo em Q 2 enfatiza que é uma quantidade que deixou o sistema). Essa diferença é dada geometricamente pelo retângulo entre as duas retas horizontais limitadas pelas isentrópicas verticais. Observe que também que o ciclo é percorrido no sentido horário e que o sistema absorve calor. Se o sentido do ciclo fosse revertido, a área compreendida pelas retas corresponderia ao calor rejeitado pelo sistema. Da primeira lei, segue-se que o trabalho realizado pelo sistema é dado por W = Q1 − Q 2 porque em um ciclo a variação da energia interna é nula, ∆E = 0 . No diagrama T-S esse valor é representado pela área compreendida pelas curvas. Temos usado o ciclo de Carnot devido à sua familiaridade, mas as discussões que seguem permanecem válidas para qualquer processo reversível. A definição de eficiência termodinâmica, η, é definida por: η= trabalho realizado W benefício . = = calor absorvido Q1 custo Do diagrama T-S para um ciclo ( ∆E = 0 ), temos W = Q1 − Q 2 = T1 ∆S − T2 ∆S = área compreendida pelas curvas. Inserindo essa relação na definição da eficiência termodinâmica, obtemos, para um ciclo de Carnot: (T1 − T2 )∆S T =1− 2 . T1 ∆S T1 OBSERVAÇÕES: (1) a quantidade de calor Q1 é usualmente denotada por Q absorvido ou Q E , e Q 2 corresponde a Q rejeitado ou Q S . (2) Convém se lembrar de que T1 se refere à temperatura na qual o calor é absorvido e T2 à temperatura na qual certa quantidade é rejeitada. Portanto, T1 > T2 . η Carnot = Exercícios 6.4 - Encontre a eficiência do ciclo esboçado na figura seguinte. Figura para o problema (6.4). 6.5 - Achar a eficiência para o seguinte ciclo. Figura para o problema (6.5) 6.6 - Compare entre si as eficiências encontradas nos problemas (6.4) e (6.5). 96 6.3 Potenciais Termodinâmicos potenciais termodinâmicas A fórmula “miniaturizada” da primeira lei é escrita como: dE = dQ - pdV ou dE = TdS - pdV (101). A relação mostra-se especialmente útil na determinação da variação da energia interna quando o sistema evolui de um estado inicial até outro estado final. Observamos que, ao analisar um ciclo, a variação dE se anula; em todos os casos a energia interna tem provado ser uma propriedade termodinâmica fundamental. Conceitualmente, a energia interna pode ser configurada como a soma da energia cinética das partículas com a energia potencial entre elas. Apesar de sua clareza conceitual, a energia interna pode se tornar inconveniente para a análise de muitos processos termodinâmicos. Nessa seção, vamos introduzir três quantidades, relacionadas diretamente com E, que mostrarão maior flexibilidade nas aplicações termodinâmicas. Essas grandezas são a entalpia (H), a energia livre de Gibbs (G) e a energia livre de Helmholtz (F). Todas elas, juntamente com E, são conhecidas como potenciais termodinâmicos. Eles fornecem envolvimento mais direto com os experimentos que se poderia ter somente com o uso da energia interna. Existe ainda outro potencial termodinâmico chamado potencial químico (μ) que não vamos tratar, embora ele seja relevante para transformações nas quais o número de partículas não se mantém constante. A forma diferencial (101) da primeira lei sugere que a entropia S e o volume V possam ser considerados como variáveis independentes e que E possa ser tratada como função de S e V E = E (S, V) . Dessa forma, a temperatura T e a pressão p se tornam variáveis dependentes cuja relação com E, S e V pode ser facilmente determinada. Do ponto de vista puramente matemático, a função E(S,V) tem sua diferencial dada por ∂E ∂E = dE dS + dV ∂S V ∂V S (102). Desde que (101) e (102) devem se iguais para todos os possíveis valores de dS e dV, então, os coeficientes correspondentes de dS e dV devem os mesmos. Portanto, ∂E T= ∂S V (103) e ∂E p = − ∂V S (104). O conteúdo importante na relação (101) é que a combinação dos parâmetros do lado direito é sempre igual à diferencial exata de uma quantidade, que nesse caso é a energia livre E do sistema. Isso implica que as variáveis T, S, p e V não podem variar de forma arbitrária: deve existir uma conexão entre elas para garantir que sua combinação determine a diferencial dE. Para obter essa conexão, basta notar que as segundas derivadas de E devem ser independentes da ordem de derivação: ∂2E ∂2E = ∂V∂S ∂S∂V (105). Alternativamente, essa igualdade pode ser escrita ∂ ∂E ∂ ∂E = ∂V S ∂S V ∂S V ∂V S (106). Usando as igualdades (103) e (104), escrevemos: ∂p ∂T = − ∂V S ∂S V (107). A ENTALPIA (variáveis independentes: S e p) A primeira lei na forma infinitesimal (“miniaturizada”) expressa por (101) exibe o efeito das variações de S e V. Entretanto, poderíamos igualmente bem considerá-la como função de S e p. O que devemos fazer é substituir termos pdV , no qual aparece dV por uma expressão equivalente onde deve aparecer dp. Isso é possível notando que d(pV) =pdV + Vdp ∴ pdV =d(pV) − Vdp (108). 97 Usamos agora (101) substituindo o termo PdV: tErModinÂMiCa dE = TdS − d(pV) + Vdp ⇒ d(E + pV) = TdS + Vdp . O termo à direita da igualdade, d(E + pV) , é escrito como dH: = dH TdS + Vdp (109) onde introduzimos a quantidade definida por: H ≡ E + pV (110). A função H(S,p) é chamada entalpia. Obviamente, suas unidades são as mesmas de energia, isto é, Joule no SI. Considerando a função H(S,p) sob o aspecto puramente matemático, então ∂H ∂H = dH dp dS + ∂S p ∂p S (111). A comparação de (109) com (111) revela que ∂H =T ∂S p (112) e ∂H = V ∂p S (113). O aspecto importante de (109) é, novamente, o fato que a combinação das variáveis à direita da igualdade é igual à diferencial exata de uma grandeza que denominamos entalpia e representamos pela letra H. As derivadas parciais “cruzadas” devem ser iguais: ∂2H ∂2H . = ∂p∂S ∂S∂p Isso pode escrito mais explicitamente usando (112) e (113): ∂T ∂V = ∂p S ∂S p (114). Essa relação é análoga à (107) e representa a conexão entre as variáveis T, S, p e V. A essa altura deveria ficar claro que tipo de procedimento estamos seguindo para obter tais relações termodinâmicas. Tudo que precisamos é fazer até o fim todas as outras possíveis mudanças nas variáveis da equação fundamental da primeira lei (98). Nosso programa (matemático) de substituição é interrompido nesse ponto para uma discussão física da grandeza H. Dissemos que esses potenciais são convenientes para diversas aplicações. A conveniência se faz sentir em processos para os quais o uso direto da relação (98) é bastante laborioso. Ao invés de considerar somente a energia interna de um sistema, podemos agregar o trabalho necessário para dar espaço a ele. Esse trabalho (mecânico) é pV, a pressão (usualmente) atmosférica do meio ambiente multiplicada pelo volume do sistema (isto é, o espaço total necessário para que o sistema seja conformado). A soma de pV com a energia interna é justamente o que definimos como entalpia: H ≡ E + pV (110). Essa quantidade é a energia total que você deve dispor para criar um sistema do nada e colocá-lo no meio ambiente. Ou, colocado de outra forma, se você de alguma maneira aniquila o sistema, a energia que você pode extrair não é somente E, mas também o trabalho realizado pelo meio para preencher o vácuo deixado quando ele colapsar. A figura 32 ilustra a situação de forma bastante hilária. Para criar um coelho do nada e colocá-lo sobre a mesa para o deleite da plateia, o mágico precisa acrescentar à energia interna do coelho, E, alguma energia extra, igual a pV, para empurrar a atmosfera para que o coelho ocupe seu espaço. Essa energia total requerida é a entalpia, H ≡ E + pV . A entalpia tem sua aplicação mais importante, talvez, nos casos envolvendo a termoquímica. Esse ramo da ciência aborda os aspectos termodinâmicos de processos que convertem calor em energia química e vice-versa. Entende-se por energia química a energia associada com as ligações de natureza elétrica que mantém os átomos ligados em uma molécula. Se você consultar um livro sobre esse tópico, encontrará inúmeras tabelas relativas a entalpia, que os termoquímicos e engenheiros usualmente chamam de entalpia (ou calor) de reação (ou de formação). Por si só, o tema é extremamente amplo e aqui cabem somente algumas considerações e aplicações simples (mas importantes). 98 potenciais termodinâmicas Figura 32 – Um mágico em seu trabalho. Inicialmente, é preciso reconhecer que diversos processos ocorrem na natureza à pressão constante (como o “aparecimento” do coelho). Na discussão subsequente, veremos a razão de se usar a letra H para a entropia. Vamos supor que em certo processo aconteça uma variação de entropia ∆H quando o volume sofre uma variação ∆V e a energia interna é acrescida de ∆E . Estamos considerando um processo isobárico. A nova entalpia é escrita como H + ∆H = (E + ∆E ) + p(V + ∆V) ∴ H + ∆H= (E + pV) + (∆E + p∆V) . O primeiro termo à direita da igualdade é H, portanto, ∆H = ∆E + p∆V (p constante) (115). Essa relação indica que a entropia pode aumentar por dois motivos: ou porque a energia interna cresce, ou porque o sistema expande e realiza trabalho sobre o meio para acomodar a nova situação. Obviamente, ambos podem ocorrer simultaneamente. Vamos escrever (89) na forma infinitesimal: dH = dE + pdV (p constante) (116). Mas essa expressão é exatamente igual à primeira lei da termodinâmica. Portanto, ( dH )p = dQ (117). A letra H (heat = calor , em inglês) é assim justificável para se representar a entalpia. A equação (117) é o calor absorvido ou rejeitado em um processo isobárico reversível. Para quantidades não infinitesimais, Q = ∆H ≡ H f − H i (118). A liquefação de gases tais como nitrogênio, oxigênio, e mesmo hélio, é conseguida usando-se um dispositivo que contém uma “válvula de estrangulamento” (o processo é conhecido como Joule – Thomson). De forma simplificada, essa válvula é constituída por um tubo de paredes adiabáticas e que oferece alta impedância ao fluxo de gás, conseguida pela colocação de um material poroso perpendicularmente às paredes do tubo. A figura 33 mostra com algum detalhe o arranjo utilizado nesse processo. A região mais escura é a parede porosa por onde o gás é forçado a passar. Figura 33 – Uma válvula de estrangulamento para liquefação de gases. O processo de estrangulamento é