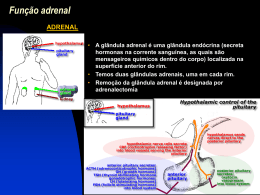
CASO CLÍNICO: Hiperplasia congênita da suprarenal (bloqueio da 21-hidroxilase) Apresentação: Johnny Emanuel Coordenação: Paulo R. Margotto Hospital Regional da Asa Sul/SES/DF www.paulomargotto.com.br Brasília, 10/8/2011 HD Johnny Emanuel ESCS! • Mãe de 43 anos, admitida na CO do HRAS dia 21/06/2011 com o diagnóstico de amniorrexe prematura às 21h30 • IG de 37 semanas, DUM = 06/10/10 • G2 P0 C1 A0 (cesárea prévia há 5 anos) • Fez 5 consultas de pré-natal com sorologias para Hep B/C e HIV e VDRL negativos em trimestre desconhecido • Tipagem sanguínea: A Rh (+) • • • • Parto cesárea por bolsa rota há 14 horas RN nascido vivo às 0h50 de 22/06 Apgar 1º / 5º min: 7/8 RN nascido com cianose central e obstrução nasal leve necessitando de reanimação por VPP sob máscara • Peso 2.885g, Comprimento: 48 cm PC: 35cm • Criança com cianose central e obstrução nasal leve, taquipneica • Ausculta Pulmonar: Roncos em ambos hemitóraces • Ausculta Cardíaca: “OK” • Abdome: Flácido, 2A 1V • Sexo indefinido, anus aparentemente perfurado • Feito credê, kanakion • Colocada em berço aquecido • Necessitou de permanecer com oxigênio sob máscara, 5L/min devido a desconforto respiratório transitório • Solicitado Rx tórax • RN corada, reativa ao exame, boca seca, acianótica, anictérica, FA aberta e normotensa, eupneica • AR: MV+, sem ruídos ou desconforto. FR: 49 • ACV: RCR, 2T, BNF sem sopros. FC: 125 • Abdome: Globoso, algo distendido, RHA+, umbigo, 2A e 1V. • Genitália: Grandes lábios proeminentes, com pequenos lábios fundidos, clitóris hipertrofiado, não palpado gônadas, uretra aparentemente estreita • Ânus pérvio, ortolani negativo. Recém Nascido a Termo, Peso Adequado para a Idade Gestacional Distúrbio Genitália de Ambígua Diferenciação Sexual • Genitália que não é possível definir um sexo fenotípico (genitália ambígua) ou • Discordância do sexo cariotípico com o sexo fenotípico • Pode ser uma emergência clínica • Tem importante impacto psico-social Genitália de aparente aspecto masculino: Genitália de aparente aspecto • Gônadas não-palpáveis; feminino: • Tamanho peniano esticado • Diâmetro clitoriano superior abaixo de -2,5DP da média de a 6 mm; tamanho peniano normal • Gônada palpável em saliência para a idade labioescrotal; • Gônadas com maior diâmetro • Fusão labial posterior; inferior a 8 mm; • Massa inguinal que possa • Presença de massa inguinal corresponder a testículos. que poderá corresponder ao útero e trompas rudimentares; • Hipospádia. • Formação da gônada bipotencial e desenvolvimento dos ductos de Miller e de Wolff • Diferenciação gonadal – Há a tendência intrínseca de desenvolvimento ovariano • Desenvolvimento da genitália interna – Desenvolvimento do ductos correspondentes ao sexo (Miller ; Wolff ) • Desenvolvimento da genitália externa – Ação androgênica ou não • Pode ser devido a: – Distúrbios genéticos – Distúrbios endócrinos – Distúrbios embriopáticos • Padrões básicos de DDS: – Criança XX virilizada – Criança XY pouco virilizada – Alterações cromossômicas/genéticas diversas • Manutenção do paciente – Dosagem de eletrólitos e glicemia com correção se necessário • Devido a alta incidência de Hiperplasia Adrenal Congênita, caso haja alguma anormalidade deve-se considerar uso de hidrocortisona – Conversa com a família sobre a criança e iniciar o planejamento de decisão do gênero • Investigação: – Anamnese – Exame físico • • • • Gônada Falo Abertura uretral Outras anomalias causadas por DDS sindrômico – Cariótipo – Dosagens hormonais como 17-OH-progesterona e outros a depender da suspeita • Cariótipo XX – Distúrbios da determinação gonadal • Hermafroditismo verdadeiro • Disgenesia gonadal pura tipo XX • Homem XX – Distúrbios da diferenciação sexual feminina • • • • Hiperplasia congênita de supra-renais (95%) Deficiência de aromatase Andrógenos maternos ingeridos e/ou produzidos Idiopática • Cariótipo XY – Distúrbios da determinação gonadal • • • • • • Hermafroditismo verdadeiro Disgenesia gonadal mista Disgenesia gonadal pura XY Disgenesia testicular Síndrome da regressão testicular Agenesia ou Hipogenesia de células de Leydig • Cariótipo XY – Distúrbios da função testicular • Deficiência ou anormalidade de LH ou de seu receptor • Síndrome da persistência dos ductos de Müller • Defeitos de síntese de Testosterona – Distúrbios dos tecidos-alvo dependentes de andrógenos • Deficiência de 5α redutase tipo 2 (SRD5A2) • Síndrome da insensibilidade androgênica (AIS) – Idiopática • Mosaicismos – Hermafroditismo verdadeiro – Disgenesia gonadal mista • Aneuploidias – Síndrome de Klinefelter e suas variantes – Síndrome de Turner e suas variantes Gônadas palpáveis bilateralmente Gônadas não palpáveis Uma gônada palpável Diagnóstico mais provável HAC 46,XX ou mosaicismos 46,XY AMH alto IPA ou Idiopático Esteróides séricos Aumentados 46,XX HAC Normais 46,XY Estímulo com hCG e AMH HV ou Homem XX AMH normal ou aumentado Deficiência na síntese de T Virilização Materna ou deficiência de aromatase AMH baixo Deficiência da conv periférica T DHT Biópisa Gonadal Cariótipo Variável HV, DGM ou PHM • A questão primordial é a determinação do sexo social – Equipe multidisciplinar – Escolha precoce com risco de inadequação futura X – Escolha tardia com participação da criança – Deve se buscar o máximo possível a função sexual satisfatória, a fertilidade e o aspecto cosmético nessa ordem de importância • Cirurgia reconstrutora da genitália – Depende da avaliação cuidadosa das estruturas presentes, visando uma técnica cirúrgica reconstrutiva mais simples – A construção de genitália externa masculina funcionante atualmente é impossível – Gonadectomia é necessária em casos que a degeneração maligna é frequente • Reposição hormonal pertinente • Indução puberal adolescente com gônadas não funcionantes • Introduzido NAN 1 complementar • Solicitado parecer para genética, respondido no mesmo dia – Exame físico genital: falus de +/- 1,5cm, eminências lábioescrotais pigmentadas aparentemente fusionadas de forma parcial, orifício uretral visível, gônadas não palpadas, ânus tópico. – Demais aspectos do exame físico dentro da normalidade – HD: DDS – Condutas sugeridas: • • • • Não registrar a criança até exames ficarem prontos Solicitado cariótipo Dosar glicose, Na+, K+, ClRealizar o teste de triagem neonatal expandido (com teste para Hiperplasia adrenal congênita) • Marcar consulta com a Endocrinologia • Não dar alta até investigação completa • Realizar USG pélvica • Criança evoluiu bem, sugando ao seio materno, aceitando a dieta complementar porém com perda ponderal • Detectado um sopro sistólico ao exame físico dia 25/06, sendo solicitado ecocardiograma • Ecocardiograma realizado dia 25/06 com laudo de Forame Oval patente e