1
UNIDADE I
INTRODUÇÃO A FISIOLOGIA: FISIOLOGIA
CELULAR E GERAL
Ø
Organização Funcional do Corpo Humano e Controle do "Meio Interno"
Ø A Célula e seu Funcionamento
Ø Controle Genético da Síntese de Proteínas, do Funcionamento e da
Reprodução Celular
2
CAPÍTULO I
Organização Funcional do Corpo Humano e
Controle do "Meio Interno"
A fisiologia tenta explicar os fatores físicos e químicos
responsáveis pela origem, desenvolvimento e progressão da
vida. Cada tipo de vida, desde o mais simples vírus até a maior
árvore ou o complexo ser humano, possui características
funcionais próprias. Portanto, o vasto campo da fisiologia pode
ser dividido cm fisiologia virai, fisiologia bacteriana, fisiologia
celular, fisiologia vegetal, fisiologia humana, e em muitas outras
áreas.
Fisiologia humana. Na fisiologia humana, estamos
interessados nas características e mecanismos específicos do corpo
humano que o tornam um ser vivo. O simples fato de que
permanecemos vivos está quase além de nosso controle, pois
a fome nos faz procurar alimento e o medo, a buscar abrigo. As
sensações de frio nos levam a produzir calor e outras forças
nos levam a procurar companhia e a reproduzir. Assim, o ser
humano é, na verdade, um autômato, e o fato de sermos seres
que sentem, que têm sentimentos e conhecimento c parte dessa
seqüência automática da vida; esses atributos especiais nos
permitem viver sob condições extremamente variáveis que, de
outra forma, impossibilitariam a vida.
AS CÉLULAS COMO AS UNIDADES
VIVAS DO CORPO
A unidade viva fundamental do corpo é a célula e cada
órgão é um agregado de muitas células diferentes, mantidas
unidas por estruturas intercelulares de sustentação. Cada tipo
de célula é especialmente adaptado para a execução de uma
função determinada. Por exemplo, os glóbulos vermelhos do
sangue, um total de 25 trilhões de células, transportam
oxigênio dos pulmões para os tecidos. Embora esse tipo de
célula talvez seja o mais abundante, é possível que existam
outros 75 trilhões de células. Todo o corpo é formado, então, por
cerca de 100 trilhões de células.
Embora as inúmeras células do corpo possam, muitas vezes,
diferir acentuadamente entre si, todas apresentam determinadas
características básicas que são idênticas. Por exemplo, em todas
as células, o oxigênio reage com carboidratos, gordura ou
proteína para liberar a energia necessária ao funcionamento
celular. Ainda mais, os mecanismos gerais para a transformação
dos nutrientes em energia são, em termos básicos, os mesmos em
todas as células e, igualmente, todas as células eliminam os
produtos finais de suas reações químicas para os líquidos onde ficam
imersas.
Quase todas as células também têm capacidade de se repro-
duzir e, sempre que células de determinado tipo são destruídas
por qualquer causa, as células remanescentes do mesmo tipo
regeneram, com muita freqüência, novas células até que seja
restabelecido seu número adequado.
O LÍQUIDO EXTRACELULAR - O MEIO
INTERNO
Cerca de 56% do corpo humano são compostos de líquidos.
Embora a maior parte desse líquido fique no interior das células
— e seja chamado de liquido intracelular —, cerca de um terço
ocupa os espaços por fora das células e é chamado de liquido
extracelular. O líquido extracelular se movimenta continuamente
por todo o corpo. É transportado rapidamente no sangue
circulante e, em seguida, misturado entre o sangue e os líquidos
teciduais por difusão através das paredes capilares. No líquido
extra-celular ficam os íons c os nutrientes necessários às células,
para manutenção da vida celular. Por conseguinte, todas as
células partilham de um mesmo ambiente, o líquido extracelular,
razão por que esse líquido extracelular é chamado de meio
interno do corpo, ou milieu intérieur, expressão criada, há
pouco mais de 100 anos, pelo grande fisiologista francês do
século XIX, Claude Bernard.
As células são capazes de viver, crescer e desempenhar suas
funções específicas enquanto estiverem disponíveis, nesse
ambiente interno, as concentrações adequadas de oxigênio,
glicose, diversos íons, aminoácidos, substâncias gordurosas e
outros constituintes.
Diferenças entre os líquidos extra e intracelulares. O líquido
extracelular contém grandes quantidades de íons sódio, cloreto
e bicarbonato, mais os nutrientes para as células, tais como
oxigênio, glicose, ácidos graxos c aminoácidos. Também contém
dióxido de carbono que está sendo transportado das células
até os pulmões para serem excretados, além de outros produtos
celulares que, igualmente, estão sendo transportados para o
rim, onde vão ser excretados.
O líquido intracelular difere, de forma significativa, do
líquido extracelular; em especial, contém grandes quantidades
de íons potássio, magnésio e fosfato, em lugar dos íons sódio e
cloreto presentes no líquido extracelular. Essas diferenças são
mantidas por mecanismos especiais de transporte de íons através
das membranas celulares. Esses mecanismos são discutidos no
Cap. 4.
3
MECANISMOS "HOMEOSTÁTICOS"
DOS PRINCIPAIS SISTEMAS
FUNCIONAIS
HOMEOSTASIA
A palavra homeostasia é usada pelos fisiologistas para
significar manutenção das condições constantes, ou estáticas, do
meio interno. Em essência, todos os órgãos e tecidos do corpo
exercem funções que ajudam a manter essas condições
constantes. Por exemplo, os pulmões fornecem oxigênio para o
líquido extracelular para repor o que está sendo consumido
pelas células; os rins mantêm constantes as concentrações iônicas
e o sistema gastrintestinal fornece nutrientes. Grande parte deste
texto está relacionado ao modo como cada órgão ou tecido
contribui para a homeostasia. Para iniciar esta discussão, serão
descritos, resumidamente, os diferentes sistemas funcionais do
corpo e seus mecanismos homeostáticos; em seguida, será
apresentada a teoria básica dos sistemas de controle que atuam
harmoniosamente entre si.
OS SISTEMAS DE TRANSPORTE DO LÍQUIDO
EXTRACELULAR - O SISTEMA CIRCULATÓRIO
O líquido extracelular é transportado para todas as partes
do corpo em duas etapas distintas. A primeira depende do
movimento do sangue ao longo do sistema circulatório, e a
segunda, do movimento de líquido entre os capilares sanguíneos
e as células. A Fig. 1.1 mostra a circulação geral do sangue.
Todo o sangue contido na circulação percorre todo o circuito
em cerca de um minuto em média, no repouso, e até seis vezes
por minuto quando a pessoa está extremamente ativa.
Conforme o sangue circula pelos capilares, ocorre troca
contínua de líquido extracelular entre a parte de plasma do
sangue e o líquido intersticial que preenche os espaços entre as
células: os espaços intercelulares. Esse processo é mostrado na
Fig. 1.2. Note que os capilares são porosos, de modo que grandes
quantidades de líquido e de seus constituintes em solução podem
difundir, nos dois sentidos, entre o sangue e os espaços
teciduais, como indicado pelas setas na figura. Esse processo
de difusão é causado pela movimentação cinética das
moléculas, tanto no plasma como no líquido intersticial. Isto é, o
liquido e as moléculas em solução estão continuamente em
movimento e saltando em todas as direções no interior do
próprio líquido e também através dos poros e pelos espaços
teciduais. Quase que nenhuma célula fica distante mais de 25 a
50 m de um capilar, o que assegura a difusão de quase todas as
substâncias do capilar para a célula dentro de poucos segundos.
Assim, o líquido extracelular, por todo o corpo, tanto o do plasma
como o do líquido contido nos espaços intercelulares, está sendo
continuamente misturado, o que garante sua homogeneidade
quase total.
ORIGEM DOS NUTRIENTES DO LÍQUIDO
EXTRACELULAR
Sistema respiratório. A Fig. 1.1 mostra que, cada vez que
o sangue circula pelo corpo, ele também flui pelos pulmões.
Nos alvéolos, o sangue capta oxigênio, ganhando, dessa forma,
o oxigênio necessitado pelas células. A membrana entre os
alvéolos e o lúmen dos capilares pulmonares tem espessura de
apenas 0,4 a 2,0 m e o oxigênio se difunde, através dessa
membrana, para o sangue exatamente da mesma maneira
como a água e os íons se difundem através dos capilares
teciduais.
Tubo gastrintestinal. Grande parte do sangue que é
bombeada pelo coração também passa pelas paredes dos órgãos
gastrintestinais. Aí, diversos nutrientes dissolvidos, incluindo
carboidratos, ácidos graxos, aminoácidos e outros, são
absorvidos para O líquido extracelular.
Fígado e outros órgãos que desempenham funções
primariamente metabólicas. Nem todas as substâncias absorvidas
do tubo gastrintestinal podem ser usadas, na forma em que foram
absorvidas, pelas células. O fígado modifica as composições
químicas dessas substâncias, transformando-as em formas mais
utilizáveis, e outros tecidos do corpo — as células adiposas, a
mucosa gastrintestinal, os rins e as glândulas endócrinas —
ajudam a modificar
Fig 1.1 Organização geral do sistema circulatório.
Fig. 1.2 Difusão de líquido através das paredes
capilares e pelos espaços intersticiais.
4
as substâncias absorvidas ou as armazenam, até que sejam
necessárias no futuro.
Sistema musculoesquelético. Algumas vezes, é levantada a
questão: como é que o sistema musculoesquelético participa nas
funções homeostáticas do corpo? A resposta a ela é óbvia e
simples. Se não fosse por esse sistema, o corpo não se poderia
deslocar para um local apropriado no tempo adequado, a fim
de obter os alimentos necessários para sua nutrição. O sistema
musculoesquelético também gera a motilidade usada na proteção
contra os ambientes adversos, sem o que todo o corpo, junto
com os demais mecanismos homeostáticos, poderia ser destruído
instantaneamente.
REMOÇÃO DOS PRODUTOS FINAIS
DO METABOLISMO
Remoção do dióxido de carbono pelos pulmões. Ao mesmo
tempo que o sangue capta oxigênio nos pulmões, o dióxido de
carbono está sendo liberado do sangue para os alvéolos, e o
movimento respiratório do ar, para dentro e para fora dos
alvéolos, transporta esse gás para a atmosfera. O dióxido de
carbono é o mais abundante de todos os produtos finais do
metabolismo.
Os rins. A passagem de sangue pelos rins remove a maioria
das substâncias que não são necessárias às células. De forma
especial, essas substâncias incluem os diferentes produtos finais
do metabolismo celular, além do excesso de íons e de água que
podem ter-se acumulado no líquido extracelular. Os rins realizam
sua função, primeiro, ao filtrarem grandes quantidades de
plasma, pelos glomérulos, para os túbulos e, em seguida,
reabsorverem para o sangue as substâncias que o corpo
necessita — como glicose, aminoácidos, quantidades
apropriadas de água e muitos íons. Contudo, a maior parte das
substâncias que não são necessárias ao corpo, especialmente os
produtos finais do metabolismo, como a uréia, é pouco
reabsorvida e, como resultado, elas passam pelos túbulos renais
para serem eliminadas na urina.
REGULAÇÃO DAS FUNÇÕES CORPORAIS
O sistema nervoso. O sistema nervoso é formado por três
constituintes principais: o componente sensorial, o sistema
nervoso central (ou componente integrativo) e o componente
motor. Os receptores sensoriais detectam o estado do corpo ou o
estado de seu ambiente. Por exemplo, os receptores, presentes
por toda a pele, denotam cada e todas as vezes que um objeto
toca a pessoa em qualquer ponto. Os olhos são órgãos
sensoriais que dá à pessoa uma imagem visual da área que a
cerca. O sistema nervoso central é formado pelo encéfalo e pela
medula espinhal. O encéfalo pode armazenar informações,
gerar pensamentos, criar ambições e determinar quais as reações
que serão executadas pelo corpo em resposta às sensações. Os
sinais apropriados são, em seguida, transmitidos, por meio do
componente motor do sistema nervoso, para a efetivação dos
desejos da pessoa.
Um grande componente do sistema nervoso é chamado de
sistema autonômico. Ele atua ao nível subconsciente e controla
muitas funções dos órgãos internos, inclusive o funcionamento
do coração, os movimentos do tubo gastrintestinal e a secreção
de diversas glândulas.
O sistema de regulação endócrina. Existem dispersas no
corpo oito glândulas endócrinas principais, secretoras de
substâncias químicas, os harmônios. Os hormônios são
transportados pelo líquido extracelular até todas as partes do
corpo, onde vão participar da regulação do funcionamento
celular. Por exemplo, os hormônios tireóideos aumentam a
velocidade da maioria das reações químicas celulares. Dessa
forma, o hormônio tiróideo deter mina a intensidade da
atividade corporal.
A insulina controla o metabolismo da glicose, os hormônios do
córtex supra-renal controlam o metabolismo iônico e protéico, e
o hormônio paratiróideo controla o metabolismo ósseo. Assim,
os hormônios formam um sistema de regulação que complementa
o sistema nervoso. O sistema nervoso, em termos gerais, regula,
principalmente, as atividades motoras e secretoras do corpo,
enquanto o sistema hormonal regula, de modo primário, as
funções metabólicas.
REPRODUÇÃO
Por vezes, a reprodução não é considerada como uma função
homeostática. Todavia, a reprodução participa da manutenção
das condições estáticas, por produzir novos indivíduos que vão
tomar o lugar dos que morreram. Isso talvez pareça um uso
permissivo do termo homeostasia, mas, na verdade, ilustra que,
em última instância, todas as estruturas do corpo, em essência,
são organizadas de forma a manter a automaticidade e a
continuidade da vida.
OS SISTEMAS DE CONTROLE DO
CORPO
O corpo humano contém literalmente milhares de sistemas
de controle. Os mais intricados deles são os sistemas genéticos
de controle, atuantes em todas as células, para regular o
funcionamento intracelular e, também, todas as funções
extracelulares. Este tópico é discutido no Cap. 3. Muitos outros
sistemas de controle atuam ao nível dos órgãos, para regular o
funcionamento de partes distintas desses órgãos; outros atuam
ao nível de todo o corpo, para regular as inter-relações entre os
órgãos. Por exemplo, o sistema respiratório, atuando em
associação com o sistema nervoso, regula a concentração de
dióxido de carbono no líquido extracelular. O fígado e o
pâncreas regulam a concentração de glicose no líquido
extracelular. Os rins regulam a concentração dos íons
hidrogênio, sódio, potássio, fosfato e muitos outros no
líquido extracelular.
EXEMPLOS DE MECANISMOS DE CONTROLE
Regulação das concentrações de oxigênio e de dióxido de
carbono no líquido extracelular. Dado que o oxigênio é uma das
principais substâncias necessárias para as reações químicas no
interior das células, é muito importante que o corpo disponha
de mecanismo especial de controle para manter uma
concentração de oxigênio constante e quase invariável no líquido
extra - celular. Esse mecanismo depende, principalmente, das
características químicas da hemoglobina, presente em todos os
glóbulos vermelhos do sangue. A hemoglobina se combina com o
oxigênio enquanto o sangue circula pelos pulmões. Em seguida,
conforme o sangue passa pelos capilares teciduais, a hemoglobina
não libera o oxigênio no líquido tecidual, caso ele já contenha
teor elevado de oxigênio, mas, se a concentração de oxigênio
estiver baixa, será liberado oxigênio em quantidade suficiente
para restabelecer a concentração tecidual adequada de
oxigênio. Dessa forma, a regulação da concentração de
oxigênio nos tecidos depende, primariamente, das características
químicas da própria hemoglobina. Essa regulação recebe o
nome de função tamponadora de oxigênio da hemoglobina.
A concentração de dióxido de carbono no líquido
extracelular é regulada de forma bastante diferente. O dióxido de
carbono é um dos principais produtos finais das reações
oxidativas das células. Se todo o dióxido de carbono formado
nas células pudesse se acumular nos líquidos teciduais, a
ação de massa
5
do próprio dióxido de carbono interromperia, em pouco tempo,
todas as reações liberadoras de energia das células. Felizmente,
um mecanismo nervoso controla a expiração do dióxido de
carbono pelos pulmões e, dessa forma, mantém concentração
constante e relativamente baixa de dióxido de carbono no líquido
extracelular. Em outras palavras, a concentração elevada de
dióxido de carbono excita o centro respiratório, fazendo com que
a pessoa respire mais freqüentemente e com maior amplitude.
Isso aumenta a expiração de dióxido de carbono e, por
conseguinte, acelera sua remoção do sangue e do líquido
extracelular, e esse processo continua até que sua concentração
retorne ao normal. Regulação da pressão arterial. Vários
sistemas distintos contribuem para a regulação da pressão arterial.
Um deles, o sistema barorreceptor, é exemplo excelente e muito
simples de um mecanismo de controle. Na parede da maioria
das grandes artérias da parte superior do corpo - e, de modo
especial, na bifurcação da artéria carótida comum e no arco
aórtico - existem numerosos receptores neurais que são
estimulados pelo estiramento da parede arterial. Quando a
pressão arterial se eleva, esses barorreceptores são estimulados
de forma excessiva, quando, então, são transmitidos impulsos
para o bulbo, no encéfalo. Aí, esses impulsos inibem o centro
vasomotor, o que, por sua vez, reduz o número de impulsos
transmitidos, pelo sistema nervoso simpático, para o coração e
para os vasos. Essa diminuição dos impulsos provoca menor
atividade de bombeamento pelo coração e maior facilidade para
o fluxo de sangue pelos vasos periféricos; esses dois efeitos
provocam o abaixamento da pressão arterial até seu valor
normal. De modo inverso, queda da pressão arterial relaxa os
receptores de estiramento, permitindo que o centro vasomotor
fique mais ativo que o usual, o que provoca a elevação da pressão
arterial ate seu valor normal.
Faixas normais de variação dos constituintes
importantes do liquido extracelular
O Quadro 1,1 enumera os constituintes mais importantes
- junto com suas características físicas - do líquido extracelular,
alem de seus valores normais, faixas normais de variação e limites
máximos que podem ser mantidos, sem morte, por curtos
períodos. Deve ser notado, de forma especial, como é estreita a
faixa normal de variação para cada um desses constituintes.
Valores fora dessa faixa são, em geral, causa ou resultado de
doença. Ainda mais importantes são os limites que, quando
ultrapassados, podem levar à morte. Por exemplo, aumento da
temperatura corporal de apenas 6 a 7°C acima da normal pode,
muitas vezes, gerar um ciclo vicioso de aumento do metabolismo
celular que, literalmente, destrói as células. Também deve ser
notada a faixa muito estreita para o equilíbrio ácido-básico do
corpo,
Quadro 1.1 Alguns constituintes importantes e as características
físicas do líquido extracelular, sua faixa normal de variação e seus
limites não letais aproximados
Limites
Valor
Faixa não-letais
normal normal aproximados Unidades
Oxigênio
40
35-45
10-1.000 mm Hg
Dióxido de carbono
40
35-45
5-80
mm Hg
Íon sódio
142
138-146 115-175
mmol/l
Íon potássio
4,2
3,8-5,0
1,5-9,0
mmol'l
Íon cálcio
1,0-1,4
0,5-2,0
mmoi'i
1,2
Íon cloreto
108
103-112
70-130
mmol/l
Íon bicarbonato
24-32
8-45
mmol/l
28
Glicose
75-95
20-1.500 mmol/l
85
Temperatura corporal 37,0
37,0
18,3-43,3
"C
Ácido-básico
7,4
7,3-7,5
6,9-8,0
pH
com valor normal do pH de 7,4 e valores letais 0,5 abaixo e
acima desse valor normal. Outro fator especialmente importante
é o íon potássio, pois, sempre que sua concentração cai até menos
de um terço da normal, a pessoa tende a ficar paralisada, devido
à incapacidade dos nervos de transmitir os sinais nervosos e,
caso chegue a aumentar até duas ou mais vezes a normal, é
muito possível que o músculo cardíaco fique gravemente
deprimido. Por outro lado, quando a concentração do íon cálcio
cai abaixo da metade da normal, a pessoa fica suscetível de
apresentar contrações tetânicas nos músculos de todo o corpo,
devido à geração espontânea de impulsos nervosos nos nervos
periféricos. Quando a concentração de glicose fica reduzida a
menos da metade da normal, a pessoa, com muita freqüência,
apresenta intensa irritabilidade mental e, por vezes, até
convulsões.
Assim, a análise desses exemplos deve levar à apreciação
extrema da importância e, até mesmo, da necessidade de grande
número de sistemas de controle, mantenedores do corpo
funcionando no estado de saúde; a ausência ou falta de um
desses controles pode resultar em doença grave e até em morte,
CARACTERÍSTICAS DOS SISTEMAS DE CONTROLE
Os exemplos antes apresentados de mecanismos de controle
homeostáticos são apenas uns poucos das muitas centenas a
milhares existentes no corpo; todos eles possuem determinadas
características comuns. Essas características comuns serão
explicadas nas páginas seguintes.
A natureza de feedback negativo da maioria dos
sistemas de controle
A maior parte dos sistemas de controle do corpo atua pelo
processo de feedback negativo, que pode ser melhor explicado
por revisão de alguns dos sistemas de controle homeostáticos
apresentados acima. Na regulação da concentração de dióxido
de carbono, uma concentração elevada de dióxido de carbono
no líquido extracelular provoca aumento da ventilação pulmonar
e isso, por sua vez, produz redução da concentração de dióxido
de carbono, dado que os pulmões conseguem excretar maior
quantidade de dióxido de carbono para fora do corpo. Em outras
palavras, a concentração elevada provoca redução dessa
concentração, o que é negativo em relação ao estímulo inicial. De
modo inverso, caso a concentração de dióxido de carbono caia
até valores muito baixos, isso vai produzir aumento por
feedback dessa concentração. Essa resposta também é negativa
em relação ao estímulo inicial.
Nos mecanismos reguladores da pressão arterial, a elevação
da pressão causa uma série de reações que resultam em redução
da pressão, ou a queda da pressão causa uma série de reações
que resultam em elevação da pressão. Nos dois casos, os efeitos
são negativos em relação ao estímulo inicial.
Por conseguinte, em termos gerais, se algum fator aumenta
ou diminui muito, um sistema de controle ativa um feedback
negativo, que consiste em uma série de alterações que fazem
com que esse fator retorne a determinado valor médio,
mantendo, assim, a homeostasia.
O "ganho" de um sistema de controle. O grau de eficácia
com que um sistema de controle mantém as condições constantes
é determinado pelo ganho do feedback negativo. Por exemplo,
admita-se que grande volume de sangue foi transfundido em
pessoa cujo sistema de controle dos barorreceptores para a
pressão não esteja atuando, e que a pressão arterial se eleve
de seu valor normal de 100 mm Hg até 175 mm Hg. Em seguida,
admita-se que esse mesmo volume de sangue seja transfundido
na mesma pessoa, quando seu sistema barorreceptor estiver
6
atuante e, nesse caso, a pressão só se eleva por 25 mm Hg.
Assim, o sistema de controle por feedback produziu "correção"
de -50 mm Hg, isto é, de 175 mm Hg para 125 mm Hg. Contudo,
ainda persiste um aumento da pressão de +25 mm Hg, o que
é chamado de "erro", e que significa que o sistema de controle
não é 100% eficaz em impedir a variação da pressão. O ganho
do sistema pode ser calculado pelo uso da seguinte relação:
Ganho =
Correção
Erro
Assim, no exemplo acima, a correção é de -50 mm Hg
e o erro que persiste é de +25 mm Hg. Por conseguinte, o
ganho do sistema barorreceptor dessa pessoa, para controle de
sua pressão arterial é —50 dividido por +25, o que é igual a 2. Isso quer dizer que um fator extrínseco que tenda a aumentar ou
a diminuir a pressão arterial só exerce efeito de cerca de dois
terços do que teria caso o sistema de controle não estivesse
atuando.
Os ganhos de outros sistemas fisiológicos de controle são
muito maiores que o do sistema barorreceptor. Por exemplo,
o ganho do sistema regulador da temperatura corporal é de cerca
de -33. Por conseguinte, pode-se ver que o sistema de controle
da temperatura corporal é muito mais eficaz que o sistema
barorreceptor.
O feedback positivo — os cicios viciosos e morte
causados por feedback positivo
Poderá ser feita a seguinte pergunta: Por que, em essência,
todos os sistemas de controle do corpo atuam por mecanismo
de feedback negativo, e não por feedback positivo? Todavia,
se for considerada a natureza do feedback positivo,
imediatamente será visto que o feedback positivo nunca leva à
estabilidade, mas, sim, à instabilidade e, muitas vezes, à morte.
A Fig. 1.3 apresenta um caso em que pode ocorrer morte
por feedback positivo. Essa figura apresenta a eficiência de
bombeamento do coração, mostrando que o coração de pessoa
normal bombeia cerca de 5 litros de sangue por minuto.
Contudo, se a pessoa perder, subitamente, 21 de sangue, a
quantidade de sangue restante no corpo fica reduzida a nível
tão baixo que chega a ser insuficiente para um bombeamento
eficaz pelo coração. Como resultado, a pressão arterial cai e o
fluxo de sangue para o músculo cardíaco, por meio dos vasos
coronários, também diminui. Isso resulta em enfraquecimento do
coração, com redução ainda maior do bombeamento, decréscimo
adicional do fluxo sanguíneo coronário e enfraquecimento ainda
maior do coração. Esse ciclo se repete indefinidamente até a
morte. Deve ser notado que cada ciclo de feedback resulta em
enfraquecimento adicional do coração. Em outras palavras, o
estímulo inicial provoca seu próprio aumento, o que é um
feedback positivo.
O feedback positivo é melhor conhecido como "ciclo
vicioso", mas, na verdade, um grau moderado de feedback
positivo pode ser compensado por mecanismos de controle por
feedback negativo do corpo, situação na qual não se
desenvolverá ciclo vicioso. Por exemplo, se a pessoa do
exemplo acima só perdesse 11, e não 2 1, os mecanismos
normais de feedback negativo de controle do débito cardíaco
e da pressão arterial poderiam anular o feedback positivo, e
a pessoa poderia se recuperar, como mostrado pela curva
tracejada da Fig. 1.3.
Fig. 1.3 Morte causada por feedback positivo quando 21 de sangue
são removidos da circulação.
do feedback positivo. Quando um vaso sanguíneo é rompido
e começa a formação do coágulo, diversas enzimas, chamadas
de fatores de coagulação, são ativadas no interior do próprio
coágulo. Algumas dessas enzimas atuam sobre outras enzimas,
ainda inativas, presentes no sangue imediatamente adjacente ao
coágulo, ativando-as e produzindo coagulação adicional. Esse
processo persiste até que a rotura do vaso fique ocluída e não
mais ocorra sangramento. Infelizmente, por vezes, esse processo
pode ficar descontrolado e produzir coágulos indesejados. Na
verdade, é isso que desencadeia a maioria dos ataques cardíacos
agudos, causados por coágulo que se forma cm placa
aterosclerótica em artéria coronária e que cresce até ocluir
completamente essa artéria.
O parto é outro exemplo de participação de feedback
positivo. Quando as contrações uterinas ficam suficientemente
intensas para empurrar a cabeça do feto contra a cérvix, o
estiramento da cérvix emite sinais, por meio do próprio
músculo uterino, até o corpo do útero, que responde com
contrações ainda mais intensas. Assim, as contrações uterinas
distendem a cérvix e o estiramento da cérvix produz mais
contrações. Quando esse processo fica suficientemente intenso, o
feto nasce. Caso não sejam suficientemente intensas, essas
contrações cessam, para reaparecer alguns dias depois.
Finalmente, outro importante uso do feedback positivo é
representado pela geração de sinais neurais. Isto é, quando a
membrana de uma fibra nervosa é estimulada, isso causa pequeno
influxo de íons sódio, através dos canais de sódio da membrana
neural, para o interior da fibra. Esses íons sódio que penetram
na fibra modificam o potencial de membrana, o que causa
abertura de mais canais, levando a maior variação do potencial,
abertura de mais canais adicionais, e assim por diante. Assim,
de um início bem pequeno, ocorre explosão do influxo de
sódio que gera o potencial de ação. Por sua vez, esse
potencial de ação excita a fibra nervosa em ponto adiante, o
que faz com que esse processo progrida ao longo de todo o
comprimento da fibra.
Contudo, vai-se aprender que, em cada um desses processos
onde o feedback positivo é útil, o próprio feedback positivo faz
parte de processo global de feedback negativo. Por exemplo,
no caso da coagulação do sangue, o processo de coagulação por
feedback positivo é um processo de feedback negativo para a
manutenção do volume normal de sangue. E o feedback positivo
que gera os sinais neurais permite que os nervos participem em
muitos milhares de sistemas de controle por feedback negativo.
7
Alguns tipos mais complexos de sistemas de controle - os
sistemas adaptativos de controle
Adiante, quando se estudar o sistema nervoso, será visto
que esse sistema contém um emaranhado de sistemas de controle
interconectados. Alguns desses sistemas são sistemas de feedback
simples, como os que foram discutidos até aqui. Contudo, muitos
não o são. Por exemplo, vários movimentos do corpo são tão
rápidos que, simplesmente, não há tempo suficiente para que
os sinais neurais trafeguem das partes periféricas do corpo até
o encéfalo e voltem para a periferia, para regular esses
movimentos. Por conseguinte, o encéfalo utiliza um princípio,
chamado de controle por feed-forward, para produzir as
contrações musculares desejadas. Então, sinais nervosos
sensoriais, originados nas partes era movimento, informam o
encéfalo de se o movimento apropriado, planejado pelo
encéfalo, foi ou não executado. Caso não tenha sido, o
encéfalo corrige os sinais de feed-forward que envia para os
músculos na próxima vez em que esse movimento vier a ser
executado. Então, mais uma vez, se for preciso correção
adicional, ela será feita para os movimentos subseqüentes. Isso
é chamado de controle adaptativo. Em determinado sentido, é
óbvio que o controle adaptativo nada mais é que um feedback
negativo retardado.
Assim, pode-se ver como são complexos alguns dos sistemas
de controle por feedback encontrados no corpo. Em termos
literais, a vida da pessoa depende de todos eles. Por conseguinte,
grande parte deste texto será dedicada à discussão desses
mecanismos protetores da vida.
RESUMO - A AUTOMATICIDADE DO
CORPO
O objetivo deste capítulo foi o de destacar, primeiro, a
organização geral do corpo e, segundo, os meios pelos quais as
diferentes partes do corpo funcionam em harmonia. Para
resumir, o corpo c, na verdade, uma ordem social com cerca de
100 trilhões de células, organizada em diferentes estruturas
funcionais, algumas das quais são chamadas órgãos. Cada
estrutura funcional contribui com sua cota para a manutenção das
condições homeostáticas do líquido extracelular, que é chamado
de ambiente interno. Enquanto as condições normais forem
mantidas no ambiente interno, as células do corpo continuarão a
viver e a funcionar adequadamente. Dessa forma, cada célula se
beneficia da homeostasia e, por sua vez, contribui com sua cota
para a manutenção dessa homeostasia. Essa interação recíproca
resulta em automaticidade contínua do corpo, que perdurará
até que um ou mais sistemas funcionais percam sua capacidade
de contribuir com sua cota de funcionamento. Quando isso
acontece, todas as células do corpo sofrem. A disfunção extrema
leva à morte, enquanto a disfunção moderada causa doença.
REFERENCIAS
Adolph, E. F.: Physiological integrations in action. The
Physiologist, 25:<Suppl.) 1. 1982. Adolph, E. F.: Physiological
adaptat ions: Hypertrophies and superfunctions.
Am. Sei., 60:608,1972. Bernard, C: Lecturea on the Phenomena of
Life Common to Animal» and Plants. Springfield, III., Charles C
Thomas, 1974. Brown, J. H. U. (ed.): Engineering Principies in
Physiology. Vols. 1 and 2.
New York, Academic Press, 1973. Bruni, C, et ai. (eds.): Systems
Theory in Immunology. New York, Springer- Verlag, L979. Bryant,
P. J., and Simpson, P.: Intrinsic and extrinsic contrai of growth in
developing organs. Q. Rev. Biol., 59:387, 1984. Burattini, R., and
Borgdorff, P.: Closed-loop baroreflex control of total pe- ripheral
resistance in the cat: Identification of gains by aid of a model.
Cardiovasc. Res., 18:715, 1984. Cannon, W. B.: The Wisdom of the
Body. New York, W. W. Norton & Co.,1932.
Frisancho, A. R.: Human Adaptation. St. Louis, C. V. Mosby Co.,
1979. Gann, D. S., et ai.: Neural interaction in control of
adrenocorticotropin. Fed.
Proc., 44:161, 1985. Guyton, A. C: Arterial Pressure and
Hypertension. Philadelphia, W. B.
Saunders Co., 1980. Guyton, A. C, andColeman, T. G.:
Quantitative analyaisof thepathophysi- ology of hypertension. Circ.
Res., 14:1-1, 1969. Guyton, A. C, et a).: Dynamics and Control of
the Body Fluids. Philadelphia, W. B. Saundera Co., 1975.
Huffaker, C. B. (ed.): Biological Control. New York, Plenum
Preás, 1974. Iberall, A. S., and Guyton, A. C. (ede.): Proc. Int.
Symp. on Dynamics and Controls in Physiological Systema.
Regulatíon and Control in Phyaiological Systems. ISA, Pittsburgh,
1973.
Jones, R. W.: Principies of Biological Regulation: An
Introduction to Feedback Systems. New York, Academic Preás,
1973. Klevecz, R. R.( et ai.: Cellular clocks and oscillators. Int. Rev.
Cytol., 86: 97,
1984. Krieger, D. T., and Aschoff, J.: Endocrine and other
biological rhythms. In DeOroot, L. H., et ai. (eds.): Endocrinology,
Vol. 3. New York, Grune & Stratton, 1979, p. 2079. Mclntosh, J. E.
A., and Mclntosh, R. P.: Mathematical Modeling and Com- puters
in Endocrinology. New York, Springer-Verlag, 1980. Milhorn, H.
T.: The Application of Control Theory to Phyaiological Systems.
Philadelphia, W. B. Saunders Co., 1966. Miller, S. L., and Orgel,
L. E.: The Origins of Life on the Earth. Englewood Cliffs, N.J.,
Prentice-Hall, 1974. Piva, F., et ai.: Regulation of hypothalamic
and pituitary function: Long, short, and ultrashort feedback loops.
In DeGroot, L. J., et ai. (eds.): Endocrinology. Vol. 1. New York,
Grune & Stratton, 1979, p. 21. Randall, J. E.: Mie roço mputers and
Physiological Simulation. 2d Ed. New York, Raven Presa, 1987.
Randall, J. E., Microcomputera and Phyaiological Simulation.
Reading, Mass., Addison-Wesley Publishing Co., 1980. Reeve, E.
B., and Guyton, A. C: Physical Bases of Circulatory Transport:
Regulation and Exchange. Philadelphia, W. B. Saunders Co.,
1967. Rusak, B., and Zucker, Li Neural regulation of circadian
rhythms. Physiol.
Rev., 59:449, 1979. Stein, J. F.: Role of the cerebellum in the
visual guidance of movement. Nature 323:217, 1986.
Sweetser, W.: Human Life {AgingandOld Age). New York, Arno
Press, 1979. Thompson, R. F.: The neurobiology of learning and
memory. Science, 233:941, 1986. Toates, F. M.: Control Theory
in Biology and Experimental Pâychology.
London, Hutchinaon Education Ltd., 1975. Weston, L.: Body
Rhythm: The Circadian Rhythms Within You. New York,
Harcourt Brace Jovanovich, 1979. Yates, F. E. (ed.): SelfOrganizing Systems. New York, Plenum Publishing Corp., 1987.
8
CAPÍTULO 2
A Célula e seu Funcionamento
Cada uma das 75 a 100 trilhões de células do corpo humano
é uma estrutura viva que pode sobreviver indefinidamente e,
em muitos casos, até se reproduzir, desde que os líquidos que
a banham contenham os nutrientes adequados. Para a
compreensão do funcionamento dos órgãos e das demais
estruturas que compõem o corpo humano, é essencial que,
primeiro, se conheça a organização básica da célula e o
funcionamento de suas partes componentes.
ORGANIZAÇÃO DA CÉLULA
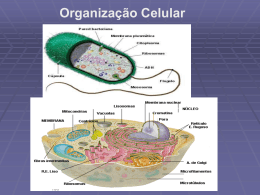
Uma célula típica, como vista ao microscópio óptico, é
apresentada na Fig. 2.1. Seus dois constituintes principais são o
núcleo e o citoplasma. O núcleo é separado do citoplasma pela
membrana nuclear, enquanto o citoplasma é separado dos
fluidos circundantes pela membrana celular.
As diferentes substâncias que compõem a célula são
chamadas, em conjunto, de protoplasma. Esse protoplasma é
formado, em sua maior parte, por cinco substâncias básicas:
água, eletrólitos, proteínas, lipídios e carboidratos.
Água. O principal meio líquido da célula é a água, presente
em concentrações que variam entre 75 e 85%. Muitas substâncias
químicas celulares estão dissolvidas na água, enquanto outras
ficam em suspensão, sob forma particulada ou membranosa. As
reações químicas ocorrem entre as substâncias químicas
dissolvidas ou nas superfícies limitantes entre as partículas ou
membranas em suspensão e a água.
Eletrólitos. Os eletrólitos mais importantes da célula são
o potássio, o magnésio, o fosfato, o sulfato, o bicarbonato, e
pequenas quantidades de sódio, cloreto e cálcio. Esses eletrólitos
serão discutidos em maior detalhe no Cap. 4, onde serão
apresentadas as relações entre os líquidos intra e extracelular.
Os eletrólitos fornecem as substâncias químicas inorgânicas
para as reações celulares. Também são necessários para a
operação de diversos mecanismos celulares de controle. Por
exemplo, os eletrólitos, atuando ao nível da membrana celular,
permitem a transmissão dos impulsos eletroquímicos nas
fibras nervosas e musculares, enquanto os eletrólitos
intracelulares determinam a velocidade de numerosas reações
catalisadas por enzimas, imprescindíveis ao metabolismo celular.
Proteínas. Após a água, a substância mais abundante na
maioria das células é a proteína que, normalmente, representa
de 10 a 20% da massa celular. Essa proteína pode ser dividida
em duas classes distintas, as proteínas estruturais e as proteínas
globulares, que são, em sua maioria, enzimas.
Para se ter idéia do que se quer dizer por proteínas estruturais,
apenas será preciso notar que o couro é formado, quase que
inteiramente, por proteína estrutural. As proteínas dessa classe
existem nas células sob forma de filamentos longos e finos que
são, em si mesmos, polímeros de muitas moléculas protéicas.
O uso mais freqüente desses filamentos intracelulares é no
mecanismo contrátil de todos os músculos. Contudo, outros
desses filamentos também ocorrem organizados nos
microtúbulos que formam os "citoesqueletos" de organetas como
os cílios e o fuso mitótico das células em mitose. No ambiente
extracelular, as estruturas fibrilares aparecem nas fibras de
colágeno e elásticas do tecido conjuntivo, dos vasos sanguíneos,
dos tendões, ligamentos etc.
Por outro lado, as proteínas globulares formam classe
inteiramente distinta de proteínas, compostas, em gerai, por
moléculas protéicas únicas ou, no máximo, por agregado de
poucas moléculas, tendo forma globular, e não fibrilar. Essas
proteínas são, em sua maioria, as enzimas celulares e, no que
diferem das proteínas fibrilares, são, com muita freqüência,
solúveis nos líquidos das células ou são parte ou aderem a
estruturas membranosas no interior das células. As enzimas
entram em contato direto com outras substâncias no interior
celular, quando catalisam as reações químicas. Por exemplo, as
reações químicas que degradam a glicose em seus componentes e,
em seguida, os combinam com o oxigênio, para gerar dióxido de
carbono e água, ao mesmo tempo que liberam energia para o
funcionamento celular, são catalisadas por várias enzimas
protéicas.
Lipídios. Os lipídios são formados por diversos tipos
diferentes de substâncias, consideradas como pertencentes a uma
mesma classe por terem a propriedade comum de serem solúveis
em solventes de gorduras. Os tipos mais importantes dos
lipídios são os fosfolipídios e o colesterol, que representam cerca
de 2% da massa celular total. A importância especial dos
fosfolipídios e do colesterol é a de que são quase insolúveis em
água e, portan-
Fig. 2.1 Estrutura de uma célula como é vista ao microscópio
óptico.
9
to, são usados na formação de barreiras membranosas,
separadoras dos diversos compartimentos intracelulares.
Além dos fosfolipídios e do colesterol, algumas células
contêm grandes quantidades de trigricerídeos, também chamados
de gordura neutra. Nas chamadas células adiposas, os
triglicerídios representam, muitas vezes, até 95% da massa
celular. A gordura armazenada nessas células representa o
principal depósito de nutriente armazenador de energia que
pode ser mobilizado e utilizado como energia sempre que o
corpo necessitar.
Carboidratos. Em geral, os carboidratos têm pequena
participação no funcionamento estrutural da célula, exceto como
parte das moléculas de glicoproteínas, mas têm participação
fundamental na nutrição celular. A maioria das células humanas
não mantém grandes depósitos de carboidratos que, em geral,
representam cerca de 1% de sua massa total. Contudo, o
carboidrato, sob forma de glicose, sempre está presente no líquido
extracelular circundante, de modo a ser facilmente disponível
para a célula. Na maioria das situações, a célula armazena
pequena quantidade de carboidrato, sob forma de glicogênio, um
polímero insolúvel da glicose e que pode ser rapidamente
utilizado para suprir as necessidades energéticas da célula.
A ESTRUTURA FÍSICA DA CÉLULA
A célula não é, simplesmente, um saco cheio de líquido,
enzimas e substâncias químicas; também contem estruturas
físicas, extremamente organizadas, muitas delas chamadas
organelas, e a natureza física de cada uma delas é tão
importante para o funcionamento celular como o são seus
constituintes químicos.
Por exemplo, sem uma das organelas, as mitocôndrias, mais de
95% do suprimento energético da célula cessaria imediatamente.
Algumas das organelas principais são mostradas na Fig. 2.2,
incluindo a membrana celular, a membrana nuclear, o retículo
endoplasmático, o aparelho de Golgi, as mitocôndrias, os
lisossomas e os centríolos.
AS ESTRUTURAS MEMBRANOSAS DAS CÉLULAS
Em essência, todas as organelas celulares são revestidas por
membranas, formadas, em sua maior parte, por lipídios e por
proteínas. Essas membranas incluem a membrana celular, a
membrana nuclear, a membrana do retículo endoplasmático e as
membranas das mitocôndrias, dos lisossomas e do aparelho de
Golgi, além de várias outras. Os lipídios dessas membranas
formam barreiras que impedem o livre deslocamento da água e
das substâncias solúveis em água entre os diferentes
compartimentos da célula. As moléculas de proteína, por sua
vez, penetram, com certa freqüência, através de toda a
espessura dessas membranas, o que interrompe a continuidade
da barreira lipídica e, por conseguinte, forma pertuitos para a
passagem de substâncias específicas através dessas membranas.
Também, muitas das proteínas das membranas são enzimas que
catalisam muitas reações químicas diferentes, que serão
discutidas adiante neste capítulo e nos subseqüentes.
A membrana celular
A membrana celular, que reveste inteiramente toda a célula,
é uma estrutura muito delgada e elástica, com espessura entre
Fig. 2.2 Reconstrução de uma célula típica, mostrando as
organelas internas no citoplasma e no núcleo.
10
7,5 e 10 nanômetros. É formada quase que exclusivamente por
proteínas e lipídios. Sua composição aproximada é de 55% de
proteínas, 25% de fosfolipídios, 13% de colesterol, 4% de outros
lipídios c 3% de carboidratos.
A barreira lipídica da membrana celular. A Fig. 2.3
apresenta a membrana celular. Sua estrutura básica é uma
bicamada lipídica, que é uma película delgada de lipídios, com a
espessura de duas moléculas, contínua por sobre toda a
superfície celular. Dispersas nessa película lipídica, existem
moléculas de proteínas globulares.
A bicamada lipídica é formada quase que inteiramente por
fosfolipídios e por colesterol. Parte das moléculas de fosfolipídios
c de colesterol é solúvel em água, isto é, hidrofílica, enquanto
outra parte só é solúvel em gordura, isto é, hidrofóbica. O radical
fosfato dos fosfolipídios é hidrofílico e os ácidos graxos são
hidrofóbicos. O colesterol contém um radical hidroxila que é
hidrossolúvel e um núcleo esteróide que ê solúvel em gordura.
Como as partes hidrofóbicas dessas moléculas são repelidas pela
água mas se atraem mutuamente, essas moléculas possuem
tendência natural para se alinharem umas às outras, como
mostrado na Fig. 2.3, com suas frações graxas ocupando a
região central da membrana e com suas regiões hidrofílicas
voltadas para sua superfície, em contato com a água que as
banha.
A bicamada lipídica da membrana representa importante
barreira, impermeável às substâncias comuns, hidrossolúveis, tais
como íons, glicose, uréia e outras. Por outro lado, as substâncias
solúveis em gordura, como o oxigênio, dióxido de carbono e
álcool, podem atravessar facilmente essa região da membrana.
Característica especial da bicamada lipídica é a de ser um
fluido, e não um sólido. Por conseguinte, partes dessa membrana
podem, literalmente, fluir de um ponto a outro, ao longo da
superfície dessa membrana. As proteínas e outras substâncias
dissolvidas ou flutuando na bicamada lipídica tendem a se difundir
para todas as áreas da membrana celular.
As proteínas da membrana celular. A Fig. 2.3 apresenta
massas globulares flutuando na bicamada lipídica. São proteínas
da membrana, a maioria das quais é formada por glicoproteínas.
São encontrados dois tipos de proteínas: as proteínas integrais,
que atravessam toda a espessura da membrana, e as proteínas
periféricas, que ficam apenas presas à superfície da membrana.
sem atravessá-la.
Muitas das proteínas integrais formam canais (ou poros)
estruturais, pelos quais podem difundir as substâncias
hidrossolúveis, especialmente os íons, entre os líquidos intra a
extracelular. Contudo, essas proteínas apresentam propriedades
seletivas que produzem difusão diferencial de algumas
substâncias mais que de outras. Outras proteínas integrais atuam
como proteínas carreadoras para o transporte de substâncias na
direção oposta à natural de sua difusão, o que é chamado de
"transporte ativo". Outras, ainda, são enzimas.
As proteínas periféricas ocorrem quase inteiramente na face
interna da membrana e, normalmente, ficam presas a uma das
proteínas integrais. Essas proteínas periféricas atuam quase que
exclusivamente como enzimas.
Os carboidratos da membrana — o "glicocálise"
celular. Os carboidratos da membrana aparecem, de modo quase
invariável, em combinação com proteínas e lipídios, sob a forma
de glicoproteínas e de glicolipídios. Na verdade, a maioria das
proteínas integrais é composta de glicoproteínas e cerca de um
décimo das moléculas lipídicas é de glicolipídios. A fração "glico"
dessas moléculas, quase que invariavelmente, proemina na face
externa da célula, chegando a ficar pendurada para fora da célula.
Muitos outros compostos carboidratos, chamados proteoglicanos,
formados principalmente por carboidratos unidos entre si por
pequenos núcleos protéicos, podem, por vezes, também ocorrer
frouxamente ligados à superfície externa da célula. Assim, toda a
superfície externa da célula é, muitas vezes, inteiramente
revestida por capa de carboidrato, chamada de glicocálice.
Os radicais carboidratos presos à superfície externa da célula
desempenham diversas funções importantes: (1) muitos deles
têm carga negativa, o que dá, à maioria das células, uma carga
global negativa em sua superfície, o que repele qualquer coisa
que também seja portadora de carga negativa; (2) o glicocálice
de muitas células se fixa ao glicocálice de outras células, o que
serve para fixar (ou unir) as células entre si; (3) muitos desses
carboidratos atuam como substâncias receptoras para a fixação
de hormônios, como a insulina, e, ao fazê-lo, ativam proteínas
integrais que, por sua vez, ativam uma cascata de enzimas
intracelulares; e (4) alguns participam de reações imunes, como
discutido no Cap. 34.
Fig. 2.3 Estrutura da membrana celular, mostrando que é composta, principalmente, de bicamada lipídica, com grande número de moléculas
de proteína protruindo através dessa bicamada. Também existem moléculas de carboidrato presas às moléculas de proteína na face externa
da membrana, além de moléculas adicionais de proteína em sua face interna. (De Lodish e Rothman: The assembly of cell membranes, Sei,
Amer., 240:48, 1979. Copyright 1979 by Scientific American Inc. Todos os direitos reservados.)
11
O CITOPLASMA E SUAS ORGANELAS
O citoplasma é cheio de partículas e organelas dispersas,
com tamanhos que vão de poucos nanômetros até muitos
micrômetros. Aparte líquida clara do citoplasma, onde ficam
dispersas essas partículas e organelas, é chamada de citosol; ele
contém muitas proteínas, eletrólitos, glicose e quantidades
diminutas de compostos lipídicos dissolvidos.
A região do citoplasma imediatamente abaixo da membrana
celular contém, com muita freqüência, um emaranhado de microfilamentos, formado, em sua maior parte, por fibrilas de actina.
Essa estrutura forma um sistema de sustentação semi-sólido, com
a consistência de gel, para a membrana celular. Essa região do
citoplasma é chamada de córtex ou de ectoplasma. A parte do
citoplasma que fica entre o córtex e a membrana nuclear é líquida
e chamada de endoplasma.
Ocorrem, dispersos no citoplasma, gotículas de gordura
neutra, grânulos de glicogênio, ribossomas, grânulos secretórios
e cinco organelas especialmente importantes: o retículo
endoplasmático, o aparelho de Golgi, as mitocôndrias, os
lisossomas e os peroxissomas.
O retículo endoplasmático
A Fig. 2.2 mostra, no citoplasma, uma rede de estruturas
tubulares e vesiculares achatadas, chamada de retículo
endoplasmático. Os túbulos e as vesículas se intercomunicam.
Por outro lado, suas paredes são formadas por membranas de
bicamada lipídica, contendo grande quantidade de proteínas,
como ocorre na membrana celular. A área total da superfície
dessa estrutura em determinadas células — como, por
exemplo, as hepáticas — pode chegar até a 30 ou 40 vezes
maior que a de toda a superfície celular.
Um detalhe da estrutura de pequena parte do retículo
endoplasmático é mostrado na Fig. 2.4. O espaço no interior
dos túbulos e das vesículas é cheio com a matriz
endoplasmática, um meio líquido que difere do encontrado por
fora do retículo endoplasmático. Micrografias eletrônicas
mostram que o espaço no interior do retículo endoplasmático
está conectado ao espaço entre as duas membranas da dupla
membrana nuclear.
As substâncias sintetizadas em outras regiões da célula
penetram nesse espaço do retículo endoplasmático e são levadas
até outras partes da célula. Por outro lado, a imensa área da
superfície desse retículo, além dos múltiplos sistemas enzimáticos
presentes em suas membranas, compõe o maquinário para
fração importante das funções metabólicas da célula.
Ribossomas e o retículo endoplasmático granular.
Existem, fixadas à superfície externa de muitos trechos do
retículo endoplasmático, pequenas partículas granulares,
denominadas ribossomas
Fig. 2.4 Estrutura do retículo endoplasmático. (Modificado de De Robertis, Saez e De Robertis: Cell Biology. 6. ed. Philadelphia, W.B.
SaundersCo., 1975.)
Nas regiões do retículo endoplasmático onde isso ocorre, esse
retículo é chamado de retículo endoplasmático granular. Os
ribossomas são formados por mistura de ácido ribonucléico
(ARN) e de proteínas e atuam na síntese de proteínas pelas
células, como discutido adiante neste capítulo e no seguinte.
O retículo endoplasmático agranular. Parte do retículo
endoplasmático não tem ribossomas fixados a ele. Essa parte é
chamada de retículo endoplasmático agranular, ou liso. O
retículo agranular atua na síntese de substâncias lipídicas e de
muitos outros processos enzimáticos das células.
O aparelho de Golgi
O aparelho de Golgi, mostrado na Fig. 2.5, é intimamente
relacionado ao retículo endoplasmático. Possui membranas
semelhantes às do retículo endoplasmático agranular. Em geral,
é formado por quatro a cinco camadas empilhadas de vesículas
fechadas, delgadas e achatadas, situadas próximo ao núcleo. Esse
aparelho é muito proeminente nas células secretoras; nelas fica
situado no lado da célula por onde são extrudadas as substâncias
secretórias.
O aparelho de Golgi funciona associado ao retículo
endoplasmático. Como mostrado na Fig. 2.5, pequenas "vesículas
de transporte", também chamadas vesículas de retículo
endoplasmático ou, simplesmente, vesículas RE, são formadas,
de forma contínua, pelo retículo endoplasmático e, em seguida,
se fundem com o aparelho de Golgi. Desse modo, as substâncias
são transferidas do retículo endoplasmático para o aparelho de
Golgi. As substâncias transferidas são, em seguida, processadas
no aparelho de Golgi, para formar lisossomas, vesículas
secretórias ou outros componentes citoplasmáticos, discutidos
adiante neste capítulo.
Os lisossomas
Os lisossomas são organelas vesiculares, formadas pelo
aparelho de Golgi e que, em seguida, ficam dispersas por todo o
citoplasma. Os lisossomas formam um sistema digestivo
intracelular que permite que a célula digira e, por conseguinte,
remova substâncias e estruturas indesejadas, em especial
estruturas estranhas ou lesadas, tais como bactérias. O
lisossoma, mostrado na Fig. 2.2, difere muito de uma célula
para outra, mas, em geral, tem diâmetro entre 250 e 750 nm. É
limitado por membrana de bicamada lipídica típica e seu interior
é cheio de pequenos grânulos, com diâmetro entre 5 e 8 nm, que
são agregados protéicos de enzimas hidrolíticas (digestivas).
Uma enzima hidrolítica
Fig. 2.5 Um típico aparelho de Golgi e sua relação com o retículo
endoplasmático e com o núcleo.
12
é capaz de degradar um composto orgânico em dois ou mais
componentes, por combinar um hidrogênio, derivado da água,
com parte desse composto, e peia combinação da hidroxila da
molécula de água com outra parte desse composto. Por exemplo,
a proteína é hidrolisada para formar aminoácidos, enquanto o
glicogênio é hidrolisado para formar glicose. Mais de 50 hidrolases
ácidas já foram identificadas nos lisossomas, e as principais
substâncias que essas organelas podem hidrolisar são as
proteínas, os ácidos nucléicos, os mucopolissacarídeos, os
lipídios e o glicogênio.
Comumente, a membrana que envolve o lisossoma impede
que as enzimas hidrolíticas de seu interior entrem em contato
com as outras substâncias no interior celular. Todavia, numerosas
e diversas condições celulares podem romper a membrana de,
pelo menos, alguns lisossomas, o que produz a liberação dessas
enzimas. Como resultado, essas enzimas degradam as substâncias
orgânicas com que entram em contato, produzindo substâncias
muito difusíveis, como aminoácidos e glicose. Algumas das
funções mais específicas dos lisossomas são discutidas adiante
neste capítulo.
Os peroxissomas
Os peroxissomas são, cm termos físicos, semelhantes aos
lisossomas, mas diferem deles por dois aspectos importantes:
primeiro, admite-se que sejam formados pelo retículo
endoplasmático liso, e não pelo aparelho de Golgi; segundo, as
enzimas em seu interior são oxidases, e não hidrolases. Diversas
dessas oxidases são capazes de combinar o oxigênio com o íon
hidrogênio para formar peróxido de hidrogênio (H2O2). O peróxido
de hidrogênio, por sua vez, é composto altamente oxidante e que
atua associado à catalase, outra enzima oxidase presente em alta
concentração nos peroxissomas, na oxidação de muitas
substâncias que, de outra forma, intoxicariam a célula. Por
exemplo, a maior parte do álcool ingerido por uma pessoa é
detoxificado pelos peroxissomas das células hepáticas por esse
mecanismo. O mecanismo oxidativo peróxido de hidrogênio
catalase também é usado para finalidades funcionais específicas
da célula, tais como a degradação de ácidos graxos a acetil-CoA
que, em seguida, é utilizado como energia pela célula.
Vesículas secretárias
Uma das funções importantes de muitas células é a secreção
de substâncias especiais. Quase todas as substâncias secretórias
desse tipo são formadas pelo sistema retículo endoplasmáticoaparelho de Golgi e são, em seguida, liberadas pelo aparelho
de Golgi no citoplasma no interior de vesículas de
armazenamento, chamadas vesículas secretórias ou grânulos
secretários. A Fig. 2.6 mostra vesículas secretórias típicas no
interior de células acinares pancreáticas, armazenando
proenzimas protéicas (enzimas que ainda não foram ativadas);
essas proenzimas vão ser, algum tempo depois, secretadas
através de membrana celular externa para o dueto pancreático e,
por meio dele, atingem o duodeno, onde vão ser ativadas e
desempenhar suas funções digestivas.
Fíg. 2.6 Grânulos secretórios nas células acinares do pâncreas.
Ainda mais, as mitocôndrias ficam concentradas nas regiões
celulares que são responsáveis pela maior fração de seu
metabolismo energético. Por outro lado, o tamanho das
mitocôndrias é muito variável, assim como sua forma; algumas
têm diâmetro de apenas poucas centenas de nanômetros, com
forma globular, enquanto outras podem ter até 1 m de
diâmetro e comprimento de 7 m, com forma filamentosa ou
ramificada.
A estrutura básica da mitocôndria é mostrada na Fig. 2.7,
onde aparece formada, em sua maior parte, por duas membranas
de dupla camada lipídica: uma membrana externa e outra
membrana interna. Muitas pregas da membrana interna formam
as cristas, sobre as quais ficam presas enzimas oxidativas. Além
disso, a cavidade interna de cada mitocôndria c cheia com matriz
contendo grande quantidade de enzimas dissolvidas, que são
necessárias para a extração de energia dos nutrientes. Essas
enzimas atuam associadas às enzimas oxidativas das cristas,
para efetuar a oxidação dos nutrientes, do que resulta a formação
de dióxido de carbono e água. A energia liberada c utilizada
na síntese de substância com alta energia, chamada trifosfato de
adenosina (ATP). Em seguida, o ATP é transportado para fora
da mitocôndria, difundindo-se por toda a célula e liberando sua
energia sempre e onde for necessário para a execução das
funções celulares. Os detalhes da síntese do ATP pelas
mitocôndrias são apresentados no Cap. 67 e algumas das
importantes funções do ATP são apresentadas adiante neste
capítulo.
As mitocôndrias são auto-replicativas, o que significa que
uma mitocôndria pode dar origem a uma segunda, a uma terceira,
e assim por diante, sempre que houver necessidade celular de
As mitocôndrias
As mitocôndrias são chamadas de "usinas" celulares. Sem
elas, as células seriam incapazes de extrair quantidades
significativas de energia dos nutrientes e do oxigênio, e, como
conseqüência, para todos os efeitos práticos, cessaria todo o
funcionamento celular. Como mostrado na Fig. 2.2, essas
organelas são encontradas disseminadas por quase todo o
citoplasma, mas seu número total varia desde menos de cem
até vários milhares, dependendo da quantidade de energia
exigida pela célula.
Fig. 2.7 Estrutura da mitocôndria. (Modificado de De Robertis, Saez
e De Robertis, Ceil Bivlogy. 6. ed. Philadelphia, W.B. Saunders Co.,
1975.)
13
quantidades aumentadas de ATP. Na verdade, as mitocôndrias
contêm ácido desoxirribonucléico (ADN) semelhante ao
encontrado no núcleo. No capítulo seguinte, será destacado
que o ADN é a substância básica do núcleo, controladora da
replicação celular. Essa substância desempenha função
semelhante na mitocôndria, porém não idêntica, visto que, no
processo de replicação mitocondrial, muitas proteínas e lipídios
que já foram formados no citoplasma são incorporados às
mitocôndrias, quando estas aumentam de volume e produzem
brotamentos, que são as novas mitocôndrias.
Estruturas filamentosas e tubulares das células
As proteínas fibrilares da célula estão, em geral, organizadas
em filamentos ou túbulos. Tais estruturas têm origem como
moléculas protéicas precursoras, sintetizadas pelos ribossomas e
que aparecem, inicialmente, dissolvidas no citoplasma. Aí, elas
polimerizam para formar filamentos. Já foi destacada a presença
freqüente de grande número de filamentos de actina na zona
externa do citoplasma, a região chamada de ectoplasma, dando
sustentação elástica à membrana celular. Também, nas células
musculares, os filamentos ocorrem organizados em mecanismo
contrátil especializado que é a base da contração muscular em
todo o corpo, como discutido em detalhe no Cap. 6.
Um tipo especial de filamento, formado por moléculas
polimerizadas de tubulina, é usado por todas as células para a
construção de estruturas tubulares, os microtúbulos. Quase
invariavelmente, eles são formados por 13 protofilamentos de
tubulina, paralelos entre si, formando círculo, compondo longo
cilindro oco, com diâmetro de cerca de 25 nm e comprimento
que varia de 1 a muitos micrômetros. Tais cilindros aparecem,
com freqüência, sob forma de feixes, o que lhes confere, em
conjunto, considerável resistência estrutural. Contudo, os
microtúbulos são estruturas rígidas, que quebram se forem
dobradas em demasia. A Fig. 2.8 mostra microtúbulos típicos,
extraídos do flagelo de um espermatozóide.
Outro exemplo de microtúbulo é a estrutura mecânica
tubular dos cílios, que lhes confere resistência estrutural, que se
irradiam desde o citoplasma celular até a ponta do cílio. Por
outro lado, os centríolos e o fuso mitótico das células em mitose
são formados por microtúbulos rígidos.
Dessa forma, uma função primária dos microtúbulos é a
de atuar como um citoesqueleto, formando estruturas físicas
rígidas para determinadas regiões celulares. Mas o citoplasma,
com freqüência se escoa (flui) na vizinhança dos
microtúbulos, o que poderia ser explicado pelo movimento dos
braços que se projetam para fora dos microtúbulos.
O NÚCLEO
O núcleo é o centro controlador da célula. De modo
resumido, o núcleo contém grande quantidade de ADN, a que se
chamou, por muitos anos, genes. Os genes determinam as
características das enzimas protéicas do citoplasma e, por esse
meio, regulam as atividades citoplasmáticas. Também controlam
a reprodução; os genes, primeiro, se reproduzem e, após isso,
a célula se divide por processo especial, chamado mitose, para
formar duas células filhas, cada uma recebendo um dos dois
conjuntos de genes. Todas essas atividades nucleares são
apresentadas em detalhes no próximo capítulo.
A imagem microscópica do núcleo não dá muitos indícios
sobre os mecanismos que usa para o desempenho de suas
atividades. A Fig. 2.9 apresenta a imagem, por microscópio
óptico, do núcleo na interfase (o período entre as mitoses), com o
material que se cora intensamente, a cromatina, presente em
todo o nucleoplasma. Durante a mitose, a cromatina fica
facilmente identificável como os cromossomas extremamente
estruturados, que podem ser observados com facilidade pelo
microscópio óptico, como discutido no Cap. 3.
O envelope nuclear
O envelope nuclear é, com freqüência, denominado
membrana nuclear. Contudo é, na verdade, formado por duas
membranas distintas, uma por dentro da outra. A membrana
externa é contínua com o retículo endoplasmático, c o espaço
entre as duas membranas nucleares também é contínuo com o
compartimento no interior do retículo endoplasmático.
O envelope nuclear é atravessado por vários milhares de
poros nucleares. Esses poros são muito grandes, com quase 10
nm de diâmetro. Contudo, grandes complexos de proteínas ficam
presos às bordas desses poros, de modo que seus orifícios centrais
Fig. 2.8 Microtúbulos dissecados do flagelo de
espermatozóide. (De Porter: Ciba Foundation
Symposium: Principies of Biomolecuhr Organizaiion.
Boston, Little, Brown & Co, 1966)
14
Fig. 2.9 Estrutura do núcleo.
têm, apenas, 9 nm de diâmetro. Mesmo assim, esses poros são
suficientemente grandes para permitir a passagem de moléculas
com peso molecular de até 44.000 com relativa facilidade;
moléculas com peso molecular abaixo de 15.000 os atravessam
com extrema rapidez.
Nucléolos
Os núcleos da maioria das células contêm uma ou mais
estruturas que se coram levemente, chamadas nucléolos. O
nucléolo, ao contrário da maioria das organelas discutidas até
aqui, não apresenta membrana limitante. Pelo contrário, é,
simplesmente, uma estrutura que contém grande quantidade de
ARN e de proteínas dos tipos encontradas nos ribossomas. O
nucléolo fica muito aumentado quando a célula está
sintetizando ativamente proteínas. Os genes de cinco
cromossomas distintos sintetizam o ARN e o armazenam no
nucléolo, a partir de ARN fibrilar frouxo que, depois, se
condensa
para
formar
as
"subunidades"
granulares dos ribossomas. Estas, por sua vez, são transportadas
através dos poros da membrana nuclear até o citoplasma, onde
se agregam para formar os ribossomas "maduros" que
desempenham papel fundamental na formação de proteínas,
tanto no citoplasma como em associação com o retículo
endoplasmático, como será discutido em mais detalhes no
capítulo seguinte.
COMPARAÇÃO DA CÉLULA ANIMAL COMAS
FORMAS PRÉ-CELULARES DE VIDA
Muitos de nós imaginam que a célula seja a forma mais simples
de vida. Todavia, a célula é organismo muito complexo e que exigiu
muitas centenas de milhões de anos para se desenvolver depois que
a forma inicial da vida, um organismo semelhante aos vírus atuais,
primeiro apareceu na terra. A Fig. 2.10 mostra as dimensões relativas
dos menores vírus conhecidos, de um vírus grande, de uma rickettsia,
de uma bactéria e de uma célula nucleada, esta célula tendo diâmetro
1.000 vezes maior que o do menor vírus e, por conseguinte, com
volume 1bilhão de vezes maior que o desse vírus. Como conseqüência, o
funcionamento e a organização anatômica da célula também são
muitíssimo mais complexos que o do vírus.
O constituinte essencial do vírus, responsável por ele ser vivo, é
o ácido nucléico, envolto por capa de proteína. Esse ácido nucléico
é formado pelos mesmos constituintes básicos (ADN e ARN)
encontrados nas células de mamíferos e será capaz de se reproduzir caso
existam condições adequadas. Assim, um vírus é capaz de propagar sua
linhagem, de geração a geração, e, portanto, é uma estrutura viva, do
mesmo modo como o são uma célula e um organismo humano.
Com a evolução da vida, outras substâncias químicas, além dos
ácidos nucléicos e simples proteínas, passaram a fazer integralmente
parte do organismo, e funções especializadas começaram a se desenvolver
em diferentes partes do vírus. Surgiram, assim, uma membrana, formada
Fig. 2.10 Comparação entre as dimensões de organismos pré-celulares
e uma célula típica do corpo humano.
a seu redor, e uma matriz fluida, por dentro dessa membrana. No interior
dessa matriz, desenvolveram-se substâncias químicas especializadas para
a execução de funções especiais; muitas enzimas protéicas surgiram,
capazes de catalisar reações químicas e, como conseqüência, de
determinar as atividades desse organismo.
Em estágios mais avançados, de modo especial, nos estágios de
rickettsia e de bactéria, organelas se desenvolveram no interior do
organismo, representadas por estruturas físicas de agregados químicos,
capazes de executar funções de forma bem mais eficiente que as
substâncias químicas dispersas por toda a matriz fluida. Finalmente, na
célula nucleada, ocorreu o desenvolvimento de organelas ainda mais
complexas, a mais importante delas sendo o próprio núcleo. O núcleo
distingue esse tipo celular de todas as outras formas mais inferiores de
vida; essa estrutura estabelece um centro de controle de todas as
atividades celulares e permite uma reprodução muito precisa de novas
células, geração após geração, cada nova célula possuindo, em
essência, a mesma estrutura de seu progenitor.
SISTEMAS FUNCIONAIS DA CÉLULA
No restante deste capítulo, serão discutidos diversos sistemas
funcionais representativos da célula, que a tornam um organismo
vivo.
INGESTÃO PELA CÉLULA - ENDOCITOSE
Se a célula vai viver e crescer, ela deverá obter nutrientes
e outras substâncias dos líquidos que a banham. A maioria das
substâncias atravessa a membrana por difusão e por transporte
ativo, discutidos em detalhe no Cap. 4. Contudo, grandes
partículas atingem o interior da célula por meio de função
especializada da membrana celular, chamada endocitose, As duas
formas principais de endocitose são a pinocitose e a fagocitose.
Pinocitose significa ingestão de vesículas extremamente pequenas,
contendo líquido extracelular. Fagocitose significa ingestão de
grandes partículas, tais como bactérias, células ou restos de tecido
em degeneração.
Pinocitose. A pinocitose ocorre continuamente na membrana
da maioria das células, mas de modo especialmente rápido em
algumas células. Por exemplo, nos macrófagos, ocorre de forma
tão rápida que cerca de 3% da membrana total dessas células
são engolfados, sob forma de vesículas, a cada minuto. Mesmo
assim, visto que as vesículas pinocíticas são muito pequenas,
com diâmetros de 100 a 200 nm, elas só podem, em geral, ser
vistas ao microscópio eletrônico.
A pinocitose representa o único meio pelo qual algumas
macromoléculas bastante grandes, tais como a maioria das
moléculas;
15
em seguida, de forma progressiva, mais e mais receptores da
membrana se fixam aos ligandos das partículas, tudo isso
ocorrendo, de modo abrupto, como o fechamento de um zíper.
3. Filamentos de actina, além de outros, também contrateis,
circundam a partícula engolfada e se contraem, em torno de
sua margem externa, o que empurra a partícula mais para dentro.
4. As proteínas contráteis, então, destacam a vesícula
fagocítica, deixando-a no interior celular, do mesmo modo pelo
qual são formadas as vesículas pinocíticas.
DIGESTÃO DE SUBSTÂNCIAS ESTRANHAS PELAS
CÉLULAS — A FUNÇÃO DOS LISOSSOMAS
Fig. 2.11 Mecanismo da pinocitose
Quase imediatamente após a chegada de vesícula pinocítica
ou fagocítica no interior celular, um ou mais lisossomas se
prendem a ela c despejam seu conteúdo de hidrolases ácidas
em seu interior, como mostrado na Fig. 2.12. Dessa forma, é
formada uma vesícula digestiva, onde as hidrolases iniciam a
hidrólise das proteínas, do glicogênio, dos ácidos nucléicos, dos
mucopo-lissacarídios e outras substâncias contidas na vesícula.
Os produtos dessa digestão são moléculas pequenas de
aminoácidos, glicose, fosfatos etc que, em seguida, difundem-se
através da membrana, para o citoplasma. O que resta da
vesícula, chamado de corpo residual, representa as substâncias
indigeríveis. Na maioria dos casos, eles são excretados, através da
membrana celular, pelo processo denominado exocitose, que é, em
essência, o oposto da endocitose.
É por isso que os lisossomas são chamados de órgãos
digestivos das células.
Regressão dos tecidos e autólise celular. Muitas vezes, os
tecidos do corpo regridem de tamanho. Por exemplo, isso ocorre
no útero, após o parto, nos músculos, durante períodos longos
de inatividade, e nas glândulas mamarias, ao término do período
de amamentação. Os lisossomas são responsáveis por grande
parte dessa regressão. Contudo, o mecanismo pelo qual a falta
de atividade de um tecido leva a aumento da atividade dos
lisossomas ainda é desconhecido.
Outro papel muito especial dos lisossomas é o da remoção
de células lesadas ou da parte do tecido onde existam células
lesadas — células lesadas por calor, por frio, por trauma, por
agentes químicos, ou por qualquer outro fator. A lesão celular
causa rotura dos lisossomas, e as hidrolases liberadas começam
imediatamente a digerir as substâncias orgânicas das cercanias.
Se a lesão for pequena, apenas uma parte da célula será removida,
seguida por seu reparo. Todavia, se a lesão for grave, toda a
célula será digerida, processo que é chamado de autólise. Desse
modo, toda a célula será removida e, comumente, uma nova
cuias de proteína podem entrar nas células. Na verdade, a
velocidade de formação das vesículas pinocíticas fica aumentada
quando essas macro moléculas se fixam à membrana celular.
A Fig. 2.11 mostra as etapas sucessivas da pinocitose, a
partir de três moléculas que se fixam à membrana celular.
Geralmente, essas moléculas se prendem a receptores na
superfície da membrana celular, que são específicos para os tipos
de proteínas que vão ser absorvidas. Esses receptores, na
maioria dos casos, ficam concentrados em pequenas depressões
da membrana celular, denominadas depressões espessadas. Na
face interna da membrana celular, por baixo dessas depressões,
existe uma malha de uma proteína fibrilar, chamada de clatrina,
além de filamentos contrateis de actina e de miosina. Uma vez
tendo ocorrido a fixação das moléculas de proteína a seus
receptores, as propriedades da superfície da membrana se
alteram, de modo que toda a depressão se invagina para dentro
da célula e as proteínas contrateis fazem com que seus bordos
se fechem, englobando as proteínas fixadas e pequena quantidade
de líquido extracelular. Imediatamente após, a porção invaginada
da membrana se solta da superfície celular, formando uma
vesícula pinocítica.
Permanece ainda como mistério o mecanismo que faz com
que a membrana celular passe pelas contorções necessárias para
formar as vesículas pinocíticas. Contudo, esse processo necessita
de energia, vinda do interior da célula; essa energia é suprida
pelo ATP, substância rica em energia, discutida adiante neste
capítulo. Por outro lado, também necessita da presença de íons
cálcio no líquido extracelular, que, provavelmente, reagem com
os filamentos contrateis, por baixo da depressão, para gerar a
força que leva à separação da vesícula da membrana celular.
Fagocitose. A fagocitose ocorre quase que do mesmo modo
que a pinocitose, exceto que envolve grandes partículas, e não
moléculas. Apenas determinados tipos celulares têm capacidade
fagocítica, de forma mais acentuada os macrófagos teciduais e
alguns glóbulos brancos.
A fagocitose tem início quando proteínas ou grandes
polissacarídios da superfície da partícula que vai ser fagocitada
— isto é, uma bactéria, uma célula morta ou qualquer outro
detrito tecidual — fixam-se a receptores na superfície do
fagócito. No caso das bactérias, elas estão, geralmente, ligadas a
anticorpos específicos, e são esses anticorpos que se prendem aos
receptores fagocíticos. Essa intermediação por anticorpos é
chamada de opsonizaçâo, e é discutida nos Caps. 33 e 34.
A fagocitose ocorre nas seguintes etapas:
1. Os receptores da membrana celular fixam-se aos ligandos
superficiais da partícula.
2. As bordas da membrana em torno desses pontos de
fixação se evaginam, dentro de fração de segundo, cercando a
Fig. 2.12 Digestão das substâncias contidas nas vesículas pinocíticas
partícula;
pelas enzimas dos lisossomas.
16
célula do mesmo tipo, formada por reprodução mitótica de célula
vizinha, toma o lugar da que foi removida.
Os lisossomas também contêm agentes bactericidas, capazes
de matar as bactérias antes que possam causar lesão à célula.
Esses agentes incluem a lisozima, que dissolve a membrana da
célula bacteriana, a lisoferrina, que fixa ferro e outros metais
imprescindíveis para o crescimento bacteriano, e ácido, em pH
de cerca de 5,0, que ativa as hidrolases e também inativa alguns
dos sistemas metabólicos bacterianos.
Os lisossomas também armazenam enzimas que podem
iniciar a digestão de agregados lipídicos e dos grânulos de
glicogênio, tornando o lipídio c o glicogênio disponíveis para a
utilização em outras regiões da célula e, até mesmo, do corpo. Na
ausência dessas enzimas, o que resulta de distúrbios genéticos
ocasionais, ocorre, muitas vezes, acúmulo de quantidades
muito grandes de lipídios ou de glicogênio nas células de muitos
órgãos, especialmente nas do fígado, o que leva à morte
precoce.
Outras funções do retículo endoplasmático. Outras funções
importantes do retículo endoplasmático — e, de novo,
especialmente do retículo liso — são:
1. Contém as enzimas que controlam a degradação do
glicogênio, quando esse composto é usado para energia.
2. Contém número muito grande de enzimas que são capazes
de detoxificar as substâncias que estão lesando as células, como
os medicamentos; esse resultado é obtido por coagulação,
hidrólise, conjugação com ácido glicurônico e por outros meios.
Funções sintéticas do aparelho de Golgi. Embora a principal
função do aparelho de Golgi seja a de processar substâncias
já formadas no retículo endoplasmático, essa estrutura também
tem capacidade para sintetizar determinados carboidratos que
não podem ser formados no retículo endoplasmático. Isso é
particularmente verdadeiro para o ácido siálico e para a
galactose. Além disso, o aparelho de Golgi pode formar polímeros
sacarídios muito grandes e fixados a quantidades muito pequenas
de proteína; os mais importantes são o ácido hialurônico e o
SÍNTESE E FORMAÇÃO DE ESTRUTURAS condroiti-nossulfato. Entre as muitas funções desses dois
CELULARES PELO RETÍCULO ENDOPLASMÁTICO E polímeros no corpo merecem destaque: (1) são os principais
componentes dos proteoglicanos secretados no muco e em outras
PELO APARELHO DE GOLGI
secreções glandulares: (2) são os principais componentes da
A grande extensão do retículo endoplasmático e do aparelho substância fundamental que preenche os espaços intersticiais,
de Golgi, especialmente nas células secretoras, já foi destacada. atuando como "recheio" entre as fibras de colágeno e as
Essas duas estruturas são formadas, principalmente, por células; e (3} são os principais componentes da matriz orgânica
membranas de bicamada lipídica, e suas paredes são literalmente das cartilagens e dos ossos.
cravejadas de enzimas protéicas que catalisam a síntese de
Processamento das secreções endoplasmáticas pelo aparelho
muitas das substâncias necessárias às células.
de Golgi — a formação de vesículas. A Fig. 2.13 resume as
Em geral, a maior parte dessa síntese começa no retículo principais funções do retículo endoplasmático e de aparelho de
endoplasmático, mas a maioria dos produtos que são aí formados Golgi. À medida que as substâncias vão sendo formadas no
é transferida para o aparelho de Golgi, onde passam por retículo endoplasmático — em especial, proteínas —, elas são
processamento adicional, antes de serem liberados no transportadas pelos túbulos até as regiões do retículo
citoplasma. Mas, primeiro, deve-se notar quais os produtos que endoplasmático liso situadas mais próximas ao aparelho de
são sintetizados em regiões especiais do retículo Golgi. Nesse ponto, pequenas vesículas de transporte se
endoplasmático e do aparelho de Golgi.
destacam, de modo contínuo, e difundem para as partes mais
Formação de proteínas pelo retículo endoplasmático profundas do aparelho de Golgi. No interior dessas vesículas
granular. O retículo plasmático granular é caracterizado pela ficam as proteínas e outros produtos sintetizados.
presença de grande número de ribossomas presos à face externa Instantaneamente, essas vesículas se fundem com o aparelho de
da membrana do retículo. Como discutido no capítulo seguinte, Golgi e despejam seu conteúdo nos espaços vesiculares dessa
as moléculas de proteína são sintetizadas no interior da estrutura estrutura. Aí são adicionados radicais adicionais de carboidrato
ribossômica. Ainda mais, os ribossomas extrudam muitas das a essas secreções. Por outro lado, é função muito importante do
moléculas de proteína sintetizadas, não para o citosol, mas, ao aparelho de Golgi a de compactar as secreções do retículo
contrário, através da parede do retículo endoplasmático, para a endoplasmático em "pacotes" muito concentrados. Conforme as
matriz endoplasmática.
secreções migram para as camadas mais externas do aparelho de
Quase tão rapidamente como as moléculas de proteína Golgi, essa compactação e o processamento continuam;
chegam à matriz endoplasmática, as enzimas da parede do finalmente, vesículas, tanto grandes como pequenas, se destacam
retículo endoplasmático as modificam. Primeiro, quase todas as continuamente do aparelho de Golgi, levando consigo
moléculas são imediatamente glicosiladas, isto é, conjugadas
com radicais de carboidratos, para formar glicoproteínas.
Portanto, essencialmente, todas as proteínas endoplasmáticas
são glicoproteínas, diferindo das proteínas formadas pelos
ribossomas no citosol, que são, em sua maioria, proteínas livres.
Segundo, as proteínas são ligadas entre si e dobradas, para formar
moléculas mais compactas.
Síntese de lipídios pelo retículo endoplasmático, em especial,
pelo retículo endoplasmático liso. O retículo endoplasmático
também sintetiza lipídios, especialmente, fosfolipídios e
colesterol. Eles são rapidamente incorporados à bicamada lipídica
do próprio retículo endoplasmático, o que permite que esse
retículo cresça continuamente. Isso ocorre, sobretudo na região
lisa do retículo endoplasmático.
Para impedir que o retículo endoplasmático cresça além dos
limites da célula, pequenas vesículas — denominadas vesículas
do retículo endoplasmático, ou vesículas transportadoras —
desprendem-se continuamente do retículo liso; será visto Fig. 2.13 Formação de proteínas, lipídios e vesículas celulares pelo
adiante que a maioria dessas vesículas migra, com muita retículo endoplasmático e pelo aparelho de Golgi,
rapidez, para o aparelho de Golgi.
17
as substâncias secretórias compactadas, e, em seguida,
difundem-se para fora da célula.
Para se ter idéia do decurso temporal desses processos:
quando uma célula glandular é imersa em solução com
aminoácidos radioativos, moléculas de proteína radioativa
recém-formadas podem ser detectadas no retículo
endoplasmático após 3 a 5 minutos; dentro de 20 minutos, essas
proteínas recém-formadas estão presentes no aparelho de Golgi
e, dentro de 1 a 2 horas, essas proteínas radioativas são
secretadas da superfície celular.
Tipos de vesículas formadas pelo aparelho de Golgi —
vesículas secretoras e lisossomas. Em célula intensamente
secretora, as vesículas formadas pelo aparelho de Golgi são, em
sua grande maioria, vesículas secretárias, contendo especialmente
as substâncias protéicas que vão ser secretadas pela superfície
celular. Essas vesículas se difundem para a superfície das células,
onde se fundem com a membrana celular e esvaziam seu
conteúdo no exterior, pelo processo chamado exocitose, que é,
em essência, o oposto da endocitose. Na maioria dos casos, a
exoeitose é estimulada pela entrada de íons cálcio na célula; esses
íons cálcio interagem com a membrana vesicular — por
mecanismo ainda não esclarecido — para provocar sua fusão
com a membrana.
Por outro lado, parte das vesículas é destinada à utilização
intracelular. Por exemplo, regiões especializadas do aparelho de
Golgi formam os lisossomas, já discutidos. Acredita-se que as
membranas dessas regiões especializadas contenham receptores
químicos que fazem com que as hidrolases ácidas se fixem a
elas. Desse modo, essas enzimas são concentradas e, em seguida,
liberadas do aparelho de Golgi sob forma de vesículas
lisossômicas.
Outro tipo de vesícula, formado por mecanismo análogo,
é o doperoxissoma. Contudo, acredita-se que este tipo de vesícula
seja formado no retículo endoplasmático liso, junto com a
formação das vesículas de transporte, e não pelo aparelho de
Golgi. Aqui, de novo, receptores especiais na membrana do
retículo endoplasmático, provavelmente, atraem e fixam as
enzimas oxidativas que vão ser liberadas, sob forma concentrada,
nos peroxissomas.
Fig. 2.14 Formação de trifosfato de adenosina (ATP) na célula,
mostrando que a maior parte do ATP é formada nas mitocôndrias.
a energia liberada na direção adequada.
Quase todas essas reações oxidativas ocorrem dentro das
mitocôndrias e a energia liberada é usada principalmente para
formar ATP. Em seguida, o ATP, e não os nutrientes originais,
é usado em toda a célula para energizar quase todas as reações
metabólicas intracelulares.
Características funcionais do ATP
A fórmula do ATP é a seguinte:
Utilização de vesículas intracelulares para recomposição das
membranas celulares. Muitas das vesículas vão, finalmente,
fundir-se com a membrana celular ou com as membranas de
quaisquer outras estruturas intracelulares, como a mitocôndria
ou o próprio retículo endoplasmático. Isso, obviamente, aumenta
a extensão dessas membranas e as recompõe, à medida que vão
sendo destruídas. Por exemplo, a membrana celular perde parte
considerável de sua substância cada vez que forma vesícula
fagocítica ou pinocítica, e são as vesículas do aparelho de Golgi
que continuamente a recompõem.
Assim, em resumo, o sistema de membranas do retículo
endoplasmático c do aparelho de Golgi representa órgão
intensamente metabólico, capaz de formar tanto novas estruturas
celulares como as substâncias secretórias que vão ser extrudadas
pela célula.
EXTRAÇÃO DA ENERGIA DOS NUTRIENTES —
A FUNÇÃO DAS MITOCÔNDRIAS
As principais substâncias de onde a célula extrai energia
são o oxigênio e um ou mais tipos de alimento — carboidrato,
gordura e proteína. No corpo humano, em termos essenciais,
os carboidratos são convertidos em glicose antes que atinjam
as células, as proteínas são convertidas em aminoácidos, e as
gorduras, em ácidos graxos. A Fig. 2.14 mostra o oxigênio e
os nutrientes — glicose, aminoácidos e ácidos graxos — entrando
todos na célula. Uma vez no interior, esses nutrientes reagem
quimicamente com o oxigênio, sob ação de diversas enzimas
— controladoras da velocidade dessas reações — e direcionam
O ATP é um nucleotídio formado pela base nitrogenada
adenina, a pentose ribose e por três radicais fosfato. Os dois
últimos radicais fosfato são ligados ao resto da molécula por
ligações chamadas de ligações fosfato de alta energia. Cada uma
dessas ligações contém cerca de 12.000 calorias de energia por
mole de ATP, nas condições físicas do corpo (nas condiçõespadrão, cerca de 7.300 cal, que é muito mais que a energia
armazenada na ligação química média de outros compostos
orgânicos, o que justifica a denominação "ligação de alta
energia". Ainda mais, a ligação fosfato de alta energia é muito
lábil, de modo que pode ser rompida instantaneamente por
demanda, sempre que for necessária energia para a promoção
de outras reações celulares.