obviamente irreversível, desde que o gás passa por estados de não-equilíbrio ao ser forçado através da parede porosa. Tais estados não podem ser descritos em todo instante pelas variáveis termodinâmicas, mas uma conclusão interessante pode ser obtida sobre os estados de equilíbrio inicial e final. Vamos analisar a situação mostrada na figura 99 tErModinÂMiCa 33. O sistema consiste de certa massa M, inicialmente à esquerda da parede porosa, ocupando um volume V1 , submetida à pressão p1 e à temperatura T1 . Depois de certo tempo, a massa M de gás tem atravessado a parede porosa e se encontra do lado direito. Nessa região, a massa M ocupa um volume V2 à pressão p 2 . Essa é a situação final do processo. Nesse processo, a diferença de energia interna do gás entre o estado final (à direita da parede porosa) e o estado inicial (à esquerda dela) é simplesmente ∆E = E 2 − E 1 = E (T2 , p 2 ) − E (T1 , p1 ) (119). Na liquefação dos gases, os aparatos utilizam uma válvula tipo agulha para o efeito de estrangulamento em substituição da parede porosa. A pressão p1 é da ordem de 200 atm enquanto p 2 permanece próxima à pressão atmosférica. Se os gases se comportassem como ideal, a variação de temperatura seria nula. Mas sob essa pressão, dificilmente a idealidade seja seguida – e esse fato é responsável pelo decréscimo da temperatura. Em gases reais, a energia interna depende da pressão e também da temperatura. O pistão à direita realiza um trabalho sobre a massa de gás enquanto o gás realiza trabalho empurrando o pistão à esquerda. O trabalho líquido nesse processo é W = p1 (Vf − Vi ) + p 2 (Vf − Vi ) ⇒W = p1 (0 − V1 ) + p 2 (V2 − 0) ∴ W = p 2 V2 − p1V1 (120). As pressões não podem ser iguais, mas são mantidas constantes. Como as paredes são adiabáticas, Q = 0 , e a primeira lei fica 0 = ∆E + W ⇒ 0 = E 2 − E1 + p 2 V2 − p1V1 ∴ E 2 + p 2 V2 = E1 + p1V1 (121). A igualdade (121) indica que no processo de estrangulamento, as entalpias, inicial e final, são iguais. Isso pode levar à conclusão errada de que o processo é isentálpico, mas ele não é – tratase de um processo irreversível. Os estados intermediários do gás durante o estrangulamento não são estados de equilíbrios. O gás sempre flui da parte à alta pressão para o lado de baixa pressão. Contudo, é essencial perceber que a entalpia é mantida constante por um processo de estrangulamento. Afirmamos anteriormente, logo após a equação (119), que para um gás ideal, o processo aqui discutido, não pode funcionar. A energia interna E, para um gás ideal é função somente da temperatura T, portanto, H =E + pV ⇒ H =BT + NkT . A constante B está ligada ao número graus de liberdade do sistema, mas o que é importante aqui é o fato de a energia interna ser diretamente proporcional à temperatura absoluta. Assim, entalpia constante implica em temperatura constante, e o processo de estrangulamento se mostra ineficaz para gases ideais. Entretanto, para gases reais o estrangulamento pode resultar em resfriamento ou em aquecimento, dependendo do valor do coeficiente de Joule – Thomson ∂T µ J T = . ∂p H A determinação desse coeficiente é realizada de forma experimental, e os detalhes não serão apresentados aqui. Para maiores informações sobre esse coeficiente e os detalhes técnicos no processo de resfriamento e liquefação de gases, o estudante pode consultar outros textos de termodinâmica e/ ou de termoquímica. Na internet, o site en.wikipedia.org/wiki/Joule-Thomson_effect traz diversas informações sobre o assunto. A ENERGIA LIVRE DE HELMHOLTZ (variáveis independentes: T e V) Podemos transformar a primeira lei (101) em uma expressão envolvendo dT ao invés de dS: dE = TdS − pdV = d(TS) − SdT − pdV ⇒ d(E − TS) = −SdT − pdV dF = −SdT − pdV (122) onde é introduzida a definição F ≡ E − TS 100 (123). A função F(T, V) é chamada de energia livre de Helmholtz. Essa é a energia total necessária para se criar o sistema, menos o calor que se pode tomar gratuitamente do meio à temperatura T. Esse calor é T ∆S = TS , onde S é a entropia final do sistema; quanto maior for a entropia de um sistema, maior a quantidade de energia que pode entrar na forma de calor. Muitos processos importantes ocorrem isotermicamente e nesses casos a energia livre de Helmholtz é a variável adequada ou conveniente em termos de energia. Para justificar essa afirmação, devemos mostrar como a variação da função F está relacionada ao trabalho realizado sobre o sistema. Para um processo reversível infinitesimal e isotérmico, a relação (122) fornece: potenciais termodinâmicas f dF = −pdV ⇒ Ff − Fi = − ∫ pdV (124). i Assim, a variação da energia livre de Helmholtz durante um processo reversível e isotérmico é igual ao trabalho realizado sobre o sistema (lembre-se de que pdV é o trabalho realizado pelo sistema, então o sinal negativo que aparece na frente dessa quantidade representa o trabalho sobre o sistema). A equação (124) pode ser escrita de forma mais reveladora: − ∆F = W (isotérmico e reversível) (125). Interpretamos isso dizendo que, em um processo reversível isotérmico, o trabalho realizado pelo sistema é igual ao decréscimo em sua energia livre de Helmholtz. O resultado se mostra coerente: o trabalho realizado (reversível e isotermicamente) pelo sistema sobre o meio acontece à custa de uma diminuição de sua energia livre. Se o processo é irreversível, temos T∆S > 0 e ao invés da igualdade (125), é substituída por uma desigualdade: − ∆F ≥ W (processo isotérmico) (126). A igualdade se verifica somente para processos reversíveis. A desigualdade vem do fato que em um processo irreversível produz mais entropia do que é requerida pela primeira lei da termodinâmica. Para obtermos relações semelhantes àquelas dadas por (103), (104), (112) e (113), considere a função F como sendo dependentes das variáveis independentes T e V. A variação dF é dada por: ∂F ∂F = dF dT + dV ∂T V ∂V T (127). Comparando essa relação com (122), podemos concluir que ∂F S = − ∂T V (128) ∂F p = − ∂V T e (129). A relação (129) é particularmente importante porque uma vez conhecida expressão funcional de F, ela nos dá a equação de estado do sistema. O exemplo seguinte mostra isso para um gás real. Exemplo 25 A energia livre de um gás é dada por F= −RTln(V − b) − a + CT , onde R é constante de gases, b, a e C são constantes. Mostre que V o gás obedece à equação de estado de van der Waals a RT . p + 2 (V − b) = V Solução A aplicação direta de (129) dá p =− ∂ a −RTln(V − b) − + CT V ∂V T = p a RT ⇒ p =− − + 2 ∴ V b V − RT a a RT − 2 ⇒ p += ∴ 2 V−b V V V−b a RT ) . p + 2 ( V − b) = V 101 tErModinÂMiCa Podemos proceder de forma inversa: dada a equação de estado, pede-se determinar a energia livre de Helmholtz. O próximo exemplo trata de um importante caso especial. Exemplo 26 Mostre que para um mol de gás ideal, sofrendo um processo reversível, a energia livre de Helmholtz é dada por = F T2 T2 Cv V dT − RT ln 2 + constante × T + constante . T V1 T1 ∫ CvdT − T ∫ T1 Solução O ponto de partida é a definição da energia livre de Helmholtz F= E − TS . Entretanto, para utilizá-la, obviamente, precisamos conhecer a energia interna E e a entropia S. Para obtêlas, vamos considerar que a capacidade térmica molar, C v , não necessariamente permaneça constante. Ela pode ser, como realmente é, função da temperatura. A energia interna, já foi tratada no volume Física Geral II, à página 123: vimos que para um gás ideal, qualquer que seja o processo, a variação infinitesimal da energia interna é dada por: dE = C v (T)dT . Então, a energia interna pode ser escrita como T ∫ C (T′)dT′ + E = E v 0 . T0 O importante aqui é fixar que esse resultado vale para qualquer processo, desde que o gás seja ideal. A entropia de um gás ideal se encontra discutida no EXEMPLO 23; a equação (IV) no dá para uma variação infinitesimal de entropia: = dS C v (T) Integrando essa expressão, temos S = T ∫ Cv (T′) T0 dT dV . +R T V dT′ V + R ln + S0 . T′ V0 Com as duas relações, para E e para S, podemos escrever a energia livre de Helmholtz. = F = F T dT′ V ′ ′ − + R ln + S0 + E 0 + F0 ∴ C (T )dT T ∫ C v (T′) ∫T v T′ V0 T0 0 T T T T0 T0 ∫ Cv (T′)dT′ − T ∫ Cv (T′) dT′ V − RT ln − S0 T + E 0 + F0 . ′ T V0 Os três últimos termos à direita podem ser escritos na forma = F T T T0 T0 ∫ Cv (T′)dT′ − T ∫ Cv (T′) dT′ V − RT ln − constante × T + constante . T′ V0 Convém observar que é essencial a troca da variável de integração: o uso de T ′ evita possível confusão com o extremo superior de integração, T. Em casos nos quais a capacidade térmica se mantém constante no intervalo de temperatura usado na integração, a expressão para F fica mais simples: F = Cv (T − T0 ) − TCv ln T V − RT ln − constante × T + constante . T0 V0 O estudante deve estar se perguntando em que situações a energia livre de Helmholtz pode ser usada. Ela é útil para variações que acontecem à temperatura e a volume constantes. Diversos experimentos de laboratórios e no cotidiano ocorrem sob essas condições. Devido ao fato que a energia livre de Helmholtz é uma função com segundas derivadas contínuas, a derivada “cruzada” fornece a igualdade: 102 ∂2F ∂2F . = ∂V∂T ∂T∂V Essa igualdade significa que: potenciais termodinâmicas ∂ ∂F ∂ ∂F = . ∂V ∂T V ∂T ∂V T Mas as derivadas entre parênteses são expressas pelas relações (128) e (129). Portanto, ∂p ∂S = ∂V T ∂T V (130). COMPLEMENTO VI.2 As propriedades da energia livre de Helmholtz são de interesse particularmente em reações químicas que acontecem isotérmica e isovolumetricamente. Entretanto, sua importância em Mecânica Estatística está estreitamente relacionada com a função de partição Z definida pela equação Z( β ) = ∑ e- βε r . r A soma é feita sobre todos estados acessíveis do sistema caracterizados pela energia ε r ; o fator β é um parâmetro definido por β = 1 kT , com k sendo a constante de Boltzmann. A relação entre a função de partição Z e com a energia livre de Helmholtz é expressa pela igualdade (I). F = −kT ln Z Essa igualdade envolve a conexão entre a representação microscópica representada pela função de partição Z e a termodinâmica macroscópica aqui na forma da energia livre de Helmholtz. Por exemplo, para um gás monoatômico ideal, formado por N partículas, a função de partição é dada por: V N 2π mkT Z( β ,V,N) = N! h 2 3N 2 (II). O fator h é a constante de Planck. Pede-se determinar a energia livre de Helmholtz. Aplicando-se ln à equação (II), temos ln Z = N ln V − ln N! + 3N 2π mk 3N ln ln T + 2 h2 2 (III). Com esse resultado, a equação (I) fica 3N 2π mk 3N F= ln ln T −kT N ln V − ln N! + + 2 2 h 2 (IV). Uma aproximação bastante útil e válida para grandes valores de N é a fórmula de Stirling: ln N ! ≈ N ln N − N . Como nesse caso o número de partículas é da ordem do número de Avogadro, a aproximação é totalmente justificável. Então, a relação (IV) pode ser escrita como 3 2π mk 3N F= ln T ∴ −kT N ln V − N ln N + N + N ln + 2 2 h 2 3 3 2π mk F =− NkT ln V − ln N + 1 + ln T + ln . 