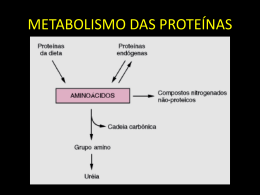
CAP3 (Contig Assembly Program) George Darmiton da Cunha Cavalcanti ([email protected]) UFPE – CIn Junho de 2001 Roteiro Introdução Arquitetura O Programa CAP3 – Entrada e Saída Pontos Fortes e Fracos do CAP3 Resultados do CAP3 – CAP3 versus PHRAP Bibliografia Introdução É um programa utilizado para montar cadeias de DNA Desenvolvido por Xiaoqiu Huang, – Department of Computer Science, Michigan Technological University Versão 3 foi desenvolvida em 1999 Atualmente encontra-se na versão 4, sendo esta comercial Arquitetura Remoção de regiões pobres Calcular sobreposição 1º Fase Remoção de falsas sobreposições Construção de Contigs 2º Fase Construção do Consenso 3º Fase Primeira Fase Composta de três etapas – Remoção de regiões pobres – Cálculo da sobreposição – Remoção de falsas sobreposições Antes dessas etapas é necessário identificar sobreposições entre fragmentos. Primeira Fase Identificação de sobreposição Criar a seqüência combinada – Os fragmentos f1, f2, ..., fn são concatenados – Caracter especial separa dois fragmentos – f1 # f2 # ... # fn Para cada fragmentos fx e o seu complemento reverso rx, encontrar o par (fx, fy) e (rx, fy) – tal que x<y e que os fragmentos tenham uma sobreposição relevante Para cada par com sobreposição uma faixa diagonal da matriz da programação dinâmica e calculado. (Smith e Waterman 1981) – Essa matriz será usada mais adiante por motivo de eficiência Primeira Fase Remoção de Regiões Pobres Remoção de regiões pobres Calcular sobreposição 1º Fase Remoção de falsas sobreposições Construção de Contigs 2º Fase Construção do Consenso 3º Fase Primeira Fase Remoção de Regiões Pobres (cont.) Posição de remoção 5’ Fragmento h Fragmento f Fragmento g Posição de remoção 3’ Primeira Fase Remoção de Regiões Pobres (cont.) O algoritmo de alinhamento local de Smith e Waterman foi generalizado para usar valores de qualidade de base q 10 log10 p Sendo p a probabilidade do erro estimado para a base m * min(q1, q2) n * min(q1, q2) -g * min(q1, q2) Primeira Fase Remoção de Regiões Pobres (cont.) Os valores que indicam a qualidade da base são usados para permitir que: – Matches em bases que possuem altos valores de qualidade recebem alta pontuação positiva; – Mismatches em bases que possuem altos valores de qualidade recebem alta pontuação negativa; – Matches e mismatches em bases que possuem baixos valores de qualidade recebem pontuações baixas positivas e negativas, respectivamente Primeira Fase Remoção de Regiões Pobres (cont.) Se os valores de qualidade de base foram informados qualpos5 crange (-y) qualpos3 crange Fragmento f Maioria dos valores de qualidade são maiores que qualcut (-c) Caso os valores de qualidade da base não sejam informados – qualpos5 = 1 e qualpos3 = tamanho de f A cobertura mínima é determinada por gdepth (-z) Primeira Fase Cálculo das Sobreposições Remoção de regiões pobres Calcular sobreposição 1º Fase Remoção de falsas sobreposições Construção de Contigs 2º Fase Construção do Consenso 3º Fase Primeira Fase Cálculo das Sobreposições (cont.) O alinhamento global é utilizado para calcular a sobreposição entre fragmentos Alinhamento global versus Alinhamento local – Utilizando o método global é possível identificar falsas sobreposições. • mostrar que algumas regiões dos fragmentos não são similares, indicando que esta sobreposição é falsa. – O alinhamento local está restrito a regiões similares. Primeira Fase Remoção de Sobreposições Falsas Remoção de regiões pobres Calcular sobreposição 1º Fase Remoção de falsas sobreposições Construção de Contigs 2º Fase Construção do Consenso 3º Fase Primeira Fase – Remoção de Sobreposições Falsas (cont.) Cada sobreposição é avaliada por 5 (cinco) medidas – 1ª Medida • Informa o comprimento mínimo para a sobreposição (-o) – 2ª Medida • Determina que o percentual de identidade não deve ser menor que o valor estabelecido pela opção –p – 3ª Medida • • • • Determina o valor de similaridade da sobreposição (-s) m * min(q1, q2) n * min(q1, q2) -g * min(q1, q2) Primeira Fase – Remoção de Sobreposições Falsas (cont.) – 4ª Medida • Se a sobreposição contiver um número grande de diferenças entre bases de altos valores de qualidade, essa sobreposição é provavelmente falsa. (-b e -d) – 5ª Medida • Se o número de diferenças em uma sobreposição for maior que o esperado, então é provável que essa sobreposição seja falsa. (-e) Caso uma dessas medidas falhe, a sobreposição é considerada falsa. Segunda Fase Construção dos Contigs Remoção de regiões pobres Calcular sobreposição 1º Fase Remoção de falsas sobreposições Construção de Contigs 2º Fase Construção do Consenso 3º Fase Segunda Fase Construção dos Contigs (cont.) 1ª Etapa – Um layout inicial é gerado • Método guloso 2ª Etapa – A qualidade