Quando o ATP libera sua energia, é liberado um radical
de ácido fosfórico e formado difosfato de adenosina (ADP). Em
18
seguida, a energia liberada dos nutrientes celulares faz com que
o ADP e o ácido fosfórico se recombinem, para gerar novo
ATP; esse processo se repete continuamente. Por essa razão,
o ATP foi chamado de moeda energética da célula, pois pode
ser gasto e refeito repetitivamente, em geral, com tempo de
renovação de apenas uns poucos minutos no máximo.
Processos químicos na formação do ATP — o papel das
mitocôndrias. Ao entrar nas células, a glicose é submetida à
ação de enzimas do citoplasma que a convertem em ácido
pirúvico (é o processo chamado de glicólise). Pequena
quantidade de ADP é convertida em ATP pela energia liberada
por essa conversão, mas essa quantidade é responsável por
menos de 5% do metabolismo energético global da célula.
De longe, a maior parte do ATP formado na célula o é
nas mitocôndrias. Os ácidos pirúvico e graxo, além da maior
parte dos aminoácidos, são convertidos no composto acetil-CoA
na matriz das mitocôndrias. Por sua vez, esse composto sofre
a ação de outra série de enzimas da matriz mitocondrial, sendo
decomposto em seqüência de reações químicas, chamadas, em
conjunto, de ciclo do ácido cítrico ou ciclo de Krebs. Essas reações
são explicadas em detalhe no Cap. 67.
No ciclo do ácido cítrico, a acetil-CoA é degradada a seus
componentes básicos, átomos de hidrogênio e dióxido de carbono.
O dióxido de carbono, por sua vez, difunde-se para fora das
mitocôndrias e, eventualmente para fora da célula.
Mas, por outro lado, os átomos de hidrogênio são
extremamente reativos e, por fim, vão combinar-se com o
oxigênio que difundiu para as mitocôndrias. Essa reação libera
quantidade muito grande de energia, que é usada pelas
mitocôndrias na conversão de grande quantidade de ADP em
ATP. Os processos dessas reações são muito complexos,
exigindo a participação de grande número de enzimas protéicas
que são parte integral das cristas membranosas que proeminam
para a matriz mitocondrial. O evento inicial é a remoção de um
elétron do átomo de hidrogênio, convertendo-o em íon
hidrogênio. O evento final é o movimento desses íons através
de grandes proteínas globulares, denominadas ATP sintetase,
que fazem protrusão, como maçanetas, das membranas das
cristas mitocondriais. A ATP sintetase é uma enzima que utiliza a
energia do movimento do íon hidrogênio para promover a
conversão de ADP em ATP, enquanto, ao mesmo tempo, os
íons hidrogênio reagem com o oxigênio para formar água.
Finalmente, o recém-formado ATP é transportado para fora das
mitocôndrias, indo para todas as regiões do citoplasma e do
nucleoplasma, onde é usado para energizar o funcionamento da
célula.
Esse processo global de formação do ATP é chamado de
mecanismo quimiosmótico para a formação de ATP. Os detalhes
químicos e físicos desse mecanismo são apresentados no Cap.
67, e muitas das funções metabólicas do ATP no corpo são
apresentadas nos Caps. 67 a 71.
Uso do ATP no funcionamento celular. O ATP é usado para
promover três categorias principais do funcionamento celular;
(1) transporte através de membranas, (2) síntese de compostos
químicos em toda a célula, e (3) trabalho mecânico. Esses três
tipos distintos de uso do ATP são mostrados nos exemplos da
Fig. 2.15: (1) fornecimento de energia para o transporte de sódio
através da membrana celular, (2) promoção da síntese de
proteínas pelos ribossomas, e (3) fornecimento da energia
necessária para a contração muscular.
Além do transporte de sódio através de membranas, a
energia do ATP é necessária, direta ou indiretamente, para o
transporte dos íons potássio, cálcio, magnésio, fosfato, cloreto,
urato, hidrogênio e muitos outros íons e diversas substâncias
orgânicas. O transporte através de membranas é tão
importante para o funcionamento celular que algumas células —
como, por exemplo, as células tubulares renais — utilizam
até 80% do ATP formado nelas exclusivamente para esse fim.
Fig. 2.15 Uso de trifosfato de adenosina para prover energia para três
processos principais do funcionamento celular: (1) transporte através
de membrana, (2) síntese de proteínas, e (3) contração muscular.
Além de sintetizar proteínas, as células também produzem
fosfolipídios, colesterol, purinas, pirimidinas e grande número
de outras substâncias. A síntese de quase todos os compostos
químicos exige energia. Por exemplo, uma só molécula de
proteína pode conter vários milhares de aminoácidos, ligados
entre si por ligações peptídicas; a formação de cada uma dessas
ligações exige a rotura de quatro ligações de alta energia; dessa
forma, muitos milhares de moléculas de ATP (ou do composto
comparável, trifosfato de guanosina [GTP]) devem liberar sua
energia para cada molécula de proteína formada. Na verdade,
algumas células chegam a utilizar até 75% do ATP formado
nelas na síntese de compostos químicos; isso é especialmente
válido durante a fase de crescimento celular.
A última utilização principal do ATP é o fornecimento de
energia para células especializadas, para produção de trabalho
mecânico. Será visto no Cap. 6 que cada contração muscular
exige o consumo de quantidades imensas de ATP. Outras células
realizam trabalho mecânico de outra forma, em especial os
movimentos ciliar e amebóide, descritos adiante neste capítulo.
A fonte de energia para todos estes tipos de trabalho
mecânico é o ATP.
Portanto, para resumir, o ATP está sempre disponível para
liberar sua energia, de forma rápida e quase explosiva, sempre
que for necessário na célula. Para repor o ATP usado pela célula,
numerosas reações químicas, distintas e mais lentas, degradam
os carboidratos, gorduras e proteínas, e a energia nelas liberada
é usada na formação de novo ATP. Cerca de 95% desse ATP
é formado nas mitocôndrias, o que explica a designação das
mitocôndrias como as "usinas" da célula.
LOCOMOÇÃO AMEBÓIDE DAS CÉLULAS
De longe, o tipo mais importante de movimento celular que
ocorre no corpo é o das células musculares especializadas que
formam os músculos esquelético, cardíaco e liso que, em
conjunto, representam quase 50% de toda a massa corporal. O
funcionamento especializado dessas células é descrito nos
Caps. 6 a 9. Todavia, existem dois outros tipos de movimento,
encontrados em outras células: a locomoção amebóide e o
movimento ciliar.
Locomoção amebóide significa o movimento de toda a célula
em relação a seu substrato, como, por exemplo, o movimento
dos glóbulos brancos através dos tecidos. Contudo, seu nome
advém do fato de as amebas se deslocarem por esse mecanismo,
representando excelente modelo para o estudo desse fenômeno.
19
Contudo, outras células se afastam da substância quimiotáxica, o que é
chamado de quimiotaxia negativa.
Mas, como é que a quimiotaxia controla a direção da locomoção
amebóide? Embora ainda não exista resposta definitiva, é sabido que
o lado da célula que fica mais exposto à substância quimiotáxica passa
por modificações de sua membrana que influenciam a protrusão de
pseudópodos.
CÍLIOS E MOVIMENTOS CILIARES
Fig. 2.16 Movimento amebóide por uma célula.
Tipicamente, o movimento amebóide começa pela protrusão de um
pseudópodo por uma das extremidades da célula. O pseudópodo se
projeta para longe da célula, fixando-se, então, sobre nova área do
tecido, e. por fim, o restante da célula se desloca em direção do
pseudópodo. A Fig. 2.16 mostra esse processo, apresentando uma célula
alongada, cuja extremidade direita é um pseudópodo. A membrana
dessa extremidade celular está continuamente se deslocando para a
frente e a membrana da extremidade esquerda a está acompanhando,
seguindo o movimento celular.
Mecanismo da locomoção amebóide. A Fig. 2.16 apresenta o princípio
geral do movimento amebóide. Basicamente, ele resulta de exocitose
contínua que forma nova membrana na extremidade anterior do
pseudópodo e de endocitose, também contínua, nas regiões média e
posterior da célula. Além disso, outro efeito é indispensável para o
movimento para a frente da célula. Esse efeito é a fixação do
pseudópodo aos tecidos circundantes, de modo que ele fica preso em
sua posição de avanço, enquanto o restante da célula é (racionado em
direção a esse ponto de fixação. Essa fixação é efetuada pelas
proteínas receptoras que revestem o interior das vesículas exocíticas.
Quando essas vesículas passam a fazer parte da membrana do
pseudópodo, elas se abrem, de modo que seu interior fica evertído para
o exterior, contactando ligandos nos tecidos circundantes. Um dos
importantes ligandos é uma proteína, denominada fihrinectina, presa às
fibras colágenas dos tecidos.
Na outra extremidade da célula, a atividade endocítica afasta os
receptores de seus ligandos, para formar vesículas endocíticas. Em
seguida, no interior da célula, essas vesículas fluem na direção do
pseudópodo, onde são usadas para formar nova membrana para esse
pseudópodo.
O que ainda permanece obscuro no processo do movimento
amebóide é a fonte de energia, responsável pelo fluxo de vesículas, da
extremidade endocítica para a ponta do pseudópodo. Parte dela poderia
resultar da contração dos filamentos de actina e de miosina no
ectoplasma das células, contraindo a célula em sua extremidade
posterior e, lateralmente, empurrando as vesículas e o citoplasma para a
extremidade do pseudópodo.
Tipos de células que apresentam locomoção amebóide. No corpo
humano, as células mais comuns que apresentam movimento amebóide
são os glóbulos brancos, que se deslocam do sangue para os tecidos,
sob forma de macrófagos ou micrófagos teciduais. Contudo, muitos tipos
de células podem apresentar locomoção amebóide em circunstâncias
específicas. Por exemplo, os fibroblastos invadem qualquer área lesada
para participar de seu reparo, e até mesmo algumas das células germinais
da pele, embora, na maioria das situações, sejam células inteiramente
sésseis, deslocam-se para uma área cortada para reparar a fenda.
Finalmente, a locomoção celular é de importância especial no
desenvolvimento do feto, pois as células embrionárias podem migrar, por
longas distâncias, desde seus locais primordiais de origem até novas áreas,
durante o desenvolvimento de estruturas especiais.
Controle da locomoção amebóide — "quimiotaxia". O fator mais
importante que, em geral, desencadeia a locomoção amebóide é o
processo chamado de quimiotaxia. Ele resulta do aparecimento de
determinadas substâncias químicas nos tecidos. O composto químico
gerador da quimiotaxia é chamado de substância quimiotáxica. A
maioria das células que apresentam locomoção amebóide se desloca em
direção ã substância quimiotáxica — isto é, de área onde sua
concentração seja baixa, para outra onde seja alta —, o que é
chamado de quimiotaxia positiva.
O segundo tipo de movimento celular — o movimento ciliar —
é semelhante ao de uma chicotada dos cílios que revestem a superfície
das células. Isso só ocorre em duas regiões do corpo humano: nas
superfícies internas das vias respiratórias e das trompas uterinas
(trompas de Falópio) no aparelho reprodutor. Na cavidade nasal e nas
vias aéreas inferiores, o movimento em chicotada dos cílios promove o
movimento da camada de muco, com velocidade de 1 cm/min, em
direção à faringe, removendo, assim, não apenas o muco dessas vias, mas
todas as partículas que ficaram retidas nesse muco. Nas trompas uterinas,
os cílios promovem o lento movimento de líquido do óstio para a
cavidade uterina; é esse movimento de líquido que leva o óvulo até o
útero.
Como mostrado na Fig. 2.17, um cílio parece um pêlo curvado,
com ponta aguda, que se projeta por 2 a 4 um da superfície celular.
Muitos cílios se projetam de cada célula — por exemplo, cerca de 200
cílios se projetam da superfície de cada célula epitelial das vias aéreas
respiratórias. O cílio é recoberto por expansão da membrana celular
e é sustentado por 11 microtúbulos: nove túbulos duplos, situados em
tomo da periferia, e dois túbulos simples, localizados em sua porção
central, como mostrado no corte transverso da figura. Cada cílio se
origina de estrutura situada imediatamente abaixo da membrana celular,
chamada de corpo basal do cílio.
O flagelo do espermatozóide tem organização semelhante à do cílio;
na verdade, tem quase que o mesmo tipo de estrutura e o mesmo tipo
de mecanismo contrátil. Todavia, o flagelo é bem mais longo e se move
em ondas quase sinusoidais, em vez de em movimentos de chicotada.
No detalhe da Fig. 2.17 é mostrado o movimento de um cílio. O
cílio se move para a frente de forma abrupta e rápida, 10 a 20 vezes
por segundo, curvando-se acentuadamente em seu ponto de emergência
da superfície celular. Em seguida, move-se para trás, bem lentamente
como em uma chicotada. O movimento rápido para frente empurra o
líquido adjacente à célula na direção do movimento do cílio, e o
movimento lento de chicotada, na direção oposta, pouco atua sobre
o líquido. Como resultado, o líquido c continuamente propelido na
direção do movimento rápido para frente. Dado que a maioria das células
ciliadas apresenta grande número de cílios em sua superfície, e dado
que todas as células têm seus cílios orientados na mesma direção, isso
representa meio muito eficaz para o deslocamento de líquido ao longo
de uma superfície.
Mecanismo do movimento ciliar. Embora nem todos os aspectos
do movimento ciliar já tenham sido esclarecidos, sabemos o que se segue.
Primeiro, os nove túbulos duplos são interligados entre si por um
complexo de pontes transversas protéicas; esse complexo total de
túbulos e de pontes transversas é chamado de axonema. Segundo,
mesmo após remoção da membrana e destruição dos outros elementos
do cílio, exceto o axonema, o cílio ainda pode mover-se em
determinadas condições. Terceiro, existem duas condições essenciais para
a continuidade do batimento do axonema, após remoção das outras
estruturas do cílio: (1) presença de ATP, e (2) condições iônicas
adequadas, incluindo, de modo especial, concentrações adequadas de
magnésio e de cálcio. Quarto, durante o movimento rápido para frente,
os túbulos da face anterior do cílio deslizam para diante, em direção
à ponta do cílio, enquanto os túbulos da face posterior permanecem
imóveis. Quinto, três braços, formados por uma proteína dotada de
atividade ATPase, chamada dineí-na, unem cada conjunto de túbulos
periféricos ao seguinte.
A partir desta informação básica, foi postulado que a liberação
de energia do ATP, ao entrar em contato com a ATPase dos braços
de dineína, faz com que esses braços "engatinhem" ao longo da superfície
dos pares de túbulos adjacentes. Se esse engatinhar ocorrer em direção
ã extremidade do cílio, nos túbulos anteriores, enquanto os posteriores
ficam estacionários, obviamente o resultado será uma curvatura.
Não é conhecido o mecanismo de controle da contração ciliar.
Contudo, os cílios de determinadas células geneticamente anormais não
contém os dois túbulos simples centrais, e esses cílios não se movem.
Portanto.
20
Fig. 2.17 Estrutura e funcionamento do cílio. (Modificado de Satir:
Cilia; Sei. Amer., 204:108. 1961. Copyright 1961 by Scientific
American Inc. Todos os direitos reservados.)
é presumido que algum sinal, talvez eletroquímico, seja transmitido ao
longo desses dois túbulos para ativar os braços de dineína.
REFERÊNCIAS
Almers, W., and Stirling, C: Diatribution of transport proteins over animal
cell membranea. J. Membr. Biol., 77:169, 1984. Baker, P. F., and Knight, D.
E.: Chemiosmotic hypotheses of exocytosis: A critique. Biosci. Rep., 4:285,
1984. Balaban, R. S.: The application of nuclear rnagnetic resonance to the
Btudy of cellular physiology. Am. J. Physiol., 246:ClO, 1984. Bershadsky,
A. D., and Vasiliev, J. M.: Cytoakeleton. New York, Plenum Publishing
Corp., 1988. Bcttger, W. J., and McKeehan, W. L.: Mecbanisms of
cellular nutrítion.
PhyBiol. Rev., 66:1, 1986. Blachley, J. E., et ai.: The harmful effects of
ethanol on ion transport and cellular respiration. Am. J. Med. ScL,
289:22,1985. Bohr, D. F.: Cell membrane in hypertension. News in
PhyBiol. Sei., 4;85, 1989.
Boyd, A., and Simon, M.: Bacterial chemotaxís. Annu. Rev. Physiol., 44:501,
1982. Brazy, P. C, and Mandei, L. J.: Does availability of inorganic
phosphate regulate cellular oxidative metabolism? News Phyaiol. Sei.,
1:100,1986. Case, R. M: The role of Ca"" storea in secretion. Cell-Calcium,
5:89, 1984. Cereijido, M., et ai.: Tight junction: Barrier between higher
organisms and environment. New Phyaiol. Sei., 4:72,1989. Chien, S. (ed.):
Molecular Biology in Physiology. New York, Raven Presa, 1988. Correia, J.
J-, and Williams, R. C, Jr: Mechanisms ofassembly and disas- sembly of
microtubules. Annu. Rev. Biophya. Bioeng., 12:211, 1983. Dean, R. T., et
ai.: Effects of exogenous aminea on mammalian cells, with
particular reference to membrane flow. Biochem. J., 217:27,1984. DeMello,
W. C. (ed.): Cell-to-Cell Communication. New York, Plenum Publishing
Corp., 1987. DeRobertis, E. D. P., and DeRobertÍB, E. M. F., Jr.: Cell and
Molecular Biology. 8th Ed. Philadelphia, Lea & Febiger, 1987.
Dowben, R. M. (ed.): Cell and Muscle Motility. New York, Plenum Publising
Corp. 1983.
Fawcett, D. W.; Bloom & Fawcett: A Textbook of Hiatology. llth Ed. Phila
delphia, W. B. Saunders Co., 19S6.
Fawcett, D. W.: The Cell. 2nd Ed. Philadelphia, W. B. Saunders Co., 1981.
Fine, R. E., and Ockleford, C. D.: Supramolecular cytology of coated veaicles.
Int. Rev. Cyto)., 91:1, 1984. Fitzgerald, P. G.: Gap junction heterogeneity in
liver, heart, and lenR. News in
Physiol. Sei,, 3:206, 1988. Frankel, R. B.: Magnetic guidance of
organisms. Annu. Rev. Biophys.
Bioeng., 13:85, 1984. (ioldstein, D. B.: The eftectB of druga on membrane
fluidity. Annu. Rev.
Pharmacol. Toxicol., 24:43, 1984. Green, H., and Barrandon, Y.: Cultured
epidennal cells and their use in the generation of epidermis. News in
Physiol. Sei., 3:53, 1988. Hoffman, E. K., and Simonsen, L. O.: Membrane
mechanisms in volume and pH regulation in vertebrate cella. Phyaiol. Rev.,
69:315, 1989. Holizman, E.: Lysosomea, New York, Plenum Publishing
Corp., 1989. Hubbard, A. L., et ai.: Biogenesis of endogenous plasma
membrane proteína in epithelian cells. Ann. Rev. Physiol., 51:755,1989.
Kornfeld, S.: Trafficking of lyaosomal enzymes. FASEB J., 1:462, 1987.
Kudlow, J. E., et ai. (eds.): Biology of Growth Factors. New York, Plenum
Publishing Corp., 1988.
Lane, M. D., et ai.: The mitochondrion updated. Science, 234:526, 1986.
Lemastera, J. J., et ai. (eds.): Integration of Mitochondrial Function. New
York, Plenum Publishing Corp., 1988. Low, M. G.: Glycosylphosphatidylinositol: A versatile anchor for cell surface proteína. FASEB J.,
3:1600, 1989. Machlin, L. J., and Bendich, A.: Freeradical tissuedamage:
Protective roleof antioxidant nutrients. FASEB J., 1:441, 1987. Macnab, R.
M., and Aizawa, S.-L: Bacterial motility and the bacterial 11 age I- lar motor.
Annu. Rev. Biophys. Bioeng., 13:51,1984. Malhotra, S. K.: The Plasma
Membrane. New York, John Wiley & Sons, 1983. McCloskey, M., and
Poo, M. M.: Protein diffusion in cell membranea: Some biological
implications. Int. Rev. Cytol., 87:19, 1984. Moore, A. L., and Beechey, R.
B. {eds.): Plant Mitochondria. New York, Plenum Publishing Corp., 1987.
Porter, K. R., and Bonneville, M. A.: Fine Structure of Cells and Tissues. 4th
Ed. Philadelphia, Lea & Febiger, 1973. Rodriguez-Boulan, E., and Salas,
P. J. I.: Externai and internai aignals for epithelial cell aurface polarization.
Ann. Rev. Physiol., 51:741,1989. Schachter, D.: Fluidity and function of
hepatocyte plasma membranea. He- patology, 4:140, 1984.
Sowers, A. E. (ed.): Cell Fusion. New York, Plenum Publishing Corp., 1987.
Thomas, K. A.: Fibroblaat growth factors. FASEB J., 1:434, 1987. van der
Laarse, W. J., et ai.: Energetics at the single cell levei. News in PhyBiol.
Sei., 4:91,1989. Wade, J. B.: Role of membrane fusion in hormonal
regulation of epithelial transport. Ann. Rev. Physiol., 48:213, 1986. Wall,
D. A., and Maack, T.: Endocytic uptake, transport, and catabolism of
proteins by epithelial cells. Am. J. Phyaiol., 24&C12, 19S5. Wheatley, D.
N.: On the possible ímportance of an intracellular circulation.
Life-Sci., 36:299, 1985. WilHngham, M. C, and Pastan, I.: Endocytosia
and exocytosia: Current concepts of veaicle traffic in animal cells. Int.
Rev. Cytol., 92:51, 1984.
21
CAPÍTULO 3
Controle Genético da Síntese de Proteínas, do Funcionamento
e da Reprodução Celular
Virtualmente todas as pessoas sabem que os genes controlam a
hereditariedade dos pais aos filhos, mas a maioria das pessoas não
compreende que esses mesmos genes controlam a reprodução e o
funcionamento dia-a-dia das células. Os genes controlam o
funcionamento celular ao determinarem quais as substâncias que serão
sintetizadas pela célula
— que estruturas, quais enzimas, quais compostos químicos.
A Fig. 3.1 mostra um esquema geral do controle genético. Cada
gene, que é um ácido nucléico, chamado de ácido desoxirribonucléico
(ADN), controla, automaticamente, a formação de outro ácido nucléico
o ácido ribonucléico (ARN), que se difunde por toda a célula e controla
a formação de proteínas específicas.
Algumas dessas proteínas são proteínas estruturais que, associadas a
diversos lipídios e carboidratos, formam a estrutura de muitas das
organelas
discutidas
no
Cap.
2.
Mas,
de
longe,
as proteínas são, em sua maioria, enzimas que catalizam as diferentes
reações químicas que ocorrem nas células. Por exemplo, as enzimas
promovem as reações oxidativas que fornecem energia para as células
e, também, promovem a síntese de diversos compostos químicos, tais
como lipídios, glicogênio, trifosfato de adenosina (ATP) etc.
Para a formação de cada proteína celular, só existe, em geral, um
par de genes em cada célula. Tem sido estimado que as células humanas
teriam mais de 100.000 desses pares de genes, o que significa que até
100.000 proteínas diferentes podem ser formadas nas diferentes células,
embora não todas por uma mesma célula, por razões que serão discutidas
adiante neste capítulo.
Fig. 3.1 Esquema geral de como os genes controlam o funcionamento
OS GENES
Grande número de genes — ligados entre si, formando uma fileira
— fica contido em moléculas de ADN, formadas por filamentos duplos
helicoidais, cujo peso molecular é medido em bilhões. Um segmento
muito curto de uma dessas moléculas é mostrado na Fig. 3.2. Essa molécula é formada por vários compostos químicos simples, dispostos segundo
um padrão regular que é explicado nos parágrafos seguintes.
As unidades básicas do ADN. A Fig. 3.3 mostra os compostos
químicos básicos que participam na formação do ADN. Esses compostos
incluem (1) ácido fosfórico. (2) um açúcar, chamado desoxirribose, e (3)
quatro bases nitrogenadas (duas purinas, adenina e guanina, e duas
pirimidinas, timina e citosina). Os dois filamentos helicoidais são
formados pelo ácido fosfórico e pela desoxirribose, representando o
arcabouço da molécula do ADN, enquanto as bases ficam entre os dois
filamentos, ligando-os.
Os nucleotídios. A primeira etapa na formação do ADN é a combinação
de uma molécula de ácido fosfórico, uma molécula de desoxirribose e uma
molécula de uma das quatro bases, para compor um nucleotídio. Dessa
forma, são formados quatro nucleotídios, um para cada uma das bases:
ácidos
desoxiadenílico,
desoxitimidílico,
desoxiguanílico
e
desoxicitidílico.
celular.
A Fig. 3.4 apresenta a estrutura química do ácido adenílico, enquanto a
Fig. 3.5 mostra os símbolos simples que representam os quatro
nucleotídios básicos do ADN.
Organização dos nucleotídios para formar o ADN. A Fig. 3.6 mostra
o modo pelo qual números múltiplos de nucleotídios se combinam para
formar o ADN. Note-se que essa combinação ocorre de modo tal que
o ácido fosfórico e a desoxirribose ocupam posições alternadas nos dois
filamentos, e esses filamentos são unidos entre si por ligações fracas
Fig. 3.2 A estrutura helicoidal de dois filamentos do gene. Os filamentos
externos são formados por ácido fosfórico e pelo açúcar desoxirribose.
As moléculas internas unindo os dois filamentos da hélice são bases
de purina e de pirimidina; elas determinam o "código" do gene.
22
Fig 3.4 O ácido desoxiadenílico, um dos que compõem o
ADN.
Fig. 3.3 As unidades básicas do ácido desoxirribonucléico (ADN).
Fig. 3.5 Combinações das unidades básicas do ADN, para formar os
nucleotídios. ({P = ácido fosfórico; D = desoxirribose.) As quatro
bases dos nucleotídios são A (adenina); T" (timina); G (guanina); e
C(citosina). Esses quatro tipos de nucleotídios formam o ADN.
entre as bases de purina e de pirimidina. Mas deve ser
cuidadosamente notado que:
1. a base purina adenina sempre se liga à base pirimidina
timina, e
2. a base purina guanina sempre se liga à base pirimidina
ciíosina.
Assim, na Fig. 3.6, a seqüência dos pares complementares de
bases é CG, CG, GC, TA, CG, TA.GC, AT e AT.
Contudo, essas bases são interligadas por pontes de
hidrogênio muito fracas, representadas na figura por linhas
tracejadas. Devido à fraqueza dessas ligações, os dois
filamentos podem separar-se facilmente, e o fazem durante o
curso de seu funcionamento na célula.
Agora, para colocar o ADN em sua perspectiva física
adequada, basta que as duas extremidades sejam apanhadas e
torcidas, para formar uma hélice. Em cada volta completa da
hélice da molécula de ADN existem 10 pares de nucleotídios,
como mostrado na Fig. 3.2.
O CÓDIGO GENÉTICO
A importância do ADN reside em sua capacidade de
controlar a formação de outras substâncias pela célula. Isso é
realizado por meio do chamado código genético. Quando os dois
filamentos da molécula de ADN são separadas, as bases de purina e
de pirimidina ficam expostas.
pois se projetam lateralmente de cada filamento. São essas bases
proeminentes que formam o código.
Estudos experimentais, realizados nos últimos anos, demonstraram
que o código genético é composto de "trincas" (triptets) sucessivas de
bases — isto é, o grupo de três bases em seqüência forma uma palavra
do código. As trincas sucessivas controlam, eventualmente, a seqüência
dos aminoácidos de uma molécula de proteína, durante sua síntese na
célula. Note-se, na Fig. 3.6, que cada filamento da molécula de ADN
tem seu próprio código genético. Por exemplo, o filamento superior
tem, da esquerda para a direita, o código genético GGC, AGA e CTT,
as trincas estando separadas por setas. Ao se acompanhar esse código
genético nas Figs. 3.7 e 3.8 será notado que essas três trincas são
responsáveis pela colocação sucessiva dos três aminoácidos, prolina,
serina e ácido glulâmico, na molécula de proteína.
ARN— O PROCESSO DE TRANSCRIÇÃO
Dado que quase todo o ADN fica no núcleo da célula e, todavia,
a maior parte do funcionamento celular ocorre no citoplasma, deve existir
algum meio para que os genes do núcleo possam controlar as reações
23
Químicas no citoplasma. Isso é realizado pela intermediação de outro
tipo de ácido nucléico, o ARN, cuja formação é controlada pelo ADN
do núcleo. Nesse processo, o código ê transferido para o ARN, o que
é chamado de transcrição. O ARN, então, difunde-se do núcleo, passando
pelos poros nucleares, para o compartimento citoplasmático, onde
controla a síntese de proteína.
Síntese de ARN
Durante a síntese do ARN, os dois filamentos do ADN se separam
durante certo tempo: em seguida, um dos filamentos é usado como
molde para a síntese das moléculas de ADN. As trincas do código do
ADN promovem a formação de trincas complementares do código no
ARN (chamadas códons); esses códons, por sua vez, controlam a
seqüência dos aminoácidos de uma proteína que vai ser,
subseqüentemente, sintetizada no citoplasma. Quando um filamento de
ADN é usado dessa maneira para a formação do ARN, o outro filamento
permanece inativo. Cada filamento de ADN em um cromossoma é
molécula tão grande que pode conter o código para 4.000 genes em
média.
As unidades básicas do ARN. As unidades básicas do ARN são
as mesmas do ADN, exceto por duas diferenças. Primeiro, o açúcar
desoxirribose não faz parte do ARN, Em seu lugar, existe outro açúcar,
com composição ligeiramente diferente, a ribose. Segundo, a timina
é substituída por outra pirimidina, o uracit.
Formação dos nucleotídios do ARN. Inicialmente, as unidades básicas
do ARN formam nucleotídios de forma idêntica à descrita acima para
a síntese do ADN. Também aqui, quatro nucleotídios são usados na
formação do ARN. Esses nucleotídios contêm as bases adenina, guanina,
áiosina e uracii. Note-se que essas são as mesmas bases do ADN, exceto
pelo uracil substituir a timina.
Ativação dos nucleotídios. A etapa seguinte na síntese do ARN
é a ativação dos nucleotídios. Isso ocorre pela incorporação de dois
radicais fosfato a cada nucleotídio, do que resulta a formação de
trifosfatos. Esses dois últimos radicais fosfato são unidos ao nucleotídio
por ligações fosfato de alia energia, derivadas do ATP da célula.
O resultado desse processo de ativação é que grandes quantidades
de energia ficam disponíveis para cada nucleotídio, e essa energia é
usada para promover as reações químicas subseqüentes, do que resulta
a formação da cadeia de ARN.
Montagem da molécula de ARN a partir de nucleotídios
ativados, usando o filamento de ADN como molde — o
processo de "transcrição".
A montagem da molécula de ARN é mostrada na Fig. 3.7, sob
a influência da enzima ARNpolimemse. Esta é uma enzima muito grande
e apresenta muitas propriedades funcionais, necessárias para a formação
da molécula de ARN. Essas propriedades funcionais são as seguintes:
1. No filamento de ADN, imediatamente antes do gene inicial.
existe uma seqüência de nucleotídeos, chamada de promotor. A ARN
polimerase contém uma estrutura complementar apropriada, que
reconhece esse promotor e se liga a ele. Esta é a etapa essencial
para a formação da molécula de ARN.
2. Uma vez que a ARN polimerase tenha-se ligado ao promotor.
ela faz com que cerca de duas voltas da hélice de ADN se destorçam
e, em seguida, se separem.
3. Após isso, a ARN polimerase passa ao longo do filamento de
ADN, temporariamente destorcendo e separando os filamentos de ADN
a cada estágio de sua passagem. Conforme passa, ela vai formando
a molécula de ARN pelas seguintes etapas;
4. Primeiro, provoca a formação de pontes de hidrogênio entre
as bases sucessivas do filamento de ADN e as bases dos nucleotídios
complementares presentes no nucleoplasma.
5. Em seguida, e um de cada vez, a ARN polimerase remove dois
dos três radicais fosfato de cada um dos nucleotídios do
ARN,
liberando Base do ARN
grandes quantidades de energia das ligações fosfato de alta energia que
são rompidas; essa energia é usada na formação de ligações covalentes
entre o radical fosfato restante no nucleotídio com a ribose da
extremidade crescente da molécula de ARN.
6. Quando a ARN polimerase atinge o fim do gene ou grupo de
genes, ela encontra nova seqüência de nucleotídios do ADN, chamada
de seqüência de terminação da cadeia; isso faz com que a ARN polimerase
se afaste do filamento de ADN. Em seguida, essa polimerase pode
prender-se a outro trecho do mesmo ou de outro filamento de ADN,
podendo
ser usada repetidamente na formação de novas moléculas de ARN.
7. Conforme o novo filamento de ARN é formado, suas pontes
de hidrogênio com o molde de ADN são rompidas, porque o filamento
complementar de ADN tem energia de ligação, o que força o afastamento
do novo filamento de ARN e promove a reunião dos dois filamentos
de ADN. Como resultado, a molécula de ARN fica solta no núcleo
plasma.
Deve ser lembrado que existem quatro tipos distintos de bases do
ADN e, também, quatro tipos distintos de bases nucleotídicas de ARN.
Ainda mais, essas bases só interagem entre si por combinações
específicas. Portanto, o código presente no filamento de ADN é
transmitido, sob forma complementar, para a molécula de ARN. As bases
dos nucleotídios de ribose se combinam com as bases dos de
desoxirribose da seguinte forma:
Base do ADN
guanina
citosina
adenina
timina
..............................................................................
............................................................................
..............................................................................
............................................................................
citosina
guanina
uracii
adenina
Após a liberação das moléculas de ARN no nucleoplasma, elas
ainda devem passar por processamento adicional, antes de ir para o
citoplasma. A razão disso é que o ARN recém-transcrito contém muitas
seqüências indesejáveis de nucleotídios de ARN. Alguns deles ocorrem
nas duas extremidades do filamento de ARN e muitos outros ficam
no meio do filamento; esse material indesejável constitui, provavelmente,
mais de 9H% de todo o filamento. Felizmente, várias enzimas do
nucleoplasma apresentam a capacidade de remover essas seqüências
indesejáveis e, em seguida, de juntar os segmentos que sobraram,
processo chamado de recomposição do ARN (ARN-splicing). Após isso,
o ARN fica pronto para ser usado na formação de proteína.
Existem três tipos distintos de ARN, cada um com papel
independente e completamente diferente na formação das proteínas.
Fig. 3.7 Combinação dos nucleotídios de ribose com um
filamento de ADN para formar uma molécula de ácido
ribonucléico (ARN) que transfere o código do ADN do gene
para o citoplasma. A ARN polimerase se desloca ao longo do
filamento de ADN e constrói a molécula de ARN.
24
Fig. 3.8 Parte da molécula de ácido ribonucléico,'mostrando três
palavras do "código", CCG, UCU e GAA, que representam os três
aminoácidos prolina, serina e ácido glutâmico.
Esses tipos são:
1. ARN mensageiro, que transporta o código genético até o
citoplasma, para o controle da formação das proteínas;
2. ARN transportador, que transforma os aminoácidos ativados até
os ribossomas, onde vão ser usados na montagem das moléculas de
proteínas; e
3. ARN ribossômico, que, junto com cerca de 75 proteínas
diferentes, formam os ribossomas, as estruturas físicas e químicas onde
ocorre
realmente a montagem das moléculas de proteína.
O ARN MENSAGEIRO — OS "CÓDONS"
As moléculas de ARN mensageiro são longos filamentos simples
de ARN que existem em suspensão no citoplasma. Essas moléculas são
formadas por centenas a milhares de nucleotídios, dispostos em filamento
único, contendo os códons que são exatamente complementares às trincas
do código dos genes. A Fig. 3.8 mostra pequeno segmento da molécula
de ARN mensageiro. Seus códons são CCG, UCU e GAA. Esses são
os códons para os aminoácidos prolina, serina e ácido glutâmico. A
transcrição desses códons é mostrada na Fig. 3.7.
Os códons do ARN para os diferentes aminoácidos. O Quadro 3.1
apresenta os códons do ARN para os 20 aminoácidos encontrados nas
moléculas de proteína. Note-se que a maioria desses aminoácidos é
representada por mais de um códon; também, existe um códon
sinalizando "comece a produzir uma molécula de proteína", e três códons
sinalizando "pare de produzir a molécula de proteína". No Quadro 3.1,
esses dois tipos de códons são designados como Cl (início da cadeia) e
CT (término da cadeia).
O ARN TRANSPORTADOR — OS "ANTICÓDONS"
Outro tipo de ARN com papel essencial na síntese de proteínas
é chamado ARN transportador, por transportar as moléculas de
aminoácidos até as moléculas de proteína ã medida que essa
proteína está
Quadro 3.1 Códons do ARN para os diferentes aminoácidos
e para o começo e fim
Aminoácido
Ácido aspártico
Ácido glutãmico
Alanína
Arginina
Asparagina
Cisteína
Fenilalanina
Glicina
Glutamina
Histidina
Isoleucina
Leucina
Usina
Metionina
Prolina
Serina
Tirosina
Treonina
Triptofano
Valina
Começo (Cl)
Fim (CT)
sendo sintetizada. Cada tipo de ARN transportador se combina,
especificamente, com um dos 20 aminoácidos que podem ser
incorporados às proteínas. O ARN transportador atua, assim, como um
carreador para o transporte de tipo específico de aminoácido até os
ribossomas, onde estão sendo formadas as moléculas de proteína. Nos
ribossomas, cada tipo específico de ARN transportador reconhece
determinado códon no ARN mensageiro, como descrito a seguir, e,
portanto, entrega o aminoácido adequado no local apropriado da cadeia
da molécula de proteína em formação.
O ARN transportador, contendo cerca de 80 nucleotídios, é
molécula relativamente pequena, em comparação com o ARN
mensageiro. Ele é uma cadeia dobrada em forma de folha de trevo,
semelhante à mostrada na Fig. 3.9. Uma das extremidades da
molécula sempre contém ácido adenílico; é nessa extremidade que o
aminoácido transportado se fixa ao radical hidroxila da ribose do ácido
adenílico. Enzima específica provoca essa fixação para cada tipo de
ARN transportador; essa mesma enzima também determina que tipo de
aminoácido vai fixar-se ao tipo respectivo de ARN transportador.
Como a função do ARN transportador c a de produzir a fixação
de aminoácido específico à cadeia em formação da proteína, é essencial
que cada tipo de ARN transportador também possua especificidade para
um códon determinado do ARN mensageiro. O código específico do
ARN transportador, que permite seu reconhecimento de um códon
específico é, de novo, uma trinca de bases nucleotídicas, chamada de
anticódon. Essa trinca fica localizada, aproximadamente, no meio da
molécula do ARN transportador (na pane mais inferior da estrutura
em forma de folha de trevo mostrada na Fig. 3.9). Durante a formação
de uma molécula de proteína, as bases do anticódon se fixam
fracamente, por meio de pontes de hidrogênio, com as bases dos códons
do ARN mensageiro. Desse modo, os aminoácidos correspondentes são
alinhados, um após outro, ao longo da cadeia de ARN mensageiro, o
que estabelece a seqüência apropriada de aminoácidos da molécula de
proteína.
O ARN RIBOSSÔMICO
O terceiro tipo de ARN na célula é ARN ribossômico; constitui
cerca de 60% dos ribossomos. O restante do ribossomo é formado por
proteína, contendo cerca de 75 tipos diferentes de proteínas, tanto
proteínas estruturais como enzimas necessárias para a produção de
moléculas de proteína.
O ribossomo é a estrutura química do citoplasma onde vai, efetivamente, ocorrer à síntese de proteínas. Contudo, sempre atua em
associarão com os dois outros tipos de ARN: o ARN transportador
carreia os aminoácidos até os ribossomos, para serem incorporados à
molécula de proteína em formação, enquanto o ARN mensageiro
fornece a informação necessária para o sequenciamento dos
aminoácidos, na ordem correta para cada tipo de proteína que vai ser
formada.
Os ribossomas de células nucleadas são formados por duas
subunidades físicas, denominadas subunidade pequena, contendo uma
molécula de ARN e 33 proteínas, e a subunidade grande, com três
ARNs e mais de 40 proteínas.
Códons do ARN
GAU
GAA
GCU
CGU
AAU
UGU
UUU
GGU
CAA
CAU
AUU
CUU
AAA
AUG
CCU
UCU
UAU
ACU
UGG
GUU
AUG
UAA
GAC
GAG
GCC
CGC
AAC
UGC
UUC
GGC
CAG
CAC
AUC
CUC
AAG
GCA
CGA
GCG
CGG
GGA
GGG
AUA
CUA
CCC
UCC
UAC
ACC
GUC
UAG
AGA
AGG
CUG
UUA
UUG
CCA
UCA
CCG
UCG
AGC
AGU
ACA
ACG
GUA
GUG
UGA
Fig. 3.9 Mecanismo de como uma molécula de proteína é formada nos
ribossomas, em associação com o ARN mensageiro e o ARN
transportador.
25
Embora só se tenha conhecimento parcial do mecanismo da síntese de
proteínas pelos ribossomas, é sabido que o ARN mensageiro e o ARN
transportador se complexam, inicialmente, com a subunidade pequena.
Em seguida, a subunidade grande fornece a maioria das enzimas que
promovem a formação das ligações peptídicas entre os aminoácidos
sucessivos. Dessa forma, os ribossomas funcionam como uma fábrica,
onde são produzidas as moléculas de proteína.
Formação dos ribossomas no nucléolo. Os genes do ADN para a
formação de ribossomas ficam localizados em cinco pares diferentes de
cromossomas no núcleo e cada cromossoma contém muitas duplicatas
desses genes, devido à grande quantidade de ARN ribossômico
que é necessária para o funcionamento celular.
À medida que o ARN ribossômico é formado, ele se acumula no
nucléolo, estrutura especializada situada ao lado dos cromossomas.
Quando grandes quantidades de ARN ribossômico estão sendo formadas,
o nucléolo aparece como uma grande estrutura, enquanto, nas células
que sintetizam quantidades muito pequenas de proteína, o nucléolo pode
ser inaparente. O ARN ribossômico é especificamente processado no
nucléolo e combinado a "proteínas ribossômicas" para formar
condensações granulares que são as subunidades primordiais dos
ribossomas. Essas subunidades são, então, liberadas pelo nucléolo e
transportadas, através dos grandes poros do envelope nuclear, para
quase todas as partes do citoplasma. Só após essas subunidades terem
chegado ao citoplasma é que são unidas para formar os ribossomas
adultos e funcionais. Por conseguinte, as proteínas não são formadas no
núcleo, pois o núcleo não contém ribossomas maduros.
FORMAÇÃO DAS PROTEÍNAS NOS RIBOSSOMAS — O
PROCESSO DE "TRADUÇÃO"
Quando uma molécula de ARN mensageiro entra em contato com
um ribossoma, ele a percorre em toda sua extensão, a partir de extremidade predeterminada da molécula de ARN, que é especificada por uma
seqüência apropriada de bases do ARN. Em seguida, como mostrado
na Fig. 3.9, enquanto o ARN mensageiro passa pelo ribossoma. é formada
a molécula de proteína — um processo chamado de tradução. Nele, o
ribossoma lê o código do ARN mensageiro, da mesma forma como
uma fita é "lida11 ao passar pela cabeça de reprodução do toca-fitas.
Então, quando é atingido o códon de término (ou de "término da
cadeia"), é sinalizado o fim da molécula de proteína que é liberada no
citoplasma.
Polirribussomas. Uma só molécula de ARN mensageiro pode formar
moléculas de proteína em diversos e diferentes ribossomas ao mesmo
tempo, com o filamento de ARN passando ao ribossoma seguinte a
medida que sai do anterior, como mostrado na Fig. 3.9. Obviamente,
as moléculas de proteína estarão em etapas diferentes de formação em
cada ribossoma. Como resultado, existem, freqüentemente, grupos de
ribossomas com cerca de 3 a 10 ribossomas presos ao mesmo tempo
à mesma molécula de ARN mensageiro, Esses grupos de ribossomas
são chamados de pofirribossomas.
Deve ser especialmente notado que um ARN mensageiro pode promover a formação de molécula de proteína em qualquer ribossoma,
por não existir qualquer especificidade do ribossoma para determinado
tipo de proteína. O ribossoma é simplesmente, a estrutura onde ocorrem
às reações químicas.
Fixação dos ribossomas ao retículo endoplasmático. No
capítulo anterior, foi notado que muitos ribossomas ficam presos ao
retículo endoplasmático.
Isso só ocorre após os ribossomas terem iniciado a formação das
moléculas de proteína. Essa fixação acontece porque as extremidades
iniciais de algumas moléculas de proteína contêm seqüências de
aminoácidos que se fixam, imediatamente, a sítios receptores
específicos do retículo endoplasmático; isso permite que essas
moléculas atravessem a parede do retículo, atingindo sua matriz. Isso
ocorre enquanto a molécula de proteína ainda está sendo formada no
ribossoma, o que puxa o ribossoma para o retículo endoplasmático, do
que resulta a aparência "granular" desse retículo.
A Fig. 3.10 apresenta a relação funcional do ARN mensageiro com
o ribossoma e, também, o modo como esse ribossoma se fixa à membrana
do retículo endoplasmático. Note-se que o processo de tradução está
ocorrendo em diversos ribossomas ao mesmo tempo, em resposta a um
só filamento de ARN mensageiro. E também deve ser notado que as
cadeias polipeptídicas recém-formadas passa através da membrana do
retículo endoplasmático para atingir sua matriz.
Todavia, também deve ser notado que, exceto em células
glandulares, formadoras de grande número de vesículas secretórias
contendo proteínas, a maioria das proteínas formadas nos ribossomas é
liberada diretamente no citosol. Essas são as enzimas e as proteínas
estruturais da célula.
Etapas químicas da síntese de proteínas. Algumas das reações
químicas que ocorrem durante a síntese de moléculas de proteína são
mostradas na Fig. 3.11. Essa figura mostra as reações representativas
para três aminoácidos distintos, AA2, AA2 e AA3. As etapas dessas
reações são as seguintes: (1) cada aminoácido é ativado por um
processo químico onde o ATP se combina com o aminoácido para
formar um complexo de monofosfato de adenostna com aminoácido,
rompendo duas ligações fosfato de alta energia; (2) o aminoácido
ativado, contendo excesso de energia, combina-se, então, com seu ARN
transportador especifico, para formar um complexo aminoácido-ARNt,
liberando, ao mesmo tempo, o monofosfato de adenosina; (3) o ARN
transportador, carreando o aminoácido complexado, entra, em seguida,
em contato com a molécula de ARN mensageiro no ribossoma, o que situa
seu aminoácido na seqüência correta para a formação da molécula de
proteína. Então, sob a influência da enzima peptidiltransferase, uma das
proteínas do ribossoma, são formadas ligações peptídicas entre os
aminoácidos sucessivos, o que, progressivamente, alonga a cadeia da
proteína. Essas etapas químicas exigem a energia de duas ligações
fosfato de alta energia, o que eleva para quatro o total de ligações
fosfato de alta energia necessárias à incorporação de um aminoácido
na cadeia de proteína. Dessa forma, a síntese de proteína é um dos
processos com maior consumo de energia da célula.
Ligação peptídica. Os aminoácidos sucessivos da cadeia de proteínas
são unidos entre si segundo a reação típica:
Nesta reação química, um radical hidroxila é removido da COOH
de um aminoácido, enquanto um hidrogênio é removido do radical NH;
do outro. O que é removido forma água e os dois sítios reativos dos
Fig. 3.10 Concepção artística da estrutura dos ribossomas e de sua relação funcional com o ARN mensageiro,
com o ARN transportador e com o retículo endoplasmático, durante a formação de moléculas de proteína.
(De Bloom e Fawcett: A Textbook of Histology. 10. ed.
Philadelphia, W.B SaundeTS Co., 1975.)
26
e o outro é chamado de regulação enzimática, responsável pelo controle
do nível da atividade das enzimas na célula.
REGULAÇÃO GENÉTICA
Fig. 3.11 Eventos químicos na formação de uma molécula de proteína.
dois aminoácidos sucessivos reagem entre si, do que resulta a formação
de uma só molécula. Esse processo é chamado de ligação peptídica.
SÍNTESE DE OUTRAS SUBSTÂNCIAS PELA CÉLULA
Os muitos milhares de enzimas protéicas formadas pelo mecanismo
descrito acima controlam, em essência, todas as demais reações químicas
que ocorrem nas células. Essas enzimas promovem a síntese de lipídios,
glicogênio, purinas, pirimidinas e centenas de outras substâncias. Muitos
desses processos sintéticos, relacionados ao metabolismo dos
carboidratos, lipídios e proteínas, são discutidos nos Caps. 67 a 69.
É por meio dessas diferentes substâncias que são realizadas muitas das
funções celulares.
CONTROLE DA FUNÇÃO GENÉTICA E DA ATIVIDADE
BIOQUÍMICA DAS CÉLULAS
Do que foi discutido até aqui, fica claro que os genes controlam
tanto o funcionamento físico como o químico das células. Contudo,
a ativação dos próprios genes também deve ser controlada; de outro
modo, algumas partes da célula poderiam crescer excessivamente ou
algumas reações químicas poderiam ocorrer de modo desmesurado,
podendo matar a célula. Felizmente, cada célula é dotada de potentes
mecanismos internos de controle por feedback que mantêm as diversas
operações funcionais da célula em ritmo e intensidade adequados entre
si. Para cada gene ou para cada pequeno grupo de genes (100.000 no
total) existe, pelo menos, um desses mecanismos de feedback.
Os menores vírus têm contribuído imensamente para a nossa
compreensão do funcionamento celular, por
terem dimensões
suficientemente pequenas para permitir que os biólogos elucidem quase
que os detalhes mais precisos de seu funcionamento, molécula a
molécula. Em um nível pouco mais alto, as bactérias também são
muito valiosas, em especial a bactéria Escherichia coli, muito
abundante nas fezes. A maior parte do que vai ser discutido nas
páginas que se seguem foi aprendida por experimentos com essas
formas mais inferiores de vida. Infelizmente, contudo, a célula nucleada é
tão complexa que só agora se está começando a compreender os
mecanismos especiais de controle que foram desenvolvidos por essa
forma mais elevada de vida. Para exemplificar a diferença de
complexidade entre a célula nucleada (chamada de eucarioto) e a
célula não-nucleada (chamada de procariota), basta mencionar que o
eucarioto do ser humano contém 1.000 vezes mais ADN que a bactéria
E. Coli.
Basicamente, existem dois métodos diferentes para o controle das
atividades bioquímicas da célula. Um deles é chamado de regulação
genética, responsável pelo controle das atividades dos próprios genes,
O opéron do procariota e seu controle da síntese bioquímica
— a função do "promotor". A síntese de produto bioquímico celular
exige, geralmente, uma série de reações, e cada uma dessas reações é
catalisada por enzima protéica especial. A formação de todas as enzimas
necessárias para os processos sintéticos é, muitas vezes, controlada por
uma seqüência de genes, localizados em série, um após outro, no
mesmo filamento de ADN cromossômico. Esse trecho do filamento de
ADN é denominado opéron, e os genes responsáveis pela formação da
enzima respectiva são chamados de genes estruturais. Na Fig. 3.12,
são mostrados três genes estruturais em um opéron e vê-se que eles
controlam a formação de três enzimas específicas, usadas em
determinado processo de síntese bioquímica.
Agora, deve ser notado na figura o segmento do filamento de ADN
designado promotor. É formado por uma série de nucleotídios que têm
afinidade específica pela ARN polimerase, como já discutido. A
polimerase deve fixar-se a esse promotor antes que possa percorrer o
filamento de ADN para sintetizar o ARN. Portanto, o promotor é o
elemento essencial na ativação do opéron.
Controle do opéron por "proteína repressora" - o "operador
repressor". Também deve ser notada na Fig. 3.12 a faixa adicional de
nucleotídios situada no meio do promotor. Essa região é chamada de
operador repressor porque uma proteína repressora pode fixar-se a ela
e impedir a fixação da ARN polimerase ao promotor, o que impede
a transcrição dos genes. Proteína reguladora desse tipo é chamada de
proteína repressora. Contudo, cada proteína reguladora repressora existe,
em geral, sob duas formas alostéricas, uma capaz de se prender ao
operador e reprimir a transcrição e outra que não se fixa. Isto é, por
exemplo, uma das formas pode ser uma proteína linear, enquanto a
outra pode ser dobrada no meio. Apenas uma dessas formas pode reprimir o operador. Por sua vez, diversas substâncias não-protéicas da célula,
como determinados metabólitos celulares, podem combinar-se com essa
proteína repressora, alterando sua forma. A substância que ao se
combinar com a proteína repressora modifica sua forma, fazendo-a
capaz de se combinar com o operador e, assim, de interromper a
transcrição, é chamada de substância repressora ou substância inibidora.
Por outro lado, a substância que ao se combinar com a proteína
repressora altera sua forma, tornando-a incapaz 6e se fixar ao operador,
é chamada de substância ativadora ou substância indutora, pois ela ativa
— ou induz — o processo da transcrição pela remoção da proteína
repressora.
Para ilustrar o controle da transcrição gênica por proteína repressora,
basta um exemplo, O sacarídio lactose não está, nas condições usuais,
disponível para a bactéria E.coli, como substrato alimentar. Por
conseguinte, geralmente a bactéria não vai sintetizar as enzimas
necessárias à degradação metabólica da lactose. Todavia, quando está
disponível, a lactose induz alteração conformacional alostérica em
proteína repressora, fazendo com que ela abandone sua fixação em
operador repressor
Fig. 3.12 Funcionamento do opéron no controle da biossíntese. Notar
que o produto sintetizado exerce feedback negativo; inibidor do
funcionamento do próprio opéron e, desse modo, controlando
automaticamente a concentração do produto sintetizado.
27
do opéron que transcreve para as enzimas metabólicas necessárias. Como
resultado, o opéron fica desreprimido e, dentro de poucos minutos,
as enzimas adequadas estão presentes na bactéria para produzir a
degradação da lactose. Em seguida, à medida que o teor de lactose na
célula começa a baixar, a intensidade da síntese enzimática começa a
diminuir até retornar ao nível adequado à disponibilidade inicial de
lactose. Assim, fica evidente a lógica da existência de tais sistemas
reguladores na célula.
Controle do opéron por uma "proteína ativadora" — o
"operador ativador". Note, agora, na Fig. 3.12, outro operador, chamado
de operador ativador, situado ao lado mas à frente do promotor. Quando
uma proteína reguladora se fixa a esse operador, ela ajuda a atrair a ARN
polimerase para o promotor, ativando. assim, o promotor. Por
conseguinte, uma proteína reguladora desse tipo é chamada de proteína
atiradora. O opéron pode ser ativado ou inibido, por meio do
operador ativador, por mecanismo exatamente oposto ao do controle
pelo operador repressor.
Controle por feedback negativo do opéron. Finalmente, deve ser
notado na Fig. 3.12 que a presença de quantidade crítica de um produto
sintetizado na célula pode provocar inibição, por feedback negativo,
do opéron responsável por sua síntese. Isso pode ocorrer por fazer com
que proteína reguladora repressora se fixe ao operador repressor ou
por fazer com que a proteína reguladora ativadora quebre sua ligação
com o operador ativador. Nos dois casos, o opéron fica inibido. Portanto,
uma vez que o produto necessário que é sintetizado atinja qualidade
suficientemente abundante, o opéron fica inativado. Por outro lado.
quando esse produto sintetizado é degradado na célula, com baixa de
sua concentração, o opéron volta a ficar ativo. Dessa forma, a
concentração desse produto é controlada automaticamente.
Outros mecanismos para o controle da transcrição pelo opéron.
Foram identificadas, com muita rapidez, nos últimos anos diversas
variantes do mecanismo básico de controle do opéron. Sem entrar em
detalhes, podemos enumerar algumas delas:
1. Um opéron é, muitas vezes, controlado por gene regulador
localizado em outro ponto do complexo genético do núcleo. Isto é, o
gene regulador causa a formação de proteína reguladora que, por sua
vez, atua como substância ativadora ou repressora, para controlar o
opéron.
2. Ocasionalmente, muitos e diferentes opérons são controlados,
ao mesmo tempo, pela mesma proteína reguladora. Em alguns casos,
a mesma proteína reguladora atua como ativadora para um opéron e
repressora para outro. Quando diversos opérons são controlados
simultaneamente dessa maneira, todos os opérons que atuam em
conjunto formam um reguton.
3. Alguns opérons são controlados não ao nível de seu ponto inicial
de transcrição no filamento de ADN, mas, pelo contrário, em ponto
mais adiante desse filamento. Por vezes, esse controle não ocorre no
próprio filamento de ADN, mas, sim, durante o processamento das
moléculas de ARN no núcleo, antes de serem liberadas no citoplasma;
ou, raramente, o controle pode ocorrer ao nível da tradução do ARN
pelos ribossomas.
4. Nos eucariotos, o ADN nuclear fica restrito a unidades estruturais
específicas, chamadas cromossomas. E, no interior de cada cromossoma,
o ADN ocorre enrolado em torno de pequenas proteínas, chamadas
historias, que, por sua vez, são ainda mais compactadas por outras
proteínas. Enquanto o ADN está nesse estado compactado, ele não
pode atuar para formar ARN. Contudo, múltiplos mecanismos de
controle estão sendo identificados, capazes de fazer com que trechos
selecionados do cromossoma sejam descompactados, região após
região, de modo a permitir a transcrição do ARN. Assim, no eucarioto,
são usadas ordens de controle ainda mais elevadas para o estabelecimento
da função celular adequada. Além disso, sinais vindos de fora da
célula, como alguns hormônios, podem ativar regiões cromossômicas
determinadas, o que produz o maquinário químico necessário para
funções específicas.
Devido à existência de até 100.000 genes diferentes em cada célula
humana, o grande número de modos como a atividade genética pode
ser controlada não é surpreendente. Os sistemas de controle genético
são especialmente importantes para a regulação das concentrações
intracelulares de aminoácidos, de derivados de aminoácidos e os
substratos intermediários do metabolismo dos carboidratos, lipídios e
proteínas.
CONTROLE DA ATIVIDADE ENZIMÁTICA
Além de controlarem o sistema regulador genético, algumas das
enzimas intracelulares podem ser, por sua vez, controladas por ativadores
ou inibidores intracelulares. Isso representa, portanto, uma segunda
categoria de mecanismos que permitem o controle das funções
bioquímicas celulares.
Inibição enzimática. Algumas das substâncias químicas formadas
nas células exercem efeito direto de feedback, ao inibirem os sistemas
enzimáticos que as sintetizam. Quase sempre, o produto sintetizado atua
sobre a primeira enzima da seqüência, e não nas enzimas subseqüentes,
em geral se fixando diretamente a essa enzima e provocando alteração
conformacional alostérica que a inativa. Pode ser facilmente reconhecida
a importância da inativação da primeira enzima: isso impede o acúmulo
de produtos intermediários que não vão ser utilizados.
Esse processo de inibição enzimática é outro exemplo de controle
por feedback negativo: é responsável pelo controle das concentrações
intracelulares de alguns aminoácidos, purinas, pirimidinas, vitaminas e
outras substâncias.
Ativação enzimática. As enzimas que normalmente estão
inativas ou que foram inativadas por alguma substância inibidora
podem ser, muitas vezes, ativadas. Exemplo disso é a ação do
monofosfato de adenosina cíclico (AMPc) produzindo a clivagem do
glicogênio, com liberação de moléculas de glicose, para formar ATP rico
em energia, como discutido no capítulo anterior. Quando a célula é
depletada da maior parte de seu ATP, começa a ser formada grande
quantidade de AMPc, como produto da degradação do ATP; a
presença desse AMPc indica que as reservas celulares de ATP
caíram a níveis muito baixos. Todavia, o AMPc ativa imediatamente
a enzima degradadora do glicogênio, a fosforilase, liberando moléculas
de glicose que são metabolizadas com muita rapidez, e sua energia é
usada para a restauração das reservas de ATP. Assim, nesse caso, o
AMPc atua como ativador enzimático e, por conseguinte, ajuda a
regular a concentração intracelular de ATP.
Outro exemplo interessante de ativação e de inibição enzimática
ocorre na formação das purinas e das pirimidinas. Essas substâncias
são demandadas pela célula, em quantidades aproximadamente iguais,
para a síntese de ADN e de ARN. Quando as purinas são formadas,
elas inibem as enzimas necessárias à formação adicional de purinas.
Todavia, elas ativam as enzimas que vão participar da formação de pirimidinas Inversamente, as pirimidinas inibem as enzimas necessárias à sua
própria formação, enquanto ativam as enzimas para formação de purinas.
Desse modo, existe contínua interação cruzada entre os sistemas de
síntese para essas duas substâncias, do que resultam quantidades quase
iguais das duas, a qualquer momento, nas células.
Para resumir, existem dois meios principais para a célula regular
as proporções e concentrações adequadas dos diferentes constituintes
celulares: (1) o mecanismo de regulação genética, e (2) o mecanismo
da regulação enzimática Os genes tanto podem ser ativados como inibidos e, de igual modo, os sistemas enzimáticos também podem ser ativados
ou inibidos. Com maior freqüência, esses sistemas reguladores atuam
por meio de sistemas de controle por feedback que, continuamente,
monitorizam a composição bioquímica das células, efetuando as
correções que forem necessárias. Mas, por vezes, substâncias vindas de
fora da célula (em especial, alguns dos hormônios que serão discutidos
adiante, neste texto) também podem controlar as reações bioquímicas
intracelulares, por ativarem ou inibirem um ou mais sistemas de controle
intracelular.
REPRODUÇÃO CELULAR
A reprodução celular é outro exemplo do papel difuso e ubíquo
que o sistema genético do ADN desempenha em todos os processos
vitais. Os genes e seus mecanismos reguladores determinam as
características de crescimento de todas as células e, também, se e quando
essas células se dividirão para formar novas células. Desse modo, o todo
importante sistema genético controla cada etapa do desenvolvimento
do ser humano, desde a célula única do óvulo fertilizado até o corpo
totalmente funcionante. Assim, se é que existe um tema central para a
vida, ele é o sistema genético do ADN.
O ciclo vital da célula. O ciclo vital de uma célula é o período
de tempo que vai de uma reprodução celular até a seguinte. Quando
as células de mamíferos não estão inibidas e se reproduzindo tão
rapidamente quanto podem, esse ciclo vital dura de 10 a 30 horas. E
terminado por uma série de eventos físicos distintivos, chamada mitose,
do que resulta a divisão dessa célula em duas novas células filhas. Os
eventos (ou etapas) da mitose estão representados na Fig. 3.13 e serão
descritos adiante.
A verdadeira fase de mitose, contudo, só dura 30 minutos,
28
Fig. 3.13 Etapas da reprodução celular. A, B e C, prófase; D,
prometáfase; E, metáfase; F, anáfase; Ge H, teldfase. (Redesenhadode
Mazia: How celta divide. Sei. Amer., 205:102, 1961. Copyright by
Scientific American Inc. Todos os direitos reservados.)
de modo que mais de 95% do ciclo vital, mesmo de células com
reprodução rápida, são representados pelo intervalo entre mitoses
sucessivas, e que é chamado de interfase. Na verdade, exceto em
condições especiais de reprodução celular acelerada, fatores inibitórios
inibem ou interrompem, quase sempre, o ciclo vital desinibido da
célula. Por conseguinte, os ciclos vitais das diferentes células do corpo
têm durações que variam entre o mínimo de 10 horas, para as células
estimuladas da medula óssea, até o máximo de toda a sobrevida do
corpo humano, para as células nervosas e musculares estriadas.
REPLICAÇÃO DO ADN, A ETAPA INICIAL
DA REPRODUÇÃO CELULAR
Como ocorre para quase todos os eventos importantes da célula,
a reprodução começa no próprio núcleo. A primeira etapa é a replicação
(duplicação) de todo o ADN nos cromossomas. Só após isso ter ocorrido
é que pode ter início a mitose.
O ADN começa a ser duplicado cerca de 5 a 10 horas antes da
mitose e termina dentro de 4 a 8 horas. O ADN só é duplicado uma
vez, de modo que o resultado final é a formação de duas réplicas precisas
de todo o ADN. Essas réplicas, por sua vez, vão ser o ADN das duas
células filhas que vão ser formadas na mitose. Após a replicação do
ADN, existe um período de 1 a 2 horas, antes que, abruptamente,
comece a mitose. Contudo, mesmo durante esse breve período, já
começam a ocorrer alterações preliminares que vão levar à mitose.
Eventos químicos e físicos da replicação do ADN. O ADN é
replicado de modo quase análogo à transcrição do ARN, a partir do
ADN, exceto por algumas diferenças importantes:
1. Os dois filamentos de ADN de cada cromossoma são replicados,
e não apenas um deles.
2. Os dois filamentos inteiros da hélice de ADN são replicados
de uma ponta a outra e não apenas pequenos trechos de cada um
como ocorre na transcrição de ARN pelos genes.
3. As principais enzimas para a replicação do ADN são um complexo
enzimático, chamado ADNpolimerase, que é comparável à ARN polime-
rase. Ele se fixa ao filamento molde de ADN e se desloca ao longo
dele, enquanto outra enzima, ADN ligase, produz ligação entre os
sucessivos nucleotídios entre si, usando ligações fosfato de alta energia
para energizar essas ligações.
4. A formação de cada novo filamento de ADN ocorre, a um só
tempo, em centenas de segmentos ao longo dos dois filamentos da hélice
até que todo o filamento seja replicado. Então, as extremidades das
subunidades são unidas entre si pela enzima ADN ligase.
5. Cada filamento recém-formado de ADN permanece fixado, por
pontes de hidrogênio fracas, ao filamento original de ADN, que foi
usado como molde. Por conseguinte, são formadas duas novas hélices
de ADN que são cópias exatas uma da outra e que ainda permanecem
enroladas entre si.
6. Dado que as hélices de ADN em cada cromossoma têm cerca
de 6 cm de comprimento e apresentam milhões de voltas em cada hélice,
seria impossível que as duas novas hélices de ADN que foram formadas
se desenrolassem uma da outra, sem a assistência de mecanismo especial.
Esse mecanismo depende de enzimas que, a determinados intervalos
cortam a hélice longitudinalmente, produzem a rotação de cada
segmento, o suficiente para provocar a separação e, em seguida,
reformam
a hélice. Desse modo, as duas novas hélices são desenroladas.
Reparo e "revisão" do ADN. Durante o período de 1 hora, ou
pouco mais, entre a replicação do ADN e o começo da mitose, ocorre
período muito ativo de reparo e "revisão" dos filamentos de ADN.
Isto é, onde quer que nucleotídios inadequados tenham sido unidos
a nucleotídios do filamento molde original, enzimas especiais removem
a área defeituosa e a substituem por nucleotídios complementares
corretos. Isso é efetuado pelas mesmas ADN polimerases e ADN
ligases que foram usadas no processo de replicação. Esse processo
de reparo é chamado de revisão do ADN.
Devido a esse reparo e revisão, o processo de transcrição quase
nunca comete erros. Quando ocorre erro, ele é chamado de mutação;
o que causará, por sua vez, a formação de proteína anormal pela célula,
muitas vezes resultando em funcionamento irregular da célula e, por
vezes, até em morte celular. Contudo, devido à precisão do processo
de transcrição, já foi calculado que cada gene humano sofre mutação
uma vez a cada 200.000 anos de vida humana. Não obstante, quando
se pensa que existem 100.000 ou mais genes no genoma humano e que
o período entre duas gerações sucessivas é de cerca de 30 anos, ainda
poderiam ser esperadas até 10 mutações na passagem do genoma do
genitor a seu filho. Felizmente, todavia, cada genoma humano é representado por dois conjuntos distintos de cromossomas com genes quase
idênticos, de modo que um gene funcional de cada par está, quase sempre,
disponível para a criança, apesar das mutações.
OS CROMOSSOMAS E SUA REPLICAÇÃO
As hélices de ADN no núcleo ficam contidas nos cromossomas.
A célula humana contém 46 cromossomas dispostos em 23 pares. A
maior parte dos genes nos dois cromossomas de cada par são idênticos
ou quase idênticos entre si, de modo que é dito que, em geral, os
diferentes genes também existem aos pares, embora, por vezes, isso não
aconteça.
No cromossoma, além do ADN, também existe grande quantidade
de proteínas, grande parte delas sendo historias, moléculas pequenas
com carga positiva. As histonas se dispõem em grande número de
estruturas, em forma de bobinas, na parte central do cromossoma. Os
segmentos sucessivos de cada hélice de ADN se enroscam,
seqüencialmente, em torno dessas bobinas. Então, durante a mitose,
essas bobinas sucessivas são empurradas, umas contra as outras, o que
permite que a molécula de ADN, extremamente longa — com
comprimento linear de 6 cm e peso molecular de cerca de 60 bilhões
—, possa assumir a forma enrolada e dobrada do cromossoma mitótico,
com comprimento de apenas alguns micrômetros, 1/10.000 do
comprimento do ADN desenrolado.
Os núcleos de histona têm provavelmente papel importante na
regulação da atividade do ADN, visto que, enquanto o ADN estiver
densamente enrolado, não pode funcionar como molde para a formação
de ARN ou para a replicação de novo ADN. Ainda mais, algumas das
proteínas reguladoras são capazes de descondensar o enrolamento do
ADN nas histonas e permitir que pequenos segmentos formem, a cada
vez, o ARN. Assim, essa é uma ordem mais superior de regulação
do que os tipos que foram discutidos antes.
Algumas proteínas não-histonas também são componentes impor-
29
tantes dos cromossomas, funcionando como proteínas estruturais
cromossômicas e, em relação ao maquinário da regulação genética, como
ativadoras, inibidoras e enzimas.
A replicação dos cromossomas, em sua totalidade, ocorre durante
poucos minutos, imediatamente após a replicação das hélices de ADN;
as novas hélices de ADN captam novas moléculas de proteína, à medida
que forem necessárias. Nessa etapa, os dois cromossomas recém
formados são chamados de cromátides. Eles permanecem
temporariamente unidos entre si (até o momento da mitose) no ponto
chamado de centro-mero, localizado próximo ao centro de cada
cromátide.
MITOSE
O processo pelo qual a célula se divide em duas novas células é
chamado de micose. Desde que cada cromossoma tenha sido replicado
para formar dois cromátides, a mitose ocorre, automaticamente, em
cerca de 1 ou 2 horas.
O aparelho mitótico. Um dos primeiros eventos da mitose ocorre
no citoplasma, durante a fase final da interfase na fase inicial da prófase.
nas ou perto das pequenas estruturas chamadas de centríolos. Como
mostrado na Fig. 3.13, dois pares de centríolos ficam próximos um do
outro, perto de um dos pólos do núcleo. (Esses centríolos, como o
ADN e os cromossomas, foram replicados na interfase, em geral pouco
antes da replicação do ADN.) Cada centríolo é estrutura cilíndrica e
pequena, com cerca de 0,4 fim de comprimento e diâmetro de 0,15
fjm, formada principalmente por nove estruturas tubulares paralelas,
dispostas em forma de cilindro. Em cada par, os centríolos ficam em
ângulo reto entre si.
Pouco antes do início da mitose, os dois pares de centríolos começam
a se afastar. Isso decorre da polimerização sucessiva da proteína dos
microtúbulos, o que os faz crescer entre os dois pares de centríolos,
do que resulta seu afastamento. Ao mesmo tempo, outros microtúbulos
crescem radialmente a partir de cada par de centríolos, formando uma
estrela cheia de pontas, denominadas áster, em cada extremidade da
célula. Algumas dessas pontas, ou espinhas, penetram no núcleo e
participam na separação dos dois conjuntos de cromátides durante a
mitose. O complexo de microtúbulos unindo os dois pares de centríolos é
chamado de fuso, e todo o conjunto de microtúbulos, mais os dois pares
de centríolos, constitui o aparelho mitótico.
Prófase. A primeira etapa da mitose, denominada próftue, é mostrada na Fig. 3.13.A, B e C. Enquanto o fuso esta se formando, os
cromossomas do núcleo, que na interfase são compostos de filamentos
frouxamente enrolados, condensam-se em cromossomas bem-definidos.
Prometáfase. Durante essa etapa (Fig. 3.13D), o envelope
nuclear se rompe. Ao mesmo tempo, um novo conjunto de microtúbulos
começa a crescer para fora, a partir de pequena região condensada de
cada cromátide, chamada de cinetócoro, situada na face externa do
centrômero, região de união dos dois cromátides. Esses novos
microtúbulos, por sua vez, fixam-se ou interagem com os microtúbulos
dos dois ásteres do aparelho mitótico, com um cromátide se fixando
ao áster de uma das extremidades celulares, enquanto o outro se fixa ao
áster da extremidade oposta.
Metáfase. Durante a metáfase (Fig. 3.13E), os dois ásteres do
aparelho mitótico são ainda mais afastados pelo crescimento adicional
do fuso mitótico. Simultaneamente, os cromátides são intensamente
tracionados, pelos microtúbulos fixados a eles, para o centro preciso da
célula, onde se alinham para formar a placa equatorial do fuso mitótico.
Anáfase. Durante essa fase (Fig. 3.13F), os dois cromátides de cada
cromossoma são afastados um do outro ao nível do centrômero. O modo
preciso de como isso é realizado pelo sistema microtubular ainda não
é conhecido; contudo, sabe-se que os microtúbulos contêm actina, além
de tubulina; a actina é uma das proteínas contrateis do músculo. Por
conseguinte, foi presumido que os microtúbulos poderiam se contrair
ou que os túbulos cromossômicos poderiam interagir de forma deslizante
com os microtúbulos do áster, gerando a forma de tração.
Independentemente do mecanismo, todos os 46 pares de cromátides são
separados, formando dois conjuntos distintos de 46 cromossomas filhos.
Um desses conjuntos é tracionado em direção a um dos ásteres
mitóticos e o outro em direção ao áster do pólo oposto da célula em
divisão.
Telófase. Na telófase (Fig. 3.13Ge H), os dois conjuntos de cromossomas filhos já estão completamente separados. Em seguida, o aparelho
mitótico se dissolve e nova membrana nuclear se forma em torno de
cada conjunto de cromossomas; essa membrana se origina de partes
do retículo endoplasmático já presentes no citoplasma. Pouco
depois, a célula se divide em duas, na região entre os dois novos
núcleos. Isso é causado por um anel contrátil de microfilamentos
(formados por actina e, provavelmente, por miosina, as duas proteínas
contrateis do músculo) que se forma na junção das duas células em
desenvolvimento e as separa.
CONTROLE DO CRESCIMENTO E DA
REPRODUÇÃO CELULARES
Todos sabemos que certas células crescem e se reproduzem, como,
por exemplo, as células hemopoéticas da medula óssea, as células das
camadas germinativas da pele e do epitélio do intestino. Contudo, muitas
outras células, tais como as musculares lisas, podem não se reproduzir
por muitos anos. Algumas células, como os neurônios e a maioria das
células musculares estriadas, não se reproduzem durante toda a vida
de uma pessoa.
Em determinados tecidos, a falta, ou número insuficiente, de alguns
tipos de células faz com que essas células cresçam c se reproduzam
muito rapidamente até que seu número volte a ser o apropriado. Por
exemplo, sete oitavos do fígado podem ser removidos cirurgicamente,
e as células do oitavo remanescente irão crescer e se reproduzir até
que a massa hepática retorne praticamente ao normal. O mesmo acontece
com quase todas as células glandulares, com o epitélio intestinal, células
da medula óssea, tecido subcutâneo e quase que qualquer outro tecido,
exceto para células muito diferenciadas, como as nervosas e musculares.
Sabe-se muito pouco sobre os mecanismos que mantêm números
adequados dos diferentes tipos de células do corpo. Contudo,
experimentos já demonstraram três modos como o crescimento pode ser
controlado. Primeiro, o crescimento, muitas vezes, é controlado por
fatores de crescimento, produzidos em outras partes do corpo. Alguns
desses fatores circulam no sangue, mas outros se originam em tecidos
adjacentes. Por exemplo, as células epiteliais de diversas glândulas,
como as do pâncreas, não crescem quando falta um fator de crescimento,
produzido pelo tecido conjuntivo subjacente da glândula. Segundo, a
maior parte das células normais pára de crescer quando elas deixam de
ter espaço para tal. Isso ocorre quando as células são mantidas em cultura
de tecidos; as células crescem até entrarem em contato com objeto
sólido, quando cessa o crescimento. Terceiro, muitas vezes, as células
mantidas em cultura de tecidos param de crescer quando quantidades
diminutas de suas próprias secreções são deixadas acumular no seu
meio de cultura. Isso também poderia representar mecanismo de
feedback negativo para o controle do crescimento.
Regulação das dimensões celulares. As dimensões celulares são
reguladas quase que inteiramente pela quantidade de ADN funcional
no núcleo. Se não ocorrer replicação do ADN, a célula cresce até
determinado tamanho e, após esse tamanho, não mais se altera. Por
outro lado, é possível, pelo uso da substância colchicina, impedir a
formação do fuso mitótico e, portanto, a mitose, embora continue a
replicação do ADN. Nesse caso, o núcleo passa a conter maior
quantidade de ADN que a normal c a célula cresce ate dimensões
proporcionalmente maiores. Admite-se que isso resulte, simplesmente, da
produção aumentada de ARN e de proteínas celulares, o que, por sua
vez, faria com que a célula crescesse até maior tamanho.
DIFERENCIAÇÃO CELULAR
Característica especial do crescimento e da divisão celular é a da
diferenciação celular, o que implica alteração das propriedades físicas
e funcionais das células, à medida que proliferam no embrião, para
formar as diferentes estruturas corporais.
A primeira — e a mais simples — teoria que buscava explicar a
diferenciação foi a de que a composição genética do núcleo sofreria
modificações, ao correr das gerações sucessivas de células, de tal modo
que uma célula filha herdasse um conjunto distinto de genes do que
o recebido pela outra célula filha.
Todavia, essa teoria mostrou-se errônea em muitos aspectos, mas,
de forma especialmente ilustrativa, pelo simples experimento seguinte.
O núcleo de célula da mucosa intestinal de rã, quando transplantado
em óvulo (também de rã) cujo núcleo original havia sido previamente
removido, pode, muitas vezes, levar ã formação de rã inteiramente
normal.
Isso demonstra que até mesmo a célula da mucosa intestinal, que
é célula relativamente bem-diferenciada, ainda contém toda a informação
30
genética necessária para o desenvolvimento de todas as estruturas
necessárias do corpo da rã.
Por conseguinte, ficou claro que a diferenciação resulta não de perda
de genes, mas, sim, da repressão seletiva de diferentes opérons genéticos.
Na verdade, micrografias eletrônicas sugerem que alguns segmentos das
hélices de ADN, enroladas em torno dos núcleos de histona, ficam tão
condensados que não mais podem desenroscar-se para formar moléculas
de ARN. Uma sugestão para a causa desse efeito é a seguinte: suponha-se
que um gene regulador no genoma comece, em determinado estágio
da diferenciação celular, a produzir proteína reguladora que produza
ativação por feedback positivo, desse mesmo gene regulador. Esse feedback positivo causaria a produção continuada dessa proteína, que, daí
para diante, seria produzida permanentemente; mas essa proteína
reguladora reprimiria outro grupo selecionado de genes. Como
resultado, os genes reprimidos nunca voltariam a funcionar.
Independentemente do mecanismo, a maioria das células adultas do
corpo humano produz entre 8.000 e 10.000 proteínas, em vez das
100.000 ou mais que, potencialmente, poderiam ser produzidas caso
todos os genes estivessem ativos.
Experimentos embriológicos também demonstram que
determinadas células do embrião controlam a diferenciação das células
adjacentes. Por exemplo, o cordamesoderma primordial é chamado de
organizador primário do embrião, por representar um foco em torno do
qual se desenvolve o resto do embrião, Ele se diferencia no eixo
mesodérmico, contendo os somitas, com disposição segmentar, e, como
resultado de induções nos tecidos circundantes, determina a formação de
praticamente todos os órgãos do corpo.
Outro exemplo de indução ocorre quando as vesículas ópticas em
desenvolvimento entram em contato com o ectoderma da cabeça, fazendo
com que se espesse — para formar a placa do cristalino — e se dobre
para dentro, dando origem ao cristalino do olho. Conseqüentemente,
grande parte do embrião se desenvolve à custa dessas induções, uma
parte do corpo agindo sobre outra e esta, por sua vez, aluando ainda
sobre outras.
Desse modo, embora nossa compreensão da diferenciação celular
ainda seja muito grosseira, conhecemos muitos mecanismos distintos
de controle pelos quais essa diferenciação poderia ocorrer.
CÂNCER
O câncer é causado em todos (ou quase todos) os casos por mutação
ou por ativação anormal de genes celulares que controlam o crescimento
e a mitose celular. Esses genes anormais são chamados de oncogenes.
Apenas fração diminuta das células do corpo que sofreram mutações
leva ao câncer. Existem diversas razões para isso. Primeiro, a maioria
das células mutantes tem menor capacidade de sobrevivência que as
células normais e, como resultado, simplesmente, elas morrem. Segundo,
apenas algumas das células mutantes que sobreviveram perdem os
controles normais de feedback que impedem o crescimento excessivo.
Terceiro, as células que são potencialmente cancerígenas são, com grande
freqüência, destruídas pelo sistema imune do corpo, antes que possam
formar um câncer. Isso ocorre do seguinte modo: a maioria das células
mutantes produz proteínas anormais em seus corpos celulares, devido a
seus genes alterados, e essas proteínas estimulam o sistema imune do
corpo, fazendo com que ele produza anticorpos ou linfócitos
sensibilizados contra as células cancerígenas, e assim as destrua. A
confirmação dessa explicação é dada pelo fato de que as pessoas cujo
sistema imune foi suprimido, como, por exemplo, as tratadas com
imunossupressores, após transplante de rim ou de coração, têm
probabilidade várias vezes maior de desenvolver câncer.
Mas, o que causa os genes alterados? Quando se leva em conta
que muitos trilhões de novas células são formadas anualmente, em cada
corpo humano, essa pergunta poderia ser melhor formulada do modo
seguinte: Por que nós não desenvolvemos literalmente milhões ou bilhões
de células cancerígenas? A resposta é dada pela incrível precisão com
que os filamentos de ADN cromossômico são replicados em cada célula
antes da mitose e, também, devido ao processo de "revisão" que corta
e repara qualquer filamento anormal de ADN antes que o processo
mitótico seja deixado prosseguir. Contudo, apesar de todas essas
precauções, é provável que uma célula recém-formada em alguns poucos
milhões de células seja portadora de características mutantes
significativas.
Dessa forma, é necessário apenas o acaso para que ocorram
mutações, de modo que se pode supor que grande número de cânceres
seja simplesmente resultado de uma infeliz ocorrência.
Todavia, a probabilidade de ocorrência de mutações pode ser
aumentada de muitas vezes quando a pessoa é exposta a determinados
fatores químicos, físicos ou biológicos. Alguns deles são os seguintes:
1. E bem conhecido que a radiação ionizante, tal como raios X,
raios gama e partículas irradiadas por substâncias radioativas, e até
mesmo a radiação ultravioleta podem predispor ao câncer. Os íons
formados nas células teciduais, por efeito dessas radiações, são muito
reativos e podem romper os filamentos de ADN, causando, assim,
muitas mutações.
2. Compostos químicos de certos tipos também apresentam muita
propensão para causar mutações. Historicamente, foi descoberto, há
muito tempo, que diversos derivados dos corantes de anilina apresentam
elevada probabilidade de causar câncer, de modo que os operários da
indústria química produtora desses compostos, caso não sejam
protegidos, têm predisposição especial para o câncer. As substâncias
químicas capazes de produzir mutações são denominadas carcinogênicas.
Os carcinógenos que, de longe, causam o maior número de mortes
em nossa sociedade atual são os presentes na fumaça dos cigarros.
Eles causam cerca de um quarto de todas as mortes por câncer.
3. Os irritantes físicos também podem causar câncer, como, por
exemplo, a abrasão continuada do revestimento do tubo digestivo por
alguns tipos de alimento. A lesão dos tecidos provoca a rápida
substituição mitótica das células. Quanto mais rápidas forem as mitoses,
maior
será a probabilidade de mutações.
4. Em muitas famílias ocorre forte tendência hereditária para o
câncer. Provavelmente, isso resulta do fato de que a maioria dos
cânceres
depende de mais de uma mutação, antes que o câncer se forme. Nas
famílias especialmente predispostas ao câncer, presume-se que um ou
mais genes do genoma herdado já sofreram mutações. Portanto, número
bem menor de mutações adicionais deve ocorrer nessas pessoas antes
que um câncer comece a crescer.
5. Em animais de experimentação, certos tipos de vírus podem
causar determinados tipos de câncer, inclusive leucemia.
Ocasionalmente, isso pode ocorrer por um de dois modos. Primeiro,
no caso dos vírus de ADN, o filamento de ADN no vírus pode
inserir-se diretamente em um dos cromossomas e, assim, causar a
mutação que leva ao câncer. No caso dos vírus de ARN, alguns deles são
portadores da enzima transcriptase reversa, que permite a transcrição
do ADN a partir do ARN. Em seguida, o ADN transcrito se insere no
genoma das células do animal, levando ao câncer. Contudo, apesar da
demonstração de que o câncer virótico pode ocorrer em animais,
ainda não foi comprovado que o câncer se propaga por esse
mecanismo nos seres humanos, nem que o câncer seja contagioso,
passando de uma pessoa a outra.
Característica invasiva da célula cancerosa. As três diferenças
principais entre uma célula normal e outra cancerosa são: (1) A célula
cancerosa não respeita os limites normais do crescimento celular; a
razão disso é que as células cancerosas não necessitam dos fatores de
crescimento, como ocorre para as células normais. (2) As células
cancerosas são muito menos aderentes entre si que as células normais.
Como resultado, tendem a vagar através dos tecidos, a entrar na circulação
e a serem transportadas para todo o corpo, onde formam ninhos para
novos e numerosos crescimentos cancerosos. (3) Alguns cânceres são
capazes de produzir fatores angiogênicos que produzem o crescimento
de vasos sanguíneos para e no câncer, o que garante o fornecimento de
nutrientes necessários para o crescimento do câncer.
Por que as células cancerosas matam? A resposta a essa
pergunta é, em geral, muito simples. O tecido canceroso compete com os
tecidos normais pelos nutrientes. Visto que as células cancerosas
continuam a proliferar indefinidamente, seu número se multiplicando
dia após dia, pode ser facilmente compreendido que as células
cancerosas, dentro de pouco tempo, exigirão toda a nutrição disponível
para o corpo. Como resultado, os tecidos normais, gradativamente
sofrem morte nutricional.
REFERÊNCIAS
Bojaj, M., and Blundell, T.: Evolution andthe tertiary strueture of proteins.