2 2 h 2 Esse resultado expressa a energia livre de Helmholtz para um gás ideal monoatômico. Exercícios 6.7 - Utilizando a expressão da energia livre de Helmholtz obtida no complemento VI.2, e a relação ∂F p = − encontrar a equação de estado para o gás ideal monoatômico. ∂V T 103 tErModinÂMiCa 6.8 - A função de partição para um conjunto de N osciladores harmônicos quânticos localizados é dada por β ω Z(T,N,V) = 2senh 2 −N . Mostre que a energia livre de Helmholtz é F(T,V,N) = N ω + NkT ln 1 − exp ( − β ω ) . 2 A definição de seno hiperbólico é sen hx ≡ e x − e −x . 2 Note que a pressão se anula para sistemas cujas partículas são localizadas. Sugestão: para obter a resposta da forma apresentada, a expressão do seno hiperbólico pode ser escrita como sen h x = ( 1 x e − e −x 2 ) e e −x −x 1 1 − e −2 x = 2 e −x . 6.9 - Um sistema paramagnético formado por N spins localizados (cada um deles com momento magnético µ ), quando submetido a um campo magnético externo B tem sua função de partição dada por N µB . Z(T,B,N) = 2cosh kT e x + e −x A função cosseno hiperbólico é definida por cosh x ≡ . Mostre que a energia livre de 2 Helmholtz pode ser escrita como F(T,B,N) = Nµ B − NkT ln 1 + e 2 µ B kT . Sugestão: o cosseno hiperbólico pode ser escrito de forma semelhante àquela usada para a função seno hiperbólico no problema anterior. A ENERGIA LIVRE DE GIBBS (variáveis independentes: T e p) A primeira lei (101) pode ser transformada em uma expressão envolvendo dT e dp ao invés de dS e dV. = dE TdS − pdV (101). Notando que a diferencial d(TS) = TdS + SdT , então = TdS d(TS) − SdT (131). Também d(pV) = pdV + Vdp e, portanto, = pdV d(pV) − Vdp (132). Substituindo as relações (131) e (132) em (101), temos: dE = d(TS) − SdT − d(pV) + Vdp . As diferenciais envolvendo os produtos de duas variáveis podem ser escritas à direita da igualdade: d [ E − TS + pV ] = −SdT + Vdp (133). O termo dentro do colchete é a definição da energia livre de Gibbs, G: G(T,p) ≡ E − TS + pV (134). dG = −SdT + Vdp (135). Então, a relação (133) se torna 104 A energia livre de Gibbs pode escrita em termos dos potenciais termodinâmicos já definidos. Como H= E + pV , temos: potenciais termodinâmicas G= H − TS . Usando a energia livre de Helmholtz, F= E − TS , escrevemos G= F + pV . Para perceber o significado da energia livre de Gibbs, chamamos à cena nosso conhecido mágico. A figura 34 mostra novamente a criação de um coelho a partir do nada. Figura 34 – O mágico em ação criando um sistema a partir do nada. Se o sistema está em um meio caracterizado por ter pressão e temperatura constantes, então, o mágico necessita realizar um trabalho para criar o coelho igual à energia livre de Gibbs. Ou, considerando o processo inverso, o trabalho que você recupera ao fazer desaparecer o coelho é dado, obviamente, pela energia livre de Gibbs. Nesse caso, onde T e p são constantes, para a criação do coelho do nada, e colocá-lo sobre a mesa, o mágico não necessita de toda entalpia H = E + pV. Certa quantidade de energia, igual a TS (lembre-se de que TS = Q), pode fluir espontaneamente para dentro do sistema (coelho); o mágico precisa fornecer como trabalho somente a diferença, G = H - TS, em seu número. Considerando T e p como variáveis independentes, a diferencial da energia livre de Gibbs. ∂G ∂G = dG dp dT + ∂T p ∂p T (136). Comparando (135) com (136), conclui-se que ∂G = −S ∂T p (137) e ∂G = V ∂p T (138). As derivadas “cruzadas” da função G (T, p) devem ser iguais porque a ordem de derivação é irrelevante para funções com derivadas contínuas. Então, ∂ 2G ∂ 2G = ∂p∂T ∂T∂p (139). Utilizando (137) e (138), a igualdade (139) fornece ∂S ∂V − = ∂ p T ∂T p (140). 105 tErModinÂMiCa Exemplo 27 Obter a variação da energia livre de Gibbs para um gás monoatômico ideal constituído por N partículas em um processo reversível. Solução A solução que apresentaremos segue uma linha diferente daquela desenvolvida no exemplo 26. O objetivo é obter G a partir das relações fundamentais envolvendo as funções termodinâmicas necessárias. Desde o início usamos E = 3 NkT e a equação de estado para um gás ideal. 2 Determinamos inicialmente a variação da entropia ocorrida no processo (reversível). A primeira lei nos dá = dE TdS − pdV Como dE = 3 NkT . Assim, a primeira lei fica NkdT , e pela equação de estado, temos p = 2 V 3 NkT 3 dT dV . NkdT = TdS − dV ⇒ dS = Nk + Nk 2 V 2 T V Integramos essa relação para obter S(T,p) − S(To ,p o )= 3 T V Nk ln + Nk ln 2 To Vo T 3 2 V = S(T,p) S(To ,p o ) + Nk ln To Vo (I). Entretanto, essa equação contém a variável V e se quisermos S(T, p) , usamos novamente a equação de estado para eliminar V e Vo : V= NkTo NkT . ∴ Vo = p po A variação de entropia pode ser escrita como: S(T,p) − S(To ,p o )= pT 3 T Nk ln + Nk ln o . 2 To pTo Podemos agrupar os termos à direita da igualdade para escrever T 5 2 p S(T,p) Nk s o (To ,p o ) + ln o = To p S(To ,p o ) O valor s o (To ,p o ) é igual a . Nk (II). A ideia é usar a definição de energia livre de Gibbs expressa em termos da energia livre de Helmholtz: G= F + pV . A função F pode ser facilmente escrita com a ajuda de (II), com a relação da energia interna de um gás ideal: T 5 2 p 3 F =E − TS ⇒ F = NkT − T So (To ,p o ) + Nk ln o ⇒ 2 To p 3 F = NkT − s o − ln 2 T To 52 po p (III). A constante s o já foi definida anteriormente. A energia livre de Gibbs pode então ser escrita usando (III) e a equação de estado do gás ideal, substituindo o termo pV = NkT : 106 3 T 5 2 p G(T,p,N) = NkT − s o − ln o + NkT ∴ 2 To p potenciais termodinâmicas 5 T 5 2 p G(T,p,N) = NkT − s o − ln o . To p 2 Esse resultado é idêntico à expressão G =∫ Cp dT − T ∫ Cp T dT + NkT ln p − constante × T 5 Nk . Essa aproximação é válida desde que a diferença de temperatura 2 não seja muito grande ( ≈ 100 K ). se considerarmos C p = O uso da energia livre de Gibbs é particularmente útil em reações químicas. O próximo exemplo trata de uma reação bastante conhecida e, portanto, os valores numéricos das funções termodinâmicas são encontrados facilmente em tabelas de referência. Exemplo 28 Queremos analisar a eletrólise, uma reação de decomposição da água em seus componentes: H 2O → H 2 + 1 O. 2 Um mol de água nos dará, ao final da reação, um mol de hidrogênio gasoso e meio mol de oxigênio gasoso. De acordo com as tabelas de referências, ∆H para essa reação (à temperatura e pressão ambientes) é 286kJ. Quando formamos hidrogênio e oxigênio a partir da água, precisamos colocar, de alguma maneira, 286kJ de energia no sistema. Dessa quantidade, uma pequena fração é usada para empurrar a atmosfera para que o sistema gasoso ocupe seu lugar: essa quantidade é dada pelo produto p∆V = p atm (Vgases − Vlíquido ) ≈ p atm × Vgases = 105 × ( 0.0224 + 0.0112 ) ≈ 3.4 kJ . O restante, 282.6kJ permanece no sistema. A pergunta que queremos responder pode ser formulada da seguinte maneira. Esses 286kJ necessários devem ser supridos na forma de trabalho, ou parte dele pode entrar na forma de calor? A resposta envolve o cálculo da variação de entropia do sistema. Entretanto, os valores de um mol de cada espécie são tabelados: SH2O = 70 J/K ; SH2 = 131J/K ; SO2 = 205 J/K . Então, a variação de entropia no processo é dada por ∆= S Sprodutos − Sreagentes= (131 + 205 2 ) − 70 ≈ 163J/K . A máxima quantidade de calor que pode entrar no sistema é dada por Q =T∆S ⇒ Q =(298 K)(163J/K) ∴ Q ≈ 49 kJ . A quantidade de energia que deve ser suprida através de trabalho elétrico é a diferença entre a energia total, 286kJ, e o calor fornecido pelo meio ambiente, 49kJ. Isto equivale à aproximadamente 237kJ. Esse valor, 237kJ, é a variação da energia livre de Gibbs para o sistema, durante o processo; ele é o trabalho mínimo requerido para que ocorra a dissociação da um mol de água em um mol de hidrogênio e meio mol de oxigênio. Podemos também aplicar ∆G para reverter a reação. Se você combinar hidrogênio com oxigênio, ambos gasosos, para produzir água de maneira controlada, podem-se, em princípio, extrair 237kJ sob a forma de trabalho elétrico para cada mol de hidrogênio consumido. Esse é o princípio de funcionamento de que é conhecido como célula de combustível, um dispositivo que poderia substituir os motores de combustão interna em futuros veículos. 