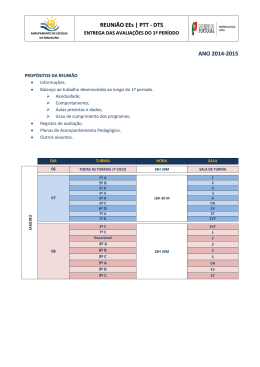
Universidade Federal de Goiás Faculdade de Farmácia RODRIGO BORGES DE OLIVEIRA AVALIAÇÃO DAS ATIVIDADES ANTINOCICEPTIVA, ANTIINFLAMATÓRIA E DEPRESSORA DO SISTEMA NERVOSO CENTRAL DAS FOLHAS DO Synadenium umbellatum PAX. (COLA-NOTA) Goiânia 2007 Livros Grátis http://www.livrosgratis.com.br Milhares de livros grátis para download. RODRIGO BORGES DE OLIVEIRA AVALIAÇÃO DAS ATIVIDADES ANTINOCICEPTIVA, ANTIINFLAMATÓRIA E DEPRESSORA DO SISTEMA NERVOSO CENTRAL DAS FOLHAS DO Synadenium umbellatum PAX. (COLA-NOTA) Dissertação apresentada no Curso de Mestrado em Ciências Farmacêuticas da Faculdade de Farmácia da Universidade Federal de Goiás, como requisito parcial para a obtenção do Título de Mestre em Ciências Farmacêuticas. Área de concentração: Fármacos Medicamentos Orientador: Prof. Dr. Luiz Carlos da Cunha Co-Orientador: Prof. Dr. Elson Alves Costa Goiânia 2007 e Aos meus pais, Onofre e Silvia, pela amizade, dedicação, incentivo, apoio, companheirismo, paciência e confiança. carinho, amor, compreensão, AGRADECIMENTOS • Ao meu orientador, Prof. Dr. Luiz Carlos da Cunha, e ao meu co-orientador, Prof. Dr. Elson Alves Costa, por toda ajuda e conhecimento dados a mim e pela paciência em me atender quando precisei. • A todos os colegas de laboratório, tanto do NEPET (núcleo de estudos e pesquisas tóxico-farmacológicas) quando do Laboratório de Farmacologia de Produtos Naturais. • Ao aluno de iniciação científica Marcus Vinícius Mariano Nascimento, por toda a ajuda técnica durante a realização dos experimentos. • A todos os colegas do Programa de Pós-Graduação em Ciências Farmacêuticas da Faculdade de Farmácia da Universidade Federal de Goiás, pela amizade, companheirismo, apoio e incentivo recebido. • À Universidade Federal de Goiás, pela bolsa concedida durante a realização deste trabalho. • Aos técnicos do NEPET e do Laboratório de Farmacologia de Produtos Naturais, por toda ajuda e aprendizado técnico. • A todos os professores do Programa de Pós-Graduação em Ciências Farmacêuticas da Faculdade de Farmácia da Universidade Federal de Goiás, que de alguma forma puderam contribuir para a realização deste trabalho. “Isto não é farmacologia. Isto é arte.” Paul Schimmel RESUMO O Synadenium umbellatum Pax. (Euphorbiacea), conhecido como “cola-nota”, “avelós”, “cancerola”, “milagrosa”, etc. é uma planta utilizada pela população Brasileira para o tratamento da inflamação, da dor, dentre outros. O extrato etanólico das folhas de Synadenium umbellatum (EES) e suas frações - frações hexânica (FH), clorofórmica (FC) e metanol/água (FM) - foram testados para confirmar o seu uso popular como analgésico e antiinflamatório e, também, baseado em observações comportamentais, foi feito um estudo da atividade depressora do sistema nervoso central (SNC). Para tal, vários modelos foram utilizados, dentre eles, o de contorção abdominal induzida por ácido acético, dor induzida pela formalina, influência do tratamento com naloxona, teste do tail flick, edema de orelha induzido por óleo de cróton, peritonite induzido por carragenina, teste do rota-rod, campo aberto e sono induzido por barbitúrico. O EES foi testado, pela via oral, nas doses de 25, 50 e 100 mg/kg, enquanto que a FH foi testada na dose de 10 mg/kg, a FC na dose de 20 mg/kg e a FM nas doses de 6, 12 e 25 mg/kg. O EES e a FM foram capazes de inibir o número contorções abdominais induzidas por ácido acético, mas não as frações FC e FH. O EES foi capaz de inibir ambas as fases da dor induzida pela formalina, além de ter revertido o efeito do tratamento com a naloxona, porém não foi capaz de prolongar o tempo de latência dos camundongos frente a um estímulo térmico. Esses resultados mostram que o EES possui atividade antinociceptiva e ainda sugerem que a ação do mesmo é no sistema opioidérgico periférico, ou como um agonista ou por induzir a liberação de peptídeos opióides periféricos. O EES e a FM possuem atividade antiinflamatória visto por suas capacidades de reduzir o edema de orelha induzido por óleo de cróton e o número de leucócitos totais migrados para a cavidade intraperitoneal. O EES e as frações FH e FC, mas não a FM, apresentaram um possível efeito depressor do SNC visto que foram capazes de aumentar o tempo parado e diminuir o número de bolos fecais no teste do campo aberto, além de potencializarem o sono induzido por barbitúrico. O teste do rota-rod mostrou que o EES e as frações não foram capazes de causar incoordenação motora ou relaxamento muscular. Nossos resultados mostram que, através do fracionamento do EES, os efeitos analgésicos e antiinflamatórios foram separados do possível efeito depressor do SNC, visto que o primeiro foi encontrado na FM enquanto o segundo foi encontrado nas frações FH e FC. O isolamento dos componentes do extrato é uma etapa a ser feita no futuro, visando separar ainda mais os efeitos e identificar os princípios ativos responsáveis pelos mesmos, além de permitir o estudo dos mecanismos de ação envolvidos nas atividades antinociceptiva, antiinflamatória e depressora do sistema nervoso central das folhas do Synadenium umbellatum. ABSTRACT Synadenium umbellatum Pax. (Euphorbiacea), known as “cola-nota”, “avelós”, “cancerola”, “milagrosa”, etc. is a plant used by Brazilian folks for the treatment of inflammation, pain, among others. The ethanolic extract of the leaves of Synadenium umbellatum (EES) and its fractions – hexane (HF), chloroformic (CF) and methanol/water fractions (MF) – were tested due to confirm its popular use as analgesic and antiinflammary and, also, based on behavioral observations, a study of the depressor activity over the central nervous system (CNS) was taken. For such, several models were used, among them, the acetic acid-induced writhing, formalininduced pain, naloxone treatment influence, tail flick test, croton oil-induced mouse ear edema, carraginin-induced peritonits, rota-rod test, open field and barbiturateinduced sleep. The EES was tested in the oral doses of 25, 50 and 100 mg/kg, while the HF was tested in the dose of 10 mg/kg, the CF in the dose of 20 mg/kg and the MF in the doses of 6, 12 and 25 mg/kg. The EES and the MF were able to inhibit the number of acetic acid-induced writhing, but not the CF and HF fractions. The EES was able to inhibit both phases of the formalin-induced pain, besides having reverted the naloxone treatment effect, but it wasn’t able to prolong mice latency time when getting thermal stimuli. These results show that EES has an antinociceptive activity and also suggests that its action is in the peripheral opioidergic system, as an agonist or by inducing the release of peripheral opioid peptides. The EES and the MF have demonstrated an antiinflammatory activity, due to their capacities of reducing the croton oil-induced ear edema and the number of total leukocytes migrated into the peritoneal cavity. The EES and the HF and CF fractions, but not the MF, have presented a possible depressor effect over the CNS once they were able to increase the stopped time and the number of fecal balls on the open field test, besides they were able to potencialize the barbiturate-induced sleep. The rota-rod test showed that the EES and the fractions weren’t able to cause motor incoordination or muscle relaxing. Our results show that, through the EES fractionment, the antinociceptive and antiinflammatory effects were separated from the possible depressor effect over the CNS, once the first one was demonstrated in MF and the second one was demonstrated in HF and CF fractions. The isolation of the compounds in the extract is a next step to be done in the future, due to separate still more the effects and identify the active substances responsible for them, besides allowing the study of the action mechanisms involved in the antinociceptive, antiinflamatory and CNS depressor activities of the leaves of Synadenium umbellatum. LISTA DE FIGURAS Figura 1 Figura 2 Figura 3 Figura 4 Figura 5 Figura 6 Fotografia da árvore do Synadenium umbellatum Pax. (cola-nota) no bairro Feliz, em Goiânia – GO. Foto tirada pelo Prof. Dr. Luiz Carlos da Cunha em novembro de 2005........................................... 36 Fluxograma do procedimento geral para a extração de clorofila e fracionamento do extrato etanólico das folhas de S. umbellatum......................................................................................... 44 Contorções abdominais induzidas pelo ácido acético (1,2 % v/v em salina, i.p.) durante 30 minutos em camundongos previamente tratados (60 min) pela via p.o. com veículo (grupo controle; C), com o extrato etanólico das folhas do S. umbellatum (EES; 25, 50 ou 100 mg/kg) ou com indometacina (INDO; 10 mg/kg). As colunas e barras verticais representam as médias ± EPM de 6 animais por grupo experimental............................................................................. 51 Contorções abdominais induzidas pelo ácido acético (1,2 % v/v em salina, i.p.) durante 30 minutos em camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com as frações hexânica (FH, 10 mg/kg), clorofórmica (FC, 20 mg/kg) e metanol/água (FM, 25 mg/kg) ou com indometacina (INDO; 10 mg/kg). As colunas e barras verticais representam as médias ± EPM de 6 animais por grupo experimental........................................ 52 Porcentagem de inibição das contorções abdominais induzidas pelo ácido acético (1,2 % v/v em salina, i.p.) durante 30 minutos em camundongos previamente tratados (60 min) pela via p.o. com EES (■) 25, 50 ou 100 mg/kg. Os símbolos e barras verticais representam as médias ± DP de 6 animais por grupo experimental....................................................................................... 53 Reatividade à aplicação intraplantar de formalina (20 µL, 3%) na pata posterior direita de camundongos, durante a primeira fase (0 – 5 min) do teste da formalina, previamente tratados (60 min) pela via p.o. com veículo (grupo controle, C; n = 10), com o EES (100 mg/kg, n = 10), com morfina (MOR; 10 mg/kg s.c., n = 6) ou com indometacina (INDO; 10 mg/kg, n = 6). As colunas e barras verticais representam as médias ± EPM............................................ 55 Figura 7 Figura 8 Figura 9 Figura 10 Figura 11 Figura 12 Reatividade à aplicação intraplantar de formalina (20 µL, 3%) na pata posterior direita de camundongos, durante a segunda fase (15 – 30 min) do teste da formalina, previamente tratados (60 min) pela via p.o. com veículo (grupo controle, C; n = 10), com o EES (100 mg/kg, n = 10), com morfina (MOR; 10 mg/kg s.c., n = 6) ou com indometacina (INDO; 10 mg/kg, n = 6). As colunas e barras verticais representam as médias ± EPM............................................ 56 Influência do tratamento prévio com o antagonista opióide não seletivo naloxona (3 mg/kg, s.c.) sobre a atividade antinociceptiva do EES (100 mg/kg, p.o.) e fentanil (100 µg/kg, s.c.) no modelo de contorções abdominais induzidas pelo ácido acético. As colunas e barras verticais representam a média ± EPM de 6 animais por grupo experimental............................................................................. 57 Latência ao estímulo térmico nociceptivo medido no teste do tail flick em camundongos antes e após tratamento com o (●) veículo p.o., com EES (■ 25, ▼50 ou ▲100 mg/kg, p.o.) ou morfina (♦ 10 mg/kg, s.c.). Nas ordenadas estão representados os tempos de latência dos camundongos ao estímulo térmico nociceptivo, em segundos. Os símbolos e barras verticais representam as médias ± EPM de 6 animais por grupo experimental........................................ 58 Edema de orelha, em mg, induzido por óleo de cróton (2,5% v/v em acetona) nos grupos previamente tratados pela via p.o. com veículo (C; n = 7), com extrato etanólico de S. umbellatum (EES 25, 50 ou 100 mg/kg, n = 7) ou com dexametasona (DEXA; 2 mg/kg, n = 6). As colunas e barras verticais representam a média ± EPM.................................................................................................... 60 Edema de orelha, em mg, induzido por óleo de cróton (2,5% v/v em acetona) nos grupos previamente tratados pela via p.o. com veículo (C; n = 7), com fração metanol/água (FM 6, 12 ou 25 mg/kg, n = 8, 8 e 7 respectivamente) ou com dexametasona (DEXA; 2 mg/kg, n = 6). As colunas e barras verticais representam a média ± EPM................................................................................... 61 Migração de leucócitos totais no modelo de peritonite induzida por carragenina (1% m/v) injetada na cavidade intraperitoneal de camundongos previamente tratados pela via p.o. com veículo (C; n = 10), com extrato etanólico de S. umbellatum (EES 25, 50 ou 100 mg/kg, n = 8, 9 e 8 respectivamente) ou dexametasona (DEXA; 2 mg/kg, n = 8). As colunas e barras verticais representam a média ± EPM.................................................................................................... 62 Figura 13 Número de quedas, no rota-rod, de camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com o extrato etanólico das folhas do S. umbellatum (EES; 25, 50 ou 100 mg/kg) ou com diazepam (DZP; 5 mg/kg). As colunas e barras verticais representam as médias ± EPM de 7 animais por grupo experimental....................................................................................... 65 Figura 14 - Número de quedas, no rota-rod, de camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com as frações hexânica (FH, 10 mg/kg), clorofórmica (FC, 20 mg/kg), metanol/água (FM, 25 mg/kg) ou com diazepam (DZP; 5 mg/kg). As colunas e barras verticais representam as médias ± EPM de 7 animais por grupo experimental......................................................... 66 Figura 15 Figura 16 Figura 17 Figura 18 Figura 19 Tempo de permanência, no rota-rod, em segundos, de camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com o extrato etanólico das folhas do S. umbellatum (EES; 25, 50 ou 100 mg/kg) ou com diazepam (DZP; 5 mg/kg). As colunas e barras verticais representam as médias ± EPM de 7 animais por grupo experimental......................................................... 67 Tempo de permanência, no rota-rod, em segundos, de camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com as frações hexânica (FH, 10 mg/kg), clorofórmica (FC, 20 mg/kg), metanol/água (FM, 25 mg/kg) ou com diazepam (DZP; 5 mg/kg). As colunas e barras verticais representam as médias ± EPM de 7 animais por grupo experimental......................... 68 Tempo parado, em segundos, no teste do campo aberto, de camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com o extrato etanólico das folhas do S. umbellatum (EES; 25, 50 ou 100 mg/kg) ou com diazepam (DZP; 5 mg/kg). As colunas e barras verticais representam as médias ± EPM de 8 animais por grupo experimental......................................................... 69 Tempo parado, em segundos, no teste do campo aberto, de camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com as frações hexânica (FH, 10 mg/kg), clorofórmica (FC, 20 mg/kg), metanol/água (FM, 25 mg/kg) ou com diazepam (DZP; 5 mg/kg). As colunas e barras verticais representam as médias ± EPM de 8 animais por grupo experimental......................... 70 Número de bolos fecais, no teste do campo aberto, deixados por camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com o extrato etanólico das folhas do S. umbellatum (EES; 25, 50 ou 100 mg/kg) ou com diazepam (DZP; 5 mg/kg). As colunas e barras verticais representam as médias ± EPM de 8 animais por grupo experimental......................................................... 71 Figura 20 Figura 21 Figura 22 Número de bolos fecais, no teste do campo aberto, deixados por camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com as frações hexânica (FH, 10 mg/kg), clorofórmica (FC, 20 mg/kg), metanol/água (FM, 25 mg/kg) ou com diazepam (DZP; 5 mg/kg). As colunas e barras verticais representam as médias ± EPM de 8 animais por grupo experimental......................... 72 Tempo de recuperação do reflexo postural (duração do sono), em minutos, no teste de potenciação do sono induzido por barbitúrico, em camundongos previamente tratados (60 min) pela via p.o. com veículo (C; n=6), com o extrato etanólico das folhas do S. umbellatum (EES; 25, 50 ou 100 mg/kg, n=8) ou diazepam (DZP; 5 mg/kg, n=6). As colunas e barras verticais representam as médias ± EPM................................................................................................. 73 Tempo de recuperação do reflexo postural (duração do sono), em minutos, no teste de potenciação de sono induzido por barbitúrico, em camundongos previamente tratados (60 min) pela via p.o. com veículo (C; n = 6), com as frações hexânica (FH, 10 mg/kg, n = 8), clorofórmica (FC, 20 mg/kg, n = 7), metanol/água (FM, 25 mg/kg, n = 7) ou diazepam (DZP; 5 mg/kg, n = 6). As colunas e barras verticais representam as médias ± EPM............................................ 74 LISTA DE TABELAS Tabela 1 Contorções abdominais induzidas pelo ácido acético (1,2 % v/v em salina, i.p.) durante 30 minutos em camundongos previamente tratados (60 min) pela via p.o. com veículo (grupo controle; C), com o extrato etanólico das folhas do S. umbellatum (EES; 25, 50 ou 100 mg/kg) (A), com as frações hexânica (FH, 10 mg/kg), clorofórmica (FC, 20 mg/kg) e metanol/água (FM, 25 mg/kg) (B) ou com indometacina (INDO; 10 mg/kg)................................................. 121 Tabela 2 Reatividade à aplicação intraplantar de formalina (20 µL, 3%) na pata posterior direita de camundongos, durante a primeira fase (0 – 5 min) (A) e segunda fase (15 – 30 min) (B) do teste da formalina, previamente tratados (60 min) pela via p.o. com veículo (grupo controle, C), com o EES (100 mg/kg), com morfina (MOR; 10 mg/kg s.c.) ou com indometacina (INDO; 10 mg/kg)......................... 122 Tabela 3 Influência do tratamento prévio com o antagonista opióide não seletivo naloxona (3 mg/kg, s.c.) sobre a atividade antinociceptiva do EES (100 mg/kg, p.o.) e fentanil (100 µg/kg, s.c.) no modelo de contorções abdominais induzidas pelo ácido acético........................ 124 Tabela 4 Latência ao estímulo térmico nociceptivo medido no teste do tail flick em camundongos antes e após tratamento por via p.o. com o veículo (A), com EES 25 (B), 50 (C) ou 100 (D) mg/kg, ou morfina (E) (10 mg/kg, s.c.)............................................................................. 125 Tabela 5 Edema de orelha, em mg, induzido por óleo de cróton (2,5% v/v em acetona) nos grupos previamente tratados pela via p.o. com veículo (C), com extrato etanólico de S. umbellatum (EES 25, 50 ou 100 mg/kg) (A), com fração metanol/água (FM 6, 12 ou 25 mg/kg) (B) ou com dexametasona (DEXA; 2 mg/kg)......................... 127 Tabela 6 Migração de leucócitos totais no modelo de peritonite induzida por carragenina (1% m/v) injetada na cavidade intraperitoneal de camundongos previamente tratados pela via p.o. com veículo (C), com extrato etanólico de S. umbellatum (EES 25, 50 ou 100 mg/kg) ou dexametasona (DEXA; 2 mg/kg)................................................... 128 Tabela 7 Número de quedas (A) e tempo de permanência (B) no rota-rod, de camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com o extrato etanólico das folhas do S. umbellatum (EES; 25, 50 ou 100 mg/kg) (1) com as frações hexânica (FH, 10 mg/kg), clorofórmica (FC, 20 mg/kg), metanol/água (FM, 25 mg/kg) (2) ou com diazepam (DZP; 5 mg/kg)................................................ 129 Tabela 8 Número de quadrados invadidos, tempo parado (segundos), número de levantadas, número de auto-limpeza e número de bolos fecais no campo aberto 60 minutos após o tratamento p.o. com veículo (C) (A), extrato etanólico de S. umbellatum (EES 25 (B), 50 (C) ou 100 mg/kg (D)), FH (10 mg/kg) (E), FC (20 mg/kg) (F), FM (25 mg/kg) (G) ou diazepam (DZP, 5 mg/kg) (H)...................................................................................................... 131 Tabela 9 Tempo de recuperação do reflexo postural (duração do sono), em minutos, no teste de potenciação do sono induzido por barbitúrico, em camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com o extrato etanólico das folhas do S. umbellatum (EES; 25, 50 ou 100 mg/kg) (A), com as frações hexânica (FH, 10 mg/kg), clorofórmica (FC, 20 mg/kg), metanol/água (FM, 25 mg/kg) (B) ou diazepam (DZP; 5 mg/kg)....................................................... 135 LISTA DE ABREVIATURAS E SIGLAS 5-HT 5-hidroxitriptamina 5-HT1 5-hidroxitriptamina 1 5-HT2 5-hidroxitriptamina 2 5-HT3 5-hidroxitriptamina 3 5-HT4 5-hidroxitriptamina 4 5-HT7 5-hidroxitriptamina 7 5-LOX 5-Lipoxigenase AA Ácido araquidônico AINE(S) Antiinflamatório(s) não-esteroidal(is) AMPc 3’5’-adenosina-monofosfato cíclico ANOVA Análise de variância C Controle (Veículo) CGRP Peptídeo relacionado ao gene de calcitocina COX Cicloxigenase COX-1 Cicloxigenase-1 COX-2 Cicloxigenase-2 CRF Fator liberador de corticotrofina DEXA Dexametasona DI50 Dose inibitória mediana DNAc Ácido desoxirribonucléico complementar DP Desvio padrão DZP Diazepam EES Extrato etanólico de Synadenium umbellatum eNOS Óxido nítrico sintase endotelial EPM Erro padrão da média FC Fração clorofórmica FH Fração hexânica FM Fração metanol/água g Grama(s) GABA Ácido gama aminobutírico GABAA Ácido gama aminobutírico A GABAB Ácido gama aminobutírico B GABAB Ácido gama aminobutírico C GMPc Monofosfato cíclico de guanosina GPCR Receptor acoplado a proteína G h Hora(s) HETE Ácido hidroeicosatetraenóico HPA Hipotálamo-hipófise-adrenal i.p. Via intraperitoneal IB4 Isolectina 4 IFN-γ Interferon-γ IL-1 Interleucina 1 IL-1β Interneucina 1β IL-8 Interleucina 8 INDO Indometacina iNOS Óxido nítrico sintase induzível IQUEGO Indústria Química do Estado de Goiás kg Kilograma(s) L Litro(s) LOX Lipoxigenase LPS Lipopolissacarídeo LT Leucotrieno LTB4 Leucotrieno B4 m/v Massa/volume MAO Monoaminaoxidase mg Miligrama(s) min Minuto(s) mL Mililitro(s) mm Milímetro(s) MOR Morfina NADPH Forma reduzida do fosfato dinucleotídeo NMDA N-metil-d-aspartato nNOS Óxido nítrico sintase neuronal NO Óxido nítrico (nitric oxide) de nicotinamida adenina NOS Óxido nítrico sintase P.A. Padrão analítico p.o. Via oral PAF Fator de agregação plaquetária PBS PG Solução salina tamponada (Phosphate Buffered Saline) Prostaglandina PGD2 Prostaglandina D2 PGE2 Prostaglandina E2 PGF2α Prostaglandina F2α PGG2 Prostaglandina G2 PGH2 Prostaglandina H2 PGI2 Prostaglandina I2 pH Potencial hidrogeniônico PL Fosfolipase PLA2 Fosfolipase A2 RIP Proteína inativadora de ribossomos RNAm Ácido ribonucléico mensageiro s Segundo(s) S. umbellatum Synadenium umbellatum s.c. Via subcutânea SNC Sistema nervoso central SP Substância P TNF Fator de necrose tumoral TNF-α Fator de necrose tumoral α TRPV1 Receptor vanilóide de potencial transitório 1 UFG Universidade Federal de Goiás UI Unidades internacionais v/v Volume/volume µg Micrograma(s) µL Microlitro(s) SUMÁRIO RESUMO.....................................................................................................................6 ABSTRACT .................................................................................................................7 LISTA DE FIGURAS ...................................................................................................8 LISTA DE TABELAS .................................................................................................12 LISTA DE ABREVIATURAS E SIGLAS ....................................................................14 1. INTRODUÇÃO ......................................................................................................19 1.1. Fisiopatologia da dor.......................................................................................19 1.2. Fisiopatologia da inflamação ..........................................................................23 1.3. Fisiopatologia da depressão do sistema nervoso central ...............................30 1.4. Os produtos naturais e o Synadenium umbellatum ........................................33 2. OBJETIVOS ..........................................................................................................38 3. MATERIAIS ...........................................................................................................40 3.1. MATERIAL BOTÂNICO ..................................................................................40 3.1.1. Coleta, identificação e herborização da planta.........................................40 3.2. MATERIAL QUÍMICO .....................................................................................40 3.2.1. Extrato, frações e medicamentos .............................................................40 3.2.2. Reagentes................................................................................................40 3.3. ANIMAIS .........................................................................................................41 4. MÉTODOS ............................................................................................................43 4.1. MÉTODOS FITOQUÍMICOS ..........................................................................43 4.1.1.Preparação do extrato etanólico ...............................................................43 4.1.2. Eliminação de clorofila e fracionamento do extrato etanólico...................43 4.2. MÉTODOS FARMACOLÓGICOS...................................................................45 4.2.1. Atividade antinociceptiva..........................................................................45 4.2.1.1. Contorções abdominais induzidas por ácido acético .........................45 4.2.1.2. Dor induzida pela formalina ...............................................................45 4.2.1.3. Influência do tratamento com naloxona .............................................46 4.2.1.4. Teste do tail flick ................................................................................46 4.2.2. Atividade antiinflamatória .........................................................................46 4.2.2.1. Edema de orelha induzido por óleo de cróton ...................................46 