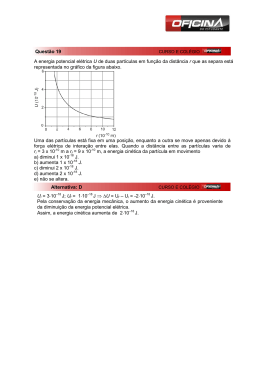
8º CONGRESSO IBEROAMERICANO DE ENGENHARIA MECANICA Cusco, 23 a 25 de Outubro de 2007 TEORIA DO TRANSPORTE PARA MISTURAS NÃO-IDEAIS Paulo Cesar Philippi 1 e Luis Orlando Emerich dos Santos Laboratório de Meios Porosos e Propriedades Termofísicas(LMPT) Departamento de Engenharia Mecânica Universidade Federal de Santa Catarina 88040-900 Florianópolis SC Brazil RESUMO As equações de balanço de quantidade de movimento e energia interna para misturas são usualmente derivadas considerando-se os fluidos que as compõem como ideais, o efeito das forças intermoleculares de atração sendo considerado apenas na equação de estado da mistura que é escrita usando-se regras de mistura empíricas. No presente artigo, as equações de transporte para misturas não ideais são derivadas partindo-se da escala molecular, onde as moléculas são consideradas como pontos materiais que seguem as leis de Newton, obtendo-se a equação de Boltzmann e as equações de transporte, através de processos sucessivos de mudança de escala e usando-se o enfoque probabilístico. Mostra-se que os efeitos de atração intermolecular resultam em termos fontes de quantidade de movimento e de energia interna até então não previstos na literatura sobre misturas não-ideais, mas que estão presentes em fenômenos físicos importantes sendo os responsáveis, e.g., pelos processos de ruptura e coalescência de gotas em dispersões. De fato, dispersões de gotículas de líquido e bolhas de gás são usualmente tratadas considerando como nula a espessura da interface entre as fases dispersa e contínua, o que acarreta dificuldades consideráveis no tratamento de misturas dispersas. Da mesma forma, deriva-se uma equação de estado para misturas não ideais com uma regra de mistura obtida formalmente, considerando o potencial atrativo entre moléculas de espécies diferentes. Palavras chaves: Misturas não-ideais, teoria do transporte, equações de balanço, dispersões 1 [email protected] INTRODUÇÃO Misturas não-ideais são de utilização freqüente em ciclos de refrigeração e em engenharia de processos. Em conseqüência, as suas propriedades termodinâmicas de equilíbrio vêm sendo intensivamente estudadas ao longo das últimas décadas e regras de mistura têm sido propostas empiricamente com base em parâmetros experimentais. Desse modo, é hoje possível o cálculo das propriedades termodinâmicas de várias misturas não-ideais de interesse tecnológico a partir de alguns poucos parâmetros associados aos componentes puros e fatores binários de interferência. A origem da não-idealidade são as forças intermoleculares de longa-distância e o objetivo do presente trabalho é estabelecer as bases de uma teoria do transporte para misturas não ideais em estados de não-equilíbrio a partir de informações moleculares elementares, levando-se em conta a forma como as moléculas interagem entre si. Considerando as dificuldades associadas a este objetivo, algumas premissas foram adotadas entre as quais se destacam: i) as moléculas são consideradas como pontos materiais, sujeitas às leis de Newton; ii) uma mistura é considerada como um sistema de partículas e o conjunto de estados iniciais possíveis para esse sistema forma um conjunto denso no espaço de fases. A obtenção das equações de transporte é feita através de processos sucessivos de mudança de escala, partindo-se da escala molecular, obtendo-se a equação de Liouville para o sistema e a partir desta a equação de Boltzmann. As equações de balanço são, enfim, recuperadas como momentos da equação de Boltzmann. ESCALA MOLECULAR Forças intermoleculares de atração Considerando-se as moléculas como eletricamente neutras, a interação entre elas a longa distância se dá basicamente através de forças polares, ou de Keesom, forças de indução, ou de Debye e forças dispersivas, ou de London. r As forças polares ocorrem apenas entre moléculas que possuem momentos dipolares