107 tErModinÂMiCa O desenvolvimento analítico, feito para analisar a eletrólise, é mostrado esquematicamente na figura E 28.1: Figura E 28.1. Fluxo de energia para a eletrólise de um mol de água. A expressão diferencial dG = −SdT + Vdp (135) estabelece a energia livre como função das variáveis termodinâmicas independentes p e T. A importância de G em química e biologia reside no fato que muitas reações químicas e biológicas se processam à temperatura e pressão constantes. Se essas reações ocorrem de forma reversível, então (135) se aplica e mostra que a energia livre de Gibbs é conservada: dG = 0 (isotérmica, isobárica e reversível). A quantidade, cujo sinal é importante, é a variação da função de estado G ∆G = G produtos − G reagentes (141). Como demonstraremos a seguir, a reação se dá de forma espontânea a temperatura e pressão constantes desde que ∆G seja negativa, isto é, se o sistema libera energia livre para o meio. Se ∆G acontece ser positiva, a reação pode ainda ocorrer, mas somente com adição de energia, talvez na forma de calor. Dessa forma, a variação em G não é espontânea e a temperatura e/ou pressão do sistema devem ser alteradas. O decréscimo em G para um processo isotérmico e isobárico pode ser estabelecido da seguinte forma: da definição de F e G, temos G = F + pV. A variação em G durante um processo isobárico é dada por ∆G = ∆F + p∆V ∴ ∆G = ∆F + p(Vf - Vi ) . Mas p( Vf − Vi ) é o trabalho mecânico realizado pelo sistema sobre o meio e, portanto, ∆G = ∆F + W (142). No desenvolvimento do tópico sobre a energia livre de Helmholtz, chegamos à conclusão que o decréscimo de F durante um processo isotérmico é sempre maior que o trabalho realizado (equação 126) − ∆F ≥ W ⇒ ∆F + W ≤ 0 . O sinal de igualdade se refere a um processo reversível. Então, a relação (142) pode ser escrita como ∆G ≤ 0 (143). Esse é o resultado que queríamos mostrar: a variação da energia livre de Gibbs não pode crescer em um processo isobárico e isotérmico. O sinal de igualdade corresponde a um processo reversível (isobárico e isotérmico), enquanto a desigualdade se refere a um processo irreversível (isobárico e isotérmico). O processo irreversível e espontâneo leva o sistema para o equilíbrio, onde a energia livre de Gibbs é um mínimo. Um estado de equilíbrio para um sistema à mesma temperatura e pressão de sua vizinhança é estável. Qualquer evolução espontânea, que poderia perturbar o equilíbrio, pode ocorrer somente se a energia G sofrer uma diminuição. Para um sistema já em um estado de energia livre G mínima, tais processos espontâneos não existem: o sistema está em equilíbrio e esse equilíbrio é estável. Certamente o equilíbrio pode ser perturbado por agentes externos, como por exemplo, uma variação da temperatura do meio ambiente. Isso vai forçar o sistema a migrar para outro estado de equilíbrio onde a função energia livre G apresente um ponto de mínimo referente a essa nova temperatura. 108 Exemplo 29 Duas reações importantes na área biológica são a oxidação da glicose e a fotossíntese. O resultado final da primeira reação pode ser representada pela reação potenciais termodinâmicas C6 H12 O6 + 6O 2 → 6CO 2 + 6H 2 O . Os valores tabelados (padrão a 298 K) de ∆G e ∆H são (por mol de glicose) ∆G = −2890 kJ/mol , ∆H = −2826 kJ/mol . Para a oxidação da glicose, ∆G é negativa e, portanto, a reação se processa espontaneamente, liberando energia livre. A fotossíntese é equivalente à reação CO 2 + H 2 O → CH 2 O + O 2 . Nesse caso os valores tabelados são (por mol) ∆G = 481 kJ/mol , ∆H = 470 kJ/mol . Como ∆G é positiva, a reação não se processa espontaneamente. A conversão de energia radiante (luz) proveniente do Sol em energia livre proporciona o processo de fotossíntese. Se estivermos interessados na variação de entropia dos processos, o cálculo é bem simples se lembrarmos que ambas as reações acontecem isotermicamente (298 K) e que ∆G = ∆H + T∆S . Para a oxidação da glicose, temos −2890 − (−2826) ∆S = ∴ ∆S =−0.21 J/mol K . 298 Para a fotossíntese, a variação da entropia é ∆S = 481 − 470 ∴ ∆S = 0.037 J/mol K . 298 Observe que no primeiro caso a variação de entropia é negativa. Consegue explicar se há ou não a violação da segunda lei da termodinâmica? RESUMO As relações que temos obtido são válidas para processos infinitesimais reversíveis de estado de equilíbrio a outro. Os principais resultados são descritos para um sistema hidrostático (somente trabalho mecânico), e o número de partículas se mantém constante. (I) A variação da energia interna é dada por dE = dQ − pdV = TdS − pdV (primeira lei) (101) onde E, T e P são todas consideradas funções das variáveis termodinâmicas independentes S e V. (II) Para uma variação infinitesimal da entalpia, temos dH =dE + pdV + Vdp = TdS + Vdp (109) onde H,T e V são funções das variáveis independentes S e p. (III) Uma variação infinitesimal na energia livre de Helmholtz é dF =dE − TdS − SdT = −SdT + Vdp (122) com F, S e p sendo funções das variáveis independentes T e V. (IV) A energia livre de Gibbs tem a diferencial dada por dG =dH − TdS − SdT = −SdT + Vdp (135) onde G, T e p são funções das variáveis T e p. 109 tErModinÂMiCa As quatro funções E, H, F e G são coletivamente chamadas de potenciais termodinâmicos. Elas foram inventadas para tornar “fácil” a termodinâmica. Cada potencial é a variável natural da energia para certas classes de processos físicos. Devemos reconhecer que se lembrar de cada uma das diferenciais resumidas acima, não é sempre imediato. Entretanto, diversas regras mnemônicas têm sido elaboradas para auxiliar. Uma delas está esboçada na figura 35. Os quatro potenciais são colocados nos lados do quadrado, enquanto as quatro variáveis, S, T, p e V ocupam os vértices desse quadrado. Note que cada um dos potenciais termodinâmicos está situado entre as duas variáveis independentes que aparecem na expressão da diferencial desse potencial. A sequência é iniciada no vértice superior esquerdo com a variável T e percorrendo o quadrado no sentido horário encontram-se F, V, E,... Figura 35 – O quadrado usado para estabelecer as diferenciais dos potenciais. Em inglês, existe para esse dispositivo uma regra para estabelecer a ordem das grandezas: “The Friendly Vagabond Usually Shares His precious Gifts”. O leitor está convidado a elaborar sua própria frase para se lembrar da sequência utilizada ao longo do quadrado. As duas diagonais orientadas determinam o sinal das grandezas pertinentes. Por exemplo, a energia E se encontra entre V e S. Podemos escrever, como primeiro passo = dE (?)dS + (?)dV Os pontos de interrogação são encontrados através das setas orientadas. O “vetor” terminando em S parte de T, portanto, a variável T deve multiplicar dS e traz um sinal positivo. O outro “vetor” parte de V e termina em p, portanto, a variável p deve multiplicar dV com o sinal negativo. Assim, temos = dE TdS − pdV . Para a energia livre de Gibbs, G está entre p e T, portanto, temos dp e dT. Uma seta parte de T (ao invés de terminar em T) e avança até S (sinal negativo); a outra parte de V e termina em p (sinal positivo). Combinando essas informações, temos = dG Vdp − SdT . O quadrado serve também, obviamente, para se encontrar as variáveis termodinâmicas em função das derivadas dos potenciais. Analisando uma vez mais a figura 35, podemos encontrar, por exemplo, a expressão (∂E ∂S)V : a energia interna E está flanqueada por S e V, e como queremos a derivada em relação a S mantendo-se V constante, vemos que o “vetor” terminando em S se inicia em T. Portanto, a derivada procurada é ∂E =T. ∂S V Se estivermos procurando o que representa ∂G ∂T à pressão constante, a figura mostra o “vetor” partindo de T e chegando a S. Se ele terminasse na variável em relação a qual estamos derivando, escolheríamos o sinal positivo, mas nesse caso deve-se escolher o sinal negativo. Assim, ∂G = −S . ∂T p 110 6.4 Relações de Maxwell Uma variedade de relações entre as variáveis termodinâmicas S, V, p e T pode ser obtida pelo fato que os potenciais termodinâmicos E, H, F, e G são todos funções de estado, isto é, possuem diferenciais exatas. Na verdade, isso já foi feito durante o desenvolvimento de cada um desses potenciais. Da função energia interna E = E (S, V ) , a derivada “cruzada” fornece a primeira relação de Maxwell: ∂p ∂T = − ∂V S ∂S V potenciais termodinâmicas (107). A função entalpia H = H ( p, V ) apresenta derivadas “cruzadas” que permitem escrever a igualdade, conhecida como segunda relação de Maxwell, ∂T ∂V = ∂p S ∂S p (114). A energia livre de Helmholtz H ( V, T ) fornece a terceira relação de Maxwell: ∂p ∂S = ∂V T ∂T V (130). Finalmente, a energia livre de Gibbs G = G ( p, T ) nos dá a quarta relação de Maxwell: ∂S ∂V − = ∂p T ∂T p (140). Essas relações são de considerável utilidade em termodinâmica porque permitem a substituição de quantidades que são difíceis, se não impossíveis, de se medir experimentalmente por quantidades cujo acesso experimental é quase direto. Por exemplo, vamos considerar a relação (107). A variação da pressão com a entropia, (∂p ∂S)V , não é diretamente mensurável, enquanto (∂T ∂V )S pode ser medido de forma bastante simples. Para obter (∂T ∂V )S , utilizamos um sistema limitado por paredes adiabáticas e realizamos um processo reversível: sob essas condições, a variação de entropia é nula, ou seja, ela se mantém constante porque = dS dQ= T 0= T 0. Essa é a condição imposta para se calcular (∂T ∂V )S . Se a variação do volume e a variação de temperatura são medidas, temos: ∆T ∆V S ∂T → quando ∆V → 0 . ∂V S 111 tErModinÂMiCa Exemplo 30 Usando a relação de Maxwell (114), mostre que TdS pode ser escrito como ∂V = TdS Cp dT − T dp . ∂T p Solução Se a entropia S é considerada como função de T e p, isto é, S = S(T, p) , temos: ∂S ∂S = dS dT + dp ∂T p ∂p T (I). Para um processo reversível, TdS é igual ao calor absorvido pelo sistema. Se essa troca de calor é efetuada a pressão constante, a capacidade térmica C p é dada por = dQ )p ( TdS ) p (= Cp dT (II). Observando (I), vemos que para um processo isobárico (dp = 0), temos ∂S dT ∂T p (I) → ( TdS ) = T p (III). Comparando (II) e (III) podemos concluir que ∂S . Cp = T ∂T p Multiplicando-se (I) por T e usando a igualdade acima, podemos escrever ∂S = TdS Cp dT + T dp . ∂p T A dependência da entropia com a pressão não é de fácil determinação experimental. Entretanto, a quarta relação de Maxwell, (114), permite substituir (∂S ∂p )T no segundo termo à direita para fornecer ∂V = TdS Cp dT − T dp ∂T p (IV). Esse resultado proposto no enunciado de problema. Essa relação é chamada, algumas vezes, de “equação TdS”. Exercícios 6.10 - Por argumentos similares que levaram à equação (IV) do exemplo 25, e usando a terceira relação de Maxwell (104) mostre a outra “equação TdS”: ∂p = TdS CV dT + T dV . ∂T V 6.11 - A dilatação térmica de um sólido foi estudada no volume Física Geral II. O coeficiente de expansão volumétrica à pressão constante, γ, foi definido como γ= 1 ∆V . V ∆T Usando essa relação no limite de ∆T → 0 e o resultado (IV) do exemplo 25, mostre que = TdS Cp dT − γ VT dp . 