Annu. Rev. Biophys. Bioeng., 13:453, 1984. Reers, R. F.(ed):CellFusion.
GeneTratisfer and Transformation. New York, Raven Press, 1984.
Bownes, M.: Differentiation of Cells. New York, Methuen, Inc., 19&5.
Bradshaw, R. A., and Prentia, S.: Oncogenes and Growth Factors. New York,
Elsevier Science PubliehkigCo., 1987. Bryant, P. J., and Simpson, P.:
Inthnsic and extrinaic control of growth in developing organs. Q. Rev.
Biol., 59:387, 1984.
31
Busch, H. (ed.}; The Cell Nucleus, Nuclear Particles. New York, Academic
Press, 1981. Cohn, W. E. (ed.): Progress in Nucleic Acid Reaearch and
Molecular Biology.
DNA: Multiproteln Interactions. New York, Academic Press, 1981.
Davidson, R.: Somatic Cell Genética. New York, Van Nostrand Reinhold,
1984. DeRerondo, A. M. {ed.): New Approaches in Eukaryotic DNA
Replication.
New York, Plenum Publishing Corp., 1983. Echols, H.: Multiple DNAprotein interactions governing high-precision DNA transactinns. Science,
233:1050, 1986.
Fojt, J. L.: Conteraplating the human genome. BioScience, 37:457, 1987.
Garvey, E. P., and Santi, D. V.: Stable amplified DNA in drug-resistant
Leishmania exista as extrachrnmosomal circles. Science, 233:535, 1986. Go,
N.: Theoretical studies of protein folding. Annu. Rev. Biophys. Bioeng.,
12:183, 1983. Hall, A.: Oncngenes — implications for human câncer: A
review. J. R. Soe.
Med., 77:410, 1984. Horton, J. D. (ed.): Development and Differentiation of
VertebrateR. New
York, Elsevier/North-Holland, 1980. Hunt, T. (ed.): DNA Makes RNA
Makes Protein. New York, Elsevier
Science Publishing Co., 1983. Klevecz, R. R., et ai.: Cellular clocks and
oscillators. Int. Rev. CytoL, 86:97, 1984.
Kornberg, A.: DNA Replication. San Francisco, W. H. Freeman, 1980.
Kucherlapati, R., and Skoultchi, A. I.: Introduction of purified genes into
mammalian cells. CRC Crit. Rev. Biochem., 16:349, 1984. Kulaev, J. S.
(ed.}: Environmental Regulationof Microbial Metabolism. New
York, Academic Press, 1985. Maçara, I. G.: Oncogenes and cellular signal
transduetion. Phyaiol. Rev., 69:797, 1989. Margalit, H., et ai.: Initiation of
DNA replication in bactéria: Analysis of an autorepressor control model. J.
Theor. Biol., 111:183, 1984. Marx, J. L.: Zipping up DNA binding proteins.
Science, 240:1732, 1988.
Nomura, M., et ai.: Regulation of the Bynthesie of riboaomes and ribosomal
components. Annu. Rev. Biochem., 53:75, 1984. Nora, J. J., and Fraser, F.
C: Medicai Genetics: Principies and Practice. 3rd
Ed. Philadelphia, Lea & Febiger, 1989. Ohiendnrf, D. H., and Matthews, B.
W.: Structural studies of protein-nucleic acid interactions. Annu. Rev.
Biophys. Bioeng., 12:259, 1983. Orcutt, B. D., et ai.: Protein and nucleic
acid sequence database systems.
Annu. Rev. Biophys. Bioeng., 12:419, 1983.
Pabo, C. O., and Sauer. R. T.: Prntein-DNA recognition. Annu. Rev. Biochem., 53:293, 1984. Raff.E.C: Genetics ofmicrotubule systems. J. Cell.
Biol., 99:1, 1984. Rigler, R., and Wintermeyer, W.: Dynamics of tRNA.
Annu. Rev. Biophys.
Bioeng., 12:475, 1983. Schimke, R. T.: Gene amplification in cultured
animal cells. Cell, 37:705,1984.
Schleif, R.: DNA looping. Science, 240:127, 1988. Schlesinger, D. H. (ed.):
Macromolecular Sequencing and Synthesis: Selectèd Methods and
Applications. New York, Alan R. Liss, Inc., 1988. Schopf, J. W.: The
evolution of the earliest cells. Sei. Am., 239:110, 1978. Scriver, C. R., et ai.
(eda.): TheMetabolicBaaiaof Inheríted Disease. 6thEd.
New York, McGraw Hill Book Co., 1989. Smith.H. S., et ai.: The biology
ofbreast câncer at the cellular levei. Biochem.
Biophys. Acta, 738:103, 19&4.
Stofiler, G., and Stoffler-Meilicke, M.: Immunoelectron microscopy of ribosomes. Annu. Rev. Biophys. Bioeng., 13:303, 1984. Thomas, K. A.:
Fibrobtast growth factors. FASEB J. 1:434, 1987. Thompson, M. W., et ai.:
Genetics in Medicine. 5th Ed. Philadelphia, W. B.
Saunders Co., 1989. Varmus, H. E.: Oncogenes and transcriptional control.
Science, 238:1337, 1987. Weiss, L., et ai.: Interactions of câncer cells wíth the
microvasculature during metastasis. FASEB J., 2:12,1988.
32
UNIDADE II
FISIOLOGIA DA MEMBRANA, DO NERVO
E DO MÚSCULO
Ø
Transporte de íons e de Moléculas Através da Membrana Celular
Ø Potenciais de Membrana e Potenciais de Ação
Ø Contração do Músculo Esquelético
Ø Excitação da Contração do Músculo Esquelético:
Ø Transmissão Neuromuscular e Acoplamento Excitação-Contração
Ø Contração e Excitação do Músculo Liso
33
CAPÍTULO 4
Transporte de Íons e de Moléculas Através da Membrana Celular
A Fig. 4.1 apresenta a composição aproximada do líquido
extracelular, situado por fora das membranas celulares, e do
líquido intracelular, que fica no interior das células. Note-se
que o líquido extracelular contém grandes quantidades de sódio,
mas apenas pequenas quantidades de potássio. Exatamente o
oposto ocorre no líquido intracelular. Ao mesmo tempo, o líquido
extracelular contém grande quantidade de cloreto, enquanto o
líquido intracelular só o tem em pequenas quantidades. Mas as
concentrações de fosfatos—em essência, todos são metabólitos
intermediários orgânicos — e de proteínas no líquido
intracelular são consideravelmente maiores que as do líquido
extracelular. Todas essas diferenças são extremamente
importantes para a vida da célula. O objetivo deste capítulo é o
de explicar como essas diferenças são produzidas pelos
mecanismos de transporte das membranas celulares.
A barreira lipídica e as proteínas de transporte da
membrana celular
A estrutura da membrana celular foi discutida no Cap. 2
e apresentada na Fig. 2.3. Ela é composta, quase que
inteiramente, da bicamada lipídica, com grande número de
moléculas de proteína flutuando no lipídio, muitas delas
atravessando toda a espessura dessa bicamada, como mostrado
na Fig. 4.2.
A bicamada lipídica não é miscível com os líquidos extra
e intracelular. Por conseguinte, ela representa barreira ao
movimento da maioria das moléculas de água e das substâncias
hidrossolúveis entre os compartimentos dos líquidos extra e
intracelulares. Contudo, como indicado pela seta à esquerda da
Fig. 4.2, algumas substâncias conseguem atravessar essa
bicamada, entrando na célula ou saindo dela, passando
diretamente pela substância lipídica.
Por outro lado, as moléculas de proteína apresentam
propriedades de transporte inteiramente diferentes. Suas
estruturas moleculares interrompem a continuidade da
bicamada lipídica e, portanto, formam via alternativa através
da membrana celular. A maioria dessas proteínas penetrantes é,
como resultado, formada por proteínas de transporte. As
diferentes proteínas vão atuar por modos distintos. Algumas
contêm espaços aquosos, ao longo de toda a sua molécula, e
permitem o livre movimento de determinados íons e moléculas;
são denominadas proteínas de canal.
Outras, chamadas de proteínas carreadoras, fixam-se às
substâncias que vão ser transportadas, e alterações
conformacionais dessas moléculas de proteína movem as
substâncias, ao longo dos interstícios da molécula, até o outro
lado da membrana.
Tanto as proteínas de canal como as proteínas carreadoras são
extremamente seletivas quanto ao tipo (ou tipos) de moléculas
ou íons que podem atravessar a membrana.
Difusão versus transporte ativo. O transporte através da
membrana celular, seja diretamente, pela bicamada lipídica, ou
por meio de proteínas, ocorre por um dos dois processos básicos,
a difusão (também chamada de "transporte passivo") e o
transporte ativo. Embora existam numerosas variantes distintas
desses dois processos básicos, como veremos adiante neste
capítulo, a difusão implica movimento molecular aleatório da
molécula da substância pelos espaços intermoleculares da
membrana ou em combinação com proteína carreadora. A
energia causadora da difusão é a energia do movimento cinético
normal da matéria. Pelo contrário, o transporte ativo implica o
movimento de íons ou outras substâncias, em combinação com
proteína carreadora, mas, além disso, contra um gradiente de
energia, como, por exemplo, de um estado de baixa
concentração para outro de alta concentração, processo que
exige outra fonte de energia além da cinética para que ocorra o
movimento. Vamos explicar em maiores detalhes a física e a
físico-química básicas desses dois processos distintos.
DIFUSÃO
Todas as moléculas e íons dos líquidos corporais, inclusive
tanto as moléculas de água como as das substâncias em solução,
estão continuamente em movimento, cada partícula seguindo
percurso próprio. O movimento dessas partículas constitui o que
os físicos chamam de calor — quanto mais intenso for essa
movimentação, maior será a temperatura — e esse movimento
nunca cessa, sob quaisquer condições, exceto na temperatura do
zero absoluto. Quando uma molécula em movimento, A, se
aproxima de outra molécula estacionária, B, as forças
eletrostáticas e internucleares da molécula A repelem a
molécula B, transferindo parte da energia do movimento para a
molécula B. Conseqüentemente, a molécula B ganha energia
cinética de movimento, ao mesmo tempo em que a molécula
A tem seu movimento lentificado, pois perdeu parte de sua
energia cinética. Assim, como mostrado na Fig. 4.3, cada
molécula de uma solução pula por entre as outras moléculas,
primeiro em determinada direção, em seguida em outra, e assim
por diante, aleatoriamente, bilhões de vezes a cada segundo.
34
Fig. 4.2 Vias de transporte através da membrana celular e os
mecanismos básicos de transporte.
lidade do oxigênio, do nitrogênio, do dióxido de carbono e dos
álcoois é muito alta, de modo que todos esses compostos são
capazes de se dissolver diretamente na bicamada lipídica e se
difundir através da membrana celular, de modo idêntico ao da
difusão em solução aquosa. Por motivos óbvios, a velocidade
Fig. 4.1 Composição química dos líquidos extras e
de difusão dessas substâncias através da membrana é diretamente
intracelulares.
proporcional às suas lipossolubilidades. Quantidades
Esse contínuo movimento de moléculas por entre as
extremamente grandes de oxigênio podem ser transportadas por
outras, nos líquidos e nos gases, é chamado difusão. Os íons se
difundem do mesmo modo como moléculas, e até mesmo esse modo; como resultado, o oxigênio chega ao interior da
partículas colóides em suspensão se difundem do mesmo modo, célula como se a membrana celular não existisse.
Transporte de água e de outras moléculas insolúveis em
exceto por sua difusão ocorrer bem mais lentamente que as lipídios. Embora a água seja extremamente insolúvel nos
substâncias moleculares, devido às suas grandes dimensões.
lipídios da membrana, ela, não obstante, atravessa facilmente a
membrana celular; em parte, ela passa, de modo direto, através
DIFUSÃO ATRAVÉS DA MEMBRANA CELULAR
da bicamada lipídica e, em sua maior parte, pelas proteínas de
A difusão através da membrana celular é dividida em dois canal. A rapidez com que a água pode atravessar a membrana
subtipos distintos, chamados de difusão simples e difusão celular é, na verdade, surpreendente. Como exemplo, a
facilitada. A difusão simples é o movimento cinético molecular quantidade total de água que se difunde, nas duas direções,
de moléculas ou íons através de pertuito da membrana ou dos através da membrana da hemácia, a cada segundo, é,
espaços intermoleculares, sem necessidade de fixação a aproximadamente, 100 vezes maior que o volume da hemácia.
A razão para a grande intensidade da difusão de água através
proteínas carreadoras da membrana. A velocidade dessa difusão é da bicamada lipídica ainda não foi determinada, mas acredita-se
determinada pela quantidade existente da substância, pela que as moléculas de água sejam suficientemente pequenas e que
velocidade do movimento cinético e pelo número de pertuitos da sua energia cinética seja grande o bastante para que elas possam,
membrana através dos quais a molécula ou íon pode passar. Por simplesmente, penetrar como projéteis na parte lipídica da
outro lado, a difusão facilitada implica a interação das membrana, antes que sua característica "hidrofóbica" consiga
moléculas ou íons com proteína carreadora que facilita sua detê-las.
Outras moléculas insolúveis em lipídios também podem
passagem através da membrana, provavelmente por se fixar
quimicamente a ela e se deslocar, através da membrana, nessa atravessar a bicamada lipídica do mesmo modo como a água,
desde que sejam suficientemente pequenas.
forma fixada.
A difusão simples pode ocorrer através da membrana por
dois percursos: pelos interstícios da bicamada lipídica ou pelos
canais aquosos de algumas proteínas de transporte, como
mostrado à esquerda da Fig. 4.2.
Difusão simples através da bicamada lipídica
Difusão de substâncias lipossolúveis. Em estudos
experimentais, os lipídios das células foram separados das
proteínas e, em seguida, reconstituídos, formando membranas
artificiais, constituídas por uma bicamada lipídica, sem qualquer
das proteínas de transporte. Por meio dessas membranas
artificiais, foram determinadas as propriedades de transporte das
bicamadas lipídicas.
Um dos fatores mais importantes que determinam com que
rapidez uma substância irá atravessar essa bicamada lipídica é
a lipossolubilidade da substância. Por exemplo, a lipossolubi-
Fig. 4.3 Difusão de uma molécula de um líquido, durante um
bilionésimo de segundo.
35
Todavia, à medida que suas dimensões aumentam, sua
capacidade de penetração cai acentuadamente. Por exemplo, o
diâmetro da molécula da uréia é apenas 20% maior que o da de
água. Contudo, sua penetração através da membrana celular é
cerca de mil vezes menor que a da água. Mesmo assim, tendose em mente a extraordinária velocidade de penetração da água,
essa velocidade ainda permite o transporte rápido da uréia
através da membrana celular. A molécula de glicose, com
diâmetro três vezes maior que o da molécula de água, atravessa a
bicamada lipídica com velocidade 100 mil vezes menor que a da
água, o que demonstra que as únicas moléculas insolúveis em
lipídios capazes de penetrar na bicamada lipídica são as de
menores dimensões.
Incapacidade de íons de se difundirem através da bicamada
lipídica. Muito embora a água e outras moléculas muito pequenas,
sem carga, possam difundir-se facilmente através da bicamada
lipídica, os íons — mesmo os mais pequenos, como os íons
hidrogênio, sódio, potássio e outros — só penetram na bicamada
lipídica com velocidades cerca de 1 milhão de vezes menores
que a da água. Por conseguinte, qualquer transporte
significativo desses íons através da membrana celular deve
ocorrer pelos canais nas proteínas, como será discutido mais
adiante. A razão para essa impenetrabilidade da bicamada
lipídica aos íons é a carga elétrica dos íons; ela impede o
movimento iônico por dois modos distintos: (1) a carga
elétrica dos íons faz com que várias moléculas de água se
prendam a esses íons, formando íons hidratados. Isso aumenta,
de muito, as dimensões dos íons, o que, por si só, impede a
penetração da bicamada lipídica; (2) o que é ainda mais
importante à carga elétrica do íon interage com as cargas da
bicamada lipídica do seguinte modo: deve ser lembrado que
cada metade da bicamada é formada por lipídios "polares",
portadores de excesso de cargas positivas, voltados para a
superfície da membrana; como resultado, quando um íon dotado
de carga tenta penetrar na barreira elétrica positiva Ou negativa,
ele é, instantaneamente, repelido. Para resumir, o Quadro 4.1
apresenta as permeabilidades relativas da bicamada lipídica a
diversos tipos de moléculas ou a íons de diferentes diâmetros.
Deve ser especialmente notada a diminuta permeância dos
íons, devida a suas cargas elétricas, e a fraca permeância da
glicose, devida a seu diâmetro molecular. Também deve ser
notado que o glicerol penetra na membrana com facilidade
quase igual à da uréia, embora seu diâmetro seja quase o dobro.
A razão disso é seu discreto grau de lipossolubilidade.
Portanto, as substâncias podem difundir-se diretamente, por
esses canais, de uma das faces da membrana até a outra.
Todavia, esses canais protéicos são distinguidos por duas
características importantes: (1) muitas vezes, eles são
seletivamente permeáveis a determinadas substâncias, e (2)
muitos desses canais podem ser abertos ou fechados por meio
de comportas.
Permeabilidade seletiva dos diferentes canais protéicos. A
maioria (mas não todos) dos canais protéicos é muito seletiva
para o transporte de um ou mais íons ou moléculas. Isso resulta
das características do próprio canal, tais como seu diâmetro,
sua forma e a natureza das cargas elétricas nas suas superfícies
internas. Como exemplo, um dos mais importantes canais
protéicos , o chamado canal de sódio, com diâmetro calculado de
apenas 0,3 por 0,5 nm, tem, em suas superfícies internas, fortes
cargas negativas, como representado pelos sinais de menos no
interior do canal protéico na parte superior da Fig. 4.4. Postulase que essas fortes cargas negativas atraiam os íons sódio, com
mais intensidade do que outros íons fisiologicamente importantes,
para o interior dos canais, devido ao menor diâmetro iônico do
sódio não-hidratado. Uma vez no interior do canal, os íons sódio
podem difundir-se em qualquer direção, segundo as leis da
difusão. Por conseguinte, o canal de sódio 6 especificamente
seletivo para a passagem dos íons sódio.
Por outro lado, outro grupo de canais protéicos é seletivo
para o transporte de potássio, como mostrado na parte inferior
da Fig. 4.4. Esses canais, com diâmetros calculados menores
que os dos canais de sódio, da ordem de 0,3 por 0,3 nm, não
contêm cargas negativas. Como resultado, não existem forças
atrativas fortes que puxem os íons para o interior dos canais,
e os íons não são retirados das moléculas de água que os hidratam.
A forma hidratada do íon potássio é muito menor que a forma
hidratada do íon sódio porque o íon sódio tem todo um conjunto
orbital de elétrons a menos que o íon potássio, o que permite
A difusão simples através dos canais das proteínas e as
"comportas" desses canais.
As proteínas de canais são consideradas como contendo pertuitos
aquosos pelos interstícios dessas moléculas protéicas. Na
verdade, a reconstrução tridimensional por computadores de
algumas dessas proteínas demonstrou a existência de canais,
em forma de tubos, que se estendem entre as duas
extremidades da molécula, nas faces extra e intracelular da
membrana.
Quadro 4.1 Relações entre os diâmetros efetivos das
diferentes substâncias para suas permeabilidades nas bicamadas
lipídicas.
Substância
Diâmetro
Molécula de água
0,3
Permeabilidade
Relativa
1,0
Molécula de uréia
0,36
0,006
Íon cloreto hidratado
0,386
0,00000001
Íon potássio hidratado
0,396
0,0000000006
Íon sódio hidratado
0,512
0,0000000002
Glicerol
0,62
0,0006
Glicerol
0,86
0,000009
Fig. 4.4 O transporte dos íons sódio e potássio pelos canais protéicos.
Também são mostradas as alterações conformacionais das
moléculas de proteína dos canais que abrem ou fecham as
"comportas" desses canais.
36
ao íon sódio atrair número bem maior de moléculas de água
do que o potássio. Por conseguinte, os íons hidratados de
potássio, menores, podem passar facilmente por esse canal mais
estreito, ao passo que os íons sódio são rejeitados, o que, de
novo, causa permeabilidade seletiva para um tipo de íon.
As comportas dos canais protéicos. A existência de comportas
nos canais protéicos representa meio de controle da
permeabilidade desses canais. Isso é mostrado nas partes
superior e inferior da Fig. 4.4, para os íons sódio e potássio.
Acredita-se que essas comportas sejam, efetivamente,
projeções em forma de comporta da molécula da proteína de
transporte, que podem ocluir a abertura do canal ou que podem
ser afastadas dessa abertura, como resultado de alteração
conformacional da forma da própria molécula protéica. Nos
canais de sódio, essa comporta abre e fecha na face externa da
membrana celular, enquanto, no canal de potássio, ela abre e
fecha na face interna.
A abertura e o fechamento das comportas são controlados
por dois modos principais:
1. Comportas voltagem-dependentes. Nesse mecanismo, a
conformação molecular da comporta depende do potencial
elétrico através da membrana celular. Por exemplo, quando
existe forte carga negativa no interior da membrana celular, os
canais de sódio permanecem fortemente fechados; por outro lado,
quando o interior da membrana celular perde sua carga
negativa, as comportas se abrem, permitindo a passagem de
quantidades imensas de sódio para o interior da célula, por
meio dos poros de sódio (até que outro grupo de comportas,
situadas nas extremidades citoplasmáticas dos canais, se feche,
como é explicado no Cap. 5). Essa é a causa básica dos
potenciais de ação dos nervos, responsáveis pelos sinais neurais.
As comportas de potássio também abrem quando o interior da
membrana celular fica carregado positivamente, mas essa
resposta é bem mais lenta que a das comportas de sódio. Esses
eventos são discutidos no capítulo seguinte.
2. Comportas ligando-dependentes. Algumas comportas dos
canais protéicos são abertas quando outra molécula se fixa à
proteína; isso produz alteração conformacional da molécula de
proteína que abre ou fecha a comporta. Elas são chamadas de
comportas ligando-dependentes, e a substância que se fixa à pro
teína é o ligando. Um dos exemplos mais importantes de com
portas ligando-dependentes é o efeito da acetilcolina sobre o
chamado canal de acetilcolina. Essa substância abre a comporta
desse canal, criando um poro com diâmetro de cerca de 0,65
nm que permite a passagem de todas as moléculas e íons positivos
com diâmetros menores que o do poro. Essa comporta é especial
mente importante na transmissão de sinais de uma célula nervosa
a outra (Cap. 45) e de uma célula nervosa à célula muscular
(Cap. 7).
O estado-aberto e o estado-fechado dos canais com
comportas. A Fig. 4.5 apresenta uma característica especialmente
importante dos canais com comportas voltagem-dependente.
Essa figura apresenta dois registros da corrente elétrica que
flui por canal de sódio isolado, quando existia gradiente de
potencial — de aproximadamente 25 milivolts — através da
membrana. Deve ser notado que o canal conduz a corrente de
modo tudo-ou-nada. Isto é, a comporta do canal se abre ou
fecha abruptamente, cada abertura ou fechamento ocorrendo
em poucos milionésimos de segundo. Isso demonstra a rapidez
com que podem ocorrer alterações conformacionais na forma
das comportas dos canais protéicos. Em determinado valor do
potencial, o canal pode permanecer fechado todo o tempo, ou
quase todo o tempo, enquanto em outro nível de voltagem ele
pode ficar aberto todo o tempo, ou quase todo o tempo.
Contudo, nas voltagens intermediárias, as comportas tendem a se
abrir e fechar intermitentemente, como mostrado no registro
superior, o que permite fluxo médio de corrente, entre o mínimo
e o máximo.
O método de fixação de placa para o registro do fluxo de
corrente iônica através de canais isolados. Pode-se questionar como é
tecnicamente possível o registro do fluxo de corrente iônica por canais
isolados, como mostrado na Fig. 4.5. Isso é conseguido pelo método de
"fixação de placas", representado na Fig. 4.5B. De modo muito simples,
uma micro-pipela, cuja ponta tenha diâmetro da ordem de 1 a 2 txm é
encostada na face externa da membrana celular. Em seguida, é feita
sucção pelo interior da pipeta, de modo a puxar a membrana
ligeiramente para o interior da ponta da pipeta. Isso cria um anel
de vedação na zona
Fig. 4.5 A, Registro do fluxo de corrente através de canal de sódio
voltagem-dependente, isolado, demonstrando o caráter de tudo-ou-nada
de abertura do canal. B, O método de "fixação de placa" para o registro
do fluxo de corrente através de canal protéico isolado; à esquerda, o
registro é feito em "placa" de membrana, ainda na célula; à direita,
o registro é feito em placa de membrana que foi removida da célula.
37
onde as bordas da pipeta entram em contato com a membrana celular.
O resultado é a formação de diminuta "placa" de membrana, através
da qual pode ser registrado o fluxo de corrente.
De modo alternativo, como mostrado à direita da Fig. 4.5B, a
pequena placa de membrana, na ponta da pipeta, pode ser removida da
célula. A pipeta com sua placa é, então, introduzida em solução salina.
Isso permite a alteração, conforme desejado, das concentrações tônicas
no interior da pipeta e na solução externa. Por outro lado, a
voltagem entre as duas faces da placa de membrana pode ser estabelecida
e mantida em qualquer valor — isto é, ela pode ser "fixada" em uma
determinada voltagem.
Felizmente, tem sido possível a obtenção de placas suficientemente
pequenas para que nelas só exista canal protéico único. Ao se variar
às concentrações dos diferentes íons e a voltagem através da membrana
celular, podem ser determinadas as características de transporte desse
canal, bem como as propriedades de suas comportas.
Difusão facilitada
A difusão facilitada também é chamada de difusão mediada
por carreador, porque uma substância transportada por esse
modo não é capaz, na maioria das vezes, de atravessar a
membrana, a não ser com a participação de proteína carreadora
específica. Isto é, o carreador facilita a difusão da substância
para o outro lado.
A difusão facilitada difere da difusão simples por um canal
aberto do seguinte modo, muito importante: embora a velocidade
da difusão por um canal aberto aumente na proporção direta
da concentração da substância difusora, na difusão facilitada à
velocidade da difusão tende a um máximo, denominado Vmáx
com o aumento da concentração da substância. Essa diferença
entre a difusão simples e a difusão facilitada é ilustrada na Fig.
4.6, mostrando que, à medida que a concentração da substância
aumenta, a velocidade da difusão simples continua a aumentar
proporcionalmente, mas também mostra a limitação da difusão
facilitada ao valor de Vmáx.
O que limita a velocidade da difusão facilitada? Uma
provável razão é o mecanismo representado na Fig. 4.7. Essa
figura mostra uma proteína carreadora com canal suficientemente
largo para transportar uma molécula específica até certo ponto,
mas não através de toda a membrana.
Fig. 4.6 Efeito da concentração de uma substância sobre a intensidade
de difusão, através de membrana, onde ocorre difusão simples, e através
de membrana onde ocorre difusão facilitada. Isso demonstra que a difusão facilitada tende a uma intensidade máxima, denominada VMÁX
Fig. 4.7 Um mecanismo proposto para a difusão facilitada.
Ela também mostra um "receptor" com capacidade fixadora nessa
proteína carreadora. A molécula que vai ser transportada entra
no canal e é fixada. Em seguida, dentro de fração de segundo,
ocorre alteração conformacional na proteína carreadora, de
modo que o canal passa a ficar aberto para o lado oposto da
membrana. Porque a força fixadora do receptor é fraca, o
movimento térmico da molécula fixada provoca sua liberação e
sua conseqüente liberação para o lado oposto. Obviamente, a
velocidade com que as moléculas podem ser transportadas por
esse mecanismo nunca pode ser maior que a velocidade com
que a molécula da proteína carreadora pode alternar-se, em seus
dois estados, por meio de alterações conformacionais. Note-se,
especialmente, que esse mecanismos permite que a molécula
transportada se "difunda" em ambas as direções através da
membrana.
Entre as mais importantes substâncias que atravessam a
membrana por difusão facilitada devem ser citadas a glicose e
a maioria dos aminoácidos. No caso da glicose, sabe-se que a
molécula carreadora tem peso molecular de cerca de 45.000;
ela é capaz de transportar vários outros monossacarídios com
estruturas semelhantes à da glicose, inclusive a manose, a
galactose, a xilose e a arabinose. Por outro lado, a insulina é
capaz de aumentar a velocidade da difusão facilitada por até 10
a 20 vezes. Esse é o principal mecanismo pelo qual a insulina
controla a utilização de glicose pelo corpo, como discutiremos
no Cap. 78.
FATORES QUE INFLUENCIAM A VELOCIDADE
EFETIVA DA DIFUSÃO
Neste ponto, já está evidente que muitas substâncias
diferentes podem difundir-se tanto através da bicamada lipídica
como por meio dos canais protéicos da membrana celular.
Contudo, deve ser claramente entendido que as substâncias que se
difundem em uma direção também podem fazê-lo na direção
oposta. Em geral, o que é importante para a célula não é a
quantidade total que se difunde nas duas direções, mas a diferença
entre as difusões nas duas direções, que é definida como a
velocidade efetiva da difusão em uma direção.
Os fatores que a influenciam são (1) a permeabilidade da
membrana, (2) a diferença de concentração da substância
difusora entre as duas faces da membrana, (3) a diferença
de pressão através da membrana, e (4) no caso dos íons, a
diferença de potencial elétrico entre as duas faces da
membrana.
38
Permeabilidade da membrana. A permeabilidade da
membrana para determinada substância é expressa como a
intensidade efetiva da difusão dessa substância, através de área
unitária da membrana, em função de diferença unitária de
concentração (quando não existem diferenças elétricas ou de
pressão). Os diversos fatores que influenciam a permeabilidade da
membrana celular são:
1. A espessura da membrana — quanto maior, mais lenta
será a difusão.
2. A lipossolubilidade — quanto maior for a solubilidade
da substância nos lipídios da membrana celular, maior será a
quantidade de substância que pode dissolver-se nessa membrana
e, conseqüentemente, que vai atravessá-la.
3. O número de canais protéicos pelos quais a substância
pode passar — a velocidade da difusão é diretamente
proporcional ao número de canais por unidades de área.
4. A temperatura — quanto maior for a temperatura, maior
vai ser o movimento térmico das moléculas e dos íons em solução,
de modo que a difusão aumenta na proporção direta com a
temperatura.
5. O peso molecular da substância difusora — isso tem efeito
complexo; a velocidade do movimento térmico de uma substância
Fig. 4.8 Efeito da diferença de concentração (A), da diferença
dissolvida é proporcional à raiz quadrada de seu peso molecular.
Por outro lado, à medida que o diâmetro molecular se aproxima de potencial elétrico (B) e da diferença de pressão (C) sobre a difusão
do diâmetro do canal, a resistência aumenta de forma muito efetiva de moléculas e de tons, através de membrana celular.
acentuada, de modo que, freqüentemente, uma membrana pode
ser centenas a milhões de vezes mais permeável às pequenas A carga positiva atrai os íons negativos, ao mesmo tempo que a
moléculas que às grandes, como é evidente pelos valores relativos carga negativa os repele. Por conseguinte, ocorre difusão
constantes do Quadro 4.1.
efetiva da esquerda para a direita. Após muito tempo, grande
O coeficiente de difusão. Outro fator que influencia a quantidade de íons negativos terá passado para o lado direito
velocidade total da difusão é a área da membrana. Como (caso se despreze, momentaneamente, o efeito perturbador dos
resultado, para a determinação da permeabilidade total de uma íons positivos da solução), criando a condição mostrada no
membrana celular, deve-se multiplicar sua permeabilidade (que painel direito da Fig. 4.8B, onde se desenvolveu diferença de
mede o movimento da substância por área unitária da concentração de direção oposta à da diferença de potencial
membrana) pela área total da membrana. Essa permeabilidade elétrico. Obviamente, essa diferença de concentração tende
total define o coeficiente de difusão; sua relação com a agora a mover os íons para a esquerda, enquanto a diferença de
permeabilidade é dada por:
potencial elétrico tende a movê-los para a direita. Quando a
diferença de concentração aumentar o suficiente, esses dois
D=PxA
efeitos se contrabalançam exatamente. Na temperatura corporal
normal (37°C), a diferença elétrica capaz de contrabalançar com
onde D é o coeficiente de difusão, P é a permeabilidade e A exatidão dada diferença de concentração de íons univalentes —
é a área total.
como o sódio (Na+), o potássio (K+) ou cloreto (Cl) — pode ser
Efeito da diferença de concentração. A Fig. 4.8A determinada pela seguinte relação, chamada de equação de
apresenta uma membrana celular, separando soluções de uma Nernst:
substância em alta concentração na face externa e baixa
concentração na interna. A velocidade com que a substância se
difunde para o interior é proporcional à concentração de suas
moléculas no exterior, pois é essa a concentração que determina
quantas moléculas irão de encontro à abertura externa dos
FEM (em milivolts) = ± 61 log C1
canais a cada segundo. Por outro lado, a velocidade com que as
moléculas se difundem para o exterior é proporcional à sua
C2
concentração no interior da membrana. Por conseguinte, é
onde
FEM
é
a
força
eletromotriz
(a
voltagem)
entre as faces
óbvio que a difusão efetiva para o interior da célula é
proporcional à concentração externa menos a concentração 1 e 2 da membrana, C, é a concentração no lado 1 e Q é a
concentração no lado 2. A polaridade da voltagem na face 1,
interna, ou seja:
nesta equação, é + para os íons negativos e — para os íons
Difusão efetiva D (Ce - Ci)
positivos. Essa relação é extremamente importante para a
compreensão da transmissão dos impulsos nervosos, razão por
onde Ce é a concentração externa, Q é a concentração interna que será discutida de modo muito mais detalhado no Cap. 5.
e D é o coeficiente de difusão da membrana para a substância.
Efeito de potencial elétrico sobre a difusão de íons.
Caso seja aplicado um potencial elétrico através da membrana,
como mostrado na Fig. 4.8B, os íons, devido à sua carga
elétrica, atravessarão a membrana, mesmo quando não existirem
diferenças de concentração para impulsioná-los. Assim, no
painel esquerdo da figura, a concentração de íons negativos é
exatamente igual nos dois lados da membrana, mas foi aplicada
carga positiva no lado direito dessa membrana e carga negativa
no lado esquerdo, criando gradiente elétrico através dela.
39
Efeito de diferença de pressão. Por vezes, pode desenvolverse diferença considerável de pressão entre as duas faces de uma
membrana. Isso ocorre, por exemplo, na membrana capilar, onde
existe pressão da ordem de 20 mm Hg maior no interior do
capilar que em seu exterior. Por pressão se entende a soma das
forças de todas as diferentes moléculas que atingem uma área
de superfície em dado instante. Portanto, quando a pressão é
maior em uma das faces de uma membrana que na outra,
isso significa que a soma das forças das moléculas que atingem
os canais dessa face da membrana é maior que a da outra face.
Isso pode resultar de maior número de moléculas atingindo a
membrana a cada segundo, ou de maior energia cinética da
molécula média que atinge a membrana. Em qualquer dos
casos, maiores quantidades de energia ficam disponíveis para
causar o movimento efetivo das moléculas da região de
maior para a de menor pressão. Esse efeito é mostrado na Fig.
4.8C, que mostra um pistão criando alta pressão em uma das faces
de membrana celular, o que provoca difusão efetiva, através da
membrana, para o outro lado.
OSMOSE ATRAVÉS DE MEMBRANAS
SELETIVAMENTE
PERMEÁVEIS
A
DIFUSÃO EFETIVA DE ÁGUA
A água é, de longe, a substância mais abundante que se
difunde através da membrana celular. Deve ser lembrado que,
regra geral, a quantidade de água que se difunde, nas duas
direções, através da membrana da hemácia, a cada segundo,
corresponde à cerca de 100 vezes o próprio volume da hemácia.
Contudo, normalmente, a quantidade que se difunde nas duas
direções é tão precisamente balanceada que não ocorre qualquer
movimento efetivo de água. Como resultado, o volume dessa
célula permanece constante. Contudo, sob certas circunstâncias,
pode desenvolver-se uma diferença de concentração para a água
através de uma membrana, exatamente do mesmo modo como
isso pode ocorrer para outras substâncias. Quando isso acontece,
ocorre, realmente, movimento efetivo de água através da
membrana celular, fazendo com que a célula murche ou inche,
na dependência da direção desse movimento efetivo. Esse
processo de movimento efetivo da água, causado por diferença
de concentração da própria água, é chamado de osmose.
Para dar um exemplo de osmose, vamos admitir as condições
mostradas na Fig. 4.9, com água pura em um dos lados da
membrana celular e solução de cloreto de sódio no outro.
Consultando-se o Quadro 4.1, vê-se que as moléculas de água
atravessam facilmente a membrana, enquanto o sódio e o cloreto
só a atravessam com grande dificuldade.
Fig. 4.9 Osmose através de membrana celular quando é colocada uma
solução de cloreto de sódio em um dos lados da membrana e água
no lado oposto.
Portanto, a solução de cloreto de sódio é, na realidade, uma
mistura de moléculas permeantes de água e de íons nãopermeantes de sódio e cloreto; a membrana é dita seletivamente
permeável (ou "semipermeável) à água, mas não aos íons sódio
e cloreto. Todavia, a presença do sódio e do cloreto deslocou
parte das moléculas de água e, portanto, reduziu a
concentração das moléculas de água a valor menor que na
água pura. Como resultado, no exemplo da Fig. 4.9, maior
número de moléculas de água atinge os canais da face esquerda,
em contato com a água pura, que à direita, onde a concentração
de água está diminuída. Assim, ocorre movimento efetivo de
água do lado esquerdo para o direito — isto é, há osmose da
água pura para a solução de cloreto de sódio.
Pressão osmótica
Se, na Fig. 4.9, fosse aplicada pressão à solução de cloreto
de sódio, a osmose da água para essa solução poderia ser
lentificada, interrompida ou invertida. A quantidade de pressão
necessária para interromper precisamente a osmose é
denominada pressão osmótica da solução de cloreto de sódio.
O princípio da oposição à osmose por uma diferença de
pressão é mostrado na Fig. 4.10, onde uma membrana
seletivamente permeável separa duas colunas de líquido, uma
contendo água e a outra contendo solução aquosa de um soluto
qualquer a que a membrana não é permeável. A osmose da água
do compartimento B para o A faz com que os níveis das colunas
líquidas fiquem progressivamente mais afastados até que,
eventualmente, se desenvolva diferença de pressão
suficientemente intensa para impedir o efeito osmótico. Nesse
ponto, a diferença de pressão entre as duas faces da membrana é
a pressão osmótica da solução contendo o soluto não-difusível.
A importância do número de partículas osmóticas (ou da
concentração molar) para a determinação da pressão osmótica.
A pressão osmótica exercida pelas partículas de uma solução
sejam elas moléculas ou íons, é determinada pelo número de
partículas por unidade de volume do líquido, e não pela massa
dessas partículas. A razão disso é que cada partícula da solução,
Fig. 4.10 Demonstração da pressão osmótica entre as
duas faces de membrana semipermeável.
40
independentemente de sua massa, exerce, em média, a mesma
quantidade de pressão sobre a membrana. Isto é, todas as
partículas estão se chocando entre si, em média com a mesma
energia. Se algumas partículas apresentarem maior energia
cinética de movimento que outras, seu impacto com as
partículas de menor energia transferirá parte de sua energia para
estas, o que diminui o teor de energia das partículas com muita
energia, ao mesmo tempo que aumenta esse teor nas partículas
com pouca energia, o que leva, ao longo do tempo, à
equalização do teor de energia de todas as partículas. Por
conseguinte, as partículas maiores, com mais massa (m) que
as partículas menores, deslocam-se com menor velocidade (v),
enquanto as partículas menores se movem com mais velocidade,
de modo que suas energias cinéticas médias (k), definidas pela
equação
K= MV2
2
serão iguais para todas as partículas. Como resultado, em média,
a energia cinética de cada molécula ou íon que atinge a membrana
vai ser aproximadamente a mesma, independente de suas
dimensões moleculares. Como conseqüência, o fator que
determina a pressão osmótica de uma solução é a concentração
dessa solução em termos do número de suas partículas (o
que é o mesmo que a concentração molar, no caso de moléculas
não-dissociadas), e não em termos da massa do
soluto."Osmolalidade" — O osmol. Como a quantidade de
pressão osmótica exercida por um soluto é proporcional à
concentração do soluto, expressa em número de moléculas ou
de íons, o uso da concentração do soluto em função de sua
massa não tem qualquer valor na determinação da pressão
osmótica. Para expressar essa concentração em termos do
número de partículas, é usada a unidade osmol, em lugar de
gramas.
Um osmol é o número de moléculas em uma molécula-grama
de soluto não-dissociado. Assim, 180 g de glicose, que
correspondem a uma molécula-grama dessa substância, são
iguais a 1 osmol, porque a glicose não se dissocia. Por outro
lado, caso o soluto se dissocie em dois íons, 1 molécula-grama
desse soluto equivale a 2 osmóis, visto que o número de
partículas osmoticamente ativas passa a ser o dobro daquele do
soluto não-dissociado. Por conseguinte, 1 molécula-grama de
cloreto de sódio, 58,5 g, é igual a 2 osmóis. Uma solução que
contenha / osmol de soluto dissolvido em 1 quilograma de
água tem osmolalidade de 1 osmol por quilograma e a solução que
contenha VIM» osmol dissolvido por quilograma tem osmolalidade
de 1 miliosmol por quilograma (1 mOsm/ kg). A osmolalidade
normal dos líquidos extra e intracelular é de cerca de 300
mOsm/kg. Relação entre a osmolalidade e a pressão osmótica. Na
temperatura normal do corpo, 37ºC, a concentração de 1 osmol
por litro produzirá - pressão osmótica de 19.300 mm Hg na
solução. De igual modo, a concentração de 1 miliosmol por litro é
equivalente à pressão osmótica de 19,3 mm Hg. Multiplicandose esse valor pela concentração de 300 miliosmóis dos líquidos
corporais, obtém-se uma pressão osmótica total calculada para
esses líquidos de 5.790 mm Hg. O valor medido, no entanto,
é, em média, de 5.500 mm Hg. A razão dessa diferença é
que muitos dos íons nos líquidos celulares tais como os de
sódio e cloreto, são fortemente atraídos uns pelos outros;
conseqüentemente, eles não conseguem deslocar-se livremente
por esses líquidos, criando todo o seu potencial osmótico. Como
resultado, em média, a verdadeira pressão osmótica dos
líquidos corporais é de cerca de 0,93 do valor calculado.
O termo "osmolaridade". Devido à dificuldade de se medir quilogramas
de água em uma solução, o que é necessário para a determinação da os-
molalidade usa-se, geralmente, outro termo, "osmolaridade",
quando a concentração osmolar é expressa como osmóis por
litro de solução, e não osmóis por quilograma de água.
Embora, em sentido estrito, sejam os osmóis por quilograma de
água os determinantes da pressão osmótica, não obstante, para
soluções diluídas, como as encontradas no corpo, as diferenças
quantitativas entre osmolaridade e osmolalidade são menores
que 1%. Visto que é muito mais fácil a medida da
osmolaridade que a da osmolalidade, ela se tornou prática
comum em quase todos os experimentos fisiológicos.
TRANSPORTE ATIVO
Do que foi discutido até agora, é evidente que nenhuma
substância pode difundir-se contra um "gradiente eletroquímico"',
que é a soma de todas as forças difusionais que agem
sobre a membrana — as forças geradas pelas diferenças de
concentração, de potencial elétrico e de pressão. Isto é,
muitas vezes é dito que as substâncias não podem difundir-se
"ladeira acima".
Contudo, é por vezes necessária grande concentração de
uma substância no líquido intracelular, embora o líquido
extracelular só contenha quantidade diminuta dela. Isso é
verdade, por exemplo, para os íons potássio. De modo inverso,
é importante a manutenção de outros íons muito baixa no
interior da célula, apesar de suas concentrações no líquido
extracelular serem muito altas. Isso é especialmente verdade
para os íons sódio. Obviamente, nenhum desses dois efeitos
poderia ocorrer pelo processo da difusão simples, pois ela tende
sempre a equilibrar as concentrações nas duas faces da
membrana. Ao contrário, alguma fonte de energia deve provocar
o movimento "ladeira acima" dos íons potássio, para o interior
da célula, e, também, o movimento dos íons sódio,
igualmente "ladeira acima", mas para fora da célula. Quando a
membrana celular transfere moléculas ou íons "ladeira acima"
contra um gradiente de concentração (ou "ladeira acima"
contra gradiente elétrico ou de pressão), o processo é
chamado de transporte ativo.
Entre as diferentes substâncias que são transportadas ativamente, através das membranas celulares, estão os íons sódio,
potássio, cálcio, ferro, hidrogênio, cloreto, iodeto, urato,
diversos e distintos açúcares e a maioria dos aminoácidos.
Transporte ativo primário e secundário. O transporte ativo
é dividido em dois tipos, segundo a fonte de energia utilizada
para o transporte. São chamados de transporte ativo primário
e de transporte ativo secundário. No transporte ativo primário,
a energia é derivada diretamente da degradação do trifosfato
de adenosina (ATP) ou de qualquer outro composto de fosfato
rico em energia. No transporte ativo secundário, a energia é
derivada, secundariamente, de gradientes iônicos que foram
criados, em primeiro lugar, por transporte ativo primário. Nos
dois casos, o transporte depende de proteínas carreadoras, que
atravessam toda a espessura da membrana, como acontece na
difusão facilitada. Contudo, no transporte ativo, a proteína
carreadora funciona de modo distinto do carreador da difusão
facilitada, pois ela é capaz de transferir energia para a
substância transportadas, a fim de que possa mover-se contra o
gradiente eletroquímico. Vamos apresentar alguns exemplos de
transporte ativo primário e secundário e explicar, com mais
detalhes, os princípios de seu funcionamento.
Transporte ativo primário – A “bomba” de sódiopotássio
Entre as substâncias que são transportadas por transporte ativo
primário estão os íons sódio, potássio, cálcio, hidrogênio,
cloreto e alguns outros. Todavia, nem todas essas substâncias
são transportadas pelas membranas de todas as células. Ainda
mais, algumas das bombas funcionam em membranas intrace-
lulares em vez de (ou além de) nas membranas da superfície
41
das células, como na membrana do retículo sarcoplasmático das
células musculares e em uma das duas membranas das
mitocôndrias. Não obstante, todas funcionam, essencialmente,
com o mesmo mecanismo básico.
O mecanismo de transporte ativo que foi estudado mais
detalhadamente é a bomba de sódio-potássio, o processo de
transporte que bombeia os íons sódio para fora, através da
membrana celular, enquanto, ao mesmo tempo, bombeia os
íons potássio de fora para dentro. Essa bomba está presente em
todas as células do corpo e é a responsável pela manutenção das
diferenças de concentração de sódio e de potássio através da
membrana celular, além de estabelecer um potencial elétrico
negativo no interior das células. Na verdade, veremos no
capítulo seguinte que essa bomba é à base do funcionamento
nervoso de transmissão de sinais nervosos por todo o sistema
nervoso.
A Fig. 4.11 apresenta os componentes básicos da bomba
Na+-K+. A proteína carreadora é um complexo de duas proteínas
globulares distintas, uma maior, com peso molecular de cerca
de 100.000, e outra menor, com peso molecular de 55.000.
Embora não seja conhecida a função da proteína menor, a maior
tem três características específicas que são importantes para o
funcionamento da bomba:
1. Apresenta três sítios de fixação para os íons sódio, situa
dos na parte da molécula que protrui para o interior da célula.
2. Tem dois sítios de fixação para os íons potássio em sua
face externa.
3. A parte interna dessa proteína, adjacente ou muito
próxima dos sítios de fixação de sódio, tem atividade de
ATPase.
Agora, para descrever o funcionamento da bomba: quando
três íons sódio se fixam na parte interna da proteína carreadora
e dois íons potássio se fixam à parte externa, a função ATPase
da proteína é ativada. Isso cliva uma molécula de ATP,
transformando-a em difosfato de adenosina (ADP) e liberando a
energia de uma ligação fosfato rica em energia. Acredita-se que
essa energia provoque alteração conformacional da molécula da
proteína carreadora, o que expulsa o sódio para o exterior e
trazendo o potássio para o interior. Infelizmente, o mecanismo
preciso dessa alteração conformacional do carreador ainda não
foi identificado.
Importância da bomba Na+-K+ para o controle do volume
celular. Uma das mais importantes funções da bomba Na+-K+ é
a de controlar o volume das células. Sem essa função da bomba, a
maioria das células iria inchar até estourar. O mecanismo para o
controle do volume é o seguinte: no interior da célula existe
grande número de proteínas e de outros compostos orgânicos
que não podem sair dela. A maior parte desses compostos tem
para impedir que isso aconteça é a bomba Na+-K+. Note-se,
de novo, que esse mecanismo bombeia dois íons Na+ para o
exterior, enquanto bombeia dois íons J+ para o interior. Por
outro lado, a membrana é bem menos permeável ao sódio que
ao potássio, de modo que, quando os íons sódio estão no
exterior, eles têm forte tendência a permanecer aí. Assim, isso
representa perda contínua e efetiva de substâncias iônicas
para fora da célula, o que produz tendência osmótica oposta
para deslocar a água para fora da célula. Ainda mais, quando a
célula começa a inchar, isso ativa, automaticamente, a bomba
Na+-K+, o que transfere mais íons para o exterior, levando água
com eles. Por conseguinte, a bomba Na+-K+, exerce papel
permanente de vigilância para a manutenção do volume
normal da célula.
A natureza eletrogênica da bomba Na+-K+. O fato de a
bomba Na+-K+, transportar três íons sódio para o exterior, em
troca de dois íons potássio transportados para o interior, implica a
efetiva transferência de uma carga positiva para o exterior, a cada
ciclo da bomba. Obviamente, isso gera positividade no exterior
da célula, mas cria déficit de íons positivos no interior celular;
isto é, ela produz negatividade nesse interior. Como resultado, a
bomba Na+-K+ é dita eletrogênica, por criar um potencial elétrico
através da membrana como conseqüência de seu bombeamento.
A bomba de cálcio
Outro mecanismo, igualmente muito importante, de
transporte ativo primário é o da bomba de cálcio. Os íons
cálcio são mantidos, normalmente, em concentrações
extremamente baixas no citosol intracelular, de cerca de
10.000 vezes menor que no líquido extracelular. Isso é
realizado por duas bombas de cálcio. Uma delas fica na
membrana celular e bombeia cálcio para fora da célula. A outra
bombeia cálcio para o interior de uma ou mais das organelas
vesiculares do interior celular, como o retículo sarcoplasmático
das células musculares e as mitocôndrias de todas as células.
Nos dois casos, a proteína carreadora atravessa toda a
espessura da membrana e também funciona como ATPase,
com a mesma capacidade de desdobrar o ATP, como tem a
ATPase das proteínas carreadoras de sódio. As duas
proteínas carreadoras diferem pelo fato de esta proteína
carreadora ter sítio de fixação para o cálcio, e não para o sódio.
Saturação do transporte ativo
O transporte ativo fica saturado de modo idêntico ao da
saturação da difusão facilitada, como mostrado na Fig. 4.6.
Quando a concentração da substância a ser transportada é
pequena, a intensidade do transporte aumenta, em proporção
direta, com o aumento da concentração. Todavia, com
concentrações muito elevadas, o transporte tende a um valor
máximo, chamado de Vmax, como ocorre na difusão facilitada. A
saturação é causada pela limitação da velocidade com que as
reações químicas de fixação, liberação e alterações
conformacionais do carreador podem ocorrer.
Energética do transporte ativo
Fig. 4.11 Mecanismo proposto para a bomba de sódiopotássio.
carga negativa e, como conseqüência, eles agregam ao seu redor
grande número de íons positivos. Todas essas substâncias atuam,
então, no sentido de provocar osmose de água para o interior
da célula; se isso não fosse impedido, a célula iria inchar,
indefinidamente, até estourar. Todavia, o mecanismo normal
A quantidade de energia necessária para transportar
ativamente uma substância através de membrana (sem levar em
conta a energia perdida com o calor nas reações químicas) é
determinada pelo grau a que a substância é concentrada durante
o transporte.
42
Tomando como base a energia necessária para aumentar a
concentração da substância por 10 vezes, será necessário
consumo de três vezes mais energia para aumentar sua
concentração por 100 vezes. Em outras palavras, a energia
necessária é proporcional ao logaritmo do grau a que é
concentrada a substância, como definido pela relação seguinte:
C1
Energia (em calorias por osmol) - 1.400 log —
C2
Isto é, em termos de calorias, a quantidade de energia necessária
para concentrar por 10 vezes 1 osmol de uma substância é de
cerca de 1.400 calorias e, por 100 vezes, 2.800 calorias. Pode-se
ver que o consumo de energia para aumentar a concentração
de substâncias no interior celular, ou para a remoção de
substâncias para o exterior, contra gradiente de concentração,
pode ser muito grande. Algumas células, como as que revestem
os túbulos renais e certas células glandulares, consomem até
90% de sua energia nesse tipo de atividade.
Transporte ativo secundário — co-transporte e
contratransporte
Quando os íons sódio são transportados para fora das células
por transporte ativo primário, forma-se, na maioria das vezes,
um gradiente de concentração de sódio muito intenso —
concentração muito elevada no exterior e muito baixa no
interior. Esse gradiente representa um reservatório de energia,
visto que o excesso de sódio, no exterior da célula, tende sempre
a se difundir para o interior. Sob condições adequadas, essa
energia de difusão do sódio pode, literalmente, puxar outras
substâncias, junto com o sódio, através da membrana. Esse
fenômeno é chamado de co-transporte; é uma das formas do
transporte ativo secundário.
Para que o sódio possa levar consigo outras substâncias, é
necessário um mecanismo de acoplamento. Isso é realizado por
meio de outro tipo de proteína carreadora da membrana celular.
Neste caso, o carreador atua como ponto de fixação para o íon
sódio e para as substâncias que vão ser co-transportadas. Uma
vez tendo acontecido a fixação dos dois, ocorre alteração
conformacional da proteína carreadora e o gradiente de energia
do sódio faz com que tanto o íon sódio como a substância cotransportada sejam transferidos juntos para o interior da célula.
No contratransporte, de novo, os íons sódio tendem a se
difundir para o interior da célula, devido a seu intenso gradiente
de concentração. Contudo, neste caso, a substância que vai ser
transportada está no interior da célula e deve ser transportada
para o exterior. Por conseguinte, o íon sódio se fixa à proteína
carreadora em sua extremidade que se projeta para fora, na
face externa da membrana, enquanto a substância que vai ser
contratransportada se fixa à projeção interna da proteína
carreadora. Uma vez tendo acontecido a fixação dos dois, ocorre
nova alteração conformacional, com a energia do íon sódio o
transferindo para o interior e levando a outra substância a se
deslocar para o exterior.
Co-transporte do sódio com glicose ou com aminoácidos. A
glicose e muitos aminoácidos são transportados para o interior
das células contra gradientes muito intensos de concentração;
o mecanismo disso é o sistema de co-transporte mostrado na
Fig. 4.12. Deve ser notado que a proteína carreadora para esse
transporte tem dois sítios de fixação em sua extremidade externa,
um para o sódio e outro para a glicose. Por outro lado, a
concentração de sódio é muito elevada no exterior e muito baixa
no interior, o que dá a energia para o transporte. É propriedade
especial dessa proteína transportadora que a alteração
conformacional que permite a transferência do sódio para o
interior só pode ocorrer quando uma molécula de glicose
também se fixa
Fig. 4.12 Mecanismo proposto para o co-transporte sódio-glicose.
a ela. Quando os dois estão fixados, a alteração conformacional
ocorre de modo automático e tanto o sódio como a glicose são
transportados, ao mesmo tempo, para o interior da célula. Esse
é, portanto um mecanismo de co-transporte sódio-glicose.
O co-transporte de sódio com aminoácidos ocorre de modo
idêntico ao da glicose, exceto pela utilização de conjunto diverso
de proteínas de transporte. Já foram identificados cinco tipos
distintos de proteínas de transporte para aminoácidos, cada um
responsável pelo transporte de um subtipo de aminoácidos com
características moleculares específicas.
O co-transporte de sódio com glicose ou aminoácidos ocorre,
de forma especial, nas células epiteliais do tubo intestinal e dos
túbulos renais, participando na absorção dessas substâncias para
o sangue, como discutiremos em outros capítulos.
Outros dois importantes mecanismos de co-transporte são
(1) o co-transporte de sódio-potássio-dois cloretos, que possibilita
a transferência de dois íons cloreto, junto com um íon sódio
e um íon potássio, para o interior da célula, todos se movendo
na mesma direção, e (2) co-transporte potássio-cloreto, que
possibilita a passagem de íons potássio e cloreto, juntos, do
interior para o exterior celular. Outros tipos de co-transporte,
encontrados, pelo menos, em algumas células, incluem os
cotrans-portes de íons iodeto, de íons ferro e de íons urato.
Contratransporte de sódio e íons cálcio e íons hidrogênio.
Dois mecanismos especialmente importantes de contratransporte
são os de contratransporte sódio-cálcio e contratransporte sódiohidrogênio, O contratransporte de cálcio existe em todas — ou
quase todas — as membranas celulares, com o íon sódio se
movendo para o interior e os íons cálcio para o exterior, ambos
fixados à mesma proteína transportadora, em sistema de contratransporte. Isso acontece adicionalmente ao transporte primário
de cálcio, encontrado em alguns tipos celulares. O
contratransporte sódio-hidrogênio existe em diversos tecidos.
Exemplo especialmente importante ocorre no túbulo proximal
dos rins, onde os íons sódio se movem do lúmen tubular para
o interior das células tubulares, ao mesmo tempo que os íons
hidrogênio são transferidos para o lúmen. Esse mecanismo é
crucial para a regulação dos íons hidrogênio nos líquidos
corporais, como é discutido em maiores detalhes no Cap. 30.
Outros mecanismos de contratransporte incluem as trocas de
cátions, de íons cálcio ou sódio, em uma das faces da membrana,
por íons magnésio ou potássio, na outra, e as trocas de ânions,
íons cloreto se movendo em uma direção, e íons bicarbonato ou
sulfato se movendo na direção oposta.
TRANSPORTE ATIVO ATRAVÉS DE LÂMINAS
CELULARES
Em muitas regiões do corpo, as substâncias devem ser
transportadas através de toda a espessura de lâminas
formadas por muitas células, e não, simplesmente, através de
uma membrana celular.
43
Fig. 4.13 Mecanismo básico para o transporte ativo através de toda
a espessura de uma lâmina celular.
Esse tipo de transporte ocorre no epitélio intestinal, no epitélio
dos túbulos renais, no epitélio de todas as glândulas exócrinas,
no epitélio da vesícula biliar, na membrana do plexo coróide
cerebral e em muitas outras membranas.
O mecanismo básico do transporte de substâncias através
de lâminas celulares é o de (1) haver transporte ativo através
da membrana celular, em uma das extremidades da célula e,
em seguida, (2) haver difusão simples ou facilitada, através da
membrana, na extremidade oposta da célula.
A Fig. 4.13 mostra o mecanismo para o transporte de íons
sódio através do epitélio do intestino, da vesícula biliar e dos
túbulos renais. Essa figura mostra que as células epiteliais são
unidas entre si, ao nível de seu pólo luminal, por meio de junções
fechadas que bloqueiam, principalmente, a difusão dos íons sódio
pelos espaços entre as células. Contudo, as superfícies luminais
dessas células são muito permeáveis aos íons sódio e à água.
Por conseguinte, os íons sódio e a água se difundem com grande
facilidade para o interior dessas células. Então, nas membranas
basais e laterais das células, os íons sódio são ativamente
transportados para o líquido extracelular. Isso gera intenso
gradiente de concentração de sódio através dessas membranas, o
que, por sua vez, provoca a osmose da água. Assim, o
transporte ativo de sódio, através das superfícies basolaterais das
células epiteliais, provoca não apenas o transporte dos íons
sódio, mas também, ao mesmo tempo, o de água.
Ainda mais, qualquer outra substância que esteja acoplada
por co-transporte ao sódio pode ser também transportada. Por
exemplo, as cargas positivas dos íons sódio puxam, em geral,
os íons cloreto, com carga negativa, junto com o sódio. De igual
modo, quando a glicose (ou aminoácidos) é co-transportada com
o sódio, através da superfície luminal da célula, a concentração
intracelular de glicose aumenta. Então, essa glicose é
transportada, por difusão facilitada, através das membranas
basolaterais dessas células, atingindo, finalmente, o líquido
extracelular, junto com os íons sódio, os íons cloreto e a água.
Esses são os mecanismos pelos quais quase todos os
nutrientes, os íons e outras substâncias são absorvidos do
intestino para o sangue; também representam o meio como essas
mesmas substâncias são reabsorvidas do filtrado glomerular
pelos túbulos renais.
REFERENCIAS
Agnew, W. S.: Voltage-regulated aodium channel molecules. Annu.
Rev. Physiol., 46:517, 1984. Almere, W., and Stirling, C: Distribution of
tranaport proteína over animal membranes. J. Membr. Biol-, 77:169,
1984.
Andreoli,T.E.,etal.
(eds.):
Physiologyof
Membrane
Disorders.2ndEd. New York, Plenum Publishing Corp,, 1986.
Auerbach, A., and Sacha, F.: Patch clamp studies of single ionic
channels.
Annu. Rev. Biophys. Bioeng., 13:269,1984. Barry, P. H., and
Diamond, J. M.: EffectB of unstirred layere on membrane
phenomena. Phyaiol. Rev., 64:763, 1984. Bíggio, G., and Costa, E.
(eds.): Chloride Channels and Their Modulation by
Neurotransmitters and Drugs. New York, Raven Press, 1988.
Blachley, J. E., et ai.: The harmful effects of ethanol on ion
transport and cellular respiration. Am. J. Med. Sei., 289:22, 1985.
Bohr, D. F.: Cell membrane in hypertension. News Physiol. Sei.,
4:85, 1989. Bretag, A. H.: Muscle chloride channels. Phyaiol. Rev.,
67:618,1987. Cereijido, M., et ai.: Tight junction: Barrier between
higher organisms and environment. News Physiol. Sei., 4:72, 1989.
Chasis, J. A., and Shohet, S. B.: Red ceílbiochemical anatomy and
membrane propertiea. Annu. Rev. Physiol., 49:237, 1987. Deuticke,
B., and Haest, C. W. M.: Lipid modulation of transport proteins in
vertebrate cell membranes. Annu. Rev. Physiol., 49:221,1987.
Dinno, M. A., and Armstrong, W. M. (eds.): Membrane Biophysics
III: Biológica! Transport. New York, Alan R. Lisa, Inc., 1988.
DiPolo, R., and Beauge, L.; The calcium purap and sodium-calcium
exchange in squid axons. Annu. Rev. Physiol., 45:313,1983.
Donowitz, M., and Welsh, M. J.: Ca>+ and cyclic AMP in regulation
of intestinal Na, K, and Cl transport. Annu. Rev. Physiol., 48:135,
1986. Ellis, D.: Na-Ca exchange in cardiac tissues. Adv.
Myocardiol., 5:295, 1985. Finkelstein, A., et ai.: Osmotic swelling
of vesicles. Annu. Rev. Physiol., 48:135, 1986. Flatman, P. W.:
Magnesium tranaport across cell membranea. J. Membr. Biol.,
80:1, 1984. Forgac, M.: Structure and function of vacuolar class of
ATP-driven proton pumps. Phyaiol. Rev., 69:765, 1989. Gadsby, D.
C: The Na/K pump of cardiac cells. Annu. Rev. Biophys. Bioeng.,
13:373, 1984. Haas, M.: Properties and diveraity of Na-KMembranes and Their
Functional Implications. New York, Plenum Publishing Corp.,
1988. HofTmann, E. K., and Simonsen, L. 0.: Membrane
mechanisins in volume and pH regulation in vertebrate cells.
Physiol. Rev., 69:315, 1989. Ive3, H. E., and Rector, F. C, Jr.:
Proton transport and cell function. J. Clin. Invest., 73:285, 1984.
Jacobson, K., et ai.: Lateral diffusion of proteína in membranes.
Annu. Rev. Phyaiol., 49:163, 1987. Jennings, M. L.: Kinetics and
mechanism of anion tranaport in red blood cells. Annu. Rev.
Physiol., 47:519, 1985.
Kaplan, J. H.: Ion movements through the sodium pump. Annu.
Rev. Physiol., 47:535, 1985. Kim, C. H., and Tedeschi, H. (eds.):
Advances in Membrane Biochemiatry and Bioenergetics. New
York, Plenum Publishing Corp., 1987. Latorre, R., et ai.: Varieties
of calcium-activated potassium channelB. Annu. Rev. Physiol.,
51:385, 1989. Lauger, P.: Dynamics of ion transport systems in
membranes. PhyBiol. Rev., 67:1296, 1987. Lear, J. D., et ai.:
Synthetic amphiphilic peptide models for protein ion channels.
Science, 240:1177, 1988. Lemer, J.: Cell membrane amino acid
transport processes in the domestic fowl (Gallus domesticus).
Comp. Biochem. Physiol., 78:205,1984. Lindermann, B.:
Fluctuation analysis of sodium channels in epthelia. Annu.
Rev. Physiol., 46:497, 1984. Macey.R. I.: Transport ofwater and
ureain red blood cells. Am. J. Physiol., 246:C195, 1984. Malhotra.
S. K.: The Plasma Membrane. New York, John Wiley & Sons,
1983.
Miller, C: Ion channels in liposomes. Annu. Rev. Physiol., 46:549,
1984. Miller, C: Integral membrane channels: Studies in model
membranea. Phys- ioi. Rev., 63:1209, 1983.
Narahashi, T.: Ion Channels. New York, Plenum Publishing
Corp., 1988. Parker, J. D., and Berkowitz, L. R.: Physiologically
instruetive genetic variants involving the human red cell membrane.
Physiol. Rev., 63:261,1983. Petersen, 0. H.: Potassium channeia and
fluid secretion. News Physiol. Sei.,1:92, 1986. Petereen, O. H., and
Petersen, C. C. H.: Thepatch-clamp technique: Recording ionic
currents through single pores in the cell membrane. News Physiol.
Sei., 1:5, 1986. Reuter, H.: Modulation of ion channels
byphosphorylation and secondmes-sengera. News Physiol. Sei.,
2:168, 1967. Reuter, H.: Ion channels in cardiac cell membranes.
44
Annu. Rev. Physiol., 46:473, 1984. Rudel, R., and Lehmann-Horn, F.:
Membrane changes in cells from myo- tonia patients. Physiol. Rev.,
65:310, 1985. Sakmann, B., and Neher, E.: Patch clamp techniques
for studying ionic channels in excitable membranes. Annu. Rev.
Physiol., 46:455, 1984. Salkoff, L. B., and Tonouye, M. A.: Genetics of
ion channels. Physiol. Rev.,66:301, 1986.
Schatzmann, H. J.: The calcium pump of the surface membrane and of
the sarcoplaaraic reticulum. Annu. Rev. PhyBÍol., 51:473, 1989.
Schatzmann, H. J.: The red cell calcium pump. Annu. Rev. Phyaiol.,
445:303, 1983. Schultz, S. G.: A cellular model for active sodium
absorption by mammalian cólon. Annu. Rev. Physiol., 46:435, 1984.
Schwartz, G. J., and Al-Awqati, Q.: Regulation of transepithelia H*
transport by exocytoaia and endocytosía. Annu. Rev. Physiol., 48:153,
1986. Smíth, P. L., and McCabe, R. D.: Mechanism and regulation of
transcellular potaasium transport by the cólon. Am. J. Physiol.,
247:G445, 1984. Stein, W. D. (ed.): The Ion Pumpa: Structure, Function,
and Regulation.
45
CAPÍTULO 5
Potenciais de Membrana e Potenciais de Ação
Existem diferenças de potencial elétrico através das membranas
de praticamente todas as células do corpo, e algumas células,
como as nervosas e musculares, são "excitáveis isto é, capazes de
autogerar impulsos eletroquímicos em suas membranas e, na
maioria dos casos, utilizar esses impulsos para a transmissão de
sinais ao longo das membranas. Em outros tipos de células, tais
como as glandulares, os macrófagos e as células ciliadas, outras
classes de variação dos potenciais de membrana têm,
provavelmente, participação ativa no controle de muitos
aspectos do funcionamento celular. Todavia, o que será discutido
neste capítulo está relacionado aos potenciais de membrana
gerados, no repouso e durante a atividade, pelas células
nervosas e musculares.
A FÍSICA BÁSICA DOS POTENCIAIS
DE MEMBRANA
OS POTENCIAIS DE MEMBRANA CAUSADOS
PELA DIFUSÃO
A Fig. 5.1A e B apresenta uma fibra nervosa sob condições
em que não ocorre transporte ativo de sódio e de potássio. Na
Fig. 5. IA, a concentração de potássio é muito elevada no interior
da membrana, enquanto é muito baixa no exterior. Vamos
admitir que, nesse caso, a membrana seja muito permeável aos
íons potássio, mas não seja permeável a qualquer outro íon.
Devido ao grande gradiente de concentração de potássio,
dirigido do interior para o exterior, existe forte tendência para
o potássio se difundir para o lado de fora. À medida que isso
acontece, esses íons transportam cargas positivas para o
exterior, o que cria estado de eletropositividade por fora da
membrana e de etetronegatividade em seu interior, causado
pelos ânions negativos que aí permanecem e que não se
difundem para fora junto com o potássio. Essa nova diferença de
potencial repele os íons positivos de potássio, na direção oposta,
do exterior para o interior. Dentro de cerca de um milissegundo,
essa variação do potencial fica suficientemente intensa para
bloquear qualquer difusão adicional de íons de potássio para o
exterior, apesar do elevado gradiente de concentração desse íon.
Nas maiores fibras nervosas normais de mamíferos, a diferença
de potencial necessária para esse efeito é da ordem de 94 mV,
com a negatividade no interior da membrana da fibra.
A Fig. 5.1B apresenta o mesmo fenômeno que a Fig. 5. IA,
mas, agora, com concentração elevada de íons sódio por fora
da membrana e muito baixa em seu interior. Esses íons também
têm carga positiva e, aqui, a membrana é extremamente
permeável aos íons sódio, mas impermeável a qualquer outro
íon. A difusão dos íons sódio para o interior resulta em potencial
de membrana com polaridade invertida, negatividade interna e
positividade interna. De novo, o potencial de membrana
aumenta o suficiente, em milissegundos, para bloquear a
continuação da difusão efetiva de íons sódio para o interior;
todavia, neste caso, nas fibras nervosas de maior calibre de
mamíferos, a diferença de potencial necessária é de 61 mV, com
a positividade no interior da fibra.
Assim, nos dois painéis da Fig. 5.1, vemos que a diferença
de concentração de íons, através de membrana seletivamente
permeável, pode, em condições adequadas, ser causa de um
potencial de membrana. Em algumas seções adiante, veremos
que muitas das alterações rápidas dos potenciais de membrana
observados durante o curso da transmissão de impulsos no
nervo e no músculo resultam da ocorrência desse tipo de
potenciais de difusão, de variação muito rápida.
Relação do potencial de difusão com a diferença de
concentração — a equação de Nernst. O nível do potencial
através da membrana, capaz de impedir, com exatidão, a difusão
efetiva de um íon, em qualquer direção, é chamado de
potencial de Nernst para esse íon. O valor desse potencial é
determinado pela proporção entre as concentrações do íon
nos dois lados da membrana — quanto maior for essa
proporção, maior será a tendência do íon a se difundir em uma
direção e, como resultado, maior será o potencial de Nernst. A
seguinte equação, chamada de equação de Nernst, pode ser
usada para o cálculo do potencial de Nernst para qualquer íon
monovalente na temperatura normal do corpo de 37°C:
FEM (em milivolts) = ± 61 log Concentração interna
Concentração externa
Ao se usar esta relação, admite-se que o potencial por fora da
membrana sempre permaneça exatamente em zero e o potencial
de Nernst que é calculado é o que vigora no interior da membrana.
Por outro lado, o sinal do potencial é positivo (+) quando o
íon em questão é negativo e negativo (-) quando esse íon é
positivo.
Por exemplo, quando a concentração de um íon positivo
46
Fig. 5.1 A, Desenvolvimento de um potencial de difusão através de
membrana celular, causado pela difusão de íons potássio, do interior
para o exterior, através de membrana que só é" seletivamente permeável
aos íons potássio. B, Desenvolvimento de um potencial de difusão quando
a membrana só é permeável aos íons sódio. Notar que o potencial interno
da membrana é negativo, pela difusão dos íons potássio, e positivo
quando a difusão é de íons sódio, devido à direção oposta dos
gradientes de concentração desses dois íons.
(digamos, o íon potássio) no interior for 10 vezes maior que
no exterior, e como o logaritmo de 10 é 1, o valor calculado
para o potencial de Nernst será de -61 mV no interior da
membrana.
Cálculo do potencial de difusão quando a
membrana é permeável a vários íons diferentes
Quando a membrana é permeável a vários e diversos íons,
o potencial de difusão que se desenvolve depende de três fatores:
(1) a polaridade da carga elétrica de cada íon; (2) a
permeabilidade da membrana (P) para cada íon; e (3) as
concentrações (C) dos íons respectivos, dentro (i) e fora (e) da
membrana. Então, pela seguinte relação, chamada de equação
de Goldman, ou de equação de Goldman-Hodgkin-Katz, podese calcular o valor do potencial de membrana vigente no interior
da membrana quando dois íons positivos monovalentes, sódio
(Na+) e potássio (K+), e um íon negativo, também monovalente,
o cloreto (Cl"), são participantes:
Vamos, agora, analisar a importância e o significado desta
equação. Primeiro, os íons sódio, potássio e cloreto são os íons
com participação mais importante no desenvolvimento dos
potenciais de membrana nas fibras nervosas e musculares, bem
como nas células neuronais do sistema nervoso central. O
gradiente de concentração de cada um desses íons, através da
membrana, ajuda na determinação da voltagem do potencial de
membrana.
Segundo, o grau de importância de cada íon, na
determinação da voltagem, é proporcional à permeabilidade da
membrana para esse íon. Assim, caso a membrana seja
impermeável aos íons potássio e cloreto, o potencial de
membrana será totalmente dependente apenas do gradiente de
concentração dos íons sódio, e o potencial resultante será
exatamente igual ao potencial de Nernst para o sódio. O mesmo
princípio permanece válido para cada um dos outros dois íons,
caso a membrana fique seletivamente permeável para apenas um
dos dois.
Terceiro, um gradiente de concentração iônica, do interior
para o exterior da membrana, vai produzir eletronegatividade
no interior dessa membrana. A razão disso é que os íons positivos
se difundem para o exterior, quando sua concentração interna
é maior que a externa. Isso carrega cargas positivas para fora,
mas deixa os ânions negativos não-difusíveis no interior. O efeito
exatamente oposto ocorre quando existe gradiente de íon
negativo. Isto é, um gradiente do íon cloreto, do exterior para
o interior, produz negatividade no interior da célula, porque os
íons cloreto, com carga negativa, se difundem para o interior,
ao mesmo tempo que os íons positivos ficam do lado de fora.
Quarto veremos adiante que as permeabilidades dos canais
de sódio e de potássio passam por variações muito rápidas,
durante a condução do impulso nervoso, enquanto a
permeabilidade dos canais de cloreto não se altera de muito
nesse processo. Por conseguinte, as variações das
permeabilidades ao sódio e ao potássio são as principais
responsáveis pela transmissão dos sinais pelos nervos, o que é o
assunto do resto deste capítulo.
MEDIDA DO POTENCIAL DE MEMBRANAS
O método para a medida do potencial de membrana é simples em
teoria, mas, muitas vezes, difícil na prática, devido às dimensões diminutas
da maioria das fibras. A Fig. 5.2 mostra pequena pipeta, cheia de solução
eletrolítica concentrada (KC1), que é impalada, através da membrana
celular, até o interior da fibra. Em seguida, outro eletródio, chamado
de "eletródio indiferente", é colocado nos líquidos intersticiais, e a
diferença de potencial entre o interior e o exterior da fibra é medida
por meio de voltímetro adequado. Esse voltímetro é, na realidade, um
aparelho eletrônico muito sofisticado, capaz de medir voltagens bastantes
reduzidas, apesar de haver resistência extremamente elevada ao fluxo
elétrico pela ponta da micropipeta. que tem, em geral, diâmetro
inferior a 1 µm e resistência que pode atingir um bilhão de ohms.
Para o registro de variações rápidas do potencial de membrana,
durante a transmissão de impulsos nervosos, o microeletródio é ligado a
um osciloscópio, como explicado adiante, neste capítulo.
A MEMBRANA CELULAR COMO UM
CAPACITADOR ELÉTRICO
Em cada uma das figuras usadas até agora, as cargas iônicas negativas
e positivas que geram o potencial de membrana foram mostradas como
dispostas em contato com a membrana, e nada se falou da sua disposição
em outras partes dos líquidos, nem do interior da fibra nervosa, ou
fora dela, no líquido intersticial. Contudo, a Fig. 5.3 mostra isso,
destacando que, exceto na região adjacente à própria membrana
celular, as cargas negativas e positivas estão precisamente em
igualdade. Isso é chamado de princípio da neutralidade elétrica; isto é,
para cada íon positivo, existe, na vizinhança, outro íon, negativo, para
neutralizá-lo; de outra forma, seriam gerados potenciais elétricos de
bilhões de volts nesses líquidos.
Quando cargas positivas são bombeadas para fora da membrana,
essas cargas positivas se alinham ao longo da face externa da membrana,
enquanto em sua face interna se alinham os ânions que permaneceram
no interior da fibra.
Fig. 5.2 Medida do potencial de membrana de fibra nervosa por meio
de micropipeta.
47
Fig. 5.3 Distribuição dos íons com cargas positivas e negativas no líquido
intersticial que banha uma fibra nervosa e no líquido no interior da
fibra; notar a disposição em dipolos das cargas negativas na superfície
interna da membrana e das cargas positivas na face externa, No painel
inferior são mostradas as variações abruptas do potencial de membrana,
ao nível da membrana, nos dois lados da fibra.
Isso cria uma camada de dipolos, de cargas positivas e negativas, entre as
faces externa e interna da membrana, mas permanece igual número de
cargas positivas e negativas nas outras regiões dos líquidos. Esse é o
mesmo efeito observado quando as placas de capacitador (ou
condensador) elétrico são eletricamente carregadas — isto é, o
alinhamento de cargas negativas e positivas nos lados opostos da
membrana dielétrica do capacitador. Por conseguinte, a bicamada
lipídica da membrana celular funciona, na realidade, como o dielétrico
do capacitador da membrana celular, do mesmo modo como mica papel
ou Mylar funcionam como dielétrico em capacitadores elétricos.
Por ser a membrana celular extremamente fina (de 7 a 10 nm),
sua capacitância é imensa, em relação à sua área — cerca de 1 microfarad
por centímetro quadrado.
A parte inferior da Fig. 5.3 mostra o potencial elétrico que vai
ser registrado em cada ponto na ou próximo da membrana de fibra
nervosa, começando do lado esquerdo da figura e progredindo para
a direita, Enquanto o eletródio estiver por fora da membrana neural,
o potencial que vai ser registrado será zero, que é o potencial do líquido
extracelular. Em seguida, quando o eletródio atravessar a camada do
dipolo elétrico na membrana celular, o potencial diminuirá,
imediatamente, até -90 mV. De novo, o potencial de membrana
permanece em valor estável enquanto o eletródio passa pelo interior da
fibra, mas volta a zero, ao passar pela região oposta da membrana.
O fato de a membrana funcionar como um capacitor tem um ponto
de importância especial: para que seja criado um potencial negativo
no interior da membrana, basta o transporte de número de íons positivos
suficientes para a produção da camada elétrica de dipolos do nível da
própria membrana. Todos os íons remanescentes no interior da fibra
ainda podem ser íons positivos e negativos. Como resultado, apenas
um número extremamente reduzido de íons precisa ser transferido para
produzir o potencial normal de —90 mV no interior da fibra nervosa
— apenas cerca de 1/5.000,000 a 1/100.000.000 das cargas positivas totais
do interior da fibra precisa ser transferido. Também, número igualmente
diminuto de íons positivos se deslocando do exterior para o interior
pode inverter o potencial de —90 mV para até +35 mV dentro de intervalo
de tempo da ordem de 1/10. 000 de segundo. Esse rápido deslocamento
de íons, dessa forma, gera os sinais nervosos que iremos discutir em
seções subseqüentes deste capítulo.
de cerca de -90 mV. Isto é, o potencial no interior da fibra é
90 mV mais negativo que o potencial do líquido intersticial,
por fora da fibra. Nos parágrafos seguintes, vamos explicar todos
os fatores que determinam o valor desse potencial, mas, antes
de fazê-lo, devemos descrever as propriedades de transporte da
membrana neural em repouso para o sódio e o potássio.
Transporte ativo dos íons sódio e potássio através da
membrana - a bomba sódio-potássio. Primeiro, devemos nos
lembrar do que foi discutido no capítulo precedente, que todas
as membranas celulares do corpo apresentam uma potente
bomba sódio-potássio e que essa bomba, continuamente,
bombeia sódio para o exterior e potássio para o interior. Devemos
nos lembrar, ainda, que essa é uma bomba eletrogênica, pois
mais cargas positivas são bombeadas para fora que para
dentro (três íons Na + para o exterior para cada dois íons K +
para o interior), deixando déficit efetivo de íons positivos no
interior; isso é o mesmo que criar carga negativa no interior da
membrana celular.
Essa bomba sódio-potássio também é causa dos imensos
gradientes de concentração para o sódio e o potássio através
da membrana neural em repouso. Esses gradientes são os
seguintes:
Na+
Na+
K+
K+
(externa):
(interna):
(externo):
(interno):
142 mEqA
14 mEq/1
4 mEq/1
140 mEq/1
As proporções entre esses dois
íons, do interior para o exterior, são:
Na+ interior / Na + exterior = 0,1
K+ interior / K+ exterior
= 35,0
Vazamento de sódio e de potássio através da membrana
neural. A direita na Fig. 5.4 é mostrada uma proteína de
canal da membrana celular, pela qual os íons sódio e potássio
podem vazar, denominada canal de "vazamento" para sódiopotássio. Na verdade, existem diversos tipos distintos de
proteínas desse tipo, com características diferentes de
vazamento. Contudo, a ênfase recai sobre o vazamento de
potássio, porque, em média, os canais são muito mais
permeáveis ao potássio que ao sódio, cerca de 100 vezes mais.
Veremos adiante que essa permeabilidade diferencial é
extremamente importante para a determinação do valor do
potencial de membrana normal em repouso.
O POTENCIAL DE MEMBRANA EM
REPOUSO DOS NERVOS
O potencial de membrana das fibras nervosas de grande
calibre, quando elas não estão transmitindo sinais nervosos, é
Fig. 5.4 As características funcionais da bomba Na+-K+ e dos canais
de "vazamento" para o potássio.
48
ORIGEM DO POTENCIAL DE MEMBRANA
EM REPOUSO NORMAL
A Fig. 5.5 apresenta os fatores importantes para o
estabelecimento do potencial de membrana em repouso normal
de -90 mV. Eles são os seguintes:
Contribuição do potencial de difusão do potássio. Na Fig.
5.5A, admitimos que o único movimento de íons através da
membrana seja por difusão de íons potássio, como mostrado
pelos canais abertos para o potássio, entre o interior da
membrana e o exterior. Devido à proporção muito alta entre as
concentrações interna e externa desse íon, da ordem de 35
para 1, o potencial de Nernst correspondente a essa
proporção é de -94 mV, dado que o logaritmo de 35 é 1,54 e
esse valor multiplicado por -61 dá -94 mV. Por conseguinte, caso
os íons potássio fossem o único fator causador do potencial de
repouso, esse potencial de repouso também seria igual a -94
mV, como mostrado na figura.
Contribuição da difusão do sódio através da membrana
neural.
A Fig. 5.5B mostra a adição da reduzida
permeabilidade
da membrana neural aos íons sódio, causada pela diminuta
difusão de íons sódio pelos canais de vazamento K+ -Na+. A
proporção entre os íons sódio do interior e do exterior é de
0,1 e isso dá um valor calculado para o potencial de Nernst
para o interior da membrana de +61 mV. Mas também é
mostrado na Fig. 5.5B o potencial de Nernst para a difusão do
potássio de -94 mV. Como eles interagem entre si e qual vai ser o
potencial resultante? Isso pode ser respondido por meio da
equação de Goldman, descrita antes. Todavia, de modo intuitivo,
pode-se ver que, se a membrana for muito permeável ao
potássio, mas pouco permeável ao sódio, é lógico que a difusão
de potássio terá contribuição muito maior para o potencial de
membrana que a difusão do sódio. Na fibra nervosa normal, a
permeabilidade da membrana ao potássio é cerca de 100 vezes
maior que para o sódio. Se for usado esse valor na equação de
Goldman. obtém-se valor para o potencial interno da membrana
de -86 mV, como mostrado à direita da figura.
Contribuição da bomba Na+- K+ Finalmente, na Fig. 5.5C, é
mostrada a contribuição adicional da bomba Na+-K+. Nessa
figura, ocorre bombeamento contínuo de três íons sódio para
o exterior, e de dois íons potássio para o interior da membrana.
O fato de serem bombeados mais íons sódio para o exterior
que de potássio para o interior resulta em perda continuada
de cargas positivas pelo interior da membrana, o que causa grau
adicional de negatividade (de cerca de - 4 mV) do interior, além
da que poderia ser explicada apenas por difusão. Por conseguinte,
como mostrado na Fig. 5.5C, o potencial de membrana efetivo,
com todos esses fatores atuando ao mesmo tempo, é de -90 mV.
Em resumo, apenas os potenciais de difusão, causados pela
difusão de potássio e de sódio, produziriam um potencial de
membrana da ordem de - 86 mV, quase que todo ele resultante
da difusão de potássio. Além disso, cerca de - 4 mV adicionais
representam a contribuição para o potencial de membrana da
bomba eletrogênica de Na + -K+, dando o potencial efetivo de
membrana de -90 mV.
O potencial de membrana em repouso nas grandes fibras
musculares esqueléticas é, aproximadamente, o mesmo que o
das fibras nervosas mais calibrosas, em torno de -90 mV.
Contudo, nas fibras nervosas mais delgadas c nas menores fibras
musculares — por exemplo, as do músculo liso —, bem como
em muitos neurônios do sistema nervoso central, o potencial de
membrana pode ser de apenas -40 a -60 mV, em vez de -90
mV.