4.2.2.2. Peritonite induzida por carragenina ...................................................47 4.2.3. Atividade no sistema nervoso central.......................................................47 4.2.3.1. Teste do rota-rod ...............................................................................47 4.2.3.2. Teste do campo aberto ......................................................................47 4.2.3.3. Potenciação do sono por barbitúrico .................................................48 4.3. ANÁLISE ESTATÍSTICA.................................................................................48 5. RESULTADOS ......................................................................................................50 5.1. RESULTADOS FITOQUÍMICOS ....................................................................50 5.1.1.Preparação do extrato etanólico ...............................................................50 5.1.2. Eliminação de clorofila e fracionamento do extrato etanólico...................50 5.2. RESULTADOS FARMACOLÓGICOS ............................................................50 5.2.1. Atividade antinociceptiva..........................................................................50 5.2.1.1. Contorções abdominais induzidas por ácido acético .........................50 5.2.1.2 Dor induzida pela formalina ................................................................54 5.2.1.3. Influência do tratamento com naloxona .............................................54 5.2.1.4. Teste do tail flick ................................................................................58 5.2.2. Atividade antiinflamatória .........................................................................59 5..2.2.1. Edema de orelha induzido por óleo de cróton ..................................59 5.2.2.2. Peritonite induzida por carragenina ...................................................59 5.2.3. Atividade no sistema nervoso central.......................................................63 5.2.3.1. Teste do rota-rod ...............................................................................63 5.2.3.2. Teste do campo aberto ......................................................................63 5.2.3.3. Potenciação do sono induzido por barbitúrico ...................................64 6. DISCUSSÃO .........................................................................................................76 7. CONCLUSÕES .....................................................................................................94 REFERÊNCIAS BIBLIOGRÁFICAS ......................................................................96 APÊNDICE ..........................................................................................................121 Introdução 19 1. INTRODUÇÃO 1.1. Fisiopatologia da dor A dor é uma experiência complexa que envolve não somente a transdução da informação gerada pelo estímulo nocivo, mas também o processamento cognitivo e emocional pelo cérebro (JULIUS e BASBAUM, 2001). A natureza altamente subjetiva da dor é um dos fatores que dificulta a sua definição e o seu tratamento clínico. Segundo a Associação Internacional para o Estudo da Dor (IASP, 1994), a dor é uma experiência sensorial e emocional associada com potenciais ou reais lesões, tem uma conotação individual e sofre influência de experiências anteriores. A dor pode ser classificada como neurogênica, nociceptiva, neuropática ou psicogênica, quando associada a lesão do tecido neuronal na periferia ou em nível central, estimulação excessiva dos nociceptores, disfunção/dano de um ou mais nervo, e a fatores psicológicos, respectivamente (MILLAN, 1999). Woolf e Salter (2000) já classificam a dor em fisiológica (quando há a ativação direta dos nociceptores), neuropática (provocada por lesões de células do sistema nervoso central) e inflamatória (provocada por danos teciduais). Em termos de duração, a dor pode ser aguda ou crônica. A dor aguda está associada com uma lesão tecidual recente, ativação de nociceptores e pode desaparecer até mesmo antes da cura do dano tecidual (CARR e GOUDAS, 1999; PARK e VASKO, 2005). Por outro lado, a dor crônica pode se perpetuar por meses ou anos, e se caracteriza em relação à persistência e alterações adaptativas, o que muitas vezes dificulta o tratamento (IADAROLA e CAUDLE, 1997; BESSON, 1999). É importante a diferenciação dos termos nocicepção e dor. Nocicepção refere-se às manifestações neurofisiológicas geradas por um estímulo nocivo, enquanto a dor envolve a percepção de um estímulo aversivo, a qual requer a capacidade de abstração e elaboração de impulsos sensoriais. Além disso, a dor não é, obrigatoriamente, proporcional ao grau da lesão (ALMEIDA et al., 2004; SNEDON, 2004). É importante salientar que sempre que qualquer tecido estiver lesado, o organismo reage para desencadear o estímulo doloroso como um mecanismo de proteção (CHENG et al., 2002; MILLAN, 1999, 2002; GUYTON e HALL, 2005). Contudo, frequentemente a dor se torna crônica e debilitante em substituição à sua função de atuar como um sistema de aviso. A transição para a fase crônica envolve mudanças na medula espinhal e encéfalo, mas ocorre 20 modulação significativa nos locais onde as mensagens da dor são iniciadas no nível do neurônio sensorial aferente primário (JULIUS e BASBAUM, 2001). Os nociceptores, que são responsáveis pela percepção do estímulo nocivo, estão amplamente distribuídos no nosso organismo. Nociceptores são terminações periféricas de neurônios sensitivos primários cujos corpos celulares estão localizados nos gânglios da raiz dorsal ou nos gânglios trigêmeos. São distribuídos em três classes principais: nociceptores térmicos (ativados por temperaturas ≥ a 45 ºC ou ≤ 15 ºC), mecânicos (ativados por pressão intensa, localizados principalmente na pele) e polimodais (ativados por estímulos mecânicos, químicos ou térmicos de alta intensidade). Quando estimulados devidamente, os nociceptores são ativados, gerando potenciais de ação que se propagam através das fibras nervosas aferentes. A sensibilização dos nociceptores causa uma redução do seu limiar de ativação e, em alguns casos, atividade espontânea. As fibras nervosas são classificadas, de acordo com seu diâmetro, estrutura e velocidade de condução em do tipo Aα, com maior diâmetro (12 – 22 µm), velocidade de condução rápida (70 – 120 m/s) e fortemente mielinizadas; do tipo Aβ, também com grande diâmetro (> 10 µm), velocidade de condução rápida (30 – 100 m/s) e fortemente mielinizadas; fibras do tipo Aδ, com diâmetro intermediário (2 – 6 µm), velocidade de condução moderada (12 – 30 m/s) e fracamente mielinizadas; e as do tipo C com menor diâmetro (0,4 – 1,2 µm), velocidade de condução lenta (0,5 – 2 m/s) e não mielinizadas. O potencial de ação é conduzido por estas fibras nervosas até atingir os tratos espinotalâmicos, espinorreticular e espinomesencefálico, transmitindo para estas regiões a informação nociceptiva. Após o processamento central da dor, estes estímulos são retransmitidos para fibras descendentes de origem cortical ou medular (GRUBB, 1998; CARR e GOUDAS, 1999; MILLAN, 1999; ALMEIDA et al., 2004; DJOUHRI et al., 2004). Algumas alterações na resposta normal podem ocorrer, como, por exemplo, o aumento da resposta a um estímulo normalmente doloroso, o qual é denominado hiperalgesia (JULIUS e BASBAUM, 2001; BASBAUM e JESSEL, 2003). Esta resposta exagerada normalmente é decorrente de importantes mudanças no processo central de sensibilização da dor. Outras alterações sensoriais também podem aparecer, entre elas, a alodinia que se refere à dor evocada por estímulo 21 inócuo (MILLAN, 1999). A hiperalgesia e a alodinia podem ter duas origens diferentes: responsividade aumentada dos neurônios da medula espinhal responsáveis pela transmissão da dor (sensibilização central), ou diminuição do limiar de ativação do nociceptor (sensibilização periférica). Com a sensibilização central, a dor pode ser produzida pela ativação de fibras sensoriais primárias não nociceptivas. Sensibilização periférica ocorre quando a terminação do nociceptor é exposta a produtos do dano tecidual ou inflamação. A bradicinina, a prostaglandina E2 (PGE2), o fator de crescimento nervoso e as interleucinas pró-inflamatórias parecem exercer papel fundamental na nocicepção periférica. A prostaglandina e a bradicinina causam alterações em receptores valinóides de potencial transitório tipo 1 (TRPV1) acoplados a canais iônicos dependentes de ligantes via ativação de 3’5’-adenosina-monofosfato cíclico (AMPc), e das proteinoquinases A e C, reduzindo o tempo de pós-hiperpolarização da membrana neural, causando, então, redução do limiar para disparo da fibra nervosa (CHUANG et al., 2001). O organismo humano produz moléculas que são capazes de modular a dor: os peptídeos opióides. A primeira evidência direta de que opióides endógenos, a exemplo de analgésicos opióides, modulam a dor em humanos foi dada pelo estudo de Levine et al. (1978), os quais usaram um modelo de dor pós-cirúrgica dentária para avaliar a analgesia mediada por opióides. Amplamente distribuídos no cérebro, os mediadores opióides mais conhecidos são a β-endorfina, a metioninaencefalina, a leucina-encefalina, a dinorfina e as endomorfinas. Os peptídeos opióides são também produzidos por muitas células não neuronais, incluindo as glândulas endócrinas e exócrinas e as células do sistema imune. Os receptores opióides pertencem à superfamília de receptores acoplados a proteínas G (GPCR), com o domínio N-terminal extracelular, sete domínios transmembranares conectados por três alças intracelulares e três extracelulares, e o domínio C-terminal intracelular. Têm cerca de 60% de identidade entre si, com maior homologia nas hélices transmembranares e maior diferença nas porções N- e Cterminais bem como nas alças extracelulares. Os receptores opióides são ativados tanto por peptídeos opióides produzidos endogenamente quanto por várias drogas opióides naturais, semi-sintéticas ou sintéticas (WALDHOER et al., 2004). Há quatro 22 subtipos de receptores opióides, cada um com seu próprio grupo de ligantes: 1) mu, µ ou MOP-R; 2) kappa, κ ou KOP-R; 3) delta, δ ou DOP-R; e 4) o receptor para orfanina FQ/nociceptina ou NOP-R (FOORD, 2005). Existe um consenso de que os opióides medeiam seus efeitos analgésicos através da ativação de receptores específicos localizados a nível espinhal, supraespinhal e periférico. A ativação desses receptores está relacionada primariamente a 3 eventos básicos: redução dos níveis de AMPc pela inibição da adenilato ciclase, bloqueio de canais de Ca+2 voltagem-dependente e aumento da corrente retificadora de K+. Com a diminuição do AMPc e da disponibilidade de Ca+2 intracelular, a analgesia produzida pelos opióides resulta da redução da liberação de neurotransmissores excitatórios como o glutamato e a substância P (SP) em fibras nociceptivas aferentes primárias (HARRISSON et al., 1998; JORDAN e DAVI, 1998; GRUBB, 1998; LAW e LOH, 1999). Estudos de co-localização têm confirmado a presença de receptores opióides em fibras C e A (PARE et al., 2001), em fibras viscerais positivas para TRPV1 (POONYACHOTI et al., 2002) e em neurônios que expressam isolectina B4 (IB4), SP ou peptídeo relacionado ao gene de calcitonina (CGRP) (MINAMI et al., 1995; WENK et al., 1999; BORGLAND et al., 2001). Opióides endógenos ou exógenos atenuam a excitabilidade de nociceptores periféricos, a propagação de potenciais de ação, a liberação de peptídeos pró-inflamatórios (SP, CGRP) de terminais sensoriais periféricos e a vasodilatação evocada pela estimulação de fibras-C. Todos esses mecanismos, em conjunto, resultam em analgesia e ações antiinflamatórias (STEIN et al., 2003). Tradicionalmente, os agonistas opióides exercem efeitos analgésicos através de ação no sistema nervoso central, como ocorre com a morfina (HERZ e TESCHEMACHER, 1971; MILLAN, 1986). Por outro lado, com a utilização de análogos quaternários dos alcalóides da morfina, como a N-metilmorfina, propõe-se que estes agentes, por apresentarem uma mínima capacidade de transpor a barreira hematoencefálica, exercem o efeito analgésico através da atuação em receptores opióides presentes nas terminações nervosas periféricas (FERREIRA e NAKAMURA, 1979a,b,c; FERREIRA et al., 1982; SMITH et al., 1982; STEIN, 1993; ANTONIJEVIC et al., 1995). Entretanto, ainda não se desenvolveu ou se isolou uma 23 droga que, administrada por via oral e com mecanismo de ação exclusivamente periférico, apresente eficácia na terapêutica analgésica. 1.2. Fisiopatologia da inflamação A resposta inflamatória é um mecanismo benéfico e fisiológico pelo qual o organismo se defende contra infecções e tenta reparar danos teciduais ou perda de função (LAWRENCE et al., 2002). Assim, o processo inflamatório agudo pode ser definido como um conjunto de alterações bioquímicas e celulares que ocorrem em resposta a estímulos inespecíficos, tais como infecções ou danos teciduais (HANSSON, 2005). As reações inflamatórias locais são caracterizadas por aumento do fluxo sangüíneo e da permeabilidade vascular, seguida de dilatação vascular e acúmulo de células do processo inflamatório, caracterizando os quatro sinais típicos da presença de inflamação: rubor (hiperemia), tumor (edema), calor (aumento da temperatura local) e dor, como descritas por Cornelius Celsus, no início da era Cristã (GILROY et al., 2004). O quinto sinal da inflamação, que é a perda da função do tecido ou órgão lesado, associado com reações crônicas foi descrito por VIRCHOW no século XIX (KALISCH, 1975). Os componentes básicos de um processo inflamatório envolvem eventos vasculares e celulares, mediadores derivados de células e da ativação plasmática, que produzem os sinais clássicos da inflamação descritos anteriormente. As alterações vasculares iniciam-se imediatamente e desenvolvem-se durante as primeiras horas após o estímulo inflamatório. Elas consistem em vasodilatação, aumento do fluxo sangüíneo, aumento da permeabilidade vascular e exsudação plasmática (WILLIAMS et al., 1983). Em condições normais a microcirculação apresenta baixíssima permeabilidade a macromoléculas. As proteínas plasmáticas circulam muito lentamente entre o sangue e os tecidos e retornam ao sangue através dos vasos linfáticos. Esta situação muda muitíssimo durante o processo inflamatório. A microcirculação torna-se permeável a macromoléculas e fluidos vindos do sangue, causando edema tecidual (GILROY et al., 2004). O processo inflamatório pode ser desencadeado por inúmeros estímulos, tais como agentes infecciosos, interação antígeno-anticorpo, traumas químicos, mecânicos ou térmicos, isquemia, dentre outros. Os eventos celulares são marcados pela saída das células circulantes da luz do vaso e a migração de leucócitos para o 24 sítio inflamatório. Esse fenômeno segue algumas fases como captura, rolamento dos leucócitos pelo endotélio, adesão firme e transmigração (MUNRO, 1993; SPRINGER, 1994; PEREIRA, 1996; WAHL et al., 1996). Todas estas etapas do processo de migração leucocitária são dependentes da expressão pelos leucócitos e pelas células endoteliais de moléculas denominadas moléculas de adesão e de mediadores quimiotáticos (SPRINGER, 1994; WEBER, 2003). A mobilização adequada dos leucócitos circulantes para o sítio inflamado é fundamental para a defesa do organismo, uma vez que estas células podem desenvolver suas ações de fagocitose e destruição de agentes patogênicos levando à resolução do processo. Os leucócitos circulantes migram seletivamente e em número significativo para o tecido inflamado no decorrer do processo. Em uma resposta inflamatória aguda, e logo nos estágios iniciais, ocorre acúmulo predominante de neutrófilos, enquanto que as células mononucleares são observadas mais tardiamente durante a fase aguda, bem como nos processos crônicos. A migração de eosinófilos também pode ocorrer em processos inflamatórios, estando principalmente associada a processos alérgicos e infecções parasitárias. Algumas das células envolvidas já estão presentes no tecido afetado tais como: células endoteliais, células mesoteliais, mastócitos, eosinófilos, macrófagos e alguns linfócitos (SIBILLE e REYNOLDS, 1990; SAMPSON, 2000; BROCHE e TELLADO, 2001; BOYTON e OPENSHAW, 2002). Diante de um agente inflamatório há a liberação de vários mediadores químicos que influenciarão a resposta e evolução da inflamação: mediadores gerados a partir do plasma; mediadores armazenados em células e liberados após a ação do agente flogístico; mediadores de natureza protéica (citocinas); mediadores lipídicos (leucotrienos – LTs –, fator de ativação plaquetária – PAF – e as prostaglandinas - PGs); e mediadores liberados pelas células do exsudato (PEREIRA, 1996). A vasodilatação, o aumento da permeabilidade vascular e a ativação leucocitária estão relacionados à ação da histamina, PGs, óxido nítrico (NO), bradicinina, LTs, C3a, C5a e PAF, sendo que esses mediadores podem ser pré-formados e armazenados, como a histamina nos mastócitos, ou formados no local do estímulo, como as PGs (DI VAIO e FREITAS, 2001; STEAVENS e LOWE, 2002). 25 Os principais eicosanóides envolvidos tanto na geração da inflamação como também da dor, são os LTs e PGs. As PGs e os LTs promovem vasodilatação, aumento da permeabilidade vascular e edema nos sítios de inflamação, enquanto que na dor podem causar hiperalgesia a estímulos mecânico, químico ou térmico (VANE e BOTTING, 1998). Os eicosanóides são produtos do processamento do ácido araquidônico (AA) que normalmente é encontrado esterificado a fosfolipídios de membrana, de onde é liberado por ação de fosfolipases (PLs), como a fosfolipase A2 (PLA2). O AA pode sofrer metabolização pelas vias das enzimas cicloxigenase (COX) e lipoxigenase (LOX) para produzir uma grande família de eicosanóides. A COX é uma enzima bifuncional, com atividade de ácido graxo (catalisando a conversão do AA em prostaglandina G2 - PGG2) e atividade de prostaglandina hidroperoxidase (catalisando a conversão da PGG2 em prostaglandina H2 - PGH2). A PGH2 é convertida, através de diferentes enzimas com especificidade celular, na prostaglandina E2 (PGE2) prostaglandina F2α (PGF2α), prostaglandina D2 (PGD2), prostaglandina I2 (PGI2) e no tromboxano A2, entre outras (VANE e BOTTING, 1998). Em 1971, Vane demonstrou que o principal mecanismo de ação dos antiinflamatórios não-esteroidais (AINES) era a propriedade de bloquear a síntese de prostanóides através da inibição da atividade da COX. Este fato implicou diretamente alguns eicosanóides como pró-inflamatórios. Vários anos se passaram até a descoberta da existência de pelo menos duas isorformas de COX envolvidas na ação não específica dos AINES, a COX-1 e a COX-2. Como a COX-2 é uma enzima expressa por células envolvidas em processos inflamatórios, foi correlacionada como sendo a maior responsável pela produção de prostanóides nos processos inflamatórios e dolorosos. As LOXs originam os LTs, o ácido hidroeicosatetraenóico (HETE) e as lipoxinas. Os LTs, potentes agentes quimiotáticos, são produzidos, predominantemente, por células inflamatórias como leucócitos polimorfonucleares, macrófagos e mastócitos. Os antiinflamatórios inibidores da COX, os AINES, estão entre as drogas mais prescritas em todo o mundo (FIORUCCI et al., 2001). Entretanto, o uso dos AINES é limitado devido aos seus efeitos colaterais, particularmente no trato gastrintestinal (TGI) e nos rins (FOSSLIEN, 1998). Vários anos se passaram até a descoberta da 26 existência de pelo menos duas isorformas de COX envolvidas na ação não específica dos AINES, a COX-1 e a COX-2. Como a COX-2 é uma enzima expressa por células envolvidas em processos inflamatórios, foi correlacionada como sendo a maior responsável pela produção de prostanóides nos processos inflamatórios e dolorosos (VANE et al., 1998) Os AINES podem ser comparados quanto à capacidade variável de inibição da COX-1 e COX-2, existindo inibidores não seletivos, como a indometacina, e inibidores seletivos para a COX-2, como o celecoxib (WARNER e MITCHELL, 2004). Os inibidores da COX-2 têm sido descritos como a classe de inibidores que possuem potente atividade antiinflamatória, sem os efeitos adversos no TGI tão comum aos AINES clássicos (GOLDENBERG, 1999). Atualmente, porém, estudos têm mostrado que a inibição exclusiva de COX-2 não está satisfazendo todas as necessidades da terapia antiinflamatória por várias razões: por comprometer a função renal (BERTOLINI et al., 2001; GAMBARO, 2002); pelo fato da COX-2 estar aumentada em pacientes com gastrites e úlceras com o objetivo de causar uma citoproteção adaptativa, visto que as PGs produzidas a partir de COX-2 são as responsáveis por essa citoproteção gástrica, assim, se as COX-2 forem inibidas, o quadro do paciente com gastrite e úlcera pode piorar devido à diminuição dessas PGs (PARENTE, 2001); por ter sido reportado que o uso desses agentes requer vigilância no que se refere a doenças cardiovasculares (MCADAM et al., 1999; MCGETTINGAN e HENRY, 2006). Além disso, as PGs produzidas pela COX-1 têm mostrado contribuírem para a resolução do processo inflamatório e hiperalgesia. Nestes casos, a eficácia antiinflamatória de inibidores seletivos da COX-2 só foi observada em doses que inibiam a COX-1 (PARENTE E PARETTI, 2003). Assim, parece que é necessário haver um equilíbrio entre a inibição de COX-1 e COX-2. O LTB4, que é produzido através da conversão do AA pela 5-LOX, é também um importante agente quimiotático para células inflamatórias tais como neutrófilos, macrófagos e eosinófilos. Através da ativação de neutrófilos, o LTB4 acaba ativando a sua degranulação que é associada com a liberação de enzimas e geração de superóxido. Ele também aumenta a adesão dos neutrófilos pelo endotélio e promove sua infiltração tecidual. Finalmente, ele parece ter um papel importante nas respostas imunológicas por aumentar a liberação de citocinas inflamatórias por macrófagos e linfócitos (SAMUELSSON et al., 1987). Assim, substâncias que inibem 27 a 5-LOX atuam como agentes antiinflamatórios por impedirem a formação dos LTs (BRAIN e WILLIAMS, 1990). Além disso, substâncias que são capazes de antagonizarem os receptores de LTs também são uma boa estratégia para a terapêutica antiinflamatória, como por exemplo, o montelucaste e o zafirlucaste. Considerando as propriedades pro-inflamatórias dos LTs e prostanóides, substâncias capazes de inibir a síntese de ambos eicosanóides (inibidores duais) deveriam não somente apresentar um perfil antiinflamatório superior como também apresentar menos efeitos colaterais que os AINES tradicionais e os que inibem seletivamente a COX-2 (CELOTTI e LAUFER, 2001). Assim, substâncias que são capazes de causar inibição dual, ou seja, inibindo tanto a 5-LOX quanto a COX-2, têm sido apontadas como moléculas promissoras na terapêutica antiinflamatória (JULEMONT et al., 2003). Outra via de ativação é a hidrólise do AA para formar o lisofosfolipídio. O lisofosfolipídio pode ser acetilado formando o PAF que é um potente lipídio bioativo que atua por ligação específica em GPCR (ISHII e SHIMIZU, 2000). O termo PAF foi denominado pelo fato deste lipídio ser o responsável pela agregação de plaquetas (ISHII e SHIMIZU, 2000), além de ser um dos mais potentes fatores quimiotáticos in vitro e in vivo, principalmente para eosinófilos e neutrófilos. O PAF apresenta várias funções patofisiológicas, sendo que alguns destes efeitos incluem ativação plaquetária, estimulação neutrofílica, contração da musculatura lisa e aumento da permeabilidade vascular com formação de edema (ISHII e SHIMIZU, 2000). Foi demonstrado que tanto a injeção intraplantar de PAF em ratos (DALLOB et al., 1987; BONNET et al., 1981) quanto a injeção intratecal em camundongos (MORITA et al., 2004) podem causar alodinia ou hiperalgesia mecânica. Contudo, o mecanismo de ação pelo qual o PAF exerce suas ações na dor ainda não está bem estabelecido. As cininas representam um grupo importante de moléculas envolvidas nas doenças inflamatórias, como na pancreatite, peritonite, artrite reumatóide, asma, disfunções do trato genito-urinário, além de dor e hiperalgesia, e inflamação neurogênica (CALIXTO et al., 2004). A produção de cininas, no sítio inflamatório, resulta em vasodilatação, extravasamento plasmático e aderência de neutrófilos, em conseqüência de uma ação direta sobre o endotélio da microvasculatura, ou ainda indireta, através da liberação de outras substâncias pró-inflamatórias. Estes 28 peptídeos exercem seus efeitos biológicos através da ativação dos receptores B1 e B2. Enquanto as cininas são os agonistas endógenos para o receptor B2, a des-Arg9BK e a des-Arg10-calidina são agonistas preferenciais para o B1. Ambos os receptores pertencem à superfamília de GPCR com sete domínios transmembranares (Gag/11 e Gai) (CALIXTO et al., 2004). O receptor B2 é constitutivo e está presente em tecidos centrais e periféricos. Estes parecem estar implicados na maioria das ações fisiológicas das cininas. O receptor B1 é geralmente ausente em tecidos normais e animais saudáveis, mas pode ser induzido e superexpresso durante uma lesão tecidual ou administração de alguns mediadores inflamatórios (SIEBECK et al., 1998). A histamina é liberada (juntamente com a serotonina, em roedores) pelos mastócitos em resposta a diversos estímulos inflamatórios (PARADA et al., 2001). Ambas as aminas vasoativas exercem um papel fundamental na inflamação, sendo capazes de causar dilatação de vênulas pós-capilares e, conseqüentemente, aumentar o fluxo sangüíneo e a permeabilidade vascular (BARNES et al., 1988). Além disso, elas estão implicadas nos processos de nocicepção em diversas condições inflamatórias (BESSON, 1997), inclusive em humanos (SCIBERRAS et al., 1987), bem como na formação de edema (KAY, 2001). A histamina e serotonina atuam sinergicamente com a bradicinina, induzindo hiperalgesia térmica (LAVICH et al., 2003). Os receptores para histamina são divididos em quatro tipos: H1, H2, H3 e H4 (DE ESCH et al., 2005). A ativação de receptores histaminérgicos H1 e H2 induz a mobilização de cálcio e acúmulo de AMPc, respectivamente. O receptor H3 está localizado em neurônios histaminérgicos e atua como um autorreceptor (SCHWARTZ et al., 1991). Diversos estudos demonstraram a participação dos receptores H1 e H2 na mediação dos processos nociceptivo e inflamatório (MALMBERG-AIELLO et al., 1998; MOBARAKEH et al., 2000; PARADA et al., 2001; OLSEN et al., 2002), Atualmente, o receptor de histamina H4 foi descoberto a partir de um estudo da informação da seqüência genética do receptor de histamina H3 humano. O gene que codifica o receptor de histamina H4 humano foi clonado por Oda et al. (2000) e Nakamura et al. (2000) a partir de feto e leucócitos, respectivamente. Sua expressão parece ser controlada por estímulos inflamatórios e é expresso em eosinófilos, além de outras células (MORSE, 2001; O’REILLY et al., 29 2002). É sabido que a histamina induz quimiotaxia em eosinófilos e que este efeito é inibido por antagonistas de receptores H4, o que torna estas moléculas um excelente alvo para o tratamento dos processos inflamatórios (O’REILLY, et al., 2002; LING et at., 2004; DE ESCH et al. 2005). A serotonina é estocada em plaquetas e mastócitos de roedores podendo ser liberada sob estímulo inflamatório (HOURANI e CUSACK, 1991). Esta amina está envolvida na nocicepção e inflamação atuando nos receptores serotoninérgicos 5HT1, 5-HT2, 5-HT3, 5-HT4 e 5-HT7 (SUFKA et al., 1992; DOAK e SAWYNOK, 1997, PARADA et al., 2001, LAVICH et al., 2003). Quando a serotonina é aplicada perifericamente, provoca dor em humanos e aumenta o comportamento de dor em diversos modelos animais. Sua administração endógena estimula uma reação inflamatória que consiste em formação de edema e rubor em humanos e ratos (MALING et al., 1974; SUFKA et al., 1992). O NO é sintetizado a partir da L-arginina e oxigênio molecular por um processo enzimático que utiliza elétrons doados pela forma reduzida do fosfato de nicotinamida adenina dinucleotídeo (NADPH). As enzimas NO sintases (NOS) convertem a L-arginina em NO-hidroxi-L-arginina que sofre uma oxidação formando L-citrulina e NO. Uma das isoformas de NOS foi originalmente caracterizada em neurônios, conhecida como NOS neuronal (nNOS ou tipo 1) enquanto a outra, foi observada em células endoteliais, a NOS endotelial (eNOS ou tipo II) (CRANE et al., 1997; COLEMAN, 2001). O terceiro tipo de NOS, sintetizada após a ativação celular, foi inicialmente descrita em macrófagos de camundongos (STUEHR e MARLETTE, 1985, 1987), e é conhecida como NOS induzível (iNOS ou tipo III) (NATHAN e XIE, 1994; STUEHR e MARLETTE, 1985, 1987). A nNOS e eNOS são ativadas em resposta a uma elevação nas concentrações de cálcio intracelular e formação do complexo cálcio-calmodulina. Por outro lado, a iNOS liga-se à calmodulina com alta afinidade mesmo em baixas concentrações de cálcio intracelular (MAYER e ANDREW, 1998). A expressão da iNOS é induzida por diversos estímulos, incluindo lipopolissacarídeos (LPS) ou citocinas como interferon- γ (IFN- γ), interleucina-1 (IL1) ou fator de necrose tumoral-α (TNF-α) (NATHAN e XIE, 1994; STUEHR e MARLETTE, 1985, 1987). 