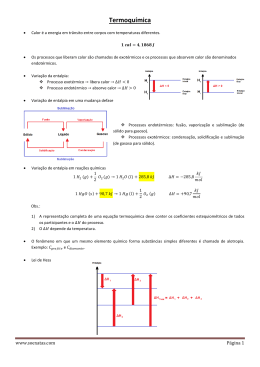
Balanço de Energia em Combustão O primeira lei da termodinâmica traduzida pelo balanço de energia permite analisar dois conceitos importantes na combustão: temperatura adiabática e poder calorífico que se descrevem de seguida. Sistema fechado W+Q=∆U=UFinal-UInicial ∆V=0 Æ W=0 e Q=∆U ∆P=0 Æ W=-P ∆V e Q=∆U+ P∆V= ∆H Sistema aberto (que troca massa com o exterior) . . . DE v2 = W + Q + ∑ m( h + gz + ) Dt 2 e −s Em regime estacionário DE/Dt=0 e desprezando as variações de energia cinética e potencial: . . . W + Q = m ∆h ou W + Q = ∆H Num sistema sem variação de volume W=0 e Q=∆H. h=u+Pv ou H=U+PV pelo que ∆h= ∆u+ ∆(Pv) ou ∆H= ∆U+ ∆(PV). ∆(PV) é muito pequeno para sólidos e líquidos e para gases pode ser relacionado por ∆(PV)=∆(nR0T) A diferença entre as variações de entalpia e de energia interna depende da variação de número de moles de espécies gasosas na reacção química. Casos extremos de evoluções na combustão Isotérmico Æ Entalpia de combustão Como a reacção de combustão é exotérmica a entalpia dos produtos é maior que a dos reagentes ou a variação de entalpia de combustão é negativa No processo isotérmico ∆H = ∆U+∆(n) R0T Ou ∆H = ∆U + (nProdutos-nReagentes)Gasosos R0T Quando se considera a reacção com condições iniciais e finais com T=25ºC, P=1 atm, definem-se as entalpias de reacção padrão ∆h 0 , ∆h 0 , ∆H 0 e de modo equivalente as energias de reacção padrão. O simetrico das entalpias e energias de reacção permitem definir o Poder Calorífico, respectivamente a pressão e a volume constante Æ Calor libertado. Definem-se ainda dois tipos de valores inferior e superior dependente de se considerar água no estado de vapor ou líquido respectivamente: m PCS = PCI + w hfg mf em base mássica para valores a pressão constante ou m PCSv = PCIv + w ufg a volume constante. mf Utiliza-se mais a pressão constante por corresponder a mais aplicações (Excepção nos motores gasolina). A diferença entre os dois valores é: PCSv = PCS + R0T0 (nP − nR ) em base molar Adiabático Æ Temperatura adiabática de combustão A temperatura final dos produtos é superior à dos reagentes sendo a entalpia semelhante. H Inicial FAdiabático FIsotérmico T A variação de entalpia de reacção não depende da quantidade de espécies inertes enquanto a temperatura adiabática de combustão depende. Vamos analisar mais este conceito posteriormente. Entalpias de formação Como à mesma temperatura reagentes e produtos têm uma diferença de entalpia, define-se uma base comum para todas as substâncias, convencionando o valor nulo para as condições de referência (T=25ºC) para algumas espécies: C(s), O2, H2, N2, S(s) etc. Para uma espécie qualquer considera-se a sua formação a partir das espécies de referência e definese a sua entalpia de formação como a entalpia de reacção para formar o elemento a partir de outras espécies às condições de referência para ambos. Valores normalmente apresentados em base molar. Exemplos: CO, CO2, H2O(gas), H2O (liq). o 1 h f CO = ∆h 0 C(s ) + O2 → CO = −110530 kJ/kmol de CO 2 h f CO2 = ∆h 0 (C(s ) + O2 → CO2 ) = −393520 kJ/kmol de CO2 o o 1 h f H2O ( g ) = ∆h 0 H 2 + O2 → H 2O( g ) = −241820 kJ/kmol H2O 2 o 1 h f H2O ( l ) = ∆h 0 H 2 + O2 → H 2O(l ) = −285830 kJ/kmol H2O 2 A diferença entre os dois primeiro representa a entalpia de combustão do monóxido de carbono: 1 ∆ h 0 CO + O2 → CO2 = −282990 kJ/kmol 2 A entalpia de formação de água líquida podia ser obtida do valor de gás - entalpia de vaporização hfg = hfg MH2O = 2442,3 × 18,02 = 44010 kJ/kmol C(s), H2, O2, N2 -110530 kJ/kmol CO -282990 kJ/kmol CO2 REFERÊNCIA -241820 kJ/kmol -393520 kJ/kmol H2O (g) hfg -285830 kJ/kmol H2O (l) A entalpia de formação dos combustíveis podem ser definidas a partir da entalpia de reacção do combustível conduzindo à formação dos produtos. Ex. Metano com poder calorífico inferior 50016 kJ/kg ou em termos molares 50016x16,04=802256 kJ/kmol ∆ h 0 (CH 4 + 2O2 → CO2 + 2H 2O(g )) = −802256 kJ/kmol 0 ∆hCH4 = hf0CO2 + 2 × hf0 H2O − 2 × hf0O2 − hf0CH4 Æ hf0CH4 = −74850kJ / kmol A entalpia de formação do combustível no estado líquido pode ser definido a partir do valor no estado gasoso, subtraindo a entalpia de vaporização. 