O POTENCIAL DE AÇÃO NEURAL
Fig, 5.5 Desenvolvimento do potencial de membrana em repouso, em
fibras nervosas, sob três condições distintas: A, quando o potencial de
membrana é causado apenas pela difusão de potássio; B quando o
potencial de membrana é causado pela difusão de íons sódio e potássio;
C, quando o potencial de membrana é causado pela difusão conjunta
de íons sódio e potássio, mais o bombeamento desses dois íons pela
bomba Na--K+.
Os sinais neurais são transmitidos por meio de potenciais
de ação, que são variações muito rápidas do potencial de
membrana. Cada potencial de ação começa por modificação
abrupta do potencial de repouso negativo normal para um
potencial positivo e, em seguida, termina com modificação
quase tão rápida para o potencial negativo. Para conduzir um
sinal neural, o potencial de ação se desloca, ao longo da fibra
nervosa, até atingir seu término. O painel superior da Fig. 5.6
apresenta as alterações que ocorrem na membrana durante o
potencial de ação, com transferência de cargas positivas para
o interior no seu início e retorno dessas cargas positivas
para o exterior ao seu fim. O painel inferior retrata
graficamente as alterações sucessivas do potencial de
membrana, durante alguns poucos décimos milésimos de
segundo, mostrando o início explosivo do potencial de ação e
sua restauração em tempo quase tão rápido.
As fases sucessivas do potencial de ação são as seguintes:
Fase de repouso. É o potencial de membrana em repouso,
antes que comece o potencial de ação. Diz-se que a membrana
está "polarizada" durante esta fase, devido ao elevado potencial
de membrana presente.
49
Fig. 5.6 Potencial de ação típico, registrado pelo método
mostrado no painel superior da figura.
Fase de despolarização. Em determinado momento, a
membrana fica, abruptamente, muito permeável aos íons
sódio, o que permite a entrada de grande número de íons
sódio para o interior do axônio. O estado "polarizado" normal
de - 90 mV é perdido, com o potencial variando rapidamente
na direção da positividade. Isso é chamado de despolarização.
Nas fibras nervosas mais calibrosas, o potencial de membrana
"ultrapassa" (overshoots) o valor zero, e fica positivo, mas, nas
fibras mais finas e em muitos neurônios do sistema nervoso
central, o potencial apenas fica próximo do valor zero, e não o
ultrapassa para ficar positivo.
Fase de repolarização. Dentro de poucos décimos milésimos
de segundo após a membrana ter ficado extremamente permeável
aos íons sódio, os canais de sódio começam a se fechar, enquanto
os canais de potássio se abrem mais que o normal. Então, a
rápida difusão dos íons potássio para o exterior restaura o
potencial de membrana negativo normal do repouso. Isso é
chamado de repolarização da membrana.
Para explicar com mais detalhes os fatores responsáveis pelos
processos de despolarização e de repolarização, precisamos,
agora, explicar as características especiais de outros dois tipos
de canais para o transporte através da membrana neural: os
canais de sódio e de potássio voltagem-dependentes.
OS CANAIS DE SÓDIO E DE POTÁSSIO
VOLTAGEM DEPENDENTES
O agente necessário para a produção da despolarização e
da repolarização da membrana neural, durante o potencial de
ação, é o canal de sódio voltagem-dependente. Contudo, o canal
de potássio voltagem dependente também tem participação
importante ao aumentar a rapidez da repolarização da
membrana. Esses dois canais voltagem - dependentes existem
juntamente com a bomba Na+ -K+ e os canais de vazamento Na+ K+.
O canal de sódio voltagem-dependente — "ativação"
e "inativação" do canal
O painel superior da Fig. 5.7 apresenta o canal de sódio
voltagem-dependente em três estados distintos. Esse canal tem
Fig. 5.7 Características dos canais de sódio e potássio voltagemdependentes, mostrando a ativação e a inativação dos canais de sódio,
mas apenas a ativação dos canais de potássio, que só ocorre quando o
potencial de membrana varia de seu valor negativo normal em repouso
para um valor positivo.
duas comportas, uma próxima à extremidade externa do canal,
chamada de comporta de ativação, e outra próxima à extremidade
interna, chamada de comporta de inativação. A esquerda é
mostrado o estado dessas duas comportas no período normal
de repouso, quando o potencial de membrana é de —90 mV.
Nesse estágio, a comporta de ativação fica fechada, o que
impede o acesso de qualquer íon sódio ao interior da fibra por
meio desses canais. Por outro lado, as comportas de inativação
estão abertas, não constituindo, nesse estágio, qualquer
impedimento à passagem de íons sódio.
Ativação do canal de sódio. Quando o potencial de membrana
fica menos negativo que durante o período de repouso, passando
de -90 mV para zero, ele passa por uma voltagem. em geral
entre -70 e -50 mV, que provoca alteração conformacional
da comporta de ativação, fazendo com que ela se abra. Isso
é chamado de estado ativado; durante ele, os íons sódio podem
literalmente jorrar por esses canais, aumentando a
permeabilidade ao sódio da membrana por até 500 a 5.000
vezes.
Inativação do canal de sódio. Na extrema direita do painel
superior da Fig. 5.7 é mostrado o terceiro estado do canal de
sódio. O mesmo aumento da voltagem que abre a comporta
de ativação também fecha a comporta de inativação. Contudo,
o fechamento da comporta de inativação só ocorre após alguns
décimos milésimos de segundo da abertura da comporta de
ativação. Isto é, a alteração conformacional que modifica o
canal de inativação para a posição fechada é um processo mais
lento, enquanto a alteração conformacional que abre a comporta
de ativação é muito rápida. Como resultado, após o canal de
sódio ter ficado aberto por alguns décimos milésimos de
segundo, ele se fecha e os íons sódio não mais podem jorrar
para o interior da membrana. A partir desse momento, o
potencial de membrana começa a variar em direção ao valor
vigente no estado de repouso da membrana, o que constitui o
processo de repolarização.
Característica muito importante do processo de inativação
50
do canal de sódio é a de que a comporta de inativação não voltará
a se abrir até que o potencial de membrana retorne até (ou bastante
próximo) o valor do potencial de membrana de repouso inicial.
Como conseqüência, não é possível nova abertura dos canais
de sódio até que a fibra nervosa se tenha repolarizado.
Os canais de potássio voltagem dependentes e sua
ativação
O painel inferior da Fig. 5.7 apresenta o canal de potássio
voltagem-dependente em dois estados distintos: durante o estado
de repouso e próximo ao término do potencial de ação. Durante
o estado de repouso, o canal de potássio fica fechado, como
mostrado à esquerda da figura, e os íons potássio são impedidos
de passar por esse canal para o exterior. Quando o potencial
de membrana começa a aumentar, a partir de -90 mV, em
direção ao zero, essa variação de voltagem provoca alteração
conformacional, abrindo o canal e permitindo aumento da difusão
do potássio por ele. Contudo, devido à lentidão com que esses
canais de potássio se abrem, eles ficam abertos apenas a partir
do momento em que os canais de sódio começam a ser inativados
e, portanto, se fechando. Assim, a diminuição do influxo de
sódio para a célula, com aumento simultâneo do efluxo de
potássio, acelera de muito o processo da repolarização, levando,
dentro de poucos décimos milésimos de segundo, à recuperação
completa do potencial de membrana de repouso.
O método de estudo para a medida do efeito da voltagem sobre a
abertura e fechamento dos canais voltagem-dependentes - a "fixação
de voltagem". Os estudos originais que levaram à nossa compreensão
quantitativa dos canais de sódio e de potássio foram tão engenhosos que
levaram à outorga de Prêmios Nobel aos cientistas responsáveis, Hodgkin
e Hux-ley. A parte fundamental desses estudos é mostrada nas Figs.
5.8 e 5.9.
A Fig. 5.8 mostra o equipamento usado nesses experimentos,
chamado de fixador de voltagem, que foi usado para medir o fluxo de
íons pelos diferentes canais. Ao se usar esse equipamento, dois
eletródios são introduzidos na fibra nervosa. Um deles destina-se à
medida da voltagem do potencial de membrana. A outra é usada para
conduzir corrente elétrica, tanto para dentro, como para fora da fibra
nervosa. Esse equipamento é usado do seguinte modo: o
experimentador escolhe a voltagem que deseja produzir no interior da
fibra nervosa. Em seguida, ajusta a parte eletrônica do equipamento
para a voltagem desejada, o que, automaticamente, faz com que
ocorra injeção de eletricidade positiva ou negativa, por meio do eletródio
de corrente, na intensidade necessária para manter a corrente, como
medida pelo eletródio de voltagem, no valor escolhido pelo
experimentador. Por exemplo, quando o potencial de membrana é
subitamente aumentado de -90 mV até zero, os canais de sódio e
potássio voltagem-dependentes se abrem.
Fig. 5.8 O método de "fixação de voltagem" para o estudo do fluxo
de íons por seus canais específicos.
e os íons sódio e potássio começam a jorrar pelos canais. Para
contrabalançar o efeito desses movimentos iônicos sobre o potencial
fixado, é injetada corrente elétrica, automaticamente, por meio do
eletródio de corrente do fixador de voltagem, para manter a voltagem
intracelular em zero. Para que isso possa ocorrer, a corrente injetada
deve ser exata-mente igual, mas com a polaridade inversa, ao fluxo
efetivo de corrente através dos canais da membrana. Para a medida de
quanto fluxo de corrente está ocorrendo a cada instante, o eletródio de
corrente é ligado a um osciloscópio que registra o fluxo de corrente,
como mostrado na tela do osciloscópio na figura. Finalmente, o
experimentador ajusta as concentrações dos íons aos valores desejados,
tanto no interior como por fora da fibra, e repete a medida. Isso pode ser
feito com bastante facilidade quando são usadas fibras nervosas muito
calibrosas, obtidas de crustáceos, em especial o axônio gigante da
lula que, por vezes, chega a ter diâmetro de 1 mm. Quando o sódio é o
único íon permeante nas soluções interna e externa que banham o axônio
da lula, esse método de fixação de voltagem só mede o fluxo de
corrente pelos canais de sódio. Quando o único íon permeante é o
potássio, só é medido o fluxo de corrente pelos canais de potássio.
Outro método para estudo do fluxo de íons por canais com
características específicas é por bloqueio de um tipo de canal de
cada vez. Por exemplo, os canais de sódio podem ser bloqueados pela
toxina tetro-dotoxina, quando aplicada à superfície externa da fibra
nervosa, onde ficam as comportas de ativação do sódio. Inversamente, o
tetraetilamônio bloqueia os poros de potássio, quando c aplicado ã
superfície interna da fibra nervosa.
A Fig. 5.9 mostra as variações típicas da condutância dos canais
de sódio e potássio voltagem-dependentes, quando se faz o potencial
de membrana variar subitamente, por meio do fixador de voltagem,
de -90 mV para +10 mV e, 2 ms depois, de volta a -90 mV. Deve
ser notada a abertura abrupta dos canais de sódio (estágio de ativação)
dentro de fração muito pequena de milissegundo após o potencial de
membrana ter atingido o valor positivo. Contudo, durante
aproximadamente o milissegundo seguinte, os canais de sódio se fecham
de forma automática (estágio de inativação).
Agora, note-se a abertura (ativação) dos canais de potássio. Eles
se abrem lentamente e só atingem o estado de abertura total após o
fechamento quase completo dos canais de sódio. Ademais, uma vez
abertos os canais de potássio, eles permanecem abertos por toda a
duração do potencial de membrana positivo c não se fecham até que o
potencial de membrana tenha retornado a valor muito negativo.
Finalmente, devemos lembrar que os canais voltagem-dependentes
passam muito rapidamente do estado aberto para o fechado, e vice-versa,
como mostrado na Fig. 4.5, Então, como as curvas da Fig. 5.9 são
tão regulares? A resposta é que essas curvas representam o fluxo de
íons sódio e potássio por literalmente milhares de canais ao mesmo
tempo. Alguns se abrem para determinada voltagem, outros em outro
valor, e assim por diante. De igual forma, alguns são inativados em
pontos distintos do ciclo que outros. Assim, as curvas mostradas
representam a soma algébrica dos fluxos iônicos por muitos canais.
Fig. 5.9 Variações típicas da condutância dos canais iônicos para o sódio
e o potássio, quando o potencial de membrana é aumentado de seu
valor normal de repouso de —90 mV para um valor positivo de +10
mV, durante 2 ms. Esta figura demonstra que os canais de sódio se
abrem (ativação) e fecham (inativação) em tempo menor que 2 ms,
enquanto, nesse período, os canais de potássio só se abrem (ativação).
51
SUMÁRIO DOS EVENTOS QUE CAUSAM
O POTENCIAL DE AÇÃO
A Fig. 5.10 apresenta, de modo sumário, os eventos
seqüenciais que ocorrem durante e logo após o potencial de
ação. Eles são os seguintes:
Na parte inferior da figura são apresentadas as variações
da condutância da membrana aos íons sódio e potássio. Durante
o período de repouso, antes do início do potencial de ação,
a condutância do potássio é mostrada como sendo de 50 a 100
vezes maior que a do sódio. Isso é causado pelo maior vazamento
de íons potássio que de íons sódio pelos canais de vazamento.
Todavia, com o início do potencial de ação, os canais de sódio
ficam instantaneamente ativados, permitindo aumento de 5.000
vezes da condutância do sódio. Em seguida, o processo de
inativação fecha os canais de sódio dentro de fração de
milissegundo. O início do potencial de ação também leva à
ativação, pela voltagem, dos canais de potássio, fazendo-os abrir
em fração de milissegundo após a abertura dos canais de
sódio. E, ao término do potencial de ação, o retorno do
potencial de membrana a seu estado negativo faz com que os
canais de potássio se fechem, voltando a seu estado original, o que
só ocorre após breve retardo.
Na parte média da Fig. 5.10 c mostrada a proporção entre
as condutâncias do sódio e do potássio, instante a instante, du-
rante todo o potencial de ação e, acima disso, o próprio potencial
de ação. Durante a fase inicial do potencial de ação, essa
proporção aumenta por mais de 1.000 vezes. Por conseguinte,
número muito maior de íons sódio está fluindo para o interior
da fibra que de íons potássio para o exterior. E isso que faz
com que o potencial de ação se torne positivo. Em seguida,
os canais de sódio começam a ficar inativados, enquanto os canais
de potássio se abrem, de modo que a proporção entre as
condutâncias varia, passando a uma condutância muito maior do
potássio que do sódio. Isso permite perda muito rápida de íons
potássio para o exterior, enquanto, em essência, não há fluxo
de íons sódio para o interior. Como conseqüência, o potencial
de ação retorna rapidamente a sua linha de base.
O pós-potencial "positivo"
Também deve ser notado na Fig. 5.10 que o potencial de membrana
fica ainda mais negativo que o potencial de membrana de repouso
original, durante alguns milissegundos, após o término do potencial de
ação. Estranhamente, isso é chamado de pós-potencial "positivo", uma
designação errônea, pois o pós-potencial positivo é, na realidade, mais
negativo que o potencial de repouso. A razão para esse pós-potencial ser
chamado de "positivo" é que, historicamente, as primeiras medidas do
potencial foram feitas na superfície externa da membrana da fibra nervosa,
e não em seu interior; quando medido dessa forma, esse pós-potencial
causa deflexão positiva no registro, e não uma deflexão negativa.
A causa do pós-potencial positivo é, em grande parte, que muitos
canais de potássio permanecem abertos após o processo de repolarização
da membrana ter-se completado. Isso permite que excesso de íons
potássio se difunda para fora da fibra nervosa, deixando déficit extra
de íons positivos no interior, o que implica negatividade.
PARTICIPAÇÃO DE OUTROS ÍONS NO POTENCIAL DE
AÇÃO
Fig. 5.10 Variações da condutância para o sódio e o potássio, durante
um potencial de ação. Note-se que a condutância para o sódio aumenta
por vários milhares de vezes, durante as fases iniciais do potencial de
ação, enquanto a do potássio só aumenta por cerca de 30 vezes, durante
as fases finais e por breve período após o término do potencial de ação.
(Estas curvas foram traçadas a partir dos resultados experimentais
publicados de Hodgkin e Huxley, mas transpostos do axônio de lula
para os potenciais de fibras nervosas calibrosas.)
Até agora, discutimos apenas a participação dos íons sódio e potássio
na geração do potencial de ação. Contudo, pelo menos três outros tipos
de íons devem ser levados em conta. Eles são:
Os íons impermeantes com carga negativa (ânions), no interior
do axônio. No interior do axônio existem muitos íons com carga
negativa que não podem passar pelos canais. Eles incluem moléculas de
proteína, muitos compostos orgânicos de fosfato, compostos sulfatados e
muitos outros. Devido a não poderem sair da fibra, qualquer déficit de
íons positivos no interior da membrana leva a excesso de íons negativos
impermeantes. Por conseguinte, esses íons negativos impermeantes são
responsáveis pelas cargas negativas no interior da fibra, sempre que
houver déficit de íons potássio com carga positiva ou de outros íons
positivos.
Íons cálcio. As membranas celulares de quase todas — se não de
todas — as células do corpo contêm uma bomba de cálcio semelhante
à bomba de sódio. Como ocorre com a bomba de sódio, essa bomba
de cálcio bombeia os íons cálcio do interior para o exterior da membrana
(ou para o retículo endoplasmático), criando gradiente de cálcio de cerca
de 10.000 vezes, deixando concentração interna de íons cálcio da ordem
de 10-7 molar, contrastando com a concentração externa de cerca de
10-3 molar. Além disso, também existem canais de cálcio voltagemdependentes. Esses canais são pouco permeáveis aos íons sódio, além de
serem permeáveis aos íons cálcio; quando abertos, tanto os íons cálcio
como os íons sódio fluem para o interior da fibra. Por isso, esses
canais são, por vezes, chamados de canais Ca+ + -Na+. Os canais de
cálcio têm ativação muito lenta, necessitando de tempo 10 a 20 vezes
maior para serem ativados que os canais de sódio. Essa é a razão de
serem, com muita freqüência, denominados canais lentos, para distinguilos dos canais de sódio, chamados de canais rápidos. Os canais de
cálcio são muito numerosos no músculo cardíaco e no músculo liso.
Na verdade, em alguns tipos de músculo liso, os canais rápidos de sódio
são bastante raros, de modo que seu potencial de ação é causado quase
que totalmente pela ativação dos canais lentos de cálcio.
Aumento da permeabilidade dos canais de sódio quando
existe déficit de íons cálcio. A concentração de íons cálcio no
líquido intersticial também exerce efeito intenso sobre o valor da
voltagem em que os canais de sódio ficam ativados.
52
. Quando existe déficit de íons cálcio, os canais de sódio são ativados
(abertos) por aumento bastante pequeno do potencial de membrana,
acima de seu nível normal de repouso. Como resultado, a fibra fica
extremamente excitável, por vezes disparando repetitivamente, sem
qualquer provocação, em vez de permanecer no estado de repouso. Na
verdade, basta que a concentração de íons cálcio baixe por 30 a 5(1%
abaixo da normal para que ocorra atividade espontânea em muitos
nervos periféricos, causando, muitas vezes, a "telania" muscular que pode
chegar a ser fatal, devido à contração tetânica dos músculos respiratórios.
O modo provável de os íons cálcio influenciarem os canais de sódio
é o seguinte. Esses íons parecem fixar-se às extremidades externas da
molécula de proteína do canal de sódio. Por sua vez, as cargas positivas
desses íons cálcio alteram o estado elétrico da própria proteína do canal
e, dessa forma, aumentam o valor da voltagem necessária para abrir
a comporta.
Íons cloreto. Os íons cloreto vazam através da membrana em repouso
de modo idêntico ao do vazamento de pequenas quantidades de íons
potássio e íons sódio. Na fibra nervosa comum, a intensidade do
vazamento de íons cloreto é apenas a metade da difusão de íons
potássio. Por conseguinte, deve ser feita a pergunta: por que não
consideramos os íons cloreto em nossa explicação do potencial de
ação? A resposta é que o íon cloreto funciona passivamente no
processo. E, também, a permeabilidade dos canais de vazamento de
cloreto não se altera de forma significativa durante o potencial de ação.
No estado normal de repouso da fibra, os -90 mV no interior
da fibra repelem a maior parte dos íons cloreto que tendem a entrar
nela. Como resultado, a concentração de íons cloreto no interior da
fibra é de apenas 3 a 4 mEq/l, enquanto a concentração externa é da
ordem de 103 mEqA. O potencial de Nemst para essa proporção entre
as concentrações do íon cloreto é exatamente igual ao potencial de
membrana de -90 mV, que seria previsto para um íon que não é
bombeado ativamente.
Durante o potencial de ação, pequenas quantidades de íon cloreto
chegam efetivamente a se difundir para o interior da fibra, devido a
perda temporária da negatividade interna. Esse movimento do cloreto
serve para alterar ligeiramente o momento de ocorrência das variações
sucessivas de voltagem durante o potencial de ação, mas não modifica
o processo fundamental.
GERAÇÃO DO POTENCIAL DE AÇÃO
Ate este ponto, explicamos a variação das permeabilidades
ao sódio e ao potássio da membrana e o desenvolvimento do
próprio potencial de ação, mas ainda não explicamos o que
produz o potencial de ação. A resposta a isso, como se segue,
é muito simples.
Um feedback positivo abre os canais de sódio. Primeiro,
enquanto a membrana da fibra nervosa permanecer sem sofrer
qualquer perturbação, nenhum potencial de ação ocorre no nervo
normal. Contudo, se algum fator produzir uma elevação inicial
suficiente do potencial de membrana, a partir do valor de -90
mV, em direção ao zero, essa elevação da voltagem irá fazer
com que muitos canais de sódio voltagem-dependentes comecem
a se abrir. Isso permite o influxo rápido de íons sódio, o que
produz elevação ainda maior do potencial de membrana, abrindo,
assim, número ainda maior de canais de sódio voltagemdependentes e resultando em jorro mais intenso de íons sódio
para o interior da fibra. Obviamente, esse processo é um ciclo
vicioso de feedback positivo que, caso esse feedback seja
suficientemente intenso, irá prosseguir até que todos os canais de
sódio voltagem-dependentes fiquem ativados (abertos). Em
seguida, dentro de fração de milissegundo, a elevação do
potencial de membrana produz o início da inativação dos canais de
sódio, além da abertura dos canais de potássio, e o potencial de
ação logo chega a seu fim.
Limiar para a geração do potencial de ação. Não
ocorrerá um potencial de ação até que a elevação do potencial
de membrana seja suficientemente grande para criar o ciclo
vicioso descrito no parágrafo anterior.
Em geral, é necessária elevação abrupta da ordem de 15 a 30
mV. Por conseguinte, aumento súbito do potencial de
membrana., em fibra calibrosa, de - 90 mV até cerca de - 65
mV será capaz, na maioria das vezes, de deflagrar o
desenvolvimento explosivo do potencial de ação. Esse nível de 65 mV é, conseqüentemente, chamado de limiar para a
estimulação.
Acomodação da membrana — falta de atividade apesar da
elevação da voltagem. Se o potencial da membrana se elevar de
forma muito lenta — durante vários milissegundos, em vez de em fração
de milissegundo —, as comportas lentas de inativação dos canais de
sódio terão tempo suficiente para se fecharem ao mesmo tempo que as
comportas de ativação estiverem se abrindo. Como resultado, a abertura
das comportas de ativação não será tão eficaz para promover o
aumento do fluxo de íons sódio, como ocorre normalmente. Portanto,
o aumento lento do potencial interno de uma fibra nervosa vai exigir
voltagem limiar mais elevada ou impede, completamente, a geração de
potencial de ação; por vezes, até mesmo com elevações da voltagem
até zero ou com voltagem positiva. Esse fenômeno é chamado de
acomodação da membrana ao estímulo.
PROPAGAÇÃO DO POTENCIAL DE AÇÃO
Nos parágrafos precedentes, discutimos o potencial de ação
como se ele ocorresse em ponto único da membrana celular.
Contudo, um potencial de ação gerado em qualquer ponto de
uma membrana excitável excita, geralmente, as regiões
adjacentes da membrana, resultando na propagação desse
potencial de ação. O mecanismo disso é mostrado na Fig. 5.11.
A Fig. 5.11A mostra uma fibra nervosa normal cm repouso,
enquanto a Fig. 5.11B mostra fibra que foi excitada em sua
região média — isto é, essa porção média, subitamente, passou a
apresentar permeabilidade aumentada para o sódio. As setas
indicam o "circuito local" do fluxo de corrente entre as regiões
despolarizadas da membrana e as áreas adjacentes da
membrana em
Fig. 5.11 Propagação dos potenciais de ação nas duas direções, em
fibra condutora.
53
repouso; cargas elétricas positivas, carreadas pelos íons sódio
que se difundem para o interior, fluem para o interior da fibra,
através da região despolarizada e, em seguida, por vários
milímetros, ao longo da parte central do axônio. Essas cargas
positivas aumentam a voltagem, por distância de 1 a 3 mm nas
fibras calibrosas, até valor acima do valor limiar da voltagem
para geração do potencial de ação. Por conseguinte, os canais
de sódio nessas novas áreas ficam imediatamente ativados e,
como mostrado na Fig. 5.11C e D, o explosivo potencial de ação
se propaga. Em seguida, essas novas áreas despolarizadas
produzem novos circuitos locais de fluxo de corrente cm pontos
mais distantes da membrana, causando mais e mais
despolarizações. Dessa forma, o processo da despolarização
trafega ao longo de toda a extensão da fibra. A transmissão do
processo de despolarização ao longo de fibra nervosa ou
muscular é chamada de impulso nervoso ou muscular.
Direção da propagação. É óbvio, como mostrado na Fig.
5.11, que uma membrana excitável não apresenta direção única
de propagação, o potencial de ação podendo passar nas duas
direções a partir do ponto estimulado — e até mesmo pelas
ramificações de uma fibra nervosa — até que toda a membrana
seja despolarizada.
O princípio do tudo-ou-nada. É igualmente óbvio que,
uma vez tendo sido produzido um potencial de ação, em algum
ponto da membrana de fibra normal, o processo de
despolarização irá se propagar, se as condições forem
adequadas, por toda a membrana e, caso as condições não
sejam adequadas, poderá não se propagar. Isso é chamado de
princípio do tudo-ou-nada e é aplicável a todos os tecidos
excitáveis normais. Todavia, ocasionalmente, o potencial de
ação poderá atingir região da membrana onde não será capaz de
gerar voltagem suficiente para estimular a área seguinte da
membrana. Quando isso ocorre, a propagação da
despolarização cessa. Por conseguinte, para que ocorra a
propagação continuada de um impulso, a proporção entre o
potencial de ação e o limiar deve ser sempre maior que 1. Isso
é chamado de fator de segurança para a propagação.
Fig. 5.12 Produção de calor por fibra nervosa, durante o repouso e
sob freqüência crescente de estimulação.
A Fig. 5.12 mostra que a fibra nervosa produz calor em excesso,
o que representa medida de seu consumo de energia, quando
aumenta a freqüência dos impulsos.
Característica especial da ATPase da bomba sódio-potássio
é que o grau de sua atividade fica fortemente estimulado quando
ocorre acúmulo excessivo de íons sódio no interior da membrana
celular.
Na verdade, a atividade de bombeamento aumenta,
aproximadamente, em proporção ao cubo da concentração de
sódio. Isto é, conforme a concentração interna de sódio
aumenta de 10 para 20 mEq/1, a atividade da bomba não fica,
simplesmente, duplicada, mas, ao contrário, aumenta por cerca
de oito vezes.
Por conseguinte, pode ser facilmente compreendido como o
processo de "recarga" da fibra nervosa pode entrar
rapidamente em ação, sempre que as diferenças de
concentração dos íons sódio e potássio, através da membrana,
RESTABELECIMENTO DOS GRADIENTES IÔNICOS começarem a "desaparecer".
DO SÓDIO E DO POTÁSSIO APÓS OS POTENCIAIS DE
O PLATÔ DE ALGUNS POTENCIAIS DE AÇÃO
AÇÃO - A IMPORTÂNCIA DO METABOLISMO
ENERGÉTICO
Em alguns casos, a membrana excitável não se repolariza
imediatamente após a despolarização, mas, pelo contrário, o
A transmissão de cada impulso ao longo da fibra nervosa potencial permanece em platô, com valor próximo ao do potencial
reduz, por quantidade infinitesimal, as diferenças de em ponta, durante muitos milissegundos antes do começo da
concentração do sódio e do potássio entre o interior e o exterior repolarização. Um desses platôs é mostrado na Fig. 5.13; pode
da membrana, devido à difusão de sódio para dentro, durante a ser facilmente notado que o platô prolonga de muito a duração
despolarização, e à difusão de potássio para fora, durante a do período de despolarização. Esse tipo de potencial de ação
repolarização.Para um só potencial de ação, esse efeito c tão ocorre nas fibras musculares do coração, onde o platô dura por
diminuto que não pode ser medido. Na verdade, de 100.000 a até dois ou três décimos de segundo e faz com que a contração
50.000.000 de impulsos podem ser transmitidos por uma fibra do músculo cardíaco dure por igual período de tempo.
nervosa — esse número depende do calibre da fibra e de
A causa do platô é uma combinação de diversos fatores
outros fatores — antes que as diferenças de concentração distintos.
tenham diminuído a ponto de interromper a condução dos
Primeiro, no músculo cardíaco, dois tipos diferentes de
potenciais de ação. Contudo, mesmo assim, com o correr do canais participam do processo de despolarização: (1) os canais de
tempo, passa a ser necessário o restabelecimento das diferenças sódio voltagem-dependentes usuais, chamados de canais rápidos,
de concentração de sódio e de potássio através da membrana. e (2) canais de cálcio voltagem-dependentes, com ativação lenta,
Isso é efetivado pela ação da bomba Na+ -K+, exatamente da e, por isso, chamados de canais lentos — estes canais permitem,
mesma maneira como foi descrita antes, neste capítulo, para o principalmente, a difusão de íons cálcio, mas também deixam
estabelecimento original do potencial de repouso. Isto é, os íons passar pequena quantidade de íons sódio.
sódio que se difundiram para o interior da célula, durante o
A ativação dos canais rápidos produz o potencial em
potencial de ação, e os íons potássio que se difundiram para o ponta do potencial de ação, enquanto a ativação lenta mas
exterior são retornados a seus locais originais pela bomba Na+- prolongada dos canais lentos é responsável, principalmente,
K+. Visto que esta bomba exige energia para operar, esse pelo platô do potencial de ação.
processo de "recarga" da fibra nervosa é metabolicamente ativo,
Um segundo fator, responsável, em parte, pelo platô, é que
usando energia do sistema do trifosfato de adenosina da célula.
os canais de potássio voltagem-dependentes são, em diversos
casos, de ativação lenta, muitas vezes não se abrindo até o fim
54
Fig. 5.13 Potencial de ação de fibra de Purkinje, mostrando o "platô"
do platô. Isso retarda o retorno do potencial de membrana a
seu valor de repouso. Mas, então, essa abertura dos canais de
potássio, ao mesmo tempo que os canais lentos começam a se
fechar, provoca o retorno rápido do potencial de ação, de seu
valor do platô até o valor negativo de repouso, explicando a
deflexão descendente rápida ao término do potencial de ação.
RITMICIDADE
DE
ALGUNS
TECIDOS
EXCITÁVEIS - A ATIVIDADE REPETITIVA
Atividade repetitiva autogerada, ou ritmicidade, ocorre
normalmente no coração, na maioria dos músculos lisos e em
muitos neurônios do sistema nervoso central. É essa atividade
rítmica que produz o ritmo cardíaco, o peristaltismo e os eventos
neuronais do tipo do controle rítmico da respiração.
Por outro lado, todos os outros tecidos excitáveis podem
apresentar atividade repetitiva caso seus limiares para
estimulação fiquem suficientemente diminuídos. Por exemplo,
até mesmo fibras nervosas e musculares esqueléticas, que são,
normalmente, muito estáveis, podem apresentar atividade
repetitiva quando imersas em solução contendo veratrina ou
quando a concentração de íons cálcio cai abaixo de um valor
crítico.
O processo de reexcitação necessário para a ritmicidade.
Para que ocorra ritmicidade, a membrana, mesmo em seu estado
natural, já deve ser suficientemente permeável aos íons sódio
(ou aos íons cálcio e sódio, pelos canais lentos de cálcio) para
permitir a despolarização automática da membrana. Assim, a
Fig. 5.14 mostra que o potencial de membrana "cm repouso" é
de apenas - 60 a - 70 mV. Essa voltagem não é suficiente
para manter os canais de sódio e de cálcio fechados. Em
outras palavras, (1) existe influxo de íons sódio e de íons cálcio;
(2) isso aumenta, ainda mais, a permeabilidade da membrana; (3)
quantidade ainda maior de íons flui para o interior; (4) a
permeabilidade aumenta mais, e assim por diante, levando ao
processo regenerativo da abertura dos canais de sódio e de
cálcio, até que seja gerado um potencial de ação. Em seguida,
após o término do potencial de ação, a membrana se repolariza.
Mas, pouco depois, recomeça o processo de despolarização, e
novo potencial de ação ocorre espontaneamente. Esse ciclo se
repete por várias vezes e resulta na excitação rítmica autogerada
do tecido excitável.
Contudo, por que a membrana não se despolariza
imediatamente após se ter repolarizado, só o fazendo após
retardo de quase um segundo, antes da geração do potencial de
ação seguinte?
Fig. 5.14 Potenciais de ação rítmicos, semelhantes aos registrados no
centro de controle do ritmo cardíaco. Notar sua relação com a
condutância para o potássio e com o estado de hiperpolarização.
A resposta a isto pode ser dada fazendo-se referência à Fig. 5.10,
que mostra que, próximo ao término de todos os potenciais de
ação, e persistindo por breve período após esse término, a
membrana fica excessivamente permeável ao potássio. Esse
efluxo excessivo de íons potássio transfere número
extremamente elevado de cargas positivas para fora da
membrana, criando, no interior da fibra, negatividade
consideravelmente maior que a que ocorreria, durante breve
período, após o potencial de ação precedente ter terminado,
deslocando, assim, o potencial de membrana para valor mais
próximo do potencial de Nernst para o potássio. Esse é o estado
chamado de hiperpolarização, que é mostrado na Fig. 5.14.
Enquanto esse estado persistir, não ocorrerá reexcitação; mas,
gradativamente, a condutância excessiva do potássio (e o estado
de hiperpolarização) diminui até desaparecer, como mostrado
nessa figura, o que permite que o potencial de membrana
aumente até atingir o limiar para excitação; então, subitamente,
aparece novo potencial de ação: esse ciclo ocorre
repetitivamente.
ASPECTOS ESPECIAIS DA TRANSMISSÃO DE SINAIS
EM TRONCOS NERVOSOS
Fibras nervosas mielínicas e amielínicas. A Fig. 5.15 apresenta
corte transversal de um típico tronco nervoso pequeno, mostrando
muitas fibras nervosas calibrosas que ocupam a maior parte da área
desse corte transverso Todavia, se essa figura for examinada com
cuidado, poderão ser notadas muitas fibras, bem mais delgadas,
intercaladas entre as mais calibrosas. Essas fibras mais calibrosas são
fibras mielínicas, enquanto as mais delgadas são amielínicas. Um tronco
nervoso típico contém cerca de duas vezes mais fibras amielínicas que
mielínicas. A Fig 5.16 mostra uma fibra mielínica típica. A pane central
dessa fibra é o axônio, c a membrana desse axônio que representa a
verdadeira membrana condutora. O interior do axônio é ocupado pelo
axoplasma, que é líquido intracelular bastante" viscoso. Circundando o
axônio existe a bainha de mielina que, muitas vezes, é bem maior que
o próprio axônio e que, a intervalos de cerca de 1 a 3 mm, ao
longo de toda a extensão do axônio, é interrompida pelos nodos de
Ranvier. A bainha de mielina é formada, em torno do axônio, pelas
células de Schwann do seguinte modo: a membrana de uma célula de
Schwann, inicialmente, circunda o axônio. Em seguida, essa célula gira
em torno do axônio por muitas voltas, depositando múltiplas camadas de
sua membrana celular, que contém a substância lipídica esfingomielina.
Essa substância é excelente isolante, capaz de diminuir o fluxo
iônico através da membrana por cerca de 5.000 vezes, ao mesmo
tempo que reduz a capacitância da membrana por 50 vezes. Contudo,
no ponto de junção entre duas células de Schwann sucessivas, ao longo
do axônio, persiste pequena região não-isolada, com apenas cerca de 2 a
3 µm de extensão, por onde os íons podem fluir, com facilidade, do
líquido extracelular para o interior do axônio. Essa região é o nodo de
Ranvier.
Condução "saltatória", de nodo a nodo, nas fibras mielínicas.
55
Fig. 5.15 Corte transverso de pequeno tronco
nervoso, contendo fibras mielínicas e amielínicas.
Muito embora os íons não possam fluir com intensidade significativa
através das espessas bainhas de mielina dos nervos mielinizados, eles
podem fluir com grande facilidade pelos nodos de Ranvier. Por
conseguinte, os potenciais de ação só podem ocorrer nos nodos. Assim,
os potenciais de ação são conduzidos de nodo para nodo, como
mostrado na Fig. 5.17; esse processo é chamado de condução saltatória.
Isto é, a corrente elétrica flui pelos líquidos extracelulares que
circundam a fibra, mas também pelo axaplasma, de nodo a nodo,
excitando seqüencialmente os sucessivos nodos. Assim, o impulso
nervoso salta ao longo da fibra, o que deu origem à designação de
"saltatória".
A condução saltatória é importante por duas razões. Primeira, por
fazer com que a despolarização salte por sobre longos trechos, ao longo
do eixo da fibra nervosa; esse mecanismo aumenta de muito a velocidade
da transmissão neural nas fibras mielinizadas por até 5 a 50 vezes.
Segundo, a condução saltatória conserva energia para o axônio, pois
apenas os nodos despolarizam, permitindo perda de íons cerca de 100
vezes menor do que a que seria necessária, caso não ocorresse condução
saltatória e, como resultado, exigindo pouca atividade metabólica para o
restabelecimento das diferenças de concentração de sódio e potássio,
através da membrana celular, após uma série de impulsos nervosos.
Outra característica da condução saltatória nas grandes fibras mielínicas é a seguinte: o excelente isolamento criado pela membrana de
mielina e a redução de 50 vezes da capacitância da membrana permitem
Flg. 5.16 Função da célula de Schwann no isolamento das fibras
nervosas. A, O enrolamento da membrana da célula de Schwann em
torno de axônio calibroso, para formar a bainha de mielina da fibra
nervosa mielínica. (Modificado de Leeson e Leeson: Hutohgy,
Philadelphia, W.B. SaundersCo., 1919.) B, Evaginação da membrana e
do citoplasma de célula de Schwann em torno de várias fibras nervosas
amielínicas.
Fig. 5.17 Condução saltatória em axônio mielínico.
56
que o processo de repolarização ocorra com transferência muito reduzida
de íons. Assim, ao término do potencial de ação, quando os canais
de sódio começam a fechar, a repolarização ocorre de modo tão rápido
que, em geral, os canais de potássio ainda não estão abertos em número
significativo. Como resultado, a condução do impulso nervoso por fibra
nervosa mielínica é efetuada, quase que inteiramente, pelas variações
seqüenciais dos canais de sódio voltagem-dependentes, com contribuição
muito pequena dos canais de potássio.
VELOCIDADE DE CONDUÇÃO NAS FIBRAS NERVOSAS
A velocidade de condução nas fibras nervosas varia desde o mínimo
de 0,5 m/s, nas fibras amielínicas mais delgadas, até cerca de 100 m/s
(o comprimento de um campo de futebol em um segundo), nas fibras
mielínicas mais calibrosas. Em termos aproximados, essa velocidade de
condução aumenta em proporção direta com o diâmetro nas fibras
mielínicas e com a raiz quadrada do diâmetro da fibra nas amielínicas.
EXCITAÇÃO — O PROCESSO DE PRODUÇÃO DO
POTENCIAL DE AÇÃO
Basicamente, qualquer fator que faça com que os íons sódio possam
fluir para o interior, através da membrana, em número significativo,
irá deflagrar a abertura regenerativa, automática, dos canais de sódio.
Isso pode resultar de perturbação mecânica da membrana, de efeitos
químicos sobre a membrana ou da passagem de eletricidade através da
membrana. Todos esses processos ocorrem em diferentes territórios do
corpo para a produção de potenciais de ação nos nervos e nos músculos:
a pressão mecânica para a excitação de terminações nervosas sensoriais
da pele, os neurotransmissores químicos para a transmissão de sinais
de um neurônio para outro, no sistema nervoso central, c a corrente
elétrica para a transmissão de sinais entre as células musculares do
coração e do intestino. Com o objetivo de compreensão do processo da
excitação, vamos começar pela discussão dos princípios da estimulação
elétrica.
Excitação de fibra nervosa por eletródio metálico com carga
negativa. O método mais comum para excitar um nervo ou músculo, no
laboratório, é o de aplicar eletricidade à superfície do nervo ou do
músculo por meio de dois pequenos eletródios, um dos quais tem
carga negativa e o outro, carga positiva. Quando isso é feito, verifica-se
que a membrana excitável é estimulada pelo eletródio negativo.
A causa desses efeitos é a seguinte. Deve ser lembrado que o
potencial se inicia com a abertura dos canais de sódio voltagem
dependentes. Ainda mais, esses canais são abertos pela diminuição da
voltagem através da membrana. A corrente negativa que passa pelo
eletródio negativo reduz, imediatamente, a voltagem fora da
membrana, reduzindo-a até bem próximo da voltagem negativa no
interior da fibra. Isso reduz a voltagem através da membrana,
permitindo a ativação dos canais de sódio, disso resultando um
potencial de ação. Inversamente, no anódio a injeção de cargas
positivas, por fora da membrana neural, aumenta a diferença de
voltagem através da membrana, e não a diminui. Isso causa estado de
"hiperpolarização", que diminui a excitabilidade da fibra.
O limiar para excitação e os "potenciais locais agudos". Estímulo
elétrico fraco pode não ser capaz de excitar uma fibra. Todavia, à medida
que o estímulo é progressivamente aumentado, é atingido um ponto
onde vai ocorrer a excitação. A Fig. 5.18 mostra os efeitos de estímulos
sucessivos com intensidades crescentes. Estímulo muito fraco {(ponto
A) faz com que o potencial de membrana varie de -90 para —85 mV,
mas essa variação não é suficiente para que se desenvolva o processo
regenerativo automático do potencial de ação. No ponto B, o estímulo
é mais intenso, mas, de novo, ainda insuficiente. Não obstante, esses
estímulos são capazes de alterar localmente o potencial de membrana
por até um milissegundo ou mais, após cada um desses estímulos fracos.
Essas variações locais do potencial são chamadas de potenciais locais
agudos, e, quando não são capazes de produzir potenciais de ação, são
referidos como potenciais subliminares agudos.
No ponto C da Fig. 5.18, o estímulo é ainda mais forte. Aí, o
potencial local apenas atingiu o valor necessário para a produção de
potencial de ação, que é chamado de valor limiar, mas o potencial de
ação só ocorre após certo tempo, que é chamado de "período latente".
No ponto D, o estímulo é ainda mais forte, o potencial local é maior
Fig. 5.18 Efeito dos estímulos sobre o potencial de membranas
excitáveis, mostrando o desenvolvimento de "potenciais subliminares
agudos", quando os estímulos ficam abaixo do valor limiar necessário para
produzir um potencial de ação.
e o potencial de ação ocorre a intervalo menor que o período latente.
Assim, essa figura mostra que até mesmo um estímulo muito fraco
sempre causa variação locai de potencial na membrana, mas que a
intensidade do potencial local deve atingir um valor limiar para que seja
produzido um potencial de ação.
O "período retratário" durante o qual novos potenciais de ação
não podem ser produzidos
Um novo potencial de ação não pode ser produzido enquanto a
membrana estiver despolarizada pelo potencial de ação precedente. A
razão disso é que, logo depois que se inicia um potencial de ação, os
canais de sódio (ou de cálcio, ou os dois) ficam inativados e qualquer
quantidade de sinal excitatório que seja aplicada a esses canais nessa
fase não irá abrir as comportas de inativação. A única condição que
as reabrirá é o retorno do potencial de ação ao valor (ou quase) do
potencial de membrana em repouso. Então, dentro de pequena fração
de segundo, as comportas de inativação dos canais se abrem, e novo
potencial de ação poderá ser produzido.
O intervalo de tempo durante o qual não pode ser produzido outro
potencial de ação, mesmo com estímulo muito forte, é chamado de
período refratário absoluto. Esse período, para as grandes fibras
mielínicas, é da ordem de 1/2. 500 de segundo. Portanto, pode ser
facilmente calculado que essa fibra poderá transmitir, no máximo, 2.500
impulsos por segundo.
Após o período refratário absoluto, existe um período refratário
relativo, com duração entre um quarto e um meio da do período absoluto.
Durante ele, estímulos mais fortes que os normais são capazes de excitar
a fibra. Essa refratariedade relativa tem duas causas: (1) durante ela,
alguns canais de sódio ainda não retornaram de seu estado de inativação,
e (2) nela, os canais de potássio ainda estão, em geral, inteiramente
abertos, produzindo estado de hiperpolarização, que dificulta a
estimulação da fibra.
INIBIÇÃO DA EXCITABILIDADE — "ESTABILIZADORES"
E ANESTÉSICOS LOCAIS
Contrastando com os fatores que aumentam a excitabilidade neural,
existem outros, chamados de fatores estabilizadores da membrana, que
podem diminuir a excitabilidade. Por exemplo, alta concentração
extracelular de íons cálcio diminui a permeabilidade da membrana, ao
mesmo tempo que também diminui sua excitabilidade. Por isso, os íons
cálcio são ditos "estabilizadores". De igual modo, baixa concentração de
íons potássio no líquido extracelular, por ter efeito direto de redução da
permeabilidade dos canais de potássio, também atua como estabilizadora,
reduzindo a excitabilidade da membrana. Ainda mais, na doença
hereditária designada como paralisia periódica familiar, a concentração
extracelular do íon potássio fica, muitas vezes, tão reduzida que a pessoa
chega, na verdade, a ficar paralisada, mas retorna ao normal,
instantaneamente, após administração venosa de potássio.
57
Anestésicos locais. Entre os mais importantes estabilizadores, são
incluídas muitas substâncias usadas na clínica como anestésicos locais,
como a procaína, a tetracaína e muitas outras. A maioria deles atua
diretamente sobre as comportas de ativação dos canais de sódio, fazendo
com que sua abertura fique dificultada e, portanto, reduzindo a
excitabilidade da membrana. Quando a excitabilidade fica tão reduzida
a ponto da proporção entre a. força do potencial de ação e o limiar de
excitabilidade (chamada de "fator de segurança") ser menor que 1, 0, o
potencial de ação não é capaz de atravessar a área anestesiada.
REGISTRO DOS POTENCIAIS DE MEMBRANA E
DE AÇÃO
O osciloscópio de raios catódicos. Antes, neste capítulo, chamamos
atenção para a rapidez com que o potencial de membrana varia no
curso do potencial de ação. Na verdade, todo o complexo do potencial
de ação, nas fibras nervosas calibrosas, ocorre em menos de 1/1. 000
de segundo. Em algumas figuras deste capítulo são mostrados medidores
elétricos para o registro dessas variações de potencial. Todavia, deve
ser entendido que qualquer sistema de medida capaz de registrar essas
variações de potencial deve ter respostas muito rápidas. Para os objetivos
práticos, o único tipo comum de sistema de medida capaz de registrar
com precisão essas variações muito rápidas do potencial de membrana
é o osciloscópio de raios catódicos.
A Fig. 5.19 apresenta os componentes básicos do osciloscópio de
raios catódicos. O tubo de raios catódicos, em si, é composto basicamente
por um canhão de elétrons e por uma superfície fluorescente contra a qual
são lançados os elétrons. No ponto atingido pelos elétrons, a superfície
fluorescente brilha. Se o feixe de elétrons é movido através da
superfície, o ponto brilhante também o faz, traçando linha fluorescente
ao longo dela.
Além do canhão de elétrons e da superfície fluorescente, o tubo
de raios catódicos também dispõe de dois conjuntos de placas
eletricamente carregadas, um desses conjuntos situado nos dois lados
do feixe de elétrons e o outro acima e abaixo dele. Circuitos de controle
eletrônico apropriados fazem variar a voltagem dessas placas, de modo
que o feixe de elétrons possa ser deslocado para cima ou para baixo,
em resposta aos sinais elétricos que vêm dos eletródios de registro nos
nervos. Também, o feixe de elétrons pode ser passado
horizontalmente ao longo da superfície fluorescente ("varredura") com
velocidade constante. Esses dois efeitos dão origem ao registro mostrado
na face do tubo de raios catódicos, com uma linha de tempo horizontal e
a variação de voltagem, nos eletródios dos nervos, no plano vertical. Deve
ser notado, na extremidade esquerda, o pequeno artefato do estímulo,
causado pelo estímulo elétrico usado para produção do potencial de
ação; em seguida, aparece o próprio potencial de ação.
Registro do potencial de ação monofásico. Em todo este capítulo.
Fig. 5.19 O osciloscópio de raios catódicos, usado para o registro de
potenciais de ação transientes,
Fig. 5.20 Registro de potenciais de ação bifásicos
as figuras apresentaram o potencial de ação "monofásico". Para o registro
desses potenciais, uma micropipeta com eletródio, mostrada no início
do capítulo, na Fig. 5.2, foi introduzida no interior da fibra. Em seguida,
à medida que o potencial de ação se propaga ao longo da fibra, foram
registradas as variações de potencial no interior da fibra, como mostrado
nas Figs. 5.6, 5.10 e 5.13.
Registro do potencial de ação bifásico. Quando se deseja registrai
os impulsos em todo um tronco nervoso, não é possível a introdução
de eletródios no interior das fibras desse tronco. Portanto, o método
usual de registro é a colocação de dois eletródios por fora das fibras.
Contudo, o registro que é obtido nessas condições é bifásico, pelas
seguintes razoes: quando um potencial de ação que se propaga ao longo
das fibras atinge o primeiro eletródio, este fica com carga negativa,
enquanto o segundo ainda não é afetado. Isso faz com que o osciloscópio
registre deflexão negativa. Em seguida, com a continuação da
propagação do potencial de ação, ocorre um momento em que a
membrana por sob o primeiro eletródio fica repolarizada, enquanto o
segundo eletródio fica negativo e o osciloscópio registra deflexão na
direção oposta. Assim, um registro como o mostrado na Fig. 5.20, obtido
por osciloscópio, apresenta variação de potencial, primeiro em uma
direção e, em seguida, na oposta.
REFERÊNCIAS
Agnew, W. S.: Voltage-regulated sodium channel molecules. Annu. Rev.
Phyaiol., 45:517, 1984. Armstrong, C. M.: Sodium channels and gating currenta.
Physiol. Rev.,61:644,1981. Auerbach, A., and Sachs, F.: Patch clamp studies of
single ionic channels.
Annu. Rev. Biophys. Bioeng., 13:269,19R4. Riggio, G., and Costa, E.: Chloride
Channels and Their Modulation by Neurotransmitters and Drugs. New York,
Raven Press, 1988. Bretag, A. H.: Muscle chloride channels. Phyaiol. Rev.,
67:1987. Byrne, J. H., and Schultz, S. G.: An Introduction to Membraní
Transportand Bioelectricity. New York, Raven Press, 1988.
Clausen, T\: Regulation of active Na+-K+ transport in skeletal muacle. Physiol.
Rev., 66:542, 1986. Cole, K. S.: Electrodiffusion modela for the membrane of
aquid giant sxon.
Physiol. Rev., 45:340, 1965. Cooper, S. A.: New peripherally-acting oral
analgesic agente. Annu. Rev.Pharmacol. Toxicol., 23:617, 1983.
DeWeer, P., et ai.: Voltage dependence of the Na-K pump. Annu. Rev. Physiol.,
50:225, 1988. DiFrancesco, D., and Noble, D.: A model of cardiac electrical activity
incorporating tonic pumps and concentration changes. Phil. Trans. R. Soe.
Lond. (BioU, 307:353, 1985. DiPolo, R., and Beauge, L.: The calcium pump and
aodium-calcium exchange in squid axons. Annu. Rev. Physiol., 45:313, 1983.
French, R. J., and Horn, R.: Sodium channel gating: Models, mimies, and
moditiers. Annu. Rev. Biophys. Bioeng., 12:319,1983. Garty, H., and Benos, DJ.: Characteriatics and regulatory mechanUms of the amiloride-blockable Na+
channel. Phyaiol. Rev,, 68:309, 198S. Grinnell, A. D., et ai. (eds.): Calcium and
Ion Channel Modulation. New York, Plenum Publishing Corp., 1988. Hille, B.:
Gating in sodium channels of nerve. Annu. Rev. Physiol., 38:139, 1976. Hodgkin,
A. L.: The Conduction of the Nervous Impulse. Springfíeld, 111- Charles C
Thomas, 1963.
Hodgkin, A. L., and Horowicz, P.: The etfect of sudden changes in ionic
concentrationa on the membrane potential of smgle muscle fibera. J. Physiol.
(Lond.), 153:370, 1960. Hodgkin, A. L., and Huxley, A. F.: Movement of sodium
and potaaaium ions during nervous activity. Cold Spr. Harb. Symp. Quant. Biol.,
17:43, 1952. Hodgkin, A. L., and Huxley, A. F.: Quantitative deacription of
membrane current and its application to conduction and excitation in nerve. J.
Physiol. (Lond), 117:500, 1952. Kapían, J. H.: lon movements through the sodium
pump. Annu. Rev. PhyBiol., 47:535, 1985.
Katz, B.: Nerve, Muscle, and Synapse. New York, McGraw-Hill, 1968. Keynes,
R. D.: lon channels in the nerve-cell membrane. Sei. Am., 240:126, 1979.
58
Kostyuk, P. C: Intracellularperfusion of nerve cells and itseffectaon membrane currents. Phyaiol. Rev., 64:435, 1984, Krueger, B. K.: Toward an
understanding of strueture and function of ion channels. FASEB J.,
3:1906,1989. Latorre, R., and Alvarez, O.: Voltage-dependent channels in
lipid bilayer membranea. Physíol. Rev., 61:77, 1981. Latorre, R., et ai.: K+
channels gated by voltage and ions. Annu. Rev. Phy8iol., 46:485, 1984.
Levitan,I. B.: Modulationof ion channels in neutrons and other cells. Annu.
Rev. Neurosci., 11:119, 1988. Malhotra, S. K.: The Plasma Membrane.
New York, John Wiley & Sons, 1983. Miller, R. J.: Multiple calcium
channels and neuronal function. Science, 235:46, 1987. Moody, W., Jr.:
Effects of intraceliular H+ on the electrical properties of excitable cells.
Annu. Rev. Neuroaci., 7:257, 1984. Naftalin, R. J.: The thermostaties and
thermodynamics of cotransport. Biochem. Biophys. Acta., 778:155, 1984.
Narahashi, T.: Ion Channela. New York, Plenuro Publishing Corp., 1988.
Requeria, J.: Calcium tranaport and regulation in nerve fibera. Annu. Rev.
Biophys. Bioeng., 12:237, 1983. Reuter, H.: Modulation of ion channeis by
phosphorylation and second mes- Bengers. News Physiol. Sei., 2:168, 1987.
Rogart, R.: Sodium channels in nerve and muscle membrane. Annu. Rev.
Physiol., 43:711, 1981. Ross, W. N.: Changes in intraceliular calcium during
neuron activity. Annu. Rev. Physiol., 51:491, 1989. Sakmann, B., and
Neher, E.: Patch clamp techniques for studying ionic channels in
excitable membranes. Annu. Rev. Physiol., 46:455, 1984. Schubert, D.:
Developmental Biology of Cultured Nerve, Muscle and Glia.
New York, John Wiley & Sons, 1964. Schwartz, W., and Passow, H.: Caa+activated K+ channels in erythrocytes and excitable cells. Annu. Rev.
Physiol., 45:359, 1983. Shepherd, G. M.: Neurobiology. New York, Oxford
University Press, 1987. Sjodi, R. A.: Ion Transport in Skeletal Muscle.
New York, John Wiley & Sons, 1982. Skene, J. H. P.: Axonal growthassociated proteins. Ann. Rev. Neurosci., 12:127, 1989. Snell, R. M. (ed.>:
Transcellular Membrane Potentials and Ionic Fluxes. New
York, Gordon Press Pubs., 1984. Sperelakis, N.: Hormonal and
neurotransmitter regulation of Ca infiux through voltage-dependent slow
channels in cardiac muscle membrane.
Membr. Biochem., 5:131, 1984. Stefani, E., and Chiarandmi, D. J.: Ionic
channels in skeletal muscle. Annu.
Rev. Phyaiol., 44:357, 1982. Swadlow, H. A., et ai.: Modulalion of impulse
conduction along the axonal
tree. Annu. Rev. Biophys. Bioeng., 9:143, 1980. Trimmer, J. A., and Agnew,
W. S.: Molecular diversity of voltage-sensitive
Na channels. Annu. Rev. Physiol., 51:401, 1989. Tsien, R. W.: Calcium
channels in excitable cell membranes. Annu. Rev.
Physiol., 45:341, 1983. Ulbricht, W.: Kinetics of drug action and
equilibrium results at the node of
Ranvier. Physiol. Rev.. 61:785. 1981. Vinores, S., and Guroff, G.: Nerve
growth factor: Mechanicsms of action.
Annu. Rev. Biophys. Bioeng., 9:223, 1980. Weiss, D.C. (ed.): Axinplasmir
Transport in Physiologyand Pathology. New
York, típringer-Verlag, 1982. Windhager, E. E., and Taylor, A.: Regulatory
role of intracellular calcium
ions in epithelial Na transport. Annu. Rev. Physiol., 45:519, 1983. Wright,
E. M.: Electrophysiology of plasma membrane vesicles. Am. J. Physiol.,
246:F363, 1984. Zigmond, R. E., andBowers, C. W.: Influenceof nerve
activity on the macromolecular content of neurons and their etfector organs.
Annu. Rev. Physiol., 43:673. 1981.
59
CAPÍTULO 6
Contração do Músculo Esquelético
Cerca de 40% do corpo são compostos por músculos
esqueléticos e quase outros 10% são formados por músculos liso e
cardíaco. Muitos dos princípios básicos da contração são
comuns a todos esses tipos de músculos, mas, neste capítulo, será
discutido principalmente o funcionamento do músculo
esquelético; o funcionamento especializado do músculo liso é
discutido no Cap. 8 e o do músculo cardíaco, no Cap. 9.
ANATOMIA FUNCIONAL DO MÚSCULO
ESQUELÉTICO
A FIBRA MUSCULAR ESQUELÉTICA
A Fig. 6.1 apresenta a organização do músculo esquelético,
mostrando que todos os músculos esqueléticos são compostos
por numerosas fibras, com diâmetros variando entre 10 e 80
µm. Por sua vez, cada uma dessas fibras é formada por diversas
subunidades, cada uma menor que a outra, também mostradas
na Fig. 6.1, que serão discutidas nos parágrafos subseqüentes.
Na maioria dos músculos, as fibras se estendem por todo
o comprimento do músculo; exceto por cerca de 2% delas, são
inervadas por terminação nervosa única, localizada perto do meio
da fibra.
O sarcolema. O sarcolema é a membrana celular da fibra
muscular. Contudo, o sarcolema é formado por uma verdadeira
membrana celular, chamada de membrana plasmática, e por
revestimento externo, composto de fina camada de material
polissacarídico, contendo numerosas fibrilas finas de colágeno. Na
extremidade da fibra muscular, esse revestimento superficial do
sarcolema se funde com uma fibra tendinosa e essas fibras
tendinosas se unem, formando feixes, até comporem um tendão
muscular que se insere no osso.
Miofíbrilas: os filamentos de actina e de miosina. Cada
fibra muscular contém de muitas centenas a vários milhares de
miofibrilas, representadas pelos numerosos círculos vazios na
vista em corte transverso da Fig. 6.1C. Cada miofibrila (Fig.
6.1D e E), por sua vez, contém, lado a lado, cerca de 1.500
filamentos de miosina e 3.000 filamentos de actina, que são grandes
moléculas poliméricas, responsáveis pela contração muscular.
Esses filamentos são apresentados, em vista longitudinal, na
microfotografia eletrônica da Fig. 6.2 e são representados,
esquematicamente, na Fig. 6.1 (partes E a L). Nesses esquemas,
os filamentos grossos são os de miosina e os finos, os de actina.
Deve ser notado que os filamentos de actina e de miosina
se interdigitam em parte, o que faz com que as miofibrilas
apresentem faixas alternadas escuras e claras. As faixas
claras só contém filamentos de actina e são chamadas de
faixas I, por serem isotrópicas à luz polarizada.
As faixas escuras contêm os filamentos de miosina além das
extremidades dos filamentos de actina e são chamadas de
faixas A por serem anisotrópicas à luz polarizada. Também
devem ser notadas as pequenas projeções laterais dos filamentos
de miosina. Elas são chamadas de pontes cruzadas: proeminam
da superfície dos filamentos de miosina, por toda sua extensão,
exceto na sua parte mais central. E a interação entre essas
pontes cruzadas e os filamentos de actina que produz a
contração.
A Fig. 6.1E também mostra que as extremidades dos
filamentos de actina estão presos ao chamado disco Z. A
partir desse disco, os filamentos se estendem, nas duas
direções, para se interdigitar com os filamentos de miosina.
O disco Z, que é formado por proteínas filamentosas diferentes
das dos filamentos de actina e de miosina, passa de miofibrila a
miofibrila, fixando-as entre si, ao longo de toda a espessura da
fibra muscular. Por conseguinte, toda a fibra muscular
apresenta faixas claras e escuras, como acontece com a
miofibrila. Essas faixas dão ao músculo esquelético e
cardíaco sua aparência "estriada".
A região de uma miofibrila (ou de toda uma fibra muscular)
situada entre duas linhas Z consecutivas é chamada de sarcômero.
Quando a fibra nervosa está em seu comprimento normal de
repouso, completamente estirada, o sarcômero tem extensão de
cerca de 2 µm. Nesse comprimento, os filamentos de actina se
sobrepõem totalmente aos filamentos de miosina e começam
a se sobrepor uns aos outros. Veremos adiante que, nesse
comprimento, o sarcômero também é capaz de gerar sua força
máxima de contração.
O sarcoplasma. As miofibrilas, no interior da fibra muscular,
ficam suspensas em uma matriz, chamada de sarcoplasma,
formada pelos constituintes intracelulares usuais. O líquido do
sarcoplasma contém grandes quantidades de potássio e de
magnésio, de fosfato e de enzimas protéicas. Também está
presente número imenso de mitocôndrias que ficam entre e
paralelas as miofibrilas, situação indicativa da grande necessidade
das miofibrilas em contração de quantidade elevada de trifosfato
de adenosina (ATP), formado nas mitocôndrias.
O retículo sarcoplasmático. Também existe no sarcoplasma
um extenso retículo endoplasmático, chamado, na fibra muscular,
de retículo sarcoplasmático. Esse retículo apresenta organização
especial, muito importante para o controle da contração
muscular, o que é discutido no capítulo seguinte. A
microfotografia eletrônica da Fig. 6.3 mostra a disposição desse
retículo sarcoplasmático c indica como pode ser extenso. Os
tipos de músculo de contração mais rápida possuem retículo
sarcoplasmático extremamente longo, indicando que essa
estrutura é importante para a produção de contração muscular
rápida, o que será também discutido adiante.
60
Fig. 6.1 Organização do músculo esquelético, do
nível macroscópico ao molecular, f, G, He /são
cortes transversais nos níveis indicados. (Desenho
de Sylvia Colard Keene; modificado de Fawcett:
Bloom and Fawcett: A lextbook ofhistotogy.
Philadelphia, W. B. Saunders Co., 1986).
O MECANISMOGERAL DACONTRAÇÃO MUSCULAR
O desencadeamento e decurso de uma contração muscular
ocorre segundo as etapas sucessivas seguintes:
1. Um potencial de ação percorre um axônio motor até
suas terminações nas fibras musculares.
2. Em cada terminação, há secreção de pequena quantidade
da substância neurotransmissora, chamada acetilcolina.
3. A acetilcolina atua sobre área localizada da membrana
da fibra muscular, abrindo numerosos canais protéicos
acetilcolina dependentes.
4. A abertura desses canais acetílcolina-dependentes
permite o influxo de grande quantidade de íons sódio para o
interior da membrana da fibra muscular, no ponto da terminação
nervosa.
Isso produz um potencial de ação na fibra muscular.
5. O potencial de ação se propaga ao longo da membrana
da fibra muscular do mesmo modo como o faz nas membranas
neurais.
6. O potencial de ação despolariza a membrana da fibra
muscular e também penetra profundamente no interior dessa
fibra. Aí, faz com que o retículo sarcoplasmático libere, para
as miofibrilas. grande quantidade de íons cálcio, que ficam
armazenadas em seu interior.
7. Os íons cálcio geram forças atrativas entre os filamentos
de actina e de miosina, fazendo com que deslizem um em direção
ao outro, o que constitui o processo contrátil.
8. Após uma fração de segundo, os íons cálcio são
bombeados de volta para o retículo sarcoplasmático, onde
permanecem
armazenados até que ocorra novo potencial de ação muscular;
termina a contração muscular.Vamos agora descrever o
mecanismo do processo contrátil, mas, no capítulo seguinte,
retornaremos aos detalhes da excitação muscular.
MECANISMOMOLECULARDACONTRAÇÃO
MUSCULAR
Mecanismo de deslizamento da contração. A Fig. 6.4
apresenta o mecanismo básico da contração muscular. Na parte
de cima, mostra o estado relaxado de um sarcômero e, na
parte de baixo, seu estado contraído. No estado relaxado, as
extremidades dos filamentos de actina derivados de dois discos Z
consecutivos se superpõem apenas discretamente, enquanto, ao
mesmo tempo, se sobrepõem completamente aos filamentos de
miosina.
61
Fig. 6.2 Microfotografia eletrônica das miofibrilas de músculo, mostrando os detalhes da organização dos filamentos de actina e de miosina.
Devem ser notadas as mitocôndrias, situadas entre as miofibrilas. (De Fawcett: The Cell. Philadelphia, W.B. Saunders Co., 1981.)
Por outro lado, no estado contraído, os filamentos de actina
foram tracionados para a parte média, de modo que ficam, nesse
estado, bem mais sobrepostos que antes. Também, os discos
Z foram puxados, pelos filamentos de actina, até as extremidades
dos filamentos de miosina. Na verdade, os filamentos de actina
podem ser tracionados tão intensamente que as extremidades
dos filamentos de miosina chegam a ficar dobradas durante as
contrações muito fortes. Assim, a contração muscular é causada
por mecanismo de deslizamento dos filamentos.
Mas, o que faz com que os filamentos de actina
deslizem, em direção central, por entre os filamentos
de miosina? Isso é o resultado de forças mecânicas
geradas pela interação das pontes cruzadas dos
filamentos de miosina com os filamentos de actina, como
discutiremos nas seções seguintes. Nas condições de
repouso, essas forças estão inibidas, mas, quando o
potencial de ação se propaga ao longo da membrana da
fibra muscular, ele provoca a liberação de grande
quantidade de íons cálcio no sarcoplasma que banha as
miofibrilas. Por sua vez, esses íon cálcio ativam as forças
Fig. 6.3. Retículo sarcoplasmático em torno das
miofribrilas mostrandos, em corte transverso, os túbulos
T ( setas). Que levam ao exterior da membrana da fibra e
que contém líquido extracelular.
62
Fig. 6.4 Os estados relaxado e contraído de unia miofibrila, mostrando
o deslizamento dos filamentos de actina (em preto) pelos espaços entre
os filamentos de miosina (em vermelho).
Entre os filamentos, dando início à contração, mas também é
necessária energia para que a contração possa seguir seu
curso. Essa energia é derivada das ligações de alta energia do
ATP, que é degradado a difosfato de adenosina (ADP), para
liberar a energia necessária.
Nas seções seguintes, vamos descrever o que se sabe sobre
os detalhes dos processos moleculares da contração. Para dar
início a essa discussão, vamos, primeiro, caracterizar os detalhes
dos filamentos de miosina e de actina.
CARACTERÍSTICAS MOLECULARES
FILAMENTOS CONTRÁTEIS
DOS
O filamento de miosina. O filamento de miosina é composto
por numerosas moléculas de miosina, cada uma com peso
molecular de cerca de 480.000. A Fig. 6.5A apresenta uma
dessas moléculas isolada; à parte B mostra o modo de organização
dessas moléculas para formar um filamento de miosina, bem
como sua interação em um dos lados com as extremidades de dois
filamentos de actina.
A molécula de miosina é formada por seis cadeias
polipeptídicas — duas cadeias pesadas, cada uma com peso
molecular de 200.000, e quatro cadeias leves, cada uma com peso
molecular de cerca de 20.000. As duas cadeias pesadas se
enrolam, de modo espiralado, uma em torno da outra, para
formar uma dupla hélice. Contudo, uma das extremidades de
cada uma dessas cadeias se dobra para formar uma massa protéica
globular, chamada de cabeça da miosina. Dessa forma, existem
duas cabeças livres, situadas uma ao lado da outra, em uma das
extremidades da molécula em dupla hélice da miosina; a porção
alongada dessa dupla hélice é chamada de cauda. As quatro
cadeias leves também fazem parte das cabeças de miosina, duas
para cada cabeça. Essas cadeias leves participam do controle
do funcionamento da cabeça, durante o processo de contração.
O filamento de miosina é formado por 200 ou mais moléculas
individuais de miosina. A parte central de um desses filamentos
é mostrada na Fig. 6.5B, com as caudas das moléculas de miosina
presas entre si para formar o corpo do filamento, enquanto muitas
cabeças das moléculas pendem para fora e para os lados desse
corpo. Também, parte da porção helicoidal de cada molécula
de miosina, junto com a cabeça, estende-se para o lado,
formando, assim, um braço que afasta a cabeça do corpo, como
mostrado na figura. Esses braços e a cabeça proeminentes
formam, em seu conjunto, as pontes cruzadas. Acredita-se que
cada ponte cruzada seja flexível em dois pontos, chamados de
dobradiças;
Fig. 6.5 A, A molécula de miosina. B, A combinação de muitas
moléculas de miosina para formar um filamento de miosina. Também são
mostradas as pontes cruzadas e a interação entre as cabeças das pontes
cruzadas e os filamentos adjacentes de actina.
um fica localizado onde o braço se afasta do corpo do filamento
de miosina e o outro, onde as duas cabeças se prendem ao braço.
O braço dobrável permite que as cabeças sejam muito afastadas
do corpo do filamento de miosina ou que sejam trazidas para
muito próximo dele. As cabeças dobráveis são consideradas como
tendo participação no próprio processo de contração, como
discutiremos nas seções seguintes.
O comprimento total de cada filamento de miosina é muito
uniforme, quase que exatamente 1,6 µm. Contudo, deve ser
notado que não existem cabeças de pontes cruzadas no centro
verdadeiro do filamento de miosina, em extensão de cerca de
0,2µm, devido ao fato de os braços dobráveis se estenderem
para as extremidades do filamento, a partir desse centro;
conseqüentemente, só existem caudas das moléculas de miosina
nesse centro, e não existem cabeças.
Agora, para completar esse quadro, o filamento de miosina
é, por sua vez, torcido, de modo que cada grupo consecutivo
de pontes cruzadas fica axialmente deslocado do grupei anterior
por 120 graus. Isso assegura que as pontes cruzadas se estendam
em todas as direções em torno do filamento.
Atividade de ATPase da cabeça de miosina. Outra
característica da cabeça de miosina, essencial para a contração
muscular, é que ela atua como uma enzima do tipo ATPase.
Como veremos adiante, essa propriedade permite que a cabeça
clive ATP e utilize a energia derivada das ligações fosfato de
alta energia desse ATP para energizar o próprio processo da
contração.
O filamento de actina. O filamento de actina também é
complexo. É formado por três constituintes protéicos: actina,
tropo-miosina e troponina.
O arcabouço do filamento de actina é uma molécula da
proteína actina-F com dois filamentos, mostrada pelos dois
filamentos mais claros da Fig. 6.6. Esses dois filamentos
formam hélice, do mesmo modo como acontece com a molécula
de miosina, mas com uma volta completa a cada 70 nm.
Cada filamento da dupla hélice da actina-F é composto de
moléculas polimerizadas de actina-G, cada uma com peso
molecular de cerca de 42.000. Existem aproximadamente 13
dessas moléculas em cada revolução de um dos filamentos da
hélice.
63
Agora, vamos discutir o papel dos íons cálcio. Em presença de
grandes quantidades de íons cálcio, o efeito inibitório da
troponina-tropomiosina sobre os filamentos de actina fica
inibido. Esse mecanismo ainda é desconhecido, mas uma
hipótese é a seguinte: quando os íons cálcio reagem com a
troponina C — e cada uma dessas moléculas pode fixar-se
fortemente a até quatro íons cálcio, mesmo quando estes estão
presentes em quantidades diminutas —, admite-se que o complexo
da troponina sofra alteração conformacional que, de algum
modo, traciona a molécula de tropomiosina e, supostamente, a
empurra mais profundamente para o fundo do sulco entre os
Fig. 6.6 O filamento de actina, formado por dois filamentos helicoidais dois filamentos de actina. Isso "descobre" os sítios ativos da
de actina-F e por moléculas de tropomiosina que se encaixam actina, o que permite o desenvolvimento da contração. Embora
frouxamente nos sulcos entre os filamentos de actina. Preso a uma das esse seja um mecanismo hipotético, ele enfatiza, todavia, que a
extremidades de cada molécula de tropomiosina existe um complexo de
relação normal entre o complexo troponina-tropomiosina e a
troponina que inicia a contração.
actina é modificada pelos íons cálcio, produzindo nova condição
que leva à contração.
Interação entre o filamento "ativado" de actina e as pontes
Em cada molécula de actina-G está fixada uma molécula de cruzadas da miosina — a teoria do "sempre em frente" da
ADP. Acredita-se que essas moléculas de ADP representem o contração. Tão logo o filamento de actina seja ativado pelos íons
sítio ativo dos filamentos de actina, com que interagem os cálcio, as cabeças das pontes cruzadas imediatamente são
filamentos de miosina para produzir a contração muscular. Os fixadas aos sítios ativos do filamento de actina e isso, de
sítios ativos dos dois filamentos de actina-F ocorrem alguma maneira, faz com que aconteça a contração. Embora
escalonados, de modo que, ao longo de todo o filamento de ainda seja desconhecido o modo preciso como essa interação
actina, existe um sítio ativo a cada 2,7 nm.
entre as pontes cruzadas e a actina produz a contração, foi
Cada filamento de actina tem comprimento aproximado de proposta uma hipótese baseada em evidências consideráveis, que
1 µm. As bases desses filamentos são fortemente fixadas nos foi chamada de teoria do "sempre em frente [walk-along] (ou
discos Z, enquanto as duas extremidades proeminam, nas duas teoria da cremalheira) da construção.
direções, para os sarcômeros adjacentes, situando-se nos espaços
A Fig. 6.7 apresenta o mecanismo postulado para o "sempre
entre as moléculas de miosina, como mostrado na Fig. 6.4.
em frente". A figura mostra as cabeças de duas pontes cruzadas
As moléculas de tropomiosina. Os filamentos de actina se fixando e se soltando dos sítios ativos do filamento de actina.
contêm outra proteína, a tropomiosina. Cada molécula de Acredita-se que, quando uma cabeça se prende a um sítio ativo,
tropomiosina tem peso molecular de 70.000 e comprimento de 40 essa fixação produz, ao mesmo tempo, profundas alterações nas
nm. Essas moléculas estão frouxamente fixadas aos filamentos forças intermoleculares entre a cabeça e o braço da ponte cruzada.
de actina-F e enroladas, de forma espiralada, ao longo dos O novo alinhamento de forças força a cabeça a se inclinar, em
lados da hélice de actina-F. No estado de repouso, acredita-se direção do braço, trazendo junto o filamento de actina. Essa
que as moléculas de tropomiosina fiquem sobrepostas aos sítios inclinação da cabeça é chamada de movimento de tensão [power
ativos dos filamentos de actina, de modo a impedir que ocorra strake]. Em seguida, imediatamente após a inclinação, a cabeça,
atração entre os filamentos de actina c de miosina, para de modo automático, solta-se do sítio ativo, voltando à sua
produção de contração. Cada molécula de tropomiosina recobre posição perpendicular normal. Nessa posição, ela se fixa a
cerca de sete sítios ativos.
novo sítio ativo, localizado cm ponto mais adiante do filamento
Troponina e sua participação na contração muscular. Ainda de actina; então, a cabeça se inclina de novo, para novo
existe outra molécula de proteína, chamada troponina, que ocorre movimento de tensão, e o filamento de actina se desloca um
fixada próximo da extremidade de cada molécula de pouco mais. Desse modo, as cabeças das pontes cruzadas se
tropomiosina. Ela é, na verdade, um complexo de três inclinam e se endireitam, seguindo sempre em frente ao longo
subunidades protéicas, frouxamente interligadas, cada uma com do filamento de actina, em direção ao centro do filamento de
participação específica no controle da contração muscular. Uma miosina.
dessas subunidades (troponina I) tem forte afinidade pela
Acredita-se
que
cada
ponte
cruzada
atue
actina, outra (troponina T) a tem pela tropomiosina e a independentemente de todas as outras, cada uma se fixando e
terceira (troponina C), pelos íons cálcio. Acredita-se que esse tracionando em ciclo contínuo, mas aleatório. Por
complexo fixe a tropomiosina a actina. Também é admitido que conseguinte, quanto maior for o número de pontes cruzadas
a forte afinidade da troponina pelos íons cálcio desencadeie o em contato com os filamentos de actina, em dado momento,
processo contrátil, como explicado na seção seguinte.
maior será, teoricamente, a força da contração.
ATP como fonte de energia para a contração - as etapas
Interação da miosina, dos filamentos de actina e químicas do movimento das cabeças da miosina. Quando o
músdos íons cálcio para a produção da contração
Inibição do filamento de actina pelo complexo
troponina-tropomiosina; ativação pelos íons cálcio. Um
filamento puro de actina, desprovido do complexo troponinatropomiosina, fixa-se fortemente a moléculas de miosina, em
presença de íons magnésio e ATP, ambos normalmente
abundantes na miofibrila. Contudo, se for adicionado o
complexo troponina-tropomiosina, essa fixação não mais ocorre.
Por conseguinte, acredita-se que os sítios ativos no filamento
normal de actina no músculo relaxado estejam inibidos ou
fisicamente recobertos pelo complexo troponina-tropomiosina.
Conseqüentemente, esses sítios não podem fixar os filamentos
de miosina para produzir a contração. Antes que a contração
possa ocorrer, o efeito inibitório do complexo troponinatropomiosina deve ser, por sua vez, inibido.
Fig. 6.7 O mecanismo "sempre em frente" para a contração do músculo
64
culo se contrai sob o efeito de uma carga, é realizado trabalho
e é necessária energia. Grandes quantidades de ATP são clivadas,
para formar ADP durante o processo da contração. Ainda mais,
quanto mais trabalho for realizado pelo músculo, maior será
a quantidade clivada de ATP, o que é chamado de efeito Fenn.
Embora ainda não seja conhecido de modo preciso como o ATP
é utilizado para fornecer a energia para a contração, foi sugerida
a seqüência que se segue para o mecanismo desse processo:
1. Antes que comece a contração, as cabeças das pontes
cruzadas fixam ATP. A atividade de ATPase das cabeças da
miosina cliva, imediatamente, o ATP, embora os produtos dessa
clivagem — ADP e Pi — permaneçam presos à cabeça. Nesse
estado, a conformação da cabeça é tal que ela se estende
perpendicularmente em direção ao filamento de actina,
embora ainda não se fixe a ele.
2. Em seguida, quando o efeito inibitório do complexo
troponina-tropomiosina for, por sua vez, inibido pelos íons
cálcio, os sítios ativos do filamento de actina ficam descobertos,
o que permite a fixação das cabeças de miosina a eles, como
mostrado na Fig. 6.7.
3. A ligação entre a cabeça da ponte cruzada e o sítio ativo
do filamento de actina provoca alteração conformacional da
cabeça, fazendo com que ela se incline em direção ao braço da
ponte
cruzada. Isso produz o movimento de tensão para tracionar o
filamento de actina. A energia que ativa o movimento de tensão
é a que já está armazenada, como uma mola "engatilhada",
pela alteração conformacional da cabeça, quando a molécula
de ATP foi clivada.
4. Uma vez tendo ocorrido a inclinação da cabeça, isso
permite a liberação do ADP e do Pi que estavam, até então,
presos à cabeça; no local de onde foi liberado o ADP, prende-se
outra molécula de ATP. Essa fixação, por sua vez, provoca o
desprendimento da cabeça da actina.
5. Após a cabeça ter-se desprendido da actina, também é
clivada a nova molécula de ATP, e a energia novamente
"engatilha" a cabeça de volta a sua posição perpendicular,
pronta para iniciar novo ciclo de movimento de tensão.
6. Então, quando a cabeça engatilhada, com sua energia
armazenada derivada do ATP clivado, fixa-se a novo sítio ativo
no filamento de actina, ela torna-se desengatilhada, gerando novo
movimento de tensão.
7. Dessa forma, o processo se repete por várias vezes, até
que os filamentos de actina puxem os discos Z até que pressionem
as extremidades dos filamentos de miosina ou até que a carga
que atua sobre o músculo seja demasiadamente grande para
impedir qualquer tracionamento adicional.
Fig. 6.8 Relação comprimento-tensão para um sarcômero isolado,
mostrando a força máxima de contração ocorrendo quando o
sarcômero tem comprimento entre 2,0 e 2,2µm. No canto superior à
direita, são mostradas as posições relativas dos filamentos de actina e de
miosina, para diferentes comprimentos do sarcômero, do ponto A ao
ponto D. (Modificado de Gordon, Huxley e Julian: The length-tension
diagram of single vertebrate striated muscle fibers. J. Physiol., 77/.-28P,
1964.)
Nesse ponto, o filamento de actina já se sobrepôs a todas as
pontes cruzadas do filamento de miosina, mas ainda não
atingiu o centro desse filamento. Com encurtamento ainda
maior, o sarcômero mantém tensão máxima até o ponto B, com
comprimento desse sarcômero de cerca de 2,0 µm. Nesse ponto,
as extremidades dos filamentos de actina começam a se sobrepor
umas às outras, além de estarem sobrepostas aos filamentos de
miosina. Quando o comprimento do sarcômero cai de 2,0 µm
para cerca de 1,6 /ira, no ponto A, a força da contração diminui.
É nesse ponto que os dois discos Z do sarcômero entram em
contato com as extremidades dos filamentos de miosina. Então,
à medida que a contração prossegue, com comprimentos do
sarcômero ainda menores, as extremidades dos filamentos de
miosina são dobradas, como mostrado na figura, e a força da
contração diminui abrupta e aceleradamente.
Esse esquema demonstra que a contração máxima ocorre
quando existe grau máximo de sobreposição entre os filamentos
de actina e as pontes cruzadas dos filamentos de miosina, e
confirma a hipótese de que, quanto maior for o número de
pontes cruzadas a puxar o filamento de actina, maior será a força
de contração.
Efeito do comprimento do músculo sobre a força de contração
GRAU DE SUPERPOSIÇÃO DOS FILAMENTOS DE
ACTINA E DE MIOSINA - EFEITO SOBRE A TENSÃO
QUE É DESENVOLVIDA PELO MÚSCULO EM
CONTRAÇÃO
A Fig. 6.8 mostra o efeito do comprimento do sarcômero
e do grau de superposição dos filamentos de actina e de miosina
sobre a tensão ativa que é desenvolvida durante a contração
de uma fibra muscular. À direita são apresentados diferentes
graus de superposição dos filamentos de actina e de miosina,
em diversos comprimentos do sarcômero. Nesse esquema, o
ponto D marca o afastamento do filamento de actina além da
extremidade do filamento de miosina, sem qualquer
superposição. Nesse ponto, a tensão desenvolvida pelo músculo
ativado é zero. Em seguida, à medida que o sarcômero se
encurta e os filamentos de actina começam a se sobrepor aos de
miosina, começa o desenvolvimento de tensão, com aumento
progressivo, até que o comprimento do sarcômero diminua
para cerca de 2,2 µm.
nos músculos íntegros. A curva superior da Fig. 6.9 é semelhante
à curva da Fig. 6.8, mas é obtida de músculos íntegros, em vez
de fibra muscular isolada. O músculo íntegro contém grande
quantidade de tecido conjuntivo; por outro lado, os sarcômeros,
em diferentes partes do músculo, não se contraem
necessariamente de modo sinerônico. Como resultado, a curva
tem dimensões algo diferentes das amostras para a fibra
muscular isolada, mas, não obstante, sua forma é a mesma.
Deve ser notado, na Fig. 6.9, que, quando o músculo está
em seu comprimento normal de repouso, que corresponde a
comprimento do sarcômero de cerca de 2 µm, sua contração
tem força máxima. Caso o músculo seja estirado até comprimento
muito acima do normal antes da contração, esse músculo vai
gerar elevada tensão de repouso, antes que ocorra a contração;
essa tensão resulta das forças elásticas do tecido conjuntivo, do
sarcolema, dos vasos sanguíneos, dos nervos etc. Contudo, o
aumento da tensão durante a contração, chamado de tensão ativa,
fica progressivamente menor, se o músculo for estirado além
65
ENERGÉTICADACONTRAÇÃOMUSCULAR
Produção de trabalho durante a contração muscular
Quando um músculo se contrai, sob ação de carga, ele realiza
trabalho. Isso significa que é transferida energia do músculo para
a carga externa, por exemplo, para elevar um objeto a uma
altura maior ou para sobrepujar resistência a movimento.
Em termos matemáticos, o trabalho é definido pela seguinte
relação:
W =C x D
Fig. 6.9 Relação entre o comprimento do músculo e a força de
contração.
onde W é o trabalho produzido, C é a carga e D é a distância
percorrida, sob ação da carga. A energia necessária para a
realização do trabalho é derivada das reações químicas, nas
células musculares, durante a contração, como descreveremos nas
seções seguintes.
Fontes de energia para a contração muscular
de seu comprimento normal — isto é, até comprimento do
sarcômero maior que cerca de 2,2µm. Na figura, isso é
mostrado pelo menor comprimento da seta.
