☰
Explorar
Assinar em
Inscrever-se
Envio
×
Baixar
Ciências
Texto Completo
Jocélio Alves de Araújo - Faculdade Adventista da Bahia
Click to add title - Portal Indicadores da UERJ
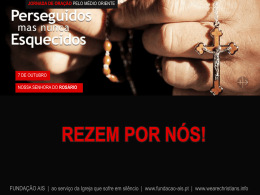
Diapositivo 1 - Fundação AIS
Acidentes com petróleo no Brasil
sindicato dos nutricionistas do estado da bahia
Ó Maria, rainha das missões
APOGEU - MÉDIO - AULA 13
Planta medicinal
Etapas do processo de extração de requisitos
REGULAMENTO DO CONCURSO Modalidade: VÍDEO