Insuficiência Aórtica discreta • Dia 27/06 foi colhido sangue para – Glicemia e eletrólitos – Gasometria – Teste de triagem neonatal – Cariótipo • No mesmo dia foi encaminhada para realização de USG renal e pélvico • Glicemia: 68mg/dL • Na+ 159, K+ 5,7; Cl- 103 • Gasometria venosa: – pH: 7,368; pCO2: 33,4; pO2: 54,9 – Hct: 41,4%; Hb 12,2; sO2: 94,4% – Na+ 153,7; K+ 3,13, Cl- 112 • USG Renal e pélvica: Rins normais, bexiga pouco repleta, estrutura tubular com “eco central” retrovesical, sugestiva de ‘útero’ porém, de avaliação prejudicada devido a pouca repleção da bexiga • Dia 28/06 foi avaliada pela genética que calculou o ânion gap em 21,9; e solicitou parecer da endocrinopediatria. • Parecer respondido no mesmo dia – Diagnóstico: “DSD” – Orientações: Dosagens séricas de 17-OHprogesterona, DHEA, S-DHEA, LH, FSH, Testosterona, diidrotestosterona e estradiol. – Retorno ao ambulatório de endocrino em 10 dias com resultados • Evoluiu com perda ponderal até dia 28/06, quando então passou a alternar dias de aumento e perda de peso com progressivo e lento ganho ponderal, sem modificações no exame físico e sempre sugando satisfatoriamente. • Dia 05/07 foi avaliada pela endocrinologia que recebendo o resultado da triagem neonatal que deu alterado apenas a 17-OHprogesterona, considerou o quadro compatível com Hiperplasia adrenal congênita e orientou a prescrição de hidrocortisona • Dia 07/07 avaliada pela genética que recebeu o resultado do cariótipo: 46,XX; e manteve a conduta • No dia 07/07 à noite apresentou desidratação intensa e foram recebidas dosagens séricas de eletrólitos com K+ de 8,6 e Na+ de 114. • Encaminhada à UTI e iniciada hidrocortisona em dose de ataque, solução de glico-insulina e reposição volêmica vigorosa. • Realizada gasometria arterial: pH 7,48, pCO2 21,4; pO2 48,2; SO2 93,4%; HCO3 18,9; BE: 8,3; • Após medidas iniciais realizadas novas dosagens de eletrólitos com resultados: K+ de 4,6 e Na+ de 118. • RN melhorou da desidratação, mas não totalmente e com diurese aumentada necessitou de várias fases rápidas. • Dia 09/07 estabilizou-se • Voltou a evoluir bem, sugando ao seio, porém sempre com ganho ponderal lento. • Eletrólitos dia 12/07: Na+ 136; K+ 4,9 • Eletrólitos dia 15/07: Na+ 133; K+ 5,5 • Dia 20/07 recebidas as dosagens sérica de 17OH-progesterona (4900 ng/dL, N=17 a 204) e da androstenediona (> 10 ng/ml, N=0,9 a 4,6) • Após mais ganho ponderal recebeu alta em reposição de glico e mineralocorticóide. • Dia 31/07 foi reinternada com hipoatividade e sonolência sem nenhum outro sintoma associado • Estava em uso de 0,03mg/kg/dia de FlorinefR e 18 mg/m2/dia de hidrocortisona • Durante a internação foram ajustadas as doses dos medicamentos: – FlorinefR 2 cps/dia – Hidrocortisona 40 mg/m2/dia Hiperplasia Adrenal Congênita Crise perdedora de Sal Hiperplasia Adrenal Congênita - HAC • Desordem autossômica recessiva na esteroidogênege no córtex adrenal levando a uma deficiência de CORTISOL • Sinônimo: síndrome androgenital • A incidência varia entre etnias, entre locais e entre os tipos de HAC • HAC clássica: 6,7/100.000 no mundo, maior entre esquimós Yupic do Alasca 357/100.000 • HAC não-clássica: 1000/100.000 no mundo, maior entre judeus Ashkenazi e eslavos Eixo Hipotálamo-hipófise normal Mineralocorticóides Desmolase Glicocorticóides Esteróides Sexuais Adrenalina Principal produto adrenal responsável