112 COMPLEMENTO VI.3 O DEMÔNIO DE MAXWELL Ao longo dos tempos têm surgido dispositivos que são construídos com objetivo único de violarem a primeira ou a segunda lei da termodinâmica, ou, em casos mais dramáticos, ambas as leis. Para as invenções que pretendem burlar a primeira lei, seus criadores atestam que tais máquinas, operando em ciclos, conseguem entregar ao usuário mais energia do que elas consomem. Esses aparatos, que violariam a primeira lei, são chamados de motos perpétuos (ou contínuos) de primeira espécie. Outros inventores aparecem frequentemente comunicando que suas criações atingem eficiência unitária, mesmo operando a temperaturas finitas e em ciclos, em total desacordo com a segunda lei. Tais dispositivos são chamados de motos perpétuos (ou contínuos) de segunda espécie. Infelizmente (ou felizmente), muitas vezes basta um análise rápida para perceber que todos eles apresentam algum ponto que faz com que as leis da termodinâmica prossigam sendo válidas. Entretanto, pessoas continuam insistindo na construção desses dispositivos, cujos nascimentos são, em geral, celebrados de forma ruidosa para dar as boas vindas às máquinas que mudariam nosso relacionamento com o Universo. Especificamente, uma multidão de pessoas habilidosas tem tentado subverter a segunda lei da termodinâmica. Muitas dessas pessoas eram malucas com, no máximo, uma compreensão muito superficial da Física. A mais notável exceção foi o esquema proposto por James Clerk Maxwell, um físico brilhante, que dentre diversas contribuições, sintetizou a teoria do eletromagnetismo. Maxwell inventou um “demônio” e, acertadamente, a criatura é conhecida como o “demônio de Maxwell”. Esse ser violaria a segunda lei da termodinâmica, forçando um fluxo de calor no sentido de um corpo frio até outro corpo quente. A figura C1 mostra os reservatórios e a criatura operando a válvula que permite o fluxo de calor do corpo mais frio para o mais quente. A proposta de Maxwell não era uma especulação ociosa. Tampouco ele não estava criando um espantalho para divertir ou embaraçar outros colegas. potenciais termodinâmicas Figura C 1– O demônio controlando o fluxo das moléculas entre dois reservatórios. 113 tErModinÂMiCa 114 Sua criação encontra-se ligada à Teoria Cinética dos Gases, que na época estava em sua infância, mas estava se desenvolvendo de forma consistente (mas não tranquila) nas mãos de Clausius, Maxwell e Boltzmann. A Teoria Cinética operava em nível microscópico, enquanto a segunda lei estava firmemente estabelecida em termos macroscópicos. Houve acirradas disputas entre duas escolas, uma denominada “energetista” e outra chamada “atomista”. A primeira delas era liderada por Ostwald com apoio de Ernest Mach; a escola atomista era representada por Boltzmann, que foi implacavelmente perseguido por defender a constituição atômica da matéria. Suspeita-se fortemente que seu suicídio tenha sido causado pela sistemática desqualificação de suas ideias e conclusões feitas pelos “energetistas”. Seus argumentos expressam que “deve-se considerar como desnecessária a ficção dos átomos, um conceito artificial, quase metafísico, carente de realidade e de significado duvidoso”. Mas esse tópico deve ser tratado adequadamente na disciplina História da Física; sua discussão aqui foi motivada para situar o leitor na polêmica envolvendo as duas escolas sobre a natureza íntima da matéria. Voltemos ao demônio de Maxwell, caracterizado pelo próprio Maxwell como “um ser cujas faculdades são tão aguçadas que ele pode seguir cada uma das moléculas em seu curso”. Mas como o demônio opera? Os dois recipientes contém gases ideais e estão isolados do meio ambiente por paredes adiabáticas. Ambos estão conectados por um tubo munido de uma válvula que pode ser controlada pela criatura. Você deve se lembrar que as moléculas dos gases apresentam uma distribuição de velocidades: algumas se movem velozmente e outras de forma mais lenta, enquanto a grande maioria tem sua velocidade próxima à velocidade média (capítulo III, seção 4). Sabemos também que a energia cinética de um gás ideal é função somente de sua temperatura. O demônio opera a válvula de tal forma que, usando suas “faculdades aguçadas”, ele permite a passagem de moléculas com altas velocidades provenientes do reservatório frio para o reservatório quente. E como não bastasse, ele pode permitir a passagem de moléculas de baixas velocidades do reservatório quente, para o reservatório frio. Essa ação demoníaca faz com que o reservatório mais frio se torne ainda mais frio e o reservatório mais quente seja aquecido. O efeito líquido do processo é a transferência de calor de uma fonte fria para outra fonte quente. O calor rejeitado pelo reservatório quente (para manter sua temperatura constante) poderia ser utilizado como em uma máquina térmica. Isto significa que se pode extrair trabalho dessa situação. Assim, temos um dispositivo – cíclico na natureza – cujo único efeito é transferir calor da fonte fria para a fonte quente. Tal demônio seria capaz, dentre outras coisas, de fritar ovos com cubos de gelo... Nada mal para um mundo cada vez mais carente de energia disponível. Entretanto, existe um pequeno probleminha: essa máquina não funciona! E por um motivo bastante simples – esquecemos do demônio. Ele não pode ser considerado como vizinhança e ter status de “extrasistema”. Sua inclusão no sistema veta qualquer tentativa de fazer esse dispositivo funcionar como planejado originalmente. A demonstração quantitativa desses argumentos não são triviais, mas a ideia persiste: é impossível construir um dispositivo que, operando em ciclos, tenha a única finalidade de converter calor integralmente em trabalho. Ou, de forma mais direta, nem o demônio consegue violar a segunda lei da termodinâmica. potenciais termodinâmicas Anotações 115 tErModinÂMiCa Anotações 116 7 A 3ª Lei da Termodinâmica 7.1 introdução 7.2 o Zero absoluto 7.3 a terceira lei da termodinâmica 7.4 história, Excessões e re-Enunciado 117 tErModinÂMiCa 7 A 3ª LEI DA TERMODINÂMICA 7.1 Introdução O objetivo desse capítulo é tratar da terceira lei da termodinâmica, entretanto, remetemos o leitor a recordar as duas primeiras. A primeira lei é o estabelecimento da conservação da energia. Ela indica a impossibilidade de se construir motos perpétuos de primeira espécie – dispositivos que, operando em ciclos, conseguem exportar mais energia do que importam. Estabelecida informalmente, essa lei pode ser expressa como “Você não pode ganhar”. A segunda lei é ainda mais pessimista sobre a possibilidade ganhar: “você não consegue nem empatar”. A segunda lei se refere à impossibilidade de se construir um dispositivo cíclico que converta completamente calor em trabalho. Em termos de eficiência térmica η= a segunda lei determina que trabalho realizado calor absorvido η <1. Por exemplo, o ciclo de Carnot tem eficiência dada por η Carnot = 1 − Tfrio . Tquente Pode parecer que se Tfrio = 0 K , então η Carnot = 1 e teríamos uma máquina que converteria calor integralmente em trabalho. Isso tem levado à seguinte proposição (informal) da segunda lei: “você pode empatar somente no zero absoluto”. 7.2 O Zero Absoluto Afinal, é possível atingir o zero absoluto? Somente a parte experimental decide se podemos ou não atingir o estado de T = 0 K. Os experimentos revelam que todas as tentativas para se reduzir a temperatura, usando variadas montagens, sofrem um ataque de rendimentos decrescentes ao se aproximar do zero absoluto. Eles se tornam menos e menos efetivos quando a temperatura decresce. A terceira lei aparece como uma decepção àqueles que aspiram conseguir empatar: "você não consegue atingir o zero absoluto". A conclusão imediata da primeira, da segunda e da terceira leis é que você não pode ganhar e nem mesmo empatar. Isso faz a termodinâmica um passatempo bastante caro. A seguir, trataremos de alguns fatos relativos a baixas temperaturas. À pressão de uma atmosfera, o hélio gasoso de liquefaz a 4.2 K. Essa temperatura pode diminuir se a pressão é reduzida, de modo que não é tão difícil atingir temperaturas ainda menores através de um bombeamento mecânico do vapor: o hélio se resfria pela evaporação [esse fenômeno acontece à temperatura ambiente com a água: o processo de evaporação diminui a temperatura do líquido remanescente. Alguns recipientes que armazenam água potável são construídos com material poroso para facilitar a evaporação, e isso torna a água mais fresca para ser ingerida]. Abaixo de 1 K, entretanto, esse procedimento é impraticável porque pequena quantidade de calor que entra no sistema faz com que a temperatura do hélio cresça significativamente, e as melhores bombas de vácuo não conseguem retirar o vapor suficientemente rápido para compensar esse aumento de temperatura. O isótopo raro hélio–3 (o núcleo do átomo contém somente um nêutron e não dois, como no hélio comum, chamado hélio–4), cujo ponto de liquefação é 3.2 K, pode ser resfriado a 0.3 K por bombeamento mecânico de seu vapor. Mas 1 K não é uma temperatura baixa suficiente? Por que seguir tentando obter temperaturas cada vez menores? Surpreendentemente, existe uma variedade de fenômenos fascinantes que ocorrem somente à temperatura de milikelvin, e outros efeitos que acontecem a temperaturas ainda mais baixas. Para estudar esses fenômenos, desenvolveram-se técnicas e arranjos experimentais igualmente fascinantes para atingir temperaturas extremamente baixas. Em 1993, pesquisadores da Universidade de Helsinki (Finlândia) reportaram temperaturas tão baixas como 10-10 K. Obviamente, isso não foi conseguido instantaneamente. Para atingir essa marca extraordinária, passaram-se mais de cem anos desde o início dos processos de liquefação de gases. É bastante comum se escutar que no zero absoluto todo movimento cessa, como se tivéssemos encontrado um estado de repouso absoluto. Por exemplo, os elétrons que circundam os átomos têm seus movimentos congelados; o próprio átomo dentro de uma rede cristalina deve se encontrar completamente imóvel, e outras afirmações similares. São ideias folclóricas – nada disso acontece. Os elétrons continuam circulando em torno do núcleo com velocidades de milhares quilômetros por segundo; os próprios átomos da rede ainda executam movimentos vibracionais 118 (muito parecidos com um pequeno oscilador, aproximadamente harmônico) embora com energia menor do que possuem à temperatura, digamos, ambiente. Esse estado é caracterizado pelo “ponto zero de energia”, mas não é, absolutamente, zero. Seu valor não nulo é uma decorrência de um dos resultados mais profundos da Mecânica Quântica, o princípio de incerteza de Heisenberg. A região de baixas temperaturas requer um tratamento adequado, e esse é um domínio no qual a Mecânica Quântica deve ser empregada. A descrição clássica está, definitivamente, fora do contexto porque suas previsões estão, via de regra, em total desacordo com os resultados experimentais. 