30 Além das funções fisiológicas do NO, incluindo vasodilatação, citotoxicidade em macrófagos e plasticidade no sistema nervoso central (SNC) (SCHUMAN e MADISON, 1994), o NO está envolvido no processamento nociceptivo central e periférico (MELLER e GEBHART, 1993). Muitos dos dados que sugerem a participação do NO na nocicepção no SNC, são baseados nos efeitos antinociceptivos de inibidores de NOS, como demonstrado em modelos em roedores, de nocicepção térmica ou mecânica (PRZEWLOCKI et al., 1993; INOUE et al., 1997) ou dor química persistente (HALEY et al., 1992; MOORE et al., 1991; BABBEDGE et al., 1993; MALMBERG e YAKSH, 1993; HAO e XU, 1996; MACHELSKA et al., 1997). Diversos trabalhos demonstraram o efeito dual do NO no mesmo modelo de nocicepção e/ou inflamação (SOUSA et al., 2001; ROCHA et al., 2002; PRADO et al., 2002). 1.3. Fisiopatologia da depressão do sistema nervoso central A capacidade de integrar informações obtidas de várias fontes externas e internas resume o papel principal do sistema nervoso central, isto é, otimizar as necessidades do organismo nas demandas do ambiente do indivíduo. Esses conceitos de integração transcendem os sistemas de transmissão individual, e enfatizam os métodos através dos quais a atividade neuronal é normalmente coordenada. Apenas com a compreensão detalhada das funções de integração, e de suas falhas em determinadas condições fisiopatológicas, podem-se desenvolver abordagens terapêuticas eficazes e específicas para os distúrbios neurológicos e psiquiátricos (GOODMAN e GILMAN, 2005). Ao examinar o efeito dos fármacos no SNC, com relação aos neurotransmissores para circuitos específicos, deve-se prestar muita atenção aos princípios gerais da organização dos neurônios. O conceito de que as sinapses representam pontos de controle modificáveis pelos fármacos dentro das suas estruturas neuronais exige a delineação explícita dos locais onde os neurotransmissores operam e o grau de especificidade com o qual estes locais são afetados (GOODMAN e GILMAN, 2005). Dentre os vários neurotransmissores responsáveis por alguma função no SNC, o ácido gama aminobutírico (GABA) é o principal responsável pela depressão central. Em 1950, o GABA foi identificado como um constituinte químico singular do cérebro, mas sua potência como um depressor do SNC não foi imediatamente 31 reconhecida. Kravitz et al. (1963) demonstraram que o GABA era o único aminoácido inibitório encontrado exclusivamente nos nervos inibitórios dos crustáceos e que a potência inibitória dos extratos desses nervos era responsável pelo seu teor de GABA. A liberação de GABA foi então relacionada à freqüência da estimulação nervosa. Registros intracelulares do músculo indicaram que a estimulação do nervo inibitório e a administração de GABA produziam aumentos idênticos na condutância de Cl- no músculo. Portanto, essas observações satisfazem completamente os critérios para identificação de um transmissor (GOODMAN e GILMAN, 2005). Os receptores que fazem parte do sistema GABAérgico são três: GABAA, GABAB e GABAC. Esses receptores são dependentes de ligante e, quando o GABA se liga ao seu sítio de ligação causa hiperpolarização do neurônio pós-sináptico. O receptor GABAB causa hiperpolarização preferencialmente pelo influxo de K+ do que pelo de Cl-. Com os receptores GABAA e GABAC ocorre o oposto, a hiperpolarização é causada pelo influxo do íon Cl- (Almeida, 2006). O receptor GABAA é o mais bem caracterizado de todos os receptores GABAérgicos e apresenta vários subtipos. O GABAA é um receptor pertencente a uma superfamília de canais iônicos dependentes de ligantes, responsável pela rápida inibição da neurotransmissão. A ligação do GABA ou de seus agonistas ao receptor GABAA causa uma mudança conformacional no canal iônico que permite o influxo do íon Cl- para dentro da célula, causando hiperpolarização. Embora a estrutura nativa do receptor em questão não tenha sido elucidada, acredita-se que o receptor GABAA seja heteropentamérico, com cinco subunidades, sendo duas cópias da subunidade α, duas da subunidade β e uma cópia da subunidade δ, γ ou ε. O subtipo α1β2γ2 representa quase 50% do total de receptores GABAA encontrados no cérebro. Estima-se que podem ocorrer mais de 100 subtipos diferentes de GABAA no SNC dos mamíferos. O GABAA tem vários sítios de ligação para vários ligantes. Os agentes terapêuticos e moduladores que se ligam a esse receptor são: barbitúricos, benzodiazepínicos e alguns neuroesteróides, tais como os derivados da progesterona e deoxicorticosterona. Todos esses compostos afetam a atividade do GABA, aumentando a abertura dos canais de cloreto. O sítio de ligação dos benzodiazepínicos se encontra entre as subunidades α e γ. Dependendo do tipo de 32 subunidade γ, o subtipo de receptor pode apresentar aumento, diminuição ou até mesmo falta de atividade de certos benzodiazepínicos (Almeida, 2006). O GABAB é um receptor metabotrópico constituído por uma proteína com sete domínios transmembranares e a este receptor encontra-se acoplada uma proteína G que, ao ser ativada pela ligação do GABA, resulta em abertura de canais de K+, Ca+2 ou na ativação da enzima adenilato ciclase. Os receptores GABAB pré-sinápticos são primariamente unidos a canais de Ca+2 e, conseqüentemente, a redução da liberação de vesículas, contendo o neurotransmissor, na fenda sináptica. Já os receptores GABAB pós-sinápticos estão unidos a canais de K+ e a sua ativação resulta em aumento da condutância de K+, causando, assim, hiperpolarização e redução da excitabilidade neuronal (Almeida, 2006). Ainda não se tem muito conhecimento a respeito do receptor GABAC. Até pouco tempo atrás ele era classificado como um subtipo do receptor GABAA. Estruturalmente, sabe-se que o receptor GABAC é ionotrópico e homoligomérico, composto de múltiplas subunidades ρ, ou seja, não se encontram as subunidades mais comuns de receptores GABA: δ, γ e ε (Almeida, 2006). O SNC central também pode ser deprimido através da ação de substâncias que se ligam aos receptores opióides, como a morfina. Como já citado anteriormente, os receptores opióides, quando ativados, ativam a adenilato ciclase inibitória, que diminui a quantidade de AMPc intracelular. Além disso, ocorre ativação da corrente de K+ reguladas pelo receptor e a supressão de Ca+2 regulados por voltagem. E são justamente essa ativação da corrente do K+ e a supressão da corrente de Ca+2 que hiperpolarizam a membrana das células e explicam o efeito depressor central dos opióides (HARRISSON et al., 1998; JORDAN e DAVI, 1998; GRUBB, 1998; LAW e LOH, 1999). Os anestésicos gerais também são capazes de levar a uma depressão do SNC. Isso pode ocorrer tanto através da desestabilização dos lipídios de membrana, que impedem a condução do impulso nervoso, quanto, principalmente, através da interação desses anestésicos com proteínas localizados no SNC. Muitos agentes anestésicos são capazes de inibirem a função de receptores excitatórios, em concentrações alcançadas durante a anestesia como os receptores do glutamato ionotrópico, da acetilcolina ou da 5-hidroxitriptamina (5-HT), assim como potencializarem a função de receptores inibitórios, como o GABAA e a glicina. É 33 agora claro que os anestésicos afetam a função de muitos canais iônicos e é provável que estes efeitos sejam responsáveis pelos seus efeitos globais sobre o SNC (RANG et al., 2004). De forma bem condensada, a depressão do SNC é alcançada através da potenciação das vias inibitórias ou através da inibição das vias excitatórias (GOODMAN e GILMAN, 2005). 1.4. Os produtos naturais e o Synadenium umbellatum As plantas medicinais são freqüentemente utilizadas com o intuito de substituir ou auxiliar as terapias convencionais no tratamento de várias doenças. Entre outros fatores, a preferência na utilização das plantas medicinais decorre da facilidade de obtenção e do baixo custo. Porém, sabe-se que as plantas medicinais apresentam ampla diversidade de metabólitos secundários com diferentes atividades biológicas (FARNSWORTH et al., 1985; SIMÕES, 2003), justificando a necessidade de um aprofundamento no conhecimento das propriedades das espécies vegetais e sua utilização na formulação de medicamentos. Apesar da preferência das grandes indústrias farmacêuticas pelo desenvolvimento de medicamentos pela via sintética, nas últimas décadas observase ainda um grande interesse do mercado pelo potencial terapêutico das plantas medicinais (CALIXTO et al., 2000; KOEHN e CARTER, 2005). Tal fato é comprovado pela evidência de que hoje cerca de 25% das drogas prescritas no mundo são obtidas direta ou indiretamente de plantas. Além disso, cerca de 49% das drogas desenvolvidas entre 1981 a 2002 foram obtidas a partir de produtos naturais, ou análogos semi-sintéticos ou ainda compostos sintéticos baseados em produtos naturais (KOEHN e CARTER, 2005). Ainda que os medicamentos derivados de plantas tenham uma boa aceitação pela população e estejam presentes no mercado farmacêutico, apenas uma pequena parcela das plantas medicinais possui dados científicos que comprovem sua eficácia e seu espectro toxicológico, assim como garantia de qualidade do produto. Considerando-se os diversos metabólitos secundários presentes nas plantas, as principais categorias de medicamentos derivados de plantas são os terpenóides, glicosídeos, alcalóides, flavonóides e outros tipos. 34 Como exemplos relevantes de medicamentos obtidos de plantas, podemos mencionar a morfina (Papaver somniferum), a digoxina (Digitalis sp.), o taxol (Taxus brevifolia), o quinino (casca da Chinchona sp.), a vincristina e a vinblastina (Catharanthus roseus), dentre outros (RATES, 2001). Assim, na terapêutica moderna, as plantas medicinais fornecem o substrato para a produção de compostos biologicamente ativos ou compostos passíveis de modificações e otimizações estruturais que dão origem às entidades químicas. Neste contexto, o Brasil é um país privilegiado, pois ocupa o primeiro lugar dentre os 17 países mais ricos do mundo em biodiversidade, detendo cerca de 23% do total de espécies existentes no planeta (RATES, 2001). A imensa variedade de espécies de plantas, animais e microrganismos existentes no ecossistema brasileiro, sem dúvida apresenta um importante diferencial para o desenvolvimento de medicamentos (KATO, 2001). A espécie botânica Synadenium umbellatum Pax. (nome em alusão às glândulas do ciátio concrescidas) (Fig. 1), pertence à ordem Geraniales e à Família Euphorbiaceae. Esta família compreende cerca de 290 gêneros e aproximadamente 7500 espécies. Os maiores centros de dispersão encontram-se nos trópicos, continentes americano e africano. São plantas de hábito variado, existindo ervas, subarbustos, árvores e também trepadeiras, com folhas alternadas inteiras ou partidas, em geral com estípulas, latescentes ou não. Flores sempre de sexo separado: flores masculinas, em geral monoclamídeas, de simetria radial com tépalas em número de 5-6; flores femininas mono ou diclamídeas, em geral pentâmeras, ovário sempre súpero, caracteristicamente tricarpelar e trilocular (cada lóculo contendo 1 ou 2 óvulos). O látex, quando possuem, pode ser incolor ou leitoso com grãos de amido em forma de fêmur muito característico (JOLY, 1977). Dentro da família Euphorbiaceae há muitas plantas com elevada toxicidade. Em levantamento recente, no Estado de Goiás, o Centro de Informação Toxicológica constatou que, dentre as 18 espécies botânicas relatadas como responsáveis por ocorrências de intoxicações por plantas tóxicas no Estado, cinco delas (28%) pertencem à família Euphorbiaceae, sendo elas a Euphorbia milii (coroa de cristo, cristo gigante), Euphorbia tirucalli (graveto do cão, figueira do diabo, dedo do diabo), Jatopha curcas (Pinhão de purga, pinhão paraguaio, pinhão bravo, purgão de 35 cavalo), Ricinus communis (carrapateira, rícino, mamoeira, palma de cristo, carrapato) e Manihot utilissima (mandioca amarga, mandioca branca, mandioca anaçunipeba). As euforbiáceas produzem albuminóides tóxicos, sendo a ricina o mais conhecido (QUER, 1962). A ricina é uma proteína inativadora de ribossomos (RIPs) do tipo II, heterodimérica, com a enzima inibidora de ribossomo (~32kDa, cadeia A) ligada por ponte dissulfeto a uma lectina galactose (~34kDa, cadeia B). Se for quebrada a ligação entre os monômeros, as partes resultantes não são tóxicas. A ricina inativa ribossomos impedindo a síntese protéica. Há RIPs tipo I que são monômeros e não citotóxicos, pois não atravessam a membrana celular (BRANDT et al., 2005). O Synadenium umbellatum é conhecido popularmente como “cola-nota”, “avelós”, ”milagrosa”, “cancerola”, etc., sendo o látex (obtido das folhas ou do caule; pH ácido ± 5,0) empiricamente utilizado na forma de solução aquosa, como segue: “Colocar 18 gotas do látex em 1 (um) litro de água e guardar na geladeira. Tomar pela manhã, à tarde e à noite um cálice de licor ou uma xicarazinha de café. Ou substituir a água pela solução bebendo várias vezes ao dia. Usar sempre para cura e prevenção” (ORTÊNCIO, 1997). No Brasil, o látex do Synadenium umbellatum tem sido utilizado popularmente para o tratamento de várias enfermidades, tais como, alergia, câncer, doença de Chagas, diabetes, gripe, hemorragias internas, impotência sexual, lepra, obesidade, úlcera nervosa, cólicas menstruais e dores no corpo (ORTÊNCIO, 1997). Várias espécies de Synadenium são conhecidas no mundo pelo seu uso como antiinflamatório, anti-câncer e analgésico. Jager (1996) demonstrou que algumas espécies do gênero Synadenium são potentes inibidores da síntese de PGs, como por exemplo, a espécie Synadenium cupulare, o que justifica o uso destas plantas como antiinflamatórios e analgésicos. Além disso, várias outras plantas pertencentes à família euforbiácea têm apresentado atividades farmacológicas, tais como a Euphorbia kansui, que possui atividade analgésica (DAISUKE e YOSHIMASA, 1975) e anti-tumoral (WU et al., 1991), e diversas espécies do gênero Phyllanthus que têm apresentado atividade antiinflamatória e analgésica (Santos, 1995). 36 Figura 1 - Fotografia da árvore do Synadenium umbellatum Pax. (cola-nota) no bairro Feliz, em Goiânia – GO. Foto tirada pelo Prof. Dr. Luiz Carlos da Cunha em novembro de 2005. Objetivos 38 2. OBJETIVOS O presente estudo teve como objetivo a obtenção de um extrato etanólico das folhas de Synadenium umbellatum, bem como a obtenção de frações (frações hexânica, clorofórmica e metanol/água) e a posterior avaliação das atividades antinociceptiva, antiinflamatória e depressora do sistema nervoso central dos mesmos visando validar o uso popular da planta no que se refere a essas atividades. Materiais 40 3. MATERIAIS 3.1. MATERIAL BOTÂNICO 3.1.1. Coleta, identificação e herborização da planta Folhas de S. umbellatum foram coletadas no bairro Feliz, Goiânia-GO, Brasil, no verão de 2005/2006. O material botânico foi identificado pelo Professor Dr. José Realino de Paula, professor adjunto de farmacognosia da Faculdade de Farmácia da Universidade Federal de Goiás (UFG). Uma exsicata foi depositada no Herbário da UFG sob número UFG-27160. As folhas foram secas a 40 °C em estufa com circulação de ar por 48 horas e trituradas em moinho de facas. 3.2. MATERIAL QUÍMICO 3.2.1. Extrato, frações e medicamentos Para todos os ensaios, uma solução de extrato etanólico de S. umbellatum a 10 mg/mL foi preparada imediatamente antes dos experimentos e, a partir desta solução, foram feitas sucessivas diluições a fim de se obter concentrações de 10, 5 e 2,5 mg/mL. Soluções das frações hexânica, clorofórmica e metanol/água também foram preparadas imediatamente antes dos experimentos, nas concentrações de 1, 2 e 2,5 mg/mL, respectivamente, sendo que diluições sucessivas foram feitas para se obter outras concentrações desejáveis, quando necessário. As soluções do extrato e das frações hexânica e clorofórmica foram sempre preparadas imediatamente antes do seu uso com salina (NaCl 0,9%) e tween-80 3%. A solução da fração metanol/água foi preparada da mesma forma, porém não requereu a utilização do tween-80. Outras drogas usadas foram indometacina (Prodome – Brasil), morfina (Cristália – Brasil), dexametasona (Hipolabor – Brasil), fentanil (Cristália – Brasil), heparina (Prodome – Brasil), naloxona (Cristália – Brasil), diazepam (Cristália – Brasil) e pentobarbital sódico (Cristália – Brasil). Todos os medicamentos foram solubilizados em salina, exceto a indometacina, que foi solubilizada em NaHCO3 5%. 3.2.2. Reagentes • Acetona (Synth – Brasil) • Ácido acético glacial (Synth – Brasil) • Bicarbonato de sódio (Synth – Brasil) • Carragenina (Sigma – USA) 41 • Celite (Synth – Brasil) • Clorofórmio (Synth – Brasil) • Cloreto de sódio (Synth – Brasil) • Etanol 95% P.A. (Synth – Brasil) • Éter etílico (Synth – Brasil) • Formaldeído (Synth – Brasil) • Fosfato de sódio dibásico (Synth – Brasil) • Fosfato de sódio monobásico (Synth – Brasil) • Metanol (Synth – Brasil) • n-Hexano (Synth – Brasil) • Óleo de cróton (Sigma – USA) • Solução de Türk (Sigma – USA) • Tween 80 (Synth – Brasil) 3.3. ANIMAIS Os animais utilizados neste estudo foram camundongos albinos (Mus musculus) tipo Swiss machos, pesando entre 25 e 35 g obtidos no Biotério da Indústria Química do Estado de Goiás (IQUEGO). Os tratamentos dos animais com o veículo (grupo controle), extrato, frações ou diferentes substâncias foram sempre realizados em concentrações adequadas para a administração de um volume constante de 10 mL/kg. As administrações antecediam 30 minutos (via subcutânea, s.c.) e 60 minutos (via oral, p.o.) os testes de atividade farmacológica. Os animais foram mantidos em condições controladas de temperatura e iluminação (ciclo claro / escuro de 12 h), com água e ração ad libitum, permanecendo no laboratório por um período de adaptação de pelo ao menos 24 horas antes dos experimentos, normalmente iniciados às 9 horas. Todos os experimentos foram desenvolvidos seguindo normas que envolvem cuidados com animais de laboratório (CIOMS, 1985). Métodos 43 4. MÉTODOS 4.1. MÉTODOS FITOQUÍMICOS 4.1.1.Preparação do extrato etanólico O extrato etanólico de S. umbellatum (EES) foi obtido por maceração em álcool etílico 95% P.A. na proporção 1:5 (m/v), sob agitação por 5 horas, seguido por filtração. A extração foi repetida por mais duas vezes para se garantir o esgotamento das substâncias extraíveis pelo álcool etílico e, em seguida, os filtrados foram misturados. O filtrado foi evaporado em rotaevaporador a 40 ºC sob pressão reduzida a fim de se concentrar o extrato. 4.1.2. Eliminação de clorofila e fracionamento do extrato etanólico A eliminação de clorofila e o fracionamento foram feitos de acordo com Ferri (1996) com algumas modificações. 45 g de EES concentrado foi dissolvido em metanol a 4 ºC e deixado em repouso a 4 ºC por 18 horas. Em seguida, a solução foi filtrada. Ao filtrado, foi adicionada água destilada a 4 ºC até que uma solução 7:3 fosse obtida. A solução resultante foi filtrada sobre Celite e particionada em nhexano (1:1) por agitação em funil de separação por 3 vezes. A fração hexânica foi reservada e a solução de metanol/água foi então particionada com clorofórmio (1:1) por agitação em funil de separação por 3 vezes. A fração clorofórmica foi separada da fração metanol/água. Em seguida, as frações hexânica (FH), clorofórmica (FC) e metanol/água (FM) foram, individualmente, submetidas à rotaevaporação a 40 °C a fim de se evaporar os solventes e concentrar as frações. Após a evaporação do metanol da fração metanol/água, a solução resultante foi liofilizada (Fig. 2). 44 Extrato etanólico de S. umbellatum (45 g) 1. 1,35 L MeOH 4 °C (18 horas) 2. Filtrar Precipitado (subst. graxas) MeOH 1. 0,58 L H2O 4 °C 2. Filtrar (Celite) Pigmentos Solução MeOH-H2O 1. Hexano (3x) Solução MeOH-H2O Fração hexânica 1. Clorofórmio (3x L) Fração MeOH-H2O Fração clorofórmica Figura 2 – Fluxograma do procedimento geral para a extração de clorofila e fracionamento do extrato etanólico das folhas de S. umbellatum. 45 4.2. MÉTODOS FARMACOLÓGICOS 4.2.1. Atividade antinociceptiva 4.2.1.1. Contorções abdominais induzidas por ácido acético Empregando-se a metodologia de Koster (1959), foram utilizados grupos experimentais de 6 camundongos que foram tratados pela via p.o. com veículo, EES (25, 50 ou 100 mg/kg), FH (10 mg/kg), FC (20 mg/kg), FM (25 mg/kg) ou indometacina (10 mg/kg). Todos os grupos receberam ácido acético 1,2% (v/v i.p.) 60 minutos após os tratamentos. O número de contorções abdominais, consideradas como contrações da parede abdominal seguidas pela extensão de ao menos uma das patas posteriores (TORNOS, 1999), foram contadas acumulativamente em 30 minutos de avaliação. Os resultados foram expressos como as médias ± EPM dos números de contorções abdominais acumuladas em 30 minutos de avaliação experimental e como a média ± DP da porcentagem de inibição das contorções, comparativamente ao grupo controle. 4.2.1.2. Dor induzida pela formalina Este modelo permite, após a injeção intraplantar de formalina, avaliar dois tipos de dor: a de origem neurogênica, que ocorre pela estimulação direta de terminações nociceptivas, e a de origem inflamatória, produzida pela liberação de mediadores inflamatórios (HUNSKAAR et al., 1985, 1986 e 1987). Foram utilizados grupos experimentais de até 10 camundongos tratados pela via p.o. com veículo, EES (100 mg/kg), indometacina (10 mg/kg) ou morfina (10 mg/kg, s.c.). 60 minutos após os tratamentos, os animais foram injetados na região intraplantar da pata posterior direita com 20 µL de formalina 3% v/v (formaldeído 1,2% v/v). Em seguida, os animais foram observados individualmente durante 30 min em caixas de acrílico com fundo especular para auxiliar a visualização. Em seguida à aplicação da formalina, foi medida a reatividade do animal considerada como o tempo, em segundos, que o animal permanece lambendo a pata injetada. O efeito antinociceptivo foi avaliado nas duas fases da dor: durante os primeiros 5 minutos (dor neurogênica) e no período de 15 a 30 minutos (dor de origem inflamatória), sendo os resultados expressos como as médias ± EPM dos tempos de reatividade nas distintas fases, comparativamente ao grupo controle experimental. 46 4.2.1.3. Influência do tratamento com naloxona Os animais foram pré-tratados pela via s.c. com salina ou com naloxona (3 mg/kg), um antagonista opióide não seletivo, 15 minutos antes dos tratamentos pela via p.o. com o veículo, EES (100 mg/kg) ou com o fentanil (100 µg/kg). Decorridos 30 min (s.c.) ou 60 min. (p.o.) dos tratamentos, os animais foram injetados com ácido acético (1,2% i.p.) e o número de contorções abdominais foi contado por 30 minutos à semelhança do item 4.2.1.1. 4.2.1.4. Teste do tail flick Segundo a metodologia descrita por D’Amour e Smith (1941), este ensaio permite o estudo de drogas com atividade opióide central, mediante a avaliação do tempo, em segundos, que o animal leva para retirar a cauda do local de incidência de um estímulo térmico doloroso. Este estímulo nociceptivo foi produzido por imersão do terço distal da cauda dos camundongos em banho-maria a 55,5 ± 0,5 ºC. Grupos experimentais de 6 animais foram previamente selecionados quanto à sua reatividade ao estímulo nociceptivo, não sendo utilizados animais cujo tempo de resposta foi superior a 7 segundos. A reatividade foi medida a cada 30 minutos, iniciando-se uma hora antes e prolongando-se por 2 horas depois da administração p.o. do veículo ou do EES (25, 50 ou 100 mg/kg). Morfina (10 mg/kg), administrada via s.c., foi utilizada como controle positivo do ensaio. O tempo máximo que se deixaram as caudas dos animais em contato com o estímulo nociceptivo térmico foi de 20 s para se evitar lesões. 