permanentes, μ . Para cada r r configuração em que duas moléculas estão orientadas de acordo com seus dipolos μ 1 e μ 2 corresponde uma energia potencial de interação e, quando a ocorrência de cada uma dessas energias potenciais é admitida seguir uma distribuição de Boltzmann, o valor médio da energia potencial é dado por, [1], Φ (12 ) 2⎛ 1 = − ⎜⎜ 3 ⎝ 4πε 0 2 2 ⎞ μ μ ⎟⎟ 6 , ⎠ r kT 2 1 (1) 2 12 onde ε 0 é a permissividade elétrica do vácuo, k é a constante de Boltzmann, T é a temperatura e r12 é a distância relativa entre os centros das moléculas. Desse modo, um aumento da temperatura reduz as ocorrências em que a energia potencial de interação é mínima, i.e., quando as moléculas estão mais fortemente atraídas e necessitam de um maior aporte de energia cinética (e.g., em um processo de colisão) para serem afastadas umas das outras. Uma molécula polar 1 induz um momento dipolar em outra molécula 2 (polar ou não polar), com a mesma orientação do primeiro. O valor deste momento é definido pela polarizibilidade α da segunda molécula e a energia potencial de interação se escreve, [1], Φ (12 ) 1⎛ 1 = − ⎜⎜ 2 ⎝ 2πε 0 2 ⎞ μ α ⎟⎟ . 6 ⎠ r 2 1 2 (2) 12 Moléculas simétricas como as dos alcanos não possuem um momento dipolar permanente e a força atrativa entre essas moléculas foi descrita pela primeira vez por London em 1930, como sendo devida às rápidas flutuações do centro geométrico de suas nuvens eletrônicas. Essas flutuações originam momentos dipolares temporários e a cada vez que um momento dipolar instantâneo é formado, ele origina, por indução, um momento dipolar nas moléculas que estão na vizinhança. A intensidade do processo de interação depende da polarizibilidade da molécula indutora porque o valor do momento dipolar que esta adquire depende da perda de controle que as cargas nucleares exercem sobre as camadas eletrônicas mais exteriores e também da polarizibilidade da segunda molécula porque, esta, é uma medida da facilidade com que se forma um dipolo sob a ação de um campo eletrostático. A energia potencial resultante foi determinada por London como sendo, [1]: Φ (12 ) 3⎛ 1 = − ⎜⎜ 2 ⎝ 4πε 0 ⎞ α α I1I 2 ⎟⎟ 6 ⎠ r I1 + I 2 2 1 (3) 2 12 onde I corresponde ao primeiro potencial de ionização da molécula. As Eqs. (1-3) podem ser reunidas em uma única equação Φ (12 ) = − B (12 ) r6 (4) 12 (12 ) (12 ) (12 ) + Bind + Bdisp sintetiza as três contribuições acima. onde o parâmetro de força B (12 ) = B (12 ) ( μ1, μ1,α 1,α 2 , I1, I 2 ) = Bpolar . . Equações do movimento Seja um sistema de N partículas N = ∑N s , onde s = 1,2,...r designa uma das r diferentes espécies presentes na s mistura e admita-se que este sistema satisfaz as leis da mecânica newtoniana, r dξ ( p ) i dt = Ns r ∑∑χ s i=1,...,Np, p=1,...,r js , ip (5) j =1 onde χ js,ip designa a força (por unidade de massa m ip ), que age na partícula i da espécie p, pela ação da partícula j da espécie s. A solução das N equações do movimento acima a partir de um estado inicial especificado pelas r r posições x i(s ) e velocidades ξ i (s ) constitui um problema de dinâmica molecular, que, presentemente, está se estabelecendo como uma disciplina do conhecimento de grande valia para a solução de problemas de nano-ciência e nano-tecnologia. Nossa escala de interesse em problemas corriqueiros de engenharia é, todavia, 1.000 ou mesmo 1.000.000 de vezes maior e precisamos, de alguma forma, fazer uma mudança de escala através de um processo de homogeneização das equações de Newton considerando a natureza estatística dos fluidos. EQUAÇÃO DE LIOUVILLE Considerando que qualquer condição inicial é possível para o sistema mecânico, desde que todas elas satisfaçam a um conjunto de restrições previamente estabelecidas (e.g., energia total do sistema) e que este conjunto de estados iniciais forme um conjunto denso no espaço de fases, ainda que não haja como saber em que estado se encontra o sistema num dado instante, é