7.3 a 3ª lei da termodinâmica A Terceira Lei da Termodinâmica Na seção anterior, a terceira lei foi enunciada em termos da impossibilidade de se atingir o zero absoluto. Existe uma forma alternativa para estabelecê-la considerando a entropia. A variação de entropia dS resultante de uma transferência reversível de calor dQ a uma temperatura T é dS = dQ . T Essa é uma definição operacional, não de entropia, mas de sua variação. Isso não deve ser motivo de pânico porque situação semelhante aconteceu no estudo de Mecânica, onde a variação da energia potencial é definida operacionalmente. A escolha da posição para o zero de energia potencial é totalmente arbitrária, entretanto, elege-se uma referência de maneira conveniente. Em termodinâmica ocorre situação semelhante com a escolha do estado de energia interna zero. A primeira lei define somente variações da energia interna de um sistema. Apesar do fato que somente dS é operacionalmente definida, nós não temos total liberdade na escolha do estado de entropia zero. A Natureza faz essa escolha para nós. Assim, a terceira lei expressa ainda uma restrição adicional sobre a entropia. Por volta de 1907, Walter Nernst percebeu a necessidade de criar uma nova lei da termodinâmica. Ele estabeleceu seu teorema do calor que, ao longo dos anos, foi aperfeiçoado, tornado-se mais abrangente devido aos resultados experimentais subsequentes, e atualmente ele tem o status de terceira lei da termodinâmica. Em sua forma mais elementar, essa lei estabelece que A entropia de um sistema se aproxima de zero quando sua temperatura tende a zero. Essa declaração deve ser qualificada para fazê-la precisa e universalmente válida – somente assim ela pode reconhecida como uma lei física. Entretanto, após as palavras de qualificação serem inseridas, o conteúdo essencial da terceira lei permanece como S→0 quando T →0. COMPLEMENTOS Muitas pessoas consideram a terceira lei como uma curiosidade de praticidade quase nula. Entretanto, para os químicos ela é extremamente útil. Existem diversos enunciados da terceira lei e nenhum deles completamente satisfatório do ponto de vista fenomenológico. (1) Teorema do calor de Nernst lim ∆S = 0 ou ∆S o = 0 . T →0 Essa forma proposta por Nernst estava baseada nas observações de medidas realizadas envolvendo reações galvânicas. Nernst pensava que esse enunciado seria universalmente verdadeiro. (2) Planck lim S = 0 ou S o = 0 . T →0 Planck supôs que essa relação se verificava para todo “corpo homogêneo”. (3) G. N. Lewis Toda substância tem uma entropia positiva finita, mas no zero absoluto, a entropia se torna zero no caso de um sólido cristalino perfeito. (4) Impossibilidade se alcançar o zero absoluto Essa é outra forma da terceira lei, que consideraremos como a mais satisfatória, embora não tenha a praticidade dos enunciados anteriores. Ele assegura que nenhum sistema pode ser esfriado ao zero absoluto. 119 tErModinÂMiCa Existem problemas com cada um dos enunciados. O Teorema do Calor de Nernst não é nem um teorema e não tem nada a ver com o calor. A declaração, devido a Planck, não está baseada em observações experimentais. Não podemos medir a entropia em si, mas somente suas variações e, além disso, a proposição é demasiadamente geral e não se aplica a todas as substâncias homogêneas. O enunciado devido a Lewis tem também seus pontos fracos e, um deles é a limitação de ser aplicado a sólidos cristalinos “perfeitos”. Entretanto, como saber se determinado sólido cristalino é “perfeito”? Vamos aceitar a forma irrestrita da terceira lei expressa pelo enunciado de Planck, S → 0 quando T → 0 , e deduzir algumas de suas consequências e, então, verificar se os resultados estão de acordo com os experimentos. Mais tarde voltaremos a comentar sobre esse ponto, abordando as limitações da terceira lei. A primeira consequência da terceira lei que vamos abordar envolve o coeficiente de expansão volumétrica de um sólido. Usando a quarta relação de Maxwell (114), ∂S ∂V − = ∂p T ∂T p (114) o coeficiente de expansão térmica γ= pode ser expresso como 1 ∂V V ∂T p γ=− 1 ∂S V ∂p T (115). A terceira lei prediz que γ tende a zero quando a temperatura se aproxima de zero. Para mostrar que (∂S ∂p )T tende a zero, note que a derivada corresponde à quantidade experimentalmente observável ∆S S(T, p + ∆p) − S(T, p) . = ∆p ∆p Se a entropia vai zero quando T se aproxima de zero, segue-se que a variação de entropia também deve tender a zero, independentemente do valor de ∆p . Assim, a razão ∆S ∆p tende a zero: ∆S ∂S → → 0 quando T → 0 . ∆p ∂p T Esse resultado permite concluir que γ tende a zero com a temperatura. A figura 36 mostra o comportamento de α ( = γ 3 ) com a temperatura para o ouro, revelando que quando a temperatura tende a zero, α vai a zero, de acordo com a terceira lei. O fato de γ ir a zero quando T tende a zero, revela que a variação de volume cessa com a temperatura. Figura 36 – Comportamento do coeficiente de expansão linear com a temperatura. A segunda consequência da terceira lei se refere ao comportamento da diferença das capacidades térmicas, C p − C v . As duas capacidades térmicas diferem porque os materiais tendem a expandir ou se contrair quando aquecidos a pressão constante. Entretanto, como γ vai a zero, a variação do volume cessa quando T → 0 , podemos esperar que ambos, C p e C v se tornem iguais quando a temperatura vai a zero. Antes de mostrar analiticamente esse fato, observam-se na figura 37 os comportamentos obtidos experimentalmente de C p e C v para o neônio (sólido) a 120 baixas temperaturas. Próximo a 12 K existe a confluência entre as duas capacidades térmicas e, a partir dessa temperatura, ambas seguem iguais até zero Kelvin. a 3ª lei da termodinâmica Figura 37 – As capacidades térmicas para o neônio sólido. A capacidade térmica a volume constante é definida por ( dQ )V = CV dT (116). De forma similar, a capacidade térmica a pressão constante é definida por ( dQ )p = Cp dT (117). Vamos considerar um sistema simples, cujo número de partículas se mantém constante, como temos feito até aqui: a massa do sistema permanece inalterada em qualquer processo realizado. As três grandezas, p, T e V servem como variáveis termodinâmicas. A equação de estado relaciona as três, portanto, somente duas delas são independentes. Vamos escolher inicialmente V e T como sendo independentes, e assim, a pressão p se torna a variável dependente, isto é, p = p(V, T ). A energia interna E do sistema pode também ser escrita em função das variáveis V e T. Uma variação infinitesimal dE é dada por ∂E ∂E = dE dT + dV ∂T V ∂V T (118). Usando (118) na primeira lei, dQ = dE + pdV , temos: ∂E ∂E dQ = dT + dV + pdV ∴ ∂T V ∂V T ∂E ∂E dQ= dT + + p dV ∂T V ∂V T (119). Primeiramente, considere um processo reversível no qual o calor é adicionado mantendo-se o volume constante. Portanto, o segundo membro à direita da igualdade de (119) se anula (dV = 0). Comparando então (116) com (119), vemos que ∂E CV dT = dT ∂T V que permite concluir que a capacidade térmica está relacionada à energia interna por ∂E CV = ∂T V (120). Na sequência, considere a adição de calor à pressão constante. A primeira lei pode ser escrita como ∂E ∂E dT + + p dV ⇒ ∂T V ∂V T ( dQ )p =Cp dT = ∂E Cp dT = CV dT + + p dV ∂V T (121). A igualdade (121) contém o termo dV que, de certa forma, dificulta a comparação entre as duas capacidades térmicas. Para eliminá-lo, podemos considerar o volume V como função das variáveis 121 tErModinÂMiCa independentes T e p. Portanto, com V = V (T, p) temos ∂V ∂V = dV dp dT + ∂T p ∂p T (122). À pressão constante, dP = 0 e ∂V dT . ∂T p ( dV )p = Substituindo essa relação em (121) resulta na igualdade ∂E ∂V Cp = CV + p + ∂V T ∂T p (123). Essa relação é bastante conveniente para analisar a diferença entre as duas capacidades térmicas. A conveniência está justamente no fato de que já conhecemos o comportamento de ∂V : essa derivada nada mais é do que o coeficiente de expansão térmica multiplicada pelo ∂T p volume, γV. Adicionalmente, sabemos também que o valor de γ se anula quando a temperatura vai a zero Kelvin. Portanto, a equação (123) se reduz a C p = C V quando T → 0 (124). Essa é, por ora, a única conclusão possível acerca do comportamento das capacidades térmicas. A seguir mostraremos que não somente elas são iguais, mas que devem se anular no limite de T →0. Para mostrar isso, escrevemos dQ = CdT, onde C simboliza uma capacidade térmica apropriada (não se esqueça de que as capacidades térmicas C p e C V são iguais a partir de certa temperatura, e que somente o extremo inferior da integral é relevante para a conclusão). A variação correspondente à entropia é dS = C dT T (125). Se integrarmos desde zero Kelvin até certa temperatura T (na qual C ainda representa a capacidade térmica do sistema), obtemos T C S(T) − S(0) = ∫0 T′ dT′ (126). Nesse ponto, temos a oportunidade de qualificar o enunciado original da terceira lei. Uma forma mais precisa é que as entropias de todos os sistemas tendem a um mesmo valor quando a temperatura se aproxima de zero. A escolha desse valor é deixada em aberto, e a comunidade científica tem elegido como sendo zero: assim S(0) = 0 . Essa escolha corresponde aos enunciados de Planck e de Lewis, citados no complemento anterior. No final desse capítulo, voltaremos a tratar dessas interpretações. Dessa forma, escolhendo S(0) = 0 , a integral em (126) fixa a entropia a uma temperatura T como T C dT′ T′ 0 S(T) = ∫ (127). O comportamento de C, quando a temperatura vai a zero, é severamente restringido pela condição S → 0 quando T → 0 . Para perceber a necessidade dessa restrição, suponha que C não se anule com T, mas ao invés, atinja um valor constante quando T → 0 . Essa hipótese permite que a constante C seja retirada do sinal de integração em (127): T dT′ T′ 0 S(T) = C ∫ Entretanto, essa integral diverge para o extremo inferior: seu valor se aproxima de ∞ quando T → 0 . Dito de outra maneira, a entropia S(T) não existe se a capacidade térmica C possuir um 122 valor não nulo quando T tende a zero. Portanto, para que a entropia tenha um valor definido, a capacidade térmica deve se anular para temperaturas próximas a zero Kelvin. A parte experimental tem mostrado essa restrição sobre C. Para sólidos eletricamente isolantes encontra-se que a capacidade térmica varia com T 3 a baixas temperaturas Cisolantes = bT 3 a 3ª lei da termodinâmica (128). Para sólidos condutores, nos quais existem elétrons livres para se moverem ao longo do material, a dependência em C inclui um termo linear em T devido à contribuição eletrônica: Ccondutores = aT + bT 3 (129). O termo cúbico na temperatura da relação (129) foi obtido por Debye considerando a contribuição de fónons (vibrações quantizadas da rede cristalina) à capacidade térmica da substância. Os dados experimentais têm confirmado essa dependência a baixas temperaturas. Exercícios 7.1 - Usando o resultado (123), mostre que para um mol de gás ideal, temos a conhecida igualdade Cp = Cv + R . 