4.2.2. Atividade antiinflamatória 4.2.2.1. Edema de orelha induzido por óleo de cróton Este experimento foi realizado de acordo com Tubaro (1985). Foram utilizados até 8 camundongos tratados pela via p.o. com o veículo, EES (25, 50 ou 100 mg/kg), FM (6, 12 ou 25 mg/kg) ou dexametasona (2 mg/kg). 60 minutos após os tratamentos, foi administrado topicamente óleo de cróton 2,5% v/v (em solução de acetona) na orelha direita e o mesmo volume de acetona na orelha esquerda dos camundongos. Após 4 horas, os animais foram sacrificados por deslocamento cervical, discos de 6 mm de diâmetro foram retirados de cada orelha e pesados em balança analítica. Os resultados da diferença de peso entre os discos das duas orelhas de cada animal foram comparados com o grupo controle experimental. 47 4.2.2.2. Peritonite induzida por carragenina Este ensaio avalia a evolução da migração leucocitária para a cavidade peritoneal, segundo protocolo originalmente descrito por Ferrándiz e Alcaraz (1991). Grupos experimentais de até 10 camundongos foram tratados previamente pela via p.o. com veículo (grupo controle), com EES (100 mg/kg) ou com dexametasona (2 mg/kg). Decorridos 60 minutos após os tratamentos, os animais foram injetados pela via i.p. com 0,25 mL de carragenina (1% m/v em salina). Após 4 horas, os animais foram sacrificados em câmara fechada com éter e injetados na cavidade peritoneal com 2 mL de PBS heparinizado (10 UI/mL de heparina). Após 60 compressões leves no abdômen, o fluído foi coletado, a amostra diluída (1:20 em Solução de Tϋrk) e a contagem do número de leucócitos totais migrados realizada em câmera de Newbauer. Os resultados foram expressos como as médias ± EPM dos números de leucócitos totais por mL. 4.2.3. Atividade no sistema nervoso central 4.2.3.1. Teste do rota-rod Este método descrito por Rosland (1990) permite avaliar a especificidade da ação nociceptiva de drogas, verificando se estas promovem incoordenação motora dos animais, seja por sedação e/ou por relaxamento muscular. Grupos de sete camundongos foram colocados no rota-rod por 1 minuto, 60 minutos após os tratamentos com veículo, EES (25, 50 ou 100 mg/kg), FH (10 mg/kg), FC (20 mg/kg), FM (25 mg/kg) ou diazepam (5 mg/kg), sendo avaliados o número de quedas e o tempo de permanência na barra giratória. O número máximo de quedas permitidas foi de 3 sendo que, após a terceira, o animal não mais era reconduzido ao rota-rod. O tempo máximo de permanência permitido no rota-rod foi de 1 minuto. 4.2.3.2. Teste do campo aberto Este modelo foi baseado na metodologia descrita por Sielgel (1946) e validado por Archer (1973), e permite uma avaliação da atividade estimulante ou depressora de um dado composto podendo ainda indicar atividades mais especificas como ação tipo ansiolítica ou ansiogênica. Foram utilizados grupos de 8 camundongos tratados previamente pela via p.o. com veículo (grupo controle), com EES (25, 50 ou 100 mg/kg), FH (10 mg/kg), FC (20 mg/kg), FM (25 mg/kg) ou com diazepam (5 mg/kg). 60 minutos após o tratamento, os animais foram colocados em 48 um campo-aberto confeccionado em acrílico e foram observados durante 5 minutos, sendo avaliados a atividade exploratória dos animais (número de quadrados invadidos), o tempo em que os animais permaneceram parados, o número de levantadas, o número de auto-limpeza e o número de bolos fecais. 4.2.3.3. Potenciação do sono por barbitúrico Foram utilizados grupos de até 8 camundongos tratados previamente pela via p.o. com veículo, com EES (25, 50 ou 100 mg/kg), FH (10 mg/kg), FC (20 mg/kg), FM (25 mg/kg) ou com diazepam (5 mg/kg). 60 minutos após os tratamentos, os animais foram injetados com pentobarbital sódico 50 mg/kg i.p. Foram cronometrados o tempo de recuperação do reflexo postural. Os resultados foram expressos como o tempo de recuperação (minutos) do sono de cada grupo experimental comparativamente ao grupo controle experimental (GONZALESTRUJANO et. al., 1998). 4.3. ANÁLISE ESTATÍSTICA Os resultados foram dados como médias ± erro padrão das médias (EPM) ou desvio padrão (DP). As diferenças estatísticas entre os grupos experimentais foram detectadas pela análise de variância (ANOVA) seguida pelo teste de Tukey. Considerou-se como valores significantes aqueles cujo P < 0,05. Para a análise dos dados foi utilizado o software GraphPad Prism 4.0. Resultados 50 5. RESULTADOS 5.1. RESULTADOS FITOQUÍMICOS 5.1.1.Preparação do extrato etanólico Foram coletados 17 kg de folhas de S. umbellatum. Após secagem, o peso das folhas foi de 1,3 kg, o que gerou 90 g de extrato, cuja coloração era verde intenso. Isso corresponde a um rendimento de 6,9%. 5.1.2. Eliminação de clorofila e fracionamento do extrato etanólico O EES (45 g) forneceu três frações: uma fração hexânica (FH), uma fração clorofórmica (FC) e uma fração metanol/água (FM). As frações FH e FC apresentaram coloração marrom escura e tiveram rendimentos de 4% e 11,5%, respectivamente em relação ao EES. A fração FM apresentou coloração castanha escura e teve um rendimento de 12,3% em relação ao EES. 5.2. RESULTADOS FARMACOLÓGICOS 5.2.1. Atividade antinociceptiva 5.2.1.1. Contorções abdominais induzidas por ácido acético O EES exibiu significante supressão do número de contorções abdominais induzidas pelo ácido acético em uma relação dose-resposta em camundongos se comparados ao grupo controle. No grupo controle, previamente tratado com veículo (60 minutos antes), a injeção de ácido acético (1,2 % i.p.) induziu 77,3 ± 2,1 contorções em 30 min. O pré-tratamento com EES (25, 50 ou 100 mg/kg) reduziu as contorções abdominais em 24,7% (58,2 ± 1,0), 39,5% (46,8 ± 0,9) e 55,0% (34,8 ± 1,5), respectivamente (Fig. 3). O pré-tratamento com FH (10 mg/kg) e FC (20 mg/kg) não alteraram o número de contorções (78,2 ± 2,9 e 75,8 ± 3,4 respectivamente) enquanto que a FM (25 mg/kg) reduziu as contorções abdominais em 59,2% (31,5 ± 1,6) (Fig. 4). O pré-tratamento com indometacina (10 mg/kg) reduziu as contorções em 39,6% (46,7 ± 0,7). A dose efetiva necessária para reduzir em 50% o efeito (DE50), determinada por interpolação gráfica da relação dose do extrato / porcentagem de inibição foi de 80,7 mg/kg (Fig. 5). Número de Contorções 51 80 70 60 50 40 30 20 10 0 * * * * C ____________________ 25 50 100 INDO EES Figura 3 - Contorções abdominais induzidas pelo ácido acético (1,2 % v/v em salina, i.p.) durante 30 minutos em camundongos previamente tratados (60 min) pela via p.o. com veículo (grupo controle; C), com o extrato etanólico das folhas do S. umbellatum (EES; 25, 50 ou 100 mg/kg) ou com indometacina (INDO; 10 mg/kg). As colunas e barras verticais representam as médias ± EPM de 6 animais por grupo experimental. * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. Os grupos tratados com as diferentes doses de EES são significativamente diferentes entre si (P < 0,05) – ANOVA, teste de Tukey. Número de Contorções 52 90 80 70 60 50 40 30 20 10 0 * * C FH 10 FC 20 FM 25 INDO Figura 4 - Contorções abdominais induzidas pelo ácido acético (1,2 % v/v em salina, i.p.) durante 30 minutos em camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com as frações hexânica (FH, 10 mg/kg), clorofórmica (FC, 20 mg/kg) e metanol/água (FM, 25 mg/kg) ou com indometacina (INDO; 10 mg/kg). As colunas e barras verticais representam as médias ± EPM de 6 animais por grupo experimental. * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. 53 % d e in ib ição 100 75 50 25 0 1.0 1.5 2.0 Log dose (mg/kg) Figura 5 - Porcentagem de inibição das contorções abdominais induzidas pelo ácido acético (1,2 % v/v em salina, i.p.) durante 30 minutos em camundongos previamente tratados (60 min) pela via p.o. com EES (■) 25, 50 ou 100 mg/kg. Os símbolos e barras verticais representam as médias ± DP de 6 animais por grupo experimental. R2 = 0,9306 54 5.2.1.2 Dor induzida pela formalina A aplicação intraplantar de formalina 3% na pata posterior direita de camundongos produziu intensa nocicepção em duas fase distintas: a primeira de 0 a 5 minutos (dor neurogênica) (Fig. 6) e a segunda de 15 a 30 minutos (dor inflamatória) (Fig. 7). Nos animais previamente tratados (60 min) pela via p.o. com veículo, a reatividade na primeira fase de nocicepção foi de 119,4 ± 12,2 s (n = 10) e na segunda fase foi de 271,5 ± 24,9 s (n = 10). O pré-tratamento com EES (100 mg/kg, p.o.) reduziu ambas as fases de nocicepção, sendo que tal redução, na primeira fase, foi de 48,7% (61,3 ± 5,4 s, n = 10) enquanto, na segunda fase, a redução foi de 73,1% (73,0 ± 12,4 s, n = 10). O grupo tratado com morfina (5 mg/kg s.c.) teve ambas as fases reduzidas, sendo que a primeira fase foi reduzida em 72,1 % (33,3 ± 15,8 s, n = 6) e a segunda fase em 84,0 % (42,7 ± 20,2 s, n = 6). O grupo tratado com indometacina (10 mg/kg, p.o.) não teve a primeira fase reduzida (106,3 ± 10,4 s, n = 6), porém teve a segunda fase reduzida em 50,9 % (133,4 ± 15,0 s, n = 6). 5.2.1.3. Influência do tratamento com naloxona A pré-administração do antagonista opióide naloxona (3 mg/kg. s.c.) no grupo tratado com veículo (p.o.) não modificou o número de contorções abdominais induzidas pelo ácido acético (1,2%, i.p.) em 30 min (67,7 ± 3,0) comparativamente ao grupo pré-administrado com salina s.c. (70,1 ± 2,6). O tratamento com o agonista opióide fentanil (100 µg/kg, s.c.) reduziu o número de contorções para 6,3 ± 1,0. No entanto, a administração de naloxona 15 min antes da injeção do fentanil, bloqueou o efeito antinociceptivo do agonista opióide nas contorções induzidas pelo ácido acético (66,7 ± 2,2). Resultado semelhante foi evidenciado com o EES (100 mg/kg, p.o.) cujo efeito (43,5 ± 1,7) foi inibido com o tratamento prévio com naloxona (68,8 ± 2,2) (Fig. 8). 55 1ª Fase (0 – 5 minutos) Tempo de reação (s) 140 120 100 80 * 60 * 40 20 0 C EES 100 MOR INDO Figura 6 - Reatividade à aplicação intraplantar de formalina (20 µL, 3%) na pata posterior direita de camundongos, durante a primeira fase (0 – 5 min) do teste da formalina, previamente tratados (60 min) pela via p.o. com veículo (grupo controle, C; n = 10), com o EES (100 mg/kg, n = 10), com morfina (MOR; 10 mg/kg s.c., n = 6) ou com indometacina (INDO; 10 mg/kg, n = 6). As colunas e barras verticais representam as médias ± EPM. * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. 56 2ª Fase (15 – 30 minutos) Tempo de reação (s) 300 250 200 * 150 100 * 50 0 C EES 100 * MOR INDO Figura 7 - Reatividade à aplicação intraplantar de formalina (20 µL, 3%) na pata posterior direita de camundongos, durante a segunda fase (15 – 30 min) do teste da formalina, previamente tratados (60 min) pela via p.o. com veículo (grupo controle, C; n = 10), com o EES (100 mg/kg, n = 10), com morfina (MOR; 10 mg/kg s.c., n = 6) ou com indometacina (INDO; 10 mg/kg, n = 6). As colunas e barras verticais representam as médias ± EPM. * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. 57 Número de contorções Salina Naloxona 75 # 50 ## * 25 * 0 Veículo EES 100 mg/kg Fentanil 100 µg/kg Figura 8 - Influência do tratamento prévio com o antagonista opióide não seletivo naloxona (3 mg/kg, s.c.) sobre a atividade antinociceptiva do EES (100 mg/kg, p.o.) e fentanil (100 µg/kg, s.c.) no modelo de contorções abdominais induzidas pelo ácido acético. As colunas e barras verticais representam a média ± EPM de 6 animais por grupo experimental. * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. # Estatisticamente diferente do grupo tratado com EES 100 mg/kg e pré-tratado com salina (P < 0,05) – ANOVA, teste de Tukey. ## Estatisticamente diferente do grupo tratado com fentanil 100 µg/kg e pré-tratado com salina (P < 0,05) – ANOVA, teste de Tukey. 58 5.2.1.4. Teste do tail flick Os tempos de latência ao estímulo térmico nociceptivo no tempo zero para o grupo controle foi de 2,29 ± 0,35 s. O tratamento prévio com o EES (25, 50 ou 100 mg/kg) não modificou a reatividade dos animais ao estímulo doloroso durante 2 horas após a administração. Em condições semelhantes, o tratamento com morfina (10 mg/kg) ampliou o tempo de latência ao estímulo térmico em 295 % (20,0 ± 0,0) 30 minutos após a sua administração e continuou causando antinocicepção durante todo o tempo avaliado (Fig. 9). 23 * Tempo de Latência (s) 20 * * * 60 90 120 18 15 13 10 8 5 3 0 -60 -30 0 30 Tempo (min) Administração dos Tratamentos Figura 9 - Latência ao estímulo térmico nociceptivo medido no teste do tail flick em camundongos antes e após tratamento com o (●) veículo p.o., com EES (■ 25, ▼50 ou ▲100 mg/kg, p.o.) ou morfina (♦ 10 mg/kg, s.c.). Nas ordenadas estão representados os tempos de latência dos camundongos ao estímulo térmico nociceptivo, em segundos. Os símbolos e barras verticais representam as médias ± EPM de 6 animais por grupo experimental. * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. 59 5.2.2. Atividade antiinflamatória 5..2.2.1. Edema de orelha induzido por óleo de cróton O EES e a FM reduziram o edema de orelha induzido pelo óleo de cróton de maneira dose-dependente. No grupo controle, previamente tratado (60 min antes) com o veículo (p.o.), a administração de óleo de cróton (2,5% v/v em solução de acetona) na orelha direita e o mesmo volume de acetona na orelha esquerda dos camundongos, gerou um edema de 21,7 ± 1,8 mg (n = 7) em 4 horas. O prétratamento com EES (25, 50 ou 100 mg/kg; p.o.) reduziu o edema em 30,0% (15,3 ± 0,4 mg, n = 7), 43,8% (12,2 ± 0,4 mg, n = 7) e 59,4% (8,8 ± 0,2 mg, n = 7), respectivamente (Fig. 10). O pré-tratamento com FM (6, 12 ou 25 mg/kg; p.o.) reduziu o edema em 19,8% (17,4 ± 0,6, n = 8), 41,0% (12,9 ± 0,9 mg, n = 8) e 61,9% (8,3 ± 0,9 mg, n = 7), respectivamente (Fig. 11). O pré-tratamento com dexametasona (2 mg/kg; p.o.) reduziu o edema em 83,9% (3,5 ± 0,9 mg, n = 6). 5.2.2.2. Peritonite induzida por carragenina O EES exibiu significante inibição da migração de leucócitos totais no modelo de peritonite induzida por carragenina de maneira dose-dependente (Fig. 12). O grupo controle, 4 horas após a administração de carragenina 1% (m/v i.p.) induziu a migração de 11,4 ± 0,3 x 106 leucócitos / mL (n = 10) para a cavidade peritoneal. O pré-tratamento com EES (25, 50 ou 100 mg/kg) reduziu a migração leucocitária em 21,9% (8,9 ± 0,5 x 106 leucócitos / mL, n = 8), 36,0% (7,3 ± 0,2 x 106 leucócitos / mL, n = 9) e 53,5% (5,3 ± 0,6 x 106 leucócitos / mL, n = 8). O prétratamento com dexametasona (2 mg/kg, n = 8) reduziu a migração leucocitária em 66,7% (3,8 ± 0,4 x 106 leucócitos / mL) respectivamente. 60 Edema (mg) 25 20 * 15 * 10 * * 5 0 C 25 50 100 DEXA ____________________ EES Figura 10 - Edema de orelha, em mg, induzido por óleo de cróton (2,5% v/v em acetona) nos grupos previamente tratados pela via p.o. com veículo (C; n = 7), com extrato etanólico de S. umbellatum (EES 25, 50 ou 100 mg/kg, n = 7) ou com dexametasona (DEXA; 2 mg/kg, n = 6). As colunas e barras verticais representam a média ± EPM. * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. Os grupos tratados com as diferentes doses de EES são significativamente diferentes entre si (P < 0,05) – ANOVA, teste de Tukey. 61 Edema (mg) 25 20 * 15 * * 10 * 5 0 C ____________________ 6 12 25 DEXA FM Figura 11 - Edema de orelha, em mg, induzido por óleo de cróton (2,5% v/v em acetona) nos grupos previamente tratados pela via p.o. com veículo (C; n = 7), com fração metanol/água (FM 6, 12 ou 25 mg/kg, n = 8, 8 e 7 respectivamente) ou com dexametasona (DEXA; 2 mg/kg, n = 6). As colunas e barras verticais representam a média ± EPM. * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. Os grupos tratados com as diferentes doses da FM são significativamente diferentes entre si (P < 0,05) – ANOVA, teste de Tukey. 62 Núm ero de Leucócitos Totais / m L (x 10 6 ) 12 11 10 9 8 7 6 5 4 3 2 1 0 * * * * C 25 50 100 ______________ DEXA EES Figura 12 - Migração de leucócitos totais no modelo de peritonite induzida por carragenina (1% m/v) injetada na cavidade intraperitoneal de camundongos previamente tratados pela via p.o. com veículo (C; n = 10), com extrato etanólico de S. umbellatum (EES 25, 50 ou 100 mg/kg, n = 8, 9 e 8 respectivamente) ou dexametasona (DEXA; 2 mg/kg, n = 8). As colunas e barras verticais representam a média ± EPM. * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. Os grupos tratados com as diferentes doses de EES são significativamente diferentes entre si (P < 0,05) – ANOVA, teste de Tukey. 63 5.2.3. Atividade no sistema nervoso central 5.2.3.1. Teste do rota-rod O EES e as frações FH, FC e FM não alteraram a atividade motora dos camundongos enquanto os animais tratados com diazepam (5 mg/kg) apresentaram diminuição da mesma. Camundongos (n = 7) tratados pela via oral (60 min antes) com veículo (p.o.), ao serem colocados na barra giratória por 1 minuto, tiveram 0,1 ± 0,1 quedas por minuto e se equilibraram por 60 ± 0,0 s no rota-rod. Os animais prétratados com EES (25, 50 ou 100 mg/kg) tiveram 0,6 ± 0,2, 0,6 ± 0,3 e 0,7 ± 0,2 quedas respectivamente, (Fig. 13) e se equilibraram por 60 ± 0,0, 60 ± 0,0 e 60 ± 0,0 s respectivamente (Fig. 15) no rota-rod. Os animais pré-tratados com FH (10 mg/kg), FC (20 mg/kg) e FM (25 mg/kg) tiveram 0,4 ± 0,2, 0,3 ± 0,2 e 0,3 ± 0,3 quedas respectivamente (Fig. 14), e se equilibraram por 60 ± 0,0, 60 ± 0,0 e 60 ± 0,0 s respectivamente (Fig. 16) no rota-rod. Os animais tratados com diazepam (5 mg/kg) apresentaram 2,0 ± 0,4 quedas e se equilibraram por 39,9 ± 9,5 s no rota-rod. 5.2.3.2. Teste do campo aberto O EES e as frações não alteraram a movimentação espontânea dos camundongos. Este efeito foi demonstrado pelo número de quadrados invadidos em 5 minutos no campo aberto. O EES e a FC, porém, da mesma maneira que o diazepam, aumentaram o tempo parado, enquanto o EES e a FH, assim como o diazepam, diminuíram o número de bolos fecais. O teste também mostrou que o EES e as frações FH, FC e FM, nas concentrações utilizadas, não alteraram o número de levantadas nem o número de auto-limpeza. O grupo diazepam (5mg/kg), porém, teve o número de levantadas diminuído, mas não apresentou alterações no número de auto-limpeza. Os resultados do tempo parado e do número de bolos fecais tanto do EES quanto das frações FH, FC e FM estão representados nas figuras 17-20. 64 5.2.3.3. Potenciação do sono induzido por barbitúrico O EES e as frações FH e FC apresentaram um possível efeito depressor do SNC em camundongos de acordo com o teste de potenciação do sono induzido por barbitúrico. Os camundongos tratados com veículo (p.o. n = 6) tiveram um tempo de recuperação do sono de 25,5 ± 3 min. Os animais tratados com EES (25, 50 ou 100 mg/kg, p.o, n = 8), tiveram os tempos de recuperação do sono aumentados em 97,6% (50,4 ± 2 min), 143,1% (62 ± 5 min) e 150,6% (63,9 ± 6 min) respectivamente (Fig. 21). Os animais tratados pela via p.o. com FH (10 mg/kg, n = 8) e FC (20 mg/kg, n = 7) tiveram os tempos de recuperação do sono aumentados em 118,0% (55,6 ± 8 min) e 89,4% (48,3 ± 3 min) respectivamente, enquanto os tratados com FM (25 mg/kg, n = 7) não tiveram o tempo de recuperação do sono aumentado (23,3 ± 3 min) (Fig. 22). Os animais tratados com diazepam (5 mg/kg, n = 6) tiveram um tempo de sono aumentado em 270,6% (94,5 ± 9 min). 65 Número de Quedas 3 * 2 1 0 C 50 100 DZP 25 _____________________ EES Figura 13 - Número de quedas, no rota-rod, de camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com o extrato etanólico das folhas do S. umbellatum (EES; 25, 50 ou 100 mg/kg) ou com diazepam (DZP; 5 mg/kg). As colunas e barras verticais representam as médias ± EPM de 7 animais por grupo experimental. * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. 66 Número de Quedas 3 * 2 1 0 C FH 10 FC 20 FM 25 DZP Figura 14 - Número de quedas, no rota-rod, de camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com as frações hexânica (FH, 10 mg/kg), clorofórmica (FC, 20 mg/kg), metanol/água (FM, 25 mg/kg) ou com diazepam (DZP; 5 mg/kg). As colunas e barras verticais representam as médias ± EPM de 7 animais por grupo experimental. * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. Tempo de permanência (s) 67 60 * 50 40 30 20 10 0 C 25 50 100 DZP _____________________ EES Figura 15 - Tempo de permanência, no rota-rod, em segundos, de camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com o extrato etanólico das folhas do S. umbellatum (EES; 25, 50 ou 100 mg/kg) ou com diazepam (DZP; 5 mg/kg). As colunas e barras verticais representam as médias ± EPM de 7 animais por grupo experimental. * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. Tempo de permanência (s) 68 60 * 50 40 30 20 10 0 C FH 10 FC 20 FM 25 DZP Figura 16 - Tempo de permanência, no rota-rod, em segundos, de camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com as frações hexânica (FH, 10 mg/kg), clorofórmica (FC, 20 mg/kg), metanol/água (FM, 25 mg/kg) ou com diazepam (DZP; 5 mg/kg). As colunas e barras verticais representam as médias ± EPM de 7 animais por grupo experimental. * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. Tempo parado (s) 69 200 175 150 125 100 75 50 25 0 * * * * C 25 50 100 DZP ____________________ EES Figura 17 - Tempo parado, em segundos, no teste do campo aberto, de camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com o extrato etanólico das folhas do S. umbellatum (EES; 25, 50 ou 100 mg/kg) ou com diazepam (DZP; 5 mg/kg). As colunas e barras verticais representam as médias ± EPM de 8 animais por grupo experimental. * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. Os grupos tratados com EES 25 mg/kg é significativamente diferente dos grupos tratados com 50 mg/kg e 100 mg/kg (P < 0,05) – ANOVA, teste de Tukey. 70 * Tem po parado (s) 200 175 150 125 100 75 50 25 0 * C FH 10 FC 20 FM 25 DZP Figura 18 - Tempo parado, em segundos, no teste do campo aberto, de camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com as frações hexânica (FH, 10 mg/kg), clorofórmica (FC, 20 mg/kg), metanol/água (FM, 25 mg/kg) ou com diazepam (DZP; 5 mg/kg). As colunas e barras verticais representam as médias ± EPM de 8 animais por grupo experimental. * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. Número de bolos fecais 71 4 3 2 * 1 0 C * * * 25 50 100 DZP ____________________ EES Figura 19 - Número de bolos fecais, no teste do campo aberto, deixados por camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com o extrato etanólico das folhas do S. umbellatum (EES; 25, 50 ou 100 mg/kg) ou com diazepam (DZP; 5 mg/kg). As colunas e barras verticais representam as médias ± EPM de 8 animais por grupo experimental. * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. Número de bolos fecais 72 4 3 2 * 1 0 C FH 10 FC 20 FM 25 * DZP Figura 20 - Número de bolos fecais, no teste do campo aberto, deixados por camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com as frações hexânica (FH, 10 mg/kg), clorofórmica (FC, 20 mg/kg), metanol/água (FM, 25 mg/kg) ou com diazepam (DZP; 5 mg/kg). As colunas e barras verticais representam as médias ± EPM de 8 animais por grupo experimental. * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. 73 Tempo de recuperação (min) 125 * 100 75 50 * * * 25 0 C __________________ 25 50 100 DZP EES Figura 21 - Tempo de recuperação do reflexo postural (duração do sono), em minutos, no teste de potenciação do sono induzido por barbitúrico, em camundongos previamente tratados (60 min) pela via p.o. com veículo (C; n=6), com o extrato etanólico das folhas do S. umbellatum (EES; 25, 50 ou 100 mg/kg, n=8) ou diazepam (DZP; 5 mg/kg, n=6). As colunas e barras verticais representam as médias ± EPM. * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. 