pelo feedback negativo sobre a liberação de CRH e ACTH Fisiopatologia Desmolase • A deficiência pode acontecer em todas as enzimas: ▫ ▫ ▫ ▫ ▫ 21-Hidroxilase ou CYP21A2 (95% dos casos) 11-β-hidroxilase ou CYP11B1(5% dos casos) 17-β-hidroxilase + 17,20-liase ou CYP17 (raro) 3-β-hidroxisteróide-desidrogenase Desmolase, entre outras mais raras • A deficiência pode ser incompleta ▫ Formas heterozigotas normal-defeito são assintomáticos ▫ Formas heterozigotas defeito1-defeito2 tem graus variados de doença Poder Mineralocorticóide Tipos clínicos de HAC • Clássica ▫ Perdedora de sal (atividade zero da CYP21) ▫ Não perdedora de sal ou virilizante simples (alguma atividade residual da CYP21) • Não clássica ( pouca atividade da CYP21) • Deficiência das outras enzimas Clássica • Virilizante simples: ▫ Manifestações do hiperandrogenismo ▫ RN femininos com DDS ▫ RN masculinos assintomáticos ou com aumento peniano e hiperpigmentação escrotal ▫ Pequena atividade da CYP21 suficiente para biossíntese de aldosterona ▫ Crescimento linear aumentado durante a infância, porém adultos menores que potencial genético Clássica • Perdedora de Sal ▫ Hiperandrogenismo igual à forma virilizante simples ▫ Atividade da CYP21 insuficiente para produzir aldosterona ▫ RN apresentam “crise perdedora de sal” devido a baixa atividade da aldosterona Não clássica • Assintomáticos ao nascimento • Sinais de hiperandrogenismo tardio • Meninas: ▫ Aumento clitoriano ▫ Pubarca precoce, acne refratária ▫ Ciclos anovolatórios, ciclos irregulares, amenorréia • Meninos: ▫ Pubarca precoce, aumento peniano ▫ Acne refratária Formas raras • Na deficiência de CYP11B1 ou CYP17 há acúmulo de Deoxicorticosterona e/ou 11-Deoxicortisol (composto S) que apresentam ação mineralocorticóide • Na deficiência de CYP11B1 há também hiperandrogenismo similar à deficiência de CYP21 ↓ Aldosterona Retenção de K, diurese excessiva de Na/Cl Diarréia e vômitos “Crise Perdedora de Sal” ↑ Mineralocorticóide Diurese excessiva de K Retenção salina Hipertensão secundária ↓ Cortisol Hipoglicemia Alteração do tono vascular/hipotensão Desidratação Acidose Metabólica Déficit pondoestatural Resumo das formas de HAC Diagnóstico Laboratorial • A alteração bioquímica mais característica é a a elevação da 17-OH-progesterona ▫ Níveis acima de 5000ng/dL são característicos das formas clássicas. (Pcte: 4900) • Para a avaliação da ação mineralocorticóide, a atividade da renina plasmática deve ser mensurada Diagnóstico Laboratorial • Nas formas não clássicas, os valores basais da 17OHP podem não estar elevados o suficiente para o diagnóstico • Então usa-se o teste de estímulo de ACTH, dosandose a 17-OHP 0 e 60 min após injeção IV de ACTH. Níveis acima de 500ng/dL basal ou 1000 ng/dL em 60 minutos são os pontos de corte • Para a deficiência de CYP11B1, os níveis de 11desoxicortisol e 11-desoxicorticosterona e de andrôgenos [DHEA(-S), androstenediona] estão aumentados Tratamento • Crise de perda de sal ▫ Reposição volêmica agressiva ▫ Administração de hidrocortisona 50mg/m2 de superfície corporal, em bolus e a mesma dose por dia dividindo em 4 ou 6 vezes(4/4-6/6h) ▫ Reposição mineralocorticóide com 9-α-fludrocortisona (Florinefe) 0,05 a 0,1 mg/dia ▫ Acrescentar NaCl à dieta em crianças pequenas Tratamento • Manutenção ▫ Reposição de glicocorticóide indefinidamente, evitando a hiperestimulação adrenal