7.2 - Usando as relações (128) e (129) calcule a entropia a baixas temperaturas para sólidos isolantes e condutores. Verifique que S(T ) → 0 quando T → 0 . 7.3 - Mostre que a entropia se anula a T = 0 desde que C ∝ T q , onde q é um número maior que zero. 7.4 História, Excessões e Re-Enunciado A terceira lei da termodinâmica foi, por muito tempo, um tópico controverso. Ela foi formulada em sua forma primitiva por Nernst (1864-1941), em 1906, e conhecida por Teorema do Calor de Nernst e, subsequentemente, foi sendo refinada e tornada mais clara, notadamente por Simon (1893-1956). Nernst sentia muito orgulho de seu empreendimento em formular o que ele chamava de “minha lei”. Ele notava que houve três pessoas associadas com a descoberta da primeira lei, duas pessoas com a segunda lei e uma com a terceira lei. Desses fatos, ele deduziu que não poderia haver mais leis da termodinâmica. Por um longo período, a terceira lei foi um tópico controverso. Por exemplo, em 1932, um artigo científico afirmava que “Nós chegamos, portanto, à conclusão bastante cruel que o Teorema do Calor de Nernst estritamente aplicado pode ou não ser verdadeiro, mas é sempre irrelevante e inútil – aplicado a “estados sólidos ideais” no zero absoluto, o teorema embora muitas vezes verdadeiro é algumas vezes falso, e falhando na generalidade ele deve ser rejeitado. Isso não é um desrespeito à idéia de Nernst que provou ser, no final, de limitada generalidade”. Fowler and Stern on Statistical Mechanics and Entropy. Como foi escrito por Ruhemanns1, “Nas controvérsias que se seguiram, os oponentes da terceira lei gradualmente se separaram em dois grupos: aqueles que sustentavam ser a lei verdadeira mas inaplicável, e aqueles que afirmavam que embora aplicável, ela era falsa”. A terceira lei, de forma alguma, é controvertida; hoje ela é mais bem entendida e a importância relativa de seus diferentes aspectos têm sido alterada Entretanto, ela permanece como um princípio valioso unificado e um guia útil na área de fenômenos de baixas temperaturas. Apesar do impressionante número de sucessos, o enunciado “não qualificado” da terceira lei não é universalmente válido. Há sistemas para os quais a entropia não tende a zero à baixa temperatura e isso nos compele a re-enunciar a terceira lei. As exceções podem ser localizadas em processos irreversíveis que, ao invés de direcionar o sistema a um estado de equilíbrio, leva-o a alcançar um estado de não-equilíbrio. 1 Low Temperature Physics, Cambridge University Press, 1937 123 tErModinÂMiCa As dificuldades resultantes não são somente com a terceira lei. Estritamente falando, a primeira e a segunda leis já não podem ser aplicadas. A razão para isso é que as propriedades termodinâmicas, tais como entropia, energia interna, são definidas operacionalmente somente para estados de equilíbrio. A termodinâmica, portanto, não pode fazer nenhuma previsão positiva sobre transformações que iniciam ou terminam em estados de não-equilíbrio. Para entender porque essas exceções não são desastrosas para a terceira lei, devemos analisar ainda outro aspecto da entropia. A entropia e a energia interna são semelhantes em um ponto importante: ambas são somas de contribuições de diversas origens. Por exemplo, considere um gás constituído por moléculas diatômicas. Além da familiar contribuição da energia cinética de translação, as moléculas podem possuir energia de rotação em torno de seu centro de massa, energia devido à vibração em relação ao ponto de equilíbrio, todas elas contribuindo para a energia interna do gás. Agora, a similaridade entre energia e entropia essa: cada mecanismo que contribui para a energia ao sistema, também o faz para sua entropia. A energia interna do sistema é a soma das energias de translação, de rotação, de vibração, etc. Semelhantemente, a entropia total é formada pela soma da entropia de translação, de rotação, etc. Essa correspondência um a um entre energia e entropia não deveria ser misteriosa, ou algo assim, porque em um processo reversível no qual nenhum trabalho mecânico é realizado, a primeira lei se reduz a dQ = dE. A variação de entropia é então dada por = dS dQ dE = . T T Como a energia total é expressa pela soma de cada contribuição, E = E translação + E rotação + E vibração + a variação da entropia pode ser escrita como dS = dE transl. dE rot. dE vib. + + + T T T (130). Podemos considerar, então, que a entropia total é composta por uma entropia de translação, por uma entropia de rotação, etc. A multiplicidade das contribuições para a energia interna e para a entropia é descrita dizendo-se que um sistema possui diferentes graus de liberdade. Para sistemas bem comportados, conhecidos também como sistemas normais, ao decrescer a temperatura reduzimos a energia e a entropia associada a cada grau de liberdade. Em particular, cada contribuição para a entropia tende a zero quando a temperatura é diminuída. Nos casos excepcionais, onde o sistema está em um estado de não-equilíbrio, nem todos os graus de liberdade estão envolvidos. Geralmente, somente as contribuições de poucos graus de liberdade persistem em não tender a zero. Para essas contribuições parciais de entropia não nulas, dizemos que elas se encontram “congeladas”; sem dúvida, uma terminologia bastante apropriada em vista da temperatura em que se encontra o sistema. Os outros graus de liberdade se comportam normalmente: suas entropias correspondentes desaparecem quando a temperatura vai a zero. Uma perspectiva útil para analisar um sistema é imaginá-lo como composto por uma coleção de subsistemas e a cada grau de liberdade associamos um subsistema. Por exemplo, ao grau de liberdade devido à energia cinética de translação podemos associar um subsistema, que é, naturalmente, parte de sistema total. Essa descrição é tão somente pictórica, mas bastante conveniente para analisar a terceira lei. Dessa maneira, as exceções à forma “não-qualificada” da terceira lei, como foi enunciada, podem ser consideradas como situações nas quais os vários subsistemas não estão todos em equilíbrio entre si. Aqueles que atingem o equilíbrio mútuo obedecem à terceira lei “não-qualificada”, e suas respectivas entropias se anulam quando a temperatura vai a zero. Entretanto, os subsistemas “rebeldes” não estão em equilíbrio: eles desafiam uma descrição termodinâmica. O conceito de subsistemas permite restabelecer a terceira lei em uma forma mais válida e universalmente aceita. Nosso primeiro enunciado (forma “não-qualificada”) “A entropia de um sistema se aproxima de zero quando a temperatura tende a zero” pode ser melhorado e seu conteúdo mais rigoroso se deve a Simon2 (1956, p. 1): “A contribuição para a entropia de um sistema, no qual cada subsistema esteja em equilíbrio termodinâmico interno, anula-se no zero absoluto”. A formulação acima da terceira lei é altamente recomendável porque ela adverte que podemos aplicar a versão “não-qualificada” da terceira lei a sistemas cujos subsistemas (= graus de liberdade) se encontram em equilíbrio interno mútuo. O sentido do enunciado original “não2 SIMON, F. E. Simon. Year Book of the Physical Society. The Physical Society, London, p. 1, 1956. 124 qualificado” é ainda mantido e, portanto, pode-se recuperar muitos dos resultados baseados nela. Para retratar a situação à qual estamos nos referindo nessa discussão, vamos abordar dois exemplos significativos. a 3ª lei da termodinâmica Exemplo 31 Discutir a entropia do estanho (Sn) a baixas temperaturas. Solução O estanho é um elemento químico que pode existir em duas diferentes estruturas cristalinas: uma delas é chamada de estanho “branco”, um sólido metálico com apreciável condutividade elétrica; a outra, conhecida como estanho “cinza” se comporta como um semicondutor. A forma cinza é estável a temperaturas abaixo de 292 K, enquanto o estanho branco é estável acima dessa temperatura. À 292 Kambas as formas podem coexistir em equilíbrio entre si, e podem fazê-lo indefinidamente em proporções arbitrárias. Pela adição de certa quantidade de calor Q o , podese transformar um mol de estanho cinza em estanho branco. Embora o estanho branco seja instável abaixo de To = 292 K , a velocidade com que se processa a transformação é bastante lenta comparada com os tempos envolvidos em medidas experimentais de interesse – o processo leva algumas horas e as medidas experimentais demandam um intervalo de, por exemplo, alguns minutos. Isso significa que se torna fácil trabalhar com o estanho branco, um metal comum, até baixas temperaturas. Queremos considerar o limite quando T → 0 ; por esse limite queremos atingir uma temperatura razoavelmente baixa (digamos, 0.1 K), mas não uma extremamente baixa (por