possível estabelecer uma probabilidade, ( r r r r r r r r f ( N ) x1(a ) ,ξ 1(a ) ,...., x N(a( ) ) ,ξ N(a( )) ,..., x1(r ) ,ξ 1(r ) ,...., x N(r() ) ,ξ N(r( )) , t a a b b ) (6) que designa a probabilidade conjunta de se encontrar, num intervalo entre o instante t e o instante t+dt, a partícula 1 r r da espécie “a” na posição x1(a ) com velocidade ξ 1(a ) e da mesma forma para todas as outras N-1 partículas do sistema. Partindo-se das equações de Newton, Eq. (5), e considerando as premissas acima é possível mostrar que a distribuição f (N ) satisfaz, [2], N v ⎞ ⎛N r ∂ t f ( N ) + ∑ ⎜⎜ ∑ ξ i (s ) .∂ xr ( ) f ( N ) + ∑ χ i(s ) .∂ ξr ( ) f ( N ) ⎟⎟ = 0 s ⎝ i =1 i =1 ⎠ (s ) (s ) s s i i (7) que constitui a equação de Liouville do sistema mecânico de partículas. Na equação acima, r r N (s ) r N(p ) r χ i(s ) = χ i(s ),e + ∑ χ ij(ss ) + ∑∑ χ ij(ap ) j ≠i p ≠ s j =1 (8) r r onde mi(s ) χ i(s ),e designa uma força externa agindo sobre a partícula i da espécie s, e mi(s ) χ ij(ap ) a força de interação molecular entre a partícula i da espécie s e a partícula j da espécie p. O conhecimento da distribuição f ( N ) constitui, ainda, uma informação por demais detalhada e uma segunda mudança de escala é conseguida através da probabilidade marginal ( r r f (1,a ) x 1(a ) ,ξ 1(a ) , t = ∫ ......∫ f (N ) ) r r r r r r r r dx 2(a )dξ 2(a ) ...dx N(a( ) ) dξ N(a( )) ...dx1(r )dξ 1(r ) ...dx N(r() ) dξ N(r( )) a a r r (9) r que é a probabilidade de se encontrar entre os instantes t e t+dt a partícula 1 da espécie a na posição x1(a ) com r velocidade ξ 1(a ) . EQUAÇÃO DE BOLTZMANN Considerando-se que as partículas de cada espécie não podem ser distinguidas entre si e que o número de partículas de cada espécie é muito grande, uma equação de evolução pode ser obtida para a distribuição f (1,a ) , integrando-se a Eq. (7), na forma r r ∂ t f (a ) + ξ .∂ xr f (a ) + χ (a ),e .∂ ξr f (a ) r r ) (a ) 2 (2,aa ) r r r r r (N ) f x,ξ , x1,ξ1,t dxr1 dξ1 = −∂ ξr .∫ ∫ χ xr(aa x r r r ) (a ) (s ) (2,as ) r r r r r .(N − ∑ ∂ ξr .∫ ∫ χ xr(as N )f x,ξ , x1,ξ 1, t dx1 dξ 1 = Ω (aa ) + ∑ Ω (as ) x ( 1 1 s ≠a ) ( ) s ≠a (10) r onde f (a ) = N ( a ) f (1,a ) é o valor esperado do número de partículas a que no instante entre t e t+dt estão na posição x r (2,as ) r r r r com velocidade ξ , f x, ξ , x 1 , ξ 1 , t é a probabilidade de se encontrar, simultaneamente uma partícula qualquer r r r da espécie a na posição x com velocidade ξ e uma partícula qualquer da espécie s na posição x 1 com velocidade r ξ1 . Os termos à direita da Eq. (10) são os termos de interação entre as partículas da espécie a e todas as outras espécies de partículas escritos como integrais no espaço físico e no espaço de velocidades. Atribuindo-se à cada partícula da espécie a uma esfera de ação de diâmetro σ as que delimita a interação separando um uma região externa à esta esfera caracterizada por um potencial de atração entre as partícula a e s, de uma região interna caracterizada por um potencial de repulsão, pode-se decompor o termo de interação Ω (as ) , [3], ( ) (as ) Ω (as ) = Ω sd + Ω ld(as ) (11) onde (as ) Ω sd = −∂ ξr . Ω (ldas ) = −∂ ξr . r( ) ( ) ( ) ( ∫ ∫ χ .(N N )f r r x1 − x ≤σ as as rr xx1 a s r( ) ( ) ( ) ( ∫ ∫ χ .(N N )f r r x1 − x >σ as as rr xx1 a s 2,as ) 2,as ) (xr,ξr, xr ,ξr , t )dxr dξr (xr,ξr, xr ,ξr , t )dxr dξr 1 1 1 1 1 1 1 1 (12) (as ) O termo Ω sd está associado às interações de curta distância onde dominam os processos de repulsão. Foi introduzido na teoria cinética por Boltzmann, [4], utilizando a hipótese do caos molecular e está na origem da sua demonstração da irreversibilidade de sistemas mecânicos, quando os estados do mesmo não podem ser assegurados com precisão, a não ser sob um enfoque estatístico. O termo Ω (ldas ) descreve o efeito dos processos de interação à longa-distância e é o termo que descreve os processos associados à não-idealidade dos fluidos e suas misturas e sobre o qual dedicaremos o restante do presente artigo. r r Admitindo independência estatística quando x 1 − x > σ as ( ) ( ) ( r r r r r r r r f (2,as ) x,ξ , x1,ξ 1, t = f (1,as ) x,ξ , t f (1,as ) x1,ξ 1, t ) (13) a Eq. (12) se escreve para o termo de longa distância ( ) ∫ ∫ χr r r Ω (ldas ) = −∂ ξr f (a ) x,ξ , t . r r x1 − x >σ as (as ) rr xx1 ( ) r r r r f ( s ) x 1 , ξ 1 , t d x 1 dξ 1 (14) r r r ) r depende apenas da distância x − x entre as partículas e como a força χ xr(as 1 x 1 r r ⎡ Ω ld(as ) = ∂ ξr f (a ) x,ξ , t .