RELAÇÃO ENTRE A VELOCIDADE DE CONTRAÇÃO E A
CARGA
Um músculo se contrai de forma extremamente rápida quando sua
contração não sofre oposição de qualquer carga - para um músculo
médio, a contração máxima é atingida dentro de 0,1 s. Contudo, quando
são aplicadas cargas, a velocidade de contração diminui progressivamente
à medida que a carga for aumentada, como mostrado na Fig. 6.10.
Quando a carga aumenta até igualar a força máxima que pode ser gerada
pelo músculo, a velocidade de contração é zero, e não ocorre contração,
apesar da ativação das fibras musculares.
Essa velocidade decrescente em função do aumento da carga ê
causada pelo fato de que a carga imposta a um músculo em
contração é uma força inversa que se opõe à força contrátil gerada
pela contração do músculo. Por conseguinte, a força efetiva
disponível para produzir a velocidade de encurtamento fica diminuída
proporcionalmente.
Fig. 6.10 Relação entre a carga e a velocidade de contração em músculo
esquelético com 8 cm de comprimento.
Já vimos que a contração muscular depende da energia
fornecida pelo ATP. A maior parte dessa energia é necessária
para pôr em ação o mecanismo de "sempre em frente" por meio
do qual as pontes cruzadas puxam os filamentos de actina, mas
pequenas quantidades são necessárias para (1) bombear cálcio
do sarcoplasma para o retículo sarcoplasmático, ao término da
contração, e (2) bombear íons sódio e potássio, através da
membrana da fibra muscular, para manter o ambiente iônico
adequado para a propagação dos potenciais de ação.
Contudo, a concentração de ATP presente na fibra muscular,
da ordem de 4 milimolar, só é suficiente para manter a contração
por, no máximo, 1 a 2 segundos. Felizmente, após o ATP ter
sido clivado a ADP, como descrito no Cap. 2, o ADP é
refosforilado, para formar novo ATP, em fração de segundo.
Existem várias fontes de energia para essa fosforilação.
A primeira fonte de energia que é utilizada para reconstituir
o ATP é o composto fosfocreatina, que contém uma ligação
fosfato de alta energia semelhante à do ATP. Essa ligação fosfato
de alta energia da fosfocreatina contém quantidade pouco maior
de energia livre que a do ATP, como discutiremos em maiores
detalhes nos Caps. 67 a 72. Como resultado, a fosfocreatina
é clivada de imediato e a energia liberada provoca a ligação
de novo íon fosfato ao ADP, para reconstituir o ATP. Todavia,
o teor de fosfocreatina também é muito reduzido — apenas cinco
vezes maior que o do ATP. Como conseqüência, a energia
combinada do ATP e da fosfocreatina armazenados no
músculo só c capaz de manter a contração máxima do
músculo por cerca de 7 a 8 segundos.
A mais importante fonte de energia a seguir, usada para
reconstituir tanto o ATP como a fosfocreatina, é o glicogênio
previamente armazenado nas células musculares. A rápida
degradação enzimática do glicogênio a ácido pirúvico e ácido
lático libera energia que é utilizada para converter ADP em
ATP e esse ATP pode ser usado diretamente para energizar à
contração muscular ou para reconstituir a fosfocreatina. A
importância desse mecanismo de "glicólise" é dupla. Primeiro, as
reações glicolíticas podem ocorrer até mesmo na ausência de
oxigênio, de modo que a contração muscular pode ser mantida,
por breve período, na falta de oxigênio. Segundo, a velocidade
com que é formado o ATP, pelo processo glicolítico, é duas
vezes e meia maior que a da formação de ATP pela reação dos
nutrientes celulares com oxigênio. Todavia, infelizmente, ocorre
acúmulo de muitos produtos finais da glicólise nas células
musculares, de modo que a glicólise, isoladamente, só pode
manter a contração muscular máxima por cerca de 1 minuto.
66
A última fonte de energia é o processo do metabolismo
oxidativo. Isso significa a combinação de oxigênio com os diversos
nutrientes celulares para formar ATP. Mais de 95% de toda
a energia utilizada pelos músculos em contrações continuadas
de longa duração derivam dessa fonte. Os nutrientes que são
consumidos são os carboidratos, as gorduras e as proteínas. Para
a atividade muscular de duração extremamente longa — de
algumas horas —, a maior proporção da energia que é
consumida deriva, em sua maior parte, das gorduras.
Os detalhes dos mecanismos desses processos energéticos
são discutidos nos Caps. 67 a 72. Além disso, a importância
dos diversos mecanismos para liberação de energia em diferentes
atividades esportivas é discutida no Cap. 81, que versa sobre
a fisiologia do esporte.
Eficiência da contração muscular. A "eficiência" de uma máquina
ou de um motor é calculada como a porcentagem da energia consumida
que é transformada em trabalho, e não em calor. A porcentagem da
energia consumida pelo músculo (a energia química dos nutrientes) que
pode ser convertida em trabalho é de menos de 20 a 25%, o restante
sendo transformado em calor. A razão para essa baixa eficiência é que
cerca da metade da energia dos nutrientes é perdida na formação de
ATP e apenas cerca de 40 a 45% da energia do próprio ATP podem
ser, posteriormente, transformados em trabalho.
Só pode ser conseguida eficiência máxima quando o músculo se
contrai com velocidade moderada. Se o músculo se contrair muito
lentamente ou sem que ocorra algum movimento, são liberadas grandes
quantidades de calor de manutenção durante o processo da contração,
mesmo se estiver sendo realizado pouco ou nenhum trabalho, o que
diminui a eficiência. Por outro lado, se a contração for muito rápida,
grande parte da energia será consumida para vencer o atrito viscoso no
interior do próprio músculo, e isso também reduz a eficiência da
contração. Comumente, a eficiência máxima é obtida quando a velocidade
da contração é de cerca de 30% da velocidade máxima.
CARACTERÍSTICAS DA CONTRAÇÃO DE TODO O
MÚSCULO
Muitas características da contração muscular podem ser
especialmente demonstradas pela produção de abalos musculares
isolados. Isso pode ser conseguido por estimulação breve do nervo que
vai para o músculo ou pela passagem de estímulo elétrico de curta
duração pelo próprio músculo, o que produz contração única e abrupta
do músculo, que dura fração de segundo.
Contrações isométrica e isotônica. Uma contração muscular é dita
isométrica quando o músculo não se encurta durante a contração e é
dita isotônica quando ele se encurta, com a tensão desenvolvida pelo
músculo permanecendo constante. Os métodos para o registro desses
dois tipos de contração muscular são mostrados na Fig. 6.11.
No método isométrico, o músculo se contrai contra um transdutor
Fig. 6.11 Sistemas para o registro de contrações isotônicas e isométricas.
de força, sem que varie seu comprimento, como mostrado à direita
da Fig. 6.11. No método isotônico, o músculo se encurta sob ação de
uma carga constante. Isso é mostrado à esquerda da figura, que apresenta
o músculo levantando um prato cheio de pesos. Obviamente, as
características da contração isotônica dependem da carga contra a qual o
músculo vai contrair-se, bem como da inércia da carga. Por outro lado, o
método isométrico só permite o registro, em sentido estrito, da
variação da força da própria contração muscular. Como resultado, é
usado com maior freqüência o método isométrico para a comparação
entre as características funcionais dos diversos tipos de músculo.
O componente elástico em série da contração muscular. Quando as
fibras musculares se contraem sob ação de uma carga, as partes do
músculo que não se contraem — os tendões, as extremidades do
sarcolema das fibras musculares por onde se fixam aos tendões e,
talvez, os braços dobráveis das pontes cruzadas — são ligeiramente
estiradas, à medida que aumenta a tensão. Conseqüentemente, o
músculo vai encurtar-se por mais de 3 a 5% para compensar o
estiramento desses elementos. Os elementos do músculo que são
estirados durante a contração formam o componente elástico em série
desse músculo.
CARACTERÍSTICAS DOS ABALOS ISOMÉTRICOS
REGISTRADOS EM DIFERENTES MÚSCULOS
O corpo apresenta músculos esqueléticos de tamanho muito variado
- desde o minúsculo músculo estapédio, no ouvido médio, com poucos
milímetros de comprimento e cerca de 1 mm de diâmetro, até o imenso
quadríceps, meio milhão de vezes maior que o estapédio. Ademais,
as fibras desses músculos podem ter diâmetro que varia do mínimo de
10 µm até o máximo de 80 µm. Finalmente, a energética da contração
muscular varia consideravelmente de um músculo para outro. Por
conseguinte, não surpreende que as características da contração muscular
difiram entre todos esses músculos.
A Fig. 6.12 mostra as contrações isométricas de três tipos distintos
de músculos esqueléticos: um músculo ocular, cuja contração dura menos
que 1/40 de segundo; e o músculo gastrocnêmio, com contração durando
cerca de1/15 de segundo; e o músculo solear com contração durando
1/5 de segundo. É interessante que as durações de contração sejam
adaptadas ao funcionamento dos músculos respectivos. Os movimentos
oculares devem ser extremamente rápidos para manter a fixação dos
olhos sobre objetos específicos, e o músculo gastrocnêmio deve
contrair-se de forma moderadamente rápida para permitir velocidade
suficiente dos movimentos das pernas, do tipo correr ou pular,
enquanto o músculo solear está relacionado, de modo prioritário, à
contração lenta para a sustentação contínua do corpo contra a ação da
gravidade.
Fibras musculares rápidas e lentas. Como discutiremos em maior
detalhe no Cap. 84, sobre a fisiologia do esporte, cada músculo do
corpo é formado por combinação das chamadas fibras musculares rápidas
e lentas, existindo outras fibras com características intermediárias entre
esses dois extremos. Os músculos que reagem muito rapidamente são
Fig. 6.12 Duração das contrações isométricas de diferentes tipos de
músculos de mamíferos, mostrando o período latente da contração
entre o potencial de ação e a contração muscular.
67
compostos por grande maioria de fibras rápidas, com número muito
pequeno de fibras do tipo lento. Inversamente, os músculos que
respondem de forma lenta, com contração longa, são compostos por
maioria de fibras lentas. As diferenças entre esses dois tipos de fibras
são as seguintes:
Fibras rápidas: (1) fibras muito maiores para uma maior força de
contração; (2) retículo sarcoplasmático extenso, para a liberação rápida
de íons cálcio, para desencadear a contração; (3) grande quantidade
de enzimas glicolíticas para a liberação rápida de energia pelo processo
glicolítico; (4) vascularização pouco extensa, pela importância secundária
do metabolismo oxidativo; (5) pequeno número de mitocôndrias,
igualmente por ser o metabolismo oxidativo secundário.
Fibras lentas: (1) fibras menores; (2) também inervado por fibras
nervosas mais finas; (3) vascularização bem mais extensa, com muitos
capilares para fornecimento de quantidades adicionais de oxigênio; (4)
número muito grande de mitocôndrias, permitindo a manutenção de
alto nível do metabolismo oxidativo; (5) as fibras contêm grande
quantidade de mioglobina, proteína contendo ferro, semelhante à
hemoglobina das hemácias. A mioglobina se combina com o oxigênio,
armazenando-o até que seja necessário, e acelera de muito o
transporte de oxigênio para as mitocôndrias. A mioglobina dá ao
músculo lento uma coloração avermelhada, razão desses músculos serem
chamados de músculos vermelhos, enquanto sua falta, nos músculos
rápidos, os faz serem chamados de músculos brancos.
A partir dessas descrições, pode-se ver que as fibras rápidas são
adaptadas para contrações musculares muito rápidas e fortes, como as
que ocorrem nos saltos e na corrida curta. As fibras lentas são adaptadas
para a atividade muscular prolongada e contínua, como a de sustentação
do corpo contra a gravidade e atividades esportivas de longa duração,
como a maratona.
MECÂNICA DA CONTRAÇÃO MUSCULAR
ESQUELÉTICA
A unidade motora
Cada motoneurônio que emerge da medula espinhal inerva
numerosas fibras musculares: esse número depende do tipo de músculo.
Todas as fibras musculares inervadas por uma só fibra nervosa motora
formam a chamada unidade motora. Em geral, os músculos pequenos, que
reagem rapidamente e cujo controle deve ser bastante precisa, têm
unidades motoras com poucas fibras musculares (até apenas duas a
três fibras nos músculos laríngeos). Por outro lado, os músculos grandes,
que não precisam de controle muito exato, como, por exemplo, o músculo
gastroenemio, podem ter unidades motoras com várias centenas de fibras
musculares. Um valor médio para todos os músculos do corpo pode ser
tomado como sendo de cerca de 100 fibras musculares em cada unidade
motora.
As fibras musculares de uma unidade motora não ficam todas
grupadas no músculo, mas, pelo contrário, ficam dispersas por todo o
músculo, em microfeixes de 3 a 15 fibras. Por conseguinte, esses
microfeixes ocorrem intercalados com outros microfeixes de diversas
unidades motoras. Essa interdigitação permite que as unidades motoras
distintas se contraiam em apoio umas às outras, e não de forma total
como se fossem segmentos isolados.
Ele é importante por permitir a gradação da força muscular, durante
uma contração fraca, em etapas pequenas; essas etapas ficam
progressivamente maiores quando são necessárias grandes imensidades
de força. A causa do princípio do tamanho é que as unidades motoras
pequenas são ativadas por fibras nervosas motoras bastante delgadas e os
pequenos motoneurônios da medula espinhal são, de longe, muito mais
excitáveis que os grandes, de modo que, naturalmente, eles são excitados
em primeiro lugar.
Outra característica importante da somação de fibras múltiplas é
que as diferentes unidades motoras são ativadas de modo assincrônico
pela medula espinhal, de modo que a contração se alterna entre diversas
unidades motoras, umas se contraindo após outras, o que permite uma
contração contínua e uniforme, mesmo sob baixas freqüências do sinal
neural.
Somacão por freqüência e tetanização. A Fig. 6.13 apresenta os
princípios da somação por freqüência e da tetanização. À esquerda são
mostrados abalos isolados ocorrendo consecutivamente, produzidos por
baixas freqüências de estimulação. Em seguida, à medida que essa
freqüência aumenta, é atingido um momento em que cada nova
contração ocorre antes do término da precedente. Como resultado, a
segunda contração é parcialmente somada à anterior, de forma que a
força total da contração aumenta progressivamente com a intensificação
da freqüência de estimulação. Quando essa freqüência atinge um nível
crítico, as contrações sucessivas são tão rápidas que, verdadeiramente,
se fundem entre si, e a contração aparece como uniforme e contínua,
como mostrado na figura. Isso é chamado de tetanização. Com
freqüências ainda mais elevadas, a força da contração atinge seu máximo,
de modo que qualquer aumento adicional da freqüência não produzirá
qualquer aumento da força contrátil. Isso decorre de que existem
suficientes íons cálcio no sarcoplasma, até mesmo no intervalo entre os
potenciais de ação, para manter o estado de contração máxima, sem
permitir o relaxamento entre os potenciais de ação.
Força máxima de contração. A força máxima das contrações
tetânicas de músculo operando em seu comprimento normal 6, em
média, de 3 a 4 kg/cm2 de músculo. Uma vez que o músculo
quadríceps pode chegar a ter 40 centímetros quadrados em sua barriga,
ele poderá exercer tensão, sobre o tendão patelar, de até 350 kg. Podese facilmente compreender como, por vezes, um músculo pode
desinserir seu tendão do osso.
Variações da força muscular no início da contração — o
fenômeno da escada (treppe). Quando um músculo começa a se contrair
após longo período de repouso, sua força inicial de contração pode ser
de apenas a metade da que será após os 10 a 50 abalos seguintes. Isto
é, a força da contração aumenta até ser atingido um platô, um fenômeno
conhecido como o efeito de escada ou treppe.
Embora ainda não sejam conhecidas» todas as causas possíveis para
o efeito de escada, acredita-se que, primariamente, seja devido a aumento
do teor de íons cálcio no citosol, decorrente da liberação desses íons
pelo retículo sarcoplasmático a cada potencial de ação e da incapacidade
de recaptação imediata desses mesmos íons.
Contrações musculares com torça diferente — somação da
força
Somação significa a adição de todas as contrações individuais dos
abalos para aumentar a intensidade da contração muscular global. A
somação pode ocorrer por dois modos distintos: (1) pelo aumento do
número de unidades motoras que se contraem a um só tempo, o que
é chamado de somação de fibras múltiplas, e (2) pelo aumento da
freqüência da contração, o que é chamado de somação por freqüência ou
tetanização.
Somação de fibras múltiplas. Quando o sistema nervoso central
envia um sinal fraco para contrair determinado músculo, as unidades
motoras com fibras pequenas e em menor número são estimuladas
preferencialmente às maiores unidades motoras. Em seguida, à medida
que aumenta a intensidade do sinal neural, são estimuladas as unidades
motoras progressivamente maiores, sendo que as unidades motoras muito
grandes chegam a desenvolver, muitas vezes, mais de 50 vezes a força
contrátil das unidades motoras menores. Isso é chamado de princípio do
tamanho.
Fig. 6.13 Somação por freqüência e tetanização.
68
Tônus do músculo esquelético
Mesmo quando os músculos estão em repouso, ainda persiste certo
grau de tensão. Isso é chamado de tônus muscular. Visto que as fibras
musculares esqueléticas não se contraem sem que exista um verdadeiro
potencial de ação para estimulá-las (exceto em algumas condições
patológicas), o tônus do músculo esquelético é totalmente dependente de
impulsos nervosos originados na medula espinhal. Esses impulsos, por
sua vez, são, em parte, controlados por impulsos transmitidos do
encéfalo para os motoneurônios anteriores correspondentes e, em parte,
por impulsos que se originam dos fusos musculares localizados no próprio
músculo. Esses dois mecanismos são discutidos em relação aos fusos
musculares e ao funcionamento da medula espinhal no Cap. 54.
Fadiga muscular
Contrações fortes e prolongadas de um músculo levam ao estado
bem conhecido de fadiga muscular. Estudos em atletas mostraram que
a fadiga muscular aumenta quase em proporção direta com a intensidade
da depleção do glicogênio muscular. Por conseguinte, a maior parte
da fadiga resulta, com muita probabilidade, simplesmente da
incapacidade dos processos contrateis e metabólicos das fibras
musculares de produzir, de modo contínuo, a mesma quantidade de
trabalho. Todavia, experimentos demonstraram que a transmissão do
sinal neural através da placa motora, que é discutida no capítulo
seguinte, pode ficar diminuída após atividade muscular prolongada, o
que diminuiria ainda mais a contração muscular.
A interrupção do fluxo sanguíneo para um músculo em contração
produz fadiga muscular quase total em um minuto ou pouco mais, devido
à perda óbvia do fornecimento de nutrientes - em especial, a falta
de oxigênio.
Os sistemas de alavanca do corpo
Obviamente, os músculos atuam pela aplicação de tensão a seus
pontos de inserção nos ossos e, estes, por sua vez, formam vários tipos
de sistemas de alavanca. A Fig. 6.14 apresenta o sistema de alavanca
ativado pelo músculo bíceps para elevar o antebraço. Se for admitido
que um músculo bíceps volumoso tenha área de seção transversa de
15 cm2, ele seria capaz de desenvolver uma força máxima de contração
da ordem de 131 kg. Quando o antebraço forma precisamente um ângulo
reto com o braço, a inserção do tendão do bíceps fica cerca de 5 cm
adiante do fulcro do cotovelo, e o comprimento total da alavanca formada
pelo antebraço é de 35 cm. Por conseguinte, a quantidade de potência
elevatória que o bíceps poderá ter, ao nível da mão, seria apenas de
um sétimo da força de 131 kg, ou seja, 19 kg. Quando o membro superior
está em sua posição de extensão completa, a inserção do bíceps fica
abem menos que 5 cm anterior ao cotovelo, e a força com que o antebraço
pode ser movido para a frente é bem menor que 19 kg.
Resumindo, a análise dos sistemas de alavanca do corpo depende
de (1) conhecimento preciso do ponto de inserção muscular, (2) sua
distância do fulcro da alavanca, (3) o comprimento do braço da alavanca,
e (4) a posição dessa alavanca. Obviamente, o corpo pode realizar muitos
e diferentes tipos de movimento, alguns exigindo grande força, outros
grandes distâncias de deslocamento. Por essa razão, existem todas as
variedades de músculos; alguns são longos e se contraem por grandes
distâncias, outros são curtos, mas têm grandes áreas de seção transversa,
e são capazes de desenvolver grandes forças contrateis durante pequenos
encurtamentos. O estudo dos diferentes tipos de músculos, dos sistemas
de alavancas e de seus movimentos é chamado de cinesiologia e é área
muito importante da fisioanatomia humana.
"Posicionamento" de parte do corpo pela contração de músculos
antagonistas nos lados opostos de uma articulação - "Coativação" dos músculos antagonistas
Virtualmente, todos os movimentos do corpo são causados pela
contração simultânea dos músculos antagonistas nos lados opostos das
articulações. Isso é chamado de co-ativação dos músculos antagonistas
e é controlado pelos mecanismos motores do encéfalo e da medula
espinhal.
A posição de cada parte distinta do corpo, como, por exemplo,
a de um membro, é determinada pelo grau relativo de contração dos
grupos de músculos antagonistas. Por exemplo, vamos admitir que um
membro seja colocado no ponto médio de sua faixa de movimento.
Para que isso seja conseguido, os músculos antagonistas são excitados
aproximadamente com igual intensidade. Deve ser lembrado que um
músculo estirado se contrai com mais força que um músculo retraído,
como aparece na Fig. 6.9, que mostra a força máxima de contração
para o comprimento total do músculo, e quase nenhuma força para
a metade do comprimento normal do músculo. Por conseguinte, o
músculo antagonista mais longo se contrai com maior força que o
músculo mais curto. Conforme o membro se move para o ponto médio,
a força do músculo mais longo diminui, ao mesmo tempo que a do
músculo mais curto aumenta, até que as duas forças fiquem
perfeitamente iguala das. É nesse ponto que cessa o movimento do
membro. Assim, ao variar a proporção entre os graus de ativação dos
músculos antagonistas, o sistema nervoso direciona o posicionamento
do membro.
Veremos no Cap. 54 que o sistema nervoso motor também possui
mecanismos adicionais muito importantes para compensar as diferentes
cargas impostas aos músculos durante esse processo de posicionamento.
REMODELAGEM DO MÚSCULO PARA ATENDER A
SUA FUNÇÃO
Todos os músculos do corpo estão sob remodelamento contínuo
para que melhor possam atender o que lhes é exigido. Seus diâmetros
são modificados, seus comprimentos são alterados, suas forças são
variadas, suas vascularizações são modificadas e, até mesmo, os tipos
de suas fibras são mudados, pelo menos, em pequeno grau. Esse
processo de remodelagem é, muitas vezes, bastante rápido,
ocorrendo dentro de poucas semanas. Na verdade, experimentos têm
demonstrado que, até mesmo em condições normais, as proteínas
contráteis do músculo podem ser totalmente substituídas uma vez a
cada 2 semanas.
Hipertrofia e atrofia musculares
Fig. 6.14 O sistema de alavanca ativado pelo músculo bíceps
Quando a massa total de um músculo aumenta, ocorre a hipertrofia
muscular. Quando essa massa diminui, o processo é chamado de atrofia
muscular.
Virtualmente, toda hipertrofia muscular é resultado da hipertrofia
das fibras musculares isoladas, o que é chamado, simplesmente, de
hipertrofia das fibras. Em geral, isso ocorre em resposta à contração do
músculo com força máxima ou quase máxima. Ocorre hipertrofia
muito mais acentuada quando o músculo é estirado durante o processo
contrátil. Bastam apenas umas poucas dessas contrações, a cada dia,
para que ocorra hipertrofia quase máxima dentro de 6 a 10 semanas.
Infelizmente, ainda é desconhecido o modo como as contrações
fortes levam a hipertrofia. Todavia, é sabido que a velocidade da síntese
das proteínas contrateis do músculo é muito maior durante o
desenvolvimento da hipertrofia que a velocidade de sua degradação,
do que resulta aumento progressivamente maior do número de
filamentos de actina e de miosina nas miofinrilas.
69
Por sua vez, as miofibrilas se dividem no interior de cada fibra muscular,
para formar novas miofibrilas. Dessa forma, é esse grande aumento do
número de miofibrilas adicionais que produz a hipertrofia das fibras
musculares.
Junto com o aumento do número de miofibrilas, os sistemas
enzimáticos que fornecem energia também aumentam. Isso é
especialmente verdade para as enzimas da glicólise, permitindo um
fornecimento rápido de energia durante as contrações» musculares fortes,
durante breves períodos.
Quando um músculo permanece inativo por longos períodos, a
velocidade de degradação das proteínas contrateis, bem como a
redução do número de miofibrilas, é maior que a velocidade com que são
repostas. Como resultado, ocorre atrofia muscular.
Ajuste do comprimento muscular. Ocorre outro tipo de hipertrofia
quando os músculos são estirados além de seu comprimento normal.
Isso faz com que sejam adicionados novos sarcômeros nas extremidades
das fibras musculares onde elas se fixam aos tendões. Na verdade, a
adição desses novos sarcômeros pode ser bastante rápida, até de vários
sarcômeros a cada minuto, demonstrando a grande rapidez desse tipo
de hipertrofia.
Inversamente, quando um músculo permanece retraído a
comprimento menor que o seu normal por longos períodos, os
sarcômeros nas extremidades das fibras desaparecem de modo
igualmente rápido. É por esses processos que os músculos são
continuamente remodelados para terem o comprimento adequado para
uma contração muscular apropriada.
Hiperplasia das Fibras musculares. Sob condições muito raras de
geração de força muscular extrema, já foi observado aumento do número
de fibras musculares, mas de apenas uns poucos pontos percentuais,
além da hipertrofia das fibras. Esse aumento do número de fibras é
chamado de hiperplasia das fibras. Quando ocorre, seu mecanismo é
o da divisão longitudinal de fibras previamente hipertrofiadas.
Remodelagem das fibras "lentas" em corredores de
maratona
Os músculos muito rápidos, de ação tipo mola, como o gastrocnêmio,
só podem manter alto nível de força contrátil por períodos muito curtos
de tempo de atividade contínua. Por conseguinte, os chamados músculos
lentos, tais como o solear, são usados para as atividades prolongadas,
tais como a corrida de maratona. Esses músculos não se hipertrofiam
tanto como os músculos rápidos. Na verdade, eles são remodelados
por outro modo. A atividade prolongada, por períodos de muitas horas
a cada dia, causa, além de hipertrofia das fibras, de discreta a moderada,
as seguintes alterações que aumentam a capacidade das fibras de
utilizarem os nutrientes:
1. Aumento da mioglobina em cada fibra, para o transporte de
oxigênio para as mitocôndrias.
2. Número muito aumentado de mitocôndrias para formar
quantidades muito maiores de ATP.
3. Quantidades aumentadas de enzimas oxidativas nessas
mitocôndrias para provocar maior intensidade do metabolismo
oxidativo, o que aumenta ainda mais a produção de ATP.
4. Intenso crescimento de capilares no próprio músculo, resultando
em menor espaçamento desses capilares por entre as fibras musculares,
de modo que o oxigênio e outros nutrientes possam ser rápida e facilmente
fornecidos durante os períodos prolongados de atividade.
Efeitos da desnervação muscular
Quando um músculo fica privado de sua inervação, ele deixa de
receber os sinais contrateis necessários para manter suas dimensões
normais. Como resultado, a atrofia começa quase imediatamente.
Após cerca de 2 meses, começam a aparecer alterações degenerativas
nas próprias fibras musculares. Se houver reinervação, ocorrerá
restauração completa da função até, nas condições usuais, 3 meses; mas,
após esse período, a capacidade de restauração funcional fica
progressivamente menor, com perda definitiva de função após 1 a 2
anos.
Nas etapas finais da atrofia de desnervação, a maior parte das fibras
musculares já está destruída e substituída por tecido fibroso e gorduroso.
As fibras remanescentes são formadas por longa membrana celular, com
fileira de núcleos de células musculares, mas desprovidas de propriedades
contráteis e sem capacidade de regeneração de miofibrilas, caso ocorra
reinervação.
Infelizmente, o tecido fibroso que toma o lugar das fibras musculares
durante a atrofia de desnervação apresenta tendência a se retrair durante
muitos meses, o que é chamado de contratura. Por conseguinte, um
dos mais importantes problemas na prática da fisioterapia é a de impedir
que os músculos atróficos venham a desenvolver contraturas debilitantes
e desfigurantes. Isso é conseguido pelo estiramento diário dos músculos
ou pelo uso de aparelhos que mantenham os músculos estirados durante
o processo da atrofia.
Recuperação da contração muscular na poliomielite:
desenvolvimento de unidades macromotoras. Quando algumas fibras
nervosas para um músculo são destruídas, com conservação de algumas,
como ocorre freqüentemente na poliomielite, as, fibras remanescentes
apresentam brotamentos de seus axônios que vão originar novos ramos
axônicos, que, por sua vez, vão formar muitas ramificações novas, que,
em seguida, inervam muitas das fibras musculares paralisadas. Disso
resulta a formação de unidades motoras muito grandes, chamadas de
unidades macromotoras, que chegam a conter número de fibras
musculares cinco vezes maior que o número normal para cada
motoneurônio da medula espinhal. Isso, obviamente, reduz a precisão
do controle que deve existir sobre os músculos, mas, não obstante,
permite que os músculos readquiram sua força.
RIGOR MORTIS
Várias horas após a morte, todos os músculos do corpo passam
para um estado de contratura que é chamado de rigor mortis; isto é, o
músculo se contrai e fica rígido, mesmo sem potenciais de ação. Essa
rigidez é causada pela perda total de ATP, que é necessário para a
separação das pontes cruzadas dos filamentos de actina durante o
processo de relaxamento. Os músculos permanecem em rigor até que as
proteínas musculares sejam destruídas, o que, em geral, é causado
por autólise por enzimas liberadas dos lisossomas, cerca de 15 a 25
horas após a morte; esse processo é mais rápido nas temperaturas
elevadas.
REFERÊNCIAS
Clausen, T.: Regulation ofactive Na*-K+transport in skeletal muscle. Physiol. Rev., 66:542, 1986. DiDonato, S., et ai. (eds.): Molecular Genetics of
Neurological and Neuromuscular Diseaae. New York, Raven Press, 1988.
Engel, A. G., and Banker, B. Q. (eds.): Myology. New York, McGraw-Hill
Book Co., 1986. Freund, H.-J.: Motor unit and muscle activity in
voluntary motor control.
Physiol. Rev., 63:387, 1983. Goldman, Y. E.: Kinetics of the actomyosin
ATPase in muacle fibers. Annu. Rev. Physiol., 49:637, 1987. Goldman, Y.
E., and Brenner, B. (eda.): General introduetion. Annu. Rev.
Physiol., 49:629, 1987. Gowitzke, B. A., et ai.: Scientific Bases of Human
Movement. Baltimore, Williams & Wilkins, 1988. Hasselbach, W., and
Oetliker, H.: Energetics andelectrogenícity of the sarcoplasmic reticulum
calcium pump. Annu. Rev. Physiol., 45:325, 1983. Haynes, D, H., and
Mandveno, A.: Computer modeling of Ca1 -Mg2 ATPase of sarcoplasmic reticulum. PhyBiol. Rev., 67:244, 1987. Homsher,
E.: Muscle enthalpy produetion and its relationship to actomyosin
ATPaae. Annu. Rev. Physiol., 49:673, 1987. Huang, C. L. H.:
Intramembrane charge movements in skeletal muscle. Physiol. Rev.,
68:1197, 1988.
Huxley, A. F.: Muscular Contraction. Annu. Rev. Phyaiol., 50:1, 1988.
Hrnrley, A. F., and Gordon, A. M.: Striation patterns in active and passive
shortening of muscle. Nature (Lond.), 193:280, 1962. Huxley, H. E., and
Faruqi, A. R.: Time-resolved x-ray diffraction studies on vertebrate striated
muscle. Annu. Rev. Biophys. Bioeng., 12:381, 1983. Ikemoto, N.:
Structure and function of the calcium pump protein of sarcoplasmic
reticulum. Annu. Rev. Physiol., 44:297,1982. Johnson, E. W. (ed.):
Practical Electromyography. Baltimore, Williama &
Wilkins, 1988. Kodama, T.: Thermodynamic analysia of muscle ATPase
mechanisms. Physiol. Rev., 65:476,1986.
Kolata, G.: Metabolic catch-22 of exercise regimens. Science, 236:146,1987.
Korn, E. D.: Actin polymerization and its regulation by proteins from nonmuscle cells. Physiol. Rev., 62:672, 1982. Korn, E. D., et ai.: Actin
polymerization and ATP hydrolysis. Science, 238:638, 1987. Laufer, R.,
et aí.: Regulation of acetylcholine receptor bíoayntheaia durin motor
endplate morphogenesis. News Physiol. Sei., 4:5, 1989.
Martonosi, A. N.: Mechanisms of Ca** release from sarcoplaamic reticulum
of skeletal muscle. Physiol. Rev., 64:1240,1984. Morgan, D. L., and
Proiske, U.: Vertebrate slow muscle: Its structure, pattern of innervation, and
mechanical properties. Physiol. Rev., 64:103,1984. Morkin, E.: Chronic
adaptations in contractile proteína: Genetic regula tion.
Annu. Rev. Physiol., 49:545, 1987. Oho, S. J.: Electromyography:
Neuromuscular Transmission Studies. Baltimore, Williams & Wilkins,
1988. Partridge, L. D.,andBenton, L. A.: Muscle, the motor. In Brooks, V.
B. (ed.):
70
Handbook of Physiology. Sec. 1, Vol. II. Bethesda, American Phyaiologícal
Society, 1981, p. 43.
Pollack, G. H.: The cross-bridge theory. Phyaiol. Rev., 63:1049, 1983.
Rash, J.: Neuromuscular Atlas. New York, Prager Publishers, 1984.
Ringel, S. P.: Neuromuscular D ia or dera: A Guide for Patient and Family.
New York, Raven Press, 1987. Rios, E., and Pizarro, G.: Voltage senso rs
and calcium channels ofexcitation contraction coupling. News Physiol. Sei.,
3:223, 1988. Rogart, R.: Sodium channels in nerve and muscle membrane.
Annu. Rev. Physiol., 43:711, 1981. Rowland, L. P.,etal. (eds.): Molecular
Geneticsin Diaeases of Brain, Nerve, and Muscle. New York, Oxford
University Presa, 1989. Rudel. R., and Lehmann-Horn, F.: Membrane
changes in cells from myotonia patients. Physiol. Rev , 65:310,1985.
Sohafer, R. C: Clinicai Biomechanics: Musculoskeletal Actions and Reactions. Baltimore, Williams & Wilkins, 1986. Schubert, D.: Developmental
Biology of Cultured Nerve, Muscle and Glia.
New York, John Wiley & Sons, 1984. Sjodin. R. A.: lon Transport in
Skeletal Muscle. New York, John Wiley & Sons, 1982.
Soderberg, G. L.: KinesioloKy. Baltimore, Williams & Wilkins, 1986.
Stcfani, E., and Chiarandini, D. J.: Ionic channels in skeletal muscle. Annu.
Rev. Physiol., 44:357, 1982. Steinbach, J. H.: Structural and functional
diversity in vertebrate skeletal muscle nicotinic acetylcholine receptors.
Annu. Rev. Physiol., 51:353, 1989. Sugi, H., and Pollack, G. H. (eds.):
Molecular Mechanism of Muscle Contraction. New York, Plenum
Publishing Corp., 1988. Swynghedauw, B.: Developmental and functional
adaptation of contractile proteins in cardiac and skeletal muscles. Physiol.
Rev., 66:710, 1986. Tnoman, D. D.; Spectroscopic probes of muscle crossbridge rotatíon. Annu.
Rev. Physiol., 49:691, 1987. Vergara, J., and Asotra, K.: The chemical
Transmission mechanism of excitation-contraction coupling in skeletal
muscle. News Physiol. Sei., 2:lfi2, 1987. Wingrad, S.: Regulation of
cardiac contractile proteins. Correlations be tween physiology and
biochemistry. Circ. Res., 55:565, 1984.
71
CAPÍTULO 7
Excitação da Contração do Músculo Esquelético: Transmissão
Neuromuscular e Acoplamento Excitação-Contração.
TRANSMISSÃO DOS IMPULSOS DOS NERVOS
PARA AS FIBRAS MUSCULARES ESQUELÉTICAS:
A PLACA MOTORA
As fibras musculares esqueléticas são inervadas por fibras
mielínicas grossas, originadas nos grandes motoneurônios da ponta
anterior da medula espinhal. Como notado no capítulo anterior,
cada uma dessas fibras nervosas em geral se ramifica
extensamente e estimula de três a várias centenas de fibras
musculares esqueléticas. A terminação nervosa forma uma
junção, chamada de placa motora (ou junção neuromuscular),
e o potencial de ação na fibra muscular se propaga nas duas
direções, dirigindo-se para as suas extremidades. Com exceção
de cerca de 2% das fibras musculares, só existe uma dessas
junções em cada fibra muscular.
Anatomia fisiológica da junção neuromuscular — a "placa
motora". A Fig. 7.1 partes A e B, apresenta uma junção
neuromuscular entre uma fibra mielínica calibrosa e uma fibra
muscular esquelética. A fibra nervosa se ramifica, próximo de sua
extremidade, formando numerosas terminações nervosas, que se
invaginam na fibra muscular, embora permaneçam
inteiramente por fora da membrana plasmática dessa fibra
muscular. Toda a estrutura resultante é revestida por uma ou
mais células de Schwann que a isolam dos líquidos
circundantes.
A Fig. 7.1 C mostra um esquema derivado de
microfotografias eletrônicas da junção entre terminação
axônica única e a membrana da fibra muscular. A
invaginação da membrana é chamada de goteira sináptica e o
espaço entre a terminação axônica e a membrana da fibra é a
fenda sináptica. A fenda sináptica tem largura de 20 a 30 nm e é
revestida por uma lâmina basal, formada por fina camada de
fibras reticulares esponjosas, através da qual se difunde o líquido
extracelular. No fundo dessa goteira existem dobras menores da
membrana muscular, chamadas de pregas subneurais, que
aumentam de muito a área da superfície sobre a qual vai atuar o
transmissor sináptico.
Na terminação nervosa existem muitas mitocôndrias que
fornecem energia, principalmente para a síntese do transmissor
excitatório acetileolina que, por sua vez, excita a fibra
muscular. A acetileolina é sintetizada no citoplasma da
terminação, sendo rapidamente absorvida para o interior de
numerosas e pequenas vesículas sinápticas; nas condições
normais, existem cerca de 300.000 dessas vesículas em cada
terminação axônica de placa motora.
Fixada à matriz da lâmina basal existe grande quantidade da
enzima acetilcolinesterase, que é capaz de destruir a acetileolina,
o que vai ser explicado adiante em maiores detalhes.
Secreção de acetileolina pelas terminações nervosas
Quando um impulso nervoso invade a junção
neuromuscular, cerca de 300 vesículas de acetileolina são
liberadas pelas terminações axônicas na goteira sináptica. A Fig.
7.2 apresenta alguns detalhes desse mecanismo, mostrando
imagem ampliada de uma goteira sináptica com a membrana
neural acima e a membrana muscular com suas fendas
subneurais abaixo.
Existem na superfície interna da membrana neural barras
densas lineares, mostradas em corte transverso na Fig. 7.2. De
cada lado de uma barra densa existem partículas protéicas que
atravessam toda a membrana, e que são consideradas como
formando canais de cálcio voltagem-dependentes. Quando o
potencial de ação se propaga por toda a terminação, esses canais
se abrem, permitindo a difusão de grande quantidade de
cálcio para o interior da terminação. Os íons cálcio, por sua vez,
exercem influência atrativa sobre as vesículas de acetileolina,
puxando-as para a membrana neural adjacente às barras densas.
Algumas dessas vesículas se fundem com a membrana neural e
esvaziam seu conteúdo de acetileolina na goteira sináptica pelo
mecanismo de exoeitose.
Embora alguns dos detalhes descritos acima ainda sejam
especulativos, sabe-se que o estímulo efetivo para fazer com
que a acetileolina seja liberada das vesículas é o influxo de íons
cálcio. Ainda mais, o esvaziamento das vesículas ocorre na
membrana adjacente às barras densas.
Efeito da acetilcolina para abrir os canais iônicos acetilcolina-
dependentes. A Fig. 7.2 mostra muitos receptores para
acetilcolina na membrana muscular; na realidade, esses
receptores são canais iônicos acetilcolina-dependentes,
localizados, em sua quase totalidade, próximo à entrada das
pregas subneurais, situadas imediatamente abaixo da área das
barras densas, onde a acetilcolina é liberada na fenda sináptica.
Cada receptor é um grande complexo protéico, com peso
molecular total de 275.000. O complexo é formado por cinco
subunidades protéicas, que atravessam toda a espessura da
membrana, uma ao lado da outra, formando um círculo que
circunda um canal tubular.
72
Fig. 7.1 Diferentes aspectos da placa motora terminal.
A, Seção longitudinal através da placa motora. B, Visão
superficial da placa motora. C, Aspecto à micrografia
eletrônica dos pontos de contato entre uma das
terminações axonais e a membrana da fibra muscular,
representando a área retangular mostrada em A. (De
Bloom e Fawcett, como modificado de R. Couteaux: A
Textbook of Histology. Philadelphia, W. B. Saunders
Company, 1975.)-
Esse canal permanece contraído até que a acetilcolina se fixe a
uma de suas subunidades. Isso provoca alteração
conformacional, abrindo o canal, como mostrado na Fig. 7.3;
no painel superior, o canal está fechado, no inferior, aberto pela
fixação de molécula de acetilcolina.
Quando aberto, o canal de acetilcolina tem diâmetro de
cerca de 0,65 nm, suficientemente grande para permitir a
passagem de todos os íons positivos importantes — sódio (Na+),
potássio (K+) e cálcio (Ca++) — com muita facilidade. Por outro
lado, os íons negativos, como os íons cloreto, não passam por
ele, devido às fortes cargas negativas presentes em sua abertura
externa.
Contudo, na prática, quantidade muito maior de íons sódio
do que de qualquer outro íon passa pelos canais de acetilcolina,
por duas razões. Primeira, só existem dois íons positivos em
concentração suficientemente alta para terem importância: os
Fig. 7.2 Liberação de acetilcolina pelas vesículas sinápticas na
membrana neural da placa motora. Notar a grande proximidade dos
locais de liberação com os receptores para acetilcolina nas bocas das
pregas subneurais.
Fig. 7.3 O canal da acetilcolina: acima, no estado fechado; abaixo, apôs
fixação de acetilcolina, uma alteração conformacional abriu o canal,
permitindo entrada de sódio em excesso na fibra muscular, excitando
a contração. Notar as cargas negativas na boca do canal que impedem
a entrada de íons negativos.
73
íons sódio, no líquido extracelular, e os íons potássio, no líquido
intracelular. Segunda, o potencial muito negativo vigente na face
interna da membrana muscular, de cerca de - 80 a - 90 mV,
puxa os íons sódio, com carga positiva, para o interior da fibra,
ao mesmo tempo que impede o efluxo dos íons potássio, quando
estes tentam sair.
Por conseguinte, como mostrado no painel inferior da Fig.
7.3, o resultado efetivo da abertura dos canais acetilcolinadependentes é o de permitir a passagem de grande número de
íons sódio para o interior da fibra, carregando com eles muitas
cargas positivas. Isso gera um potencial local no interior da fibra,
chamado de potencial da placa, que leva a um potencial de ação
na membrana muscular, produzindo, assim, a contração
muscular.
Destruição da acetilcolina liberada pela acetilcolineslerase.
A acetilcolina, uma vez que tenha sido liberada na fenda
sináptica, continua a ativar os receptores para a acetilcolina,
enquanto persistir na fenda. Contudo, ela é rapidamente
removida por dois mecanismos. (1) A maior parte da
acetilcolina é destruída pela enzima acetilcolinesterase, que, em
sua maior parte, ocorre fixada à lâmina basal, a fina camada
esponjosa de tecido conjuntivo que enche a fenda sináptica
entre o terminal pré-sináptico e a membrana muscular póssináptica. (2) Pequena quantidade de acetilcolina se difunde
para fora da fenda sináptica, não mais sendo disponível para
atuar sobre a membrana da fibra muscular.
Todavia, no intervalo de tempo extremamente breve em
que a acetilcolina permanece na fenda sináptica — no máximo
de uns poucos milissegundos -, a acetilcolina é quase sempre
suficiente para excitar a fibra muscular. Então, a rápida remoção
dessa acetilcolina impede a reexcitação muscular, após a fibra
ter-se recuperado do primeiro potencial de ação.
O "potencial da placa" e a excitação da fibra muscular esquelética.
O influxo abrupto dos íons sódio para o interior da fibra muscular,
conseqüente à abertura dos canais de acetilcolina, faz com que o potencial
de membrana, na área localizada da placa motora, varie, em direção
à positividade. por até 50 a 75 mV, gerando um potencial locai, chamado
de potencial da placa. Deve ser lembrado do Cap. 5 que o aumento
súbito do potencial de membrana por mais de 15 a 30 mV é suficiente
para desencadear o feedback positivo, efeito da ativação dos canais de
sódio, o que leva à compreensão de que o potencial de placa causado
pela estimulação por acetilcolina é, em condições normais, mais que
suficiente para desencadear um potencial de ação na fibra muscular.
A Fig. 7.4 mostra como o potencial de placa pode produzir um
potencial de ação. Essa figura mostra três potenciais de placa distintos.
Os potenciais da placa A e C são fracos demais para provocar um
potencial de ação, mas, não obstante, produzem os pequenos potenciais
locais mostrados na figura. Como contraste, o potencial da placa B é
bem mais forte e provoca a ativação de canais de sódio em número
suficiente para que o efeito auto-regenerativo do influxo crescente de
íons sódio, para o interior da fibra, inicie um potencial de ação. O
pequeno potencial da placa no ponto A foi produzido pelo
envenenamento da fibra muscular com curare, substância que impede a
ação excitatória da acetilcolina sobre os canais de acetilcolina, ao
competir com a própria acetilcolina pela fixação ao seu receptor. O
outro pequeno potencial da placa, no ponto C, resultou da aplicação de
toxina botulínica, uma toxina bacteriana que reduz a liberação de
acetilcolina pelas terminações nervosas.
O "fator de segurança" para a transmissão na placa motora fadiga da junção. Comumente, cada impulso que chega à placa motora
provoca um potencial de placa que é de três a quatro vezes maior que
o necessário para estimular a fibra muscular. Por conseguinte, diz-se
que a placa motora normal tem um fator de segurança muito alto.
Todavia, a estimulação artificial da fibra nervosa, com freqüências
acima de 100 por segundo, durante vários minutos, costuma diminuir o
número de vesículas de acetilcolina liberadas a cada impulso, de modo que
muitos desses impulsos deixam de atingir a fibra muscular. Isso é
chamado de fadiga da placa motora, e é análoga à fadiga das sinapses no
sistema nervoso central. Sob as condições normais de funcionamento, a
fadiga da placa motora só ocorreria raríssimas vezes e, assim mesmo, nos
níveis mais extenuantes da atividade muscular.
Fig. 7.4 Potenciais de placa motora. A, Potencial de placa terminal
reduzido, registrado num músculo curarizado, demasiado fraco para
deflagrar um potencial de ação. B, Potencial normal de placa motora
produzindo um potencial de ação no músculo. C, Potencial de placa
motora reduzido causado pela toxina botulínica que diminui a liberação de
acetilcolina na placa, novamente muito fraco para deflagrar um
potencial de ação no músculo.
Biologia molecular da formação e da liberação de
acetilcolina
Uma vez que a placa motora é suficientemente grande para ser
estudada com facilidade, ela é uma das poucas sinapses do sistema
nervoso em que a maior parte dos detalhes da transmissão química já
foi elucidada. Nessa junção, a formação e a liberação da acetilcolina
ocorrem nas seguintes etapas;
1. Numerosas vesículas pequenas, com diâmetro de cerca de 40
nm, são formadas no aparelho de Golgi, no corpo celular do
motoneurônio da medula espinhal. Essas vesículas são, então,
transportadas pela "torrente" do axoplasma pela parte central do
axônio, desde o corpo celular até a placa motora, na extremidade da
fibra muscular. Cerca de 300.000 dessas vesículas são coletadas nas
terminações nervosas de uma só placa terminal.
2. A acetilcolina é sintetizada no citosol das terminações das fibras
nervosas, mas é, em seguida, transportada através de suas membranas
para o interior das vesículas, onde fica armazenada de forma extrema
mente concentrada, com cerca de 1.000 moléculas de acetilcolina em
cada vesícula.
3. Em condições de repouso, ocasionalmente uma vesícula se funde
com a membrana superficial da terminação nervosa, liberando seu
conteúdo de acetilcolina na goteira sináptica. Quando isso ocorre,
aparece um potencial miniatura de placa, com amplitude de 1 mV e
duração de poucos milissegundos, restrito à área localizada da fibra
muscular, devido à ação desse "pacote" de acetilcolina.
4. Quando um potencial de ação invade a terminação nervosa, ele
induz ã abertura de muitos canais de cálcio na membrana dessa
terminação, visto que ela contém muitos canais.de cálcio voltagemdependentes.
Como resultado, a concentração de íons cálcio, no interior da terminação,
aumenta por cerca de 100 vezes, o que, por sua vez, intensifica a fusão
das vesículas de acetilcolina com a membrana da terminação por cerca
de 10.000 vezes. Quando uma vesícula se funde, sua superfície externa
atravessa a membrana celular, do que resulta a exoeitose da acetilcolina
para a goteira sináptica. Em geral, para cada potencial de ação, ocorre
rotura de 200 a 300 vesículas. Em seguida, ainda na goteira sináptica,
a acetilcolina é degradada, pela acetilcolinesterase, em íon acetato e
em colina; essa colina é ativamente reabsorvida pela terminação neural,
para ser reutilizada na síntese de acetilcolina. Toda essa seqüência leva
de 5 a 10 ms.
5. Após cada vesícula ter liberado seu conteúdo de acetilcolina,
a membrana da vesícula passa a fazer parte da membrana celular.
Contudo, o número de vesículas disponíveis na terminação neural é
suficiente para permitir a transmissão de apenas alguns milhares de
impulsos nervosos. Por conseguinte, para a continuidade do
funcionamento da placa motora, as vesículas devem ser recuperadas da
membrana celular. Essa
74
recuperação é realizada pelo processo de endocitose, que foi explicado
no Cap. 2. Dentro de poucos segundos após o término do potencial
de ação, "depressões revestidas" aparecem na superfície da membrana
da terminação neural, induzidas pelas proteínas contrateis do citosol,
em especial pela proteína catrina, que fica fixada por baixo da membrana
nas áreas das vesículas originais. Dentro de 20 segundos, essas proteínas
se contraem c fazem com que essas depressões passem para o interior
da terminação, formando novas vesículas. Dentro de mais poucos
segundos, a acetílcolina é transportada para o interior dessas vesículas,
que ficam, assim, prontas para novo ciclo de liberação de acetilcolina.
Substâncias que atuam sobre a transmissão na placa
motora.
Substâncias que estimulam a fibra muscular por ação
semelhante à da acetilcolina. Muitos compostos distintos, incluindo a
metacolina, o carbacol e a nicotina, têm o mesmo efeito sobre a
fibra muscular que a acetilcolina. A diferença entre esses compostos
e a acetilcolina é que eles não são degradados pela acetilcolinesterase,
ou o são apenas muito lentamente, de modo que, quando aplicados à
fibra muscular, sua ação persiste por muitos minutos, podendo durar
várias horas. Esses compostos atuam por produzir áreas localizadas de
despolarização na placa motora, onde ficam localizados os receptores
para a acetilcolina. Então, a cada vez que a fibra muscular fica
repolarizada em algum outro ponto, essas áreas despolarizadas, em
virtude de seu vazamento de íons, induzem novos potenciais de ação,
o que leva a estado de espasmo.
Substâncias que bloqueiam a transmissão na placa motora.
Um grupo de compostos, conhecidos como substâncias curare miméticas, pode impedir a passagem dos impulsos da placa motora para
o músculo. Assim, a D-tubocurarina atua sobre a membrana por competir
com a acetilcolina pelos receptores da membrana, de modo que a
acetilcolina não pode aumentar a permeabilidade dos canais de
acetilcolina o suficiente para desencadear uma onda de despolarização.
Substâncias que estimulam a placa motora por inativação da acetilcolinesterase. Três compostos particularmente bem conhecidos, a neostigmina, a fisostigmina e o diisopropil-fluorofosfato, inativam a acetilcolinesterase, de modo que a colinesterase presente normalmente nas
sinapses não hidrolisa a acetilcolina liberada na placa motora. Como
resultado, a quantidade de acetilcolina aumenta a cada impulso nervoso
sucessivo, de forma que quantidades excessivas de acetilcolina podem
ficar acumuladas e, então, estimular repetitivamente a fibra muscular.
Isso provoca espasmos musculares, até mesmo quando só uns poucos
impulsos atingem o músculo; isso pode levar à morte por espasmo
laríngeo, que sufoca a pessoa.
A neostigmina e a fisostigmina se fixam à acetilcolinesterase,
inativando-a por várias horas, após o que elas são deslocadas da
acetilcolinesterase, que volta a ficar ativa. Por outro lado, o diisopropilfluorofosfato, que tem uso militar potencial como um gás "dos nervos"
muito potente produz inativação da acetilcolinesterase por várias
semanas, o que o torna um composto particularmente letal.
Miastenia gravis
A doença miastenia gravis, com incidência de uma entre cada 20.000
pessoas, faz com que o paciente fique paralisado pela incapacidade das
placas motoras de transmitir sinais da fibra nervosa para as fibras
musculares. Em situações patológicas, foram demonstrados anticorpos
contra as proteínas carreadoras acetileolina-dependentes na maioria dos
pacientes. Por conseguinte, acredita-se que a miastenia gravis é, na
maior parte dos casos, doença auto-imune em que os pacientes
desenvolveram anticorpos contra seus próprios canais iônicos
acetileolina-dependentes.
Independentemente da causa, os potenciais de placa que aparecem
nas fibras musculares são por demais fracos para estimular com
intensidade adequada as fibras musculares. Se a doença for
suficientemente grave, o paciente morre de paralisia — de modo
especial por paralisia dos músculos respiratórios. Contudo, a doença
pode ser controlada pelo uso de neostigmina ou de qualquer outro
composto com ação anticolines-terasica. Isso permite o acúmulo de
maiores quantidades de acetilcolina na fenda sináptica. Dentro de
minutos, algumas dessas pessoas paralisadas podem começar a atuar de
forma quase normal.
O POTENCIAL DE AÇÃO MUSCULAR
Quase tudo o que foi discutido no Cap. 5 sobre o
desencadeamento e a propagação de potenciais de ação nas
fibras nervosas também é inteiramente aplicável às fibras
musculares esqueléticas, exceto por diferenças quantitativas.
Alguns dos aspectos quantitativos característicos dos potenciais
musculares são os seguintes:
1. Potencial de repouso da membrana: aproximadamente,
- 80 a - 90 mV nas fibras esqueléticas - o mesmo que nas
fibras mielínicas mais calibrosas.
2. Duração do potencial de ação: de 1 a 5 ms no músculo
esquelético — cerca de cinco vezes maior que nas fibras nervosas
mielínicas mais calibrosas.
3. Velocidade de condução: de 3 a 5 m/s — cerca de 1/18
da medida nas grossas fibras nervosas mielínicas que inervam
o músculo esquelético.
Propagação do potencial de ação para o interior da
fibra muscular por meio do sistema de túbulos
transversos
A fibra muscular esquelética é tão grossa que os potenciais
de ação que se propagam por sua membrana superficial produzem
fluxo de corrente quase nulo na profundidade dessas fibras.
Contudo, para que ocorra contração, essas correntes elétricas
devem penetrar ate a vizinhança imediata de todas as
miofibrilas. Isso é conseguido pela transmissão dos potenciais de
ação pelos túbulos transversos (túbulos T) que atravessam toda a
espessura da fibra muscular, de um lado a outro. Os potenciais
de ação nos túbulos T, por sua vez, fazem com que o retículo
sarcoplasmático libere íons cálcio na vizinhança imediata de
todas as miofibrilas, e esses íons cálcio, então, induzem à
contração. Esse processo global é chamado de acoplamento
excitação-contração. Vamos, agora, descrevê-lo com muito
mais detalhe.
ACOPLAMENTO EXCITAÇÃO-CONTRAÇÃO
O sistema túbulo transverso-retículo sarcoplasmático
A Fig. 7.5 mostra diversas miofibrilas envoltas pelo sistema
túbulo T-retículo sarcoplasmático. Os túbulos transversos são
muito delgados e seu percurso é transversal as miofibrilas.
Começam na membrana celular e atravessam, de um lado a
outro, toda a espessura da fibra muscular, até sua face
oposta. Não é mostrado na figura que esses túbulos se ramificam
e se interconectam, para formar verdadeiros planos de túbulos T,
interligados por entre todas as diferentes miofibrilas. Também,
deve ser notado que, onde os túbulos T se originam da
membrana celular, eles ficam abertos para o exterior.
Conseqüentemente, eles se comunicam com o líquido que banha
a fibra muscular, contendo líquido extracelular em seus lumens.
Em outras palavras, os túbulos T são extensões internas da
membrana celular. Por conseguinte, quando um potencial de
ação se propaga pela membrana de uma fibra muscular, ele
também se propaga, por meio dos túbulos T, para a
profundidade interior da fibra muscular. As correntes do
potencial de ação, em torno desses túbulos T, induzem a
contração muscular.
A Fig. 7.5 também mostra o retículo sarcoplasmático,
representado em vermelho. Ele é composto por duas estruturas:
(1) os longos túbulos longitudinais, com percurso paralelo ao
das miofibrilas e que terminam em (2) grandes câmaras,
chamadas de cisternas terminais, acopladas aos túbulos
transversos. Quando
75
Fig. 7.5 Sistema retículo sarcoplasmático-túbulo
transverso. Observem-se lúbuhs longitudinais que
terminam em grandes cisternas. As cisternas, por
sua vez, entram em contato com os túbulos
transversos. Observe-se também que os túbulos
transversos se comunicam com o exterior da
membrana celular. Esta ilustração foi desenhada a
partir do músculo de rã. que apresenta um túbulo
transverso por sarcômero, localizado na linha Z. Um
arranjo semelhante é encontrado em músculo
cardíaco de mamífero, sendo que o músculo
esquelético de mamífero, no entanto, apresenta dois
túbulos transversos por sarcômero localizados nas
junções A-I. (De Bloom e Fawcett: A Textbook of
Histology. Philadelphia. W. B. Saunders Company.
1986. Modificado de Pea-chey: J. Cell BioL,
25:209, 1965. Desenhado por Sylvia Colard
Keene.)
a fibra muscular é cortada longitudinalmente, para
microfotografias eletrônicas, a imagem resultante mostra esse
acoplamento das cisternas aos túbulos transversos, o que dá ao
conjunto a aparência de uma tríade, com um pequeno túbulo
central e uma grande cisterna de cada lado. Isso é mostrado na
Fig. 7.5 e também aparece na microfotografia eletrônica da Fig.
6.3.
Nos músculos de animais inferiores, como a rã, existe rede
única de túbulos T para cada sarcômero localizada ao nível
do disco Z, como mostrado na Fig, 7.5. O músculo cardíaco
também apresenta esse tipo de sistema de túbulos T. Contudo,
no músculo esquelético de mamíferos, existem duas redes de
túbulos T para cada sarcômero, localizados próximo das duas
extremidades dos filamentos de miosina, que são os locais onde
são geradas as forças mecânicas efetivas da contração muscular.
Dessa forma, o músculo esquelético de mamífero é otimamente
organizado para a excitação rápida da contração muscular.
das a esse túbulo T. Nesses pontos, cada cisterna projeta pés
de juntura que se prendem à membrana do túbulo T,
presumivelmente facilitando a passagem de algum sinal do
túbulo T para a cisterna. É possível que esse sinal seja a
corrente elétrica do próprio potencial de ação. Contudo,
também existem razões para se acreditar que esse sinal poderia
ser químico ou mecânico. Qualquer que seja a natureza desse
sinal, ele provoca a abertura rápida de muitos canais de cálcio
nas membranas das cisternas e nos túbulos longitudinais do
retículo sarcoplasmático que as continuam. Esses canais
permanecem abertos por poucos milissegundos; durante esse
período, os íons cálcio, responsáveis pela contração muscular,
são liberados no sarcoplasma que banha as miofibrilas. Os íons
cálcio que são, assim, liberados pelo retículo sarcoplasmático se
difundem até as miofibrilas adjacentes, onde vão se fixar
fortemente a troponina C, como descrito no capítulo anterior, e
isso induz à contração muscular.
A bomba de cálcio para a remoção dos tons cálcio do líquido
LIBERAÇÃO DE ÍONS CÁLCIO PELO RETÍCULO
SARCOPLASMÁTICO
Uma das características especiais do retículo sarcoplasmático
é que ele contém íons cálcio em concentrações muito elevadas,
e muitos desses íons são liberados quando o túbulo T adjacente
é excitado.
A Fig. 7.6 mostra que o potencial de ação do túbulo T
produz fluxo de corrente através das pontas das cisternas acopla-
sarcoplasmático. Uma vez tendo ocorrido a liberação de íons
cálcio pelo retículo sarcoplasmático e sua difusão até as
miofibrilas, a contração muscular vai ocorrer enquanto os íons
cálcio permanecerem em concentração elevada no líquido
sarcoplasmático. Todavia, uma bomba de cálcio continuamente
ativa situada nas paredes do retículo sarcoplasmático bombeia os
íons cálcio do líquido sarcoplasmático para o interior dos túbulos
sarcoplas-máticos. Essa bomba é capaz de concentrar os íons
cálcio no interior do retículo sarcoplasmático por cerca de
10.000 vezes.
76
Fig. 7.6 Acoplamento excitação-contraçáo no músculo,
mostrando um potencial de ação que causa a liberação de
íons cálcio do retículo sarcoplasmático e a sua recaptação por
uma bomba de cálcio.
Além disso, existe no interior desse retículo uma proteína,
chamada 3e calsequestrina, que pode fixar quantidade de
cálcio 40 vezes maior que a existente no estado iônico, o que
gera aumento da capacidade de armazenamento do cálcio de 40
vezes. Assim, essa transferência maciça de cálcio para o retículo
sarcoplasmático produz depleção quase total dos íons cálcio no
líquido que banha as miofibrilas. Por conseguinte, exceto
imediatamente após um potencial de ação, a concentração de
íons cálcio nas miofibrilas é mantida em valor extremamente
baixo.
O "pulso" excitatório de íons cálcio. A concentração normal
(de menos de 10- 7 molar) dos íons cálcio no citosol que banha
as miofibrilas é pequena demais para induzir à contração. Por
conseguinte, no estado de repouso, o complexo troponina-tropomiosina mantém os filamentos de actina inativos, mantendo o
estado relaxado do músculo.
Por outro lado, a excitação completa do sistema túbulo Tretículo sarcoplasmático provoca liberação de quantidade
suficiente de íons cálcio para aumentar sua concentração no
líquido miofibrilar até 2 x 10 -4 molar, que é cerca de 10
vezes maior que o teor necessário para induzir a contração
máxima do músculo (aproximadamente, 2 X 10-5 molar).
Imediatamente após, a bomba de cálcio volta a depletar os
íons cálcio. Esse "pulso" de cálcio na fibra muscular
esquelética típica dura cerca de 1/20 de segundo, embora sua
duração possa ser várias vezes maior em certos tipos de fibras
musculares esqueléticas e também várias vezes menor em outros
(no músculo cardíaco, esse pulso de cálcio dura até 1/3 de
segundo, devido à longa duração do potencial de ação
cardíaco). É durante esse pulso de cálcio que ocorre a
contração. Caso a contração deva continuar por período mais
prolongado, deve ser desencadeada uma série desses pulsos, por
seqüência continuada de potenciais de ação repetidos, como
discutido no capítulo anterior.
REFERÊNCIAS
Ver referências dos Caps. 5 e 6.
77
CAPÍTULO 8
Contração e Excitação do Músculo Liso
CONTRAÇÃO DO MÚSCULO LISO
Nos dois capítulos anteriores, a discussão versou sobre o
músculo esquelético. Vamos, agora, passar para o músculo liso,
formado por fibras bem menores — em geral, com diâmetro
de 2 a 5 µm e comprimento de apenas 20 a 500 µm — em
relação ao músculo esquelético, com fibras de diâmetro 20 vezes
maior e comprimento milhares de vezes maior. Não obstante,
muitos dos princípios básicos da contração se aplicam tanto ao
músculo liso como ao músculo esquelético. O que é mais
importante, essencialmente as mesmas forças atrativas entre os
filamentos de miosina e de actina geram a contração no músculo
liso, como fazem no músculo esquelético, mas a organização
física interna das fibras do músculo liso é inteiramente diferente,
como será mostrado adiante.
TIPOS DE MÚSCULO LISO
O músculo liso encontrado em um órgão é diferente do
presente nos demais por vários aspectos: dimensões físicas,
organização em feixes ou camadas, resposta a diversos tipos de
estímulos, características de inervação e de função. Todavia, para
simplificar, o músculo liso é geralmente dividido em dois tipos
principais, mostrados na Fig. 8.1: o músculo liso multiunitário e
o músculo liso de uma só unidade.
Músculo liso multiunitário. Esse tipo de músculo liso é
formado por fibras independentes de músculo liso. Cada fibra
atua de modo completamente independente das demais e, muitas
vezes, é inervada por terminação nervosa única, como acontece
com as fibras musculares esqueléticas. Ademais, as superfícies
externas dessas fibras, como as das fibras musculares esqueléticas
é revestida por fina camada de uma substância "semelhante à
membrana basal", uma mistura de fibrilas de colágeno e de
proteoglicanos que participa do isolamento da fibra de suas
vizinhas.
A característica mais importante das fibras do músculo liso
multiunitário é que cada fibra pode contrair-se
independentemente das outras, e elas são controladas, em
grande parte, por sinais neurais. Isso contrasta com o controle
predominante nos músculos lisos viscerais, por estímulos nãoneurais. Característica adicional é a de que só muito raramente
esses músculos apresentam contrações espontâneas.
Alguns exemplos de músculo liso multiunitário são as fibras
musculares lisas do músculo ciliar do olho, a íris do olho, a
membrana nictitante que recobre o olho em alguns animais
inferiores e os músculos piloeretores que produzem a ereção
dos pêlos, quando estimulados pelo sistema nervoso simpático.
Músculo liso de uma só unidade. A expressão "de uma só
unidade" gera confusão por não significar fibras musculares
únicas. Ao contrário, ela define grande massa de centenas a
milhões de fibras musculares que se contraem juntas, como uma
só unidade. Essas fibras ocorrem em geral em feixes ou camadas
e suas membranas celulares são aderentes entre si, em diversos
pontos, de modo que a força gerada por uma fibra muscular pode
ser transmitida à seguinte. Além disso, as membranas celulares
são unidas por muitas junções abertas, o que permite o fluxo
de íons de uma célula a outra, de modo que o potencial de
ação se propaga de uma fibra para a seguinte, fazendo com que
todas as fibras musculares se contraiam a um só tempo. Esse
tipo de músculo liso também é chamado de músculo liso
sincicial, devido as interconexões entre suas fibras. Dado que
esse tipo de músculo é encontrado na parede da maioria das
vísceras do corpo - inclusive no intestino, vias biliares, ureteres,
útero e muitos vasos sanguíneos —, ele também é, muitas vezes,
referido como músculo liso visceral.
O PROCESSO CONTRÁTIL NO MÚSCULO LISO
A base química para a contração do músculo liso
O músculo liso contém tanto filamentos de actina como de
miosina, ambos com características químicas semelhantes às dos
filamentos de actina e de miosina do músculo esquelético.
Todavia, ele não contém troponina, de modo que o mecanismo
para o controle da contração é inteiramente diferente. Isso será
discutido em detalhe em seção subseqüente deste capítulo.
Estudos químicos mostraram que a actina e a miosina
extraídas do músculo liso interagem entre si de modo quase
idêntico ao da actina e miosina extraídas do músculo
esquelético. Ainda mais, o processo contrátil é ativado pelos íons
cálcio e o trifosfato de adenosina (ATP) é degradado a difosfato
de adenosina (ADP) para o fornecimento de energia para a
contração.
Por outro lado, existem diferenças importantes entre a
organização física do músculo liso e a do músculo esquelético,
bem como diferenças no acoplamento excitação-contração, no
controle do processo da contração pelos íons cálcio, na
duração da contração e na quantidade de energia necessária
para o processo contrátil.
A base física da contração do músculo liso
O músculo liso não apresenta a disposição estriada dos
filamentos de actina e de miosina encontrada nos músculos
esqueléticos. Durante muito tempo foi impossível identificar,
mesmo em microfotografias eletrônicas, qualquer organização
específica na célula muscular lisa que pudesse explicar sua
contração.
78
Fig. 8.1 O músculo liso multiunitário e o de uma só unidade.
Contudo, usando-se métodos especiais de preparação para
microscopia eletrônica, foi possível a sugestão da organização
física mostrada na Fig. 8.2. Ela mostra grande número de
filamentos de actina presos aos chamados corpos densos. Alguns
desses corpos densos estão fixados à membrana celular. Outros
ocorrem dispersos no interior da célula e são mantidos em
seus lugares por malha de proteínas estruturais que os
interconecta. Deve ser notado na Fig. 8.2 que alguns dos
corpos densos fixados à membrana de células adjacentes estão
interligados por pontes de proteínas intracelulares. É
principalmente por meio dessas interligações que a força da
contração é transmitida de uma célula para a seguinte.
Espalhados entre os numerosos filamentos de actina existem
alguns filamentos de miosina. Esses filamentos de miosina têm
diâmetro mais de duas vezes maior que o dos filamentos de
actina. Quando vistos em corte transverso, em microfotografias
eletrônicas, podem ser contados até 15 vezes mais filamentos
de actina que de miosina. Parte dessa diferença é causada pelo
fato de que a proporção entre os comprimentos dos filamentos
de actina e de miosina, no músculo liso, é bem maior que a
encontrada no músculo esquelético. Por conseguinte, a
probabilidade de se ver um excesso de filamentos de actina é bem
maior. Não obstante, fica-se impressionado com a raridade dos
filamentos de miosina em relação aos de actina.
A direita da Fig. 8.2 é mostrada a estrutura sugerida das
unidades contrateis isoladas das células musculares lisas, com
grande número de filamentos de actina se irradiando de dois
corpos densos; esses filamentos se sobrepõem a filamento único
de miosina, situado a meia distância entre os dois corpos densos.
É óbvio que essa unidade contrátil é semelhante à unidade
contrátil do músculo esquelético, sem, contudo, apresentar a
regularidade estrutural característica deste último; na verdade,
os corpos densos do músculo liso desempenham o mesmo papel
dos discos z do músculo esquelético
Comparação entre as contrações do músculo liso e do
músculo esquelético
Embora o músculo esquelético se contraia com muita
rapidez, a maioria das contrações do músculo liso resulta em
contrações tônicas prolongadas, algumas vezes perdurando por
horas e até por dias. Por conseguinte, pode ser previsto que as
características tanto físicas como químicas das contrações dos
músculos.
Fig. 8.2 A estrutura física do músculo liso. A fibra na parte superior
esquerda da figura mostra filamentos de actina irradiando de "corpos
densos". O detalhe à direita da fibra inferior apresenta as inter-relações
entre os filamentos de miosina e os de actina.
lisos difiram das contrações dos músculos esqueléticos. Algumas
dessas diferenças são as seguintes:
Ciclos lentos das pontes cruzadas. A duração dos ciclos das
pontes cruzadas no músculo liso — isto é, sua fixação a actina,
em seguida seu desligamento dessa actina, e nova fixação para
outro ciclo — é muito, mas muito maior no músculo liso que
no músculo esquelético; na verdade, a freqüência desses ciclos
no músculo liso é, no máximo, de 1/100 a 1/300 da do músculo
esquelético. Contudo, admite-se que a fração de tempo em que
as pontes cruzadas permanecem presas aos filamentos de actina,
que é o principal fator determinante da força de contração, é
muito maior no músculo liso. Uma razão possível para esses
lentos ciclos é a de que as cabeças de miosina conteriam menos
atividade de ATPase que no músculo esquelético, de modo que
a degradação do ATP, energizadora dos movimentos das cabeças,
ficaria muito reduzida, com diminuição correspondente da
freqüência dos ciclos.
Energia necessária para manter a contração do músculo liso.
Apenas de 1/10 a 1/300 da energia consumida na manutenção
79
de uma mesma tensão de contração é necessária no músculo
liso, em relação ao músculo esquelético. Também isso é
considerado como resultando da longa fixação de cada ciclo e de
apenas uma molécula de ATP ser necessária para cada ciclo,
independente de sua duração.
Essa economia na utilização de energia pelo músculo liso
é extremamente importante para a economia global de energia
do corpo, dado que órgãos como os intestinos, a bexiga urinária
e outras vísceras devem manter sua contração muscular tônica
durante todo o dia.
Lentidão do desenvolvimento da contração e do relaxamento
do músculo liso. Um tecido muscular liso típico começa a se
contrair dentro de 50 a 100 ms após ter ficado excitado, atinge
sua contração máxima após 1/2 s e começa a apresentar declínio
de sua força de contração decorridos outros 1 a 2 s, o que dá
um tempo total de contração de 1 a 3 s. Isso corresponde à
duração cerca de 30 vezes maior que a medida para um músculo
esquelético médio. Contudo, devido à multiplicidade de tipos
de músculos lisos, a contração de alguns pode durar apenas 0,2
s, enquanto a de outros pode ser de até 30 s.
O lento início da contração do músculo liso c sua prolongada
contração são, provavelmente, causados pela lentidão da fixação
e do desligamento das pontes cruzadas. Além disso, o início
da contração, em resposta aos íons cálcio, chamado de mecanismo
de acoplamento excitação-contração, é muito mais lento que no
músculo esquelético, como será discutido adiante.
Força da contração muscular. Apesar do número
relativamente pequeno de filamentos de miosina no músculo liso
e da duração prolongada dos ciclos das pontes cruzadas, a força
máxima de contração que pode ser desenvolvida pelo músculo
liso é, muitas vezes, bem maior que a do músculo esquelético chegando até a 4 a 6 kg/cm2 da área da seção transversa do
músculo liso, em comparação a 3 a 4 kg para o músculo
esquelético. Postula-se que essa grande força atrativa seja
resultante do prolongado período de fixação das pontes cruzadas
da miosina aos filamentos de actina.
Encurtamento percentual do músculo liso durante a
contração. Característica do músculo liso, que muito o distingue
do músculo esquelético, é a capacidade de se encurtar por
porcentagem muito maior de seu comprimento que o músculo
esquelético, enquanto mantém quase que a força total da
contração. O músculo esquelético tem comprimento utilizável
para encurtamento de cerca de um terço de seu comprimento
estirado, enquanto o músculo liso pode, muitas vezes, encurtarse efetiva-mente por mais de dois terços de seu comprimento
estirado. Isso permite ao músculo Uso o desempenho de funções
especialmente importantes nas vísceras ocas, permitindo que o
intestino, a bexiga, os vasos sanguíneos e outras estruturas
internas do corpo variem seus diâmetros luminais desde valores
muito grandes até quase zero.
Por que essa diferença entre o músculo liso e o músculo
esquelético? A resposta a esta pergunta não é conhecida em
sua totalidade, mas parecem existir duas razões prováveis.
Primeira, é possível que algumas unidades contrateis do músculo
liso apresentem superposição ótima entre os filamentos de
actina e de miosina para determinados comprimentos do
músculo, enquanto outras unidades a teriam em comprimentos
diferentes desse músculo, não existindo sincronia entre todas as
unidades contrateis, como acontece normalmente no músculo
esquelético. Como resultado, pode ser atingido alto grau de
encurtamento. Segunda, os filamentos de actina são muito mais
longos no músculo liso que no esquelético. Como resultado,
esses filamentos podem ser puxados por sobre os filamentos de
actina por distância muito maior, no caso do músculo Uso em
contração, do que pode ser durante a contração do músculo
esquelético.
O mecanismo de “tranca” para a manutenção de contrações
muito prolongadas do músculo liso. Uma vez que se tenha
desenvolvido uma contração total no músculo liso, o grau de
ativação desse músculo pode ser, em geral, reduzido a nível
bem abaixo do inicial sem que, todavia, o músculo perca sua
força total de contração. Ainda mais, a energia consumida
para manter essa contração é, freqüentemente, minúscula algumas vezes, de apenas 1/300 da energia necessária para
manter uma contração contínua comparável no músculo
esquelético. Isso é chamado de mecanismo de “tranca”. Esse
mesmo efeito ocorre em grau mínimo no músculo esquelético,
muitíssimo menor que no músculo liso.
A importância do mecanismo de tranca é a de que ele permite
a manutenção de contração tônica prolongada, no músculo liso,
por horas e horas, com consumo mínimo de energia. Por outro
lado, quase nenhum sinal excitário, de fontes neurais ou
hormonais, é necessário.
A causa do fenômeno de tranca é, fora de qualquer dúvida,
relacionada ao prolongado período de fixação das pontes cruzadas
de miosina aos filamentos de actina. Entretanto, desconhece-se
por que esse mecanismo é mais evidente em certos tipos de
músculo liso que em outros, bem como por que sua intensidade
pode variar.
Relaxamento por estresse do másculo liso. Outra
característica muito importante do músculo liso, em especial do
tipo visceral de músculo liso encontrado em muitos órgãos
ocos, é sua capacidade de retornar quase que a sua força original
de contração segundos ou minutos após ter sido alongado ou
encurtado. Por exemplo, aumento súbito do volume de líquido
contido na bexiga urinária provoca aumento substancial e
imediato da pressão vesical. Contudo, durante os 15 s seguintes,
apesar do contínuo estiramento da parede da bexiga, a pressão
retorna quase que a seu valor inicial. Se, em seguida, o volume
for novamente aumentado, o mesmo efeito torna a ocorrer.
Quando o volume é abruptamente diminuído, a pressão cai,
de início, a valores muito baixos, retornando após alguns
segundos a seu valor original. Esse fenômeno é chamado de
relaxamento por estresse. Sua importância óbvia é a de permitir
que órgãos ocos mantenham aproximadamente a mesma pressão
no interior de seus lumens, independentemente do comprimento
de suas fibras musculares.
O fenômeno do relaxamento por estresse está,
provavelmente, relacionado ao fenômeno da tranca. Quando o
músculo é inicialmente estirado, o fenômeno da tranca resiste à
alteração do comprimento. Todavia, com os ciclos sucessivos das
cabeças de miosina, durante os segundos a minutos
subseqüentes, as cabeças se desligam é voltam a se prender em
pontos mais afastados dos filamentos de actina. Por conseguinte,
em função do tempo, o comprimento do músculo se modifica,
embora a tensão do músculo retorne até quase seu valor inicial,
visto que o número das pontes cruzadas de miosina, causadoras
da força contrátil, permanece muito próximo do que havia
antes.
REGULAÇÃO DA CONTRAÇÃO PELOS ÍONS CÁLCIO
Como é válido para o músculo esquelético, o fator
desencadeante na maioria das contrações do músculo liso é um
aumento da concentração intracelular de íons cálcio. Esse
aumento pode ser causado por estimulação da fibra nervosa para a
fibra muscular lisa, por estimulação hormonal, por estiramento
da fibra e, até mesmo, por alteração do ambiente químico da
fibra.
Todavia, o músculo liso não contém troponina, a proteína
reguladora que é ativada pelos íons cálcio para promover a
contração do músculo esquelético. Pelo contrário, a contração do
músculo liso é ativada por mecanismo inteiramente diferente,
que é o seguinte:
Combinação dos íons cálcio com a "calmodulina" — ativação
80
da miosina quinase e fosforilação da cabeça da miosina. Em
lugar da troponina, as células do músculo liso contêm grande
quantidade de outra proteína reguladora, denominada
calmodulina. Embora essa proteína seja semelhante à
troponina, por reagir com quatro íons cálcio, ela difere no
modo como desencadeia a contração. A calmodulina faz isso
por ativar as pontes cruzadas de miosina. Essa ativação e a
contração subseqüente ocorrem nas seguintes etapas:
1. Os íons cálcio se fixam a calmodulina.
2. A combinação calmodulina-cálcio se fixa, então, e ativa
a miosina quinase, uma enzima fosforilativa.
3. Uma das cadeias leves de cada cabeça de miosina, chama
da de cadeia regulatória, fica fosforilada, em resposta à miosina
quinase. Quando essa cadeia não está fosforilada, não ocorre
o ciclo de fixação-desligamento da cabeça. Mas quando a cadeia
regulatória está fosforilada, a cabeça adquire a capacidade de
se fixar ao filamento de actina e seguir por todo o processo
do ciclo, o que resulta em contração muscular.
Término da contração — o papel da "miosina fosfatase".
Quando a concentração de cálcio cai abaixo de seu nível crítico,
todas as etapas descritas acima se invertem de modo automático,
exceto pela fosforilação da cabeça de miosina. Essa inversão
exige ação de outra enzima, a miosina fosfatase, que remove
o fosfato da cadeia leve regulatória. Então, o ciclo é interrompido
e cessa a contração. O tempo necessário para o relaxamento
da contração muscular é, portanto, determinado, em grande parte,
pela quantidade de miosina fosfatase ativa na célula.
abordaremos, primeiro, o controle neural da contração do
músculo liso, seguido pelo controle hormonal e pelos outros
meios de controle.
AS
JUNÇÕES
MÚSCULO LISO
NEUROMUSCULARES
DO
Anatomia fisiológica das junções neuromusculares do
músculo liso. As junções neuromusculares do tipo encontrado
nas fibras do músculo esquelético não são encontradas no
músculo liso. Em seu lugar, fibras nervosas autonômicas, que
inervam o músculo liso, em geral se ramificam difusamente
por sobre uma lâmina de fibras musculares, como mostrado na
Fig. 8.3. Na maioria dos casos, essas fibras não fazem contato
direto com as fibras musculares lisas, mas formam junções difusas
que secretam seus transmissores no líquido intersticial, a alguns
nanômetros e até a alguns micrômetros de distância das células
musculares; então a substância transmissora se difunde para as
células. Ademais, onde existem muitas camadas de células
musculares, as fibras nervosas, muitas vezes, só inervam a
camada mais externa, e a excitação muscular passa dessa
camada mais externa até as mais internas pela propagação do
potencial de ação pela massa muscular ou por difusão
subseqüente da substância transmissoras.
Os axônios que inervam as fibras musculares lisas também
não têm os botões terminais do tipo encontrado nas placas
motoras das fibras musculares esqueléticas. Em seu lugar, a
maioria dos finos terminais axônicos apresenta múltiplas
Um possível mecanismo para a regulação do
varicosidades ao longo de sua extensão. Nesses pontos, as
fenômeno de tranca
células de Schwann são interrompidas, de modo que a substância
transmissora pode ser secretada através da parede dessas
Devido à importância do fenômeno de tranca no músculo varicosidades. Nessas varicosidades existem vesículas,
liso e por ser esse fenômeno o determinante da manutenção semelhantes às da placa motora dos músculos esqueléticos,
a longo prazo do tônus de muitos órgãos contendo músculo liso, contendo a substância transmissora. Contudo, contrastando com
muitas tentativas já foram feitas para explicar o fenômeno da as vesículas das junções dos músculos esqueléticos, que só
tranca. Entre os muitos mecanismos propostos, um dos mais contêm acetileolina, as vesículas das terminações das fibras
simples é o que se segue.
nervosas autonômicas contêm acetileolina em algumas e, em
Quando a miosina quinase e a miosina fosfatase são muito outras, norepinefrina.
intensamente ativadas, a freqüência dos ciclos das cabeças de
Em alguns casos, e de forma particular no tipo multiunitário
miosina fica muito aumentada, e também aumenta a velocidade de músculo liso, as varicosidades jazem diretamente sobre a
da contração. Em seguida, à medida que a ativação das enzimas membrana da fibra muscular, com separação dessa membrana
declina, a freqüência dos ciclos diminui, mas, ao mesmo tempo, de apenas 20 a 30 nm — a mesma distância da fenda
a menor ativação também faz com que as cabeças de miosina sináptica das junções do músculo esquelético. Essas junções por
fiquem fixadas aos filamentos de actina por proporção contato atuam de modo idêntico ao das junções
crescentemente maior da duração do ciclo. Por conseguinte, o neuromusculares do músculo esquelético, e o período latente
número de cabeças fixadas ao filamento de actina, em da contração dessas fibras musculares lisas é
determinado instante, permanece muito grande. Visto que é o consideravelmente menor que o das fibras estimuladas pelas
número de cabeças fixadas à actina o determinante da força de junções difusas.
contração, a tensão é mantida, ou "trancada"; contudo, muito
pouca energia é usada, porque o ATP não é degradado a ADP,
exceto nas raras ocasiões em que uma cabeça é desligada.
CONTROLE NEURAL E HORMONAL
CONTRAÇÃO DO MÚSCULO LISO
DA
Embora o músculo esquelético seja ativado exclusivamente
pelo sistema nervoso, o músculo liso pode ser estimulado a se
contrair por múltiplos tipos de sinais: por sinais neurais, por
estimulação hormonal, e por vários outros meios. A razão
principal para essa diferença é que a membrana do músculo
liso contém muitos tipos distintos de proteínas receptoras,
capazes de desencadear o processo contrátil. Outras proteínas
receptoras são capazes de inibir a contração do músculo liso, o que
representa outra diferença do músculo esquelético. Portanto,
nesta seção.
Fig. 8.3 Inervação do músculo liso.
81
Substâncias transmissoras excitatórias e inibitórias na junção
neuromuscular do músculo liso. A acetilcolina e a norepinefrina
são duas substâncias distintas sabidamente secretadas pelos
nervos autonômicos que inervam o músculo liso. A acetilcolina
é uma substância transmissora excitatória para os músculos
lisos de determinados órgãos, embora também seja substância
inibitória para as fibras musculares lisas de outros órgãos.
Quando a acetilcolina excita uma fibra muscular, a norepinefrina
em geral a inibe. Inversamente, quando a acetilcolina inibe
uma fibra, a norepinefrina em geral a excita.