do layout corrente é avaliada – O número de restrições satisfeitas e não satisfeitas é calculado para cada sobreposição – Restrições não satisfeitas são particionadas em grupos • cada grupo possui restrições associadas com uma sobreposição não usada ou com um par de contigs Segunda Fase Construção dos Contigs (cont.) 3ª Etapa – O grupo com o maior número de restrições não satisfeitas é selecionado – Caso 1 • grupo associado a uma sobreposição não usada – Caso 2 • grupo associado a um par de contigs – Se nenhuma correção for feita • o processo é repetido com os grupos restantes – Caso contrário • a 2ª etapa é repetida para o novo layout Terceira Fase Construção do Consenso Remoção de regiões pobres Calcular sobreposição 1º Fase Remoção de falsas sobreposições Construção de Contigs 2º Fase Construção do Consenso 3º Fase Terceira Fase Construção do Consenso (cont.) A soma ponderada dos valores de qualidade é calculada para cada base – Os valores de qualidade são divididos em dois grupos, um para cada sentido (5’ 3’ - 3’ 5’) – cada grupo é ordenado em ordem decrescente – pesos: w1=1, wi = 0.5, para i>1 – Ex: 20+, 40-, 30+ e 10• grupo1: 30+ e 20+ grupo2: 40- e 10• soma ponderada = 30(1) + 40(1) + 20(0.5) + 10(0.5) = 85 Terceira Fase Construção do Consenso (cont.) Cálculo da média do valor de qualidade qs d qi qi k 1i k e ci d 1i k e ci d qd qi k 1i k e ci qn qi k 1i k Terceira Fase Construção do Consenso (cont.) Match Mism atch Deletion Insertion A 20 C 10 A 30 - 15 A 40 T 15 C 20 C 25 - 10 C 30 G 10 - 30 - 25 - 40 C 15 - 20 - 10 - 15 - 30 - 25 A 25 T 10 - 15 C 15 q s A 13 qs T 14 qd 5 qn 20 score 13m score 10n score 5 g score 15g Parâmetros de Entrada do CAP3 CAP3 recebe um arquivo com as seqüências de fragmentos no formato FASTA – Uso: cap3 arquivo_de_fragmentos [opções] Arquivos opcionais – arquivo contendo os quality values no formato FASTA, usando extensão .qual – arquivo contendo restrições forward-reverse, usando extensão .con. • Pode ser gerado usando o programa FORMCON • Formato: ReadA ReadB MinDistance MaxDistance Opções Valor das penalidades Parâmetro descrição -g N gap N>0 (6) -m N match N>0 (2) -n N mismatch N<0 (-5) Faixa Diagonal Parâmetro descrição -a N N>10 (20) Resposta do Programa Consenso no formato ace – arquivo com extensão .ace Consenso – arquivo com extensão .contigs Quality values do consenso – arquivo com extensão .contigs.qual Fragmentos não são usados na montagem – arquivo com extensão .singlets Resposta do Programa (cont.) Informações adicionais sobre a montagem – arquivo com extensão .info Satisfação das restrições – arquivo com extensão .results Pontos Fortes do CAP3 Uso de forward-reverse constraints para corrigir erros de montagem – Objetivo: localizar e corrigir erros no layout da seqüência e ligar contigs separados por gaps – Dois fragmentos devem estar em direções opostas na molécula de DNA e a uma determinada distância. – O algoritmo usado no CAP3 é tolerante a restrições erradas Pontos Fortes do CAP3 (cont.) Geração do resultado da montagem no formato ace para Consed – Consed – ferramenta gráfica para editar seqüências CAP3 pode ser usado no GAP4 do pacote Staden. – GAP4(Genome Assembly Program) é uma ferramenta gráfica do pacote de ferramentas Staden Pontos Fortes do CAP3 (cont.) Uso de base quality values – Usados no alinhamento de fragmentos e na construção do consenso. – Melhora a qualidade na geração do consenso Remoção de regiões pobres, 5’ e 3’ – Objetivo • Utilizar apenas regiões ‘boas’ do fragmento na montagem. Pontos Fracos do CAP3 A remoção de regiões ditas pobres, pode excluir áreas importantes no processo de alinhamento Tempo de processamento A ferramenta não possui interface gráfica (entretanto pode ser usada no pacote GAP4) Resultados do CAP3 Conjunto de dados BAC (Bacterial Artificial Chromossome) Dados GenBank Nº Nº de frag Tam médio frag Tam seq 203 AC004669 1812 598 89.779 216 AC004638 2353 614 124.645 322F16 AF111103 4297 1011 159.179 526N18 AF123462 3221 965 180.182 Dados Tempo (min) Nº de contigs Tam seq CAP Nº de <> 203 37 1 90.292 0 216 154 1 132.057 17 322F16 127 1 157.982 11 526N18 73 2 180.128 10 CAP3 versus PHRAP Conjunto de dados BAC – PHRAP, normalmente, produz cadeias mais longas de contigs – CAP3 produz menos erros no consenso Caso os valores de qualidade da base não estejam disponíveis – CAP3 é uma boa escolha já que trata redundância Bibliografia Xiaoqiu Huang, Anup Madan. CAP3: A DNA Sequence Assembly Program. Genome Research 9:869-877, 1999. Xiaoqiu Huang. Na Improved Sequence Assembly Program. Genomics 33, 21-31, 1996. Site oficial na Internet. http://genome.cs.mtu.edu/cap3/cap3.html Staden Package WWW site. – http://www.mrc-lmb.cam.ac.uk/pubseq/staden_home.html – http://www.mrc-lmb.cam.ac.uk/pubseq/contig.html
Download