Hidrocortisona 10 a 25mg/m2 Menos interferência no crescimento Indisponibilidade forma oral exceto manipulando Prednisona/prednisolona 4-5 mg/m2 Mais comercialmente disponível Uso aceito amplamente para pós-púberes Tratamento • Em caso de stress, deve ser aumentada a dose do glicocorticóide • Paciente deve receber do médico e sempre levar consigo uma carta de urgência avisando da doença e da necessidade desse aumento Tratamento • A correção cirúrgica inicial (clitoroplastia e introitoplastia) deve acontecer até cerca de 24 meses, para evitar prejuízos à identidade sexual. • Algumas vezes, principalmente se houver algum grau de hipoplasia vaginal, há necessidade de novas intervenções cirúrgicas, na época da puberdade. Acompanhamento • Consultas periódicas verificando crescimento do clitóris e pênis e aparecimento/aumento de pilificação pubiana, que podem indicar excesso de produção de andrógenos. • O crescimento em altura deve ser mantido dentro dos padrões normais para a idade e padrão familiar ▫ Excesso de glicocorticóide diminui a velocidade de crescimento e a falta aumenta • Controle com exames complementares ▫ 17-OHP, androstenediona a cada três a seis meses ▫ idade óssea anualmente. Prognóstico • Ótimo prognóstico se detectada e tratada precocemente, sem alterações significativas de fertilidade, altura final, desenvolvimento puberal e psicossexual ▫ No sexo feminino o sucesso da reconstrução cirúrgica influencia bastante esse prognóstica • O diagnóstico tardio ou o tratamento irregular, pode levar a alteração da idade óssea, baixa estatura final e virilização, com distúrbios menstruais e de fertilidade. Informações adicionais • No teste do pezinho ampliado há a triagem para a HAC por deficiência de CYP21 • Em gestação de risco pode se realizar a biópsia de vilo corial após a 10ª semana de gestação visando determinar o sexo e estudar o gene possível de ser defeituoso (CYP21) • O tratamento pré-natal é a dexametasona do diagnóstico da gestação até o resultado da biópsia de vilo corial e após apenas para as meninas afetadas • Gadelha, MM; Margotto, PR; Barbosa, ME. Distúrbios do desenvolvimento sexual e hiperplasia congênita de supra-renal in Assistência ao recém-nascido de risco, ESC, Brasília, 2011, no prelo • Damiani D et al. Genitália Ambígua: Diagnóstico Diferencial e Conduta. Arq Bras Endocrinol Metab 2001;45/1:37-48 • UpToDate, Waltham, MA, 2011. Consultem também: Hiperplasia congênita das Supras-renais Autor(es): Mariana de Melo Gadelha Distúrbios do desenvolvimento sexual e hiperplasia congênita da supra-renal Autor(es): Mariana de Melo Gadelha, Maristela Estevão Barbosa, Paulo R. Margotto Caso Clínico: Disgenesia gonadal mista Autor(es): Romina S. Heredia Garcia Silva, Maria Teresinha de Oliveira Cardoso, Paulo R. Margotto Distúrbios do desenvolvimento sexual e hiperplasia congênita de supra-renal Autor(es): Mariana de Melo Gadelha, Maristela Estevão Barbosa, Paulo R. Margotto Desordens da diferenciação sexual Autor(es): Mariana de Melo Gadelha Hiperplasia Adrenal Congênita: Revisão de Literatura Autor(es): Hannah Peixoto Schechtman Clicar aqui! E nos casos de HIPOSPÁDIA? Afecções urológicas comuns na criança Autor(es): Maurícia Cammarota Sempre que houver hipospádia associada à criptorquia com gônada não palpável – avaliação genética OBRIGADO Ddo Johnny Emunuel e Dr. Paulo R. Margotto ESCS!
Download