Por que essas respostas diferentes? A resposta é que tanto
a acetilcolina como a norepinefrina excitam ou inibem o músculo
liso por primeiro se fixarem a uma proteína receptora na superfície
da membrana da célula muscular. Por sua vez, esse receptor
controla a abertura ou fechamento de canais iônicos ou controla
qualquer outro meio para a ativação ou inibição da fibra muscular
lisa. Além disso, algumas dessas proteínas receptoras são
receptores excitatórios, enquanto outras são receptores
inibitórios. Assim, é o tipo de receptor que determina se o
músculo liso será excitado ou inibido e também determina qual
dos dois transmissores, a acetilcolina ou a norepinefrina, será
eficaz na produção da excitação ou da inibição. Esses receptores
são discutidos em mais detalhes no Cap. 60, em relação ao
funcionamento do sistema nervoso autonômico.
POTENCIAIS DE MEMBRANA E POTENCIAIS
DE AÇÃO NO MÚSCULO LISO
Potenciais de membrana do músculo liso. O valor, em termos
quantitativos, do potencial de membrana do músculo liso é
variável de um tipo de músculo liso para outro, além de também
depender das condições momentâneas do músculo. Contudo,
no estado normal de repouso, o potencial de membrana é, em
geral, de - 50 a - 60 mV, isto é, cerca de 30 mV menos negativo
que no músculo esquelético.
Potenciais de ação no músculo liso de uma só unidade. No
músculo liso de uma só unidade, os potenciais de ação ocorrem
pelo mesmo modo como no músculo esquelético. Contudo, na
maioria, se não em todos os tipos de músculo liso multiunitário,
não ocorrem, normalmente, potenciais de ação, como discutido
em seção subseqüente.
Os potenciais de ação do músculo liso visceral ocorrem sob
duas formas: (1) potenciais em ponta [spikes] e (2) potenciais
de ação com platôs.
Potenciais em ponta. Potenciais de ação em ponta típicos,
semelhantes aos registrados no músculo esquelético, ocorrem
na maioria dos tipos de músculo liso de uma só unidade. A
duração desse tipo de potencial de ação é de cerca de 10 a 50
ms, como mostrado na Fig. 8.4A e B. Tais potenciais de ação
podem ser induzidos por muitos modos, como, por exemplo,
por estimulação elétrica, pela ação de hormônios sobre o músculo
liso, pela ação de substâncias transmissoras das fibras nervosas
ou pela geração espontânea da própria fibra muscular, como
descrito adiante.
Potenciais de ação com platôs. A Fig. 8.4C apresenta um
potencial de ação com platô. O início desse potencial de ação
é semelhante ao de típico potencial de ação em ponta. Contudo,
em vez da rápida repolarização da membrana da fibra muscular,
essa repolarização é retardada por várias centenas a vários
milhares de milissegundos. A importância do platô é que ele
pode ser o responsável por contrações por períodos prolongados
que ocorrem em alguns tipos de músculo liso, como o do
ureter, do útero, sob certas condições, e de alguns tipos de
músculo liso vascular. (Também, esse tipo de potencial de ação
é encontrado nas fibras do músculo cardíaco que apresenta
período prolongado de contração, como será discutido nos dois
próximos capítulos.)
Fig. 8.4 A, Um típico potencial de ação (potencial em agulha) de um
músculo liso deflagrado por um estímulo externo. B, Uma série de
potenciais de ação em agulha produzidos por ondas elétricas rítmicas
lentas que ocorrem espontaneamente nos músculos lisos da parede
intestinal. C, Potencial de ação com platô em uma fibra muscular lisa
do útero.
A importância dos canais de cálcio na geração do potencial
de ação do músculo liso. A membrana da célula muscular lisa
contém número muito maior de canais de cálcio voltagem-dependentes do que a fibra muscular esquelética, mas número
muitíssimo menor de canais de sódio voltagem-dependentes.
Como resultado, o sódio tem participação mínima, se é que
a tem, na geração do potencial de ação na maioria dos
músculos lisos. Em seu lugar, o fluxo de cálcio para o interior
da fibra é o principal responsável pelo potencial de ação. Isso
ocorre do mesmo modo auto-regenerativo dos canais de sódio
das fibras nervosas e das fibras musculares esqueléticas.
Contudo, os canais de cálcio abrem com lentidão muito maior do
que o fazem os canais de sódio. Isso explica, em grande parte,
os lentos potenciais de ação das fibras musculares lisas.
Outra característica importante da entrada de cálcio para
o interior da célula, durante o potencial de ação, é que esse
mesmo cálcio atua diretamente sobre o mecanismo contrátil do
músculo liso, para desencadear a contração, como discutido
acima. Dessa forma, o cálcio desempenha duas funções a um só
tempo.
Potenciais ondulatórios lentos no músculo liso de uma só unidade
e a geração espontânea de potenciais de ação. Alguns músculos
lisos são auto-excitatórios. Isto é, surgem os potenciais de ação
no próprio músculo liso, sem que atue um estímulo extrínseco.
Em geral, isso está associado a um ritmo ondulatório lento básico
do potencial de membrana. Processo ondulatório lento típico
desse tipo no músculo liso visceral do intestino é mostrado na
Fig. 8.4B. A própria onda lenta não é um potencial de ação. Não
é um processo auto-regenerativo que se propaga
progressivamente ao longo das membranas das fibras
musculares. É, todavia, uma propriedade local das fibras do
músculo liso que compõem a massa muscular.
82
A causa desse ritmo ondulatório lento é desconhecida; uma
das propostas já formuladas é a de que as ondas lentas seriam
causadas por aumento e diminuição do bombeamento de sódio
através da membrana, para fora da célula; o potencial de
membrana ficaria mais negativo quando o sódio fosse bombeado
rapidamente e menos negativo quando a bomba de sódio
ficasse menos ativa. Outra proposta é a de que as
condutâncias dos canais iônicos aumentariam e diminuiriam
ritmicamente.
A importância das ondas lentas repousa no fato de que
podem desencadear potenciais de ação. Por si mesmas, as ondas
lentas são incapazes de provocar contrações musculares, mas
quando o potencial da onda lenta se eleva acima do nível de
aproximadamente - 35 mV (o limiar aproximado para a indução
de potenciais de ação na maioria dos músculos lisos viscerais),
aparece um potencial de ação que se propaga pela massa
muscular, quando, então, ocorre contração. A Fig. 8.4B mostra
esse efeito, apresentando um ou mais potenciais de ação no
pico de cada onda lenta. Esse efeito, obviamente, pode
promover uma serie de contrações rítmicas da massa de músculo
liso. Por conseguinte, as ondas lentas são, muitas vezes, referidas
como ondas marcapasso. No Cap. 62 será mostrado que esse tipo
de atividade controla as contrações rítmicas do intestino.
Excitação do músculo liso visceral pelo estiramento. Quando
o músculo liso visceral (de uma só unidade) é suficientemente
estirado, na maioria das vezes podem ser gerados potenciais de
ação espontâneos. Isso resulta de combinação dos potenciais das
ondas lentas normais com uma redução da negatividade do
potencial de membrana, causada pelo próprio estiramento. Essa
resposta ao estiramento permite que um órgão oco que seja
excessivamente distendido se contraia de modo automático e,
como resultado, possa resistir ao estiramento. Por exemplo,
quando o intestino é hiperdistendido por seu conteúdo, uma
contração local automática muitas vezes provoca onda
peristáltica que desloca o conteúdo da área de intestino
hiperdistendida.
Despolarização do músculo liso multiunitário sem potenciais
de ação. As fibras musculares lisas do músculo liso multiunitário
normalmente só se contraem, na maior parte das vezes, em
resposta a estímulos neurais. As terminações nervosas secretam
acetileolina para alguns tipos de músculos lisos multiunitários
e norepinefrina para outros. Nos dois casos, essas substâncias
transmissoras causam despolarização da membrana da célula
muscular lisa e essa resposta causa, por sua vez, a contração.
Contudo, com muita freqüência, não são gerados potenciais de
ação. A razão disso é que as fibras são por demais pequenas
para gerarem um potencial de ação. (Quando potenciais de ação
são induzidos no músculo liso visceral de uma só unidade, cerca
de 30 a 40 fibras musculares lisas devem despolarizar-se ao mesmo
tempo antes que seja gerado um potencial de ação propagado.)
Todavia, mesmo sem um potencial de ação nas fibras musculares
lisas multiunitárias, a despolarização local, chamada de
"potencial de junção", causada pela substância neural
transmissora, propaga-se "eletrotonicamente" por toda a fibra, o
que é necessário para desencadear a contração muscular.
CONTRAÇÃO DO MÚSCULO LISO SEM POTENCIAIS
DE AÇÃO - O EFEITO DOS FATORES TECIDUAIS
LOCAIS E DOS HORMONIOS
Provavelmente, 50% ou mais de todas as contrações
musculares lisas são induzidos, não por potenciais de ação,
mas por fatores estimulatórios atuando diretamente sobre o
mecanismo contrátil do músculo liso.
Os dois tipos de fatores não-neurais e não-dependentes de
potenciais de ação estimulatórios ativos mais freqüentemente
envolvidos são (1) fatores teciduais locais c (2) diversos
hormônios.
Contração do músculo liso em resposta a fatores teciduais
locais. No Cap. 17 será discutido o controle da contração das
arteríolas, metarteríolas e esfíncteres pré-capilares. Nessas
estruturas, as menores quase não possuem inervação. Todavia,
seu músculo liso é extremamente contrátil, respondendo
rapidamente a variações das condições locais do líquido
intersticial que as banha. Desse modo, um potente sistema de
controle por feedback regula o fluxo sanguíneo para essa área
localizada de tecido. Alguns dos fatores controladores
específicos são os seguintes:
1. Falta de oxigênio nos tecidos locais provoca o relaxa
mento do músculo liso e produz, conseqüentemente,
vasodilatação.
2. Excesso de dióxido de carbono provoca vasodilatação.
3. Aumento da concentração do íon hidrogênio também
provoca aumento da vasodilatação.
Outros fatores, como a adenosina, o ácido lático, aumento
dos íons potássio, redução da concentração de íons cálcio e
redução da temperatura corporal, também produzem
vasodilatação local.
Efeitos de hormônios sobre a contração do músculo liso. A
maioria dos hormônios circulantes influencia a contração do
músculo liso pelo menos em algum grau, e alguns exercem
efeitos muito potentes. Entre os hormônios circulantes mais
importantes com influência sobre a contração, então
norepinefrina, epinefrina, acetileolina, angioíensina, vasopressina,
ocitocina, serotonina e histamina,
Um hormônio provoca contração do músculo liso quando
a membrana de suas células contém receptores excitatórios
hormônio-dependentes para esse hormônio. Contudo, caso o
hormônio produza inibição, em lugar da contração, os receptores
da membrana da célula muscular lisa serão receptores
inibitórios, e não excitatórios.
Excitação ou inibição do músculo liso causada por hormônios
ou por fatores teciduais locais. Alguns receptores para hormônios,
na membrana da célula muscular lisa, abrem canais de sódio
ou de cálcio, despolarizando a membrana pelo mesmo mecanismo
da estimulação neural. Ocasionalmente, mas não sempre, podem
ser produzidos potenciais de ação, ou os potenciais rítmicos
preexistentes podem ser acentuados. Contudo, em muitos casos
ocorre despolarização sem potenciais de ação; não obstante, até
mesmo essa despolarização está associada a influxo de íons
cálcio para o desencadeamento da contração.
A ativação de outros receptores da membrana inibe a
contração. Isso é efetivado pelo fechamento dos canais de
sódio ou de cálcio, o que impede a entrada desses íons
positivos, ou pela abertura de canais de potássio, o que permite a
saída dos íons positivos de potássio para o exterior; nos dois
casos, ocorre aumento da negatividade no interior da célula
muscular, um estado chamado de hiperpolarização.
Algumas vezes, a contração ou a inibição é desencadeada
por hormônios sem que ocorra qualquer alteração do potencial
de membrana. Nesses casos, o hormônio em geral ativa um
receptor da membrana que não abre qualquer canal iônico, mas,
ao invés, promove uma alteração interna na fibra muscular, tal
como a liberação de íons cálcio retículo sarcoplasmático, o que
induz a contração. Ou, para inibir a contração, são conhecidos
outros mecanismos receptores que ativam a enzima adenil
ciclase ou a guanil ciclase na membrana celular; parte dessa
enzima proemina para o interior da célula e causa formação de
AMP cíclico ou de GMP cíclico, que são chamados de segundos
mensageiros. Por sua vez, o AMP cíclico ou GMP cíclico
exercem muitos efeitos, um dos quais é o de alterar o grau de
fosforilação de diversas enzimas que, indiretamente, inibem a
contração.
83
De modo especial, são afetadas a bomba que transporta os
íons cálcio do sarcoplasma para o retículo sarcoplasmático e a
bomba da membrana celular que transporta os íons cálcio
para fora da célula; esses efeitos reduzem a concentração
intracelular de íons cálcio, inibindo, assim, a contração.
Infelizmente, não se sabe como a maior parte dos fatores
teciduais locais que não são hormônios, como, por exemplo,
a falta de oxigênio, o excesso de dióxido de carbono, ou variações
da concentração de íons hidrogênio, excitam ou inibem a
contração do músculo liso. Contudo, os mecanismos possíveis
incluem variações do potencial de membrana da célula, variações
da permeabilidade da membrana celular, alterações do
mecanismo contrátil intracelular ou alguma combinação desses
mecanismos.
FONTE DOS ÍONS CÁLCIO QUE CAUSAM A
CONTRAÇÃO, TANTO ATRAVÉS DA MEMBRANA
CELULAR QUANTO POR LIBERAÇÃO PELO
RETÍCULO SARCOPLASMÁTICO
Embora o processo contrátil do músculo liso, como no
músculo esquelético, seja ativado pelos íons cálcio, a fonte dos
íons cálcio difere, pelo menos parcialmente, no músculo liso; a
diferença é que o retículo sarcoplasmático, de onde é
virtualmente derivado todo o íon cálcio para a contração do
músculo esquelético, é, muitas vezes, apenas rudimentar na
maior parte do músculo liso. Assim, na maioria dos tipos de
músculo liso, quase todos os íons cálcio responsáveis pela
contração entram para o interior da célula muscular, vindos do
líquido extracelular, durante o potencial de ação. Existe
concentração relativamente alta de íons cálcio no líquido
extracelular, maior que 10-3M, em comparação com a menor
que 10-7 M no sarcoplasma da célula, e, como foi destacado
antes, o potencial de ação do músculo liso é causado, em sua
maior parte, pelo influxo de íons cálcio para o interior da célula
muscular. Visto que as fibras musculares lisas são
extremamente diminutas (em comparação com as dimensões das
fibras musculares esqueléticas), esses íons cálcio podem
difundir-se para todas as regiões do músculo liso e induzir o
processo contrátil. O tempo necessário para essa difusão é, em
geral, de 200 a 300 ms e é chamado de período latente, antes
que se inicie a contração; esse período latente é cerca de 50
vezes maior que o da contração do músculo esquelético.
Todavia, ainda pode entrar cálcio adicional para o interior
da fibra muscular lisa, por meio dos canais de cálcio ativados
por hormônio, e esse cálcio também induz a contração. Em geral,
a abertura desses canais não causa um potencial de ação e, por
vezes, quase nenhuma alteração do potencial de repouso da
membrana, visto que a bomba de sódio, na membrana celular,
transporta quantidade suficiente de íons sódio para manter um
potencial de membrana quase normal. Mesmo assim, a contração
continua enquanto esses canais de cálcio estiverem abertos, dado
que são os íons cálcio, e não a variação do potencial de
membrana, que causam a contração. Esse é um meio pelo qual
ocorre à contração do músculo liso, sem alteração significativa
do potencial de membrana da célula.
Papel do retículo sarcoplasmático.
musculares
lisas
contêm
retículo
Algumas
Fig. 8.5 Túbulos sarcoplasmáticos de fibra muscular lisa, mostrando suas
inter-relações com as invaginações da membrana celular, denominadas
cavéolos.
Em geral, quanto mais extenso for o retículo sarcoplasmático
na fibra do músculo liso, maior será a rapidez com que ela se
contrai, presumivelmente porque o influxo de cálcio, através
da membrana celular, é muito mais lento que a liberação interna
de íons cálcio pelo retículo sarcoplasmático.
Efeito da concentração extracelular de íons cálcio sobre a
contração do músculo liso. Embora a concentração de íons cálcio
no líquido extracelular tenha efeito quase nulo sobre a força
de contração do músculo esquelético, isso não é verdade para
a maioria dos músculos lisos. Quando a concentração de íons
cálcio no líquido extracelular cai até valor baixo, a contração
do músculo liso, em geral, quase cessa. Na verdade, após vários
minutos de imersão em meio com baixo cálcio, até mesmo o
retículo sarcoplasmático das fibras musculares lisas perde seu
conteúdo de cálcio. Por conseguinte, a força de contração do
músculo liso é muito dependente da concentração de íons cálcio
no líquido extracelular. Será mostrado no capítulo seguinte que
isso também é válido para o músculo cardíaco.
células
sarcoplasmático
moderadamente desenvolvido. A Fig. 8.5 mostra um exemplo
apresentando diversos túbulos sarcoplasmáticos situados próximo
à membrana celular. Pequenas invaginações da membrana,
chamadas de cavéolos, entram em contato com as superfícies
desses túbulos. Admite-se que os cavéolos representem análogo
rudimentar do sistema de túbulos T do músculo esquelético.
Quando um potencial atinge as invaginações dos cavéolos, isso
parece excitar a liberação de íons cálcio pelos túbulos
sarcoplasmáticos, do mesmo modo como os potenciais de ação,
nos túbulos T do músculo esquelético, também produzem
liberação de íons cálcio.
A bomba de cálcio. Para que ocorra o relaxamento dos
elementos contrateis do músculo liso, é necessário que sejam
removidos os íons cálcio. Essa remoção é realizada por bombas
de cálcio que transportam os íons cálcio para fora da fibra
muscular lisa, lançando-os de volta para o líquido extracelular ou
transportando-os para o interior do retículo sarcoplasmático.
Contudo, essas bombas têm funcionamento muito lento, em
comparação com a bomba de ação rápida do retículo
sarcoplasmático do músculo esquelético. Por conseguinte, a
duração da contração do músculo liso é, muitas vezes, da
ordem de segundos, e não de centésimos a décimos de segundo,
como ocorre no músculo esquelético.
84
REFERENCIAS
Bennett, M. R.: Development of neuromuscular synapses. Physiol. Rev,,
63:915, 1983. Blaustein, M. P., and Hamlyn, J. M.: Sodium tranaport
inhibition, cell calcium, and hypertension. The natriuretic hormone/Na+Ca exchange/ hypertension hypothesís. Am. J. Med., 77:45, 1984. Bohr, D.
F., and Webb, R. C: Vascular amooth muscle function and its
changes in hypertension. Am. J. Med., 77:3, 1984. Bolton, T. R.:
Mechanisma of action of transmitters and other substances on smooth
muscle. Physiol- Rev., 59:606, 1979. Borgstrom, P., et ai.: An evaluation of
the metabolic interaction with myogenic vascular reactivity during blood
flow autoregulation. Acta Physiol.
Scand., 122:275, 1984. Bulbring, E. (ed.): Smooth Muscle. An Assessment
of Current Knowledge. Austin, University of Texas Press, 1981. Butler, T.
M., and Siegman, M. J.: High-energy phosphate metabolism in
vascular smcoth muscle. Annu. Rev. Phyaiol., 47:629, 1985. Campbell, J.
H., and Campbell, G. R.: Endothelial cell influences on vascular
smooth muscle phenotype. Annu. Rev. Physiol., 48:295, 1986. Dowben,
R. M. (ed.): Cell and Muscle Motility. New York, Plenum Publishing Corp.,
1983. Eisenberg, E., and Greene, L. E.: The relation of muscle
biochemistry to muscle physiology. Annu. Rev. Physiol., 42:293, 1980.
Fleming, W. W.: The electromagnetic Na+, K+ -pump in smooth muscle:
Physiologic and pharmacologic significance. Annu. Rev. Pharmacol. Toxicol., 20:129, 1980. Furchgott, R. F.: The roleof endothelium in the responses
of vascular smooth muscle to drugs. Annu. Rev. Pharmacol. Toxicol.,
24:175, 1984. Gabella, G-: Structural apparatus for force transmiasion in
smooth muscle.
Physiol. Rev., 64:455, 1984. Hai, C.-M., and Murphy, K- A.: Ca**,
crossbridge phosphorylation, and contraction. Annu. Rev. Phyaiol., 51:285,
1989. Hartshorne, D. J., and Gorecka, A.: Biochemistry of the contractile
proteins of amooth muscle. In Bohr, D. F., et ai. (eds.): Handbook of
Physiology. Sec. 2, Vol. II. Baltimore, Williams & Wilkins, 1980, p. 83.
Hartshorne, D. J., and Siemankowski, R. R.: Regulation of smooth muscle
actomyosin. Annu. Rev. Phyaiol., 4ò:òli>, 1981. Hertzberg, E. L., et ai.: Gap
junctional communication. Annu. Rev. Phyaiol., 43:479, 1981. Hesa, G. P.,
et ai.: Acetylcholine receptor-controlled ion translocation: Chemical
kinetic investigations of the mechanísm. Annu. Rev. Biophys.
Bioeng., 12:443, 1983. Hirat, G. D. S., and Edwarda, F. R.: Sympathetic
neuroeffector transmission in arteries and arterioles. Physiol. Rev.,
69:546,1989. Homsher, E.: Muscle enthalpy production and its relatinnahip
to actomyosin ATPaae. Annu. Rev. Phyaiol., 49:673, 1987.
Johansson, B., and Somlyo, A. P.: Electrophyaiology and escitation-contrac tion
cmipltng. In Bohr, D. F., et ai. (eds.): Handbook of Physiology. Sec. 2,
Vol. II. Baltimore, Williams & Wilkins, 1980, p. 301. Kamm. K. E., and Stull, J.
T.: Regulation of smooth muscle contractile elements by second messengers.
Annu. Rev. Physiol., 51:299, 1989. Kito, S., et ai. (eds.): Neuroreceptors and
Signal Tranaduction New York,
Plenum Publishing Corp., 1988. Lambert, J. J., et ai.: Drug-induced modification
of ionic conductance at the neuromuscular junction. Annu. Rev. Pharmacol.
Toxicol., 23:505, 1983. I.owenstein, W. R.: Junctional intercellular
communication: The cell-to-cell merabrane channel. Physiol. Rev., 61:829, 1981.
McKinney, M., and Richelson, E.: The coupling of neuronal muscahnic
receptor to responses. Annu. Rev. Pharmacol. Toxicol., 24:121, 1984. Morgan, D.
L., and Proske, U.: Vertebrate slow muscle: Ita structure, pattórn of innervation,
and mechanical properties. Physiol. Rev., 64:103, 1984. Murphy, R. A.; Muscle
cells of hollow organa. News Physiol. Sei., 3:124,1988. Paul, R. J.: Smooth muscle
energeties. Annu. Rev. Physiol., 51:331. 1989. Paul, R. J.: Chemical energética of
vascular smooth muscle. In Bohr, D. F., et ai. (eds.): Handbook of Physiology. Sec.
2, Vol. II. Baltimore, Williams & Wilkins, 1980, p. 201. Proaser, C. L.: Evolution
and diversity of nonstriated musclea. In Bohr, D. F.. et ai. (eds.): Handbook of
Physiology. Sec. 2, Vol. II. Baltimore, Williams &
Wilkins. 1980, p. 635. Purves, D., and Lichtman, J. W.: Specific connections
between nerve cells.
Annu. Rev. Physiol., 45:553, 1983. Putney, J. W., Jr., et ai.: How do inositol
phosphates regulate calcium signaling? FASEB J., 3:1899, 1989. RasmiiRsen, H.,
et ai.: Protein kinase C in the regulation of smooth muscle contraction. FASEB J.,
1:177, 1987. Rosenthal, W.,et ai.: Control of voltage dependent Ca^channels by
G protein-coupled receptors. FASEB J., 2:2784, 1988. Rowland, L. P., et ai.
(eds.): Molecular Genetics in Diseases of Brain, Nerve, and Muscle. New York,
Oxford University Press, 1989. Schneider, M. F.: Membrane charge movement
and depolarization-contraction coupling. Annu. Rev. Physiol., 43:507, 1981.
Seidel, C. L., and Schildmeyer, L. A.: Vascular smooth muscle adaptation to
increased load. Annu. Rev. Physiol., 49:489,1987. Somlyo, A. P.: Ultrastructure of
vascular smooth muscle. In Bohr, D. F., et ai. (eds.J: Handbook of Physiology. Sec.
2, Vol. II. Battimore, Williams &
Wilkins, 1980, p. 33. Spray. D. C, and Bennett, M. V. L.: Physiology and
pharmacology of gap junctions. Annu. Rev. Physiol., 47:281, 1985. van
Rreemen, C, and Saida, K.: Cellular mechanisms regulating (Ca1*), smooth
muscle. Annu. Rev. Physiol., 51:315, 1989. Vanhoutte, P. M-: Calcium-entry
blockers, vascular smooth muscle and sya temíc hypertension. Am. J. Cardiol.,
55:17B, 1985.
(Ver também Caps. 5 e 6.)
85
UNIDADE III
O CORAÇÃO
Ø
Ø
Ø
Ø
Ø
Ø
Ø
O Músculo Cardíaco; O Coração como Bomba
Excitação Rítmica do Coração
O Eletrocardiograma Normal
Interpretação Eletrocardiográfica das Anormalidades
Coronárias e do Músculo Cardíaco: Análise Vetorial
Arritmias Cardíacas e sua Interpretação
Eletrocardiográfica
86
CAPÍTULO 9
O Músculo Cardíaco; O Coração como Bomba
Neste capítulo, iniciamos a discussão do coração e do sistema
circulatório. O coração, ilustrado na Fig. 9.L, é constituído, na
realidade, por duas bombas distintas: o coração direito, que
bombeia o sangue pelos pulmões, e o coração esquerdo, que
bombeia o sangue pelos órgãos periféricos. Cada um desses
corações distintos, por sua vez, é uma bomba pulsátil de duas
câmaras composta de um átrio e um ventrículo. O átrio funciona
principalmente como reservatório de sangue e como via de
entrada para o ventrículo, mas, também, bombeia fracamente
para ajudar a levar o sangue até o ventrículo. O ventrículo, por
sua vez, é a principal fonte da força que impulsiona o sangue pela
circulação pulmonar ou pela periférica.
Mecanismos especiais no coração mantêm a ritimicidade car-
díaca e transmitem potenciais de ação para todo o músculo
cardíaco, de modo a produzir o batimento rítmico do coração.
Esse sistema de controle rítmico é explicado no Cap. 10. No
presente capítulo, explicamos como o coração opera como
bomba, começando pelas características especiais do próprio
coração.
FISIOLOGIA DO MÚSCULO CARDÍACO
O coração é constituído de três tipos principais de músculo
cardíaco: músculo atrial, músculo ventricular e fibras musculares
condutoras e excitatórias especializadas. Os tipos atrial e
ventricular de músculo contraem-se da mesma maneira que o
músculo esquelético, exceto que a duração da contração é
muito maior. Por outro lado, as fibras condutoras e excitatórias
especializadas contraem-se apenas fracamente, por conterem
poucas fibrilas contrateis; em vez disso, elas apresentam
ritmicidade e velocidades variáveis de condução, proporcionando
um sistema excitatório para o coração e um sistema de
transmissão para a condução controlada do sinal excitatório
cardíaco por todo o coração.
ANATOMIA FISIOLÓGICA DO MÚSCULO CARDÍACO
A Fig. 9.2 ilustra imagem histológica típica do músculo cardíaco,
mostrando as fibras musculares cardíacas dispostas em retículo,
recombinando-se a seguir e, depois, dispersando-se novamente. Nota-se
imediatamente, por essa figura, que o músculo cardíaco é estriado, da
mesma
Fig. 9.2 A natureza interligada, "sincicial", do músculo cardíaco.
Fig. 9.1 Estrutura do coração e trajeto do fluxo sanguíneo pelas câmaras
cardíacas.
87
forma que o músculo esquelético típico. Além disso, o músculo cardíaco
tem miofibrilas típicas, que contêm filamentos de actina e miasma quase
idênticos aos encontrados no músculo esquelético, e esses filamentos
interdigitam-se e deslizam uns sobre os outros durante o processo de
contração da mesma maneira como ocorre nos músculos esqueléticos
(ver Cap. 6).
Músculo cardíaco como um sincício. As áreas escuras anguladas que
cruzam as fibras musculares cardíacas na Fig. 9.2 são denominadas
discos intercalados; elas são, na realidade, membranas celulares que
separam células musculares cardíacas individuais umas das outras. Isto
é, as fibras musculares cardíacas são constituídas por muitas células
individuais ligadas em série umas às outras. Entretanto, a resistência
elétrica através dos discos intercalados é apenas 1/400 da resistência
através da membrana externa da fibra muscular cardíaca, porque as
membranas celulares fundem-se umas às outras e formam junções
“comunicantes” (junções abertas) muito permeáveis, que possibilitam
difusão relativamente livre dos íons. De um ponto de vista funcional,
portanto, os íons movem-se com facilidade ao longo dos eixos das fibras
musculares cardíacas, de modo que os potenciais de ação vão de uma
célula muscular cardíaca para outra, passando pelos discos intercalados
com apenas ligeira dificuldade. Por essa razão, o músculo cardíaco é um
sincício de muitas células musculares cardíacas, as quais se encontram
tão interligadas que, quando uma dessas células é excitada, o potencial de
ação dissemina-se para todas elas, passando de uma célula para outra e
por todas as interligações do retículo.
O coração é constituído por dois sincícios distintos: o sincício atrial,
que constitui as paredes dos dois átrios, e o sincício ventricular, que
as paredes dos dois ventrículos . Os átrios são separados dos ventrículos
por um tecido fibroso que circunda as aberturas valvulares entre os
átrios e os ventrículos. Normalmente, os potenciais de ação só podem
ser conduzidos do sincício atrial para o ventricular por meio do sistema
de condução especializado, o feixe A-V, que é discutido com detalhes
no capítulo seguinte. Essa divisão da massa muscular do coração em
dois sincícios funcionais distintos possibilita que os átrios se contraiam
um pouco antes da contração ventricular, o que é importante para a
eficácia do bombeamento cardíaco.
POTENCIAIS
CARDÍACO
DE
AÇÃO
NO
MÚSCULO
O potencial de membrana em repouso do músculo cardíaco
normal é de aproximadamente -85 a -95 milivolts (mV) e de
cerca de -90 a —100 mV nas fibras de condução especializadas,
as fibras de Purkinje, que são discutidas no capítulo seguinte.
O potencial de ação registrado no músculo ventricular,
mostrado pelo traçado inferior da Fig. 9.3, é de 105 mV, o
que quer dizer que o potencial de membrana se eleva de seu
valor normalmente muito negativo para um valor ligeiramente
positivo, de +20 mV. A parte positiva é designada como
potencial de ultrapassagem. Em seguida, após a ponta inicial, a
membrana permanece despolarizada por cerca de 0,2 s. no
músculo atrial, e 0,3 s, no músculo ventricular, formando o
platô, conforme ilustrado na Fig. 9.3, seguido por repolarização
abrupta ao final do platô. A presença desse platô no potencial
de ação faz a contração muscular durar 3 a 15 vezes mais no
músculo cardíaco que no músculo esquelético.
Neste ponto, devemos fazer as perguntas: Por que o
potencial de ação do músculo cardíaco é tão demorado, e por
que ele tem um platô, enquanto o do músculo esquelético não
tem? As respostas biofísicas básicas a estas questões foram
apresentadas no Cap. 5, mas merecem ser novamente
resumidas. Pelo menos duas diferenças importantes entre as
propriedades da membrana do músculo cardíaco e do músculo
esquelético são responsáveis pelo prolongado potencial de ação e
pelo platô do músculo cardíaco.
Em primeiro lugar, o potencial de ação do músculo
esquelético é causado quase que integralmente pela súbita
abertura de um grande número de canais rápidos de sódio, que
permitem a entrada, na fibra muscular esquelética, de um
número enorme de íons sódio. Esses canais são denominados
canais "rápidos"
Fig. 9.3 Potenciais de ação rítmicos de uma fibra de Purkinje e de uma
fibra muscular ventricular, registrados por meio de microeletródios.
porque permanecem abertos apenas por alguns décimos
milésimos de segundo, fechando-se, então, abruptamente. Ao
término deste fechamento, há o processo de repolarização, e o
potencial de ação termina dentro de outro décimo milésimo de
segundo, aproximadamente. No músculo cardíaco, por outro lado,
o potencial de ação é causado pela abertura de dois tipos de
canais: (1) os mesmos canais rápidos de sódio do músculo
esquelético e (2) outra população inteira dos chamados canais
lentos de cálcio, também denominados canais de cálcio-sódio.
Essa segunda população de canais difere dos canais rápidos de
sódio por abrir-se lentamente; porém, o que é mais importante,
eles permanecem abertos por alguns décimos de segundo.
Durante esse período, grande quantidade tanto de íons sódio
como de íons cálcio flui por esses canais para o interior da
fibra muscular cardíaca e isso mantém a despolarização por
período prolongado, ocasionando o platô do potencial de ação.
Além disso, os íons cálcio que penetram no músculo durante
esse potencial de ação têm um papel importante ajudando a
excitar o processo de contração muscular, o que é outra
diferença entre os músculos cardíaco e esquelético, como
discutiremos mais adiante neste capítulo.
A segunda diferença funcional importante entre o músculo
cardíaco e o músculo esquelético, que ajuda a explicar tanto
o prolongado potencial de ação como seu platô, é esta:
imediatamente após o início do potencial de ação, a
permeabilidade da membrana do músculo cardíaco ao potássio
diminui por cerca de cinco vezes, efeito que não ocorre no
músculo esquelético. É possível que essa menor permeabilidade
ao potássio seja causada, de alguma forma, pelo influxo excessivo
de cálcio pelos canais de cálcio, que acabamos de mencionar.
Entretanto, independentemente da causa, a menor
permeabilidade ao potássio diminui muito a saída de íons
potássio, durante o platô do potencial de ação, impedindo,
portanto, a recuperação precoce. Quando os canais lentos de
cálcio-sódio se fecham ao fim de 0,2 a 0,3 s, cessando o influxo
de íons cálcio e sódio, a permeabilidade da membrana ao
potássio aumenta bem rapidamente, e a rápida perda de
potássio pela fibra faz o potencial de membrana retornar a seu
nível de repouso, terminando, assim, o potencial de ação.
Velocidade de condução no músculo cardíaco. A velocidade
de condução do potencial de ação, tanto nas fibras musculares
atriais como nas ventriculares, é de cerca de 0,3 a 0,5 m/s, ou
cerca de 1/250 da velocidade em fibras nervosas muito grossas e
cerca de 1/10 da velocidade em fibras musculares esqueléticas. A
velocidade da condução no sistema de condução especializado
varia de 0,02 a 4 m/s em diferentes partes do sistema, como
88
é explicado no capítulo seguinte.
Período refratário do músculo cardíaco. O músculo
cardíaco, como todos os tecidos excitáveis, é refratário a
reestimulação durante o potencial de ação. Por esta razão, o
período refratário do coração é o intervalo de tempo, como é
mostrado à esquerda na Fig. 9.4, durante o qual um impulso
cardíaco normal não pode reexcitar uma área já excitada do
músculo cardíaco. O período refratário normal do ventrículo
é de 0,25 a 0,3 s, que é aproximadamente a duração do
potencial de ação. Há um período refratário relativo adicional
de cerca de 0,5 s, durante o qual o músculo é mais difícil de
excitar que o normal mas, ainda assim, pode ser excitado,
conforme ilustra a contração prematura inicial no segundo
exemplo da Fig. 9.4.
O período refratário do músculo atrial é muito mais curto
que o dos ventrículos (cerca de 0,15 s) e o período refratário
relativo tem mais de 0,03 s. Assim sendo, a freqüência rítmica
de contração dos átrios pode ser muito mais rápida que a dos
ventrículos.
CONTRAÇÃO DO MÚSCULO CARDÍACO
Acoplamento excitação-contração — função dos íons cálcio
e dos túbulos T. O termo "acoplamento excitação-contração"
indica o mecanismo pelo qual o potencial de ação faz
contraírem-se as miofibrilas musculares. Isso é discutido no
Cap. 7, em relação ao músculo esquelético. Entretanto,
novamente, há diferenças quanto a este mecanismo no músculo
cardíaco, que tem efeitos importantes sobre as características da
contração muscular cardíaca.
Como ocorre com os músculos esqueléticos, ao se propagar
pela membrana do músculo cardíaco, o potencial de ação também
se dissemina para o interior da fibra muscular cardíaca, pelas
membranas dos túbulos T. Os potenciais de ação dos túbulos
T, por sua vez, atuam sobre as membranas dos túbulos
sarcoplasmáticos longitudinais, causando a liberação instantânea
de quantidade muito grande de íons cálcio do retículo
sarcoplasmático para o sarcoplasma muscular. Em mais alguns
milésimos de segundo , esses íons cálcio difundem-se até as
miofibrilas e catalisam as reações químicas que promovem o
deslizamento dos filamentos de actina e miosina uns pelos
outros; isso produz, por sua vez, a contração muscular.
Até aqui, este mecanismo de acoplamento excitaçãocontração é o mesmo que para o músculo esquelético, mas há um
segundo efeito que é bem diferente. Além dos íons cálcio libera-
dos no sarcoplasma pelas cisternas do retículo sarcoplasmático,
grande quantidade de íons cálcio extra também se difunde dos
túbulos T para o sarcoplasma por ocasião do potencial de ação.
Na verdade, sem esse cálcio extra dos túbulos T, a força de
contração do músculo cardíaco seria consideravelmente reduzida,
porque o retículo sarcoplasmático do músculo cardíaco não é
tão desenvolvido quanto o dos músculos esqueléticos e não
armazena cálcio suficiente para proporcionar contração completa.
Por outro lado, os túbulos T do músculo cardíaco têm diâmetro
5 vezes maior que o dos túbulos dos músculos esqueléticos e
volume 25 vezes maior; da mesma forma, há no interior dos
túbulos T grande quantidade de mucopolissacarídeos
eletronegativamente carregados que fixam abundante reserva de
íons cálcio, mantendo-os sempre disponíveis para a difusão
para dentro da fibra muscular cardíaca ao ocorrer o potencial de
ação dos túbulos T.
A força de contração do músculo cardíaco depende, em
grande parte, da concentração de íons cálcio nos líquidos
extracelulares. A razão disto é que as extremidades dos túbulos T
abrem-se diretamente no exterior das fibras musculares cardíacas,
possibilitando ao mesmo líquido extracelular do interstício do
músculo cardíaco também fluir pelos túbulos T. Por
conseguinte, tanto a quantidade de íons cálcio no sistema de
túbulos T como a disponibilidade de íons cálcio para causar a
contração do músculo cardíaco dependem diretamente da
concentração de íons cálcio no líquido extracelular.
A título de contraste, a força de contração do músculo
esquelético dificilmente é afetada pela concentração extracelular
de íons cálcio, porque sua contração é causada quase que
inteiramente pelos íons cálcio liberados pelo retículo
sarcoplasmático no interior da própria fibra muscular
esquelética.
Ao final do platô do potencial de ação, o influxo de íons
cálcio para o interior das fibras musculares é interrompido
subitamente e os íons cálcio presentes no sarcoplasma são
rapidamente bombeados de volta tanto para o retículo
sarcoplasmático como para os túbulos T. Em conseqüência, a
contração cessa até que ocorra novo potencial de ação.
Duração da contração. O músculo cardíaco começa a se contrair
alguns milissegundos após o início de um potencial de ação, continuando
a contrair-se por alguns milissegundos após o término desse potencial.
Por esta razão, a duração de contração do músculo cardíaco é função
principalmente da duração do potencial de ação — cerca de 0,2 s no
músculo atrial e 0,3 s no músculo ventricular.
Efeito da freqüência cardíaca sobre a duração das
contrações. Quando a freqüência cardíaca aumenta, a duração
de cada fase do ciclo cardíaco, incluindo tanto a fase de
contração como a de relaxamento, obviamente diminui. A
duração do potencial de ação e do período de contração
(sístole) também diminui, mas não tanto quanto a fase de
relaxamento (diástole). Na freqüência cardíaca normal de 72
batimentos por minuto, o período de contração é cerca de 0,40 de
todo o ciclo. Numa freqüência cardíaca três vezes maior, esse
período compreende 0,65 de todo o ciclo, o que quer dizer que o
coração batendo a um ritmo muito rápido não fica relaxado por
tempo suficientemente longo para possibilitar o enchimento
completo das câmaras cardíacas antes da próxima contração.
O CICLO CARDÍACO
Fig. 9.4 Contração do coração, mostrando a duração dos períodos
refratário e refratário relativo, efeito de contração prematura precoce e
efeito de contração prematura mais tardia. Observe que as contrações
prematuras não causam a somação das contrações, como ocorre nos
músculos esqueléticos.
O período do início de um batimento cardíaco até o início
do batimento seguinte é denominado ciclo cardíaco. Cada ciclo
é iniciado pela geração espontânea de um potencial de ação
no nodo sinusal, ou sinoatrial, como é explicado no próximo
capítulo. Esse nodo está localizado na parede superior lateral
do átrio direito, próximo à abertura de veia cava superior, e
o potencial de ação passa rapidamente por ambos os átrios e,
daí, pelo feixe A-V até os ventrículos. Contudo, devido ao arranjo
especial do sistema de condução dos átrios para os ventrículos,
89
há um retardo de mais de 1/10 de segundo na passagem do impulso
cardíaco dos átrios para os ventrículos. Isso possibilita aos átrios
contraírem-se antes dos ventrículos, bombeando o sangue para
os ventrículos antes das muito potentes contrações ventriculares.
Os átrios atuam, portanto, como bombas de reforço para os
ventrículos e eles proporcionam, então, a principal fonte de força
para o movimento do sangue ao longo do sistema vascular.
SÍSTOLE E DIÁSTOLE
O ciclo cardíaco consiste em um período de relaxamento,
denominado diástole, durante o qual o coração se enche de
sangue, seguido por um período de contração, denominado
sístole.
A Fig. 9.5 ilustra os diferentes eventos durante o ciclo
cardíaco. As três curvas superiores mostram as alterações da
pressão na aorta, no ventrículo esquerdo e no átrio esquerdo,
respectivamente. A quarta curva mostra as alterações do volume
ventricular, a quinta, o eletrocardiograma, e a sexta um
fenocardiograma , que é um registro dos sons produzidos
pelo coração — principalmente pelas válvulas cardíacas
enquanto bombeia. É particularmente importante que o leitor
estude de forma detalhada o diagrama desta figura e fique
conhecendo as causas de todos os eventos ilustrados. Eles são
explicados adiante.
RELAÇÃO DO ELETROCARDIOGRAMA COM O
CICLO CARDÍACO
O eletrocardiograma da Fig. 9.5 mostra as ondas P, Q, R,
S e T, que são discutidas nos Caps. 11, 12 e 13. Elas são
voltagens elétricas geradas pelo coração e registradas pelo
eletrocardiógrafo a partir da superfície corporal. A onda P é
causada pela despolarização que se difunde pelos átrios, sendo
isso seguido pela contração atrial que ocasiona ligeira elevação na
curva da pressão atrial imediatamente após a onda P.
Aproximadamente 0,16 s após o início da onda P, aparecem
às ondas QRS, em conseqüência da despolarização dos
ventrículos, que iniciam a contração dos ventrículos e fazem a
pressão ventricular começar a subir, o que também é ilustrado
na figura
O complexo ORS começa, portanto, pouco antes do início da
sístole ventricular.
Finalmente, observa-se no eletrocardiograma a onda T
ventricular. Ela representa a fase de repolarização dos
ventrículos, ocasião em que as fibras musculares dos
ventrículos começam a se relaxar. A onda T ocorre, pois,
pouco antes do fim da contração ventricular.
Função dos átrios como bombas. Normalmente, o sangue
flui de modo contínuo das grandes veias para os átrios; cerca
de 75% do sangue fluem diretamente através dos átrios para
os ventrículos antes mesmo que os átrios se contraiam. Aí, então,
a contração atrial causa enchimento adicional dos ventrículos
da ordem de 25%. Assim sendo, os átrios funcionam
simplesmente como bombas de reforço que aumentam em
até 25% a eficácia do bombeamento ventricular. Entretanto, o
coração pode continuar a operar de modo bastante satisfatório,
em condições normais de repouso, mesmo sem esses 25% de
eficácia extra, por já ter normalmente a capacidade de
bombear 300 a 400% mais sangue do que o corpo necessita. Por
essa razão, quando os átrios funcionam de forma insuficiente, a
diferença tem pouca probabilidade de ser notada, a não ser
que a pessoa se exercite; nessas condições, surgem,
ocasionalmente, sinais agudos de insuficiência cardíaca.
Alterações de pressão nos átrios — as ondas a, c e v. Na curva
de pressão atrial da Fig. 9.5 podem ser notadas três elevações principais
da pressão, denominadas ondas de pressão atrial a, c, e v.
A onda a é causada pela contração atrial. Normalmente, a pressão
atrial direita eleva-se 4 a 6 mm Hg durante a contração atrial. enquanto
a pressão atrial esquerda eleva-se cerca de 7 a 8 mm Hg.
A onda c ocorre quando os ventrículos começam a se contrair;
ela é causada, em parte, pelo pequeno refluxo de sangue para os átrios
ao início da contração ventricular, mas, provavelmente, em sua maior
parte, pela protrusão das válvulas A-V em direção aos átrios, devido
ao aumento da pressão nos ventrículos.
Fig. 9.5 Os eventos do ciclo cardíaco,
mostrando alterações da pressão atrial
esquerda, pressão ventricular esquerda,
pressão aórtica, volume ventricular, o
eletrocardiograma e o fonocardiograma.
90
A onda v ocorre ao final da contração ventricular; ela decorre do
lento acúmulo de sangue nos átrios enquanto as válvulas A-V estão
fechadas durante a contração ventricular. Ao término dessa contração,
as válvulas A-V se abrem, possibilitando que o sangue flua rapidamente
para os ventrículos e fazendo desaparecer a onda v.
FUNÇÃO DOS VENTRÍCULOS COMO BOMBAS
Enchimento dos ventrículos. Durante a sístole ventricular,
grande quantidade de sangue acumula-se nos átrios, por estarem
fechadas às válvulas A-V. Por esta razão, logo que termina a
sístole c as pressões ventriculares caem novamente para seus
baixos valores diastólicos, as válvulas A-V abrem-se e
possibilitam ao sangue fluir rapidamente para os ventrículos,
como é mostrado pela elevação da curva do volume ventricular
na Fig. 9.5. Este é denominado período de enchimento rápido dos
ventrículos. As pressões atriais caem até uma fração de
milímetro das pressões ventriculares, porque os orifícios normais
das válvulas A-V são tão grandes que não oferecem praticamente
qualquer resistência ao fluxo sanguíneo.
O período de enchimento rápido dura aproximadamente
o primeiro terço da diástole. Durante o terço médio da diástole,
apenas pequena quantidade de sangue flui normalmente para
os ventrículos; este sangue continua a chegar das veias para os
átrios e a passar através deles para os ventrículos.
Durante o último terço da diástole, os átrios se contraem
e dão um impulso adicional ao influxo de sangue para os
ventrículos; isto responde por cerca de 25% do enchimento dos
ventrículos durante cada ciclo cardíaco.
Esvaziamento dos ventrículos durante a sístole. Período de
contração isovolúmica (isométrica). Imediatamente após o início
da contração ventricular, a pressão ventricular eleva-se
abruptamente, como é mostrado na Fig. 9.5. fazendo fecharemse as válvulas A-V. Um período adicional de 0,02 a 0,03 s é,
então, necessário para o ventrículo acumular pressão suficiente
para forçar as válvulas semilunares (aórtica e pulmonar) a se
abrirem contra as pressões na aorta e na artéria pulmonar.
Durante este período há, portanto, contração dos ventrículos,
mas não há qualquer esvaziamento. Este período é denominado
período de contração isovolúmica ou isométrica, indicando-se
com estes termos que a tensão está aumentando no músculo mas
não há encurtamento das fibras musculares. (Isto não é totalmente
verdadeiro, porque há encurtamento do ápice para a base e
alongamento circunferencial)
Período de ejeção. Quando a pressão no ventrículo esquerdo
se eleva ligeiramente acima de 80 mm Hg (e a pressão ventricular
direita, ligeiramente acima de 8 mm Hg), as pressões ventriculares
forçam, então, as válvulas semilunares a se abrirem.
Imediatamente, o sangue começa a jorrar para fora dos
ventrículos, com cerca de 70% do esvaziamento ocorrendo
durante o primeiro terço do período de ejeção e os 30%
restantes, durante os dois terços seguintes. Assim, o primeiro
terço é denominado período de ejeção rápida e os dois terços
finais, período de ejeção lenta.
Por uma razão bem peculiar, a pressão ventricular cai para
um valor ligeiramente abaixo da pressão na aorta durante o
período de ejeção lenta, apesar do fato de ainda haver sangue
saindo do ventrículo esquerdo. A razão é que o sangue que
flui para fora do ventrículo gera um momento [momentum]. À
medida que este momento diminui, durante a última parte da
sístole, sua energia cinética é convertida em pressão na aorta,
o que torna a pressão arterial ligeiramente maior que a pressão
no interior do ventrículo.
Período de relaxamento isovolúmico (ísométrico). Ao final da
sístole, o relaxamento ventricular se inicia subitamente,
possibilitando
a
rápida
diminuição
das
pressões
intraventriculares. Imediatamente, as elevadas pressões nas
grandes artérias distendidas fazem o sangue refluir para os
ventrículos, o que força as válvulas aórtica e pulmonar a se
fecharem.
Por mais 0,03 a 0,06 s, o músculo ventricular continua a se
relaxar, mesmo que o volume ventricular não se altere,
ocasionando o período de relaxamento isovolúmico ou
isométrico. Durante este período, as pressões intraventriculares
caem rapidamente de volta a seus valores diastólicos, muito
baixos. Então, as válvulas A-V se abrem para iniciar novo
ciclo de bombeamento ventricular.
Volume diastólico final, volume sistólico final e débito
sistólico. Durante a diástole, o enchimento dos ventrículos
aumenta normalmente ate cerca de 110 a 120 ml o volume de
cada ventrículo. Esse volume é conhecido como volume
diastólico final. Em seguida, com o esvaziamento dos
ventrículos durante a sístole, seu volume cai por cerca de 70
ml, que c designado como o débito sistólico. O volume
restante em cada ventrículo, cerca de 40 a 50 ml, é
denominado volume sistólico final. A fração do volume
diastólico final que é ejetada e designada como fração de ejeção geralmente igual à aproximadamente 60%.
Quando o coração se contrai vigorosamente, o volume
sistólico final pode cair para até 10 a 20 ml. Por outro lado,
quando grande quantidade de sangue flui para os ventrículos
durante a diástole, seus volumes diastólicos finais podem tomarse grandes, até 150 a 180 ml em corações normais- F. o débito
sistólico pode, por vezes, aumentar até aproximadamente o
dobro do normal, tanto pelo aumento do volume diastólico
final como pela diminuição do volume sistólico final.
FUNÇÃO DAS VÁLVULAS
As válvulas atrioventriculares. As válvulas A-V (tricúspide e
mitral) impedem o refluxo de sangue dos ventrículos para os
átrios durante a sístole e as válvulas semilunares (as válvulas
aórtica e pulmonar) impedem o refluxo das artérias aorta e
pulmonar para os ventrículos durante a diástole. Todas essas
válvulas, que são ilustradas na Fig. 9.6, fecham-se e abrem-se
passivamente. Isso quer dizer que elas se fecham quando um
gradiente retrógrado de pressão empurra o sangue para trás e
abrem-se quando um gradiente de pressão anterógrado força o
sangue a seguir avante. Por razões anatômicas óbvias, o
fechamento das delgadas e tênues válvulas A-V não requer quase
nenhum refluxo, enquanto o das muito mais pesadas válvulas
semilunares requer um refluxo bastante forte por alguns
milissegundos.
Função dos músculos papilares. A Fig. 9.6 também ilustra os
músculos papilares que se fixam aos folhetos das válvulas
Fig. 9.6 Válvulas mitral e aórtica
91
A-V pelas cordas tendíneas. Os músculos papilares se contraem
quando as paredes ventriculares o fazem, mas, contrariamente
ao que seria de esperar, eles não ajudam as válvulas a se fecharem.
Em vez disso, eles puxam os folhetos das válvulas para dentro,
em direção aos ventrículos, para impedir que eles façam
demasiada protrusão para trás, em direção aos átrios, durante a
contração ventricular. Quando uma corda tendínea se rompe
ou um dos músculos papilares fica paralisado, a válvula faz
protrusão bem para trás, por vezes de forma tão excessiva que
vaza muito e causa incapacidade cardíaca grave ou até mesmo
letal.
As válvulas aórtica e pulmonar. Há diferenças entre a
operação das válvulas aórtica e pulmonar e a das válvulas A-V.
Em primeiro lugar, as elevadas pressões nas artérias ao final da
sístole forçam as válvulas semilunares a se fecharem, em
comparação com o fechamento muito mais suave das válvulas AV. Segundo, devido à sua menor abertura, a velocidade de
ejeção do sangue pelas válvulas aórtica e pulmonar é bem
maior do que pelas válvulas A-V, muito maiores. Assim,
também devido ao fechamento súbito e à rápida ejeção, as bordas
das válvulas semilunares estão sujeitas a desgaste mecânico
muito maior que as válvulas A-V, que também são sustentadas
por cordas tendíneas. Pela anatomia das válvulas aórtica e
pulmonar, como é ilustrado na Fig. 9.6, fica evidente que elas
estão adaptadas para suportar esse trauma físico extra.
A CURVA DE PRESSÃO AÓRTICA. Quando o ventrículo
esquerdo se contrai, a pressão ventricular eleva-se rapidamente
até que a válvula aórtica se abra. Depois disso, a pressão no
ventrículo eleva-se muito menos, como ilustra a Fig. 9.5, pois o
sangue flui imediatamente para fora do ventrículo e para a
aorta.
A entrada de sangue nas artérias faz com que a parede
dessas artérias se distenda e a pressão se eleve. Ao final da
sístole, então, após o ventrículo esquerdo parar de ejetar sangue
e a válvula aórtica se fechar, a retração elástica das artérias
mantém pressão elevada nas artérias até mesmo durante a
diástole. A chamada incisura ocorre na curva de pressão
aórtica ao se fechar à válvula aórtica. Ela é causada por um
curto período de refluxo de sangue imediatamente antes do
fechamento da válvula, seguido, então, pela cessação súbita do
refluxo. Depois do fechamento da válvula aórtica, a pressão na
aorta cai lentamente durante toda a diástole, porque o sangue
armazenado nas artérias elásticas distendidas flui continuamente
de volta para as veias por meio dos vasos periféricos. Antes dos
ventrículos se contraírem novamente, a pressão aórtica
geralmente cai para cerca de 80 mm Hg (pressão diastólica), o
que constitui dois terços da pressão máxima de 120 mm Hg
(pressão sistólica), ocorrendo na aorta durante a contração
ventricular. A curva de pressão na artéria pulmonar é
semelhante à da aorta, exceto pelo fato das pressões serem
apenas cerca de um sexto das aórticas, como é discutido no
Cap. 14.
RELAÇÃO ENTRE OS SONS CARDÍACOS E O
BOMBEAMENTO CARDÍACO
Ao se auscultar o coração com estetoscópio, não se ouve a
abertura das válvulas, pois esse é um processo de
desenvolvimento relativamente lento e que não produz
qualquer ruído. Entreta nto, ao se fecharem às válvulas, seus
folhetos e os líquidos circundantes vibram sob a influência das
súbitas diferenças de pressão que ocorrem, produzindo sons que
se propagam pelo tórax em todas as direções.
Quando os ventrículos começam a se contrair, ouve-se um
som que é causado pelo fechamento das válvulas A-V. A
vibração é de tom baixo e mantém-se por período relativamente
longo, sendo conhecida como primeira bulha cardíaca. Quando
as válvulas aórtica e pulmonar se fecham, ouve-se um estalido
relativamente rápido, pois essas válvulas se fecham com extrema
rapidez e as regiões circunvizinhas vibram apenas por curto
período.
Esse som é conhecido como segunda bulha cardíaca.
Ocasionalmente, pode-se ouvir um som atrial quando os átrios
batem devido a vibrações associadas ao fluxo de sangue para os
ventrículos. Assim, também uma terceira bulha cardíaca ocorre,
por vezes, aproximadamente ao final do primeiro terço da
diástole, supostamente causada pelo sangue fluindo em
turbilhão para os ventrículos já quase cheios. As causas exatas
dos sons cardíacos são discutidas de modo mais completo no Cap.
23. em relação à ausculta.
PRODUÇÃO DE TRABALHO PELO CORAÇÃO
O trabalho sistólico e o trabalho-minuto. O trabalho
sistólico do coração ê a quantidade de energia que o coração
converte em trabalho durante cada batimento cardíaco enquanto
bombeia sangue para as artérias. O trabalho-minuto é a
quantidade total de energia convertida no período de 1 minuto;
obviamente, isso equivale ao trabalho sistólico multiplicado pela
freqüência cardíaca. O trabalho do coração é de dois tipos. Em
primeiro lugar, a maior parte é empregada para mover o
sangue das veias de baixa pressão para as artérias de alta
pressão. Isto é denomi nado trabalho externo ou trabalho de
volume-pressão. Segundo, menor proporção da energia c
empregada para acelerar o sangue até sua velocidade de ejeção
através das válvula aórtica e pulmonar. Este é o componente de
energia cinética do fluxo sanguíneo do trabalho cardíaco.
Trabalho externo (trabalho de volume-pressão). O trabalho
realizado pelo ventrículo esquerdo para elevar a pressão do
sangue durante cada batimento cardíaco (o trabalho externo
sistólico do ventrículo esquerdo, é igual ao débito sistólico
multiplicado pela pressão média de ejeção do ventrículo
esquerdo, a menos pressão média de entrada do ventrículo
esquerdo, durante o enchimento ventricular). Quando a pressão é
expressa em dinas por ce ntímetro quadrado e o débito
sistólico em mililitros, o trabalho externo é expresso em ergs.
O trabalho externo do ventrículo direito é normalmente
cerca de um sexto do trabalho do ventrículo esquerdo, devido ã
diferença na pressão sistólica contra a qual os dois ventrículos
têm de bombear.
A energia cinética do fluxo sanguíneo. O trabalho adicional
de cada ventrículo necessário para criar a energia cinética do
fluxo sanguíneo é proporcional à massa de sangue ejetada,
multiplicada pelo quadrado da velocidade de ejeção. Isto é,
mv2
Energia cinética = -----2
Ouando a massa é expressa em gramas de sangue ejetados e
a velocidade, em centímetros por segundo, o trabalho o é em
ergs.
Comumente, o trabalho ventricular esquerdo necessário
para criar a energia cinética do fluxo sanguíneo é apenas cerca
de 1% do trabalho total do ventrículo, sendo, pois, ignorado no
cálculo do trabalho sistólico total. Em certas condições
anormais, como a estenose aórtica, em que o sangue flui com
grande velocidade pela válvula estenosada, mais de 50% do
trabalho total podem ser necessários para criar a energia cinética
do fluxo sanguíneo.
Análise gráfica do bombeamento ventricular
A Fig. 9.7 apresenta um diagrama que é particularmente útil
para explicar à mecânica de bombeamento do ventrículo
esquerdo. Os dois componentes mais importantes do diagrama
são as duas curvas pretas contínuas designadas como "pressão
diastólica" e "pressão sistólica". Essas duas curvas são curvas de
volume-pressão. A curva da pressão diastólica é determinada
enchendo-se o coração com quantidades cada vez maiores de
sangue e medindo-se, então, a pressão diastólica imediatamente
antes que ocorra a contração ventricular, que é a pressão
diastólica final do ventrículo. A curva de pressão sistólica é
determinada impedindo-se qualquer descarga de sangue do
coração e medindo-se a pressão sistólica máxima que é obtida
durante a contração ventricular para cada volume de enchimento.
Fica muito claro que a pressão diastólica não aumenta
muito ate que o volume ventricular se eleve acima de 150 ml,
aproximadamente. Até este volume, portanto, o sangue pode
fluir facilmente do átrio para o ventrículo.
92
Fig. 9.7 Relação entre o volume ventricular esquerdo e a pressão
intraventricular durante a diástole e a sístole. Também c mostrado,
pelas linhas vermelhas fortes, o "diagrama de volume-pressão" que
ilustra as alterações do volume e pressão intraventriculares durante o
ciclo cardíaco.
Acima de 150 ml, a pressão diastólica passa a aumentar rapidamente, em
parte porque o tecido fibroso do coração não se distende mais e, em parte,
porque o pericárdio que circunda o coração fica distendido praticamente
até o seu limite. Durante a contração ventricular, a pressão sistólica
aumenta rapidamente com volumes ventriculares crescentes, atingindo
seu máximo com o volume ventricular de 150 a 170 ml. Em seguida,
quando o volume aumenta ainda mais, a pressão sistólica em algumas
condições até diminui, conforme ilustra a curva da pressão sistólica
descendente, porque nesses volumes muito grandes os filamentos de
actina e miosina das fibras musculares cardíacas encontram-se de fato
separados o suficiente para que a força de contração das fibras fique
menos que ótima.
Observe especialmente na figura que a pressão sistólica máxima
para o ventrículo esquerdo normal não estimulado está entre 250 e 300
mm Hg, mas isso varia muito com a força do coração. Para o ventrículo
direito normal, ela fica entre 60 e 80 mm Hg.
O diagrama de volume-pressão durante o ciclo cardíaco;
trabalho cardíaco. As curvas vermelhas na Fig. 9.7 formam uma alça
denominada diagrama de volume-pressão do ciclo cardíaco para o
ventrículo esquerdo. Ele é dividido em quatro fases distintas:
Fase I: Período de enchimento. Esta fase do diagrama de volumepressão inicia-se com volume ventricular de cerca de 45 ml e pressão
diastólica muito próxima de 0 mm Hg. Quarenta e cinco mililitros são
a quantidade de sangue que permanece no ventrículo após o batimento
cardíaco anterior, sendo designada como volume sistólico final. Quando
o sangue venoso pulmonar flui do átrio para o ventrículo, o volume
aumenta normalmente para cerca de 115 ml, designados como volume
diastólico final, um aumento de 70 ml. Por esta razão, o diagrama de
volume-pressão durante a fase I estende-se ao longo da linha marcada
"I", aumentando o volume para 115 ml e elevando a pressão diastólica
para cerca de 5 mm Hg.
Fase II: Período de contração isovolúmica. Durante a contração
isovolúmica, o volume do ventrículo não se altera. Contudo, a pressão
no interior do ventrículo se eleva até se igualar à pressão na aorta,
um valor de pressão de cerca de 80 mm Hg. como é mostrado pela
linha "II".
Fase III: Período de ejeção. Durante a ejeção, a pressão sistólica
eleva-se ainda mais, devido à contração ainda maior do coração. Ao
mesmo tempo, o volume do ventrículo diminui porque o sangue agora
flui para fora do ventrículo e para a aorta. Em vista disso, a curva
marcada "III" registra as alterações no volume e na pressão sistólica
durante este período de ejeção.
Fase IV: Período de relaxamento isovolumétrico. Ao final do
período de ejeção, as válvulas semilunares dos ventrículos fecham-se
e a pressão ventricular cai novamente até o nível da pressão diastólica,
A linha marcada "IV" registra esta diminuição da pressão
intraventricular sem qualquer alteração do volume.
Assim, o ventrículo retorna a seu ponto de partida, com cerca de 45 ml
de sangue residuais no ventrículo e pressão atrial de fato muito
próxima de 0 mm Hg.
Trabalho calculado a partir do diagrama de volume-pressão.
Os leitores bem treinados nos princípios básicos da física devem
reconhecer que a área subtendida por este diagrama de volumepressão, a parte direita da área sombreada designada como TE,
equivale ao trabalho externo efetivo do ventrículo, durante seu ciclo de
contração- Em estudos experimentais da contração cardíaca, este
diagrama é, portanto, empregado para o cálculo do trabalho cardíaco.
Quando o coração bombeia grande quantidade de sangue, o
diagrama de trabalho passa a ter área muito maior. Isto quer dizer
que ele se estende bem para a direita, porque o ventrículo agora se
enche mais de sangue durante a diástole, eleva-se muito mais porque o
ventrículo se contrai com pressão maior e, geralmente, estende-se mais
para a esquerda porque o ventrículo se contrai até um volume menor especificamente , quando o ventrículo é estimulado a maior atividade pelo
sistema nervoso simpático.
Os conceitos de "pré-carga" e "pós-carga". Ao se avaliar as
propriedades contrateis do músculo, é importante especificar o grau de
tensão sobre o músculo quando ele começa a se contrair, o que é
designado como pré-carga, e também se especificar a carga contra a qual
o músculo exerce sua força contrátil. que é designada como pós-carga.
Para a contração cardíaca, a pré-carga é geralmente considerada
como sendo o volume de sangue no ventrículo ao final da diástole,
ou seja, o volume diastólico final. Entretanto, por vezes, essa pré-carga
é expressa como a pressão diastólica final quando o ventrículo fica cheio
de sangue.
A pós-carga do ventrículo é a pressão na artéria que sai do mesmo.
Na Fig. 9.7, ela corresponde à pressão sistólica descrita pela curva Fase
III do diagrama de volume-pressão. (Por vezes, a pós-carga é considerada
muito livremente como sendo a resistência na circulação, e não a pressão.)
A importância dos conceitos de pré-carga e pós-carga é que, em
muitos estados funcionais anormais do coração ou da circulação, o grau
de enchimento do ventrículo (a pré-carga), a pressão arterial contra
a qual o ventrículo tem de se contrair (a pós-carga), ou ambos, alteram-se
muito em relação ao normal.
A ENERGIA QUÍMICA PARA A CONTRAÇÃO CARDÍACA:
UTILIZAÇÃO DE OXIGÉNIO PELO CORAÇÃO
O músculo cardíaco, assim como os músculos esqueléticos, utiliza
energia química para gerar o trabalho da contração. Essa energia deriva
principalmente do metabolismo oxidativo dos ácidos graxos e, em menor
grau, de outros nutrientes, especialmente ácido lático e glicose. Como
conseqüência, a intensidade do consumo de oxigênio é uma excelente
medida da energia química liberada enquanto o coração realiza seu
trabalho. As diferentes reações liberadoras dessa energia são discutidas
nos Caps. 67 e 68.
Estudos experimentais em corações isolados mostraram que o
consumo de oxigênio pelo coração e. portanto, a energia química
despendida durante a contração estão diretamente relacionados à área
sombreada total da Fig. 9.7. Essa parte sombreada consiste no trabalho
externo.TE, conforme explicado antes, e numa outra parte, chamada de
energia potencial, designada como EP. A energia potencial constitui o
trabalho adicional que poderia ser realizado pela contração do
ventrículo, se este se esvaziasse de todo o sangue em sua câmara a cada
contração.
Infelizmente, é impossível medir-se a área sombreada total da Fig.
.7 em animais ou seres humanos vivos. Em vez disto, também se verificou
experimentalmente que o consumo de oxigênio c praticamente
proporcional à tensão que ocorre no músculo cardíaco durante a
contração multiplicada pelo período de tempo que a contração persiste,
denominado o índice tensão-tempo. Como a tensão é muito alta, quando
a pressão sistólica é elevada, é utilizada uma quantidade
correspondente maior de oxigênio. Da mesma forma, quantidade muito
maior de energia química é despendida mesmo com pressão sistólica
normal quando o ventrículo está anormalmente dilatado, porque a tensão
do músculo cardíaco durante a contração é proporcional à pressão vezes
o diâmetro do ventrículo. Isto é particularmente importante na
insuficiência cardíaca, pois o ventrículo encontra-se, então, dilatado e,
paradoxalmente, a quantidade de energia química necessária para
dada quantidade de trabalho tem de ser maior do que nunca, ainda que o
coração já esteja insuficiente.
93
Eficiência da contração cardíaca. Durante a contração muscular, a
maior parte da energia química é convertida em calor, e parte muito
menor, em trabalho. A proporção entre o trabalho e o gasto de energia
química é denominada eficiência da contração cardíaca ou, simplesmente,
eficiência do coração, A eficiência máxima do coração normal está entre
20 e 25%. Na insuficiência cardíaca, isto pode cair até para 5 a 10%.
REGULAÇÃO DO BOMBEAMENTO CARDÍACO
Quando a pessoa está em repouso, o coração bombeia apenas
4 a 6 I de sangue a cada minuto. Entretanto, durante exercício
intenso, o coração pode ser solicitado a bombear até quatro
a sete vezes essa qualidade. A presente seção discute os meios
pelos quais o coração pode adaptar-se a aumentos tão extremos
do débito cardíaco.
Os dois meios básicos pelos quais o volume bombeado pelo
coração é regulado são (1) a regulação intrínseca do
bombeamento pelo coração em resposta a alterações do volume
de sangue que flui até o coração e (2) o controle do coração pelo
sistema nervoso autonômico.
REGULAÇÃO INTRÍNSECA DO BOMBEAMENTO
CARDÍACO — O MECANISMO DE FRANKSTARUNG
No Cap. 20, vemos que a quantidade de sangue bombeada
pelo coração a cada minuto é determinada pela intensidade do
fluxo sanguíneo das veias para o coração, o que é denominado
retorno venoso. Isso quer dizer que cada tecido periférico do
corpo controla seu próprio fluxo sanguíneo, e a soma de todos
os fluxos sanguíneos locais por todos os tecidos periféricos volta
ao átrio direito por meio das veias. O coração, por sua vez,
bombeia, automaticamente, esse sangue que chega para as
artérias sistêmicas, de modo que ele possa fluir novamente pelo
circuito.
Essa capacidade intrínseca de adaptação do coração à
alteração no volume de sangue que entra é denominada
mecanismo de Frank-Starling do coração, em homenagem a
Frank e Starling, dois grandes fisiologistas de quase 100 anos
atrás. Basicamente, o mecanismo de Frank-Starling indica que
quanto mais o coração se enche, durante a diástole, maior vai ser
a quantidade de sangue bombeada para a aorta. Outra forma de
expressar isto é: Dentro dos limites fisiológicos, o coração
bombeia todo o sangue que chega até ele, sem permitir acúmulo
excessivo de sangue nas veias.
Qual é a explicação do mecanismo de Frank-Starling?
Quando quantidade extra de sangue flui para os ventrículos, o
músculo cardíaco propriamente dito c distendido até maior
comprimento. Isso, por sua vez, faz o músculo contrair-se com
mais força, porque os filamentos de actina e miosina são então
trazidos a grau de superposição mais próximo do ótimo para
a geração de força. Assim sendo, devido ao aumento de sua
ação de bombear, o ventrículo bombeia automaticamente o
sangue extra para as; artérias. Essa capacidade do músculo
distendido até seu comprimindo ótimo de contrair-se com maior
força é característica de todos os músculos estriados,
conforme explicado no Cap. 6, e não somente do músculo
cardíaco.
Além do importante efeito de distensão do músculo cardíaco,
ainda outro fator aumenta o bombeamento cardíaco quando seu
volume está aumentado. A distensão da parede atrial direita
aumenta diretamente a freqüência cardíaca por até 10 a 20%;
isso também ajuda a aumentar a quantidade de sangue bombeada
a cada minuto, embora sua contribuição seja muito menor que
a do mecanismo de Frank-Starling.
Ausência de efeito de alterações da carga da pressão arterial
Fig. 9,8 Constância do débito cardíaco mesmo face a amplas alterações
da pressão arterial. É somente quando a pressão arterial se eleva acima
da faixa operacional normal da pressão que a carga de pressão faz o
coração começar a falhar.
sobre o débito cardíaco. Uma das conseqüências mais importantes
do mecanismo de Frank-Starling do coração é que, dentro de
limites razoáveis, as alterações da pressão arterial contra a qual
o coração bombeia quase não têm efeito sobre a intensidade
com que o sangue é bombeado a cada minuto (o débito cardíaco).
Esse efeito é ilustrado na Fig. 9.8, que é uma curva extrapolada
para seres humanos a partir de dados obtidos em cães nos quais
a pressão foi alterada progressivamente pela constrição da aorta,
sendo o débito cardíaco medido simultaneamente. O significado
deste efeito é o seguinte: independentemente da carga de pressão
arterial até um limite razoável, o fator importante para a
determinação da quantidade de sangue bombeada pelo
coração ainda é a intensidade da entrada de sangue no coração.
Curvas de função ventricular
Uma das melhores maneiras de expressar a capacidade
funcional de bombear sangue dos ventrículos é por curvas de
função ventricular, como é mostrado nas Figs. 9.9 e 9.10. A
Fig. 9.9 ilustra um tipo de curva de função ventricular
denominada curva do trabalho sistólico. Observe que, à medida
que a pressão atrial aumenta, o trabalho sistólico também
aumenta, até atingir o limite da capacidade cardíaca.
A Fig. 9.10 apresenta outro tipo de curva de função
ventricular denominada curva do débito-minuto ventricular. Essas
duas curvas representam a função dos dois ventrículos do
coração humano, com base em dados extrapolados de animais
inferiores. Ao se elevar à pressão atrial, o respectivo débito
por minuto do volume ventricular também aumenta.
Fig. 9.9 Curvas de função ventricular esquerda e direita em um cão,
mostrando o trabalho sistólico ventricular em função das pressões atriais
médias esquerda e direita. (Curvas reconstruídas a partir de dados em
Sarnoff: Physiol. Rev. 35:101, 1955.)
94
Fig. 9.10 Curvas aproximadas do débito ventricular esquerdo e direito
para o coração humano, conforme extrapolado a partir de dados obtidos
em cães.
Assim, as curvas de função ventricular são outra maneira
de expressar o mecanismo de Frank-Starling do coração. Isto
é, quando os ventrículos se enchem sob pressões atriais mais
elevadas, o volume ventricular e a força da contração cardíaca
aumentam, fazendo o coração bombear maior quantidade de
sangue para as artérias.
CONTROLE DO CORAÇÃO PELOS NERVOS
SIMPÁTICOS E PARASSIMPÁTICOS
A capacidade de bombeamento do coração é muito
controlada pelos nervos simpáticos e parassimpáticos (vago),
que suprem abundantemente o coração, como ilustra a Fig. 9.11.
A quantidade de sangue bombeada pelo coração a cada
minuto, o débito cardíaco, pode muitas vezes ser aumentada
em mais de 100% pela estimulação simpática. Em contraste,
ela pode ser reduzida a zero ou a quase isto pela estimulação
vagal (parassimpática).
Excitação do coração pelos nervos simpáticos. Uma forte
estimulação simpática pode aumentar a freqüência cardíaca de
seres humanos para 200 e, em raros casos, até mesmo 250
batimentos por minuto, em pessoas jovens. A estimulação
simpática aumenta igualmente a força com que o coração se
contrai, aumen-
Fig. 9.11 Os nervos cardíacos
tando também, como conseqüência, tanto o volume de sangue
bombeado como a pressão de ejeção. Assim, a estimulação
simpática pode freqüentemente aumentar o débito cardíaco por
até duas a três vezes.
A inibição do sistema nervoso simpático pode ser utilizada
para diminuir, em grau moderado, o bombeamento cardíaco,
da seguinte maneira: em condições normais, as fibras nervosas
simpáticas para o coração descarregam, continuamente, a baixa
freqüência, que mantém o bombeamento em cerca de 30% do
que é observado sem qualquer estimulação simpática. Por esta
razão, quando a atividade do sistema nervoso simpático é
reduzida a um nível abaixo do normal, isso diminui tanto a
freqüência cardíaca como a força de contração ventricular,
diminuindo, assim, o nível de bombeamento cardíaco até 30%
abaixo do normal.
Estimulação parassimpática (vagal) do coração. Uma forte
estimulação vagal do coração pode, de fato, fazer cessar por
alguns segundos os batimentos cardíacos, mas depois o coração
geralmente "escapa", batendo, daí em diante, com freqüência
de 20 a 30 batimentos por minuto. Além disso, a forte estimulação
parassimpática diminui em até 20 a 30% a força de contração
do coração. Esta não é uma grande diminuição, porque as fibras
vagais distribuem-se principalmente para os átrios e pouco para
os ventrículos onde ocorre à contração motriz do coração. Apesar
disso, a grande diminuição da freqüência cardíaca, associada à
ligeira diminuição da contração cardíaca, pode reduzir em até
50% ou mais o bombeamento ventricular — especialmente
quando o coração está operando sob grande carga de trabalho.
Efeito da estimulação simpática ou parassimpática sobre a
curva de função cardíaca. A Fig. 9.12 apresenta quatro curvas
distintas de função cardíaca. Elas são iguais às curvas de função
ventricular das Figs. 9.9 e 9.10, exceto por representarem a função
de todo o coração, e não de um ventrículo individual; elas
mostram a relação entre a pressão atrial direita na entrada do
coração e o debito cardíaco para a aorta.
As curvas da Fig. 9.12 demonstram que, a qualquer pressão
atrial direita, o débito cardíaco aumenta com o aumento da
estimulação simpática e diminui com o aumento da estimulação
parassimpática. Deve-se recordar que as alterações do débito
cardíaco ocasionadas pela estimulação nervosa são causadas
tanto por alterações da freqüência cardíaca como por alterações
da força contrátil do coração, pois ambas afetam o débito
cardíaco.
Fig. 9.12 Efeito sobre a curva do débito cardíaco de graus diferentes
de estimulação simpática e parassimpática.
95
EFEITO DA FREQUÊNCIA CARDÍACA SOBRE A
FUNÇÃO DO CORAÇÃO COMO UMA BOMBA
Em geral, quanto mais vezes o coração bate por minuto,
mais sangue ele pode bombear, mas há importantes limitações
a este efeito. Após a freqüência cardíaca elevar-se acima de
um nível crítico, por exemplo, a força do próprio coração diminui,
presumivelmente devido ao uso excessivo de substratos
metabólicos pelo músculo cardíaco. Além disso, o período de
diástole entre as contrações fica tão reduzido que o sangue não
tem tempo para fluir adequadamente dos átrios para os
ventrículos. Por estas razões, quando a freqüência cardíaca é
aumentada artificialmente pela estimulação elétrica, o coração
tem sua capacidade máxima de bombear grande quantidade de
sangue na freqüência cardíaca entre 100 e 150 batimentos por
minuto. Por outro lado, quando sua freqüência é aumentada por
estimulação simpática, ele atinge sua capacidade máxima de
bombear sangue nas freqüências entre 170 e 220 batimentos por
minuto. A razão para essa diferença é que a estimulação
simpática aumenta não só a freqüência como também a força
cardíaca. Ao mesmo tempo, ela diminui a duração da contração
sistólica e possibilita mais tempo para o enchimento durante a
diástole.
AVALIAÇÃO DA CONTRATILIDADE CARDÍACA
Embora seja muito fácil determinar-se a freqüência cardíaca
simplesmente pela contagem do pulso, sempre foi difícil determinar-se
à força de contração cardíaca, comumente designada como contratilidade
cardíaca. Muito freqüentemente, a alteração da contratilidade é
exatamente o oposto da alteração da freqüência cardíaca. Na verdade,
esse efeito ocorre quase que invariavelmente nas cardiopatias e doenças
debilitantes.
Uma das maneiras pelas quais a contratilidade cardíaca pode ser
determinada com grande precisão é o registro de uma ou mais curvas
de função cardíaca. Entretanto, isto so pode ser feito com facilidade
em animais experimentais. Por essa razão, muitos fisiologistas clínicos
têm procurado métodos de avaliação da contratilidade cardíaca de
maneira simples. Um desses métodos é a determinação da chamada
dP/dt.
dP/dt como medida da contratilidade. A dP/dt significa velocidade
da alteração da pressão ventricular em função do tempo. O registro
de dP/dt é gerado por um computador que diferencia a onda da pressão
ventricular, dando, portanto, um registro da velocidade da alteração
da pressão ventricular. A Fig. 9.13 mostra dois registros distintos da
onda de pressão ventricular, assim como registros simultâneos (em cor)
da dP/dt. Na parte superior da figura, o coração estava batendo
normalmente e na parte inferior o coração havia sido estimulado pelo
isoproterenol, um composto que tem basicamente o mesmo efeito sobre o
coração que tem a estimulação simpática.
Observe no registro superior que, ao mesmo tempo que a pressão
ventricular está aumentando com maior velocidade, o registro da dP/dt
também atinge seu valor mais alto. Por outro lado, no momento em
que a pressão ventricular está caindo mais rapidamente, o registro da
dP/dt atinge seu nível mais baixo. Quando a pressão ventricular não
está nem se elevando nem caindo, o registro da dP/dt está no valor
zero.
Estudos experimentais mostraram que a velocidade da elevação da
pressão ventricular, a dP/dt, correlaciona-se em geral muito bem com
a força de contração do ventrículo. Este efeito é ilustrado pela
comparação do registro da dP/dt da parte superior da Fig. 9.13 com a
parte inferior. Assim, a dP/dt máxima é freqüentemente utilizada como
meio de comparação da contratilidade do coração em diferentes estados
funcionais.
Infelizmente, o valor quantitativo da dP/dt máxima também é
afetado por outros fatores que não estão relacionados à contratilidade
cardíaca. Esse valor fica aumentado, por exemplo, tanto pela maior
pressão de entrada para o ventrículo esquerdo (a pressão diastólica final
ventricular), que é a pré-carga do ventrículo, como pela pressão
contra a qual o coração está bombeando sangue, denominado pós-carga.
Portanto, muitas vezes é difícil utilizar-se a dP/dt como medida de
contratilidade ao se comparar o coração de uma pessoa ao de outra,
pois um desses fatores pode diferir. Por essa razão, outras medidas
quantitativas também têm sido empregadas em tentativas de avaliação da
contratilidade cardíaca. Uma delas tem sido a utilização da dP/dt dividida
pela pressão instantânea no ventrículo, ou (dP/dt)/P.
EFEITO DOS ÍONS POTÁSSIO E CÁLCIO SOBRE A
FUNÇÃO CARDÍACA
Na discussão dos potenciais de membrana no Cap. 5 foi ressaltado
que os íons potássio têm efeito acentuado sobre os potenciais de
membrana e os potenciais de ação, e no Cap. 6 observou-se que os
íons cálcio têm papel particularmente importante no desencadeamento
do processo contrátil muscular. Deve-se esperar, portanto, que as
concentrações desses dois íons nos líquidos extracelulares também
tenham efeitos importantes sobre o bombeamento cardíaco.
Efeito dos íons potássio. O excesso de potássio no líquido extracelular
faz o coração ficar extremamente dilatado e flácido e lentifica a freqüência
cardíaca. Quantidades muito grandes também podem bloquear a
condução do impulso cardíaco dos átrios para os ventrículos pelo feixe
A-V. A elevação da concentração de potássio até apenas 9 a 12
m.Eq/1 - duas a três vezes o valor normal - pode causar um
enfraquecimento tal do coração e um ritmo tão anormal que isso pode
causar a morte.
Esses efeitos são causados parcialmente pelo fato de que uma
concentração elevada de potássio no líquido extracelular causa
diminuição do potencial de membrana em repouso nas fibras musculares
cardíacas, conforme explicado no Cap. 5. Quando o potencial de
membrana diminui, a intensidade do potencial de ação também diminui,
o que torna progressivamente mais fraca a contração do coração.
Efeito dos íons cálcio. O excesso de íons cálcio causa efeitos quase
que exatamente opostos aos dos íons potássio, fazendo o coração entrar
em contração espástica. Isso é causado pelo efeito direto dos íons cálcio
na excitação do processo contrátil cardíaco, conforme explicado antes.
Inversamente, deficiência de íons cálcio causa flacidez cardíaca, de modo
semelhante ao efeito do potássio. Entretanto, com os níveis normalmente
regulados dentro de limites estreitos, é raro que esses efeitos cardíacos
das concentrações anormais de cálcio causem problemas clínicos.
EFEITO DA TEMPERATURA SOBRE O CORAÇÃO
Fig. 9.13 Registros simultâneos da pressão ventricular e da dP/dt. A
mostra resultados obtidos em coração normal e B mostra resultados
de um coração estimulado por isoproterenol. (Modificado de Mason
et ai., in Sodcman e Sodeman [eds.j. Pathologic Physiology 6. ed. Phila
delphia, W.B. Saunders Co., 1979.)
O aumento da temperatura, que ocorre quando se tem febre- causa
grande aumento da freqüência cardíaca, por vezes até o dobro do normal.
A diminuição da temperatura causa grande redução da freqüência
cardíaca, caindo até alguns batimentos por minuto quando a pessoa está
próxima da morte por hipotermia, na faixa de 15,5 a 21°C. Esses
efeitos decorrem, presumivelmente, do calor causando maior
permeabilidade da membrana muscular aos íons, ocasionando a
aceleração do processo de auto-excitação.
96
Muitas vezes, a força contrátil do coração é aumentada
temporariamente por aumento moderado da temperatura, mas a elevação
prolongada da temperatura exaure os sistemas metabólicos do coração e
causa fraqueza.
REFERÊNCIAS
Brunwald, E., and Rosa, J., Jr: Control of cardiac performance. In Berne,
R. M., et ai, (eda.): Handbook of Physiology. Sec. 2, Vol. I. Baltimore,
Williams & Wilkina, 1979, p. 533. Cooper, G-, IV: Cardiocyte adaptation to
chronically altered load. Annu. Rev. Physiol., 49:501, 1987. Cowley, A. W., Jr.,
andGuyton, A. C: Hean rate as a determinant of cardiac output in dogs with
arteriovenous fistula. Am. J. Cardiol., 28:321, 1971. EIlis, D,: Na-Ca exchange in
cardiac tisaue. Adv. Myocardiol., 5:295, 1985. Fabiato, A., and Fabiato, F.:
Calcium and cardiac excitation-contraction coupling. Annu. Rev. Phyaiol.,
41:473, 1979. FitzGerald, P. G.: Gap junction heterogeneity in liver, heart, and
lens. News Physiol. Sei., 3:206, 1988. Fozzard, H. A., et ai.: The Heart and
Cardiovascular Syatem: Scientific Foundations. New York, Raven Press, 1986.
Gadsby, D. C: The Na/K pumpof cardiac cella. Annu. Rev. Biophys. Bioeng.,
13:373, 1984.
Gevers, W.: Protein metabolism Df the heart. J. Mol. CelL Cardiol., 16:3,1984.
Gilmour, R. F-, Jr, and Zipes,D. P-: Slow inwardcurrent and cardiac arrhythmias.