74 Tempo de recuperação (min) 125 * 100 75 * 50 * 25 0 C FH 10 FC 20 FM 25 DZP Figura 22 - Tempo de recuperação do reflexo postural (duração do sono), em minutos, no teste de potenciação de sono induzido por barbitúrico, em camundongos previamente tratados (60 min) pela via p.o. com veículo (C; n = 6), com as frações hexânica (FH, 10 mg/kg, n = 8), clorofórmica (FC, 20 mg/kg, n = 7), metanol/água (FM, 25 mg/kg, n = 7) ou diazepam (DZP; 5 mg/kg, n = 6). As colunas e barras verticais representam as médias ± EPM. * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. Discussão 76 6. DISCUSSÃO Os resultados do presente estudo demonstram que o EES possui efeito antinociceptivo e antiinflamatório analisados em vários modelos de nocicepção e inflamação em camundongos. Além do mais, o estudo também demonstrou, através da realização de vários modelos comportamentais, um possível efeito depressor do sistema nervoso central. Atualmente, várias doenças ainda permanecem com sua etiologia desconhecida ou apresentam múltiplos fatores que contribuem para a permanência da doença no organismo. Algumas dessas doenças podem apresentar um caráter inflamatório e/ou doloroso persistente, sendo tratadas com a utilização de alguns medicamentos classicamente utilizados na inflamação e/ou dor como os AINES, glicocorticóides, opióides, e derivados, na tentativa de melhorar a sintomatologia geral do paciente (GUPTA e DUBOIS, 2001). Assim, a descoberta de novas substâncias com atividade analgésica e/ou antiinflamatória é ainda um aspecto altamente desejável e de enorme importância para a utilização clínica. Várias evidências demonstram que um grande número de medicamentos utilizados na terapêutica é derivado direta ou indiretamente de produtos naturais, especialmente de plantas superiores, sendo uma fonte inesgotável de possibilidades para a descoberta de novos fármacos (KOEHN e CARTER, 2005). Entretanto, várias dificuldades são encontradas por aqueles que desejam estudar as ações das plantas medicinais em sistema biológico, como por exemplo, a escolha de modelos experimentais, a obtenção de extratos padronizados e a dificuldade de obtenção, isolamento e identificação das substâncias ativas. Além disso, muitas doenças estão associadas a múltiplos fatores, dificultando a escolha de um alvo específico para o estudo (COOKSON, 2004). A fim de se estudar a atividade antinociceptiva e antiinflamatória do EES e de suas frações, escolhemos como um dos modelos experimentais, o modelo das contorções abdominais em camundongos. Este modelo baseia-se na contagem das contrações da parede abdominal seguidas de torção do tronco e extensão de ao menos um dos membros posteriores (writhing) como respostas reflexas à irritação peritoneal e à peritonite produzidas pela injeção intraperitoneal de ácidos fracos ou outros agentes inflamatórios. Introduzido por Siegmund et al. (1957a) utilizando-se a 77 fenilquinona, o teste foi posteriormente padronizado com outros agentes. Acetilcolina, ácidos acético ou clorídrico diluídos (ECKHARDT et al., 1958; KOSTER et al., 1959; NIEMEGEERS et al., 1975), bradicinina (EMELE e SHANAMAN, 1963), adrenalina (MATSUMOTO e NICKANDER, 1967), trifosfato de adenosina, triptamina (COLLIER et al., 1968), e ocitocina (MURRAY e MILLER, 1960) têm sido usados. Modificações têm sido feitas na concentração, temperatura e volume da solução injetada, e condições e maneiras de monitorar as condições comportamentais, de modo a simplificar o teste e aumentar a sua sensibilidade (LINÉE e GOURET, 1972; HARADA et al., 1979). O modelo também é usado em macacos (PEARL et al., 1969). O ácido acético atua indiretamente causando a liberação de mediadores endógenos envolvidos na modulação da nocicepção, incluindo a bradicinina, serotonina, histamina, PGs, entre outros, que causariam a estimulação dos neurônios nociceptivos e, consequentemente, a indução da dor (WHITTLE, 1964; BERKENKOPF e WEICHMAN, 1988; CHAU, 1989). Além disso, recentemente, Ribeiro et al. (2000) mostraram que a nocicepção induzida pelo ácido acético depende da liberação de citocinas, como a IL-1β, TNF-α e IL-8, a partir de macrófagos e basófilos residentes na cavidade abdominal, e que em conjunto com outros mediadores podem induzir a nocicepção característica observada neste modelo. O modelo das contorções abdominais induzidas por ácido acético é um modelo relativamente simples e com pouca especificidade, mas de fácil observação, rápido e com boa sensibilidade a várias drogas analgésicas, antiinflamatórias esteroidais e não-esteroidais, bem como a drogas semelhantes à morfina e outros analgésicos que atuam centralmente ou mesmo perifericamente. Além disso, os resultados obtidos com as várias classes de analgésicos neste modelo mostram boa correlação com a ação analgésica encontrada em outros modelos pré-clínicos, bem como em estudos clínicos (KOSTER et al., 1959; TABER, 1964; BLUMBERG et al., 1965; BLANE, 1967; SIEGMUND, 1957a,b; CHAU, 1989). Entretanto, devido à baixa especificidade da resposta antinociceptiva neste modelo, ensaios complementares são necessários para a interpretação dos resultados, pois uma variada gama de compostos essencialmente não analgésicos como anti-histamínicos, parassimpaticomiméticos, estimulantes do sistema nervoso central, inibidores da 78 monoaminaoxidase (MAO), antagonistas serotoninérgicos, relaxantes musculares, e neurolépticos podem também inibir o writhing (HENDERSHOT e FORSAITH, 1959; CHERNOV et al., 1967; PEARL et al., 1968; LOUX et al., 1978; RATES e BARROS, 1994). Os resultados do presente estudo demonstraram que, no modelo das contorções abdominais induzidas pelo ácido acético, o EES nas doses aqui utilizadas (25, 50 e 100 mg/kg) e a fração FM (25 mg/kg) obtidos das folhas do S. umbellatum apresentaram atividade antinociceptiva quando administrados pela via oral. As frações FH e FC não apresentaram redução no número de contorções abdominais, indicando que os princípios ativos responsáveis pelas atividades antinociceptiva e antiinflamatória foram arrastados pelos solventes de maior polaridade utilizados no fracionamento: uma mistura metanol/água 7:3. Além disso, o EES foi efetivo de maneira dose-dependente. Como controle positivo do ensaio foi utilizado indometacina, um antiinflamatório não-esteroidal, também conhecido como um dos mais potentes inibidores da COX in vitro (VANE, 1971). Como esperado, ele foi capaz de inibir o número de contorções abdominais induzidas pelo ácido acético. A analgesia moderada produzida pelo ácido acetilsalicílico e pela indometacina é explicada pela inibição da atividade da COX e redução da síntese de PGs (VANE, 1971). Entretanto, nos testes padrões em animais nos quais se induz uma reação inflamatória, tem sido difícil a separação entre a influência da atividade antiinflamatória desses compostos e entre um efeito analgésico singular (HUNSKAAR et al., 1986). Com o objetivo de melhor caracterizar a atividade antinociceptiva do EES, utilizamos o teste da formalina em camundongos, um modelo químico de nocicepção, que fornece uma resposta específica comparativamente ao modelo do ácido acético (SHIBATA et al., 1989; TJØLSEN et al., 1992), além de ser considerado o modelo que mais se aproxima da dor clínica (TJØSEN e HOLE, 1997). Descrita originalmente em gatos e ratos por Dubuisson e Dennis (1977), o teste da formalina foi posteriormente adaptado para camundongos (HUNSKAAR et al., 1985). A grande maioria dos estudos realizados com esse modelo utiliza roedores (TJØLSEN et al., 1992), porém, ocasionalmente, outras espécies de animais podem ser utilizadas, tais como, primatas (ALREJA et al., 1984), coelhos 79 (CARLI et al., 1981), cobaias (TAKAHASHI et al., 1984), crocodilos (KANUI et al., 1990) e aves (HUGHES e SUFKA, 1991). O ensaio consiste na injeção intraplantar de formaldeído na pata posterior do animal, a qual desencadeia intensa nocicepção por estimulação direta dos nociceptores. A nocicepção causada pela formalina é caracterizada por vigorosas lambidas, mordidas e batidas na pata injetada com o irritante. A aplicação do agente irritante na pata posterior torna a resposta nociceptiva mais específica, pois o animal, durante o grooming, utiliza mais frequentemente as patas anteriores (TJØLSEN et al., 1992). Seguindo as recomendações de Hunskaar et al. (1985) e Murray et al. (1988) somente as lambidas (licking) foram contatas em nossos experimentos visto que as mordidas e batidas são difíceis de serem contadas em camundongos devido ao tamanho pequeno do animal e seus rápidos movimentos. O teste da formalina possui uma característica importante em roedores, que é a apresentação de duas fases distintas de nocicepção, que parecem envolver diferentes mediadores (DUBUISSON e DENNIS, 1977; HUNSKAAR et al., 1985; HUNSKAAR e HOLE, 1987; ROSLAND, 1991; CORRÊA e CALIXTO, 1993; SEGUIN et al., 1995; SANTOS E CALIXTO, 1997). A primeira fase da nocicepção inicia-se imediatamente após a injeção de formalina, estendendo-se pelos primeiros 5 minutos, o que se acredita ser devido à estimulação química direta dos nociceptores (DEBUISSON e DENNIS, 1977; HUNSKAAR et al., 1985), predominantemente das fibras aferentes do tipo C e, em parte, as do tipo Aδ (HEAPY et al., 1987). A segunda fase desse modelo ocorre entre 15-30 minutos após a injeção de formalina e está relacionada com a liberação de vários mediadores pró-inflamatórios (HUNSKAAR e HOLE, 1987). As pesquisas indicam que vários mediadores químicos como a SP, NO, bradicinina e aminoácidos excitatórios, como o glutamato, estão envolvidos com a primeira fase visto que são esses mediadores que fazem a estimulação direta de nociceptores periféricos que integram as fibras aferentes primárias do tipo C e parte das do tipo Aδ (DUBUISSON e DENNIS, 1977). A ativação desses nociceptores, bem como de fibroblastos, mastócitos, neutrófilos e plaquetas, promove a liberação de muitos mediadores inflamatórios, como histamina, serotonina, taquicininas (SP, neurocinina A, neurocinina B) e glutamato (FUJIMAKI et al., 1992; SANTOS e 80 CALIXTO, 1997; CAO et al., 1998; OMOTE et al., 1998; BEIRITH et al., 2002) que, agindo de forma conjunta e/ou sinérgica, sensibilizam as vias nervosas periféricas e centrais de condução do sinal doloroso, dando origem à segunda fase (ou fase inflamatória, mais tardia e duradoura) da resposta nociceptiva à formalina (HUNSKAAR e HOLE, 1987). Entre as duas fases, há uma interfase, na qual o comportamento nociceptivo não é observado, ou seja, há um período de quiescência da resposta nociceptiva. A duração da interfase é determinada pela ativação dos sistemas monoaminérgicos descendentes inibitório e excitatório, que modulam os sinais nociceptivos ao nível do corno dorsal da medula (HENRY et al., 1999). A primeira fase do teste da formalina é inibida por agonistas opióides de ação central, como a morfina e o fentanil, e por antagonistas de receptores de bradicinina B1 e B2, além de antagonistas de receptores NMDA e de receptores valinóides. Agonistas opióides de ação exclusivamente periférica e antiinflamatórios esteroidais e não esteroidais não são capazes de inibir a primeira fase da formalina, porém, junto ainda com os agonistas opióides de ação central, são capazes de inibir a segunda fase (HUNSKAAR e HOLE, 1987; SHIBATA et al., 1989; OLUYOMI, 1992; CORRÊA e CALIXTO, 1993; STEIN, 1993). O pré-tratamento pela via oral com o EES na dose que produziu efeito máximo no modelo das contorções abdominais (100 mg/kg) inibiu fortemente tanto a primeira quanto a segunda fase da formalina. Como esperado, a indometacina, um AINE, inibiu somente a segunda fase, enquanto a morfina, um agonista opióide de ação central e periférica, inibiu ambas as fases da formalina. Desta forma, estes resultados reforçam a hipótese de que esta planta é dotada de importante atividade antinociceptiva e/ou antiinflamatória. Através do teste das contorções abdominais induzidas pelo ácido acético para o estudo do mecanismo da atividade antinociceptiva do EES foi verificado que o prétratamento com naloxona, um antagonista opióide não seletivo, inibiu a atividade antinociceptiva do EES. Esses resultados indicam que os princípios ativos presentes nas folhas do S. umbellatum interagem com o sistema opioidérgico. A fim de diferenciar entre uma ação opióide central ou periférica, foi realizado o teste do tail flick. Este modelo algesiométrico térmico foi introduzido por D’Amour e 81 Smith (1941) e, à semelhança do teste da placa quente (WOOLFE e MACDONALD, 1944), mede o tempo de latência do animal a um estímulo térmico. Estes ensaios são sensíveis a drogas opióides tipo morfina (JANSSEN et al., 1963), cuja atividade analgésica é mediada por receptores µ, κ e δ distribuídos no sistema nervoso central (BESSON e CHAOUCH, 1987; STEIN e SHIPPENBERG, 1989). O tratamento dos animais com EES, nas mesmas doses utilizadas no teste das contorções abdominais induzidas por ácido acético, não alterou a latência dos camundongos ao estímulo térmico doloroso, embora a morfina, um analgésico opióide utilizado como controle positivo do ensaio, tenha aumentado o tempo de latência. Esses resultados excluem um mecanismo de antinocicepção dependente de atividade opióide no sistema nervoso central, e nos leva a sugerir que os princípios ativos presentes nas folhas do S. umbellatum possivelmente atuam como agonistas opióides periféricos ou por induzirem a liberação de peptídeos opióides endógenos perifericamente. O fato do EES não ter modificado o tempo de latência no teste de tail flick pode ser explicado pelo fato deste modelo ser sensível somente a drogas de atividade supraespinhal (YAKSH e RUDY, 1977; SMITH et al., 1982, 1985). Tradicionalmente, o efeito analgésico dos agonistas opióides é explicado por ação no sistema nervoso central, como ocorre com a morfina e o fentanil (HERZ e TESCHEMACHER, 1971; MILLAN, 1986). Entretanto, mecanismos periféricos na atividade analgésica da morfina passaram a ser evidenciados e caracterizados através da aplicação tópica desses agentes (KAYSER et al., 1990) ou com a utilização de análogos quaternários de agonistas ou antagonistas opióides, por exemplo, os alcalóides N-metilmorfina e N-metilnalorfina. Estes agentes, com mínima capacidade de atravessarem a barreira hematoencefálica, parecem exercer o efeito analgésico através da interação com receptores opióides presentes nas terminações nociceptivas periféricas (FERREIRA e NAKAMURA, 1979a,b,c; FERREIRA et al., 1981; SMITH et al., 1982). Pelo fato dos agonistas opióides periféricos terem mínimo acesso ao SNC, eles podem ser muito úteis na prática clínica uma vez que eles são desprovidos de efeitos colaterais centrais típicos, como os causados por opióides semelhantes à morfina (como depressão respiratória e uso abusivo). Similarmente, o risco de efeitos adversos poderia ser reduzido com a 82 combinação de opióides periféricos e opióides semelhantes à morfina (SEVOSTIANOVA et al., 2005). Bentley et al. (1981) utilizando o modelo das contorções abdominais em camundongos, determinaram que a ação analgésica da morfina e de encefalinas dependiam, parcialmente, da atuação nas terminações de nervos sensoriais do peritônio. Estudos com a N-metilmorfina no mesmo modelo de nocicepção (writhing) ou na hiperalgesia produzida pela injeção intraplantar de PGE2 em ratos mostram que o efeito analgésico deste alcalóide opióide quaternário era revertido pelo prétratamento com naloxona ou com N-metilnalorfina (LORENZETTI e FERREIRA, 1982; MAGNAN et al., 1982; SMITH et al., 1982). A utilização de modelos de inflamação ocorre na quase totalidade dos estudos que caracterizam a atividade antinociceptiva opióide periférica (STEIN, 1993). Aparentemente, a ruptura do perineuro durante o processo inflamatório, além de induzir hiperalgesia, favorece o acesso e o efeito do opióide no terminal nervoso sensitivo (OLSSON, 1990, ANTONIJEVIC et al., 1995). Outros autores mostraram que, na inflamação, o número de receptores opióides está aumentado (supraregulação) nos terminais aferentes periféricos (HASSAN et al., 1993; SCHÄFER et al., 1995). Por outro lado, a diminuição do pH no local inflamatório aumenta a eficácia do agonista opióide em inibir a adenilato ciclase (SELLEY et al., 1993). Estudos in vitro e in vivo demostraram que receptores opióides modulam o eixo hipotálamo-hipófise-adrenal (HPA) e a liberação do fator liberador de corticotrofina (CRF) na porção endócrina do hipotálamo (PFEIFFER e PFEIFFER, 1984; NIKOLARAKIS et al., 1987). Por outro lado, a glândula adrenal armazena opióides endógenos, predominantemente encefalinas, que são liberadas junto com as catecolaminas durante o estímulo de estresse (NORTH e EGAN, 1983). Ferreira et al. (1995) mostraram que drogas com mecanismo farmacológico semelhante aos opióides periféricos podem agir através da liberação de opióides das glândulas adrenais. Além da possibilidade das adrenais liberarem peptídeos opióides, células do sistema imunológico também são capazes de fazê-lo. Durante a inflamação, leucócitos secretam peptídeos opióides que se ligam a receptores em neurônios 83 sensoriais periféricos e medeiam a antinocicepção. Esta secreção pode ser induzida por estresse ou injeção local de CRF (MOUSA et al., 2003). Em humanos, peptídeos opióides liberados localmente diminuem a intensidade da dor e o consumo voluntário de medicamentos analgésicos em condições de estresse pós-cirúrgico (STEIN et al., 1993). O mecanismo pelos quais os opióides induzem analgesia periférica envolve a participação dos receptores µ, κ e δ (FERREIRA e NAKAMURA, 1979a,b; FERREIRA et al., 1984; STEIN et al., 1988, 1989, 1990; KAYSER et al., 1991). Estudos sugerem que a analgesia promovida por ativação destes receptores possa ser mediada pela geração de NO a partir da L-arginina e ativação de guanilato ciclase, uma vez que os receptores opióides periféricos são acoplados a proteína G e acoplados à via da L-arginina-NO-GMPc (FERREIRA et al., 1991a,b, 1995; DUARTE et al., 1992; GRANADOS-SOTO et al., 1997; NOZAKI-TAGUCHI e YAMAMOTO, 1998). A morfina demonstrou exercer sua ação nociceptiva periférica através da ativação de canais de K+ sensíveis a ATP (RODRIGUES e DUARTE, 2000). Foi demonstrado que a ação antinociceptiva periférica do doador de óxido nítrico nitroprussiato de sódio e dibutiril GMPc está associada com canais de potássio sensíveis a ATP, estabelecendo, assim, uma ligação entre a participação da via do óxido nítrico/GMPc na analgesia induzida por certas drogas e a ativação de canais de potássio sensíveis a ATP (SOARES et al., 2000, 2001). A maioria dos leucócitos que contêm peptídeos opióides na fase inicial da inflamação é do tipo polimorfonuclear (PMN), enquanto que os monócitos predominam durante a fase mais tardia (RITTNER et al., 2001). Tanto esses leucócitos circulantes quanto o endotélio vascular expressam moléculas de adesão, que promovem a migração de leucócitos para o local da inflamação. No entanto, este importante passo na resolução do processo inflamatório pode ser bloqueado por anticorpos anti-moléculas de adesão, prejudicando assim a antinocicepção mediada por opióides (STEIN et al., 2003; MACHELSKA et al., 1998, 2002, 2004). Por outro lado, o recrutamento de leucócitos polimorfonucleares mediado por quimiocinas intensifica os processos de rolamento, diapedese e extravasamento de células, através do aumento da expressão das moléculas de adesão (ZHANG et al., 2001). 84 Muitos estudos em animais têm demonstrado efeitos antinociceptivos proeminentes dos opióides em modelos de dor persistente com componentes de inflamação tecidual (JORIS et al., 1987). Além disso, tem sido mostrado que esses efeitos antiinflamatórios são devidos, principalmente, à ativação de receptores opióides periféricos (STEIN et al., 1989), os quais, como dito anteriormente, sofrem supra-regulação durante a inflamação e são ativados por peptídeos opióides endógenos produzidos por células do sistema imunológico que migram para o tecido inflamado (STEIN et al., 1989; PRZEWLOCKI et al., 1992). O fato de o EES atuar no sistema opioidérgico periférico explica a inibição da segunda fase da formalina, porém não explica a inibição da primeira. A inibição da primeira fase da formalina poderia ser justificada, dentre muitas possibilidades, pela ação dos aminoácidos excitatórios (glutamato e aspartato) ou SP no sistema nervoso periférico Essas substâncias apresentam papel importante no processo de sensibilização do corno dorsal da medula espinhal, uma vez que a estimulação das fibras aferentes primárias induzindo a liberação desses transmissores no local (MILLAN, 1999, 2002). Outros estudos mostram que os aminoácidos excitatórios, quando injetados pela via intraplantar ou intratecal, promovem um comportamento nociceptivo (AANONSEN e WILSON, 1987; BEIRITH et al., 2002). O glutamato é o neurotransmissor encontrado em maior concentração nas sinapses excitatórias nos cérebros de mamíferos e possui um papel fisiológico (memória e aprendizado) e patológico (doenças neurodegenerativas e na dor aguda e crônica). Entretanto, apresenta uma importância mais relevante nas condições crônicas que envolvem a patogênese da dor neuropática (GAVIRAGHI, 2000). O glutamato atua em receptores localizados no sistema nervoso periférico através da interação com receptores NMDA e não NMDA por um mecanismo que depende da ativação da via L-arginina-óxido nítrico, causando, portanto, dor (BEIRITH, 2002). Assim, a administração espinhal de antagonista de SP e/ou glutamato é capaz de bloquear a facilitação do processo nociceptivo produzido pela estimulação direta ou prolongada das fibras C (DICKENSON e SULLIVAN, 1987a,b; CODERRE e MELZACK, 1991; MALMBERG e YAKSH, 1992; MAO et al., 1992; REN et al., 1992; YAMAMOTO e YAKSH, 1992). 85 Antagonistas de bradicinina (B1 e B2) também têm mostrado capazes de inibir a primeira fase da formalina (HUNKAAR e HOLE, 1987). A SP, neurocininas A e B podem estimular diferentes tipos de receptores taquicinérgicos (MAGGI et al., 1987; LAVIELLE et al., 1988; QUIRION e DAM, 1988). Quaisquer moléculas que antagonizam esses receptores são capazes de inibir a primeira fase da formalina. A segunda fase da formalina pode ser inibida também por agentes antiinflamatórios. Assim, a investigação dessa atividade também foi realizada através de um modelo que mede a atividade antiedematogênica e um outro que quantifica a inibição da migração de leucócitos para o foco inflamatório. O tecido conjuntivo possui uma população transitória e uma residente, as quais são influenciadas por processos inflamatórios e infecciosos e está intimamente associado a todos os tecidos do corpo, refletindo suas condições fisiológicas (BRITO et al., 1998; GAHMBERG et al., 1999; CUMMINGS, 1999; GARTNER e HIATT, 1999; BORINI e GUIMARÃES, 1999; CATÃO-DIAS e SINHORINI, 1999). Assim, uma agressão a um tecido adjacente ao tecido conjuntivo desencadeia a liberação de mediadores que atraem, para o local lesado, células da população transitória que, em cooperação com as células residentes, buscam inibir o processo inflamatório (VELO et al., 1973; KUHNS et al., 1992; FAITLWICZ, 1993; DOWNEY et al., 1995; MARINHO et al., 1997; BRITO et al., 1998; FAIÇAL e UEHARA, 1998; GAHMBERG et al., 1999; CUMMINGS, 1999; GARTNER e HIATT, 1999). A migração celular até o tecido injuriado ocorre por um complexo ciclo de eventos, ainda não totalmente esclarecidos, mas estudados há muitos anos, que permite às células se locomoverem do espaço vascular para o extravascular e dentro do espaço intersticial, exercendo suas funções de defesa em um padrão comportamental conhecido (DRUBIN e NETSONT, 1996; LAUFFENBURGER e HORWITZ, 1996; MITCHISON e CRAMER, 1996; ASSOIAN, 1997; BRITO et al., 1998; CATÃO-DIAS e SINHORINI, 1999; CUMMINGS, 1999; GAHMBERG et al., 1999; HYNES et al., 1999; BOROJEVIC, 1999; SMILENOV et al., 1999; SERVANT, 2000; JIN, 2000). Para a verificação da atividade antiedematogênica do EES e da fração FM foi utilizado o teste do edema de orelha induzido por óleo de cróton. Neste ensaio, a 86 intensidade do edema, induzida pela aplicação tópica de um agente irritante na cavidade auricular externa, é avaliada como parâmetro de atividade antiinflamatória de substâncias administradas anteriormente ao agente irritante. O óleo de cróton é um irritante que provoca a migração de leucócitos causando edemas intervasculares (SWINGLE et al., 1981). Ele possui na sua composição o acetato de tetradecanoilforbol que é um dos responsáveis por sua ação irritante e formação de edema e está relacionado ao aumento da permeabilidade vascular e exsudação plasmática (PUIGNERO et al., 1998; LAPA et al., 2003). Assim, devem-se considerar, ao interpretar os resultados deste ensaio, fatores que influenciam a formação do edema, tais como a perfusão vascular do tecido, pressão sanguínea sistêmica e nível de glicocorticóides, pois a intensidade do edema é o parâmetro observado quanto à ação antiinflamatória analisada (RATES e BARROS, 1994; SOUCCAR e LAPA, 1997). Na avaliação pelo teste de edema de orelha induzido pelo óleo de cróton, o tratamento prévio dos camundongos com EES e com a FM, em diferentes doses, bem como a dexametasona, controle positivo do teste, foram capazes de reduzir a formação do edema de maneira dose-dependente. A FM foi a fração escolhida para ser avaliada quanto à atividade antiinflamatória visto que ela foi a única das frações capaz de inibir o número de contorções abdominais induzidas pelo ácido acético. Contudo, os resultados obtidos não dão suporte para se afirmar em qual das fases do processo inflamatório se deu a intervenção do EES e da FM, pois um processo inflamatório caracteriza-se por três fases distintas (FREIRE, 1992), coexistentes a partir de um dado momento: uma fase aguda, onde ocorre vasodilatação, aumento da permeabilidade vascular e extravasamento líquido, tendo como principais mediadores a histamina, a serotonina, as cininas e os produtos do AA; uma fase tardia, onde ocorre quimiotaxia e migração celular; e uma fase crônica, caracterizada por degeneração e fibrose. Assim, não ficou evidente o mecanismo de ação do efeito antiedematogênico gerado pelo EES e pela FM, o qual pode ter várias explicações, dentre elas, uma ação inespecífica gerando vasoconstricção, feedback negativo por ligação a receptores autacóides, inibição da liberação de enzimas lisossômicas e estabilização das membranas celulares; ou por mecanismos de inibição da migração celular, se foi por uma ação direta sobre a cascata do AA, seja inibindo a COX e acil-hidrolases, 87 seja inibindo a fosforilação do complexo PLA2/anexina 1 em PLA2 ativada; ou se foi por ligação a receptores citoplasmáticos que enviam um sinal para o núcleo e promovem um mecanismo de ação gênica, ou mesmo através de outros mecanismos antiinflamatórios pouco estudados ou desconhecidos (FAIÇAL e UEHARA, 1998). A presença de alguns mecanismos de ação gênica, bem como da degeneração e fibrose estão descartados para tratamentos agudos, como os realizados por nós, entretanto não se pode desconsiderar a sua presença em tratamentos crônicos e, portanto, esta possibilidade deverá ser investigada futuramente. A redução da quimiotaxia é uma importante resposta observada em tratamentos nos quais se utilizam antiinflamatórios. A migração leucocitária está relacionada à ação do AA produzido através da ação da LOX sobre o mesmo previamente liberado pela PLA2 (BRAIN e WILLIANS, 1990; YOKOMIZO et al., 1995; BRITO et al., 1998; FAIÇAL e UEHARA, 1998; BORINI e GUIMARÃES, 1999; GAHMBERG et al., 1999; WAGNER e ROTH, 2000). A composição celular da inflamação está relacionada ao tipo de estímulo inflamatório e ao ambiente em que ela se desenvolve, estando os neutrófilos, por exemplo, associados a respostas rápidas a infecções e danos tissulares, e os eosinófilos a inflamações alérgicas, enquanto monócitos e linfócitos são normalmente recrutados na inflamação crônica (ADAMS e LLOYD, 1997). Os neutrófilos são as primeiras células leucocitárias a migrarem através do endotélio para o sítio inflamatório (WITKO-SARSAT et al., 2000). A inibição da COX pode resultar em migração leucocitária devido à conseqüente inibição da liberação de diversas interleucinas (ILs) e TNF, porém é mais provável que uma inibição de LOX ou de PLA2 resulte em uma redução na migração leucocitária, visto que um dos agentes quimiotáticos, como