exemplo, 10−6 K ), tal que as orientações aleatórias dos spins nucleares dos átomos do estanho possam ser afetadas. Portanto, quando T → 0 , S branco (T ) → S o S cinza (T ) → S o , e isto significa que S branco (0) = S cinza (0) (I). A relação (I) expressa exatamente o conteúdo da terceira lei: a entropia tende a um valor único independente de qualquer parâmetro do sistema (nesse caso, independente do parâmetro representado pela estrutura cristalina do estanho). O estanho branco tem uma estrutura tetragonal, enquanto o estanho cinza apresenta uma estrutura cúbica; isso é que se conhece como formas alotrópicas (mesmo elemento com diferentes estruturas). O que queremos mostrar é como a terceira lei pode ser combinada com o conhecimento da capacidade térmica para calcular o calor de transformação (calor latente) do estanho cinza para = T= o estanho branco à temperatura de transição T o ( 292 K) . Suponha que desejamos calcular a entropia S branco (To ) de um mol de estanho branco. Isso pode ser feito de dois processos reversíveis diferentes indo desde T = 0 até a temperatura To . (1) Aqueça um mol de estanho branco reversivelmente iniciando em T = 0 e terminando em T = To . A entropia final é dada por To = Sbranco (To ) Sbranco (0) + ∫ 0 Cbranco (T) dT T (II). (2) Usando um mol de estanho cinza, a partir de zero Kelvin, aqueça-o reversivelmente até à temperatura To . Em seguida, transforme-o reversivelmente (à temperatura fixa To ) em estanho branco; a esse último processo está associada uma variação de entropia dada por L cinza →branco To ≡ Q o T o , onde L cinza →branco é o calor latente de transformação de uma forma à outra. Portanto, a entropia para o processo global é To Sbranco (To ) = Scinza (0) + ∫ 0 Como (II) e (III) são iguais, temos To Ccinza (T) Q dT + o T To (III). T o C (T) C (T) Q = Scinza (0) + ∫ cinza Sbranco (0) + ∫ branco dT dT + o ∴ T T To 0 0 To T o C (T) Q C (T) Sbranco (0) −= Scinza (0) ∫ cinza dT + o − ∫ branco dT T To 0 T 0 (IV). 125 tErModinÂMiCa escrever Entretanto, pela relação (I), o lado esquerdo da igualdade (IV) se anula. Assim, podemos Qo = To To T o Cbranco (T) C (T) − dT ∫0 T ∫0 cinzaT dT (V). Nesse estágio, as medidas experimentais das capacidades térmicas de ambas as estruturas cristalinas, e as integrações numéricas, permitem estabelecer que a primeira integral tem valor 51.4 J/K, e a segunda integral vale 44.1 J/K. Então, Qo = 7.3 J K . To = = K )(292 K) 2131 J . Esse valor concorda bastante Isso permite estabelecer o valor Qo (7.3J bem com aquele de 2240 J obtido através de medidas diretas do calor de transformação para o processo. Os resultados estabelecem que S branco (0) = S cinza (0) são iguais dentro da incerteza experimental e, portanto, o estanho obedece à terceira lei em sua forma “não-qualificada”. Exemplo 32 Analisar a variação de entropia do glicerol (a glicerina é o nome comercial do glicerol com pureza em torno de 95%). Solução Glicerol é uma substância que apresenta propriedades singulares: ela pode existir como um líquido super-resfriado mesmo em baixas temperaturas, a despeito de seu ponto de solidificação ser 292 K (trata-se apenas de uma coincidência esse valor ser igual à temperatura de transformação de Sncinza →branco no exemplo anterior). Embora o líquido super-resfriado estar em um estado de não-equilíbrio, é possível determinar experimentalmente sua capacidade térmica. Observa-se que as capacidades térmicas da fase cristalina (glicerol sólido) e a do líquido super-resfriado diferem significativamente na região compreendida entre seu ponto de solidificação até a temperatura de 180K. Abaixo dessa temperatura ambas, C líquido super -resfriado e C sólido , são praticamente iguais e tendem a zero quando a temperatura se aproxima de zero Kelvin. O mesmo desenvolvimento utilizado no exemplo anterior pode ser repetido aqui. (1) Um mol de glicerol líquido super-resfriado é aquecido reversivelmente desde zero Kelvin até a temperatura de solidificação (ou de fusão), 292 K. A variação de entropia é dada por = Slíquido (292) Slíquido (super-resf..) (0) + 292 ∫ Clíquido (super-resf..) (T) 0 T dT (I). (2) Aquece-se reversivelmente a mesma massa de glicerol sólido desde zero Kelvin até 292 K. A seguir, fornecemos certa quantidade de calor Q o = L sólido→l 'quido para ocorrer a fusão. Nessa última etapa, a temperatura permanece constante (transição de fase) e L sólido→l 'quido se refere ao calor latente de fusão da substância. Então, a variação de entropia envolvida no processo global pode ser escrita como Slíquido (292) = Ssólido (0) + 292 ∫ 0 Csólido (T) Q dT + o T 292 (II). Porém, as relações (I) e (II) são iguais pela terceira lei. Portanto, Slíquido (super-resf..) (0) + 292 ∫ 0 Clíquido (super-resf..) (T) T dT = Ssólido (0) + 292 ∫ 0 Csólido (T) Q dT + o (III). T 292 As integrações podem efetuadas numericamente com auxílio de dados experimentais das capacidades térmicas das duas fases e o resultado é 292 Clíquido (super-resf..) (T) Csólido (T) − dT ≈ 41.2 J K T T ∫ 0 126 (IV). O valor experimental do calor latente de fusão dividido pela temperatura 292 K fornece o valor Qo ≈ 62.5 J K 292 (V). a 3ª lei da termodinâmica Utilizando esses valores, a expressão (III) fica Slíquido (super-resf..) (0) − Ssólido (0) ≈ 62.5 J K − 41.2 J K = 21.5 J K (VI). A diferença de entropia dada por (VI) está muito acima de incertezas experimentais. Portanto, a entropia do líquido super-resfriado e a entropia do sólido diferem no zero absoluto: essa diferença reflete a maior desordem do estado líquido (super-resfriado) do que aquela encontrada na fase cristalina. O líquido super-resfriado preserva o excesso de desordem – e ela aparece experimentalmente como uma diferença de entropia “congelada”. PROBLEMA PROPOSTO (7.4) As capacidades térmicas para as duas formas, α e β, de uma substância hipotética são dadas por C α = 0.376 T + 0.0125 T 3 [J K ] e C β = 0.0418 T [J K ] . A forma β sofre uma transição de fase para a forma α à 20 K e o calor latente da transição vale 794 J/mol. Determine se essa substância obedece ou não a versão “não-qualificada” da terceira lei, isto é, se as duas formas, α e β, têm a mesma entropia a zero Kelvin. Suponha que os dados do calor latente são confiáveis dentro de uma incerteza de 2%. COMPLEMENTOS A INATINGIBILIDADE DO ZERO ABSOLUTO Nosso prévio enunciado da terceira lei foi simplesmente que “Você não pode atingir o zero absoluto”. Isso é uma consequência do fato que a entropia tende a zero quando temperatura se aproxima do zero absoluto. Em linguagem leiga, isso é um ataque fatal de retornos decrescentes que impede qualquer esquema para se reduzir a temperatura a zero Kelvin. Existem demonstrações bastante gerais da inatingibilidade do zero absoluto, entretanto, iremos considerar um exemplo particular para ilustrar a ideia. A ilustração pode ser construída de forma idêntica ao seguinte enigma: um viajante dirige seu automóvel de uma cidade A a outra B distante 1500 km e, no primeiro dia de viagem, ele percorre metade da distância; no dia seguinte, ele percorre metade da distância que faltava, e assim sucessivamente, dia após dia. Quantos dias ele precisa para completar sua jornada? A resposta é que a viagem nunca será completada. A mesma situação acontece com os métodos designados para diminuir a temperatura ao zero absoluto. Esses expedientes invariavelmente levam à variação de temperatura proporcional à temperatura em si. Qualquer método otimizado para abaixar a temperatura faz uso de uma transformação adiabática reversível. Assim, se o sistema realiza certa quantidade positiva de trabalho adiabaticamente, ele faz isso as expensas de sua energia interna – e um decréscimo de E diminui sua temperatura. A reversibilidade é também desejável porque processos irreversíveis geram entropia e sabemos que entropia e temperatura tendem a zero juntamente. A produção de entropia em processos irreversíveis se opõe, portanto, à redução da temperatura. Para caracterizar nosso modelo, vamos supor que a entropia seja diretamente proporcional ao produto da temperatura pelo volume S(T, V) = aTV . Com o sistema inicialmente no estado descrito por T = To e V = Vo , executamos uma série de processos de “dois passos”, como indicados do diagrama TS da figura I. 127 tErModinÂMiCa Figura I Compressão isotérmica reduz o volume e a entropia. A expansão isentrópica abaixa a temperatura. As duas retas no diagrama representam S = aTV para V = Vo e para V = Vo 2 . Primeiramente, o sistema é comprimido isotermicamente a partir de Vo até Vo 2 : esse processo reduz sua entropia como indicada pela reta vertical. A compressão é seguida, então, por uma expansão adiabática (e isentrópica) que leva o sistema a seu volume inicial, representada pela reta horizontal. O ponto notável após essa etapa isentrópica é que a entropia se manteve constante. Portanto, o aumento do volume de Vo 2 para Vo deve ser acompanhado por um decréscimo da temperatura de To para To 2 . Isso é facilmente verificado usando-se a relação S(T, V) = aTV . Repetindo-se novamente o processo a partir desse último estágio, a nova temperatura atinge o valor To 4 . E, assim, ele poderia continuar ad infinitum. Cada etapa “vertical-horizontal” no diagrama TS reduz a temperatura pela metade. Aqui nos lembramos da jornada do motorista que nunca consegue chegar à cidade B em número finito de tentativas: é exatamente dessa maneira que, por qualquer processo finito, não se consegue atingir o zero absoluto, não importando quão próximo dele pode-se chegar. 128 a 3ª lei da termodinâmica Anotações 129 tErModinÂMiCa Anotações 130 8 Referências RUHEMANN’S, M. Low temperature Physics. Cambridge: Cambridge University Press, 1937. p. 11. SIMON, F. E. Simon. Year Book of the Physical Society. The Physical Society, London , p. 1, 1956. Referências Complementares ZEMANSKY, M. W. Heat and Thermodynamics. 5th Edition - McGran-Hill, 1968. DUGDALE, J. S. Entropy and Its Physical Meaning. - Taylo e Francis, 1996. YUNUS A. CENGEL and MICHAEL A. Boles. Thermodynamics, An Engineering Aproach. SCHROEDER, D. V. An Introducing To Thermal Physics, Addison-Wesley, 2000. 131
Baixar