⎢− ⎣⎢ ( ) r (as ) ∫χ rr xx1 r r x1 − x >σ as r r ⎤ n (s ) (x1, t )dx1 ⎥ ⎦⎥ (15) ) (16) onde ( r r r r n (s ) (x1, t ) = ∫ f (s ) x1,ξ 1, t dξ 1 r é o valor esperado do número de partículas s que ocupam a posição x1 no instante t, por unidade de volume do espaço físico. r Quando a distribuição de partículas s numa esfera centrada em x é uniforme, a força resultante, Fld(as ) = − r (as ) ∫χ rr xx1 r r x1 − x >σ as r r n (s ) (x1, t )dx1 (17) r que as partículas s exercem sobre uma partícula a em x , é nula. Desse modo, fazendo-se uma expansão em séries de r r r r r Taylor de n (s ) (x1, t ) em torno de n (s ) (x, t ) e definindo η ≡ x1 − x r r ∂ n (s ) 1 ∂ 2 n (s ) n (s ) (x1, t ) = n (s ) (x, t ) + α ηα + ∂η α ∂η β + ... ∂η α 2 ∂η α ∂η β (18) apenas o segundo termo (os termos de ordem ímpar) apresenta uma contribuição diferente de zero para a força r ) r Fld(as ) . Por outro lado, a força elementar χ xr(as entre duas partículas a e s, pode ser escrita como x 1 r χ xr(asxr ) = − 1 1 ∇φ (as ) m (a ) (19) com a energia potencial φ (as ) dada pela Eq. (4). Usando-se as Eqs. (4), (17)-(19), o termo de interação de longa distância é finalmente obtido na forma Ω (ldas ) = − 8π B (as ) ∇n (s ) .∂ ξr f (a ) 3σ as3 m (a ) (20) Uma interpretação física de Ω ld(as ) pode ser obtida quando a distribuição f (a ) é uma distribuição de equilíbrio local, r uma distribuição de Maxwell-Boltzmann, escrita em termos da velocidade do centro de massa da mistura, u Ω ld(as ) = ( 16π B (as ) (a ) (s ) r r f ∇n . ξ − u 3σ as3 m (a ) ) (21) que quantifica o número de partículas da espécie a que são reorientadas para a direção de ∇n (s ) em função do processo de interação com as partículas da espécie s (veja, também, Santos et al., [5-6]). Uma conseqüência importante da presente análise é a demonstração formal de que os termos de interação de longa distância atuam apenas em regiões do espaço físico onde há gradientes de concentração. Nessas regiões enquanto que o termo Ω (ldaa ) tende a reorientar as partículas a para o próprio componente a, caracterizando um efeito de separação entre as espécies, os termos Ω (ldas ) com s ≠ a tendem a misturar as partículas entre si sobrepondo a sua ação às dos processos de dispersão, cuja origem são os potenciais de repulsão de curta distância. Desse modo, dois fluidos a e b são (ab ) imiscíveis quando os efeitos Ω (ldaa ) e Ω (ldbb ) predominam sobre os efeitos Ω (ldab ) de longa distância e Ω sd de curta distância. Propriedades do termo de interação de longa distância Ainda que os termos de interação Ω (ldas ) não tenham qualquer efeito sobre a massa da espécie a, i.e., que ∑ ∫ m ( )Ω ( s as ) ld r dξ = 0 s (22) o processo de interação entre as partículas afeta a quantidade de movimento das partículas a. Deste modo, ∫m com a ( ps ) = r r (aa ) (as ) ⎞ (a ) ⎛ (aa ) (a ) (as ) (s ) ⎞ ⎜ Ω ld + ∑ Ω ld ⎟ξ dξ = n ⎜ a ∇n + ∑ a ∇n ⎟ s ≠a s ≠a ⎝ ⎠ ⎝ ⎠ (a ) ⎛ (23) 8πB ( ps ) . A equação acima traduz a variação da quantidade de movimento das partículas a sob o efeito 3σ ps3 dos processos de interação a-a e a-s, com s ≠ a. Da mesma forma, r ∑ ∫ m ( )ξ Ω ( p p ,s r dξ = ∑ n ( p )a ( ps )∇n (s ) ps ) ld p ,s (24) associado à variação da quantidade de movimento da mistura como um todo, em uma unidade de volume do espaço físico, devida aos processos de interação entre as moléculas e cujo efeito é, em geral, diferente de zero. Considere-se agora o efeito das forças atrativas sobre a energia cinética por unidade de volume da espécie a: 1 ∫2m r (aa ) (as ) ⎞ 2 (a ) r ⎛ (aa ) (a ) (as ) (s ) ⎞ ⎜ Ω ld + ∑ Ω ld ⎟ξ dξ = n ua .