por exemplo, o leucotrieno B4 (LTB4), teria a sua produção diminuída. O aumento da quantidade de leucócitos no sítio inflamatório pode ser interpretado como uma ação pró-inflamatória, resultando em altas concentrações de enzimas lisossômicas e aumento no dano tissular (HIGGS et al., 1980). Mikami e Miyasaka (1983) demonstraram que, na pleurisia induzida por carragenina em ratos, o efeito antiexsudativo dos antiinflamatórios não esteroidais estaria associado principalmente à redução de PGE2 e, parcialmente, à redução da atividade de 88 enzimas lisossomais, enquanto a atividade antiinflamatória dos antiinflamatórios esteroidais seria explicada pela redução da migração leucocitária. A ação dos glicocorticóides pode estar relacionada à regulação da expressão de genes envolvidos na resposta inflamatória, havendo uma interação dos glicocorticóides com receptores específicos, formando dímeros que migram para o núcleo e se ligam a elementos de resposta aos esteróides, exercendo efeitos repressores ou indutores (BIOLA e PALLARDY, 2000; HANG et al., 2004; NORMAN et al., 2004). Podem, por exemplo, interagir com os genes responsáveis pela produção de diversas enzimas responsáveis pela formação de PGs, inibindo a transcrição do RNA mensageiro (RNAm) destas enzimas (NEWTON et. al., 1998), com o gene da anexina 1, induzindo sua expressão (WALLNER et al., 1986), além de interferirem na síntese e liberação de ILs e TNF (UTSUNOMIYA et al., 1994). A anexina 1 possui atividade inibidora da PLA2 intra e extracelular podendo ser considerada como segundo mensageiro responsável pela ação dos glicocorticóides no processo inflamatório (FAROOQUI et al., 1999). As ações da anexina 1 estão relacionadas à uma inibição direta da PL assim como uma inibição de sua ativação, além da inibição de outras enzimas relacionadas à inflamação, como a óxido nítrico sintase (NOS) e COX-2 (PARENTE e SOLITO, 2004). Para avaliar a atividade do EES na migração de leucócitos totais para o local da inflamação, utilizamos o teste de peritonite induzido por carragenina. A carragenina, um polissacarídeo sulfatado, desencadeia uma inflamação aguda envolvendo liberação seqüencial de vários mediadores pró-inflamatórios, principalmente a histamina, a serotonina, as cininas, as PGs e os tromboxanos (DI ROSA et al., 1971; DAMAS et al., 1990). Este modelo de inflamação aguda permite a quantificação de leucócitos que migram para a cavidade peritoneal sob a ação de agentes quimiotáticos, principalmente LTs e ILs, sendo sensível à ação de antiinflamatórios esteroidais (VINEGAR et al., 1973; HIGGS et al., 1980; MIKAMI e MIYASAKA, 1983; BROOKS e DAY, 1991). A administração oral de EES nas mesmas doses utilizadas no modelo das contorções abdominais reduziu de forma dose-dependente o número de leucócitos que migraram para a cavidade peritoneal, seguindo o perfil da dexametasona, 89 controle positivo do teste. Esse resultado, somado ao resultado positivo no teste de edema de orelha induzido por óleo de cróton sugere fortemente que o EES possui uma ação antiinflamatória, assim como a FM, de acordo com o modelo antiedematogênico. Essa ação antiinflamatória é capaz de reduzir a migração de leucócitos para o foco inflamatório, mostrando a possível interferência do EES nos mecanismos quimiotáticos. Durante os nossos experimentos, verificamos uma mudança no comportamento dos animais após a administração do EES, o que nos levou a estudar uma possível atividade depressora no SNC. Além disso, considerando que os modelos utilizados para a avaliação da atividade antinociceptiva sempre exigem respostas motoras dos animais, resultados falso positivos poderiam ser obtidos caso ocorresse incoordenação motora por sedação e/ou relaxamento muscular. O efeito do EES e das frações FH, FC e FM foi estudado no teste do rota-rod. Neste teste, o pré-tratamento dos animais com o EES e com as frações FH, FC e FM nas doses utilizadas no modelo das contorções abdominais induzidas pelo ácido acético não modificou o número de quedas, nem o tempo de permanência dos camundongos no rota-rod, excluindo as influências motoras. Este resultado indica também que o efeito antinociceptivo do EES e da FM não parece ser acompanhado de sedação ou relaxamento muscular. No teste do campo aberto, cinco parâmetros foram avaliados: o número de quadrados invadidos, o tempo parado, o número de levantadas, o número de autolimpeza e o número de bolos fecais. O EES, nas doses utilizadas, não alterou o número de quadrados invadidos, o que confirma a não influência do EES na atividade locomotora dos animais. O tempo parado, porém, foi diminuído, o que é sugestivo de uma atividade depressora do SNC, sendo que este efeito pode vir a ser um efeito ansiolítico, que ainda deve ser melhor investigado. O número de bolos fecais diminuído pode também indicar um efeito depressor do SNC, como também pode ter sido provocado por uma atividade anticolinérgica ou até mesmo por uma diminuição do peristaltismo provocada por agonistas opióides. Não houve alteração no número de levantadas e no número de auto-limpeza. 90 O teste do campo aberto também foi realizado com as frações FH, FC e FM. Nenhuma das frações foi capaz de alterar o número de quadrados invadidos, nem o número de levantadas, nem o número de auto-limpeza. Esse resultado é condizente ao observado com o EES, e também mostrou que as frações não alteraram a atividade locomotora dos animais. O tempo parado só foi aumentado pela FC, enquanto o número de bolos fecais só foi diminuído pela FH. Estes resultados condizem com o observado no mesmo experimento realizado com o EES, visto que os únicos parâmetros alterados, tanto para o EES como para as frações, foram o tempo parado e o número de bolos fecais. Ainda para verificar a atividade depressora do SNC, utilizou-se o modelo da potenciação do sono induzido por barbitúrico. Neste modelo, substâncias que deprimem o SNC, em geral, aumentam a duração do sono produzido pelo pentobarbital, ligante de sítios localizados nos receptores GABAérgicos do tipo A (CHWEH et al., 1987, LANCEL, 1999). Os benzodiazepínicos apresentam esse efeito, evidenciado principalmente pelo prolongamento da duração do sono induzido por agentes hipnóticos, o que foi bem observado nos resultados obtidos, quando o diazepam promoveu aumento do tempo de sono induzido por barbitúrico. Os nossos experimentos mostraram que o EES (25, 50 e 100 mg/kg) e as frações FH e FC, mas não a fração FM, foram capazes de potencializar o sono induzido por barbitúrico, sugerindo um efeito depressor do SNC do EES e das frações FH e FC. Esse efeito depressor do SNC observado no EES e nas frações FH e FC ainda foram confirmados pelos dados obtidos através do teste do campo aberto. A dose das frações administradas foi o equivalente a aproximadamente o dobro da maior dose utilizada com o EES (100 mg/kg), entretanto, em termos percentuais, o EES na dose de 100 mg/kg foi capaz de potencializar a ação do pentobarbital mais do que as frações FH e FC isoladamente, sugerindo que o efeito das frações FH e FC sejam aditivos. Os efeitos depressores do SNC das frações FH e FC condizem com as características químicas das substâncias nelas presentes, visto que o n-hexano arrasta substâncias de baixa polaridade e o clorofórmio arrasta substâncias de polaridade intermediária. Assim, como é sabido, para uma ação no SNC, as substâncias têm que penetrar a barreira hematoencefálica e, geralmente, somente 91 moléculas de baixa polaridade tendem a atravessá-la, como o que parece ter acontecido com as frações FH e FC. Além disso, a fração FM, que possui moléculas de alta polaridade, não apresentou efeito depressor do SNC. Uma possibilidade que não pode ser descartada é a de que a potenciação do sono induzido pelo EES e pelas frações FH e FC pode ter ocorrido devido a uma interferência de ordem farmacocinética, o que pode levar a um resultado falso positivo na avaliação do efeito depressor do SNC nesse modelo utilizado (VIEIRA, 2001). As enzimas hepáticas, envolvidas no processo de metabolização de diversos compostos, são importantes para a metabolização dos barbitúricos no organismo animal (PACIFICI, 1995), sendo também usadas na metabolização de outros xenobióticos como os constituintes de plantas ditas medicinais. A este respeito, há alguns trabalhos mostrando a interação farmacocinética entre espécies de plantas, como Smilax sp, Eucalyptus globulus, Blupeurum falcatum, Piper methysticum, Stachytarpheta cayennensis, entre outras, com fármacos tais como diazepam, álcool etílico e barbitúricos (BLUMENTHAL, 2000; BLUMENTHAL et al., 1998; CUPP, 1999; HU et al., 2005; VIEIRA, 2001). Assim, a potenciação do sono induzido por barbitúrico poderia ser resultado de uma interferência, competição ou inibição do sistema microssomal hepático, o qual metaboliza o pentobarbital sódico. A administração de Hypericum perforatum, por exemplo, afeta o sistema micromossomal hepático P450, aumentando a atividade da isoenzima CYP3A4, o que reduz, possivelmente, a atividade de fármacos que são substratos para essa enzima e que são administrados simultaneamente com a mesma, incluindo anti-histamínicos não-sedativos, contraceptivos orais, alguns antiretrovirais, antiepilépticos, bloqueadores de canais de cálcio, ciclosporina, alguns quimioterápicos, antibióticos macrolídeos, e antifúngicos (ROBY et al., 2000). Alguns compostos presentes no EES e nas frações FH e FC poderiam estar contribuindo para esse efeito potencializador do sono induzido por barbitúrico. A possibilidade de interferência farmacocinética do EES e das frações FH e FC sobre as enzimas hepáticas deve ser melhor investigada para se compreender seus efeitos farmacológicos, mas principalmente pela questão de segurança farmacológica, já que o uso concomitante com muitos outros fármacos é muito comum, e pode interferir com seus efeitos terapêuticos. Uma maneira possível para 92 descartar essa possível interferência farmacocinética seria a realização do teste de potenciação do sono induzido por éter, visto que essa substância não sofre metabolização do sistema enzimático P450. Faz-se necessário, portanto, investigar de modo mais profundo a possível atividade depressora do SNC do EES e das frações FH e FC e tentar comprovar ou descartar uma possível atividade ansiolítica e uma possível capacidade de inibição enzimática. Além disso, uma maior purificação das frações se faz necessária, a fim de se tentar isolar e identificar os princípios ativos responsáveis pelas ações farmacológicas encontradas neste estudo. Conclusões 94 7. CONCLUSÕES 1. O EES possui atividade antinociceptiva e antiinflamatória, além de uma possível atividade depressora do SNC. 2. A atividade antinociceptiva do EES envolve a ação do sistema opióide periférico, sendo essa ação através de atividade agonista opióide periférica ou através da indução da liberação de peptídeos opióides periféricos. 3. A atividade antiinflamatória do EES envolve mecanismos capazes de reduzir a formação de edemas e o número de leucócitos totais que migram para o foco inflamatório. 4. A FM é a fração responsável pelas atividades antinociceptiva e antiinflamatória. 5. As frações FH e FC são as frações responsáveis pelo possível efeito depressor do SNC. 6. Este estudo respalda o uso popular do Synadenium umbellatum como analgésico e antiinflamatório. Referências Bibliográficas Bibliográficas 96 REFERÊNCIAS BIBLIOGRÁFICAS AANONSEN, L. M.; WILCOX, G. L. Nociceptive action of excitatory amino acids in the mouse: effects of spinally administered opioids, phencyclidine and sigma agonists. J Pharmacol Exp Ther, v. 243, n. 1, p.9-19, 1987. ADAMS, D. H.; LLOYD, A. R. Chemokines: leucocyte recruitment and activation cytokines. Lancet, v. 349, n. 9050, p.490-5, 1997. ALMEIDA, T. F.; ROIZENBLATT, S.; TUFIK, S. Afferent pain pathways: a neuroanatomical review. Brain Res, v. 1000, n. 1-2, p.40-56, 2004. ALMEIDA, R. N. Psicofarmacologia: fundamentos práticos. Rio de Janeiro: Guanabara Koogan, 2006. 969 p. ALREJA, M.; MUTALIK, P.; NAYAR, U.; MANCHANDA, S. K. The formalin test: a tonic pain model in the primate. Pain, v. 20, n. 1, p.97-105, 1984. ANTONIJEVIC, I.; MOUSA, S. A.; SCHAFER, M.; STEIN, C. Perineurial defect and peripheral opioid analgesia in inflammation. J Neurosci, v. 15, n. 1 Pt 1, p.16572, 1995. ARCHER, J. Tests for emotionality in rats and mice: a review. Anim Behav, v. 21, n. 2, p.205-35, 1973. ASSOIAN, R. K. Anchorage-dependent cell cycle progression. J Cell Biol, v. 136, n. 1, p.1-4, 1997. BABBEDGE, R. C.; HART, S. L.; MOORE, P. K. Anti-nociceptive activity of nitric oxide synthase inhibitors in the mouse: dissociation between the effect of LNAME and L-NMMA. J Pharm Pharmacol, v. 45, n. 1, p.77-9, 1993. BARNES, P. J.; CHUNG, K. F.; PAGE, C. P. Inflammatory mediators and asthma. Pharmacol Rev, v. 40, n. 1, p.49-84, 1988. BASBAUM, A. I.; JESSELL, T. M. The perception of pain. In: Kandel, E. R.; Schwartz, J. H.; Jessell, T. M. (Ed.). Principles of Neral Science. New York: McGraw Hill, 2000. p.472-491. BEIRITH, A.; SANTOS, A. R.; CALIXTO, J. B. Mechanisms underlying the nociception and paw oedema caused by injection of glutamate into the mouse paw. Brain Res, v. 924, n. 2, p.219-28, 2002. BENTLEY, G. A.; NEWTON, S. H.; STARR, J. Evidence for an action of morphine and the enkephalins on sensory nerve endings in the mouse peritoneum. Br J Pharmacol, v. 73, n. 2, p.325-32, 1981. 97 BERKENKOPF, J. W.; WEICHMAN, B. M. Production of prostacyclin in mice following intraperitoneal injection of acetic acid, phenylbenzoquinone and zymosan: its role in the writhing response. Prostaglandins, v. 36, n. 5, p.693709, 1988. BERTOLINI, A.; OTTANI, A.; SANDRINI, M. Dual acting anti-inflammatory drugs: a reappraisal. Pharmacol Res, v. 44, n. 6, p.437-50, 2001. BESSON, J. M. The complexity of physiopharmacologic aspects of pain. Drugs, v. 53 Suppl 2, p.1-9, 1997. BESSON, J. M. The neurobiology of pain. Lancet, v. 353, n. 9164, p.1610-5, 1999. BESSON, J. M.; CHAOUCH, A. Peripheral and spinal mechanisms of nociception. Physiol Rev, v. 67, n. 1, p.67-186, 1987. BIOLA, A.; PALLARDY, M. Mode of action of glucocorticoids. Presse Med, v. 29, n. 4, p.215-23, 2000. BLANE, G. F. Blockade of bradykinin-induced nociception in the rat as a test for analgesic drugs with particular reference to morphine antagonists. J Pharm Pharmacol, v. 19, n. 6, p.367-73, 1967. BLUMBERG, H.; WOLF, P. S.; DAYTON, H. B. Use of writhing test for evaluating analgesic activity of narcotic antagonists. Proc Soc Exp Biol Med, v. 118, p.7636, 1965. BLUMENTHAL, M. Interactions between herbs and conventional drugs: introductory considerations. Herbal Gram, v. 49, p.52-63, 2000. BLUMENTHAL, M.; BUSSE, W. R.; GOLDBERG, A.; GRUENWALD, J.; HALL, T.; RIGGINS, C. W.; RISTER, R. S. The complete German Commission E Monographs: therapeutic guide in herbal medicines, American Botanical Council. Boston: Integrative Medicine Communications, 1998 BONANNO, G.; RAITERI, M. Multiple GABAB receptors. Trends Pharmacol Sci, v. 14, n. 7, p.259-61, 1993. BONNET, J.; LOISEAU, A. M.; ORVOEN, M.; BESSIN, P. Platelet-activating factor acether (PAF-acether) involvement in acute inflammatory and pain processes. Agents Actions, v. 11, n. 6-7, p.559-62, 1981. BORGLAND, S. L. Acute opioid receptor desensitization and tolerance: is there a link? Clin Exp Pharmacol Physiol, v. 28, n. 3, p.147-54, 2001. BORINI, P.; GUIMARAES, R. C. Indicators of inflammation and cellular damage in chronic asymptomatic or oligosymptomatic alcoholics: correlation with alteration of bilirubin and hepatic and pancreatic enzymes. Rev Hosp Clin Fac Med Sao Paulo, v. 54, n. 2, p.53-60, 1999. 98 BORINI, P.; GUIMARAES, R. C. Indicators of inflammation and cellular damage in chronic asymptomatic or oligosymptomatic alcoholics: correlation with alteration of bilirubin and hepatic and pancreatic enzymes. Rev Hosp Clin Fac Med Sao Paulo, v. 54, n. 2, p.53-60, 1999. BOROJEVIC, R. Extracellular matrix: understanding the complexity. Braz J Med Biol Res, v. 32, n. 5, p.497-9, 1999. BOWERY, N. G. GABAB receptor pharmacology. Annu Rev Pharmacol Toxicol, v. 33, p.109-47, 1993. BOYTON, R. J.; OPENSHAW, P. J. Pulmonary defences to acute respiratory infection. Br Med Bull, v. 61, p.1-12, 2002. BRANDT, N. N.; CHIKISHEV, A. Y.; SOTNIKOV, A. I.; SAVOCHKINA, Y. A.; AGAPOV, I. I.; TONEVITSKY, A. G. Ricin, ricin agglutinin, and the ricin binding subunit structural comparison by Raman spectroscopy. Journal of Molecular Structure, v. 735–736, p.293–298, 2005. BRAIN, S. D.; WILLIAMS, T. J. Leukotrienes and inflammation. Pharmacol Ther, v. 46, n. 1, p.57-66, 1990. BRITO, G. A.; FALCAO, J. L.; SARAIVA, S. N.; LIMA, A. A.; FLORES, C. A.; RIBEIRO, R. A. Histopathological analysis of rat mesentery as a method for evaluating neutrophil migration: differential effects of dexamethasone and pertussis toxin. Braz J Med Biol Res, v. 31, n. 10, p.1319-27, 1998. BROCHE, F.; TELLADO, J. M. Defense mechanisms of the peritoneal cavity. Curr Opin Crit Care, v. 7, n. 2, p.105-16, 2001. BROOKS, P. M.; DAY, R. O. Nonsteroidal antiinflammatory drugs - differences and similarities. N Engl J Med, v. 324, n. 24, p.1716-25, 1991. CALIXTO, J. B.; BEIRITH, A.; FERREIRA, J.; SANTOS, A. R.; FILHO, V. C.; YUNES, R. A. Naturally occurring antinociceptive substances from plants. Phytother Res, v. 14, n. 6, p.401-18, 2000. CALIXTO, J. B.; MEDEIROS, R.; FERNANDES, E. S.; FERREIRA, J.; CABRINI, D. A.; CAMPOS, M. M. Kinin B1 receptors: key G-protein-coupled receptors and their role in inflammatory and painful processes. Br J Pharmacol, v. 143, n. 7, p.803-18, 2004. CAO, Y. Q.; MANTYH, P. W.; CARLSON, E. J.; GILLESPIE, A. M.; EPSTEIN, C. J.; BASBAUM, A. I. Primary afferent tachykinins are required to experience moderate to intense pain. Nature, v. 392, n. 6674, p.390-4, 1998. CARLI, G.; FARABOLLINI, F.; FONTANI, G. Effects of pain, morphine and naloxone on the duration of animal hypnosis. Behav Brain Res, v. 2, n. 3, p.373-85, 1981. CARR, D. B.; GOUDAS, L. C. Acute pain. Lancet, v. 353, n. 9169, p.2051-8, 1999. 99 CATÃO-DIAS, L. J.; SINHORINI, I. L. Influence of low environmental temperature on inflammation in Bullfrog (Rana catesbeiana): qualitavive and quantitative evaluation. Braz. J. Vet. Res. Anim. Sci., v. 36, n. 2, p.0-0, 1999. CELOTTI, F.; LAUFER, S. Anti-inflammatory drugs: new multitarget compounds to face an old problem. The dual inhibition concept. Pharmacol Res, v. 43, n. 5, p.429-36, 2001. CHAU, T. T. Analgesic testing in animals models. Pharmacol. Meth. Control Inflamm. p.195-212, 1989. CHENG, H. Y.; PITCHER, G. M.; LAVIOLETTE, S. R.; WHISHAW, I. Q.; TONG, K. I.; KOCKERITZ, L. K.; WADA, T.; JOZA, N. A.; CRACKOWER, M.; GONCALVES, J.; SAROSI, I.; WOODGETT, J. R.; OLIVEIRA-DOS-SANTOS, A. J.; IKURA, M.; VAN DER KOOY, D.; SALTER, M. W.; PENNINGER, J. M. DREAM is a critical transcriptional repressor for pain modulation. Cell, v. 108, n. 1, p.31-43, 2002. CHERNOV, H. I.; WILSON, D. E.; FOWLER, W. F.; PLUMMER, A. J. Non-specificity of the mouse writhing test. Arch Int Pharmacodyn Ther, v. 167, n. 1, p.171-8, 1967. CHUANG, H. H.; PRESCOTT, E. D.; KONG, H.; SHIELDS, S.; JORDT, S. E.; BASBAUM, A. I.; CHAO, M. V.; JULIUS, D. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature, v. 411, n. 6840, p.957-62, 2001. CHWEH, A. Y.; SWINYARD, E. A.; WOLF, H. H. Hypnotic action of pentobarbital in mice: a possible mechanism. Exp Neurol, v. 97, n. 1, p.70-6, 1987. CIOMS - International Guiding Principles for Biomedical Research Involving Animals. Altern Lab Anim, v. 12, n. 4, 1985. CLARK, R. A.; GALLIN, J. I.; KAPLAN, A. P. The selective eosinophil chemotactic activity of histamine. J Exp Med, v. 142, n. 6, p.1462-76, 1975. CODERRE, T. J.; MELZACK, R. Central neural mediators of secondary hyperalgesia following heat injury in rats: neuropeptides and excitatory amino acids. Neurosci Lett, v. 131, n. 1, p.71-4, 1991. COLEMAN, J. W. Nitric oxide in immunity and inflammation. Int Immunopharmacol, v. 1, n. 8, p.1397-406, 2001. COLLIER, H. O.; DINNEEN, L. C.; JOHNSON, C. A.; SCHNEIDER, C. The abdominal constriction response and its suppression by analgesic drugs in the mouse. Br J Pharmacol Chemother, v. 32, n. 2, p.295-310, 1968. COOKSON, W. The immunogenetics of asthma and eczema: a new focus on the epithelium. Nat Rev Immunol, v. 4, n. 12, p.978-88, 2004. 100 CRANE, B. R.; ARVAI, A. S.; GACHHUI, R.; WU, C.; GHOSH, D. K.; GETZOFF, E. D.; STUEHR, D. J.; TAINER, J. A. The structure of nitric oxide synthase oxygenase domain and inhibitor complexes. Science, v. 278, n. 5337, p.425-31, 1997. CUMMINGS, R. D. Structure and function of the selectin ligand PSGL-1. Braz J Med Biol Res, v. 32, n. 5, p.519-28, 1999. CUPP, M. J. Herbal remedies: adverse effects and drug interactions. Am Fam Physician, v. 59, n. 5, p.1239-45, 1999. DAISUKE, U.; YOSHIMASA, H. The structure of kansuinine A, a new miltoxygenated diterpene from Euphorbai kansui. Tetrahedron Lett, v. 21, n., p.1697-1700, 1975. DALLOB, A.; GUINDON, Y.; GOLDENBERG, M. M. Pharmacological evidence for a role of lipoxygenase products in platelet-activating factor (PAF)-induced hyperalgesia. Biochem Pharmacol, v. 36, n. 19, p.3201-4, 1987. DAMAS, J.; BOURDON, V.; REMACLE-VOLON, G.; ADAM, A. Kinins and peritoneal exudates induced by carrageenin and zymosan in rats. Br J Pharmacol, v. 101, n. 2, p.418-22, 1990. D'AMOUR, F. E.; SMITH, J. A method for determining loss of pain sensation. J. Pharmacol. Exp. Ther., v. 72, n., p.74-79, 1941. DE ESCH, I. J.; THURMOND, R. L.; JONGEJAN, A.; LEURS, R. The histamine H4 receptor as a new therapeutic target for inflammation. Trends Pharmacol Sci, v. 26, n. 9, p.462-9, 2005. DI ROSA, M.; GIROUD, J. P.; WILLOUGHBY, D. A. Studies of the mediators of acute inflammatory response induced in rats in different sites by carrageenin and turpentine. J. Phathol., v. 104, p.15-29, 1971. DI VAIO, M. A. V.; FREITAS, A. C. C. Inflamação, tratamento e avanços recentes na terapia de doenças inflamatórias. Rev Ciênc Biol Saúde, v. 2, n. 1, p.37-67, 2001. DICKENSON, A. H.; SULLIVAN, A. F. Peripheral origins and central modulation of subcutaneous formalin-induced activity of rat dorsal horn neurones. Neurosci Lett, v. 83, n. 1-2, p.207-11, 1987a. DICKENSON, A. H.; SULLIVAN, A. F. Subcutaneous formalin-induced activity of dorsal horn neurones in the rat: differential response to an intrathecal opiate administered pre or post formalin. Pain, v. 30, n. 3, p.349-60, 1987b. DJOUHRI, L.; LAWSON, S. N. Abeta-fiber nociceptive primary afferent neurons: a review of incidence and properties in relation to other afferent A-fiber neurons in mammals. Brain Res Rev, v. 46, n. 2, p.131-45, 2004. 101 DOAK, G. J.; SAWYNOK, J. Formalin-induced nociceptive behavior and edema: involvement of multiple peripheral 5-hydroxytryptamine receptor subtypes. Neuroscience, v. 80, n. 3, p.939-49, 1997. DOWNEY, G. P.; FUKUSHIMA, T.; FIALKOW, L. Signaling mechanisms in human neutrophils. Curr Opin Hematol, v. 2, n. 1, p.76-88, 1995. DRUBIN, D. G.; NELSON, W. J. Origins of cell polarity. Cell, v. 84, n. 3, p.335-44, 1996. DUARTE, I. D. G.; SANTOS, I. R.; LORENZETTI, B. B.; FERREIRA, S. H. Analgesia by direct antagonism of nociceptor sensitisation involves the arginine-nitric oxidecGMP pathway. European Journal of Pharmacology, v. 217, p.225-227, 1992. DUBUISSON, D.; DENNIS, S. G. The formalin test: a quantitative study of the analgesic effects of morphine, meperidine, and brain stem stimulation in rats and cats. Pain, v. 4, n. 2, p.161-74, 1977. ECKHARDT, E. T.; CHEPLOVITZ, F.; LIPO, M.; GOVIER, W. M. Etiology of chemically induced writhing in mouse and rat. Proc Soc Exp Biol Med, v. 98, n. 1, p.186-8, 1958. EMELE, J. F.; SHANAMAN, J. Bradykinin writhing: a method for measuring analgesia. Proc Soc Exp Biol Med, v. 114, p.680-2, 1963. FAIÇAL, S.; UEHARA, M. H. Efeitos sistêmicos e síndrome de retirada em tomadores crônicos de glicocorticóides. Rev. Assoc. Med. Bra., v. 44, p.01, 1998. FAITLWICZ, A. R. Neutrófilos: novos agentes nos seus mecanismos de ação e aplicações clínicas. Revista da Associação Médica Brasileira, v. 39, p.243248, 1993. FARNSWORTH, N. R.; AKERELE, O.; BINGEL, A. S.; SOEJARTO, D. D.; GUO, Z. Medicinal plants in therapy. Bull World Health Organ, v. 63, n. 6, p.965-81, 1985. FAROOQUI, A. A.; LITSKY, M. L.; FAROOQUI, T.; HORROCKS, L. A. Inhibitors of intracellular phospholipase A2 activity: their neurochemical effects and therapeutical importance for neurological disorders. Brain Res Bull, v. 49, n. 3, p.139-53, 1999. FERRANDIZ, M. L.; ALCARAZ, M. J. Anti-inflammatory activity and inhibition of arachidonic acid metabolism by flavonoids. Agents Actions, v. 32, n. 3-4, p.2838, 1991. FERREIRA, S. H.; DUARTE, I. D.; LORENZETTI, B. B. The molecular mechanism of action of peripheral morphine analgesia: stimulation of the cGMP system via nitric oxide release. Eur J Pharmacol, v. 201, n. 1, p.121-2, 1991a. 102 FERREIRA, S. H.; DUARTE, I. D.; LORENZETTI, B. B. Molecular base of acetylcholine and morphine analgesia. Agents Actions Suppl, v. 32, p.101-6, 1991b. FERREIRA, S. H.; LORENZETTI, B. B.; DEVISSAGUET, M.; LESIEUR, D.; TSOUDEROS, Y. S14080, a peripheral analgesic acting by release of an endogenous circulating opioid-like substance. British Journal of Pharmacology, v. 114, n., p.303-308, 1995. FERREIRA, S. H.; LORENZETTI, B. B.; RAE, G. A. Is methylnalorphinium the prototype of an ideal peripheral analgesic? Eur J Pharmacol, v. 99, n. 1, p.23-9, 1984. FERREIRA, S. H.; MOLINA, N.; VETTORE, O. Prostaglandin hyperalgesia, V: a peripheral analgesic receptor for opiates. Prostaglandins, v. 23, n. 1, p.53-60, 1982. FERREIRA, S. H.; NAKAMURA, M. II - Prostaglandin hyperalgesia: the peripheral analgesic activity of morphine, enkephalins and opioid antagonists. Prostaglandins, v. 18, n. 2, p.191-200, 1979a. FERREIRA, S. H.; NAKAMURA, M. III - Prostaglandin hyperalgesia: relevance of the peripheral effect for the analgesic action of opioid-antagonists. Prostaglandins, v. 18, n. 2, p.201-8, 1979b. FERREIRA, S. H.; NAKAMURA, M. Pathogenesis and pharmacology of pain. In: Wessman, G.; Samuelsson, B.; Paoletti, R. (Ed.). Advances in Inflammatory Research. New York: Raven Press, 1979c. p.317-329. FERRI, P. H. Química de produtos naturais: métodos gerais. In: Di Stasi, L. C. (Ed.). Plantas medicinais: arte e ciência. Um guia de estudo interdisciplinar. Minas Gerais: Universidade Federal de Viçosa, 1996. p.129-156. FIORUCCI, S.; MELI, R.; BUCCI, M.; CIRINO, G. Dual inhibitors of cyclooxygenase and 5-lipoxygenase. A new avenue in anti-inflammatory therapy? Biochem Pharmacol, v. 62, n. 11, p.1433-8, 2001. FOORD, S. M.; BONNER, T. I.; NEUBIG, R. R.; ROSSER, E. M.; PIN, J. P.; DAVENPORT, A. P.; SPEDDING, M.; HARMAR, A. J. International Union of Pharmacology. XLVI. G protein-coupled receptor list. Pharmacol Rev, v. 57, n. 2, p.279-88, 2005. FOSSLIEN, E. Adverse effects of nonsteroidal anti-inflammatory drugs on the gastrointestinal system. Ann Clin Lab Sci, v. 28, n. 2, p.67-81, 1998. FRANKS, N. P.; LIEB, W. R. Molecular and cellular mechanisms of general anaesthesia. Nature, v. 367, n. 6464, p.607-14, 1994. 