Am. J. Cardiol., 55:89B, 1985. Guyton, A. C: Determination of cardiac output by
equating venous return curves with cardiac response curves. Physiol. Rev.,
35:123, 1955. Guyton, A. C, et ai.: Circulatory Physiology: Cardiac Output and Its
Regulation. 2nd Ed. Phíladelphia, W. B. Saunders Co., 1973. Hotfman, B. B.,
and Leflíowitz, R. J.: Adrenergic receptora in the heart. Annu. Rev. Physiol.,
44:475, 1982.
Hurst, J. W., et ai.: The Heart. New York, McGraw-Hill Book Co., 1990.
Jacobus, W. E.: Respiratory control and the integration of heart high-energy
phosphate metabolism by mitochondrial creatine kinase. Annu. Rev.
Physiol., 47:707, 1985. Johnson, E. A.: Force-interval relationship of cardiac
muscle. In Berne,
R. M., et ai. (eds.): Handbook of Physiology. Sec. 2, Vol. I. Baltimore,
Williams & Wilkins, 1979, p. 475. Manfredi, J. P., and Holmes, E. W.: Purine
salvage pathwaya in myocardium.
Annu. Rev. Physiol., 47:691, 1985. Matsubara, I.: X-ray diffraction studies of the
heart. Annu. Rev. Biophys. Bioeng., 9-81, 1980.
Mela-Riker, L. M., and Bukoaki, R. D.: Regulation of mitochondrial activity
in cardiac cells. Annu. Rev. Physiol., 47:645, 1985. Morgan, H. E., et ai.:
Biochemical mechamsms of cardiac hypertrophy.
Annu. Rev, Physiol., 49:533, 1987. Nozawa, T., et ai.: Relation between
oxygen consumption and pressure-volume área of in situ dog heart. Am. J.
Phyaiol., 253:H31, 1987. Page, E., and Shibata, Y.: Permeable junctions
between cardiac cella. Annu.
Rev. Physiol., 43:431, 1981. Parmley, W. W., and Talbot, W.: Heart as a
pump. In Berne, R. M., et ai.
(eds.): Handbook of Physiology. Sec. 2, Vol. I. Baltimore, Williams &
Wilkina, 1979, p. 429. Rovetto, M. J.: Myocardial nucleotide tranaport. Annu.
Rev. PhyBÍol., 47:605, 1985. Ruegg, J. C: Dependence of cardiac
contractílity on myohbnllar calcium
sensitivity. News Physiol. Sei., 2:179, 1987. Sarnoff, S. J.: Myocardial
contractility aa described by ventricular function curves. Physiol. Rev.,
35:107, 1955. Sommer, J. R., and Johnson, E. A.: Ultrastructure of
cardiac muscle. In Berne, R. M., et ai. (eds.): Handbook of PhyBiology.
Sec. 2, Vol. I. Baltimore, Williams & Wilkins, 1979, p. 113. Starling, E.
H.: The Linacre Lecture on the Law of the Heart. London,
Longmans Green & Co., 1918. Suga, H ,etal.: Prospective prediction of
Ojconsumptionfrompressure-volume área in dog hearts. Am. J. Phyaiol.,
252:H1258,1987. Suga, H., et ai.: Criticai evaluation of left ventricular
systolic pressure volume área as predictor of oiygen consumption rate.
Jap. J. Physiol., 30:907, 1980. Sugden, P. H.: The effects of hormonal
factors on cardiac protein turnover.
Adv. Myocardiol., 5:105,1985. Sugimoto, T., et ai.: Effect of maximal work
load on cardiac function. Jap. Heart J., 14:146, 1973. Sugimoto, T., et ai.:
Quantitative effect of low coronary pressure on left ventricular
performance, Jap. Heart J., 9:46,1968. Swynghedauw, B.: Developmental
and functional adaptation of contractile proteins in cardiac and skeletal
muscles. Physiol. Rev., 66:710,1986. Vary, T. C, et ai.: Control of energy
metabolism of heart muscle. Annu. rev. Physiol., 43:419, 1981.
Winegrad, S.: Regulation of cardiac contractile proteins. Correlations between physiology and biochemistry. Circ. Res., 55:565, 1984. Winegrad, S.:
Calcium release from cardiac Barcoplasmic reticulum. Annu.
Rev. Physiol., 44:451, 1982.
(Ver também Cap. 10.)
97
CAPÍTULO 10
Excitação Rítmica do Coração
O coração é provido de um sistema especializado (1) para
a geração de impulsos rítmicos, para causar a contração rítmica
do músculo cardíaco, e (2} para a condução rápida desses impulsos
por todo o coração. Quando esse sistema funciona normalmente,
os átrios se contraem cerca de um sexto de segundo antes da
contração ventricular, o que possibilita maior enchimento dos
ventrículos antes que eles bombeiem o sangue pelos pulmões
e pela circulação periférica. Outra importância especial do
sistema é que ele possibilita que todas as partes dos ventrículos se
contraiam simultaneamente, o que é essencial para a geração
efetiva de pressão nas câmaras ventriculares.
Infelizmente, porém, este sistema rítmico e condutor do
coração é muito suscetível a danos por doenças cardíacas,
especialmente a isquemia dos tecidos cardíacos decorrente do
fluxo sanguíneo coronário insuficiente. A conseqüência é,
muitas vezes, um ritmo cardíaco muito bizarro ou uma seqüência
anormal da contração das câmaras cardíacas, e a eficácia do
bombeamento cardíaco muitas vezes fica gravemente afetada, a
ponto de causar a morte do paciente.
O SISTEMA ESPECIALIZADO DE EXCITAÇÃO
E ONDUÇÃO DO CORAÇÃO
A Fig. 10.1 apresenta o sistema especializado de excitação
e condução do coração que controla as contrações cardíacas.
A figura mostra (A) o nodo sinusal (também denominado nodo
sinoatrial ou S – A) , no qual é gerado o impulso rítmico normal;
(B) as vias internodais que conduzem o impulso do nodo sinusal
para o nodo A-V; (C) o nodo A-V (também denominado nodo
atrioventricular), no qual o impulso dos átrios sofre retardo antes
de passar para os ventrículos; (D) o feixe A-V, que conduz o
impulso dos átrios para os ventrículos; e (E) os feixes esquerdo
e direito das fibras de Purkinje, que conduzem o impulso cardíaco
a todas as partes dos ventrículos.
O NODO SINUSAL
O nodo sinusal é uma pequena tira achatada e elíptica de
músculo especializado, com aproximadamente 3 mm de largura,
15 mm de comprimento e 1 mm de espessura; está localizado
na parede superior lateral do átrio direito, imediatamente abaixo
e lateral à abertura da veia cava superior. As fibras deste nodo
quase não têm filamentos contrateis e têm 3 a 5 µm de diâmetro,
em contraste com o diâmetro de 10 a 15 µm fibras musculares
atriais circunvizinhas. No entanto, as fibras sinusais são
contínuas com as fibras atriais, de modo que qualquer potencial
de ação que se inicia no nodo sinusal espalha-se imediatamente
para os átrios.
Fig. 10.1 O nodo sinusal e o sistema de Purkinje do coração, mostrando
também o nodo A-V, as vias internodais atriais e os ramos ventriculares.
Ritmicidade automática das fibras sinusais
Muitas fibras cardíacas têm a capacidade de auto-excitação,
um processo que pode ocasionar contrações rítmicas automáticas.
Isto é particularmente verdadeiro para as fibras do sistema
especializado de condução do coração; a parte desse sistema que
apresenta o maior grau de auto-excitação são as fibras do nodo
sinusal. Em vista disso, o nodo sinusal controla normalmente
a freqüência de batimento de todo o coração, como é discutido
com detalhes mais adiante neste capítulo. Primeiramente, porém,
vamos descrever essa ritmicidade automática.
Mecanismos da ritmicidade do nodo sinusal. A Fig. 10.2
apresenta potenciais de ação registrados em fibra do nodo sinusal
por três batimentos cardíacos e. para fins de comparação, um
potencial de ação de fibra muscular ventricular, mostrado à
direita. Observe que o potencial da fibra nodal sinusal entre as
descargas tem negatividade de apenas -55 a -60 mV, em
comparação com -85 a -90 mV para a fibra ventricular. A causa
dessa negatividade reduzida é que as membranas celulares das
fibras sinusais são naturalmente permeáveis a íons sódio.
98
Antes de tentar explicar a ritmicidade das fibras do nodo
sinusal, recordemos primeiro, das discussões dos Caps. 5 e 9,
que, no músculo cardíaco, três tipos diferentes de canais iônicos
na membrana contribuem de forma importante para ocasionar
as alterações de voltagem do potencial de ação. São eles (1) os
canais rápidos de sódio; (2) os canais lentos de cálcio-sódio, e
(3) os canais de potássio. A abertura dos canais rápidos de
sódio por alguns décimos milésimos de segundo é responsável
pelo início muito rápido e semelhante a uma ponta do potencial
de ação observado no músculo ventricular devido ao rápido
influxo de íons sódio positivos para o interior da fibra. Em
seguida, o platô do potencial de ação ventricular é causado
principalmente pela abertura mais lenta dos canais lentos de
cálcio-sódio, que dura alguns décimos de segundo. Finalmente,
a crescente abertura de canais de potássio e a difusão de uma
grande quantidade de íons de potássio positivos para fora da fibra
fazem o potencial de membrana voltar a seu nível de repouso.
Contudo, há uma diferença na função desses canais na fibra
do nodo sinusal devido à negatividade muito menor do potencial
"de repouso" -apenas -55 mV. Nesse nível de negatividade, os
canais rápidos de sódio ficam em grande parte "inativados", o
que significa que eles foram bloqueados. A causa disto é que, em
qualquer ocasião em que o potencial de membrana permanece
menos negativo que cerca de -60 mV por mais de alguns
milissegundos, as comportas de inativação no interior da
membrana celular que fecham esses canais fecham-se e
permanecem fechadas. Por esta razão, somente os canais lentos
de cálcio-sódio podem abrir-se (ou seja, podem ficar
"ativados") e, portanto, causar o potencial de ação. O potencial
de ação tem, pois, desenvolvimento mais lento que o do
músculo ventricular e também se recupera por diminuição lenta
do potencial, em vez da recuperação abrupta que ocorre no caso
da fibra ventricular.
Auto-excitação das fibras do nodo sinusal. Os íons sódio
tendem naturalmente a vazar para dentro das fibras do nodo
sinusal por múltiplos canais na membrana, e esse influxo de
cargas positivas também causa elevação do potencial de
membrana. Assim, como é ilustrado na Fig. 10.2, o potencial "de
repouso" eleva-se gradualmente entre cada dois batimentos
cardíacos. Ao atingirem a voltagem limiar, de cerca de -40 mV,
os canais de cálcio-sódio são ativados, levando à entrada muito
rápida tanto de íons cálcio como sódio e causando, assim, o
potencial de ação. Assim sendo, é basicamente a permeabilidade
intrínseca das fibras do nodo sinusal aos íons sódio que causa
sua auto-excitação.
Por que essa permeabilidade aos íons sódio não faz as fibras
do nodo sinusal ficarem despolarizadas todo o tempo? A resposta
é que ocorrem dois eventos durante o potencial de ação. Em
primeiro lugar, os canais de cálcio-sódio são inativados (ou seja,
eles se fecham) dentro de cerca de 100 a 150 ms após sua abertura
e, segundo, mais ou menos ao mesmo tempo, abre-se um número
muito maior de canais de potássio. Por esta razão, agora o influxo
de íons cálcio e sódio pelos canais de cálcio-sódio cessa
simultaneamente, enquanto uma grande quantidade de íons
potássio positivos difunde-se para fora da célula, pondo fim,
portanto, ao potencial de ação. Além disso, os canais de potássio
permanecem abertos por mais alguns décimos de segundo,
levando um grande excesso de cargas positivas de potássio para
fora da célula, o que causa, temporariamente, considerável
excesso de negatividade dentro da fibra, isto é denominado
hiperpolarização. Essa hiperpolarização provoca inicialmente
redução do potencial de membrana em "repouso" para cerca de
-55 a -60 mV ao final do potencial de ação.
Por fim, temos de explicar por que o estado de
hiperpolarização também não é mantido indefinidamente. A
razão é que, durante os décimos de segundo subseqüentes ao fim
do potencial de ação, um número cada vez maior dos canais de
potássio começa a se fechar. Então, os íons sódio que vazam
para o interior superam novamente o fluxo para fora dos íons
potássio, o que faz o potencial "de repouso" elevar-se,
atingindo, finalmente, o nível limiar de descarga, em potencial
de cerca de -40 mV. Então, todo o processo tem início de novo:
auto-excitação, recuperação do potencial de ação,
hiperpolarização após o fim do potencial de ação, elevação do
potencial "de repouso" e, depois, mais uma vez, reexcitação, para
iniciar outro ciclo. Este processo continua indefinidamente por
toda a vida da pessoa.
VIAS INTERNODAIS E TRANSMISSÃO
IMPULSO CARDÍACO PELOS ÁTRIOS
DO
As extremidades das fibras do nodo sinusal fundem-se às
fibras musculares atriais circundantes e os potenciais de ação
que se originam no nodo sinusal dirigem-se para adiante, por
meio dessas fibras. Desse modo, o potencial de ação se propaga
por toda a massa muscular atrial, e acaba por chegar também
ao nodo A-V. A velocidade de condução no músculo atrial é
de aproximadamente 0,3 m/s. A condução é, porém, mais rápida
em vários pequenos feixes de fibras musculares atriais. Um deles,
denominado feixe interatrial anterior, passa para o átrio esquerdo
pelas paredes anteriores dos átrios e conduz o impulso cardíaco
com velocidade de aproximadamente 1 m/s. Além disso, três
outros pequenos feixes atravessam as paredes atriais e terminam
no nodo A-V, também conduzindo o impulso cardíaco com essa
velocidade rápida. Esses três pequenos feixes, ilustrados na Fig.
10.1, são denominados, respectivamente, vias internodais
anterior, média e posterior. A causa da velocidade de condução
mais rápida nesses feixes é a presença de certo número de fibras
de condução especializadas misturadas ao músculo atrial. Essas
fibras são semelhantes às fibras de Purkinje dos ventrículos, de
condução muito rápida, que são discutidas a seguir.
O NODO A -V E O RETARDO NA CONDUÇÃO DE
IMPULSOS
Fig. 10.2 Descarga rítmica de uma fibra do nodo sinusal e comparação
do potencial de ação do nodo sinusal com o de uma fibra muscular
ventricular.
Felizmente, o sistema de condução é organizado de tal forma
que o impulso cardíaco não passa dos átrios para os ventrículos
de modo demasiado rápido; isso dá tempo para os átrios lançarem
seu conteúdo nos ventrículos antes que se inicie a contração
ventricular. São principalmente o nodo A-V e suas fibras de
condução associadas que retardam essa transmissão do impulso
cardíaco dos átrios para os ventrículos.
99
O nodo A-V está localizado na parede septal posterior do
átrio direito, imediatamente atrás da válvula tricúspide e
adjacente à abertura do seio coronário, como c mostrado na
Fig. 10.1. A Fig. 10.3 mostra esquematicamente as diferentes
partes desse nodo e suas conexões com as fibras da via
internodal atrial e o feixe A-V. A figura também mostra os
intervalos aproximados em frações de segundo entre a gênese do
impulso cardíaco no nodo sinusal e seu aparecimento em
diferentes pontos do sistema nodal A-V. Note que o impulso,
após passar pela via internodal, chega ao nodo A-V
aproximadamente 0,03 após sua origem no nodo sinusal. Há,
então, um retardo adicional de 0,09 no nodo A-V propriamente
dito, antes do impulso passar à parte penetrante do feixe A-V. Um
retardo final, de mais de 0,04 s, ocorre principalmente neste feixe
A-V penetrante que é composto por múltiplos pequenos
fascículos que atravessam o tecido fibroso que separa os átrios
dos ventrículos.
Portanto, o retardo total no sistema nodal A-V e do feixe
A-V é de aproximadamente 0,13 s. Cerca de um quarto desse
tempo ocorre nas fibras de transição, fibras muito delgadas que
ligam as fibras das vias internodais atriais ao nodo A-V (ver
Fig. 103). A velocidade de condução nessas fibras é tão pequena
quanto 0,02 a 0,05 m/s (cerca de 1/12 da velocidade no músculo
cardíaco normal), o que retarda muito a entrada do impulso
no nodo A-V. Após entrar no nodo propriamente dito, a
velocidade de condução nas fibras nodais ainda é bastante baixa,
apenas 0,05 m/s, cerca de um oitavo da velocidade de condução
no músculo cardíaco normal. Essa baixa velocidade de condução
também é aproximadamente a mesma da parte penetrante do
feixe A-V.
Causa da condução lenta. A causa da condução
extremamente lenta tanto nas fibras de transição como nas
fibras nodais e nas fibras do feixe A-V penetrante é. em parte,
que seu tamanho é consideravelmente menor do que o das fibras
musculares atriais normais. Entretanto, a maior parte da
condução lenta é provavelmente causada por dois fatores
totalmente diferentes. Em primeiro lugar, todas essas fibras têm
potenciais de membrana em repouso que são muito menos
negativos que o potencial de repouso normal do restante do
músculo cardíaco.
Segundo, muito poucas junções abertas unem as fibras
sucessivas na via, de modo que há grande resistência à
condução de íons excitatórios de uma fibra para outra. Assim,
havendo tanto baixa voltagem impulsionando os íons como
grande resistência ao seu movimento, é fácil ver-se por que
cada fibra sucessiva demora a ser excitada.
TRANSMISSÃO NO SISTEMA DE PURKINJE
As fibras de Purkinje saem do nodo A-V para os ventrículos
pelo feixe A-V. Exceto pela parte inicial dessas fibras, no ponto
onde elas penetram na barreira fibrosa atrioventricular, elas têm
características funcionais bem opostas às das fibras do nodo A-V;
são fibras muito grandes, maiores ainda que as fibras musculares
normais dos ventrículos, e transmitem potenciais de ação com
velocidade de 1,5 a 4,0 m/s., velocidade cerca de 6 vezes a
verificada no músculo cardíaco habitual e 150 vezes a medida
em algumas fibras de transição. Isso possibilita a transmissão
quase que imediata do impulso cardíaco por todo o sistema
ventricular.
A transmissão muito rápida dos potenciais de ação pelas
fibras de Purkinje é considerada como sendo causada pela maior
permeabilidade das junções abertas nos discos intercalados entre
as sucessivas células cardíacas que constituem as fibras de
Purkinje. Nesses discos, os íons são facilmente transmitidos de
uma célula para a seguinte, aumentando, assim, a velocidade de
transmissão. As fibras de Purkinje também têm muito poucas
miofibrilas, o que quer dizer que elas mal se contraem durante a
transmissão de impulsos.
Condução em sentido único pelo feixe A-V. Uma
característica especial do feixe A-V é a incapacidade dos
potenciais de ação, exceto em estados anormais, fazerem o
trajeto retrógrado no feixe dos ventrículos para os átrios. Isso
impede a reentrada de impulsos cardíacos dos ventrículos para
os átrios por essa via, possibilitando apenas a condução
anterógrado dos átrios para os ventrículos.
Além disso, deve-se recordar que o músculo atrial c separado
do músculo ventricular por barreira fibrosa contínua, parte da
qual é ilustrada na Fig. 10.3. Essa barreira atua normalmente
como isolante, impedindo a passagem de impulsos cardíacos entre
os átrios e os ventrículos por qualquer outra via que não a
condução anterógrada pelo próprio feixe A-V. (Entretanto, em
raros casos, uma ponte muscular anormal atravessa, de fato, a
barreira fibrosa em outro ponto que não o feixe A-V. Nessas
condições, os impulsos cardíacos podem, então, reentrar nos
átrios a partir dos ventrículos e causar arritmia cardíaca grave.)
Distribuição das fibras de Purkinje nos ventrículos. Após
Fig. 10.3 Organização do nodo A-V. Os números representam o
intervalo de tempo a partir da origem do impulso no nodo sinusal. Os
valores foram extrapolados para os seres humanos.
passar através do tecido fibroso atrioventricular, à parte distai
do feixe A-V desce pelo septo ventricular, em direção ao ápice
cardíaco, por 5 a 15 mm, como é mostrado nas Figs. 10.1 e
10.3. O feixe se divide, então, nos ramos esquerdo e direito,
situados por sob o endocárdio dos dois lados respectivos do septo.
Cada ramo desce até o ápice do ventrículo, dividindo-se em
ramos menores que circundam cada câmara ventricular e voltam
em direção à base do coração. As fibras de Purkinje terminais
penetram cerca de um terço do trajeto pela massa muscular
e depois, tornam-se contínuas com as fibras musculares
cardíacas.
Do momento em que o impulso cardíaco chega aos ramos
até atingir as terminações das fibras de Purkinje, o tempo total
transcorrido é de apenas 0,03 s; assim sendo, uma vez tendo
entrado no sistema de Purkinje, o impulso cardíaco dissemina-se
quase que imediatamente para toda a superfície endocárdica do
músculo ventricular.
100
TRANSMISSÃO DOS IMPULSOS CARDÍACOS
NO MÚSCULO VENTRICULAR
Após alcançar as extremidades das fibras de Purkinje, o
impulso é, então, transmitido, através da massa muscular do
ventrículo, pelas próprias fibras musculares ventriculares. A
velocidade de transmissão é agora de apenas 0,3 a 0,5 m/s, um
sexto da verificada nas fibras de Purkinje.
O músculo cardíaco envolve o coração numa espiral dupla,
com septos fibrosos entre as camadas espirais; por esta razão,
o impulso cardíaco não segue necessariamente direto para fora,
rumo à superfície do coração, mas faz um ângulo em direção
à superfície segundo as direções das espirais. Devido a isto, a
transmissão da superfície endocárdica para a superfície epicárdica
do ventrículo requer até mais 0,03 s, tempo aproximadamente
igual ao necessário para a transmissão por todo o sistema de
Purkinje. Assim, o tempo total para a transmissão do impulso
cardíaco dos remos iniciais até a última das fibras musculares
ventriculares no coração normal é de cerca de 0,06 s.
RESUMO DA DIFUSÃO DO IMPULSO CARDÍACO
PELO CORAÇÃO
A Fig. 10.4 mostra, de forma resumida, a transmissão do
impulso cardíaco pelo coração humano. Os números na figura
representam os intervalos de tempo, em centésimos de segundo,
transcorridos entre a origem do impulso cardíaco no nodo sinusal
e seu aparecimento em cada ponto respectivo do coração.
Observe que o impulso difunde-se com velocidade moderada
pelos átrios, mas é retardado em mais de 0,1 na região do nodo
A-V, antes de aparecer no feixe A-V do septo ventricular. Após
ter atingido esse feixe, ele se difunde rapidamente para toda a
superfície endocárdica dos ventrículos pelas fibras de Purkinje.
Daí, o impulso novamente se difunde lentamente através do
músculo ventricular até as superfícies epicárdicas.
É extremamente importante que o leitor aprenda em detalhe
Fig. 10.4 Transmissão do impulso cardíaco pelo coração, mostrando
o tempo de aparecimento (cm frações de segundo) do impulso em
diferentes partes do coração.
O trajeto do impulso cardíaco pelo coração e o tempo de seu
aparecimento em cada parte distinta do coração, pois o
conhecimento quantitativo desse processo é essencial para a
compreensão da eletrofisiologia, que é discutida nos três capítulos
subseqüentes.
CONTROLE DA EXCITAÇÃO E CONDUÇÃO NO
CORAÇÃO
O NODO SINUSAL COMO MARCAPASSO DO
CORAÇÃO
Na discussão anterior sobre a gênese e transmissão do
impulso cardíaco pelo coração, notamos que o impulso se origina
normalmente no nodo sinusal. Entretanto, isso muitas vezes não
ocorre, em condições anormais, pois outras partes do coração
podem apresentar contrações rítmicas da mesma forma que as
fibras do nodo sinusal; isto é particularmente verdadeiro para
as fibras de Purkinje do nodo A-V.
As fibras nodais A-V, quando não estimuladas por alguma
fonte externa, descarregam com freqüência rítmica intrínseca
de 40 a 60 vezes por minuto, e as fibras de Purkinje descarregam
com freqüência entre 15 e 40 vezes por minuto. Essas freqüências
contrastam com a freqüência normal de 70 a 80 vezes por minuto
do nodo sinusal.
Portanto, a pergunta que temos de fazer é; por que é o
nodo sinusal que controla a ritmicidade do coração, e não o
nodo A-V ou as fibras de Purkinje? A resposta a isto decorre
do fato de que a freqüência de descarga do nodo sinusal é
consideravelmente maior que a do nodo A-V ou das fibras de
Purkinje. A cada descarga do nodo sinusal, seu impulso é
conduzido tanto para o nodo A-V como para as fibras de
Purkinje, descarregando suas membranas excitáveis. Em seguida,
tanto esses tecidos como o nodo sinusal recuperam-se do potencial
de ação e ficam hiperpolarizados. Contudo, o nodo sinusal perde
sua hiperpolarização muito mais rapidamente do que qualquer
um dos outros dois, emitindo novo impulso antes que qualquer
um deles possa atingir seu próprio limiar de auto-excitação. O
novo impulso descarrega novamente tanto o nodo A-V como as
fibras de Purkinje. Esse processo continua indefinidamente, com
o nodo sinusal sempre excitando esses tecidos potencialmente
auto-excitáveis antes que sua auto-excitação possa de fato
ocorrer.
O nodo sinusal controla, portanto, o batimento do coração
porque sua freqüência de descarga rítmica é maior do que a
de qualquer outra parte do coração. Portanto, o nodo sinusal
é o marcapasso normal do coração.
Marcapassos anormais - o marcapasso ectópico.
Ocasionalmente, alguma outra parte do coração apresenta
freqüência de descarga rítmica que é mais rápida que a do nodo
sinusal. Isso ocorre muitas vezes, por exemplo, no nodo A-V
ou nas fibras de Purkinje. Em qualquer desses casos, o
marcapasso cardíaco passa do nodo sinusal para o nodo A-V ou
para as fibras excitáveis de Purkinje. Em condições mais raras,
um ponto no músculo atrial ou ventricular desenvolve
excitabilidade excessiva e torna-se o marcapasso.
Um marcapasso em outro local que não o nodo sinusal é
denominado marcapasso ectópico. Obviamente, o marcapasso
ectópico ocasiona uma seqüência anormal de contrações nas
diferentes partes do coração.
Outra causa da mudança do marcapasso é o bloqueio da
transmissão de impulsos do nodo sinusal para outras partes do
coração, que ocorre mais freqüentemente no nodo A-V ou na
parte penetrante do feixe A-V a caminho dos ventrículos. Quando
ocorre bloqueio A-V, os átrios continuam a bater na freqüência
normal do ritmo do nodo sinusal, enquanto um novo marcapasso
101
se instala no sistema de Purkinje dos ventrículos e impulsiona
o músculo ventricular com nova freqüência entre 15 e 40
batimentos por minuto. Entretanto, após bloqueio súbito, o
sistema de Purkinje só começa a emitir seus impulsos rítmicos
15 a 30 s depois, porque até esse ponto ele estava
"hiperestimulado" pelos rápidos impulsos sinusais, encontrandose, por conseguinte, em estado de supressão. Durante esses 5 a
30 s, os ventrículos não bombeiam sangue algum e a pessoa
desmaia após os primeiros 4 a 5 segundos, devido à falta de
fluxo sanguíneo para o cérebro. Esse retardo da seqüência dos
batimentos cardíacos é denominado síndrome de Stokes-Adams.
Quando demasiado longo, esse intervalo pode levar à morte.
PAPEL DO SISTEMA DE PURKINJE NA CAUSA
DA CONTRAÇÃO SINCRÔNICA DO MÚSCULO
VENTRICULAR
Ficou claro pela descrição anterior do sistema de Purkinje
que o impulso cardíaco chega a quase todas as partes dos
ventrículos em intervalo de tempo muito curto, excitando a
primeira fibra muscular ventricular apenas 0,06 s antes da
excitação da última fibra muscular ventricular. Isso faz com que
todas as partes do músculo ventricular de ambos os ventrículos
comecem a se contrair quase que exatamente ao mesmo tempo.
O bombeamento efetivo pelas duas câmaras requer esse tipo
sinerônico de contração. Se o impulso cardíaco trafegasse muito
lentamente pelo músculo ventricular, grande parte da massa
ventricular iria contrair-se antes da contração do resto, caso em
que o efeito global de bombeamento ficaria bastante reduzido.
De fato, em certos tipos de cardiopatias, algumas das quais são
discutidas nos Caps. 12 e 13, há transmissão lenta e a eficácia do
bombeamento ventricular diminui, talvez. 20 a 30%.
CONTROLE DA RITMICIDADE E CONDUÇÃO
CARDÍACAS PELOS NERVOS SIMPÁTICOS E
PARASSIMPÁTICOS
O coração é suprido tanto de nervos simpáticos como
parassimpáticos, conforme ilustrado na Fig. 9.11 do capítulo
anterior. Os nervos parassimpáticos (os vagos) distribuem-se
principalmente para os nodos sinusal e A-V, em menor escala
para o músculo dos dois átrios e menos ainda para o músculo
ventricular. Os nervos simpáticos, por outro lado, distribuem-se a
todas as partes do coração, com forte representação para o
músculo ventricular, assim como para todas as outras áreas.
Efeito da estimulação parassimpática (vagal) sobre o ritmo
cardíaco e a condução cardíaca - escape ventricular. A
estimulação dos nervos parassimpáticos para o coração (os
vagos) faz com que o hormônio acetileolinu seja liberado nas
terminações vagais. Esse hormônio tem dois efeitos principais
sobre o coração. Em primeiro lugar, ele diminui a freqüência do
ritmo do nodo sinusal e, segundo, ele diminui a excitabilidade das
fibras juncionais A-V entre a musculatura atrial e o nodo A-V,
lentificando, assim, a transmissão do impulso cardíaco para os
ventrículos. Uma estimulação vagal muito forte pode fazer
cessar totalmente a contração rítmica do nodo sinusal ou
bloquear por completo a transmissão do impulso cardíaco pela
junção A-V. Em qualquer dos casos, não são mais transmitidos
impulsos rítmicos para os ventrículos. Estes param de bater por
4 a 10 segundos, mas, então, algum ponto das fibras de
Purkinje, geralmente na parte septal ventricular do feixe A-V,
desenvolve um ritmo próprio e produz contrações ventriculares
com freqüência de 15 a 40 batimentos por minuto. Esse
fenômeno é denominado escape ventricular.
Mecanismos dos efeitos vagais. A acetileolina liberada nas
terminações nervosas vagais aumenta muito a permeabilidade
da membrana das fibras ao potássio, o que possibilita o vazamento
rápido de potássio para o exterior. Isso causa negatividade
aumentada no interior das fibras, um efeito denominado
hiperpolarização, que torna o tecido excitável muito menos
excitável, como foi explicado no Cap. 5.
No nodo sinusal, o estado de hiperpolarização diminui o
potencial de membrana "em repouso" das fibras do nodo sinusal
para um nível de negatividade consideravelmente inferior ao valor
normal, um nível de até - 65 a -75 mV, em vez do nível normal
de - 55 a - 60 mV. Como conseqüência, a elevação do potencial de
membrana em repouso, ocasionada pelo vazamento de sódio,
requer um intervalo muito maior até atingir o potencial limiar
de excitação. Evidentemente, isto torna bem mais lenta a
freqüência da ritmicidade dessas fibras nodais. Assim também,
quando a estimulação vagal é suficientemente forte, é possível
fazer cessar totalmente a auto-excitação rítmica desse nodo.
No nodo A-V, o estado de hiperpolarização torna difícil
às diminutas fibras juncionais excitar as fibras nodais, pois elas
só podem gerar pequena quantidade de corrente durante o
potencial de ação. Por essa razão, o fator de segurança para a
transmissão dos impulsos cardíacos pelas fibras juncionais e até
para as fibras nodais diminui. Uma redução moderada desse
fator simplesmente retarda a condução dos impulsos, mas uma
diminuição do fator de segurança abaixo da unidade (o que
significa um nível tão baixo que o potencial de ação de uma
fibra não pode causar potencial de ação na parte subseqüente da
fibra) bloqueia totalmente a condução.
Efeito da estimulação simpática sobre o ritmo cardíaco
e a condução cardíaca. A estimulação simpática produz efeitos
basicamente opostos dos ocasionados pela estimulação vagal,
da seguinte maneira: em primeiro lugar, ela aumenta a freqüência
de descarga do nodo sinusal; depois ela aumenta a velocidade
da condução e, também, o nível de excitabilidade em todas as
partes do coração; por fim, ela aumenta muito a força de
contração de toda a musculatura cardíaca, tanto atrial como
ventricular, conforme discutido no capítulo anterior.
Em suma, a estimulação simpática aumenta a atividade
global do coração. A estimulação máxima pode quase triplicar a
freqüência dos batimentos cardíacos e até duplicar a força de
contração do coração.
Mecanismo do efeito simpático. A estimulação dos nervos
simpáticos libera o hormônio norepinefrina nas terminações
nervosas simpáticas. O mecanismo exato pelo qual esse
hormônio atua sobre as fibras musculares cardíacas ainda é algo
duvidoso, mas a opinião atual é a de que ele aumenta a
permeabilidade da membrana das fibras ao sódio e ao cálcio.
No nodo sinusal, um aumento da permeabilidade ao sódio
ocasiona um potencial de repouso mais positivo e uma elevação
mais rápida do potencial de membrana até o nível limiar de autoexcitação. ambos, evidentemente, capazes de acelerar o início da
auto-excitação, aumentando, portanto, a freqüência cardíaca.
No nodo A-V, a maior permeabilidade ao sódio torna mais
fácil ao potencial de ação excitar a parte subseqüente da fibra
de condução, diminuindo, assim, o tempo de condução dos átrios
para os ventrículos.
O aumento da permeabilidade aos íons cálcio é. pelo menos,
parcialmente responsável pelo aumento da força contrátil do
músculo cardíaco sob influência da estimulação simpática,
porque os íons cálcio têm papel muito importante no
desencadeamento do processo contrátil das miofibrilas.
REFERÊNCIAS
Akera, T., and Brody, T. M.: Myocardial rnembranea: Regulation and func tion of
the sodium pump. Annu. Rev. Physiol., 44:37.% 1982.
Brown, H. R: Electrophysiology of the einoatrial node. Phyaiol. Rev., 62:505, 1982.
102
Brutsaert, D. L., and Paulus, W. J.: Contraction and relaxation of the heart
asmuscle and pump. In Guyton, A. C, and Young, D. B. (eds.):
InternationalReview of Physiology: Cardiovascular Physiology III. Vol. 18.
Baltimore, TJniversity Park Press, 1979, p. 1. DiFranceaco, D., and Noble,
D.: A mudei of cardiac eléctrica] activity incorporating ionic pumps anil
concentration changes. Phil. Trans. R. Soe. Lond. (Biol.), 307:353, ;985.
Ellis, D.: Na-Ca exchange in cardiac tissues. Adv. Myocardiol., 5:295,1985.
Farah, A. E., et ai.: Positive inotropic agents. Annu. Rev. Pharmacol. Toxicol-, 24:275, 1985. Fozzard, H. A.: Heart: Excitation-contraction coupling.
Annu. Rev. Physol., 39:201,1977. Fozzard, H. A., et ai.: The Heart and
Cardiovascular System: Scientific Foundations. New York, Raven Presa,
1986. Geddes, L. A.: Cardiovascular Medicai Devices. New York, John
Wiley & Sons, 1984. Gilmour, R. F., Jr., and Zipes, D. P.: Slow inward
current and cardiac arrhythmias. Am. J. Cardiol., 55:89B, 1985. Glitsch, H.
G.: Electrogenic Na pumping in the heart. Annu. Rev. Physiol., 44:389,
1982. Gravanis, M. B. (ed.): Cardiovascular Pathophysiology. New York,
McGraw Hill Rook Co, 1987. Guyton, A. C, and Sattertield, J.: Factors
concerned in electrical defihrillation of the heart, particularly through the
unopened chest. Am. J. Physiol., 167:81, 1951. Herbette, L., et ai.: The
interaction of drugs with the sarcoplasmic reticulum.Annu. Rev. Pharmacol.
Toxicol., 22:413, 1982. Hondeghem, L. M., and Katzung, B. G.:
Antiarrhythmic agents: The modulated receptor mechanism of action of
sodium andcalcium channel-blocking drugs. Annu. Rev. Pharmacol.
Toxicol., 24:387, 1984. Irisawa, H.: Comparative physiology of the cardiac
pacemaker mechanism.
Physiol. Rev., 58:461, 1984. Josephson, M. E., and Singh, B. N.: Use of ca lei
um antagonists in ventricular dysfunction. Am. J. Cardiol., 55:8lB, 1985.
Langer, G. A.: Sodium-calcium exchange in the heart. Annu. Rev. Phyaiol.,
44:435, 1982.
Latorre, R., et ai.: K+ channels gated by voltage and ions. Annu. Rev. Physiol., 46:485, 1984. Lazdunski, M., and Renaud, J. F.: The action of
cardiotoxina on cardiac plasma membranes. Annu. Rev. Physiol, 44:463,
1982. Levy, M. N., and Martin, P. J.: Neural control of the heart. In Berne, R.
M., et ai. (eds.): Handbook of Physiology. Sec. 2, Vol. I. Baltimore, Williams
& Wilkins, 1979, p. 581. Levy, M. N, et ai.: Neural regulation of the heart
beat. Annu. Rev. Physiol.,
43:443, 1981. Loewenstein, W. R.: Junctional intercellular communícation:
the cell-to-cell membrane channel. Physiol. Rev., 61:829, 1981. Mazzanti,
M, and DeFeiice, L. J.: K channel kineticsduringthe spontaneoua heart beat
in embryonic chick ventricle cells. Biophys. J., 54:1139, 1988. Mazzanti, M,
and DeFeiice, L. J.: Na channel kineties duhng the spontaneous heart beat in embryonic chick ventricle cells. Biophys. J., 52:95,1987.
Mazzanti, M, and DeFeiice, L. J.: Regulation of the Na-conducting Ca channel
during the cardiac action potential. Biophys. .1, 51:115, 1987. McAnulty, J.,
and Rahimtoola, S.: Prognosis in bundle branch hlock. Annu.Rev. Med,
32:499, 1981. McDonald, T. F.: The slow inward calcium current in the heart.
Annu. Rev. Physiol., 44:425, 1982. Meijler, F. L.: Atrioventricular
conduetíon versus heart size from mouse to whale. J Am. Coll. Cardiol.,
5:363, 1985. Meijler, F. L., and Janse, M. J.: Morphology and
eleçtrophysiology of the mammalian atrioventricular node. Physiol. Rev.,
68:608, 1988. Orrego F.: Calcium and the mechanism of action of digital is.
Gen. Pharmacol, 15:273, 1984. Reuter, H.: Ion channels in cardiac cell
membranes. Annu. Rev. Physiol., 44:473, 1984. Sheridan, J. D.,
andAtkinson, M. M.: Physiological roles ofpermeablejunctions: Some
possibilities. Annu. Rev. Physiol., 47:337,1985. Spear, J. F., and Moore, E.
N.: Mechanisms of cardiac arrhythmias. Annu Rev. Physiol., 44:485, 1982.
Sperelakis, N.: Hormonal and neurotransmitter regulation of Ca in
Huxthrough voltage-dependent slow channels in cardiac muscle
membrane.
Membr. Biochem, 5:131, 1984. Vasselle, M.: Electrogenesis of the plateau
and pacemaker potential. Annu.Rev. Physioi- 41:425, 1979. Verrier, R. L.,
and Lown, B.: Behavioral stress and cardiac arrhythmias.Annu. Rev.
Physiol, 46:155, 1984.
(Ver também o Cap. 9.)
103
CAPÍTULO 11
O Eletrocardiograma Normal
À medida que o impulso cardíaco se propaga pelo coração, correntes
elétricas se difundem para os tecidos que circundam o coração e uma
pequena proporção delas percorre todo o trajeto até a superfície do
corpo. Quando são colocados eletródios na pele em lados opostos do
coração, os potenciais elétricos gerados por essas correntes podem ser
registrados; o registro é conhecido como eletrocardiograma. Um
eletrocardiograma normal com dois batimentos cardíacos é ilustrado na
Fig. 11.1.
CARACTERÍSTICAS DO ELETROCARDIOGRAMA
NORMAL
O eletrocardiograma normal (Fig. 11.1) é composto pela onda P,
pelo "complexo QRS" e pela onda T. O complexo QRS é com freqüência
constituído por três ondas distintas, a onda Q, a onda R e a onda S.
A onda P é causada por potenciais elétricos gerados quando os
átrios se despolarizam antes da contração. O complexo QRS é causado
por potenciais gerados quando os ventrículos se despolarizam antes da
contração, ou seja, quando a onda de despolarização se difunde pelos
ventrículos. Tanto a onda P como os componentes do complexo QRS.
portanto, são ondas de despolarização.
A onda T é causada por potenciais gerados enquanto os ventrículos
se recuperam do estado de despolarização. Esse processo no músculo
ventricular ocorre 0,25 a 0,35 s após a despolarização, sendo esta onda
conhecida como onda de repolarização.
O eletrocardiograma é, pois, constituído tanto por ondas de
despolarização como de repolarização. Os princípios da despolarização e
da repolarização são discutidos no Cap. 5. Entretanto, a distinção
entre as ondas de despolarização e as ondas de repolarização é tão
importante que precisa ser melhor esclarecida, da maneira que se segue.
Fig. 1 1. 1 O eletrocardiograma normal.
ambos os eletródios encontram-se agora em áreas com igual negatividade.
A onda completa é uma onda de despolarização porque decorre da
propagação da despolarização por toda a extensão da fibra muscular.
A Fig. 11.2C mostra o processo de repolarização na fibra muscular,
que já chegou à metade da fibra, da esquerda para a direita. Nesse
ONDAS DE DESPOLARIZAÇÃO VERSUS ONDAS DE
REPOLARIZAÇÃO
A Fig. 11.2 apresenta uma fibra muscular em quatro diferentes
estágios de despolarização e repolarização. Durante o processo de
despolarização, o potencial negativo normal no interior da fibra é
perdido e o potencial de membrana até se inverte; ou seja, ele fica
ligeiramente positivo internamente e negativo externamente.
Na Fig. 11.2A, o processo de despolarização, representado pelas
cargas vermelhas positivas do lado interno e as negativas do lado de
fora, está indo da esquerda para a direita e a primeira metade da fibra
já se despolarizou, enquanto a metade restante ainda está polarizada.
Por esta razão, o eletródio esquerdo sobre a fibra ainda está em área
de negatividade, enquanto o eletródio direito encontra-se numa área
de positividade; isto faz o medidor ter um registro positivo. À direita
da fibra muscular é mostrado o registro do potencial entre os eletródios.
captado por um aparelho de registro de alta velocidade nesse estágio
específico da despolarização. Note que, quando a despolarização chega
à marca da metade do caminho, o registro se eleva ao valor máximo.
Na Fig. 11.2B, a despolarização já se estendeu por toda a fibra
muscular c o registro da direita já retornou ao valor basal zero, porque
Fig. 11.2 Registro da onda de despolarização e da onda de repolarização
de uma fibra muscular cardíaca.
104
ponto, o eletródio da esquerda encontra-se em área de positividade,
enquanto o da direita está em área de negatividade. Isto é o contrário
da polaridade na Fig. 11.2A. Por conseguinte, o registro, conforme é
mostrado à direita, fica negativo.
Na Fig. 11.2D, a fibra muscular já se repolarizou inteiramente e
ambos os eletródios encontram-se em áreas de positividade, de modo
que não é registrado qualquer potencial entre eles. Assim sendo, no
registro à direita, o potencial volta novamente ao nível zero. Esta onda
negativa completada é uma onda de repolarização porque decorre da
propagação do processo de repolarização pela fibra muscular.
Relação do potencial de ação monofásico do músculo cardíaco
para com as ondas QRS e T. O potencial de ação monofásico do
músculo ventricular, discutido no capítulo anterior, dura normalmente
entre 0,25 e 0,35 s. A parte superior da Fig. 11.3 mostra um potencial
de ação monofásico registrado por um microeletródio inserido dentro de
fibra muscular ventricular única. A deflexão para cima deste potencial
de ação é ocasionada pela despolarização, e seu retorno ao nível
basal é causado pela repolarização.
Note, na parte inferior da figura, o registro simultâneo do
eletrocardiograma desse mesmo ventrículo, que mostra a onda QRS
aparecendo ao início do potencial de ação monofásico e a onda T
aparecendo ao final do mesmo. Observe, em especial, que absolutamente
nenhum potencial é registrado no eletrocardiograma quando o músculo
ventricular está inteiramente polarizado ou totalmente despolarizado.
Somente quando o músculo está parcialmente polarizado ou
parcialmente despolarizado é que correntes fluem de uma para outra
parte dos ventrículos e, portanto, também para a superfície do corpo para
produzir o eletrocardiograma.
RELAÇÃO
ENTRE
A
CONTRAÇÃO
ATRIAL
E
VENTRICULAR E AS ONDAS DO ELETROCARDIOGRAMA
Antes que possa ocorrer a contração muscular, a despolarização
tem de se propagar pelo músculo para dar início aos processos químicos
da contração. A onda P ocorre, portanto, imediatamente antes do início
da contração dos átrios e a onda QRS imediatamente antes do início
da contração dos ventrículos. Os ventrículos permanecem contraídos por
até alguns milissegundos após ter havido a repolarização, ou seja, até
depois do final da onda T.
A onda de repolarização ventricular é a onda T do eletrocardiograma
normal. Comumente, o músculo ventricular começa a repolarizar-se em
algumas fibras aproximadamente 0,20 s após o início da onda de
despolarização, mas, em muitas outras, somente após 0,35 s. O processo
de repolarização estende-se, pois, por longo período, cerca de 0,15 s.
Por esta razão, a onda T no eletrocardiograma normal é com
freqüência uma onda prolongada, mas sua voltagem é
consideravelmente menor que a do complexo QRS, em parte devido à
sua longa duração.
Os átrios se repolarizam aproximadamente 0,15 a 0,20 s após a
onda P. Entretanto, isso ocorre exatamente no momento em que a onda
QRS está sendo registrada no eletrocardiograma. Em vista disso, a onda
de repolarização atrial. conhecida como onda T atrial, é em geral
totalmente obscurecida pela onda QRS, muito maior. Por esta razão, a
onda T atrial só muito raramente é observada ao eletrocardiograma.
CALIBRAÇÃO DA VOLTAGEM E DO TEMPO
NO ELETROCARDIOGRAMA
Todos os registros eletrocardiográficos são feitos com as linhas
apropriadas de calibragem no papel de registro. Essas linhas ou já são
marcadas no papel, como ocorre quando é utilizado um aparelho de
registro a tinta, ou são registradas no papel ao mesmo tempo que o
eletrocardiograma é registrado, como é o caso dos tipos fotográficos de
eletrocardiógrafos.
Como é mostrado no Fig. 11.1, as linhas horizontais de calibração
são dispostas de tal forma que 10 pequenas divisões para cima ou para
baixo, no eletrocardiograma-padrão, representam 1 milivolt (mV), com
a positividade na direção ascendente e a negatividade na direção
descendente.
As linhas verticais no eletrocardiograma são linhas para a calibração
do tempo. Cada 2,5 cm na direção horizontal equivalem a 1 segundo,
sendo este segmento, por sua vez, dividido geralmente em cinco partes
por linhas verticais escuras; os intervalos entre essas linhas representam
0,20 s. Esses intervalos são. então, divididos em cinco intervalos menores
por linhas finas, e cada um deles representa 0,04 s.
Voltagens normais no eletrocardiograma. A voltagem das ondas no
eletrocardiograma normal depende da maneira pela qual os eletródios
são aplicados à superfície do corpo. Quando um eletródio é colocado
diretamente sobre o coração e o segundo sobre outra parte do corpo,
a voltagem do complexo QRS pode ser de até 3 a 4 mV. Até mesmo
esta voltagem é muito pequena, em comparação com o potencial de
ação monofásico de 110 mV, registrado diretamente na membrana do
músculo cardíaco. Ao serem registrados eletrocardiogramas, a partir
de eletródios nos dois braços ou em um braço e numa perna, a voltagem
do complexo QRS 6 geralmente de aproximadamente 1 mV da parte
superior da onda R até a parte inferior da onda S; a voltagem da onda
P fica entre 0,1 e 0,3 mV; e a da onda T, entre 0,2 e 0,3 mV.
O intervalo P-Q ou P-R. O tempo que transcorre do início da
onda P ao início da onda QRS é o intervalo entre o início da contração
do átrio e o início da contração ventricular. Esse período é denominado
intervalo P-Q. O intervalo P-Q normal é de aproximadamente 0,16 s.
Este intervalo é ocasionalmente também designado como intervalo P-R,
porque a onda Q muitas vezes está ausente.
O intervalo Q-T. A contração do ventrículo dura quase do início
da onda Q ao fim da onda T. Este intervalo é denominado intervalo
Q-T, sendo habitualmente de 0,35 s.
A freqüência do coração determinada eletrocardiograficamente. A
freqüência dos batimentos cardíacos pode ser facilmente determinada
eletrocardiograficamente, porque o intervalo temporal entre dois
batimentos sucessivos é a recíproca da freqüência cardíaca. Quando o
intervalo entre dois batimentos, determinado pelas linhas de calibragem
do tempo, é de 1 s, a freqüência cardíaca é de 60 batimentos por
minuto. O intervalo normal entre dois complexos QRS sucessivos é de
aproximadamente 0,83 s. Isto corresponde a uma freqüência cardíaca de
60/0,83 vezes por minuto, ou 72 batimentos por minuto.
MÉTODOS
PARA
O
ELETROCARDIOGRAMAS
REGISTRO
DE
As correntes elétricas geradas pelo músculo cardíaco, durante cada
batimento cardíaco, por vezes apresentam alterações de potenciais e
polaridade em menos de 0,01 s. É essencial, portanto, que qualquer
aparelho para registro de eletrocardiogramas seja capaz de responder
rapidamente a essas alterações dos potenciais elétricos. Em geral, dois
tipos diferentes de aparelhos de registro são utilizados para este fim,
da seguinte forma:
O APARELHO DE PENA
Fig 11.3 Acima: Potencial de ação monofásico de uma fibra muscular
ventricular durante a função cardíaca normal, mostrando a
despolarização rápida e, depois, a repolarização ocorrendo
lentamente durante o estágio de platô, porém muito rapidamente
próximo do final. Abaixo: Eletrocardiograma registrado
simultaneamente.
Muitos aparelhos eletrocardiográficos clínicos modernos empregam
registrador de inscrição direta que faz o registro escrito do
eletrocardiograma diretamente por pena inscritora em folha de papel
móvel.
105
A pena é freqüentemente um tubo fino ligado a um tinteiro em uma
de suas extremidades, com sua extremidade de registro ligada a um
potente sistema eletromagnético que é capaz de mover a pena para
cima e para baixo com alta velocidade. À medida que o papel se move
para a frente, a pena registra o eletrocardiograma. O movimento da
pena é, por sua vez, controlado por meio de amplificadores eletrônicos
apropriados, ligados aos eletródios eletrocardiográficos no paciente.
Outros sistemas de registradores inscritores utilizam um papel
especial que não requer tinta no estilete de registro. Um desses papéis
fica negro ao ser exposto ao calor; o estilete propriamente dito é muito
aquecido pelas correntes elétricas que fluem por sua extremidade. Outro
tipo enegrece quando correntes elétricas fluem da extremidade da pena
e através do papel para um eletródio atrás do mesmo. Isso produz uma
linha preta em todos os pontos do papel tocados pela agulha.
FLUXO DE CORRENTE EM TORNO
CORAÇÃO DURANTE 0 CICLO CARDÍACO
DO
REGISTRO DE POTENCIAIS ELÉTRICOS A PARTIR DE UMA
MASSA
DE
MÚSCULO
CARDÍACO
SINCICIAL
PARCIALMENTE DESPOLARIZADO
A Fig. 11.4 mostra uma massa sincicial de músculo cardíaco que
foi estimulada em seu ponto mais central. Antes da estimulação, as
partes externas de todas as células musculares estavam positivas e as
internas, negativas. Entretanto, por razões apresentadas no Cap. 5, na
discussão dos potenciais de membrana, logo que uma área do sincício
cardíaco fica despolarizada, cargas negativas vazam para a parte externa
das fibras musculares despolarizadas, tornando eletronegativa esta
superfície representada pelos sinais negativos na figura, relativamente à
superfície restante do coração que ainda se encontra polarizada da
maneira normal, representada pelos sinais positivos. Por esta razão, um
medidor ligado por seu terminal negativo à área de despolarização e
por seu terminal positivo a uma das áreas ainda polarizadas, ilustradas à
direita da figura, registra positivamente.
Duas outras possíveis colocações de eletródios e leituras do medidor
são também mostrados na Fig. 11.4. Elas devem ser estudadas
cuidadosamente e o leitor deve ser capaz de explicar as causas das
respectivas leituras do medidor. Obviamente, como o processo de
despolarização espalha-se em todas as direções pelo coração, as
diferenças de potencial mostradas na figura duram apenas alguns
milissegundos e as medidas da voltagem real só podem ser feitas por
um aparelho de registro com alta velocidade.
FLUXO DE CORRENTES ELÉTRICAS EM TORNO DO
CORAÇÃO NO TÓRAX
A Fig. 11.5 mostra o músculo ventricular no interior do tórax. Até
mesmo os pulmões, embora cheios principalmente de ar, conduzem
eletricidade em grau surpreendente e os líquidos de outros tecidos
circundando o coração conduzem eletricidade com facilidade ainda
maior.
Fig. 11.5 Fluxo de corrente no tórax em torno de um coração
parcialmente despolarizado.
O coração encontra-se, pois, suspenso de fato em meio condutor. Quando
uma parte dos ventrículos fica eletronegativa relativamente ao restante,
uma corrente elétrica flui da área despolarizada para a polarizada por
grandes vias, como é mostrado na figura.
Deve-se recordar da discussão do sistema de Purkinje no Cap. 10
que o impulso cardíaco chega inicialmente aos ventrículos pelo septo
e, logo depois, atinge as superfícies endocárdicas do resto dos ventrículos,
como é mostrado pelas áreas coloridas e os sinais negativos na Fig.
11.5. Isto gera uma eletronegatividade no interior dos ventrículos e eletropositividade nas paredes externas dos ventrículos e a corrente flui através
dos líquidos que circundam os ventrículos seguindo trajetórias elípticas,
como é mostrado na figura. Caso se faça uma média algébrica de todas
as linhas de fluxo de corrente (as linhas elípticas), verifica-se que o
fluxo médio de corrente, na direção do negativo para o positivo, é da
base para o ápice do coração. Durante a maior parte do restante do
processo de despolarização, a corrente continua a fluir nesta direção.
enquanto a despolarização se propaga da superfície endocárdica para
fora através da massa ventricular. Contudo, imediatamente antes da
despolarização ter completado seu trajeto pelos ventrículos, a direção
do fluxo de corrente se inverte por cerca de 1/100 segundo, fluindo,
então, do ápice para a base, porque a última parte do coração a se
despolarizar são as paredes externas dos ventrículos próximas à base
do coração.
Assim, no coração normal, acorrente flui principalmente na direção
da base para o ápice durante quase todo o ciclo de despolarização,
exceto bem ao fim deste. Quando um medidor é ligado à superfície
do corpo, portanto, como é mostrado na Fig. 11.5, o eletródio mais
próximo da base vai ser positivo e o registrador vai apresentar onda
positiva no eletrocardiograma.
DERIVAÇÕES ELETROCARDIOGRÁFICAS
AS TRÊS DERIVAÇÕES BIPOLARES DOS MEMBROS
A Fig. 11.6 mostra as conexões elétricas entre os membros e o
eletrocardiógrafo para o registro de eletrocardiogramas pelas chamadas
derivações bipolares-padrão dos membros. O termo "bipolar" significa
que o eletrocardiograma é registrado a partir de dois eletródios
específicos sobre o corpo, neste caso, nos membros. Assim, uma
“derivação” não é um fio individual ligado ao corpo, mas uma
combinação de dois fios e seus eletródios, formando um circuito
completo com o eletrocardiógrafo. O eletrocardiógrafo, em cada caso, é
ilustrado por medidores mecânicos no diagrama, embora o aparelho seja,
de fato, um registrador de alta velocidade em papel móvel.
Fig. 11.4 Potenciais instantâneos desenvolvidos na superfície
de uma massa muscular cardíaca que foi despolarizada em seu
centro
Derivação I. No registro da derivação I dos membros o terminal
negativo do eletrocardiógrafo é ligado ao braço direito e o terminal
positivo, ao braço esquerdo. Assim, quando o ponto no tórax em que o
braço direito se fixa ao tórax apresenta-se eletronegativo
relativamente ao ponto de união do braço esquerdo, o eletrocardiógrafo
registra positivamente - ou seja, acima da linha de voltagem zero no
eletrocardiograma. Quando ocorre o contrário, o eletrocardiograma
registra abaixo da linha.
106
Fig. 11.7 Eletrocardiogramas normais registrados a partir das três
derivações eletrocardiográficas-padrão.
Fig. 11.6 Arranjo convencional dos eletródios para o registro das
derivações eletrocardiográficas-padrão. O triângulo de Einthoven está
superposto ao tórax.
Derivação II. No registro da derivação II dos membros, o terminal
negativo do eletrocardiógrafo está ligado ao braço direito e o terminal
positivo, à perna esquerda. Assim, quando o braço direito apresenta-se
negativo em relação à perna esquerda, o eletrocardiógrafo registra
positivamente.
Derivação III. No registro da derivação III dos membros, o terminal
negativo do eletrocardiógrafo está ligado ao braço esquerdo, e o terminal
positivo, à perna esquerda. Isto quer dizer que o eletrocardiógrafo registra
positivamente quando o braço esquerdo encontra-se negativo
relativamente à perna esquerda.
Triângulo de Einthoven. Na Fig. 11.6, um triângulo denominado
triângulo de Einthoven está desenhado em torno da área do coração.
Este é um meio esquemático de mostrar que os dois braços e a perna
formam os vértices de um triângulo circundando o coração. Os dois
vértices na parte superior do triângulo representam os pontos em que
os dois braços se ligam eletricamente aos líquidos em volta do coração
c o vértice inferior é o ponto em que a perna se liga aos líquidos.
Lei de Einthoven. A lei de Eintohoven diz, simplesmente, que,
se a qualquer instante forem conhecidos os potenciais elétricos de duas
quaisquer das três derivações eletrocardiográficas bipolares dos
membros, a terceira poderá ser determinada matematicamente a partir
das duas conhecidas pela simples soma das mesmas (note, porém, que
os sinais negativos e positivos das diferentes derivações têm de ser
observados ao se fazer essa soma).
Suponhamos, por exemplo, conforme mostra a Fig. 11.6, que,
momentaneamente, o braço direito é 0,2 mV negativo em relação ao
potencial médio do corpo, o braço esquerdo é 0,3 mV positivo e a
perna esquerda é 1,0 mV positiva. Observando-se os medidores na
figura, pode-se ver que a derivação I registra diferença de potencial
de 0,5 mV, porque esta é a diferença entre os - 0,2 mV no braço
direito e os + 0,3 mV no braço esquerdo. Da mesma forma, a
derivação III registra potencial positivo de 0,7 mV e a derivação II
registra potencial
positivo de 1,2 mV, porque essas são as diferenças de potencial
instantâneas entre os respectivos pares de membros.
Note. agora, que a soma das voltagens nas derivações I e III equivale
à voltagem da derivação II. Ou seja, 0,5 mais 0,7 é igual a 1.2.
Matematicamente, este princípio, denominado Lei de Binthoven, é
verdadeiro para qualquer instante em que o eletrocardiograma esteja
sendo registrado.
Eletrocardiogramas normais registrados a partir das três derivações
bipolares dos membros. A Fig. 11.7 mostra registros simultâneos do
eletrocardiograma nas derivações I, II e III. Essa figura deixa claro
que os eletrocardiogramas nessas três derivações são muito semelhantes
entre si, pois todos eles registram ondas P positivas e ondas T positivas
e a parte principal do complexo QRS também é positiva em cada
eletrocardiograma.
Pela análise dos três eletrocardiogramas pode-se mostrar, com
medidas cuidadosas, que, a qualquer instante, a soma dos potenciais
nas derivações I e III é igual ao potencial na derivação II, ilustrando,
assim, a validade da lei de Einthoven.
Como os registros de todas as derivações bipolares são semelhantes
uns aos outros, não importa muito qual é a derivação registrada quando
se quer diagnosticar as diferentes arritmias cardíacas, pois o diagnóstico
das arritmias depende principalmente das relações temporais entre as
diferentes ondas do ciclo cardíaco. Por outro lado, quando se deseja
diagnosticar lesões do músculo ventricular ou atrial ou do sistema de
condução, é de fato muito importante quais são as derivações registradas,
pois as anormalidades do músculo cardíaco alteram acentuadamente os
padrões eletrocardiográficos em algumas derivações e podem, apesar
disto, não afetar outras derivações.
A interpretação eletrocardiográfica desses dois tipos de condições
- miopatias cardíacas e arritmias cardíacas - é discutida separadamente nos
dois capítulos que se seguem.
DERIVAÇÕES TORÁCICAS (DERIVAÇÕES PRECORDIAIS)
Muitas vezes, os eletrocardiogramas são registrados com um
eletródio colocado na superfície anterior do tórax por sobre o
coração em um dos seis pontos vermelhos distintos da Fig. 11 .S. Esse
eletródio liga-se ao terminal positivo do eletrocardiógrafo, enquanto o
eletródio negativo, denominado eletródio indiferente, é normalmente
ligado, por resistências elétricas. ao braço direito, braço esquerdo e
perna esquerda, todos ao mesmo tempo, como também é mostrado na
figura. Geralmente, seis derivações torácicas - padrão diferentes são
registradas a partir da parede anterior do tórax, sendo o eletródio
torácico colocado, respectivamente, nos seis pontos ilustrados no
diagrama. Os diferentes registros obtidos pelo método ilustrado na Fig.
11.8 são conhecidos como derivações V2+ V3+ V4+ V5+ , e V6+
107
Fig. 11.10 Eletrocardiogramas normais registrados a partir das três
derivações unipolares aumentadas dos membros
Nas derivações V, e V2, os registros de QRS do coração normal
são em grande parte negativos porque, como é mostrado na Fig. 11.8,
o eletródio torácico nessas derivações está mais próximo da base que
do ápice do coração, que é a direção da eletronegatividade durante
a maior parte do processo de despolarização. Por outro lado, os
complexos QRS nas derivações V4, V, r Vft são, em grande parte,
positivos, porque o eletródio torácico nessas derivações está mais
próximo do ápice, que é a direção da eletropositividade durante a maior
parte da despolarização.
Fig. 11.8 Conexões do corpo ao eletrocardiógrafo para o registro das
derivações precordiais.
DERIVAÇÕES UNIPOLARES AUMENTADAS
DOS MEMBROS
Outro sistema de derivações em uso geral é o da derivação unipolar
aumentada dos membros. Nesse tipo de registro, dois dos membros
são ligados, por resistências elétricas, ao terminal negativo do
eletrocardiógrafo, enquanto o terceiro membro está ligado ao terminal
positivo. Quando o terminal positivo está no braço direito, a derivação é
conhecida como a derivação AvR ; quando no braço esquerdo, derivação
AvL; e quando na perna esquerda, derivação aVF.
Registros normais das derivações unipolares aumentadas dos
membros são mostrados na Fig. 11.10. Eles são todos semelhantes aos
registros das derivações-padrão dos membros, exceto que o registro da
derivação aVR está invertido.
REFERÊNCIAS
Ver referências do Cap. 13.
Fig. 11.9 Eletrocardiogramas normais registrados a partir das seis
derivações precordiais.
A Fig. 11.9 apresenta os eletrocardiogramas do coração normal
registrados a partir dessas seis derivações torácicas-padrão. Como as
superfícies cardíacas estão próximas à parede torácica, cada derivação
precordial! registra principalmente o potencial elétrico da musculatura
cardíaca imediatamente abaixo do eletródio. Por conseguinte,
anormalidades relativamente pequenas dos ventrículos, sobretudo na
parede ventricular anterior, causam muitas vezes alterações acentuadas
nos eletrocardiogramas registrados a partir das derivações torácicas.
108
CAPÍTULO 12
Interpretação Eletrocardiográfica das Anormalidades
Coronárias e do Músculo Cardíaco: Análise Vetorial
A discussão da transmissão de impulsos pelo coração, no Cap. 10,
deixou claro que qualquer alteração do padrão dessa transmissão pode
causar potenciais elétricos anormais em torno do coração, e, por
conseguinte, alterar a forma das ondas do eletrocardiograma. Por esta
razão, quase todas as anormalidades graves do músculo cardíaco podem
ser detectadas pela análise do contorno das diferentes ondas nas
diferentes derivações eletrocardiográficas. Este é o tema deste capítulo.
PRINCÍPIOS DA ANÁLISE
ELETROCARDIOGRAMAS
VETORIAL
DOS
USO DE VETORES PARA REPRESENTAR POTENCIAIS
ELÉTRICOS
Antes que se possa saber como as anormalidades cardíacas afetam
o contorno das ondas no eletrocardiograma, devemos, inicialmente,
familiarizar-nos com o conceito de vetores e a análise vetorial
aplicados aos potenciais elétricos do coração e em seu redor.
Em diversas ocasiões, no capítulo anterior, foi ressaltado que as
correntes fluem no coração numa direção específica em determinado
momento do ciclo cardíaco. Um vetor é uma seta apontada para a direção
do potencial elétrico gerado pelo fluxo de corrente, com a ponta da seta
voltada para a direção positiva. Assim também, por convenção, a
seta é desenhada em tamanho proporcional à voltagem do potencial.
O vetor "resultante" no coração em um momento qualquer. A Fig.
12.1 mostra, através da área sombreada e dos sinais negativos, a
despolarização do septo ventricular e de partes das paredes endocárdicas
laterais dos dois ventrículos. As correntes elétricas fluem entre essas áreas
despolarizadas, no interior do coração, para as áreas nãodespolarizadas, no lado externo do coração, como indicam as setas
elípticas. As correntes também fluem pelo interior das câmaras
cardíacas, diretamente das áreas despolarizadas para as áreas
polarizadas. Embora pequena quantidade de corrente flua para cima
dentro do coração, uma quantidade consideravelmente maior flui para
baixo, por fora dos ventrículos, em direção ao ápice. Por esta razão, o
vetor somado do potencial gerado nesse momento específico,
denominado vetor instantâneo médio, é representando como passando
pelo centro dos ventrículos, na direção da base para o ápice do
coração. Além disso, como essas correntes são quantitativamente
consideráveis, o potencial é grande, de modo que o vetor é relativamente
longo.
DENOTANDO A DIREÇÃO DE UM VETOR EM
TERMOS DE GRAUS
Quando um vetor é horizontal e dirigido para o lado esquerdo do
indivíduo, diz-se que ele se estende na direção de 0 grau, como é mostrado
na Fig. 12.2. A partir deste ponto de referência zero, a escala de vetores
gira no sentido horário; quando se estende de cima para baixo, o vetor
tem direção de 90°; quando vai da esquerda para a direita do indivíduo,
o vetor Tem direção de 180° e quando se estende para cima, tem direção
de -90 ou +270°.
No coração normal, a direção média do vetor do coração durante
a difusão da onda de despolarização pelos ventrículos, denominado vetor
médio do QRS, é de aproximadamente 59°, o que é ilustrado pelo vetor
A, que passa pelo centro da Fig. 12.2, na direção de 59º. Isto indica
que, durante a maior parte da onda de despolarização, o ápice do coração
permanece positivo relativamente à base, como é discutido mais adiante
no capítulo.
“EIXO” DE CADA UMA DAS DERIVAÇÕES BIPOLARES
E UNIPOLARES DOS MEMBROS
No capítulo anterior foram descritas as três derivações bipolares
e as três derivações unipolares dos membros. Cada derivação é, na
verdade, um par de eletródios ligados ao corpo, nos lados opostos do
coração, e a direção do eletródio negativo para o positivo é
denominada eixo da derivação. A derivação I é registrada a partir de dois
eletródios colocados, respectivamente, nos dois braços. Como os
eletródios estão na direção horizontal, com o eletródio positivo para
a esquerda, o eixo da derivação I é de 0".
Ao registrar-se a derivação II, os eletródios são colocados no braço
direito e na perna esquerda. O braço direito liga-se ao tronco no canto
superior direito e a perna esquerda, ao canto inferior esquerdo. Por
esta razão, a direção desta derivação é de aproximadamente 60º.
Por uma análise semelhante, pode-se ver que a derivação III tem
seu eixo com aproximadamente 120º; a derivação AvR, 210°; AvF, 90°; e
aVL, -30º. A direção do eixo de todas essas diferentes derivações é
mostrada no diagrama da Fig. 12.3, que é conhecido como sistema
hexagonal de referência. As polaridades dos eletródios são representadas
pelos sinais de mais e menos. O leitor tem de aprender esses eixos e
suas polaridades, especialmente no que concerne às derivações bipolares
dos membros I, II e III, para compreender o restante deste capítulo.
ANÁLISE VETORIAL DOS POTENCIAIS REGISTRADOS
NAS DIFERENTES DERIVAÇÕES
Agora que já discutimos as convenções para a representação de
potenciais no coração por meio de vetores e segundo os eixos das
derivações, torna-se possível reunir-se os dois para determinar o
potencial que vai ser registrado em cada derivação para um dado vetor
no coração.
A Fig. 12.4 mostra um coração parcialmente despolarizado; o vetor
109
Fig. 12.1 Um vetor médio através do coração parcialmente
despolarizado.
A representa a direção instantânea média do fluxo de corrente no coração e
seu potencial. Neste caso, a direção do potencial é de 55º e a voltagem do
potencial vai ser considerada como sendo de 2 mV. Pela base do vetor
A é traçado o eixo da derivação I na direção 0". Para determinar-se à
parte da voltagem do vetor A que vai ser registrada na derivação I,
traça-se uma perpendicular do ponto do vetor A até o eixo da derivação I, e
o chamado vetor projetado (B) é traçado ao longo do eixo. A ponta deste
vetor aponta para a extremidade positiva do eixo da derivação I, o que
indica que o registro que está sendo momentaneamente obtido na
derivação I do eletrocardiograma vai ser positivo. A voltagem registrada
vai ser igual ao comprimento de B dividido pelo de A vezes 2 mV, ou
aproximadamente 1 mV.
A Fig. 12.5 apresenta outro exemplo de análise vetorial. Neste
exemplo, o vetor A representa o potencial elétrico em determinado
momento da despolarização ventricular em outro coração, no qual o lado
esquerdo fica despolarizado pouco mais rapidamente que o direito.
Neste caso, o vetor tem a direção de 100" e a voltagem é novamente
de 2 mV. Para determinar o potencial registrado de fato na derivação I,
traçamos uma perpendicular ate o eixo da derivação I e encontramos o
vetor projetado B. O vetor B é muito curto e, desta vez, está na
direção negativa, indicando que, nesse momento específico, o registro na
derivação I vai ser negativo (abaixo da linha zero) e a voltagem
registrada vai ser pequena. Esta figura mostra que, quando o vetor do
coração está em direção quase perpendicular ao eixo da derivação, a
voltagem
Fig. 12.3 Eixos das três derivações bipolares e das três derivações
unipolares.
registrada no eletrocardiograma nessa derivação é muito baixa. Por outro
lado, quando o vetor do coração tem quase que o mesmo eixo da derivação,
praticamente toda a voltagem do vetor vai ser registrada.
Análise vetorial de potenciais nas três derivações bipolares - padrão
dos membros. Na Fig. 12.6, o vetor A representa o potencial elétrico
instantâneo de um ventrículo parcialmente despolarizado. Para se
determinar o potencial registrado nesse momento no eletrocardiograma
de cada uma das três derivações bipolares-padrão dos membros, são
traçadas perpendiculares até todas as linhas que representam as diferentes
derivações, como é mostrado na figura. O vetor projetado B mostra o
potencial registrado, naquele momento, na derivação I; o vetor projetado
C mostra o potencial na derivação II; e o vetor projetado D mostra o
potencial na derivação III. Em cada um deles, o registro
eletrocardiográfico é positivo - ou seja, acima da linha zero - porque os
vetores projetados apontam para direções positivas ao longo do eixo de
todas as derivações. O potencial na derivação I é aproximadamente
metade do potencial real no coração, representado pelo vetor A; na
derivação II, ele é quase exatamente igual ao potencial no coração; e na
derivação III ele é cerca de um terço do potencial no coração.
Pode-se fazer uma análise idêntica para determinar os potenciais
nas derivações aumentadas dos membros, exceto que os eixos respectivos
dessas derivações (ver Fig. 12.3) são utilizados em lugar dos eixos das
derivações bipolares-padrão dos membros usadas na Fig. 12.6.
ANÁLISE VETORÍAL DO ELETROCARDIOGRAMA
NORMAL
VETORES
QUE
OCORREM
DURANTE
A
DESPOLARIZAÇÃO DOS VENTRÍCULOS - O COMPLEXO
QRS
Quando o impulso cardíaco penetra nos ventrículos pelo feixe A-V,
a primeira parte dos ventrículos a se despolarizar é a superfície
endocárdica esquerda do septo. Essa despolarização se difunde
rapidamente.
Fig. 12.2 Vetores traçados para representar as direções de potenciais
em vários corações diferentes.
Fig. 12.4 Determinação de um vetor projetado B ao longo do eixo da
derivação I quando o vetor A representa o potencial instantâneo nos
ventrículos.
110
Fig. 12.5 Determinação do vetor projetado B ao longo do eixo da
derivação I quando o vetor A representa o potencial instantâneo nos
ventrículos.
envolvendo ambas as superfícies endocárdicas do septo, o que é ilustrado
pela parte sombreada do ventrículo na Fig. 12.7A. Em seguida, a
despolarização se difunde ao longo das superfícies endocárdicas dos dois
ventrículos, como e mostrado na Fig. 12.7B e C. Finalmente, ela se
difunde pelo músculo ventricular até a parte externa do coração, como é
mostrado progressivamente na Fig. 12.7 C, D e E.
A cada estágio da despolarização dos ventrículos na Fig. 12.7, partes
A e E, o potencial elétrico instantâneo é representado por um vetor
superposto ao ventrículo em cada figura. Cada um desses vetores é
analisado pelo método descrito na seção anterior, para a determinação
das voltagens que vão ser registradas a cada momento em cada uma
das três derivações-padrão do eletrocardiograma. Em cada etapa da
figura é mostrado, à direita, o desenvolvimento progressivo do complexo
QRS. Lembre-se que um vetor positivo em uma derivação faz com que
o registro no eletrocardiograma fique acima da Unha zero, enquanto um
vetor negativo vai fazer o registro ficar abaixo da linha.
Antes de passar a outras considerações da análise vetorial, ê essencial
que esta análise dos sucessivos vetores normais apresentada na Fig. 12,7
seja compreendida. Cada uma dessas análises deve ser estudada com
detalhe pelo procedimento anteriormente apresentado. Segue-se um
resumo sucinto desta seqüência.
Na Fig. 12.7A, o músculo ventricular apenas começou a se
despolarizar, representando um instante cerca de 0,01 segundo após o
início da despolarização. Nesse momento, o vetor é curto porque apenas
pequena parte dos ventrículos - o septo - está despolarizada. Todas as
voltagens eletrocardiográficas apresentam-se portanto baixas, como é
registrado ã direita do músculo ventricular para cada uma das derivações.
A voltagem na derivação II é maior que a das derivações I e III, porque
o vetor do coração estende-se, em grande parte, na mesma direção
do eixo da derivação II.
Na Fig. 12.7B, que representa aproximadamente 0,02 s após o início
da despolarização, o vetor do coração é longo porque grande parte
Fig. 12.6 Determinação dos vetores projetados nas derivações I, II e
III, quando o vetor A representa o potencial instantâneo nos ventrículos.
dos ventrículos já se despolarizou. A voltagem aumentou, pois, em todas
as derivações eletrocardiográficas.
Na Fig. 12.7C, cerca de 0,035 s após o início da despolarização,
o vetor do coração está ficando mais curto e as voltagens
eletrocardiográficas registradas são menores porque a parte externa do
ápice do coração está agora eletronegativa, neutralizando grande parte
da negatividade das superfícies endocárdicas do coração. Da mesma
forma, o eixo do vetor está se deslocando para o lado esquerdo do
coração, porque o ventrículo esquerdo demora um pouco mais a
despolarizar-se que o direito. A proporção entre a voltagem na
derivação I e na derivação III está aumentando.
Na Fig. 12.7D, cerca de 0,05 s após o início da despolarização,
o vetor do coração aponta para a base do ventrículo esquerdo e é curto
porque apenas parte diminuta do músculo ventricular ainda está
polarizada. Devido ã direção do vetor nesse momento, as voltagens
registradas nas derivações II e III são ambas negativas - ou seja, abaixo
da linha.
Na Fig. 12.7E, cerca de 0,06 s após o início da despolarização,
toda a massa muscular do ventrículo está despolarizada, de modo que
não há absolutamente qualquer corrente fluindo em torno do coração
e não é gerado potencial elétrico, O vetor torna-se zero e a voltagem
de todas as derivações torna-se zero.
Assim, os complexos QRS completam-se nas três derivações
bipolares - padrão dos membros, O complexo QRS tem, por vezes,
ligeira depressão negativa em seu início em uma ou mais de suas
derivações, o que não é mostrado na Fig. 12.7; essa é a onda Q.
Quando ocorre, ela é causada pela despolarização inicial do lado
esquerdo do septo, antes do lado direito, o que cria um vetor fraco da
esquerda para a direita por uma fração de segundo, antes que ocorra
o vetor habitual do ápice para a base. A grande deflexão positiva
mostrada na Fig. 12.7 é a onda R e a deflexão negativa final é a onda S.
O ELETROCARDIOGRAMA DURANTE A REPOLARIZAÇÃO
- A ONDA T
Após o músculo ventricular se despolarizar, decorre
aproximadamente 0,15 s antes que se inicie repolarização suficiente para
ser observada no eletrocardiograma; a repolarização prossegue, então,
por todo o músculo ventricular até completar-se acerca de 0,35 s após
o início do complexo ORS. É este processo de repolarização que
causa a onda T no eletrocardiograma.
Como o septo e outras áreas endocárdicas do músculo ventricular
despolarizam-se primeiro, parece lógico que essas áreas também
deveriam repolarizar-se primeiro, mas isto não ocorre habitualmente,
porque o septo e outras áreas endocárdicas têm período mais longo de
contração, repolarizando-se, pois, mais lentamente que as superfícies
externas do coração. Assim sendo, a maior parte do músculo ventricular a
se repolarizar primeiro é a situada sobre toda a superfície externa do
coração, em especial próximo ao ápice. As áreas endocárdicas, por outro
lado, normalmente repolarizam-se por último. A razão desta seqüência
anormal de repolarização é considerada como sendo que a pressão
elevada nos ventrículos durante a contração reduz muito o fluxo
sanguíneo coronário para o endocárdio, lentificando, assim, o processo
de despolarização nas áreas endocárdicas.
Como as superfícies externa e apical dos ventrículos se repolarizam
antes das superfícies interna e basal, a extremidade positiva do vetor
cardíaco durante a repolarização se dirige para o ápice do coração.
Portanto, a direção predominante do vetor através do coração durante a
repolarização ventricular ê da base para o ápice, que também é a direção
predominante do vetor durante a despolarização. Como conseqüência,
a onda T nas derivações bipolares normais dos membros é positiva, o
que é também a polaridade da maior parte dos complexos QRS normais.
Na Fig. 12.8, cinco etapas na repolarização ventricular são denotadas
pelo aumento progressivo das áreas brancas — as áreas repolarizadas.
Hm cada etapa, o vetor se estende da base para o ápice, até desaparecer
na última etapa. Inicialmente, o vetor é relativamente pequeno, porque
a área de repolarização é pequena. Posteriormente, o vetor torna-se
mais e mais forte, devido ao maior grau de repolarização. Finalmente,
o vetor volta a ser fraco, porque as áreas de despolarização que ainda
persistem tornam-se tão pequenas que o fluxo de corrente total começa
a diminuir. Essas alterações mostram que o vetor é maior quando
aproximadamente metade do coração está no estado polarizado e o
restante ainda está despolarizado.
111
Fig. 12.7 A, os vetores ventriculares e os complexos QRS 0,01 s após o início da despolarização ventricular. B. 0,02 s após o início da despolarização.
C, 0,035 segundo após o início da despolarização. D, 0,05 s após o início da despolarização. E, Após completar-se a despolarização dos ventrículos
0,06 s após o inicio.
As alterações nos eletrocardiogramas das três derivações-padrão
dos membros durante o processo de despolarização estão anotadas sob
cada um dos ventrículos, mostrando os estágios progressivos da
Tepolarização. A onda T do eletrocardiograma é gerada por
aproximadamente 0,15 s, o tempo necessário para que todo o processo
ocorra.
relativamente ao restante dos átrios. O vetor da repolarização atrial,
portanto, tem direção inversa relativamente ao vetor de despolarização.
(Veja novamente que este é o contrário do efeito que ocorre nos
ventrículos.) Assim sendo, como é notado à direita da Fig. 12.9, a
chamada
DESPOLARIZAÇÃO DOS ÁTRIOS — A ONDA P
A despolarização dos átrios começa pelo nodo sinusal e se difunde
em todas as direções pelos átrios. O ponto de eletronegatividade original
nos átrios, portanto, é aproximadamente no ponto de entrada da veia
cava superior, onde se encontra o nodo sinusal, e a direção do potencial
elétrico no átrio, ao início da despolarização, é na direção mostrada
na Fig. 12.9. Além disso, o vetor geralmente permanece nesta direção
durante todo o processo de despolarização.
Assim, o vetor do fluxo de corrente durante a despolarização dos
átrios aponta quase que na mesma direção que nos ventrículos. E, como
essa direção é quase a mesma dos eixos das derivações bípolares-padrão
dos membros 1, 11 e III, os eletrocardiogramas registrados a partir dos
átrios durante o processo de despolarização são geralmente positivos
em todas essas três derivações, como ilustra a Fig. 12.9. O registro
da despolarização atrial é conhecida como a onda P.
Repolarização dos átrios — a onda T atrial. A difusão da
onda de despolarização pelo músculo atrial é muito mais lenta que nos
ventrículos. A musculatura em torno do nodo sinusal, portanto,
despolariza-se muito antes da musculatura nas partes distais dos átrios.
Devido a isso, a área que se repolariza primeiro nos átrios ê a região
do nodo sinusal, a área que originalmente se despolarizou em primeiro
lugar, uma situação totalmente diferente da verificada nos ventrículos.
Assim, quando se inicia a repolarização, a região em torno do nodo
sinusal torna-se positiva
Fig. 12.8 Geração da onda T durante a repolarização dos ventrículos,
mostrando a análise vetorial da primeira etapa da repolarização. O tempo
total do início da onda T até seu final é de cerca de 0,15 s.
112
Fig. 12.9 Despolarização dos átrios e geração da onda P.
mostrando o vetor pelos átrios e os vetores resultantes nas três
derivações-padrão. À direita estão as ondas P e T atriais.
onda T atrial aparece cerca de 0,15 s após a onda P atrial, mas essa
onda T está do lado oposto da linha de referência zero em relação
à onda P — ou seja, ela é normalmente negativa, e não positiva nas
três derivações bipolares-padrão dos membros. No eletrocardiograma
normal, essa onda T aparece aproximadamente ao mesmo tempo que
o complexo QRS ventricular. Por esta razão, ela c quase sempre
totalmente obscurecida pelo maior complexo QRS, embora, em alguns
estados anormais, realmente participe do eletrocardiograma registrado.
O VETORCARDIOGRAMA
Observou-se nas discussões anteriores que o vetor do fluxo de corrente
pelo coração se altera rapidamente à medida que o impulso se difunde
pelo miocárdio. Ele se altera em dois aspectos: primeiro, o vetor aumenta
e diminui de comprimento devido ao aumento e diminuição da voltagem
do vetor. Segundo, o vetor muda de direção devido a alterações na
direção média do potencial elétrico do coração. O chamado
vetorcardiagrama mostra essas alterações nos vetores em diferentes
momentos durante o ciclo cardíaco, como é mostrado na Fig. 12.10.
No vetor cardiograma da Fig. 12.10, o ponto 5 é o ponto de referência
zero, sendo este ponto a extremidade negativa de todos os vetores.
Enquanto o coração está em repouso, a extremidade positiva do vetor
também permanece no ponto zero. porque não há qualquer potencial
elétrico. Entretanto, logo que a corrente começa a fluir pelo coração,
a extremidade positiva do vetor sai do ponto de referência zero.
Quando o septo começa a se despolarizar, o vetor se estende para
baixo em direção ao ápice do coração.-mas é relativamente fraco, geran-
Fig. 12.10 Os vetorcardiograma QRS e T.
do, assim, a primeira parte do vetorcardiograma, como é mostrado pela
extremidade positiva do vetor 1. Quando uma parte maior do coração
se despolariza, o vetor fica cada vez mais forte, em geral oscilando
ligeiramente para um dos lados. Assim, o vetor 2 da Fig. 12.10 representa
o estado de despolarização do coração cerca de 0,02 s após o vetor
1. Após mais 0,02 s o vetor 3 representa o potencial do coração, e
o vetor 4 ocorre após ainda outro 0,01 s. Finalmente, o coração se
despolariza totalmente e o vetor volta novamente a zero, como é
mostrado no ponto 5.
A figura elíptica produzida pelas extremidades positivas dos vetores
é denominada vetorcardiograma do QRS.
Os vetorcardiogramas podem ser registrados instantaneamente num
osciloscópio, ligando-se eletródios acima e abaixo do coração às placas
verticais do osciloscópio e ligando-se eletródios de cada lado do coração
as placas horizontais. Quando o vetor se altera, o ponto de luz no
osciloscópio segue a trajetória da extremidade positiva do vetor em
alteração, inscrevendo, assim, o vetorcardiograma na tela do
osciloscópio.
O vetorcardiograma “T” A mudança dos vetores no coração não
ocorre apenas durante o processo de despolarização, pois os vetores
que representam o fluxo de corrente em torno dos ventrículos também
reaparecem durante a repolarização. Por esta razão, um segundo e menor
vetorcardiograma - o vetorcardiograma T - é inscrito durante a
repolarização da massa muscular; ele é representado à direita na Fig.