⎜ a ∇n + ∑ a ∇n ⎟ s ≠a s ≠a ⎝ ⎠ ⎝ ⎠ (a ) ⎛ (25) Da mesma forma, 1 ⎛ ∑ ∫ 2 m ( ) ⎜⎜ Ω ( p p ⎝ pp ) ld r r ⎞ + ∑ Ω (ldps ) ⎟⎟ξ 2dξ = ∑ a ( ps )n ( p )u p .∇n (s ) s≠p p ,s ⎠ (26) que constitui a variação da energia cinética dos componentes da mistura devida ao processo de interação intermolecular de longa distância. Da mesma forma como a quantidade de movimento, a energia cinética não é, portanto, conservada em misturas não-ideais, constituindo uma fonte de energia potencial intermolecular. EQUAÇÕES DE BALANÇO As equações macroscópicas de transporte podem ser obtidas como momentos cinéticos da equação de Boltzmann, Eq. (10). Conservação da massa Multiplicando a equação de Boltzmann por m(a) e integrando no espaço de velocidades r ∂ρ (a ) + ∇.(ρ (a )u (a ) ) = 0 ∂t (27) onde ρ (a ) = n (a ) m (a ) . Esta equação apresenta a mesma forma que a equação de conservação da massa para misturas ideais e pode ser reescrita na forma r r ∂ρ (a ) + ∇.(ρ (a )u ) = −∇. j (a ) ∂t (28) r r r onde j (a ) = ρ (a ) (u (a ) − u ) representa a densidade de fluxo difusivo da substância a. Observe-se, todavia, que para r misturas não ideais o fluxo j (a ) não é puramente difusivo e apresenta uma componente associada à força de atração intermolecular entre as moléculas a e as outras moléculas da mistura, incluindo as próprias moléculas a (Santos et al., [6]). Somando-se a Eq. (29) para todos os componentes da mistura r ∂ρ + ∇.(ρu ) = 0 ∂t (29) Equação de balanço da quantidade de movimento r r Multiplicando, agora a Eq. (10) por m (a )ξ (a ) , considerando a força externa como a gravitacional g e somando-se a equação resultante sobre todos os componentes, obtém-se ∂ (ρuα ) ∂ + .(ρuα u β + Pαβ ) = S (Π αβ ) ∂t ∂x β onde r Pαβ = ∑ ∫ m ( p ) (ξ α − uα )(ξ β − u β )f ( p )dξ (30) (31) p é o tensor pressão Pαβ = Pδ αβ + τ αβ . O termo r S (Π αβ ) = ρg + ∑ n ( p )a ( ps )∇n (s ) (32) p ,s constitui um termo fonte de quantidade de movimento, que é gerada pela ação gravitacional e pela ação de forças de atração intermoleculares. Ainda que este último termo não esteja presente em derivações clássicas da equação de balanço da quantidade de movimento, o seu efeito é não nulo, inclusive para substâncias puras em regiões onde ocorrem gradientes de densidade, quando r S (Π αβ ) = ρg + ρaM ∇ρ com a M = (33) aN av2 , onde N av é o número de Avogadro e M é a massa molecular. M2 Equação de balanço da energia cinética 1 (a ) 2 m ξ e somando-se a equação resultante sobre todos os componentes, obtém-se 2 a equação de balanço da energia cinética ρ ec da mistura que pode ser decomposta de acordo com Multiplicando-se a Eq. (10) por ρe c = 1 ρu 2 + ρe c ,f 2 (34) onde 1 ρu 2 constitui a energia cinética advectiva e 2 ρe c ,f = ∑ ∫ p ( ) 1 (p ) r r 2 (p ) r m ξ − u f dξ 2 (35) r é a energia cinética peculiar ou de flutuação da mistura com relação à velocidade u do centro de massa da mistura. Apresenta-se, neste trabalho, apenas a equação de balanço para a energia cinética peculiar da mistura, que é uma das componentes da energia interna termodinâmica, ∂ ( ρ ec , f ) ∂t r + ∇.ε c , f = S ( Ec , f ) (36) A densidade de fluxo r r r (37) r (38) ε c ,f = ε c ,f ,adv + ε c ,f ,dif onde r ε c ,f ,adv = e c ,f u é o fluxo advectivo e r ε c ,f ,dif = 1 ∑ ∫ 2m (ξ p β ( ) r r r 2 − u β ) ξ − u f ( p ) dξ p (39) corresponde ao fluxo difusivo de energia cinética peculiar. O termo fonte de energia cinética peculiar na Eq. (36) se escreve S (E c ,f ) = ∑ a p ,s ( ps ) r (s ) r j .