103 FREIRE, S. M. F. Atividades analgésica, antiinflamatória e cardiovascular da Scoparia dulcis (Vassorinha). Estudos químicos e farmacológicos. 1992. 120 p. Dissertação (Mestrado em Farmacologia) - Universidade Federal de São Paulo - Escola Paulista de Medicina, São Paulo, 1992. FUJIMAKI, H.; KAWAGOE, A.; BISSONNETTE, E.; BEFUS, D. Mast cell response to formaldehyde. 1. Modulation of mediator release. Int Arch Allergy Immunol, v. 98, n. 4, p.324-31, 1992. GAHMBERG, C. G.; VALMU, L.; TIAN, L.; KOTOVUORI, P.; FAGERHOLM, S.; KOTOVUORI, A.; KANTOR, C.; HILDEN, T. Leukocyte adhesion - a fundamental process in leukocyte physiology. Braz J Med Biol Res, v. 32, n. 5, p.511-7, 1999. GAMBARO, G. Strategies to safely interfere with prostanoid activity while avoiding adverse renal effects: could COX-2 and COX-LOX dual inhibition be the answer? Nephrol Dial Transplant, v. 17, n. 7, p.1159-62, 2002. GARTNER, L. P.; HIATTI, J. L. Tratado de Histologia. Rio de Janeiro: Guanabara Koogan, 1999 GAVIRAGHI, G. Excitatory amino acid receptors. Pharm Acta Helv, v. 74, n. 2-3, p.219-20, 2000. GILROY, D. W.; LAWRENCE, T.; PERRETTI, M.; ROSSI, A. G. Inflammatory resolution: new opportunities for drug discovery. Nat Rev Drug Discov, v. 3, n. 5, p.401-16, 2004. GOLDENBERG, M. M. Celecoxib, a selective cyclooxygenase-2 inhibitor for the treatment of rheumatoid arthritis and osteoarthritis. Clin Ther, v. 21, n. 9, p.1497513, 1999. GONZALES-TRUJANO, M. E.; NAVARRETE, C. A.; REYES, B.; HONG, E. Some pharmacological effects of ethanol extract of leaves of Annona diversifolia on the centrous nervous system in mice. Phytother. Res., v. 2, p.600-602, 1998. GOODMAN; GILMAN. The Pharmacological Basis of Therapeutics. Ed. 11: McGraw-Hill, 2005. 1984 p. GRANADOS-SOTO, V.; RUFINO, M. O.; GOMES LOPES, L. D.; FERREIRA, S. H. Evidence for the involvement of the nitric oxide-cGMP pathway in the antinociception of morphine in the formalin test. Eur J Pharmacol, v. 340, n. 2-3, p.177-80, 1997. GRUBB, B. D. Peripheral and central mechanisms of pain. Br J Anaesth, v. 81, n. 1, p.8-11, 1998. GUPTA, R. A.; DUBOIS, R. N. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer, v. 1, n. 1, p.11-21, 2001. 104 GUYTON, A. C.; HALL, J. E. Tratado de Fisiologia Médica. Ed. 11. Rio de Janeiro: Guanabara Koogan, 2005. 1118 p. HALEY, J. E.; DICKENSON, A. H.; SCHACHTER, M. Electrophysiological evidence for a role of nitric oxide in prolonged chemical nociception in the rat. Neuropharmacology, v. 31, n. 3, p.251-8, 1992. HANG, H. P.; DALE, M. M.; RITTER, J. M.; MOORE, P. K. Farmacologia. Ed. 5. Rio de Janeiro: Elsevier, 2004. 904 p. HANSSON, G. K. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med, v. 352, n. 16, p.1685-95, 2005. HAO, J. X.; XU, X. J. Treatment of a chronic allodynia-like response in spinally injured rats: effects of systemically administered nitric oxide synthase inhibitors. Pain, v. 66, n. 2-3, p.313-9, 1996. HARADA, T.; TAKAHASHI, H.; KAYA, H.; INOKI, R. A test for analgesics as an indicator of locomotor activity in writhing mice. Arch Int Pharmacodyn Ther, v. 242, n. 2, p.273-84, 1979. HARRISON, C.; SMART, D.; LAMBERT, D. G. Stimulatory effects of opioids. Br J Anaesth, v. 81, n. 1, p.20-8, 1998. HASSAN, A. H.; ABLEITNER, A.; STEIN, C.; HERZ, A. Inflammation of the rat paw enhances axonal transport of opioid receptors in the sciatic nerve and increases their density in the inflamed tissue. Neuroscience, v. 55, n. 1, p.185-95, 1993. HEAPY, C. G.; JAMIESON, A.; RUSSEL, N. J. W. Afferent C-fiber and A-delta activity in models of inflammation. Br. J. Pharmacol., v. 90, p.164, 1987. HENDERSHOT, L. C.; FORSAITH, J. Antagonism of the frequency of phenylquinone-induced writhing in the mouse by weak analgesics and nonanalgesics. J Pharmacol Exp Ther, v. 125, n. 3, p.237-40, 1959. HENRY, J. L.; YASHPAL, K.; PITCHER, G. M.; CODERRE, T. J. Physiological evidence that the 'interphase' in the formalin test is due to active inhibition. Pain, v. 82, n. 1, p.57-63, 1999. HERZ, A.; TESCHEMACHER, H. J. Activities and sites of antinociceptive action of morphine-like analgesics and kinetics of distribution following intravenous, intracerebral and intraventricular application. Adv. Drug Res., v. 6, p.79-119, 1971. HIGGS, G. A.; EAKINS, K. E.; MUGRIDGE, K. G.; MONCADA, S.; VANE, J. R. The effects of non-steroid anti-inflammatory drugs on leukocyte migration in carrageenin-induced inflammation. Eur J Pharmacol, v. 66, n. 1, p.81-6, 1980. HOURANI, S. M.; CUSACK, N. J. Pharmacological receptors on blood platelets. Pharmacol Rev, v. 43, n. 3, p.243-98, 1991. 105 HU, Z.; YANG, X.; HO, P. C.; CHAN, S. Y.; HENG, P. W.; CHAN, E.; DUAN, W.; KOH, H. L.; ZHOU, S. Herb-drug interactions: a literature review. Drugs, v. 65, n. 9, p.1239-82, 2005. HUGHES, R. A.; SUFKA, K. J. Morphine hyperalgesic effects on the formalin test in domestic fowl (Gallus gallus). Pharmacol Biochem Behav, v. 38, n. 2, p.247-51, 1991. HUNSKAAR, S.; BERGE, O. G.; HOLE, K. Dissociation between antinociceptive and anti-inflammatory effects of acetylsalicylic acid and indomethacin in the formalin test. Pain, v. 25, n. 1, p.125-32, 1986. HUNSKAAR, S.; FASMER, O. B.; HOLE, K. Formalin test in mice, a useful technique for evaluating mild analgesics. J Neurosci Methods, v. 14, n. 1, p.69-76, 1985. HUNSKAAR, S.; HOLE, K. The formalin test in mice: dissociation between inflammatory and non-inflammatory pain. Pain, v. 30, n. 1, p.103-14, 1987. HYNES, R. O.; BADER, B. L.; HODIVALA-DILKE, K. Integrins in vascular development. Braz J Med Biol Res, v. 32, n. 5, p.501-10, 1999. IADAROLA, J. M.; CAUDLE, R. M. Good pain, bad pain. Science, v. 278, n. 5336, p.239-40, 1997. IASP. Classification of chronic pain: descriptors of chronic pain syndromes and definitions of pain terms. Ed. 2. Seatle: IASP Press, 1994. INOUE, T.; MASHIMO, T.; SHIBUTA, S.; YOSHIYA, I. Intrathecal administration of a new nitric oxide donor, NOC-18, produces acute thermal hyperalgesia in the rat. J Neurol Sci, v. 153, n. 1, p.1-7, 1997. ISHII, S.; SHIMIZU, T. Platelet-activating factor (PAF) receptor and genetically engineered PAF receptor mutant mice. Prog Lipid Res, v. 39, n. 1, p.41-82, 2000. JAGER, A. K.; HUTCHINGS, A.; VAN STADEN, J. Screening of Zulu medicinal plants for prostaglandin-synthesis inhibitors. J Ethnopharmacol, v. 52, n. 2, p.95100, 1996. JANSSEN, P. A.; NIEMEGEERS, C. J.; DONY, J. G. The inhibitory effect of fentanyl and other morphine-like analgesics on the warm water induced tail withdrawl reflex in rats. Arzneimittelforschung, v. 13, p.502-7, 1963. JIN, T.; ZHANG, N.; LONG, Y.; PARENT, C. A.; DEVREOTES, P. N. Localization of the G protein betagamma complex in living cells during chemotaxis. Science, v. 287, n. 5455, p.1034-6, 2000. JOLY, A. B. Botânica: introdução à taxonomia vegetal. . Ed. 4. São Paulo, 1977. 106 JORDAN, B.; DEVI, L. A. Molecular mechanisms of opioid receptor signal transduction. Br J Anaesth, v. 81, n. 1, p.12-9, 1998. JORIS, J. L.; DUBNER, R.; HARGREAVES, K. M. Opioid analgesia at peripheral sites: a target for opioids released during stress and inflammation? Anesth Analg, v. 66, n. 12, p.1277-81, 1987. JULÉMONT, F.; DOGNÉ, J. M.; LAECKMANN, D.; PIROTTE, B.; LEVAL, X. D. Recent developments in 5lipoxygenase inhibitors. Expert Opinion on Therapeutic Patents, v. 13, n. 1, p.1-13, 2003. JULIUS, D.; BASBAUM, A. I. Molecular mechanisms of nociception. Nature, v. 413, n. 6852, p.203-10, 2001. KALISCH, P. A. An overview of research on the history of leprosy. Part 1. From Celsus to Simpson, Circa. 1 A.D. Part 2. From Virchow to Moller-Christense, 1845-1973. Int J Lepr Other Mycobact Dis, v. 43, n. 2, p.129-44, 1975. KANUI, T. I.; HOLE, K.; MIARON, J. O. Nociception in crocodiles-capsaicin instillation, formalin and hot plate tests. Zool. Sci., v. 7, p.537-540, 1990. KATO, M. J. Global phytochemistry: the Brazilian approach. Phytochemistry, v. 57, n. 5, p.621-3, 2001. KAY, A. B. Allergy and allergic diseases. First of two parts. N Engl J Med, v. 344, n. 1, p.30-7, 2001. KAYSER, V.; CHEN, Y. L.; GUILBAUD, G. Behavioural evidence for a peripheral component in the enhanced antinociceptive effect of a low dose of systemic morphine in carrageenin-induced hyperalgesic rats. Brain Res, v. 560, n. 1-2, p.237-44, 1991. KAYSER, V.; GOBEAUX, D.; LOMBARD, M. C.; GUILBAUD, G.; BESSON, J. M. Potent and long lasting antinociceptive effects after injection of low doses of a mu-opioid receptor agonist, fentanyl, into the brachial plexus sheath of the rat. Pain, v. 42, n. 2, p.215-25, 1990. KOEHN, F. E.; CARTER, G. T. The evolving role of natural products in drug discovery. Nat Rev Drug Discov, v. 4, n. 3, p.206-20, 2005. KOSTER, R.; ANDERSONS, M.; DEBBER, E. J. Acetic acid analgesic screening. Federation Proceedings, v. 18, p.418-420, 1959. KRAVITZ, E. A.; KUFFLER, S. W.; POTTER, D. D. Gamma-aminobutyric acid and other blocking compounds in Crustacea. Iii. Their Relative Concentrations in Separated Motor and Inhibitory Axons. J Neurophysiol, v. 26, p.739-51, 1963. KUHNS, D. B.; DECARLO, E.; HAWK, D. M.; GALLIN, J. I. Dynamics of the cellular and humoral components of the inflammatory response elicited in skin blisters in humans. J Clin Invest, v. 89, n. 6, p.1734-40, 1992. 107 LANCEL, M. Role of GABAA receptors in the regulation of sleep: initial sleep responses to peripherally administered modulators and agonists. Sleep, v. 22, n. 1, p.33-42, 1999. LAPA, A. J.; SOUCCAR, C.; LIMA-LANDMAN, M. T. R.; CASTRO, M. S. A.; LIMA, T. C. M. D. Métodos de avaliação da atividade farmacológica de plantas medicinais. Sociedade Brasileira de Plantas Medicinais. 2003. LAUFFENBURGER, D. A.; HORWITZ, A. F. Cell migration: a physically integrated molecular process. Cell, v. 84, n. 3, p.359-69, 1996. LAVICH, T. R.; CORDEIRO, R. S.; CALIXTO, J. B.; E SILVA, P. M.; MARTINS, M. A. Combined action of vasoactive amines and bradykinin mediates allergen-evoked thermal hyperalgesia in rats. Eur J Pharmacol, v. 462, n. 1-3, p.185-92, 2003. LAVIELLE, S.; CHASSAING, G.; PLOUX, O.; LOEUILLET, D.; BESSEYRE, J.; JULIEN, S.; MARQUET, A.; CONVERT, O.; BEAUJOUAN, J. C.; TORRENS, Y.; ET AL. Analysis of tachykinin binding site interactions using constrained analogues of tachykinins. Biochem Pharmacol, v. 37, n. 1, p.41-9, 1988. LAW, P. Y.; LOH, H. H. Regulation of opioid receptor activities. J Pharmacol Exp Ther, v. 289, n. 2, p.607-24, 1999. LAWRENCE, T.; WILLOUGHBY, D. A.; GILROY, D. W. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat Rev Immunol, v. 2, n. 10, p.787-95, 2002. LENÉE, P.; GOURET, C. Test des crampes abdominales provoqués chez la souris par la phénylbenzoquinone. J Pharmacol (Paris), v. 3, p.513–515, 1972. LEVINE, J. D.; GORDON, N. C.; JONES, R. T.; FIELDS, H. L. The narcotic antagonist naloxone enhances clinical pain. Nature, v. 272, n. 5656, p.826-7, 1978. LING, P.; NGO, K.; NGUYEN, S.; THURMOND, R. L.; EDWARDS, J. P.; KARLSSON, L.; FUNG-LEUNG, W. P. Histamine H4 receptor mediates eosinophil chemotaxis with cell shape change and adhesion molecule upregulation. Br J Pharmacol, v. 142, n. 1, p.161-71, 2004. LORENZETTI, B. B.; FERREIRA, S. H. The analgesic effect of quaternary analogues of morphine and nalorphine. Braz J Med Biol Res, v. 15, n. 4-5, p.285-90, 1982. LOUX, J. J.; SMITH, S.; SALEM, H. Comparative analgetic testing of various compounds in mice using writhing techniques. Arzneimittelforschung, v. 28, n. 9, p.1644-7, 1978. 108 MACHELSKA, H.; BRACK, A.; MOUSA, S. A.; SCHOPOHL, J. K.; RITTNER, H. L.; SCHAFER, M.; STEIN, C. Selectins and integrins but not platelet-endothelial cell adhesion molecule-1 regulate opioid inhibition of inflammatory pain. Br J Pharmacol, v. 142, n. 4, p.772-80, 2004. MACHELSKA, H.; CABOT, P. J.; MOUSA, S. A.; ZHANG, Q.; STEIN, C. Pain control in inflammation governed by selectins. Nat Med, v. 4, n. 12, p.1425-8, 1998. MACHELSKA, H.; LABUZ, D.; PRZEWLOCKI, R.; PRZEWLOCKA, B. Inhibition of nitric oxide synthase enhances antinociception mediated by mu, delta and kappa opioid receptors in acute and prolonged pain in the rat spinal cord. J Pharmacol Exp Ther, v. 282, n. 2, p.977-84, 1997. MACHELSKA, H.; MOUSA, S. A.; BRACK, A.; SCHOPOHL, J. K.; RITTNER, H. L.; SCHAFER, M.; STEIN, C. Opioid control of inflammatory pain regulated by intercellular adhesion molecule-1. J Neurosci, v. 22, n. 13, p.5588-96, 2002. MAGGI, C. A.; GIULIANI, S.; SANTICIOLI, P.; REGOLI, D.; MELI, A. Peripheral effects of neurokinins: functional evidence for the existence of multiple receptors. J Auton Pharmacol, v. 7, n. 1, p.11-32, 1987. MAGNAN, J.; PATERSON, S. J.; TAVANI, A.; KOSTERLITZ, H. W. The binding spectrum of narcotic analgesic drugs with different agonist and antagonist properties. Naunyn Schmiedebergs Arch Pharmacol, v. 319, n. 3, p.197-205, 1982. MALING, H. M.; WEBSTER, M. E.; WILLIAMS, M. A.; SAUL, W.; ANDERSON, W., JR. Inflammation induced by histamine, serotonin, bradykinin and compound 4880 in the rat: antagonists and mechanisms of action. J Pharmacol Exp Ther, v. 191, n. 2, p.300-10, 1974. MALMBERG, A. B.; YAKSH, T. L. Hyperalgesia mediated by spinal glutamate or substance P receptor blocked by spinal cyclooxygenase inhibition. Science, v. 257, n. 5074, p.1276-9, 1992. MALMBERG, A. B.; YAKSH, T. L. Spinal nitric oxide synthesis inhibition blocks NMDA-induced thermal hyperalgesia and produces antinociception in the formalin test in rats. Pain, v. 54, n. 3, p.291-300, 1993. MALMBERG-AIELLO, P.; LAMBERTI, C.; IPPONI, A.; BARTOLINI, A.; SCHUNACK, W. Evidence for hypernociception induction following histamine H1 receptor activation in rodents. Life Sci, v. 63, n. 6, p.463-76, 1998. MAO, J.; PRICE, D. D.; MAYER, D. J.; LU, J.; HAYES, R. L. Intrathecal MK-801 and local nerve anesthesia synergistically reduce nociceptive behaviors in rats with experimental peripheral mononeuropathy. Brain Res, v. 576, n. 2, p.254-62, 1992. 109 MARINHO, S. F.; PACIULLO, V. H. A.; FONSECA, M. O.; KHOURY, Z.; YAMIN, M. A.; MINKOVES, R.; R., A. M. O. S.; CAVALLARI, M. M. Meningite neutrofílica persistente em paciente com síndrome de imunodeficiência adquirida. Rev. Soc. Bras. Med. Trop., v. 30, p.03, 1997. MATSUMOTO, S.; NICKANDER, R. Epinephrine-induced writhing in mice. Fed. Proc., v. 26, p.619, 1967. MAYER, B.; ANDREW, P. Nitric oxide synthases: catalytic function and progress towards selective inhibition. Naunyn Schmiedebergs Arch Pharmacol, v. 358, n. 1, p.127-33, 1998. MCADAM, B. F.; CATELLA-LAWSON, F.; MARDINI, I. A.; KAPOOR, S.; LAWSON, J. A.; FITZGERALD, G. A. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci U S A, v. 96, n. 1, p.272-7, 1999. MCGETTIGAN, P.; HENRY, D. Cardiovascular risk and inhibition of cyclooxygenase: a systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase 2. Jama, v. 296, n. 13, p.1633-44, 2006. MELLER, S. T.; GEBHART, G. F. Nitric oxide (NO) and nociceptive processing in the spinal cord. Pain, v. 52, n. 2, p.127-36, 1993. MIKAMI, T.; MIYASAKA, K. Effects of several anti-inflammatory drugs on the various parameters involved in the inflammatory response in rat carrageenin-induced pleurisy. Eur J Pharmacol, v. 95, n. 1-2, p.1-12, 1983. MILLAN, M. J. Multiple opioid systems and pain. Pain, v. 27, n. 3, p.303-47, 1986. MILLAN, M. J. The induction of pain: an integrative review. Prog Neurobiol, v. 57, n. 1, p.1-164, 1999. MILLAN, M. J. Descending control of pain. Prog Neurobiol, v. 66, n. 6, p.355-474, 2002. MINAMI, M.; MAEKAWA, K.; YABUUCHI, K.; SATOH, M. Double in situ hybridization study on coexistence of mu-, delta- and kappa-opioid receptor mRNAs with preprotachykinin A mRNA in the rat dorsal root ganglia. Brain Res Mol Brain Res, v. 30, n. 2, p.203-10, 1995. MITCHISON, T. J.; CRAMER, L. P. Actin-based cell motility and cell locomotion. Cell, v. 84, n. 3, p.371-9, 1996. MOBARAKEH, J. I.; SAKURADA, S.; KATSUYAMA, S.; KUTSUWA, M.; KURAMASU, A.; LIN, Z. Y.; WATANABE, T.; HASHIMOTO, Y.; YANAI, K. Role of histamine H(1) receptor in pain perception: a study of the receptor gene knockout mice. Eur J Pharmacol, v. 391, n. 1-2, p.81-9, 2000. 110 MOORE, P. K.; OLUYOMI, A. O.; BABBEDGE, R. C.; WALLACE, P.; HART, S. L. LNG-nitro arginine methyl ester exhibits antinociceptive activity in the mouse. Br J Pharmacol, v. 102, n. 1, p.198-202, 1991. MORITA, K.; MORIOKA, N.; ABDIN, J.; KITAYAMA, S.; NAKATA, Y.; DOHI, T. Development of tactile allodynia and thermal hyperalgesia by intrathecally administered platelet-activating factor in mice. Pain, v. 111, n. 3, p.351-9, 2004. MORSE, K. L.; BEHAN, J.; LAZ, T. M.; WEST, R. E., JR.; GREENFEDER, S. A.; ANTHES, J. C.; UMLAND, S.; WAN, Y.; HIPKIN, R. W.; GONSIOREK, W.; SHIN, N.; GUSTAFSON, E. L.; QIAO, X.; WANG, S.; HEDRICK, J. A.; GREENE, J.; BAYNE, M.; MONSMA, F. J., JR. Cloning and characterization of a novel human histamine receptor. J Pharmacol Exp Ther, v. 296, n. 3, p.1058-66, 2001. MOUSA, S. A.; BOPAIAH, C. P.; STEIN, C.; SCHAFER, M. Involvement of corticotropin-releasing hormone receptor subtypes 1 and 2 in peripheral opioidmediated inhibition of inflammatory pain. Pain, v. 106, n. 3, p.297-307, 2003. MUNRO, J. M. Endothelial-leukocyte adhesive interactions in inflammatory diseases. Eur Heart J, v. 14 Suppl K, p.72-7, 1993. MURRAY, C. W.; PORRECA, F.; COWAN, A. Methodological refinements to the mouse paw formalin test: an animal model of tonic pain. J Pharmacol Methods, v. 20, n. 2, p.175-86, 1988. MURRAY, W. J.; MILLER, J. W. Oxytocin-induced "cramping" in the rat. J Pharmacol Exp Ther, v. 128, p.372-9, 1960. NAKAMURA, T.; ITADANI, H.; HIDAKA, Y.; OHTA, M.; TANAKA, K. Molecular cloning and characterization of a new human histamine receptor, HH4R. Biochem Biophys Res Commun, v. 279, n. 2, p.615-20, 2000. NATHAN, C.; XIE, Q. W. Nitric oxide synthases: roles, tolls, and controls. Cell, v. 78, n. 6, p.915-8, 1994. NEWTON, R.; SEYBOLD, J.; KUITERT, L. M.; BERGMANN, M.; BARNES, P. J. Repression of cyclooxygenase-2 and prostaglandin E2 release by dexamethasone occurs by transcriptional and post-transcriptional mechanisms involving loss of polyadenylated mRNA. J Biol Chem, v. 273, n. 48, p.32312-21, 1998. NIEMEGEERS, C. J.; VAN BRUGGEN, J. A.; JANSSEN, P. A. Suprofen, a potent antagonist of acetic acid-induced writhing in rats. Arzneimittelforschung, v. 25, n. 10, p.1505-9, 1975. NIKOLARAKIS, K.; PFEIFFER, A.; STALLA, G. K.; HERZ, A. The role of CRF in the release of ACTH by opiate agonists and antagonists in rats. Brain Res, v. 421, n. 1-2, p.373-6, 1987. 111 NORMAN, A. W.; MIZWICKI, M. T.; NORMAN, D. P. Steroid-hormone rapid actions, membrane receptors and a conformational ensemble model. Nat Rev Drug Discov, v. 3, n. 1, p.27-41, 2004. NORTH, R. A.; EGAN, T. M. Actions and distributions of opioid peptides in peripheral tissues. Br Med Bull, v. 39, n. 1, p.71-5, 1983. NOZAKI-TAGUCHI, N.; YAMAMOTO, T. Involvement of nitric oxide in peripheral antinociception mediated by kappa- and delta-opioid receptors. Anesth Analg, v. 87, n. 2, p.388-93, 1998. ODA, T.; MORIKAWA, N.; SAITO, Y.; MASUHO, Y.; MATSUMOTO, S. Molecular cloning and characterization of a novel type of histamine receptor preferentially expressed in leukocytes. J Biol Chem, v. 275, n. 47, p.36781-6, 2000. OLSEN, U. B.; ELTORP, C. T.; INGVARDSEN, B. K.; JORGENSEN, T. K.; LUNDBAEK, J. A.; THOMSEN, C.; HANSEN, A. J. ReN 1869, a novel tricyclic antihistamine, is active against neurogenic pain and inflammation. Eur J Pharmacol, v. 435, n. 1, p.43-57, 2002. OLSSON, Y. Microenvironment of the peripheral nervous system under normal and pathological conditions. Crit Rev Neurobiol, v. 5, n. 3, p.265-311, 1990. OLUYOMI, A. O.; HART, S. L.; SMITH, T. W. Differential antinociceptive effects of morphine and methylmorphine in the formalin test. Pain, v. 49, n. 3, p.415-8, 1992. OMOTE, K.; KAWAMATA, T.; KAWAMATA, M.; NAMIKI, A. Formalin-induced release of excitatory amino acids in the skin of the rat hindpaw. Brain Res, v. 787, n. 1, p.161-4, 1998. O'REILLY, M.; ALPERT, R.; JENKINSON, S.; GLADUE, R. P.; FOO, S.; TRIM, S.; PETER, B.; TREVETHICK, M.; FIDOCK, M. Identification of a histamine H4 receptor on human eosinophils-role in eosinophil chemotaxis. J Recept Signal Transduct Res, v. 22, n. 1-4, p.431-48, 2002. ORTÊNCIO, W. B. Medicina popular do Centro-Oeste. Ed. 2. Brasília: Theasaurus, 1997. 464 p. PACIFICI, G. M.; FRACCHIA, G. N. Advances in drug metabolism in man. Ed. 1. Bélgica: European Communities, 1995. PARADA, C. A.; TAMBELI, C. H.; CUNHA, F. Q.; FERREIRA, S. H. The major role of peripheral release of histamine and 5-hydroxytryptamine in formalin-induced nociception. Neuroscience, v. 102, n. 4, p.937-44, 2001. PARE, M.; ELDE, R.; MAZURKIEWICZ, J. E.; SMITH, A. M.; RICE, F. L. The Meissner corpuscle revised: a multiafferented mechanoreceptor with nociceptor immunochemical properties. J Neurosci, v. 21, n. 18, p.7236-46, 2001. 112 PARENTE, L. Pros and cons of selective inhibition of cyclooxygenase-2 versus dual lipoxygenase/cyclooxygenase inhibition: is two better than one? J Rheumatol, v. 28, n. 11, p.2375-82, 2001. PARENTE, L.; PERRETTI, M. Advances in the pathophysiology of constitutive and inducible cyclooxygenases: two enzymes in the spotlight. Biochem Pharmacol, v. 65, n. 2, p.153-9, 2003. PARENTE, L.; SOLITO, E. Annexin 1: more than an anti-phospholipase protein. Inflamm Res, v. 53, n. 4, p.125-32, 2004. PARK, K. A.; VASKO, M. R. Lipid mediators of sensitivity in sensory neurons. Trends Pharmacol Sci, v. 26, n. 11, p.571-7, 2005. PEARL, J.; ACETO, M. D.; HARRIS, L. S. Prevention of writhing and other effects of narcotics and narcotic antagonists in mice. J Pharmacol Exp Ther, v. 160, n. 1, p.217-30, 1968. PEARL, J.; MICHEL, C. R.; BOHNET, E. A. Effects of morphine and nalorphine on the phenylquinone-induced syndrome in monkeys. Psychopharmacologia, v. 14, n. 4, p.266-70, 1969. PEREIRA, F. E. L.; BOGLIOLO, L. Inflamações. In: Brasileiro Filho, G., Barbosa, A., Pitella, A., Pereira, Fel. (Ed.). Bogliolo Patologia. 5a edição. Rio de Janeiro: Guanabara Koogan, 1996. p.111-143. PFEIFFER, A.; PFEIFFER, D. G. Central mu- and kappa-opiate receptors may mediate opiate-induced release of GH and ACTH in rats. Acta. Endocrinol., v. 105, p.40-41, 1984. POONYACHOTI, S.; KULKARNI-NARLA, A.; BROWN, D. R. Chemical coding of neurons expressing delta- and kappa-opioid receptor and type I vanilloid receptor immunoreactivities in the porcine ileum. Cell Tissue Res, v. 307, n. 1, p.23-33, 2002. PRADO, W. A.; SCHIAVON, V. F.; CUNHA, F. Q. Dual effect of local application of nitric oxide donors in a model of incision pain in rats. Eur J Pharmacol, v. 441, n. 1-2, p.57-65, 2002. PRZEWLOCKI, R.; HASSAN, A. H.; LASON, W.; EPPLEN, C.; HERZ, A.; STEIN, C. Gene expression and localization of opioid peptides in immune cells of inflamed tissue: functional role in antinociception. Neuroscience, v. 48, n. 2, p.491-500, 1992. PRZEWLOCKI, R.; MACHELSKA, H.; PRZEWLOCKA, B. Inhibition of nitric oxide synthase enhances morphine antinociception in the rat spinal cord. Life Sci, v. 53, n. 1, p.PL1-5, 1993. 113 PUIGNERO, V.; TURULL, A.; QUERALT, J. Arachidonic acid (AA) and tetradecanoylphorbol acetate (TPA) exert systemic effects when applied topically in the mouse. Inflammation, v. 22, n. 3, p.307-14, 1998. QUER, P. F. Plantas medicinales: el Dioscórides renovado. Barcelona: Editorial Labor, 1962 QUIRION, R.; DAM, T. V. Multiple neurokinin receptors: recent developments. Reg. Peptides, v. 22, p.18-25, 1988. RANG, H. P.; DALE, M. M.; RITTER, J. M.; MOORE, P. K. Farmacologia. 5 Ed. Rio de Janeiro: Elsevier, 2004. 904 p. RATES, S. M. K. Plants as source of drugs. Toxicon, v. 39, n. 5, p.603-13, 2001. RATES, S. M. K.; BARROS, H. M. T. Modelos animais para a avaliação da dor: métodos para triagem de novos analgésicos. Rev. Bras. Farm., v. 75, n. 2, p.314, 1994. REN, K.; HYLDEN, J. L.; WILLIAMS, G. M.; RUDA, M. A.; DUBNER, R. The effects of a non-competitive NMDA receptor antagonist, MK-801, on behavioral hyperalgesia and dorsal horn neuronal activity in rats with unilateral inflammation. Pain, v. 50, n. 3, p.331-44, 1992. RIBEIRO, R. A.; VALE, M. L.; THOMAZZI, S. M.; PASCHOALATO, A. B.; POOLE, S.; FERREIRA, S. H.; CUNHA, F. Q. Involvement of resident macrophages and mast cells in the writhing nociceptive response induced by zymosan and acetic acid in mice. Eur J Pharmacol, v. 387, n. 1, p.111-8, 2000. RITTNER, H. L.; BRACK, A.; MACHELSKA, H.; MOUSA, S. A.; BAUER, M.; SCHAFER, M.; STEIN, C. Opioid peptide-expressing leukocytes: identification, recruitment, and simultaneously increasing inhibition of inflammatory pain. Anesthesiology, v. 95, n. 2, p.500-8, 2001. ROBY, C. A.; ANDERSON, G. D.; KANTOR, E.; DRYER, D. A.; BURSTEIN, A. H. St John's Wort: effect on CYP3A4 activity. Clin Pharmacol Ther, v. 67, n. 5, p.4517, 2000. ROCHA, J. C. S.; PEIXOTO, M. E.; JANCAR, S.; CUNHA, F. Q.; RIBEIRO, R. A.; ROCHA, F. A. Dual effect of nitric oxide in articular inflammatory pain in zymosaninduced arthritis in rats. Br J Pharmacol, v. 136, n. 4, p.588-96, 2002. RODRIGUES, A. R.; DUARTE, I. D. The peripheral antinociceptive effect induced by morphine is associated with ATP-sensitive K(+) channels. Br J Pharmacol, v. 129, n. 1, p.110-4, 2000. ROSLAND, J. H. The formalin test in mice: the influence of ambient temperature. Pain, v. 45, n. 2, p.211-6, 1991. 