0 Ex: hf CH OH ( g ) = −200670 kJ / kmol , hfg = 37900 kJ / kmol 3 hf 0 CH3OH ( l ) = hf0CH3OH ( g ) − hfg CH OH = −238570 kJ / kmol 3 O poder calorífico do combustível no estado líquido também pode ser obtido a partir do valor correspodnente no estado gasoso. A entalpia para qualquer temperatura para uma espécie é obtida somando à entalpia de formação a parcela ‘sensível’ (h-h298). Existem tabelas com valores de entalpia sensível em relação à referência (298K) ou com valor nulo correspondente a 0K, quando são obtidos de polinómios (a6i=0) h/R0= a1iT+ a2iT2/2+ a3iT3/3+ a4iT4/4+ a5iT5/5+ a6i cp/R0= a1i+ a2iT+ a3iT2+ a4iT3+ a5iT4 Cálculo da temperatura adiabática de combustão A temperatura adiabática de combustão pode ser obtida a partir de um balanço de energia, calculando a entalpia dos produtos como igual à entalpia dos reagentes. A partir da aproximação polinomial da entalpia em função da temperatura pode-se calcular a temperatura. Ex. Temperatura adiabática estequiometrica de Metano à referência (298K) com oxigénio: de combustão temperatura de H Pr od (CO2 + 2H 2O(g )) = H Re ag . (CH 4 + 2O2 ) = −74850 kJ ( H Pr od (CO2 + 2H 2O(g )) = hf0 + ∆h ) CO2 ( ( + 2 × hf0 + ∆h ) H 2O ( g ) H Pr od (CO2 + 2H 2O(g )) = −877160 + ∆hCO2 + 2 × ∆hH 2O ( g ) ) Função da temperatura De onde se pode calcular TAdiabático= Se considerarmos a combustão com ar em vez de oxigénio o valor da temperatura adiabática vai ser diferente: H Pr od (CO2 + 2H 2O(g ) + 7,52N 2 ) = H Re ag . (CH 4 + 2O2 + 7,52N 2 ) = −74850 kJ ( H Pr od (CO2 + 2H 2O(g ) + 7,52N 2 ) = hf0 + ∆h ) CO2 ( ( + 2 × hf0 + ∆h ) H 2O ( g ) + 7,52∆hN2 H Pr od (CO2 + 2H 2O(g ) + 7,52N 2 ) = −877160 + ∆hCO2 + 2 × ∆hH 2O ( g ) + 7,52∆hN2 De onde se pode calcular TAdiabático=2226K ) Balanço de energia em sistemas de combustão H Inicial ∆H0 Calor trocado Final T Num caso geral a temperatura dos reagentes e a dos produtos são ambas diferenças do estado de referência Nestes casos para quantificar o calor fornecido (normalmente<0) Q=∆H tem de se quantificar as várias parcelas desde a situação inicial à final: Q = H Pr od − H Re ag = ∑ n& (h i =Pr od i f 0 + ∆h ) i − ∑ n& (h j =Re ag j 0 f + ∆h ) j que também se pode escrever em termos mássicos & ∑ x i hf0 + ∆h i − ∑ x j hf0 + ∆h Q=m j =Re ag i =Pr od & ∑ x i ∆hi − ∑ x j ∆h j + ∑ x i hfi0 Q=m j =Re ag i =Pr od i =Pr od ( ) ( ) j − ∑x h j =Re ag j 0 fj Entalpia de reacção0 = - Poder Calorífico No caso de combustão completa (Comb. Gasoso) pode-se obter: [ ] & Gases c p Re ag (TRe ag − T0 ) − c p Pr od (TPr od − T0 ) + m & Combustível PC QTransf = m Para o caso de combustão incompleta ou quando ocorre mudança de fase de reagentes e/ou produtos tem de se quantificar a variação de entalpia correspondente e incluir na expressão para o calor transferido. O rendimento de um equipamento de uma caldeira ou câmara de combustão é normalmente representado como a razão entre o calor obtido e a entrada de energia no sistema. A entrada de energia é quantificada pelo produto do caudal de combustível pelo PCS (Poder Calorífico Superior) e a entalpia do ar de combustão (pode ser pré-aquecido) Existem dois métodos para determinar o rendimento de câmaras de combustão: Método directo e indirecto. Método Directo: Representa a razão entre o calor obtido na caldeira (do lado da água/vapor) em relação à entrada total de energia. Método Indirecto: Representa 1 menos as perdas a dividir pela entrada total de energia. As parcelas principais das perdas são: - Entalpia sensível dos produtos de combustão - Entalpia latente nos produtos de combustão (vapor de água nos produtos) - Perda de vaporização da humidade do ar - Perda por inqueimados sólidos e gasosos - Perdas de calor para o ambiente - Perdas do circuito de água/vapor e outras ... Existem normas para o cálculo da eficiência de caldeiras que apresentam esquemáticos (DIN, ASME)
Baixar