∇n (s ) − P∇.u − τ αβ ∂ β uα (s ) m (40) r que compreende além da contribuições do trabalho de compressão - P∇.u e do termo de dissipação viscosa −τ αβ ∂ β u α , presentes em misturas ideais, um termo de conversão de energia cinética peculiar em energia potencial por ação dos potenciais de atração intermoleculares, representados por a ( ps ) . Energia potencial intermolecular r Para uma única molécula da espécie a na posição x , a energia potencial intermolecular devida à interação desta molécula com as moléculas que as rodeiam pode ser escrita como, Φ (as ) = −B (as ) r ∫ η >σ as r r r n (s ) (x + η )dη r6 η (41) r r Fazendo-se a expansão de n (s ) (x + η ) em séries de Taylor, apenas os termos de ordem par dão uma contribuição não nula e se nos limitarmos ao termo de ordem zero Φ (as ) = − 2 B (as ) (s ) r a (as ) (s ) r π 3 n (x ) = − n (x ) 3 σ as 4 (42) r Desse modo, a energia potencial intermolecular total correspondente a um volume unitário centrado em x se escreve r a ( ps ) ( p ) r (s ) r Φ (x ) = −∑ n (x )n (x ) 4 p,s (43) Para uma substância pura Ψ = N av a Φ a = −N av n = − VDW 4 n v (44) que corresponde à energia potencial para uma substância cuja equação de estado é a de van der Waals, onde, como N a usualmente, v = av é o volume molar, Ψ é a energia potencial molar e aVDW = N av2 . n 4 Para uma mistura binária de componentes a e b, Ψ = N av ( bb ) (ab ) ⎛ a (aa ) (a ) 2 Φ (x ) + 2 a x (a ) x (b ) + a (x (b ) )2 ⎞⎟⎟ = − aVDW ,mist = −N av n⎜⎜ n 4 4 v ⎝ 4 ⎠ (45) onde x (s ) corresponde à fração molar do componente s, (aa ) (ab ) (a ) (b ) (bb ) (x (a ) ) + 2aVDW (x (b ) ) aVDW ,mist = aVDW x x + aVDW 2 2 (46) Na expressão acima, as constantes de força de van der Waals estão relacionadas com os parâmetros de interação a ( ps ) 2 ( ps ) moleculares através de aVDW N av . = 4 Desse modo, a Eq. (46) pode ser considerada como uma regra de mistura, mas a sua derivação formal a partir da escala molecular possibilita esclarecer a natureza física dos parâmetros de van der Waals. Assim o presente (ab ) resultado associa o parâmetro cruzado aVDW ao parâmetro de força intermolecular a (ab ) . Este parâmetro é usualmente considerado heuristicamente como uma média geométrica dos parâmetros de força dos componentes individuais da mistura, multiplicado por um fator de interferência experimental, [1]. O que se mostra aqui é que essa constante de interferência está diretamente relacionada com um parâmetro de força intermolecular entre moléculas de espécies diferentes, possível de ser avaliado a partir de propriedades moleculares elementares. A equação de balanço para a energia potencial se escreve r ∂Φ + ∇.(Φu ) = S (Φ ) ∂t (47) onde o termo fonte de energia potencial é dada pela taxa de variação da energia cinética devida à interação intermolecular , Eq. (26), com o sinal oposto, S (Φ ) = −∑ a ( ps ) n ( p )u ( p ) ∇n (s ) p ,s (48) Energia interna termodinâmica A energia interna termodinâmica é a soma da energia cinética peculiar Eq. (35) e a energia potencial intermolecular, Eq. (43), ρe = ρe c ,f + Φ No equilíbrio, a distribuição de velocidades das moléculas é uma distribuição de Maxwell-Boltzmann (49) f (p ) m (p ) = n ⎜⎜ ⎝ 2πkT (p ) ⎛ ⎞ ⎟⎟ ⎠ D/2 ( ⎛ m (p ) r r exp⎜⎜ − ξ −u ⎝ 2kT ) ⎞⎟⎟ 2 (50) ⎠ e, da Eq. (35), ρe c ,f = D nkT 2 (51) Influência das forças intermoleculares no processo de difusão de espécies em misturas binárias Sem perda de generalidade a influência das forças intermoleculares no processo de difusão de espécies será analisada, apenas, para misturas binárias em problemas isotérmicos. Para o presente caso, com os termos de interação de longa-distância dados pela Eq. (20) e usando um modelo com um único tempo de relaxação para os termos de interação de curta-distância, uma análise de Chapman-Enskog da Eq. (10) mostra que, para misturas nãoideais, a densidade de fluxo de massa de um componente a genérico da mistura pode ser escrita como n (a )m (a ) ∇n n m (a ) + n (b )m (b ) ⎛ n (b )m (b ) n (a )m (a ) (aa ) (a ) (a ) (bb ) (b ) (b ) ⎞ ⎟ + τ m (a ) (a ) + ⎜⎜ − τ m (a ) (a ) ( b ) ( b ) a n ∇n ⎟ ( b ) ( b ) a n ∇n + n m +n m n m n m ⎝ ⎠ r (a ) j = −τ m kT∇n (a ) + τ m kT + τm (a ) ⎛ n (b )m (b ) n (a )m (a ) (ab ) (a ) (b ) (ab ) (b ) (a ) ⎞ ⎟ + ⎜⎜ − τ m (a ) (a ) ( b ) ( b ) a n ∇n ⎟ (a ) ( b ) ( b ) a n ∇n + n m n m n m +n m ⎝ ⎠ (a ) (52) onde r (a ) j ideal = −τ m kT∇n (a ) + τ m kT n (a ) m (a ) ∇n n m (a ) + n ( b ) m ( b ) (a ) (53) é o fluxo difusivo de massa do componente a para misturas de gases ideais, quando as moléculas não se atraem entre si com forças de origem eletrostática. O parâmetro τ m é um tempo de relaxação, associado à difusividade binária D ab , i.e., ao processo de interação de curta-distância, ou colisão, entre as moléculas a e b. O termo r (a ) j ld ,sep = τ m ⎛ n (b )m (b ) n (a )m (a ) (aa ) (a ) (a ) (bb ) ( b ) (b ) ⎞ ⎟ + ⎜⎜ − τ m (a ) (a ) (a ) ( b ) ( b ) a n ∇n ( b ) ( b ) a n ∇n ⎟ n m +n m n m +n m ⎝ ⎠ (a ) (54) corresponde ao fluxo de massa da espécie a associado às forças atrativas a-a e b-b e produzem um efeito de separação entre as espécies a e b. O termo r (a ) j ld ,mist = τ m ⎛ n (b )m (b ) n (a )m (a ) (ab ) (a ) (b ) (ab ) (b ) (a ) ⎞ ⎜ ⎟ a n n τ ∇ + − m (a ) (a ) (b ) (b ) (a ) (a ) ( b ) ( b ) a n ∇n ⎟ ⎜ n m +n m n m +n m ⎝ ⎠ (55) corresponde ao fluxo de massa da espécie a associado à interação intermolecular entre as moléculas a e as moléculas b e tende a misturar os componentes entre si. Mostra-se, também, com a análise de Chapman-Enskog, que a pressão na mistura é dada pela equação de van der Waals, P = n m RT − aVDW v m2 (56) com nm=n/N , v m = vN , com aVDW dado pela Eq. (46). Ainda que não previsto na teoria clássica do transporte para misturas, o termo dado pela Eq. (54) é de grande importância para misturas não-ideais, sendo o responsável, e.g., pelo processo de separação entre dois líquidos imiscíveis e pela estabilização da interface de separação em processos de coalescência, [6]. Desse modo, para que r ) associado ao processo de dois líquidos a e b não se misturem, quando em contato, é necessário que o termo j ld(a,sep atração de longa distancia entre as moléculas da mesma espécie, equilibre o efeito conjugado do termo dispersivo, r ) , dado pela Eq. (55). Estes dois últimos efeitos Eq. (53), e do termo de atração entre as moléculas a e b, j ld(a,mist tendem a misturar os componentes da mistura entre si. A Fig. 1 apresenta uma representação para os perfis de concentração das espécies n(a) e n(b) e n através de uma r interface de separação plana e estacionária entre dois fluidos imiscíveis, soluções das equações j x( a ) = 0 e r(b) j x = 0 em condições temperatura e pressão constantes, onde x é a coordenada normal à interface, [6]. Quando as forças de atração intermolecular permitem o equilíbrio de fluxos, as concentrações n(a) e n(b) adaptam-se de acordo com a temperatura e pressão e de acordo com os valores do parâmetro de difusão τ m , e dos parâmetros de força a (aa ) , a (bb ) e a (ab ) . Fig.1: Perfis de concentração numa interface plana estacionária, separando dois fluidos imiscíveis. As concentrações das espécies a e b são indicadas pelos níveis de cinza. Ainda que a pressão mantém-se constante através da interface, a concentração volumétrica total n sofre uma queda, devida aos processos de interação intermolecular, [6]. CONCLUSÃO A teoria do transporte para misturas não-ideais constitui ainda um campo aberto, especialmente quando os seus componentes se encontram na fase líquida, quando os processos de interação intermolecular são importantes e modificam não somente as relações entre as variáveis termodinâmicas de equilíbrio, mas também as relações entre fluxos e potenciais. No que se refere aos aspectos puramente termodinâmicos, o presente trabalho mostrou que quando a mistura é considerada como um sistema de pontos materiais regidos pelas leis de Newton e sujeitos a processos de interação intermolecular de longa distância, a sua equação de estado pode ser obtida formalmente como a equação de van der Waals, com uma regra de misturas quadrática. No que