114 ROSLAND, J. H.; HUNSKAAR, S.; HOLE, K. Diazepam attenuates morphine antinociception test-dependently in mice. Pharmacol Toxicol, v. 66, n. 5, p.3826, 1990. SAMPSON, A. P. The role of eosinophils and neutrophils in inflammation. Clin Exp Allergy, v. 30 Suppl 1, p.22-7, 2000. SAMUELSSON, B.; DAHLEN, S. E.; LINDGREN, J. A.; ROUZER, C. A.; SERHAN, C. N. Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science, v. 237, n. 4819, p.1171-6, 1987. SANTOS, A. R. S. Efeito antinociceptivo de extratos e princípios isolados de plantas do gênero Phylanthus (quebra-pedra). 1995. 89 p. Dissertação (Mestrado em Farmacologia) - Curso de Pós-Graduação em Farmacologia, Universidade Federal de Santa Catarina, Florianópolis, 1995. SANTOS, A. R.; CALIXTO, J. B. Further evidence for the involvement of tachykinin receptor subtypes in formalin and capsaicin models of pain in mice. Neuropeptides, v. 31, n. 4, p.381-9, 1997. SCHÄFER, M.; IMAI, Y.; UHL, G. R.; STEIN, C. Inflammation enhances peripheral mu-opioid receptor-mediated analgesia, but not mu-opioid receptor transcription in dorsal root ganglia. Eur J Pharmacol, v. 279, n. 2-3, p.165-9, 1995. SCHUMAN, E. M.; MADISON, D. V. Nitric oxide and synaptic function. Annu Rev Neurosci, v. 17, p.153-83, 1994. SCHWARTZ, J. C.; ARRANG, J. M.; GARBARG, M.; POLLARD, H.; RUAT, M. Histaminergic transmission in the mammalian brain. Physiol Rev, v. 71, n. 1, p.151, 1991. SCIBERRAS, D. G.; GOLDENBERG, M. M.; BOLOGNESE, J. A.; JAMES, I.; BABER, N. S. Inflammatory responses to intradermal injection of platelet activating factor, histamine and prostaglandin E2 in healthy volunteers: a double blind investigation. Br J Clin Pharmacol, v. 24, n. 6, p.753-61, 1987. SEGUIN, L.; LE MAROUILLE-GIRARDON, S.; MILLAN, M. J. Antinociceptive profiles of non-peptidergic neurokinin1 and neurokinin2 receptor antagonists: a comparison to other classes of antinociceptive agent. Pain, v. 61, n. 2, p.325-43, 1995. SELLEY, D. E.; BREIVOGEL, C. S.; CHILDERS, S. R. Modification of G proteincoupled functions by low-pH pretreatment of membranes from NG108-15 cells: increase in opioid agonist efficacy by decreased inactivation of G proteins. Mol Pharmacol, v. 44, n. 4, p.731-41, 1993. SERVANT, G.; WEINER, O. D.; HERZMARK, P.; BALLA, T.; SEDAT, J. W.; BOURNE, H. R. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science, v. 287, n. 5455, p.1037-40, 2000. 115 SEVOSTIANOVA, N.; DANYSZ, W.; BESPALOV, A. Y. Analgesic effects of morphine and loperamide in the rat formalin test: interactions with NMDA receptor antagonists. Eur J Pharmacol, v. 525, n. 1-3, p.83-90, 2005. SHIBATA, M.; OHKUBO, T.; TAKAHASHI, H.; INOKI, R. Modified formalin test: characteristic biphasic pain response. Pain, v. 38, n. 3, p.347-52, 1989. SIBILLE, Y.; REYNOLDS, H. Y. Macrophages and polymorphonuclear neutrophils in lung defense and injury. Am Rev Respir Dis, v. 141, n. 2, p.471-501, 1990. SIEBECK, M.; SCHORR, M.; SPANNAGL, E.; LEHNER, M.; FRITZ, H.; CHERONIS, J. C.; WHALLEY, E. T. B1 kinin receptor activity in pigs is associated with preexisting infection. Immunopharmacology, v. 40, n. 1, p.49-55, 1998. SIEGMUND, E. A.; CADMUS, R. A.; LU, G. A method for evaluating both nonnarcotic and narcotic analgesics. Proc Soc Exp Biol Med, v. 95, n. 4, p.729-31, 1957a. SIEGMUND, E. A.; CADMUS, R. A.; LU, G. Screening analgesics, incluiding aspirintype compound, based upon the antagonism of chemically induced "writhing" in mice. J. Pharmacol. Exp. Ther., v. 119, n. 184-193, 1957b. SIELGEL, P. S. A simple electronic device for the measurement of gross bodily activity of small animals. J. Psychol., v. 21, p.227-236, 1946. SIMÕES, C. M. O.; SCHENKEL, E. P.; GOSMANN, G.; MELLO, J. C. P.; MENTZ, L. A.; PETROVICK, P. R. Farmacognosia: da planta ao medicamento. Ed. 5. Porto Alegre: Editora da UFRGS, 2004. 1102 p. SMILENOV, L. B.; MIKHAILOV, A.; PELHAM, R. J.; MARCANTONIO, E. E.; GUNDERSEN, G. G. Focal adhesion motility revealed in stationary fibroblasts. Science, v. 286, n. 5442, p.1172-4, 1999. SMITH, T. W.; BUCHAN, P.; PARSONS, D. N.; WILKINSON, S. Peripheral antinociceptive effects of N-methyl morphine. Life Sci, v. 31, n. 12-13, p.1205-8, 1982. SMITH, T. W.; FOLLENFANT, R. L.; FERREIRA, S. H. Antinociceptive models displaying peripheral opioid activity. Int J Tissue React, v. 7, n. 1, p.61-7, 1985. SNEDDON, L. U. Editorial. Brain Res Brain Res Rev, v. 46, n. 2, p.121, 2004. SOARES, A. C.; LEITE, R.; TATSUO, M. A.; DUARTE, I. D. Activation of ATPsensitive K(+) channels: mechanism of peripheral antinociceptive action of the nitric oxide donor, sodium nitroprusside. Eur J Pharmacol, v. 400, n. 1, p.67-71, 2000. SOARES, A. C.; DUARTE, I. D. Dibutyryl-cyclic GMP induces peripheral antinociception via activation of ATP-sensitive K(+) channels in the rat PGE2induced hyperalgesic paw. Br J Pharmacol, v. 134, n. 1, p.127-31, 2001. 116 SOUCCAR, C.; LAPA, A. J. Analgesic and anti-inflamatory screening of two Brazilian medicinal plants: a positive and a false-positive result. Ciência e Cultura J. Braz. Assoc. Advancem. of Sci., v. 49, n. 5/6, p.417-21, 1997. SOUSA, A. M.; PRADO, W. A. The dual effect of a nitric oxide donor in nociception. Brain Res, v. 897, n. 1-2, p.9-19, 2001. SPRINGER, T. A. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell, v. 76, n. 2, p.301-14, 1994. STEAVENS, A.; LOWE, J. Patologia. 2 edição. Barueri, SP: Molone, 2002. 671 p. STEIN, C. Peripheral mechanisms of opioid analgesia. Anesth Analg, v. 76, n. 1, p.182-91, 1993. STEIN, C.; HASSAN, A. H.; PRZEWLOCKI, R.; GRAMSCH, C.; PETER, K.; HERZ, A. Opioids from immunocytes interact with receptors on sensory nerves to inhibit nociception in inflammation. Proc Natl Acad Sci U S A, v. 87, n. 15, p.5935-9, 1990. STEIN, C.; MILLAN, M. J.; SHIPPENBERG, T. S.; HERZ, A. Peripheral effect of fentanyl upon nociception in inflamed tissue of the rat. Neurosci Lett, v. 84, n. 2, p.225-8, 1988. STEIN, C.; MILLAN, M. J.; SHIPPENBERG, T. S.; PETER, K.; HERZ, A. Peripheral opioid receptors mediating antinociception in inflammation: evidence for involvement of mu, delta and kappa receptors. J Pharmacol Exp Ther, v. 248, n. 3, p.1269-75, 1989. STEIN, C.; SCHAFER, M.; MACHELSKA, H. Attacking pain at its source: new perspectives on opioids. Nat Med, v. 9, n. 8, p.1003-8, 2003. STUEHR, D. J.; MARLETTA, M. A. Mammalian nitrate biosynthesis: mouse macrophages produce nitrite and nitrate in response to Escherichia coli lipopolysaccharide. Proc Natl Acad Sci U S A, v. 82, n. 22, p.7738-42, 1985. STUEHR, D. J.; MARLETTA, M. A. Induction of nitrite/nitrate synthesis in murine macrophages by BCG infection, lymphokines, or interferon-gamma. J Immunol, v. 139, n. 2, p.518-25, 1987. SUFKA, K. J.; SCHOMBURG, F. M.; GIORDANO, J. Receptor mediation of 5-HTinduced inflammation and nociception in rats. Pharmacol Biochem Behav, v. 41, n. 1, p.53-6, 1992. SWINGLE, K. F.; REITER, M. J.; SCHWARTZMILLER, D. H. Comparison of croton oil and cantharidin induced inflammations of the mouse ear and their modification by topically applied drugs. Arch Int Pharmacodyn Ther, v. 254, n. 1, p.168-76, 1981. 117 TABER, R. I.; GREENHOUSE, D. D.; IRWIN, S. Inhibition of phenylquinone-induced writhing by narcotic antagonists. Nature, v. 204, p.189-90, 1964. TAKAHASHI, H.; OHKUBO, T.; SHIBATA, M.; NARUSE, S. A modified formalin test for measuring analgesia in mice. Jpn. J. Oral Biol., v. 26, p.543-548, 1984. TJØLSEN, A.; BERGE, O. G.; HUNSKAAR, S.; ROSLAND, J. H.; HOLE, K. The formalin test: an evaluation of the method. Pain, v. 51, n. 1, p.5-17, 1992. TJØLSEN, A.; HOLE, K. Animals models of analgesia. In: Dickenson, A. H.; Besson, J. M. (Ed.). The Pharmacology of Pain. Berlin: Springer, 1997. p.1-20. TORNOS, M. P.; SAENZ, M. T.; GARCIA, M. D.; FERNANDEZ, M. A. Antinociceptive effects of the tubercles of Anredera leptostachys. J Ethnopharmacol, v. 68, n. 13, p.229-34, 1999. TUBARO, A.; DRI, P.; DELBELLO, G.; SILLI, C.; LOGIA, R. D. The crotón oil ear test revisited. Agents and Actions, v. 17, p.47-49, 1985. UTSUNOMIYA, I.; NAGAI, S.; OH-ISHI, S. Differential effects of indomethacin and dexamethasone on cytokine production in carrageenin-induced rat pleurisy. Eur J Pharmacol, v. 252, n. 2, p.213-8, 1994. VANE, J. R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol, v. 231, n. 25, p.232-5, 1971. VANE, J. R.; BAKHLE, Y. S.; BOTTING, R. M. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol, v. 38, p.97-120, 1998. VANE, J. R.; BOTTING, R. M. Mechanism of action of antiinflammatory drugs. Int J Tissue React, v. 20, n. 1, p.3-15, 1998. VELO, G. P.; DUNN, C. J.; GIROUD, J. P.; TIMSIT, J.; WILLOUGHBY, D. A. Distribution of prostaglandins in inflammatory exudate. J Pathol, v. 111, n. 3, p.149-58, 1973. VIEIRA, R. A. Avaliação dos efeitos centrais do extrato aquoso de Stachytarpheta cayennensis Vahl., 2001. Dissertação (Mestrado em Farmacologia) - Curso de Pós-Graduação em Farmacologia, Universidade Federal de Santa Catarina, Florianópolis, 2001. VINEGAR, R.; TRUAX, J. F.; SELPH, J. L. Some quantitative temporal characteristics of carrageenin-induced pleurisy in the rat. Proc Soc Exp Biol Med, v. 143, n. 3, p.711-4, 1973. WAGNER, J. G.; ROTH, R. A. Neutrophil migration mechanisms, with an emphasis on the pulmonary vasculature. Pharmacol Rev, v. 52, n. 3, p.349-74, 2000. 118 WAHL, S. M.; FELDMAN, G. M.; MCCARTHY, J. B. Regulation of leukocyte adhesion and signaling in inflammation and disease. J Leukoc Biol, v. 59, n. 6, p.789-96, 1996. WALDHOER, M.; BARTLETT, S. E.; WHISTLER, J. L. Opioid receptors. Annu Rev Biochem, v. 73, p.953-90, 2004. WALLNER, B. P.; MATTALIANO, R. J.; HESSION, C.; CATE, R. L.; TIZARD, R.; SINCLAIR, L. K.; FOELLER, C.; CHOW, E. P.; BROWING, J. L.; RAMACHANDRAN, K. L.; ET AL. Cloning and expression of human lipocortin, a phospholipase A2 inhibitor with potential anti-inflammatory activity. Nature, v. 320, n. 6057, p.77-81, 1986. WARNER, T. D.; MICHELL, J. A. Cyclooxygenases: new forms, new inhibitors, and lessons from the clinic. The FASEB Journal, v. 18, n. 790-804, 2004. WEBER, C. Novel mechanistic concepts for the control of leukocyte transmigration: specialization of integrins, chemokines, and junctional molecules. J Mol Med, v. 81, n. 1, p.4-19, 2003. WENK, H. N.; HONDA, C. N. Immunohistochemical localization of delta opioid receptors in peripheral tissues. J Comp Neurol, v. 408, n. 4, p.567-79, 1999. WHITTLE, B. A. Release of a kinin by intraperitoneal injection of chemical agents in mice. Int J Neuropharmacol, v. 3, p.369-78, 1964. WILLIAMS, T. J. Interactions between prostaglandins, leukotrienes and other mediators of inflammation. Br Med Bull, v. 39, n. 3, p.239-42, 1983. WITKO-SARSAT, V.; RIEU, P.; DESCAMPS-LATSCHA, B.; LESAVRE, P.; HALBWACHS-MECARELLI, L. Neutrophils: molecules, functions and pathophysiological aspects. Lab Invest, v. 80, n. 5, p.617-53, 2000. WOOLF, C. J.; SALTER, M. W. Neuronal plasticity: increasing the gain in pain. Science, v. 288, 5472, p.1765-9, 2000. WOOLFE, G.; MACDONALD, A. D. The evaluation of the analgesic action of pethidine hydrochloride. J. Pharmacol. Exp. Ther., v. 80, n., p.300-307, 1944. WU, T. S.; LIN, Y. M.; HARUNA, M.; PAN, D. J.; SHINGU, T.; CHEN, Y. P.; HSU, H. Y.; NAKANO, T.; LEE, K. H. Antitumor agents, 119. Kansuiphorins A and B, two novel antileukemic diterpene esters from Euphorbia kansui. J Nat Prod, v. 54, n. 3, p.823-9, 1991. YAKSH, T. L.; RUDY, T. A. Studies on the direct spinal action of narcotics in the production of analgesia in the rat. J Pharmacol Exp Ther, v. 202, n. 2, p.411-28, 1977. 119 YAMAMOTO, T.; YAKSH, T. L. Spinal pharmacology of thermal hyperesthesia induced by constriction injury of sciatic nerve. Excitatory amino acid antagonists. Pain, v. 49, n. 1, p.121-8, 1992. YOKOMIZO, T.; UOZUMI, N.; TAKAHASHI, T.; KUME, K.; IZUMI, T.; SHIMIZU, T. Leukotriene A4 hydrolase and leukotriene B4 metabolism. J Lipid Mediat Cell Signal, v. 12, n. 2-3, p.321-32, 1995. ZHANG, X. W.; LIU, Q.; WANG, Y.; THORLACIUS, H. CXC chemokines, MIP-2 and KC, induce P-selectin-dependent neutrophil rolling and extravascular migration in vivo. Br J Pharmacol, v. 133, n. 3, p.413-21, 2001. Apêndice 121 APÊNDICE A – Tabelas de dados Tabela 1 – Contorções abdominais induzidas pelo ácido acético (1,2 % v/v em salina, i.p.) durante 30 minutos em camundongos previamente tratados (60 min) pela via p.o. com veículo (grupo controle; C), com o extrato etanólico das folhas do S. umbellatum (EES; 25, 50 ou 100 mg/kg) (A), com as frações hexânica (FH, 10 mg/kg), clorofórmica (FC, 20 mg/kg) e metanol/água (FM, 25 mg/kg) (B) ou com indometacina (INDO; 10 mg/kg). A (EES) Animal C EES 25 EES 50 EES 100 INDO 1 80 57 47 36 45 2 78 55 49 39 48 3 80 57 50 32 47 4 83 62 45 38 46 5 74 60 46 35 45 6 69 58 44 29 49 Média ± EPM 77,3 ± 2,1 58,2 ± 1,0* 46,8 ± 0,9* 34,8 ± 1,5* 46,7 ± 0,7* B (Frações) Animal C FH 10 FC 20 FM 25 INDO 1 80 69 72 28 45 2 78 74 76 30 48 3 80 81 63 28 47 4 83 81 82 38 46 5 74 75 73 31 45 6 69 89 89 34 49 Média ± EPM 77,3 ± 2,1 78,2 ± 2,9 75,8 ± 3,4 31,5 ± 1,6* 46,7 ± 0,7* * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. Os grupos tratados com as diferentes doses de EES são significativamente diferentes entre si (P < 0,05) – ANOVA, teste de Tukey. 122 Tabela 2 – Reatividade à aplicação intraplantar de formalina (20 µL, 3%) na pata posterior direita de camundongos, durante a primeira fase (0 – 5 min) (A) e segunda fase (15 – 30 min) (B) do teste da formalina, previamente tratados (60 min) pela via p.o. com veículo (grupo controle, C), com o EES (100 mg/kg), com morfina (MOR; 10 mg/kg s.c.) ou com indometacina (INDO; 10 mg/kg). A (Primeira fase) Animal C EES 100 MOR INDO 1 95 67 4 119 2 116 48 0 122 3 162 48 20 93 4 165 73 97 102 5 115 55 15 137 6 78 26 64 65 7 93 78 8 91 68 9 189 84 10 90 66 Média ± EPM 119,4 ± 12,2 61,3 ± 5,4* 33,3 ± 15,8* 106,3 ± 10,4 123 B (Segunda fase) Animal C EES 100 MOR INDO 1 250 78 13 61 2 405 113 0 149 3 360 31 50 162 4 233 87 136 145 5 121 22 15 150 6 326 3 42 133 7 252 96 8 226 97 9 269 105 10 273 98 Média ± EPM 271,5 ± 24,9 73,0 ± 12,4* 42,7 ± 20,2* 133,3 ± 15,0* * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. 124 Tabela 3 – Influência do tratamento prévio com o antagonista opióide não seletivo naloxona (3 mg/kg, s.c.) sobre a atividade antinociceptiva do EES (100 mg/kg, p.o.) e fentanil (100 µg/kg, s.c.) no modelo de contorções abdominais induzidas pelo ácido acético. Salina Naloxona Salina Naloxona Salina Naloxona Animal Veículo Veículo EES 100 mg/kg EES 100 mg/kg Fentanil 100 µg/kg Fentanil 100 µg/kg 1 68 57 42 68 3 62 2 74 71 37 59 4 72 3 59 64 47 70 7 63 4 76 68 41 72 10 68 5 69 67 46 69 7 74 6 75 79 48 75 7 61 Média ± EPM 70,1 ± 2,6 67,7 ± 3,0 43,5 ± 1,7* 68,8 ± 2,2 # 6,3 ± 1,0* ## 66,7 ± 2,2 * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. # Estatisticamente diferente do grupo tratado com EES 100 mg/kg e pré-tratado com salina (P < 0,05) – ANOVA, teste de Tukey. ## Estatisticamente diferente do grupo tratado com fentanil 100 µg/kg e pré-tratado com salina (P < 0,05) – ANOVA, teste de Tukey. 125 Tabela 4 – Latência ao estímulo térmico nociceptivo medido no teste do tail flick em camundongos antes e após tratamento por via p.o. com o veículo (A), com EES 25 (B), 50 (C) ou 100 (D) mg/kg, ou morfina (E) (10 mg/kg, s.c.). A (Controle) Imersão das caudas (min) Animal -60 -30 0 1 3,66 2,20 3,14 2 2,20 4,27 3,10 3 1,25 2,94 4 3,86 5 6 Média ± EPM 30 60 90 120 2,53 3,14 2,28 3,53 1,68 3,60 3,75 6,38 1,59 1,62 2,46 3,28 3,96 2,60 1,86 4,85 2,57 3,72 3,21 3,23 1,77 1,16 2,18 3,88 5,27 5,78 3,16 2,47 2,88 3,73 2,64 1,25 2,97 2,89 ± 0,40 2,71 ± 0,36 2,29 ± 0,35 2,79 ± 0,31 3,58 ± 0,66 3,40 ± 0,39 3,61 ± 0,69 60 90 120 Tempo (s) B (EES 25 mg/kg) Imersão das caudas (min) Animal -60 -30 0 30 1 3,08 2,45 2,16 4,09 3,45 3,74 3,73 2 1,07 1,57 1,15 1,67 3,52 4,41 4,37 3 3,49 2,25 2,01 3,00 2,33 1,43 4,00 4 1,70 1,50 3,03 4,28 3,62 3,18 3,45 5 2,02 2,02 2,87 2,29 1,26 1,56 2,45 6 0,90 1,48 1,32 4,41 3,07 3,65 1,25 Média ± EPM 2,04 ± 0,43 1,88 ± 0,17 2,09 ± 0,31 3,37 ± 0,35 3,26 ± 0,50 3,01 ± 0,44 2,73 ± 0,50 60 90 120 Tempo (s) C (EES 50 mg/kg) Imersão das caudas (min) Animal -60 -30 0 30 Tempo (s) 1 1,89 3,63 3,22 5,39 4,48 2,41 1,98 2 2,26 3,54 2,07 2,78 3,77 5,70 3,14 3 1,37 1,11 2,29 2,39 3,14 4,33 3,97 4 1,22 2,74 0,78 6,88 1,87 1,74 1,32 5 1,53 2,77 2,37 3,29 2,91 2,39 4,48 6 1,63 1,41 1,13 4,44 0,94 4,31 1,26 Média ± EPM 1,65 ± 0,15 2,53 ± 0,43 1,98 ± 0,36 3,47 ± 0,54 3,78 ± 0,48 3,00 ± 0,83 2,97 ± 0,67 126 D (EES 100 mg/kg) Imersão das caudas (min) Animal -60 -30 0 1 2,97 2,56 2,65 2 2,48 3,16 3,44 3 1,35 2,73 4 3,92 3,06 5 0,99 6 Média ± EPM 30 60 90 120 5,71 4,85 2,49 3,14 2,88 3,49 3,87 5,86 3,54 2,67 3,32 2,05 2,80 1,26 1,37 5,02 1,95 3,46 3,74 3,86 1,55 2,43 4,46 4,59 1,20 1,43 3,48 1,86 3,00 1,62 1,36 2,15 ± 0,48 2,78 ± 0,32 3,04 ± 0,39 3,76 ± 0,51 3,43 ± 0,55 2,63 ± 0,57 2,82 ± 0,59 60 90 120 Tempo (s) E (Morfina 10 mg/kg) Imersão das caudas (min) Animal -60 -30 0 1 3,58 2,96 2,42 20,00 13,78 13,21 9,65 2 1,28 2,52 4,10 20,00 20,00 20,00 20,00 3 1,18 1,84 0,91 20,00 13,65 14,95 19,03 4 2,44 3,50 4,64 20,00 20,00 20,00 20,00 5 2,14 1,62 2,70 20,00 20,00 18,90 13,54 6 4,34 2,49 ± 0,51 2,42 2,48 ± 0,28 2,69 2,91 ± 0,54 20,00 20,00 ± 0,00* 20,00 17,91 ± 1,32* 20,00 17,84 ± 1,22* 20,00 17,04 ± 1,80* Média ± EPM 30 Tempo (s) * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. 127 Tabela 5 – Edema de orelha, em mg, induzido por óleo de cróton (2,5% v/v em acetona) nos grupos previamente tratados pela via p.o. com veículo (C), com extrato etanólico de S. umbellatum (EES 25, 50 ou 100 mg/kg) (A), com fração metanol/água (FM 6, 12 ou 25 mg/kg) (B) ou com dexametasona (DEXA; 2 mg/kg). A (EES) Animal C EES 25 EES 50 EES 100 DEXA 1 30,0 14,6 11,2 9,1 5,9 2 22,1 15,4 11,6 8,7 6,3 3 23,6 15,3 12,5 8,0 1,9 4 14,3 14,9 11,3 8,3 2,8 5 23,3 14,3 13,8 8,7 3,2 6 18,7 14,9 13,7 9,7 0,8 7 19,9 17,5 11,3 9,0 Média ± EPM 21,7 ± 1,8 15.3 ± 0,4* 12,2 ± 0,4* 8,8 ± 0,2* 3,5 ± 0,9* B (Frações) Animal C FM 6 FM 12 FM 25 DEXA 1 30,0 16,2 13,8 4,5 5,9 2 22,1 15,3 17,3 6,7 6,3 3 23,6 17,5 10,0 9,4 1,9 4 14,3 16,7 10,1 8,9 2,8 5 23,3 16,3 12,9 7,9 3,2 6 18,7 20,0 13,4 7,9 0,8 7 19,9 18,0 10,8 12,5 19,8 14,8 17,4 ± 0,6* 12,9 ± 0,9* 8 Média ± EPM 21,7 ± 1,8 8,3 ± 0,9* 3,5 ± 0,9* * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. Os grupos tratados com as diferentes doses de EES ou FM são significativamente diferentes entre si (P < 0,05) – ANOVA, teste de Tukey. 128 Tabela 6 – Migração de leucócitos totais (x 106) no modelo de peritonite induzida por carragenina (1% m/v) injetada na cavidade intraperitoneal de camundongos previamente tratados pela via p.o. com veículo (C), com extrato etanólico de S. umbellatum (EES 25, 50 ou 100 mg/kg) ou dexametasona (DEXA; 2 mg/kg). Animal C EES 25 EES 50 EES 100 DEXA 1 12,0 8,8 6,9 6,4 4,0 2 11,3 7,2 8,8 3,6 3,4 3 10,8 9,5 6,8 5,5 6,5 4 10,4 10,5 7,3 4,4 4,1 5 13,7 7,4 7,9 3,8 3,1 6 10,6 8,9 6,6 7,4 2,9 7 11,3 8,2 6,8 7,5 3,3 8 11,3 10,8 7,2 3,9 3,3 9 11,8 10 11,1 Média ± EPM 11,4 ± 0,3 5,3 ± 0,6* 3,8 ± 0,4* 7,0 8,9 ± 0,5* 7,3 ± 0,2* * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. Os grupos tratados com as diferentes doses de EES são significativamente diferentes entre si (P < 0,05) – ANOVA, teste de Tukey. 129 Tabela 7 – Número de quedas (A) e tempo de permanência (B) no rota-rod, de camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com o extrato etanólico das folhas do S. umbellatum (EES; 25, 50 ou 100 mg/kg) (1) com as frações hexânica (FH, 10 mg/kg), clorofórmica (FC, 20 mg/kg), metanol/água (FM, 25 mg/kg) (2) ou com diazepam (DZP; 5 mg/kg). A1 (Número de quedas - EES) Animal C EES 25 EES 50 EES 100 DZP 1 0 1 0 1 3 2 0 0 2 1 2 3 0 0 0 1 1 4 1 1 1 1 1 5 0 0 0 0 3 6 0 1 1 1 1 7 0 1 0 0 3 Média ± EPM 0,1 ± 0,1 0,6 ± 0,2 0,6 ± 0,3 0,7 ± 0,2 2,0 ± 0,4* A2 (Número de quedas - Frações) Animal C FH 10 FC 20 FM 25 DZP 1 0 0 0 0 3 2 0 1 1 2 2 3 0 0 1 0 1 4 1 1 0 0 1 5 0 0 0 0 3 6 0 0 0 0 1 7 0 1 0 0 3 Média ± EPM 0,1 ± 0,1 0,4 ± 0,2 0,3 ± 0,2 0,3 ± 0,3 2,0 ± 0,4* 130 B1 (Tempo de permanência - EES) Animal C EES 25 EES 50 EES 100 DZP 1 60 60 60 60 14 2 60 60 60 60 60 3 60 60 60 60 60 4 60 60 60 60 60 5 60 60 60 60 14 6 60 60 60 60 60 7 60 60 60 60 11 Média ± EPM 60 ± 0,0 60 ± 0,0 60 ± 0,0 60 ± 0,0 39,9 ± 9,5* B2 (Tempo de permanência - Frações) Animal C FH 10 FC 20 FM 25 DZP 1 60 60 60 60 14 2 60 60 60 60 60 3 60 60 60 60 60 4 60 60 60 60 60 5 60 60 60 60 14 6 60 60 60 60 60 7 60 60 60 60 11 Média ± EPM 60 ± 0,0 60 ± 0,0 60 ± 0,0 60 ± 0,0 39,9 ± 9,5* * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. 131 Tabela 8 - Número de quadrados invadidos, tempo parado (segundos), número de levantadas, número de auto-limpeza e número de bolos fecais no campo aberto 60 minutos após o tratamento p.o. com veículo (C) (A), extrato etanólico de S. umbellatum (EES 25 (B), 50 (C) ou 100 mg/kg (D)), FH (10 mg/kg) (E), FC (20 mg/kg) (F), FM (25 mg/kg) (G) ou diazepam (DZP, 5 mg/kg) (H). A (Controle) Animal Número de quadrados invadidos Tempo parado (segundos) Número de levantadas Número de auto-limpeza Número de bolos fecais 1 34 26 11 51 2 2 42 42 11 15 6 3 31 29 13 65 1 4 54 23 20 73 1 5 49 45 24 46 2 6 40 16 10 23 2 7 41 22 20 58 6 8 59 26 14 53 5 Média ± EPM 43,8 ± 3,4 28,6 ± 3,5 15,4 ± 1,9 48,0 ± 7,0 3,1 ± 0,8 B (EES 25 mg/kg) Animal Número de quadrados invadidos Tempo parado (segundos) Número de levantadas Número de auto-limpeza Número de bolos fecais 1 53 66 17 26 2 2 58 59 19 83 2 3 49 109 19 42 1 4 52 48 18 73 1 5 48 88 15 35 0 6 67 48 10 74 2 7 51 55 5 93 0 8 34 65 14 46 1 Média ± EPM 51,5 ± 3,3 67,2 ± 7,5* 14,6 ± 1,7 59,9 ± 8,7 1,1 ± 0,3* 132 C (EES 50 mg/kg) Animal Número de quadrados invadidos Tempo parado (segundos) Número de levantadas Número de auto-limpeza Número de bolos fecais 1 62 74 11 35 2 2 18 173 4 23 2 3 33 136 1 21 0 4 56 125 6 0 0 5 43 69 12 71 0 6 54 106 18 20 1 7 51 152 20 58 1 8 45 127 15 36 1 Média ± EPM 45,2 ± 5,0 120,3 ± 12,7* 10,9 ± 2,4 33,0 ± 8,0 0,9 ± 0,3* D (EES 100 mg/kg) Animal Número de quadrados invadidos Tempo parado (segundos) Número de levantadas Número de auto-limpeza Número de bolos fecais 1 19 179 2 60 0 2 63 30 19 84 0 3 55 123 19 46 2 4 85 93 3 40 0 5 48 105 11 73 1 6 25 52 12 32 2 7 59 103 32 45 2 8 30 145 18 43 2 Média ± EPM 48,0 ± 7,9 103,8 ± 16,9* 14,5 ± 3,5 52,9 ± 6,3 1,1 ± 0,4* 133 E (FH 10 mg/kg) Animal Número de quadrados invadidos Tempo parado (segundos) Número de levantadas Número de auto-limpeza Número de bolos fecais 1 61 113 16 50 2 2 30 21 20 75 0 3 54 5 19 37 0 4 37 10 15 37 2 5 22 15 3 112 1 6 36 5 13 62 2 7 62 10 17 0 0 8 57 5 13 75 0 Média ± EPM 44,9 ± 5,5 23,0 ± 13,1 14,5 ± 1,9 56,0 ± 11,8 1,1 ± 0,4* F (FC 20 mg/kg) Animal Número de quadrados invadidos Tempo parado (segundos) Número de levantadas Número de auto-limpeza Número de bolos fecais 1 28 77 5 62 4 2 6 134 1 25 0 3 38 31 10 37 2 4 39 159 16 50 0 5 55 26 15 12 2 6 41 211 8 37 4 7 36 77 12 25 3 8 52 101 15 12 5 Média ± EPM 36,9 ± 5,4 102,0 ± 22,4* 10,2 ± 1,9 32,5 ± 6,2 2,5 ± 0,7 134 G (FM 25 mg/kg) Animal Número de quadrados invadidos Tempo parado (segundos) Número de levantadas Número de auto-limpeza Número de bolos fecais 1 38 26 16 25 0 2 15 0 2 75 3 3 52 108 26 50 3 4 16 10 2 62 4 5 44 31 10 62 3 6 11 15 3 50 1 7 62 41 17 25 2 8 0 139 14 12 1 Média ± EPM 29,8 ± 7,8 46,2 ± 17,7 11,25 ± 3,1 45,1 ± 7,8 2,1 ± 0,5 H (Diazepam 10 mg/kg) Animal Número de quadrados invadidos Tempo parado (segundos) Número de levantadas Número de auto-limpeza Número de bolos fecais 1 53 163 4 44 0 2 2 229 6 0 0 3 27 176 1 14 0 4 42 169 9 30 0 5 73 152 10 32 0 6 34 213 4 22 0 7 30 187 10 38 0 8 58 180 2 43 0 Média ± EPM 39,0 ± 7,7 183,6 ± 9,1* 5,8 ± 1,3* 27,8 ± 5,4 0,00 ± 0,00* * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. 135 Tabela 9 – Tempo de recuperação do reflexo postural (duração do sono), em minutos, no teste de potenciação do sono induzido por barbitúrico, em camundongos previamente tratados (60 min) pela via p.o. com veículo (C), com o extrato etanólico das folhas do S. umbellatum (EES; 25, 50 ou 100 mg/kg) (A), com as frações hexânica (FH, 10 mg/kg), clorofórmica (FC, 20 mg/kg), metanol/água (FM, 25 mg/kg) (B) ou diazepam (DZP; 5 mg/kg). A (EES) Animal C EES 25 EES 50 EES 100 DZP 1 32 52 49 51 110 2 25 46 60 59 95 3 26 53 55 55 60 4 18 43 50 50 114 5 17 45 54 58 108 6 35 55 74 57 80 7 60 67 96 8 49 87 85 50,4 ± 2* 62,0 ± 5* 63,9 ± 6* 94,5 ± 9* Média ± EPM 25,5 ± 3 B (Frações) Animal C FH 10 FC 20 FM 25 DZP 1 32 40 53 16 110 2 25 81 42 15 95 3 26 45 49 25 60 4 18 36 44 23 114 5 17 49 40 35 108 6 35 40 49 16 80 7 53 61 33 8 101 48,3 ± 3* 23,3 ± 3 Média ± EPM 25,5 ± 3 55,6 ± 8* 94,5 ± 9* * Estatisticamente diferente do grupo controle (P < 0,05) – ANOVA, teste de Tukey. Livros Grátis ( http://www.livrosgratis.com.br ) Milhares de Livros para Download: Baixar livros de Administração Baixar livros de Agronomia Baixar livros de Arquitetura Baixar livros de Artes Baixar livros de Astronomia Baixar livros de Biologia Geral Baixar livros de Ciência da Computação Baixar livros de Ciência da Informação Baixar livros de Ciência Política Baixar livros de Ciências da Saúde Baixar livros de Comunicação Baixar livros do Conselho Nacional de Educação - CNE Baixar livros de Defesa civil Baixar livros de Direito Baixar livros de Direitos humanos Baixar livros de Economia Baixar livros de Economia Doméstica Baixar livros de Educação Baixar livros de Educação - Trânsito Baixar livros de Educação Física Baixar livros de Engenharia Aeroespacial Baixar livros de Farmácia Baixar livros de Filosofia Baixar livros de Física Baixar livros de Geociências Baixar livros de Geografia Baixar livros de História Baixar livros de Línguas Baixar livros de Literatura Baixar livros de Literatura de Cordel Baixar livros de Literatura Infantil Baixar livros de Matemática Baixar livros de Medicina Baixar livros de Medicina Veterinária Baixar livros de Meio Ambiente Baixar livros de Meteorologia Baixar Monografias e TCC Baixar livros Multidisciplinar Baixar livros de Música Baixar livros de Psicologia Baixar livros de Química Baixar livros de Saúde Coletiva Baixar livros de Serviço Social Baixar livros de Sociologia Baixar livros de Teologia Baixar livros de Trabalho Baixar livros de Turismo
Baixar