se refere à influência do volume das moléculas, o modelo de Enskog para gases densos pode ser utilizado para introduzir a correção necessária na Eq. (56), levando-se em conta que duas moléculas não podem ocupar a mesma posição no espaço. Essa correção é bastante conhecida na literatura e pode ser encontrada em [7]. A formação de pontes de hidrogênio entre as moléculas constituintes da mistura contribui para aumentar a energia potencial média entre dipolos permanentes, Eq. (1), uma vez que privilegia certas orientações, afastando a distribuição P (Φ (12 ) ) de uma distribuição de Boltzmann. Esse efeito é dependente da temperatura e um estudo detalhado neste sentido poderia explicar a dependência do parâmetro de interação, a, com a temperatura. Da mesma forma, como as moléculas não são pontos materiais, a relação a(T) deve depender da forma, i.e., dos momentos de inércia das moléculas, o que justificaria a introdução de um fator acêntrico em grande parte das equações de estado obtidas empiricamente. O presente trabalho mostrou, também, que a origem da condição de miscibilidade ou imiscibilidade entre dois líquidos reside nas forças intermoleculares de longa distância entre as moléculas da mesma espécie e de espécies diferentes. Um aumento da temperatura tende a aumentar os efeitos de dispersão, Eq. (53), e reduzir os efeitos de atração eletrostática entre as moléculas, contribuindo para que estes dois líquidos se misturem. Uma redução da temperatura aumenta a importância das forças eletrostáticas de separação, Eq. (54). REFERÊNCIAS 1. J. M. Prausnitz, R. N. Lichtenthaler, E.G. Azevedo, Molecular thermodynamics of fluid-phase equilibria, Prentice-Hall, Englewood Cliffs (1986). 2. C. Cercignani, Mathematical methods in kinetic theory, Plenum Press, New York (1990). 3. X. He e G. D. Doolen, Thermodynamic foundations of kinetic theory and lattice Boltzmann models for multiphase flows, J. Stat. Phys., 107, 309-328 (2002). 4. L. Boltzmann, Lectures on Gas Theory, Dover Publications, Reprint edition (1995) 5. L. O. E. dos Santos, P. C. Philippi, Lattice-gas model based on field mediators for immiscible fluids, Phys. Rev. E 65 (4): art. no. 046305 (2002) 6. L. O. E. Santos, P. C. Facin, e P. C. Philippi, Lattice-Boltzmann model based on field mediators for immiscible fluids, Phys. Rev. E 68, 056302 (2003) 7. S. Chapman e T.G. Cowling, The mathematical theory of non-uniform gases, Cambridge University Press, Cambridge (1970). NOMENCLATURA a(ab),B(ab) ec ec,f e r F f(N) f(1,a) f(2,ab) f(a) r g I r j k M m n nm N P Pαβ r q r r T r u v=V/n vm=V/nm parâmetros de interação molecular energia cinética total por unidade de massa energia cinética peculiar por unidade de massa energia interna termodinâmica por unidade de massa Força por unidade de volume probabilidade conjunta definida pela Eq. (6) α,β,γ αp r εc r ε c ,f densidade de fluxo de energia cinética densidade de fluxo de energia cinética peculiar probabilidade marginal definida pela Eq. (9) probabilidade a duas partículas definidas abaixo da Eq. (10) função distribuição de partículas aceleração gravitacional εo σas permissividade elétrica do vácuo diâmetro da esfera de influência entre as partículas a e s termo de interação energia potencial intermolecular potencial de ionização ξ μ densidade de fluxo de massa constante de Boltzmann massa molecular massa da molécula número de partículas por unidade de volume número de moles por unidade de volume número de Avogrado pressão termodinâmica tensor pressão densidade de fluxo de calor vetor distância intermolecular temperatura absoluta velocidade macroscópica volume molecular volume molar ξ índices para componente de vetores polarizibilidade molecular velocidade molecular r força por unidade de massa das moléculas χ r Ω Φ r Παβ ρ τ ταβ velocidade molecular momento dipolar molecular tensor tensão massa específica tempo de relaxação tensor tensão viscosa
Download