12.10. Assim, também, um vetorcardiograma P, menor ainda, é inscrito
durante a despolarização atrial.
O EIXO ELÉTRICO MÉDIO DO QRS VENTRICULAR
O vetorcardiograma da onda de despolarização ventricular
(vetorcardiograma do QRS) mostrado na Fig. 12.10 é o de um coração
normal. Observe por esse vetorcardiograma que a direção
preponderante dos vetores ventriculares é normalmente para o ápice
do coração — ou seja, durante a maior parte do ciclo de
despolarização ventricular, a direção de potencial elétrico é da base
para o ápice dos ventrículos. Essa direção preponderante do potencial
durante a despolarização é denominada eixo elétrico médio dos
ventrículos, ou vetor médio do QRS. O eixo elétrico médio dos
ventrículos normais é de 59º. Entretanto, em certas condições
patológicas do coração, essa direção se altera — acentuadamente — por
vezes, até mesmo para o pólo oposto do coração.
DETERMINAÇÃO DO EIXO ELÉTRICO A PARTIR
DE ELETROCARDIOGRAMAS DE DERIVAÇÕESPADRÃO
Clinicamente, o eixo elétrico do coração é em geral determinado
a partir dos eletrocardiogramas das derivações bipolares-padrão dos
membros, e não do vetorcardiograma. A Fig. 12.11 mostra um método
para se fazer isto. Após o registro das derivaçÕes-padrão, determina-se
o potencial máximo e a polaridade do registro em duas das derivações.
Na derivação 1 da figura, o registro é positivo, e, na derivação III,
o registro é em grande parte positivo, mas é negativo durante outra
parte do ciclo. Quando qualquer parte do registro é negativa, o potencial
negativo ê subtraído do potencial positivo para a determinação do poten-
Fig. 12.11 Representação do eixo elétrico médio do coração a partir
de duas derivações eletrocardiográficas.
113
cial efetivo para essa derivação, ilustrado pelas setas à direita dos
complexos QRS das derivações I e III. (Para serem ainda mais precisos,
alguns cardiologistas subtraem a área da onda negativa da área da onda
positiva.) Após subtrair-se a parte negativa da onda QRS na
derivação III da parte positiva, cada potencial efetivo é representado
graficamente nos eixos das derivações respectivas, com a base do
potencial no ponto de interseção dos eixos, como é mostrado na Fig.
12.11.
Quando o potencial efetivo da derivação I é positivo, ele é
representado graficamente na direção positiva, ao longo da linha que
representa a derivação I. Por outro lado, quando negativo, é
representado na direção negativa. Assim, também para a derivação III,
o potencial efetivo é colocado com sua base no ponto de interseção e,
quando positivo, é representado na direção positiva ao longo da linha
que representa a derivação III. Quando negativo, ele é representado na
direção negativa.
Para se determinar o vetor real do potencial elétrico médio
ventricular, traçam-se linhas perpendiculares a partir das pontas dos dois
potenciais efetivos das derivações I e III, respectivamente. O ponto de
intersecção dessas duas linhas perpendiculares representa, por análise
vetorial, a ponta do vetor médio do QRS real nos ventrículos; e o
ponto de interseção dos eixos das duas derivações representa a
extremidade negativa do vetor real. Assim sendo, o vetor médio do
QRS é traçado entre esses dois pontos. O potencial médio aproximado
gerado pelos ventrículos durante a despolarização é representado pelo
comprimento do vetor, e o eixo elétrico médio é representado pela
direção do vetor. Assim, a orientação do eixo elétrico médio dos
ventrículos normais, conforme determinado na Fig. 12.11, é de 59°.
CONDIÇÕES VENTRICULARES ANORMAIS CAUSANDO
DESVIOS DO EIXO
Embora o eixo elétrico médio dos ventrículos seja em média de
cerca de 59°, esse eixo pode deslocar-se para a esquerda, mesmo em
coração normal, até aproximadamente 20º. ou para a direita, até
aproximadamente 100°. As causas das variações anormais são
principalmente diferenças anatômicas na distribuição do sistema de
Purkinje ou na própria musculatura dos diferentes corações. Contudo,
algumas condições podem ocasionar desvios do eixo até mesmo além
desses limites normais, da seguinte forma:
Alterações na posição do coração. Evidentemente, se o próprio
coração estiver posicionado em ângulo para a esquerda, o eixo elétrico
médio do coração também vai desviar-se para a esquerda. Esse desvio
ocorre (1) durante a expiração, (2) quando a pessoa está deitada,
porque o conteúdo abdominal empurra o diafragma para cima, e (3),
com muita freqüência, em pessoas obesas e de compleição avantajada,
cujo diafragma normalmente faz pressão para cima sobre o coração
todo o tempo.
Da mesma forma, o desvio do coração em ângulo para a direita
faz o eixo elétrico ventricular médio desviar-se para a direita. Esta
condição ocorre (1) durante a inspiração, (2) quando a pessoa fica
de pé, e (3) normalmente em pessoas altas e magras, cujo coração pende
para baixo.
Hipertrofia de um ventrículo. Quando um dos ventrículos se
hipertrofia muito, o eixo do coração se desvia em direção ao ventrículo
hipertrofiado, por duas razões. Em primeiro lugar, há quantidade muito
maior de músculo do lado hipertrofiado, c isto possibilita excessiva
geração de potencial elétrico nesse lado. Segundo, é necessário mais
tempo para a onda de despolarização passar pelo ventrículo
hipertrofiado que pelo ventrículo normal. Por conseguinte, o ventrículo
normal se despolariza - ou seja, torna-se negativo - consideravelmente
antes do ventrículo hipertrofiado, e isso causa forte vetor do lado
normal do coração para o lado hipertrofiado, que ainda permanece
positivamente carregado. Assim, o eixo desvia-se em direção ao
ventrículo hipertrofiado.
Análise vetorial do desvio do eixo para a esquerda em
conseqüência da hipertrofia do ventrículo esquerdo. A Fig. 12.12
mostra as três derivações bipolares-padrão dos membros de um
eletrocardiograma no qual a análise da direção do eixo revela desvio
desse eixo para a esquerda, com o eixo elétrico médio apontando na
direção de -15°. Este é um típico eletrocardiograma decorrente do
aumento da massa muscular do ventrículo esquerdo. Nesse caso, o
desvio do eixo foi causado por hipertensão (pressão arterial elevada), que
fez o ventrículo esquerdo hipertrofiar-se para bombear sangue contra a
elevada pressão arterial sistêmica.
Fig. 12.12 Desvio do eixo para a esquerda nas cardiopatias
hipertensivas. Note, também, o ligeiro alargamento do complexo QRS.
Entretanto, um quadro semelhante de desvio para a esquerda
ocorre quando o ventrículo esquerdo se hipertrofia como conseqüência
de estenose da válvula aórtica regurgitação valvular aórtica ou qualquer
das várias cardiopatias congênitas em que o ventrículo esquerdo
aumenta de tamanho enquanto o lado direito do coração permanece
relativamente normal.
Análise vetorial dos desvios do eixo para a direita, em
conseqüência da hipertrofia do ventrículo direito. O eletrocardiograma
da Fig. 12.13 mostra um grande desvio do eixo para a direita, com eixo
elétrico de aproximadamente 170°, o que é 111º para a direita do eixo
elétrico médio normal de 59º dos ventrículos. O desvio para a direita do
eixo ilustrado nessa figura foi provocado pela hipertrofia do ventrículo
direito devido a estenose pulmonar. Contudo, desvios do eixo para a
direita também podem ocorrer em outras cardiopatias congênitas que
causam a hipertrofia do ventrículo direito, tais como a tetralogia de
Fallot ou o defeito do septo interventricular. Também a hipertrofia do
ventrículo direito, em conseqüência do aumento da resistência vascular
pulmonar, pode causar desvio do eixo para a direita.
Bloqueio de ramo. Normalmente, as duas paredes laterais dos
ventrículos se despolarizam quase ao mesmo tempo, porque os dois
ramos do sistema de Purkinje, o direito e o esquerdo, transmitem o
impulso cardíaco para as superfícies endocárdicas das duas paredes
ventriculares quase ao mesmo tempo. Como conseqüência, os potenciais
gerados pelos dois ventrículos praticamente neutralizam um ao outro.
No entanto, quando um dos ramos maiores é bloqueado, o impulso
cardíaco difunde-se pelo ventrículo normal muito antes de difundir-se
pelo outro. Por esta razão, a despolarização dos dois ventrículos não é
nem mesmo próxima uma da outra e os potenciais de despolarização
não se neutralizam uns aos outros. Nessas condições, há desvios do eixo,
da seguinte maneira:
Análise vetorial do desvio do eixo para a esquerda nos bloqueios do
ramo esquerdo. Quando o ramo esquerdo é bloqueado, a despolarização
cardíaca se difunde pelo ventrículo direito duas a três vezes mais rápido
que pelo esquerdo. Como conseqüência, grande parte do ventrículo
esquerdo permanece polarizada muito tempo após o ventrículo direito
ter-se despolarizado totalmente. Assim, o ventrículo direito fica
eletronegativo, enquanto o ventrículo esquerdo permanece eletropositivo
durante a maior parte do processo de despolarização, e um vetor muito
forte projeta-se do ventrículo direito para o esquerdo. Em outras
palavras, há grande desvio para a esquerda porque a extremidade
positiva do vetor aponta para o ventrículo esquerdo. Isso é mostrado na
Fig. 12.14, na qual se vê um desvio típico para a esquerda do eixo,
conseqüência de bloqueio do ramo esquerdo. Note que o eixo é de
aproximadamente -50º.
114
Fig. 12.14 Desvio do eixo para a esquerda devido a um bloqueio do
ramo esquerdo. Note o grande alargamento do complexo QRS.
Fig. 12.13 Eletrocardiograma de alta voltagem na estenose pulmonar
com hipertrofia do ventrículo direito. Também é visto intenso desvio
do eixo para a direita, assim como ligeiro alargamento do complexo
QRS.
Devido à lentidão da condução de impulsos quando o sistema de
Purkinje está bloqueado, os desvios decorrentes do bloqueio de ramo
também aumentam muito a duração do complexo QRS, o que se pode
ver observando a excessiva largura das ondas ORS na Fig. 12.14. Esse
alargamento do complexo QRS diferencia essa condição dos desvios
do eixo causados por hipertrofia.
Análise vetorial dos desvios do eixo para a direita no bloqueio
do ramo direito. Quando o ramo direito está bloqueado, o ventrículo
esquerdo despolariza-se bem mais rapidamente que o direito, de modo
que o esquerdo fica eletronegativo enquanto o direito permanece
eletropositivo. Por esta razão surge um vetor muito forte, com sua
extremidade negativa para o ventrículo esquerdo e a positiva para o
ventrículo direito. Em outras palavras, há grande desvio para a direita.
O desvio do eixo para a direita,'causado pelo bloqueio do ramo
direito, é mostrado e seu vetor é analisado na Fig. 12.15, que apresenta
eixo de aproximadamente 115º e alargamento do complexo QRS devido
ao bloqueio da condução. Note também o grande alargamento do
complexo ORS.
o aumento da massa muscular do coração, que decorre normalmente
da hipertrofia do músculo em resposta a uma carga excessiva sobre uma
ou outra parte do coração. Como exemplo, o ventrículo direito se
hipertrofia quando tem de bombear sangue por uma válvula pulmonar
estenosada e o ventrículo esquerdo se hipertrofia quando a pessoa tem
pressão arterial elevada. A maior quantidade de músculo possibilita
geração de maior quantidade de eletricidade em torno do coração.
Como conseqüência, os potenciais elétricos registrados nas derivações
eletrocardiográficas são consideravelmente maiores que o normal, como
é mostrado nas Figs. 12.12 e 12.13.
DIMINUIÇÃO DA VOLTAGEM DO
ELETROCARDIOGRAMA
Há três causas principais de diminuição de voltagem do
eletrocardiograma. São elas, em primeiro lugar, anormalidades do
próprio músculo cardíaco, que impedem a geração de grande quantidade
de corrente; segundo, condições anormais em torno do coração, de
tal modo que a corrente não pode ser conduzida com facilidade do
coração até a superfície do corpo; e terceiro, a rotação do ápice do
coração de modo a apontar para a parede anterior do tórax, de tal
forma que a corrente elétrica do coração flui ântero-posteriormente no
tórax, e não nu plano frontal do corpo, o que diminui a voltagem nas
derivações dos membros.
CONDIÇÕES QUE CAUSAM VOLTAGENS ANORMAIS
DO COMPLEXO QRS
AUMENTO DA VOLTAGEM NAS DERIVAÇÕES
BIPOLARES-PADRÃO DOS MEMBROS
Normalmente, as voltagens das três derivações bipolares-padrão dos
membros, medidas do pico da onda R à parte inferior da onda S, variam
entre 0,5 e 2,0 mV, com a derivação III registrando, geralmente, a
voltagem mais baixa, e a derivação II, a mais alia. Entretanto, essas
relações não são invariavelmente verdadeiras, nem mesmo no coração
normal. Em geral, quando a soma das voltagens de todos os complexos
QRS das três derivações-padrão é superior a 4 mV, considera-se que
o paciente tem um eletrocardiograma de alta voltagem.
A causa dos complexos QRS de alta voltagem é mais comumente
Fig. 12.15 Desvio do eixo para a direita devido a um bloqueio do ramo
direito. Note o grande alargamento do complexo QRS.
115
Diminuição da voltagem causada por miopatias cardíacas. Uma
das
causas mais comuns de diminuição da voltagem do complexo QRS é
uma série de infartos do miocárdio antigos, com conseqüente diminuição
da massa muscular. Isso também faz com que a onda de despolarização
atravesse lentamente os ventrículos e impeça que grandes partes do
coração fiquem maciçamente despolarizadas de uma só vez. Por
conseguinte, essa condição causa alargamento moderado do complexo
QRS, juntamente com a diminuição da voltagem. A Fig. 12.16 mostra o
típico eletrocardiograma de baixa voltagem com alargamento do
complexo QRS que se encontra com freqüência após múltiplos
pequenos infartos do coração que ocasionaram bloqueios locais e perda
de massa muscular por todo o ventrículo.
Diminuição da voltagem causada por condições circundando o
coração. Uma das causas mais importantes da diminuição da voltagem
nas derivações eletrocardiográficas é a presença de liquido no
pericárdio. Como o líquido extracelular conduz correntes elétricas com
grande facilidade, grande parte da eletricidade que flui para fora do
coração é conduzida de uma parte do coração para outra pela efusão
pericárdica. Assim, essa efusão provoca efetivamente um "curtocircuito" dos potenciais elétricos gerados no coração. A efusão pleural
em menor grau, também pode "colocar em curto" a eletricidade em
torno do coração, de modo que as voltagens na superfície do corpo e
nos eletrocardiogramas diminuem.
O enfisema pulmonar pode reduzir os potenciais
eletrocardiográficos, mas por processo diferente do da efusão
pericárdica. No enfisema pulmonar, a condução de correntes elétricas
pelos pulmões fica consideravelmente reduzida, devido à excessiva
quantidade de ar nos pulmões. Ocorre, igualmente, que a cavidade
torácica aumenta de volume e os pulmões tendem a envolver o coração
em grau maior do que o normal. Por esta razão, os pulmões atuam como
um isolante, impedindo a difusão da voltagem elétrica do coração para a
superfície do corpo, e isso causa diminuição dos potenciais
eletrocardiográficos nas diversas derivações.
PADRÕES PROLONGADOS E BIZARROS DO
COMPLEXO QRS
ALARGAMENTO
DO
COMPLEXO
QRS
EM
CONSEQUÊNCIA DE HIPERTROFIA OU DILATAÇÃO
DO CORAÇÃO
O complexo QRS dura enquanto o processo de despolarização
continua a se difundir pelos ventrículos — ou seja, enquanto parte dos
ventrículos está despolarizada e parte polarizada. Assim, a causa do
alargamento do complexo QRS é sempre um prolongamento da
condução do impulso pelos ventrículos. Tal prolongamento ocorre com
freqüência quando um dos ventrículos ou ambos estão hipertrofiados ou
dilatados, devido à via mais longa que o impulso tem, então, de
percorrer. O complexo QRS normal dura 0,06 a 0,09 s, enquanto na
hipertrofia ou dilatação do ventrículo esquerdo ou direito o complexo
QRS pode estar alargado até 0,10 a 0,12 s.
Fig. 12.16 Eletrocardiograma de baixa voltagem, com evidência de lesões
localizadas em todo o ventrículo, causadas por infarto do miocárdio antigo.
ALARGAMENTO DO COMPLEXO QRS RESULTANTE DE
BLOQUEIOS AO SISTEMA DE PURKINJE
Quando as fibras de Purkinje são bloqueadas, o impulso cardíaco
tem de ser conduzido pelo próprio músculo ventricular, diminuindo a
velocidade de condução de impulsos para aproximadamente um terço
a um quarto do normal. Por esta razão, quando ocorre bloqueio completo
de um dos ramos, a duração do complexo QRS fica geralmente
aumentada para 0,14 s ou mais.
Em geral, considera-se o complexo QRS como anormalmente longo
quando dura mais de 0,09 s, e, quando dura mais de 0,12 s, o alargamento
é quase que certamente causado pelo bloqueio patológico do sistema
de condução em algum ponto dos ventrículos, como é mostrado pelos
eletrocardiogramas para o bloqueio de ramo nas Figs. 12.14 e 12.15.
CONDIÇÕES CAUSADORAS DE COMPLEXOS QRS
BIZARROS
Os padrões bizarros do complexo QRS são causados mais
freqüentemente por duas condições: primeiro, a destruição do músculo
cardíaco em diversas áreas por todo o sistema ventricular, com
substituição desse músculo por tecido cicatricial, e. segundo, bloqueios
locais da condução de impulsos pelo sistema de Purkinje.
Por vezes, podem ocorrer bloqueios locais em múltiplos pontos dos
ventrículos. Como conseqüência, a condução dos impulsos cardíacos
fica muito irregular, causando rápidas alternâncias, entre os desvios do
eixo para a esquerda e para a direita. Isso causa picos duplos ou até
mesmo triplos em algumas das derivações eletrocardiográficas, tais como
as apresentadas na Fig. 12.14.
CORRENTE DE LESÃO
Muitas anormalidades cardíacas diferentes, especialmente as que
lesam o próprio músculo cardíaco, fazem muitas vezes com que parte
do coração permaneça parcial ou totalmente despolarizada iodo o tempo.
Quando isso ocorre, a corrente flui entre as áreas patologicamente
despolarizadas e as normalmente polarizadas. Isto é denominado
corrente de lesão. Note, especialmente, que à parte lesada do coração fica
negativa porque é essa parte que está despolarizada e emite cargas
negativas para os líquidos circundantes, enquanto o restante do coração
está positivo.
Algumas das anormalidades que podem causar corrente de lesão
são: (1) traumas mecânicos, que fazem as membranas ficarem tão
permeáveis que a repolarização total não pode ocorrer; (2) processos
infecciosos que lesam as membranas musculares; e (3) a isquemia de
áreas locais do músculo, causada pela oclusão coronária, que é, de
longe, a causa mais comum de correntes de lesão no coração. Durante
uma isquemia, não há simplesmente quantidade suficiente de nutrientes
do suprimento sanguíneo coronário disponíveis para o músculo cardíaco
para a manutenção da função cardíaca.
EFEITO DA CORRENTE DE LESÃO SOBRE O
COMPLEXO QRS
Na Fig. 12.17, a área sombreada na base do ventrículo esquerdo
apresenta um infarto recente. Por esta razão, durante o intervalo T-P
— ou seja, quando o músculo ventricular normal está polarizado —
uma corrente negativa flui da base do ventrículo esquerdo para o resto
dos ventrículos. O vetor do potencial dessa "corrente de lesão", ilustrado
no primeiro e último corações da figura, tem direção de aproximadamente
125°, com sua base, a extremidade negativa, apontando para o músculo
lesado. Como é mostrado nas partes inferiores da figura, antes mesmo
que o complexo QRS se inicie, este vetar causa deflexão inicial na
derivação í abaixo de linha de potencial zero, porque o vetor projetado
da corrente de lesão na derivação 1 aponta para a extremidade
negativa do eixo da derivação I. Na derivação II, o registro está acima
da linha, porque o vetor projetado aponta para o terminal positivo da
derivação II. Na derivação III, o vetor projetado do fluxo de
corrente também é na mesma direção que a polaridade da derivação
III, de modo que o registro é positivo. Além disso, como o vetor da
corrente de lesão encontra-se quase que exatamente ao longo do eixo
da derivação III. o potencial da corrente de lesão na derivação III é
muito maior do que em qualquer um dos dois registros.
116
Fig. 12.17 Efeito de uma corrente de lesão sobre o eletrocardiograma.
Como o coração passa, então, por seu processo normal de
despolarização, o septu é o primeiro a se despolarizar, e a
despolarização se difunde por ele até o ápice e de volta em direção às
bases dos ventrículos. A última parte dos ventrículos a se despolarizar
totalmente é à base do ventrículo direito, pois a base do ventrículo
esquerdo já está total e permanentemente despolarizada. Por análise
vetorial, como é mostrado na figura, o eletrocardiograma gerado pela
passagem da onda de despolarização pelos ventrículos pode ser
graficamente construído, como é mostrado na Fig. 12.17.
Quando o coração se encontra totalmente despolarizado ao final
do processo de despolarização, como é mostrado pelo penúltimo estágio
da Fig. 12.17, todo o músculo ventricular está no estado negativo. POR
esta razão, nesse instante, não aparece no eletrocardiograma
absolutamente nenhuma corrente fluindo em torno da musculatura dos
ventrículos, porque agora tanto o músculo cardíaco lesado como o
músculo em contração estão totalmente despolarizados.
Em seguida, quando ocorre a repolarização, todo o coração acaba
por se repolarizar, exceto a área de despolarização permanente na base
lesada do ventrículo esquerdo. Assim, a repolarização causa retorno
da corrente de lesão em cada derivação, como se nota bem à direita
da Fig. 12.17.
O PONTO J — O POTENCIAL DE REFERÊNCIA ZERO
PARA A ANÁLISE DE CORRENTES DE LESÃO
Seria de se esperar que os aparelhos para o registro de
eletrocardiogramas pudessem determinar quando- não há corrente
fluindo em torno do coração. Contudo, há muitas correntes errantes no
corpo, tais como as correntes resultantes de "potenciais cutâneos" e de
diferenças nas concentrações iônicas nas distintas partes do corpo. Em
vista disso, quando dois eletródios estão ligados entre os braços ou
entre um braço e uma perna, essas correntes errantes tornam
impossível determinar-se o nível referência zero exato no
eletrocardiograma. Por essas razões, o seguinte procedimento tem de
ser empregado para a determinação do nível potencial zero: primeiro,
nota-se o ponto exato em que a onda de despolarização acaba de
completar sua passagem pelo coração, que ocorre bem ao final do
complexo QRS. Exatamente nesse ponto todas as partes dos
ventrículos estão despolarizadas, de modo que nenhuma corrente flui em
torno do coração. Até mesmo a corrente de lesão desaparece nesse ponto.
Por este motivo, o potencial do eletrocardiograma, nesse instante, está
exatamente na voltagem zero. Esse ponto é conhecido como o "ponto J"
no eletrocardiograma, como é mostrado nas Figs. 12.17 e 12.18.
Para a análise do eixo elétrico do potencial de lesão causado por
uma corrente de lesão, é traçada uma linha horizontal através do
eletrocardiograma ao nível do ponto J, sendo essa linha horizontal a
linha de potencial zero no eletrocardiograma a partir da qual devem ser
medidos todos os potenciais causados por correntes de lesão.
Fig. 12.18 O ponto "J" como a voltagem de referência zero do
eletrocardiograma. Também é mostrado um método para a representação
gráfica do eixo de uma corrente de lesão (abaixo).
Uso do ponto J para representar graficamente o eixo de um potencial
de lesão. A Fig. 12.18 ilustra os eletrocardiogramas registrados a partir
das derivações I e III, ambos mostrando correntes de lesão. Em outras
palavras, o ponto J de cada um desses dois eletrocardiogramas não está
na mesma linha que o segmento T-P. Uma linha horizontal foi traçada
através do ponto J, representando o nível de potencial zero em cada
um dos dois registros. O potencial da corrente de lesão em cada derivação
é a diferença entre o nível do segmento T-P do eletrocardiograma (que
é registrado entre batimentos cardíacos quando existe corrente de lesão)
e a linha de potencial zero, como é mostrado pelas setas. Na derivação
I, o potencial registrado causado pela corrente de lesão está acima da
linha de potencial zero, sendo, portanto, positivo. Por outro lado, na
derivação III, o segmento T-P está abaixo do da linha potencial zero;
por esta razão, o potencial da corrente de lesão na derivação III é
negativo.
Na parte inferior da Fig. 12.18, os potenciais da corrente de lesão
nas derivações 1 e III são representados graficamente, nas coordenadas
dessas derivações e o vetor resultante do potencial de lesão para toda a
massa ventricular é determinado pelo método já descrito. Neste caso, o
vetor da corrente de lesão estende-se do lado direito dos ventrículos para
a esquerda e ligeiramente para cima, com eixo de aproximadamente -30º.
Caso se coloque o vetor da corrente de lesão diretamente sobre os
ventrículos, a extremidade negativa do vetor aponta para a área lesada ,
permanentemente despolarizada, dos ventrículos. No caso ilustrado na
Fig. 12.18, a área lesada seria na parede lateral do ventrículo direito.
O fenômeno do desvio do segmento S-T. À parte do
eletrocardiograma que ocorre entre o final do complexo QRS e o
início da onda T é denominada segmento S-T. O ponto J encontra-se
bem no inicio desse segmento. Portanto, cada vez que ocorre uma
corrente de lesão numa das derivações eletrocardiográficas, verifica-se
também que o segmento S-T e o segmento T-P do eletrocardiograma não
estão nos mesmos níveis de potencial no registro. Na verdade, é o
segmento T-P, e não o segmento S-T, que é deslocado do eixo zero.
Entretanto, muitas pessoas estão condicionadas a considerar o segmento
T-P do eletrocardiograma como o nível potencial de referência, e não o
ponto J. Por esta razão, quando se evidencia uma corrente de lesão no
eletrocardiograma, parece que o segmento S-T está desviado de seu nível
normal no eletrocardiograma, e isto é denominado desvio do segmento ST. Obviamente, quando se vê um desvio do segmento S-T no
eletrocardiograma, sabe-se imediata-
117
mente que ele apresenta as características de uma corrente de lesão.
De fato, muitos eletrocardiografistas não falam absolutamente de
correntes de lesão mas, simplesmente, de desvios do segmento S-T, o
que significa a mesma coisa.
ISQUEMIA
CORONÁRIA
CORRENTES DE LESÃO
COMO
CAUSA
DE
O fluxo sanguíneo insuficiente para o músculo cardíaco diminui
o metabolismo do músculo por três razões diferentes: falta de oxigênio,
acúmulo excessivo de dióxido de carbono e falta de nutrientes suficientes.
Por conseguinte, a repolarização das membranas não pode ocorrer em
áreas de isquemia miocárdica grave. Com freqüência, o músculo cardíaco
não morre porque o fluxo sanguíneo é suficiente para manter a vida
do músculo, ainda que não seja adequado para causar a repolarização
das membranas. Enquanto existir esse estado, continua a fluir corrente
de lesão durante a diástole.
Isquemia extrema do músculo cardíaco ocorre após oclusão
coronária, e forte corrente de lesão flui, a partir da área infartada dos
ventrículos, durante a intervalo T-P entre os batimentos cardíacos, como
é mostrado nas Figs. 12.19 e 12.20. Por esta razão, uma das mais
importantes características diagnosticas dos eletrocardiogramas
registrados após trombose coronária aguda é a corrente de lesão.
Infarto agudo da parede anterior. A Fig. 12.19 mostra o
eletrocardiograma nas três derivações bipolares-padrão dos membros e
em derivação torácica, registrada em paciente com infarto agudo da parede
cardíaca anterior. A característica diagnostica mais importante desse
eletrocardiograma é a intensa corrente de lesão na derivação torácica. Se
traçarmos uma linha de potencial zero pelo ponto J desse
eletrocardiograma, será encontrado um forte potencial de lesão
negativo durante o intervalo T-P, o que indica que o eletródio
torácico sobre a frente do coração está em área de potencial
fortemente negativo. Em outras palavras, a extremidade negativa do
vetor do potencial de lesão está contra a parede torácica. Isto quer dizer
que a corrente de lesão está se originando da parede anterior dos
ventrículos, o que diagnostica essa condição como um infarto da parede
anterior.
Analisando-se as correntes de lesão nas derivações I e III, encontra-se
um potencial negativo, causado pela corrente de lesão, na derivação I, e
um potencial positivo para a corrente de lesão na derivação III. Isto
que dizer que o vetor resultante para a corrente de lesão no coração é de
aproximadamente +150°, com a extremidade negativa do vetor Fig. 12.19 Corrente de lesão em infarto agudo da parede anterior. Note
apontando para o ventrículo esquerdo e a extremidade positiva para a intensa corrente de lesão na derivação V2
o ventrículo direito. Assim, nesse eletrocardiograma específico,
acorrente de lesão parece estar vindo principalmente do ventrículo
esquerdo, assim como da parede anterior do coração. Seria de se da parte do coração que está afetada. Ao se fazer essas análises vetoriais,
suspeitar, pois, que esse infarto da parede anterior fosse provavelmente deve-se recordar sempre que a extremidade positiva do vetor do
causado por trombose do ramo descendente anterior da artéria coronária potencial de lesão aponta para o músculo cardíaco normal e a
esquerda. Infarto da parede posterior. A Fig. 12.20 mostra as três extremidade negativa aponta para a parte anormal do coração que está
derivações bipolares-padrão dos membros e uma derivação torácica de emitindo a corrente de lesão.
um paciente com infarto da parede posterior. A principal característica
Recuperação de trombose coronária aguda. A Fig. 12.21 mostra
diagnostica desse eletrocardiograma também é a derivação torácica.
Se se traçar à linha de referência de potencial zero através do ponto J uma derivação torácica V3 de um paciente com infarto posterior agudo,
dessa derivação, ficará logo claro que, durante o intervalo T-P, o mostrando a alteração no eletrocardiograma dessa derivação do dia do
potencial da corrente de lesão é positivo. Isto quer dizer que a ataque até 1 semana depois, a seguir 3 semanas depois e, finalmente.
1 ano após. Por esse eletrocardiograma pode-se ver que a corrente de
extremidade positiva do vetor está na parede torácica e a extremidade
negativa (lesada) dirige-se para longe da mesma parede. Em outras
palavras, a corrente de lesão está vindo do lado oposto do coração
relativamente à parte adjacente à parede torácica, sendo esta a razão
pela qual esse tipo de eletrocardiograma é à base do diagnóstico dos
infartos da parede posterior.
Analisando-se as correntes de lesão nas derivações I e III da Fig.
12.20, fica imediatamente claro que o potencial de lesão é negativo
em ambas as derivações. Por análise vetorial, como é mostrado na figura,
verifica-se que o vetor do potencial de lesão é de aproximadamente 95º, com a extremidade negativa do vetor apontando para baixo e
a extremidade positiva, para cima. Assim, como o infarto é na parede
posterior do coração, como indica a derivação torácica, e na parte apical
do coração, como indicam as correntes de lesão nas derivações II e
III, seria de se suspeitar que esse infarto esteja próximo do ápice, na
parede posterior do ventrículo esquerdo.
Infartos em outras partes do coração. Utilizando-se os mesmos
procedimentos ilustrados nas duas discussões anteriores de infartos da
parede anterior e posterior, c possível determinar-se a localização de
qualquer área infartada que emita corrente de lesão independentemente
Fig. 12.20 Corrente de lesão em infarto agudo da parede apical
posterior.
118
Ela é causada por isquemia relativa do coração. Não é sentida qualquer
dor enquanto a pessoa está totalmente imóvel, mas logo que o paciente
sobrecarrega o coração a dor aparece.
Ocorre freqüentemente corrente de lesão durante um ataque de
angina de peito grave, pois, por vezes a insuficiência coronária relativa
toma-se aí intensa o suficiente para impedir a repolarização adequada
das membranas em algumas áreas do coração durante a diástole.
ANORMALIDADES DA ONDA T
Fig. 12.21 Recuperação do miocárdio após um infarto moderado da
parede posterior, ilustrando o desaparecimento da corrente de lesão
(derivação V3).
lesão é forte imediatamente após o ataque agudo (segmento T-P
deslocado positivamente do ponto J e do segmento S-T). mas,
aproximadamente 1 semana, a corrente de lesão diminui
consideravelmente e após 3 semanas desapareceu totalmente. Depois
disso, o eletrocardiograma não se alterou muito durante o ano que se
segue. Este é o padrão habitual de recuperação após infarto cardíaco
agudo moderado, quando o fluxo sanguíneo colateral coronário é
suficiente para restabelecer a nutrição apropriada a maior parte da
área infartada.
Por outro lado, quando todos os vasos coronários de todo o coração
estão bastante esclerosados, pode não ser possível aos vasos coronários
adjacentes suprir a área infartada com sangue suficiente para a
recuperação. Por esta razão, em alguns pacientes com infarto
coronário, a área infartada nunca volta a desenvolver suprimento
sanguíneo coronário adequado, parte do músculo cardíaco morre e
persiste insuficiência coronária relativa indefinidamente nessa área do
coração. Caso não morra c não seja substituído por tecido cicatricial, o
músculo emite continuamente uma corrente de lesão, enquanto existir a
isquemia relativa, especialmente em períodos de exercício quando o
coração fica sobrecarregado.
infarto do miocárdio antigo recuperado. A Fig. 12.22 mostra as
derivações I e III após infarto anterior e infarto posterior, conforme
aparecem nessas derivações aproximadamente 1 ano após o episódio
agudo. Estas são o que se poderia chamar as configurações "ideais"
do complexo QRS nesses tipos de infarto do miocárdio recuperado.
Geralmente surge uma onda O no início do complexo QRS, na derivação
1 no infarto anterior, devido à perda de massa muscular na parede anterior
do ventrículo esquerdo, enquanto no infarto posterior a onda Q surge
no início do complexo QRS na derivação III, devido à perda do músculo
na parte apical posterior do ventrículo.
Essas configurações não são certamente as encontradas em todos
os casos de infarto cardíaco antigo anterior e posterior. A perda local
de músculo e as áreas locais de bloqueio da condução podem causar
as seguintes anormalidades do complexo QRS: padrões bizarros (as ondas
Q proeminentes, por exemplo), diminuição da voltagem e alargamento.
Correntes de lesão na angina do peito. "Angina do peito" significa,
simplesmente, dor nas regiões peitorais da parte superior do tórax,
irradiando-se geralmente para o pescoço e ao longo do braço
esquerdo.
I
Fig. 12.22 Eletrocardiogramas de antigos infartos da parede anterior
e da parede posterior, mostrando a onda Q na derivação I no infarto
antigo da parede anterior, e a onda Q na derivação III no infarto antigo
da parede posterior.
Antes neste capítulo foi ressaltado que a onda T é normalmente
positiva em todas as derivações bipolares-padrão dos membros e que
isso é causado pela repolarização do ápice e das superfícies externas
dos ventrículos antes das superfícies endocárdicas. Essa direção segundo
a qual a repolarização se difunde por sobre o coração é o inverso da
direção em que ocorre a despolarização. (Se os princípios básicos da
onda T ascendente nas derivações-padrão ainda não são conhecidos,
o leitor deve familiarizar-se com a discussão anterior, mais detalhada,
sobre isto antes de passar às seções seguintes.)
A onda T torna-se anormal quando a seqüência normal da
repolarização não ocorre. Vários fatores podem alterar esta seqüência de
repolarização, como se segue.
EFEITO DA CONDUÇÃO LENTA DA ONDA DE
DESPOLARIZAÇÃO SOBRE A ONDA T
Voltando à Fig. 12.14, observe que o complexo QRS está
consideravelmente alargado. A razão desse alargamento é o retardo da
condução no ventrículo esquerdo, como conseqüência de bloqueio do
ramo esquerdo. O ventrículo esquerdo se despolariza aproximadamente
0,08 s após a despolarização do ventrículo direito, o que produz forte
vetor médio do QRS para a esquerda. O período refratário das massas
musculares ventriculares esquerda e direita não difere muito um do
outro. Por esta razão, o ventrículo direito começa a se repolarizar muito
antes do esquerdo; isso causa positividade no ventrículo direito e
negatividade no esquerdo. Em outras palavras, o eixo médio da onda T
apresenta um desvio para a direita, o que é o contrário do eixo elétrico
médio do complexo QRS nesse mesmo eletrocardiograma. Assim,
quando a condução do impulso pelos ventrículos sofre grande retardo, a
onda T é quase sempre de polaridade oposta à do complexo QRS.
Na Fig. 12.15 e em várias figuras do capítulo seguinte, a condução
também não ocorre pelo sistema de Purkinje. Como conseqüência, a
velocidade da condução fica muito identificada e, em cada caso, a
onda T é de polaridade oposta à do complexo QRS, quer seja a
condição que cause esse retardo da condução um bloqueio do ramo
esquerdo, um bloqueio do ramo direito, uma contração ventricular
prematura ou outra coisa.
DESPOLARIZAÇÃO PROLONGADA EM PARTES DO
MÚSCULO VENTRICULAR COMO CAUSA DE
ANORMALIDADES DA ONDA T
Se o ápice dos ventrículos tivesse um período de despolarização
anormalmente longo, ou seja, um potencial de ação prolongado, a
repolarização dos ventrículos não se iniciaria no ápice como o faz
normalmente. Em vez disso, a base dos ventrículos iria repolarizar-se
antes do ápice e o vetor da repolarização apontaria do ápice para a
base do coração, o contrário do vetor de repolarização habitual.
Como conseqüência, a onda T em todas as três derivações-padrão seria
negativa, e não positiva como de hábito. Assim, o simples fato de que o
músculo apical do coração apresenta um prolongado período de
despolarização seria suficiente para causar alterações acentuadas na
onda T, até mesmo a ponto de alterar toda sua polaridade, como é
mostrado na Fig. 12.23.
Uma isquemia leve é certamente a mais comum das causas de maior
duração da despolarização do músculo cardíaco, e, quando a isquemia
ocorre em apenas uma área do coração, o período de despolarização
dessa área ocorre desproporcionalmente ao de outras partes. Como
conseqüência, pode haver alterações nítidas na onda T. A isquemia
pode ser conseqüente à oclusão coronária crônica e progressiva, à
oclusão coronária aguda ou à insuficiência coronária relativa, ocorrendo
durante o exercício.
119
Fig. 12.24 Onda T bifásica causada por toxicidade digitálica
Fig. 12.23 Onda T invertida resultante de isquemia leve do ápice dos
ventrículos.
Um meio de se detectar uma leve insuficiência coronária é fazer
o paciente exercitar-se e, então, registrar-se o eletrocardiograma
imediatamente depois, observando-se se ocorrem ou não alterações nas
ondas T. As alterações da onda T não precisam ser específicas, pois
qualquer alteração na onda T em qualquer derivação - inversão, por
exemplo, ou onda bifásica - é freqüentemente evidência suficiente de
que alguma parte do músculo ventricular aumentou seu período de
despolarização desproporcionalmente ao resto do coração, e isso é
provavelmente causado por insuficiência coronária relativa.
Todas as outras condições que podem causar correntes de lesão,
incluindo pericardites, miocardites e traumas mecânicos do coração,
também podem causar alterações de onda T. Ocorre corrente de lesão
quando o período de despolarização de algum músculo é tão longo que o
músculo não se repolariza totalmente antes que o próximo ciclo cardíaco
se inicie. Assim sendo, uma corrente de lesão é, de fato, uma forma
exacerbada de onda T anormal, pois ambas decorrem do aumento do
período de despolarização de uma ou mais partes do músculo cardíaco,
sendo a diferença apenas de grau.
Efeito do digital sobre a onda T. Como é discutido no Cap. 22,
o digital é um medicamento que pode ser usado durante a insuficiência
coronária relativa para aumentar a força de contração do músculo
cardíaco. Entretanto, o digital também aumenta o período de
despolarização
do músculo cardíaco. Ele geralmente aumenta esse período quase na
mesma proporção em todo o músculo ventricular ou na maior parte
*dele, mas, quando são administradas doses excessivas de digital, o
período de despolarização de uma parte do coração pode aumentar
desproporcionalmente ao de outras partes. Como conseqüência, podem
ocorrer alterações inespecíficas, tais como a inversão da onda T ou
ondas T bifásicas, em uma ou mais das derivações
eletrocardiográficas. Uma onda T bifásica causada pela administração
excessiva de digital é apresentada na Fig. 12.24. Também há pequena
quantidade de corrente de lesão. Isso decorre, provavelmente, da
despolarização contínua de parte do músculo ventricular.
As alterações de onda T durante a administração de digitálicos são
os primeiros sinais da toxicidade digitálica. Caso se administre ao paciente
quantidade ainda maior do medicamento, podem surgir fortes correntes
de lesão. Da mesma forma, o digital pode bloquear a condução dos
impulsos cardíacos para diversas partes do coração, de modo que podem
resultar daí várias arritmias. É clinicamente desejável impedir-se os
efeitos do digital de ir além do estágio de leves anormalidades da
onda T. Por esta razão, o eletrocardiógrafo é utilizado de rotina no
acompanhamento dos pacientes digitalizados.
REFERÊNCIAS
Ver as referências do Cap. 13.
120
CAPÍTULO 13
Arritmias Cardíacas e Sua Interpretação
Eletrocardiográfica
Alguns dos tipos mais perturbadores de disfunção cardíaca não ocorrem
como conseqüência de anormalidade do músculo cardíaco, mas, sim.
de um ritmo cardíaco anormal. Por vezes, a freqüência cardíaca é
demasiado rápida ou lenta para bombear quantidade de sangue
adequada; por vezes, o intervalo entre os batimentos cardíacos é
demasiado curto para que os ventrículos se encham; e por vezes o
batimento dos átrios é totalmente descoordenado em relação ao dos
ventrículos, de modo que os átrios não funcionam mais como
preparadores dos ventrículos,
O objetivo do presente capítulo é o de discutir as arritmias cardíacas
mais comuns e seu efeito sobre o bombeamento cardíaco, assim como
seu diagnóstico pela eletrocardiografia. As causas das arritmias cardíacas
são geralmente uma das seguintes anormalidades no sistema de
ritmicidade — condução do coração ou de suas combinações:
1. Ritmicidade anormal do marcapasso.
2. Deslocamento do marcapasso do nodo sinusal para outras partes
do coração.
3. Bloqueios em diferentes pontos da transmissão de impulsos
através do coração.
4. Vias anormais de transmissão de impulsos pelo coração.
5. Geração espontânea de impulsos anormais em praticamente toda
e qualquer parte do coração.
RITMO SINUSAL ANORMAL
TAQUICARDIA
O termo “taquicardia” significa freqüência cardíaca elevada,
definida geralmente como acima de 100 batimentos por minuto. O
eletrocardiograma registrado em paciente com taquicardia é apresentado
na Fig. 13.1. Esse eletrocardiograma é normal, exceto que a freqüência
dos batimentos cardíacos, determinada pelo intervalo de tempo entre os
complexos GRS, é de aproximadamente 150 por minuto, em vez dos
72 por minuto normais.
As três causas gerais de taquicardia são o aumento da temperatura
corporal, a estimulação do coração pelos nervos simpáticos e
condições tóxicas do coração.
A freqüência cardíaca aumenta aproximadamente 15 batimentos
por minuto para cada centígrado de aumento da temperatura corporal
até a temperatura corporal de cerca de 41ºC; acima disto, a freqüência
cardíaca reduz-se de fato devido ao progressivo enfraquecimento do
músculo cardíaco produzido pela febre. A febre causa taquicardia porque
a elevação da temperatura aumenta o metabolismo do nodo sinusal,
o que, por sua vez, aumenta diretamente sua excitabilidade e freqüência
rítmicas.
Muitos fatores podem fazer o sistema nervoso simpático excitar
o coração como discutimos em múltiplos pontos deste texto. Quando
um paciente perde sangue e entra em estado de choque ou semichoque,
por exemplo, a estimulação reflexa do coração aumenta sua freqüência
até 150 a 180 batimentos por minuto. Assim, também o simples
enfraquecimento do miocárdio aumenta geralmente a freqüência
cardíaca, porque o coração enfraquecido não bombeia sangue para a
árvore arterial de maneira normal, e isso evoca reflexos que aumentam a
freqüência do coração.
BRADICARDIA
O termo "bradicardia" significa uma freqüência cardíaca baixa
definida geralmente como abaixo de 60 batimentos por minuto. A
bradicardia é mostrada no eletrocardiograma na Fig. 13.2.
Bradicardia em atletas. O coração de um atleta é consideravelmente
mais forte que o da pessoa normal, fato que lhe possibilita bombear
maior débito sistólico por batimento. A excessiva quantidade de sangue
bombeada para a árvore arterial a cada batimento desencadeia reflexos
circulatórios ou outros efeitos que ocasionam a bradicardia.
Estimulação vagal como causa de bradicardia. Qualquer reflexo
circulatório que estimule o nervo vago pode fazer a freqüência cardíaca
diminuir consideravelmente. devido ao efeito inibitório que os sinais
nervosos parassimpáticos têm sobre a função cardíaca. Talvez o exemplo
mais notável disto ocorra em pacientes com a síndrome do seio carotídio.
Nesses pacientes, um processo artenosclerótico na região do seio
carotídeo na artéria carótida causa sensibilidade excessiva dos
receptores da pressão (barorreceptores) localizados na parede arterial;
como conseqüência, uma leve pressão no pescoço evoca forte reflexo
barorreceptor, causando intensa estimulação vagal do coração e
bradicardia extrema. De fato, por vezes este reflexo é tão potente que
até faz parar o coração.
ARRITMIA SINUSAL
A Fig. 13.3 apresenta um registro cardiotacométrico da freqüência
cardíaca durante a respiração normal e a respiração profunda. O
cardiotacômetro é um instrumento que registra, pela altura dos
potenciais em ponta sucessivos, a duração do intervalo entre cada dois
complexos QRS no eletrocardiograma. Note. por esse registro, que a
freqüência cardíaca aumenta e diminui aproximadamente 5% durante as
diversas fases do ciclo respiratório em repouso. Entretanto, durante a
respiração profunda, como é mostrado à direita da Fig. 13.3, a
freqüência cardíaca, ainda que normalmente, aumenta e diminui por
até 30% a cada ciclo respiratória.
A arritmia sinusal pode ocorrer como conseqüência de, qualquer
um dos muitos reflexos circulatórios ou outros efeitos nervosos que
afetam a potência dos sinais nervosos simpáticos e parassimpáticos
para o nodo sinusal. No tipo respiratório de arritmia sinusal mostrado
na Fig. 13.3, isto decorre principalmente do "derrame" de sinais do
centro respiratório bulbar para o centro vasomotor durante os ciclos
inspiratório e expiratório da respiração.
121
Fig. 13.1 Taquicardia sinusal (derivação I).
Fig. 13.3 Arritmia sinusal conforme detectada por cardiotacômetro. À
esquerda está o traçado obtido enquanto o indivíduo estava respirando
normalmente; à direita, enquanto ele respirava profundamente.
Os sinais de derrame causam aumentos e reduções alternados do
número de impulsos transmitidos para o coração pelos nervos simpáticos
e pelo nervo vago.
quando a condução pelo nodo e feixe A-V diminui nessa escala, a
condução cessa totalmente. Assim, quando o intervalo P-R de um
paciente está se aproximando desses limites, um pequeno aumento
adicional na gravidade da condição vai bloquear a condução de
impulsos em vez de simplesmente retardar mais a condução.
Um dos meios para a determinação da gravidade de algumas
cardiopatias —febre reumática aguda, por exemplo — é a medida do
intervalo P-R.
Bloqueio de segundo grau. Quando a condução pela junção AV é lentificada até o intervalo P-R estender-se por 0,25 a 0,45 s, os
potenciais de ação que passam pelo nodo A-V são por vezes
suficientemente fortes para passar pelo nodo A-V e, em outras
ocasiões, não o são. Muitas vezes, o impulso passa para os
ventrículos após a contração atrial e deixa de fazê-lo na primeira ou
segunda contrações subseqüentes, alternando, pois, entre a condução e a
não-condução. Neste caso, os átrios batem com freqüência
consideravelmente maior que a dos ventrículos e diz-se que há "falha
dos batimentos" ventriculares. Essa condição é denominada bloqueio
cardíaco de segundo grau incompleto.
A Fig. 13.6 mostra intervalos P-R de 0,30 s, ilustrando também
uma falha do batimento como conseqüência da ausência de condução
dos átrios para os ventrículos.
Por vezes, a falha atinge os batimentos cardíacos alternados de
forma constante, de modo que se estabelece um ritmo 2:1 no coração,
batendo os átrios duas vezes para cada batimento dos ventrículos. Por
vezes, também ocorrem outros ritmos, tais como 3:2 ou 3:1.
Bloqueio A-V completo (bloqueio de terceiro grau). Quando a
condição que causa a condução deficiente no nodo ou feixe A-V se torna
extremamente grave, há um bloqueio do impulso dos átrios para os
ventrículos. Nesse caso, as ondas P se dissociam totalmente dos
complexos QRS-T, como é mostrado na Fig. 13.7. Note que a
freqüência rítmica atrial nesse eletrocardiograma é de
aproximadamente 100 batimentos por minuto, enquanto a freqüência
dos batimentos ventriculares é de menos de 40 por minuto. Além
disso, não há qualquer relação entre o ritmo dos átrios e o dos
ventrículos, pois os ventrículos “escaparam” ao controle dos átrios e estão
batendo com seu ritmo próprio e natural.
Síndrome de Stokes-Adams
escape ventricular. Em alguns
pacientes com bloqueio A-V, o bloqueio total vai e volta — ou seja,
são conduzidos impulsos dos átrios para os ventrículos por um dado
período e, então, de súbito, não é transmitido absolutamente qualquer
impulso. A duração do bloqueio total pode ser de alguns segundos, alguns
minutos ou algumas horas ou podem passar semanas ou até mais
tempo antes que a condução retorne. Essa condição ocorre
particularmente em corações com isquemia fronteiriça (borderline).
Imediatamente apôs o início do bloqueio da condução A-V, os
ventrículos param totalmente de se contrair por 5 a 30 s, devido ao
fenômeno denominado supressão por estimulação excessiva, o que quer
dizer que a excitabilidade dos ventrículos foi suprimida por terem sido
estimulados pelos átrios com freqüência maior que seu ritmo natural.
Finalmente, alguma parte do sistema de Purkinje além do bloqueio —
geralmente na parte distai do nodo A-V, além do ponto bloqueado
neste nodo ou no feixe A-V — começa a descarregar ritmicamente
com freqüência de 15 a 40 vezes por minuto e a agir como
marcapasso ventricular. Isto é denominado escape ventricular.
RITMOS ANORMAIS CONSEQUENTES AO BLOQUEIO
DA CONDUÇÃO DE IMPULSOS
BLOQUEIO SINOATRIAL
Em raros casos, o impulso do nodo sinusal é bloqueado antes de
chegar ao músculo atrial. Este fenômeno é mostrado na Fig. 13.4, que
mostra a súbita cessação das ondas P, com conseqüente parada do átrio.
Entretanto, o ventrículo adquire um novo ritmo, geralmente originado
no nodo A-V, de modo que o complexo QRS-T ventricular não se altera.
BLOQUEIO ATRIOVENTRICULAR
O único meio pelo qual os impulsos podem passar normalmente
dos átrios para os ventrículos é pelo feixe A-V, também conhecido como
feixe de His. As diferentes condições que podem diminuir a condução
de impulsos por este feixe ou bloqueá-la totalmente são;
O A isquemia das fibras do nodo A-V ou do feixe A-V
freqüentemente retarda ou bloqueia a condução dos átrios para os
ventrículos.
A insuficiência coronária pode causar a isquemia do nodo e do feixe
A-V, da mesma maneira como pode causar a isquemia do miocárdio.
2. A compressão do feixe A-V por um tecido cicatricial ou por
partes calcificadas do coração pode deprimir ou bloquear a condução
dos átrios para os ventrículos.
3. A inflamação do nodo A-V ou do feixe A-V pode diminuir a
condutividade entre os átrios e os ventrículos. A inflamação ocorre
freqüentemente devido a diferentes tipos de miocardite, tais como os
que ocorrem na difteria e na febre reumática.
4. A estimulação extrema do coração pelos nervos vagos bloqueia
em raros casos a condução dos impulsos através do nodo A-V. Essa
excitação vagal ocorre ocasionalmente como conseqüência da forte
estimulação dos barorreceptores em pessoas portadoras da síndrome
do seio carotídeo, que foi discutida anteriormente em relação a
bradicardia.
Bloqueio cardíaco incompleto. Prolongamento do intervalo P-R ( ou
P-Q)— “bloqueio de primeiro grau”. O período de tempo que transcorre
normalmente entre o início da onda P e o início do complexo QRS
é de aproximadamente 0,16 s quando o coração está batendo com sua
freqüência normal. Esse intervalo P-R em geral diminui com batimentos
cardíacos mais rápidos e aumenta com batimentos mais lentos. Em geral,
quando o intervalo P-R aumenta acima do valor de cerca de 0.20 s em
coração batendo na freqüência normal, diz-se que ele está prolongado
e diz-se do paciente que ele tem um bloqueio cardíaco incompleto do
primeiro grau. A Fig. 13.5 mostra um eletrocardiograma com
prolongamento do intervalo P-R, sendo esse intervalo, neste caso, de
aproximadamente 0,30 s. Portanto, o bloqueio de primeiro grau é
definido como um retardo da condução dos átrios para os ventrículos,
mas não como um bloqueio real da condução.
O intervalo P-R raramente aumenta acima de 0,35 a 0,45 s, pois.
Fig. 13.2 Bradicardia sinusal (derivação III).
Fig. 13. 4 Bloqueio do nodo S-A com rit mo nodal A-V
(derivação I I I ) .
122
Fig. 13.5 Prolonga ment o do inte rvalo P-R
(der ivação I I ) .
Como o cérebro não pode permanecer ativo por mais de 3 a 5
s sem suprimento sanguíneo, os pacientes geralmente desmaiam por
alguns segundos após ocorrer um bloqueio completo, porque o coração,
nessas condições, não bombeia sangue algum por 5 a 30 s até os
ventrículos "escaparem". Após o escape, até mesmo ventrículos batendo
lentamente em geral bombeiam sangue suficiente para possibilitar a
recuperação rápida do desmaio e, em seguida, para manter a pessoa.
Esses episódios periódicos de desfalecimento são conhecidos como
síndrome de Stokes-Adams.
Ocasionalmente, o intervalo de parada ventricular ao início de um
bloqueio completo é tão longo que se torna prejudicial à saúde do paciente
ou causa a sua morte. Por conseguinte, muitos são agora providos de
um marcapasso artificial, que é um pequeno estimulador elétrico a
bateria, implantado sob a pele, cujos eletródios são ligados geralmente ao
ventrículo direito. Esse marcapasso proporciona impulsos rítmicos
contínuos que assumem o controle dos ventrículos. As baterias são
substituídas mais ou menos uma vez a cada 5 anos.
BLOQUEIO INTRAVENTRICULAR INCOMPLETO ALTERNÂNCIA ELÉTRICA
Muitos dos mesmos fatores que podem causar o bloqueio A-V
também podem bloquear a condução de impulsos nas partes
periféricas do sistema de Purkinje. Em certos casos, há bloqueio
incompleto, de modo que algumas vezes os impulsos são transmitidos e
outras não, causando bloqueio dos impulsos durante alguns ciclos
cardíacos e não durante outros. O complexo QRS pode apresentar-se
consideravelmente anormal, durante os ciclos em que os impulsos são
bloqueados. A Fig. 13.8 mostra a condição conhecida como alternância
elétrica, que decorre de bloqueio intraventricular parcial cm batimentos
alternados. Esse eletrocardiograma também mostra uma taquicardia,
que é provavelmente a razão pela qual ocorreu o bloqueio, pois, quando
a freqüência cardíaca é muito rápida, pode ser impossível a partes do
sistema de Purkinje recuperar-se do período refratário de modo
suficientemente rápido para responder a cada batimento cardíaco
sucessivo. Assim, também, muitas condições que deprimem o coração,
tais como isquemia, miocardite e toxicidade digitálica, podem causar
bloqueio intraventricular, com a conseqüente alternância elétrica.
CONTRAÇÕES PREMATURAS
A contração prematura é uma contração do coração antes do
momento em que seria esperada a contração normal. Esta condição
também é freqüentemente denominada extra-sístole, batimento prematuro
ou batimento ectópico.
Causas das contrações prematuras. Muitas contrações prematuras
decorrem de focos ectópicos no coração, que emitem impulsos anormais
em ocasiões irregulares durante o ritmo cardíaco. Entre as possíveis
causas de foco ectópico estão (1) áreas locais de isquemia; (2) pequenas
placas calcificadas em diferentes pontos do coração, que comprimem
o músculo cardíaco adjacente de tal modo que algumas das fibras são
Fig. 13.6 Bloqueio atrioventricular incompleto de segundo grau
(derivação V3).
Fig. 13.7 Bloqueio atrioventricular completo (derivação II).
irritadas; e (3) irritações tóxicas do nodo A-V. sistema de Purkinje ou
miocárdio causadas por medicamentos, nicotina ou cafeína. O
desencadeamento mecânico das contrações prematuras também é
freqüente durante o cateterismo cardíaco, ocorrendo muitas vezes
grande número de contrações prematuras quando o cateter penetra no
ventrículo direito e comprime o endocárdio.
Em pessoas com cardiopatia isquêmica, o ritmo ectópico é
ocasionalmente considerado como sendo causado por um sinal
reentrante, da maneira que se segue. O batimento cardíaco normal
excita uma área de tecido isquêmico pela qual o impulso cardíaco passa
com extrema lentidão Em seguida, após terminar a contração do
músculo cardíaco normal, o sinal lento do tecido isquêmico escapa
novamente para o músculo cardíaco normal, causando, deste modo, uma
segunda contração numa etapa posterior do batimento cardíaco.
CONTRAÇÕES ATRIAIS PREMATURAS
A Fig. 13.9 é um eletrocardiograrna onde aparece uma só contração
atrial prematura. A onda P deste batimento ocorre demasiado cedo
no ciclo cardíaco, e o intervalo P-R está mais curto, indicando que
a origem ectópica do batimento é próxima do nodo A-V. Além disso,
o intervalo entre a contração prematura e a primeira contração
subseqüente está ligeiramente prolongado, o que é denominado pausa
compensatória. A razão disso é que a contração prematura origina-se
no átrio, a alguma distância do nodo sinusal, e o impulso teve de passar
por quantidade considerável do músculo atrial antes de se descarregar
no nodo sinusal. Por conseguinte, o nodo sinusal descarregou-se muito
tardiamente no ciclo prematuro, e isso faz com que o batimento cardíaco
subseqüente também tenha aparecimento tardio.
As contrações atriais prematuras ocorrem freqüentemente em
pessoas sadias, sendo de fato encontradas muitas vezes em atletas ou
outros indivíduos cujo coração certamente está em condições saudáveis.
No entanto, leves condições tóxicas conseqüentes a fatores tais como
fumar em excesso, ingestão de café em demasia, alcoolismo e o uso de
diversos medicamentos também podem provocar essas contrações.
Déficit de pulso. Quando o coração se contrai antes da hora, os
ventrículos ainda não se encheram normalmente de sangue e o débito
sistólico durante essa contração fica diminuído ou, por vezes, quase
ausente. Por esta razão, a onda de pulso que vai até a periferia após
uma contração prematura pode ser tão fraca que o pulso não pode
ser sentido na artéria radial. Assim, há um déficit no número de pulsações
sentidas no pulso radial relativamente ao número de contrações do coração.
Pulso bigeminado. Por vezes, cada batimento alternado do coração
pode ser uma contração prematura. Isto faz o paciente ter um pulso
bigeminado, ou seja, duas pulsações bem próximas, depois um intervalo
diastólico mais longo, depois novamente duas pulsações, e assim por diante.
CONTRAÇÕES PREMATURAS DO NÓDO A-V OU DO FEIXE
A-V
A Fig. 13.10 mostra uma contração prematura originando-se do
nodo A-V ou do feixe A-V. A onda P está ausente do registro da contra-
Fig. 13.8 Bloqueio intraventricular parcial alternância elétrica ( derivação III)
123
Fig. 13.9 Contração atrial prematura (derivação I).
ção prematura. Em vez disso, ela está superposta ao complexo QRS-T
da contração prematura, porque o impulso cardíaco vai de volta até
os átrios, ao mesmo tempo que segue adiante até os ventrículos; essa
onda P distorce o complexo, mas ela própria não pode ser distinguida
como tal.
Em geral, as contrações prematuras do nodo A-V têm o mesmo
significado e as mesmas causas das contrações atriais prematuras.
CONTRAÇÕES PREMATURAS VENTRICULARES
O eletrocardiograma da Fig. 13.11 mostra uma série de contrações
prematuras ventriculares (CPVs) alternando-se com contrações normais.
A maioria das CPVs desse tipo decorre provavelmente de um sinal
reentrante de área isquêmica do músculo ventricular, como foi descrito
antes. Isso causa vários efeitos no eletrocardiograma, da seguinte
maneira:
1. O complexo QRS está em geral consideravelmente alargado.
A razão é que o impulso é conduzido em sua maior parte pelo músculo
de condução lenta do ventrículo, e não pelo sistema de Purkinje.
2. O complexo QRS tem voltagem muito alta, pela seguinte razão:
ao atravessar o coração, o impulso normal passa por ambos os ventrículos
quase simultaneamente; como conseqüência, os dois lados do coração
se neutralizam parcialmente. Entretanto, quando ocorre a CPV, o
impulso segue apenas em uma direção, de modo que não há esse
efeito de neutralização, e todo um lado do coração se despolariza
enquanto o outro ainda está inteiramente polarizado; isso ocasiona
intensos potenciais elétricos.
3. Após quase todas as CPVs, a onda T tem potencial oposto ao
do complexo QRS, porque a condução lenta do impulso faz com que
a área que se despolarizou primeiro também se repolarize primeiro.
Em conseqüência, a direção do fluxo de corrente no coração, durante
a repolarização, é oposta àquela durante a despolarização, e o potencial
da onda T é o inverso daquele do complexo QRS. Isso não ocorre
cora a onda T normal, como explicado no Cap. 11.
Algumas CPVs têm origem relativamente benigna e decorrem de
fatores simples como cigarros, café, falta de sono, diversos estados tóxicos
leves e, até mesmo, de irritabilidade emocional. Por outro lado, grande
proporção das CPVs decorre de impulsos errantes ou sinais reentrantes
originados em torno das margens de áreas infartadas ou isquêmicas do
coração. Assim sendo, a presença dessas CPVs não deve ser deixada
de lado. As estatísticas mostram que as pessoas com número significativo
de CPVs, têm chance muito acima do normal de vir a apresentar fibrilação
ventricular espontânea letal, presumivelmente iniciada por uma das
próprias CPVs. Isto é particularmente verdadeiro quando as CPVs
ocorrem durante o período vulnerável de ocorrência da fibrilação,
imediatamente ao final da onda T, quando os ventrículos estão saindo do
estado refratário, como é explicado mais adiante no capítulo.
Análise vetorial da origem da contração prematura ventricular
ectópica. No Cap. 12 foram explicados os princípios da análise
vetorial. Aplicando-se esses princípios, pode-se determinar, pelo
eletrocardiograma aa Fig. 13.11, o ponto de origem das CPVs como se
segue. Note que os potenciais das contrações prematuras nas
derivações II e III
Fig. 13.11 Contrações prematuras ventriculares (CPVs) ilustradas pelos
grandes complexos QRS-T anormais (derivações II e III). O eixo das
contrações prematuras é representado graficamente de acordo com os
princípios de análise vetorial explicados no Cap. 12.
são ambos fortemente positivos. Representando-se graficamente esses
potenciais sobre os eixos das derivações II e III e resolvendo-se por
análise vetorial para o vetor médio do QRS no coração, verifica-se que
esse vetor da contração prematura tem sua extremidade negativa
(origem) na base do coração e sua extremidade positiva voltada para
o ápice. Portanto, a primeira parte do coração a se despolarizar
durante a contração prematura encontra-se próximo à base do coração,
que é, pois, a localização do foco ectópico.
TAQUICARDIA PAROXÍSTICA
Anormalidades de qualquer parte do coração, incluindo os átrios,
o sistema de Purkinje e os ventrículos, podem, ocasionalmente, causar
uma descarga rítmica de impulsos que se difunde em todas as direções
pelo coração. Isto é supostamente causado com mais freqüência por
vias reentrantes que estabelecem auto-reexcitação local repetida. Devido
ao ritmo rápido do foco irritável, este foco torna-se o marcapasso do
coração. O termo “paroxística” significa que a freqüência cardíaca
torna-se em geral muito rápida em paroxismos, que se iniciam
subitamente c duram alguns segundos, alguns minutos, algumas horas
ou, por vezes, muito mais tempo. Os paroxismos terminam, então, em
geral tão subitamente quanto haviam se iniciado, e o marcapasso volta a ser
o nodo sinusal. A taquicardia paroxística pode cessar freqüentemente ao
ser evocado um reflexo vagal. Um tipo estranho de reflexo vagal por
vezes evocado com esta finalidade é o que ocorre quando é aplicada
pressão dolorosa sobre os olhos. Também a pressão sobre os seios
carotídeos pode ocasionalmente evocar um reflexo vagal suficiente para
fazer cessar a taquicardia. Diversos medicamentos também podem ser
usados, entre os quais estão com freqüência a quinidina e a lidocaína, que
deprimem o aumento normal na permeabilidade da membrana do
músculo cardíaco ao sódio durante a geração do potencial de ação,
bloqueando, assim, freqüentemente, a descarga rítmica da região focal
que está causando o ataque paroxístico.
TAQUICARDIA PAROXÍSTICA ATRIAL
A Fig. 13.12 mostra, no meio do registro, um súbito aumento da
freqüência dos batimentos cardíacos, de aproximadamente 95 para cerca
de 150 batimentos por minuto. A observação cuidadosa do
eletrocardiograma evidencia uma onda P invertida antes de cada um dos
complexos QRS-T durante o paroxismo de batimentos cardíacos
rápidos, e essa onda P está parcialmente superposta ã onda T normal
do batimento anterior.
Fig. 13.10 Contração nodal A-V prematura (derivação III).
124
Fig. 13.12 Taquicardia paroxística atrial — início na metade do traçado
(derivação I).
Isso indica que a origem dessa taquicardia paroxística é no átrio, mas,
como a onda P é anormal, a origem não é próxima do nodo sinusal.
Taquicardia paroxística nodal A-V. A taquicardia paroxística
decorre muito freqüentemente de um ritmo aberrante que envolve o
nodo A-V. Uma das causas postuladas desse tipo de taquicardia é um
sinal reentrante local que chega ao nodo A-V como se segue. Acredita-se
que algumas das fibras do nodo A-V conduzem muito mais que outras.
Então, após o impulso cardíaco ter chegado aos ventrículos, um sinal
reentrante volta pelas fibras anteriormente não excitadas, iniciando,
assim, um novo impulso do nodo A-V. Desse modo, há nova contração
ventricular, seguida pela reentrada, mais uma vez. por meio das
fibras do nodo A-V de recuperação rápida e a continuação do ciclo
repetitivo.
Independentemente da causa, a taquicardia do nodo A-V geralmente
causa complexos QRS-T praticamente normais, mas ondas P ausentes
ou obscurecidas.
A taquicardia paroxística atrial ou a do A-V, ambas denominadas
taquicardia supraventriculares, ocorrem geralmente em pessoas jovens e
sadias em todos os demais aspectos, e essas pessoas em geral perdem a
predisposição à taquicardia após a adolescência. Em geral, a taquicardia
supraventricular assusta muito o indivíduo e pode causar fraqueza
durante os paroxismos, mas só raramente provoca danos permanentes
pelos ataques.
TAQUICARDIA PAROXÍSTICA VENTRICULAR
A Fig. 13.13 mostra um paroxismo típico e curto de taquicardia
ventricular. O eletrocardiograma da taquicardia paroxística ventricular
tem a aparência de uma série de batimentos ventriculares prematuros,
ocorrendo um após o outro sem qualquer batimento normal entre eles.
A taquicardia paroxística ventricular é geralmente uma condição
grave, por duas razões. Em primeiro lugar, esse tipo de taquicardia
não costuma ocorrer, a não ser que consideráveis lesões isquêmicas
estejam presentes nos ventrículos. Segundo, a taquicardia ventricular
inicia muito freqüentemente a fibrilação ventricular, devido à
estimulação rápida e repetida do músculo ventricular, como discutimos na
seção seguinte.
acontece, muitas pequenas partes do músculo ventricular se contraem
ao mesmo tempo, enquanto número igual de outras partes se relaxa.
Assim, nunca ocorre contração coordenada de todo o músculo cardíaco
de uma só vez, o que é necessário para o ciclo de bombeamento do
coração. Por esta razão, apesar do fluxo maciço de sinais estimulatórios
por todos os ventrículos, a câmara ventricular não se dilata nem se
contrai, permanecendo cm estágio intermediário de contração parcial,
não bombeando absolutamente qualquer sangue, nem em quantidade
desprezível. Assim sendo, após iniciada a fibrilação, a perda de
consciência ocorre dentro de 4 a 5 segundos por ausência de fluxo
sanguíneo para o cérebro, ocorrendo a morte irreversível dos
tecidos em todo o corpo dentro de alguns minutos.
Múltiplos fatores podem desencadear fibrilação ventricular - com
um batimento cardíaco normal em um segundo e, um segundo após,
os ventrículos em fibrilação. São particularmente capazes de desencadear
a fibrilação (1) choque elétrico súbito ao coração ou (2) isquemia do
músculo cardíaco, seu sistema especializado de condução, ou ambos.
Em qualquer desses casos, pode ser estabelecido um padrão instantâneo
de sinais de reentrada, de modo que os impulsos contrateis circulam
repetidamente pelo músculo cardíaco. Esse fenômeno também é
freqüentemente denominado movimento circular.
O FENÔMENO DE “REENTRADA” - MOVIMENTOS
CIRCULARES
COMO
BASE
DA
FIBRILAÇÃO
VENTRICULAR
Após ter passado por toda a extensão dos ventrículos, o impulso
cardíaco normal não tem, então, qualquer outro lugar para ir, porque
todo o músculo ventricular está, nesse momento, refratário e não pode
conduzir os impulsos para adiante. Portanto, esse impulso se desvanece
e o coração aguarda novo sinal de início do nodo sinusal.
Entretanto, em algumas circunstâncias essa seqüência normal de
eventos não ocorre. Vamos, pois, explicar de modo mais completo as
condições básicas que podem desencadear a reentrada e levar aos
movimentos circulares da fibrilação ventricular.
A Fig. 13.14 mostra várias pequenas liras de músculo cardíaco
cortadas em forma de círculos. Quando uma dessas tiras é estimulada
na posição das 12 horas, de modo que o impulso segue apenas uma
direção. o impulso difunde-se progressivamente em torno do círculo
até voltar para a posição das 12 horas. Quando as fibras musculares
originalmente estimuladas ainda estão no estado refratário, o impulso
cessa, pois o músculo refratário não pode transmitir um segundo
impulso. Contudo, há três condições diferentes que podem fazer esse
impulso continuar seu percurso em torno do círculo, ou seja, causar a
"reentrada" do impulso no músculo que já havia sido excitado.
Em primeiro lugar, quando a via em torno do círculo é longa, até
o impulso retornar ã posição de 12 horas, o músculo originalmente
estimulado não vai mais estar refratário, e o impulso vai continuar em
torno do círculo mais e mais vezes.
Segundo, quando a extensão da via permanece constante mas a
velocidade de condução diminui o suficiente, vai transcorrer maior
intervalo antes que o impulso retorne á posição de 12 horas.
FIBRILAÇÃO VENTRICULAR
A mais grave de todas as arritmias cardíacas é a fibrilação ventricular.
quase que invariavelmente fatal quando não tratada de pronto.
A fibrilação ventricular é conseqüente a impulsos cardíacos, que
ficaram descontrolados no interior da massa ventricular, estimulando,
primeiro, uma parte do músculo ventricular, depois outra parte, depois
outra e, finalmente, outra, e voltando por elas para reexcitar o mesmo
músculo ventricular mais e mais vezes, sem cessar jamais. Quando isso
Fig. 13.13 Taquicardia ventricular paroxística (derivação III).
Fig. 13.14 O movimento circular, mostrando a anulação dos impulsos
na via curta e a propagação contínua dos impulsos na via longa.
125
A esta altura, o músculo originalmente estimulado pode ter saído do
estado refratário e o impulso pode continuar em torno do círculo
novamente.
Terceiro, o período refratário do músculo pode ficar muito encurtado.
Nesse caso, o impulso também poderia continuar a fazer a volta no
círculo.
Todas essas três condições ocorrem em diferentes estados
patológicos do coração humano, da seguinte forma: (1) uma via longa
ocorre freqüentemente, em corações dilatados; (2) a redução da
velocidade de condução ocorre freqüentemente em conseqüência do
bloqueio do sistema de Purkinje, isquemia do músculo, potássio
sanguíneo elevado e muitos outros fatores; (3) período refratário mais
curto ocorre freqüentemente em resposta a vários medicamentos,
como a epinefrina, ou após estimulação elétrica repetida. Assim, em
muitos distúrbios cardíacos, a reentrada pode causar padrões anormais
de contração cardíaca ou ritmos cardíacos anormais que ignoram
completamente os efeitos de marcapasso do nodo sinusal.
O mecanismo de “reação em cadeia” da fibrilação
Na fibrilação ventricular são vistas muitas ondas contrateis,
pequenas e distintas, difundindo-se ao mesmo tempo em direções
diferentes pelo músculo cardíaco. É evidente, então, que os impulsos
reentrantes na fibrilação não são simplesmente um só impulso movendose em círculo, como é mostrado na Fig. 13.14. Em vez disso, eles
degeneraram a uma série de múltiplas frentes de onda que têm a
aparência de uma "reação em cadeia". Uma das melhores maneiras de
explicar esse processo na fibrilação é descrever o desencadeamento da
fibrilação por um choque elétrico ocasionado por corrente elétrica
alternada de 60 ciclos.
Fibrilação causada por corrente alternada de 60 ciclos. Num
ponto central dos ventrículos do coração A na Fig. 13.15, um
estímulo de 60 ciclos c aplicado por meio de um eletródio estimulador.
O primeiro ciclo do estímulo elétrico faz com que uma onda de
despolarização se difunda em todas as direções, deixando todo o
músculo por sob o eletródio no estado refratário. Após cerca de 0,25
s, este músculo começa a sair do estado refratário. Entretanto, algumas
partes do músculo saem desse estado antes das outras. Esse estado de
coisas é mostrado no coração A por muitas manchas claras, que
representam o músculo cardíaco excitável, e manchas escuras, que
representam o músculo ainda refratário. Novos estímulos pelo eletródio
podem agora fazer com que os impulsos sigam em algumas direções
pelo coração, mas não em todas elas. Assim, no coração A, alguns
impulsos percorrem uma curta distância até chegarem a uma área
refratária e serem bloqueados. Outros impulsos, porém, passam por
entre as áreas refratárias e continuam seu trajeto por áreas excitáveis
do músculo. Vários eventos transcorrem, então, em rápida sucessão,
todos eles ocorrendo simultaneamente e acabando por levar ao estado de
fibrilação. São eles os seguintes:
Em primeiro lugar, o bloqueio dos impulsos numa direção, com
a transmissão efetiva em outras direções, cria uma das condições
necessárias ao desenvolvimento de um sinal reentrante - ou seja, a
transmissão
Fig. 13.15 A.Desencadeamento de fibrilação num coração quando
estão presentes áreas esparsas de musculatura refratária. B, Propagação
contínua de impulsos fibrilatórios no ventrículo em fibrilação.
de algumas das ondas de despolarização em torno do coração em apenas
uma direção. Como conseqüência, essas ondas não se encontram com
outras ondas seguindo na direção oposta, e, portanto, não se anulam
no lado oposto do coração, podendo continuar cm torno dos ventrículos.
Segundo, a estimulação rápida do coração produz duas alterações
no músculo cardíaco propriamente dito, ambas as quais predispõem ao
movimento circular: (1) a velocidade de condução pelo coração diminui,
o que possibilita um tempo maior para os impulsos fazerem o percurso
em torno do coração. (2) O período refratário do músculo é reduzido,
permitindo a reentrada do impulso no músculo cardíaco antes excitado
num intervalo muito mais curto que o normal.
Terceiro, uma das características mais importantes da fibrilação é
a divisão de impulsos, como é mostrado no coração A. Ao chegar a
uma área refratária no coração, as ondas de despolarização passam em
torno da área nas duas direções. Assim, um só impulso transforma-se
em dois. Quando cada um deles chega à outra área refratária, também
se divide para formar dois outros impulsos. Desse modo, muitas novas
e diferentes ondas estão sendo continuamente formadas no coração por
uma reação em cadeia progressiva, até que, por fim, existem muitas
pequenas ondas de despolarização seguindo em muitas direções
diferentes ao mesmo tempo. Além disso, esse padrão irregular do
trajeto dos impulsos gera uma via circular para o percurso desses
impulsos, aumentando de muito a extensão da via condutiva, que é uma das
condições que mantêm a fibrilação. Isso também ocasiona um padrão
irregular contínuo de áreas refratárias esparsas no coração. Pode-se ver
prontamente que foi iniciado um círculo vicioso. Mais e mais impulsos
são formados, esses ocasionam mais e mais regiões esparsas do músculo
refratário e as áreas refratárias esparsas causam a divisão adicional dos
impulsos. Portanto, cada vez que uma área individual do músculo cardíaco
sai do estado refratário, há sempre um impulso por perto para reentrar
nela.
O coração B na Fig. 13.15 mostra o estado final na fibrilação. Podem
ser vistos aí muitos impulsos seguindo em todas as direções, alguns
dividindo-se e aumentando o número dos impulsos, enquanto outros
são totalmente bloqueados pelas áreas refratárias.
Período vulnerável para o surgimento da fibrilação
ventricular. O período do ciclo cardíaco durante o qual é provável que
haja áreas simultâneas do músculo cardíaco em estado refratário e em
estado não-refratário é exatamente quando o coração se recupera do
ciclo cardíaco anterior — ou seja, justamente ao final da contração
cardíaca. Esse momento do ciclo é, portanto, considerado como sendo o
período vulnerável dos ventrículos no que concerne a fibrilação. De fato,
um só choque elétrico durante esse período vulnerável pode levar
freqüentemente ao bizarro padrão de impulsos difundindo-se
unidirecionalmente em torno das áreas refratárias do músculo,
desencadeando, pois, a fibrilação.
O eletrocardiograma na fibrilação ventricular
Na fibrilação ventricular o eletrocardiograma é extremamente
bizarro, como se vê na Fig. 13.16, e, normalmente, não apresenta
tendência a qualquer tipo de ritmo regular. Nas fases iniciais da
fibrilação ventricular, massas musculares relativamente grandes
contraem-se ao mesmo tempo, e isso causa ondas fortes, porém
irregulares, no eletrocardiograma. Contudo, após apenas alguns
segundos, as grandes contrações dos ventrículos desaparecem e o
eletrocardiograma passa a um padrão de ondas muito irregulares de
baixa voltagem. Assim, não há um padrão eletrocardiográfico repetido
que possa ser atribuído a fibrilação ventricular, exceto que os potenciais
elétricos se alteram constante e espasmodicamente porque as correntes no
coração fluem primeiro em uma direção e depois em outra, raramente
repetindo qualquer ciclo específico.
A voltagem das ondas no eletrocardiograma, na fibrilação
ventricular, é geralmente de cerca de 0,5 mV ao início da
condição, mas
Fig. 13.16 Fibrilação ventricular (derivação II).
126
diminui rapidamente, de tal forma que após 20 a 30 s ela é de apenas
0,2 a 0,3 mV. Pequenas voltagens de 0,1 mV ou menos podem ser
registradas por 10 minutos ou mais após o início da fibrilação ventricular.
Como já foi dito. a fibrilação ventricular é letal se não for interrompida
por alguma terapia heróica, como eletrochoque imediato no coração,
como é explicado na seção que se segue.
A falta de fluxo sanguíneo para o cérebro por mais de 5 a 10 minutos
causa em geral distúrbios mentais permanentes ou até mesmo a destruição
total do cérebro. Ainda que o coração seja revivido, a pessoa pode
morrer pelos efeitos das lesões cerebrais ou viver com distúrbios mentais
permanentes.
FIBRILAÇÃO ATRIAL
Desfibrilação dos ventrículos por eletrochoque
Embora a corrente alternada fraca, quase que invariavelmente, faça
os ventrículos entrarem em fibrilação, a corrente elétrica muito forte
passando pelos ventrículos por curto período pode interromper a
fibrilação, fazendo com que todo o músculo ventricular passe ao estado
refratário a um só tempo. Isso é feito pela aplicação de corrente
intensa por eletródios colocados dos dois lados do coração. A corrente
penetra em muitas das fibras dos ventrículos, estimulando, portanto, ao
mesmo tempo praticamente todas as partes dos ventrículos e tornando-as
refratárias. Todos os impulsos cessam e o coração permanece, então, em
repouso por 3 a 5 s, após o que ele começa a bater novamente, em
geral com o nodo sinusal ou alguma outra parte do coração passando a
ser o marca-passo. Ocasionalmente, porém, o mesmo foco reentrante
que originou a fibrilação dos ventrículos ainda está presente e a
fibrilação imediatamente começa de novo.
Ao serem aplicados os eletródios diretamente nos dois lados do
coração, a fibrilação pode ser em geral interrompida com 110 V de
corrente alternada de 60 ciclos aplicados por 0,1 s ou 1.000 V de corrente
direta aplicados por alguns milésimos de segundo. Quando aplicado
através da parede torácica, como é mostrado na Fig. 13.17, o
procedimento habitual é o de carregar-se um grande capacitor elétrico
com até vários milhares de volts e depois fazer-se esse capacitor
descarregar em alguns milésimos de segundo através dos eletródios e do
coração. Em nosso laboratório, o coração de um cão anestesiado foi
desfibrilado 130 vezes através da parede torácica, e o animal permaneceu
em condições perfeitamente normais.
Bombeamento manual do coração (ressuscitarão
cardiopulmonar) como método auxiliar para a
desfibrilação
A não ser quando desfibrilado dentro de 1 minuto do início da
fibrilação, o coração fica em geral demasiado fraco para ser revivido
apenas por desfibrilação, devido à falta de nutrição pelo fluxo sanguíneo
coronário. No entanto, ainda é possível reviver-se o coração por
bombeamento manual preliminar e posterior a desfibrilação. Desse
modo, há aporte de pequena quantidade de sangue à aorta c renovação do
suprimento sanguíneo coronário. Apôs. alguns minutos, então, é possível
freqüentemente a desfibrilação elétrica. De fato, corações em fibrilação já
foram bombeados manualmente por até 90 minutos antes da desfibrilação.
A técnica de bombear o coração sem abrir o tórax consiste em
pressões intermitentes muito fortes sobre a parede torácica, juntamente
com a respiração artificial. Isso é denominado ressuscitarão
cardiopulmonar, ou simplesmente RCP.
Fig. 13.17 Aplicação de corrente elétrica ao tórax para fazer cessar a
fibrilação ventricular.
Lembre-se que, exceto pela conexão pelo feixe A-V, a massa
muscular atrial é totalmente separada da dos ventrículos, isolados um da
outra por tecido fibroso. Assim sendo, a fibrilação ventricular ocorre
amiúde de forma totalmente independente da fibrilação atrial. Também,
a fibrilação ocorre com freqüência nos átrios independentemente da
fibrilação ventricular, sendo denominada fibrilação atrial; isto é
mostrado à direita na Fig. 13.18.
O mecanismo da fibrilação atrial é idêntico ao da fibrilação
ventricular, exceto que o processo ocorre na massa muscular atrial, e
não na ventricular. Uma causa muito freqüente de fibrilação atrial é a
dilatação atrial conseqüente de lesões das valvas cardíacas, que impede
os átrios de se esvaziarem adequadamente nos ventrículos, ou a
insuficiência ventricular, com acúmulo excessivo de sangue nos átrios.
As paredes atriais dilatadas proporcionam as condições ideais de via
condutiva longa e também uma condução lenta, ambas predisponentes a
fibrilação atrial.
Características do bombeamento atrial durante a fibrilação
atrial. Pelas mesmas razões pelas quais os ventrículos não bombeiam
sangue durante a fibrilação ventricular, os átrios também não bombeiam
sangue na fibrilação atrial. Como conseqüência, eles se tornam inúteis
como bombas preparatórias dos ventrículos. Ainda assim, o sangue flui
passivamente pelos átrios é para dentro dos ventrículos, e a eficácia do
bombeamento ventricular diminui por apenas 20 a 30%. Por esta razão,
em contraste com o caráter letal da fibrilação ventricular, uma pessoa
pode viver meses ou mesmo anos com fibrilação atrial, embora com
reduzida eficiência do bombeamento cardíaco global.
O eletrocardiograma na fibrilação atrial. A Fig. 13.19 mostra o
eletrocardiograma durante a fibrilação atrial. Inúmeras pequenas ondas
e despolarização difundem-se em todas as direções pelos átrios durante
fibrilação atrial. Como as ondas são fracas e por serem muitas delas,
qualquer momento, de polaridade oposta, elas em geral neutralizam-se
umas às outras quase totalmente. Por esta razão, não se pode ver no
eletrocardiograma as ondas P dos átrios, nem registro nítido de
ondas de alta freqüência e voltagem muito baixa. Por outro lado, os
complexos QRS-T são inteiramente normais, a não ser que haja alguma
patologia ventricular, mas sua ocorrência é muito irregular pelas razões
que se seguem.
Irregularidade do ritmo ventricular durante a fibrilação atrial.
Quando os átrios estão fibrilando, os impulsos chegam ao nodo A-V
rapidamente mas também irregularmente. Como o nodo A -V não deixa
passar um segundo impulso por cerca de 0,35 s após o impulso anterior,
pelo menos 0,35 s têm de transcorrer entre uma e outra contração
ventricular, havendo um intervalo adicional porém variável de 0 a 0,6 s
antes que um dos impulsos fibrilantes irregulares chegue ao nodo AV. Assim, o intervalo entre as contrações ventriculares sucessivas varia
do mínimo de 0.35 s ao máximo de cerca de 0,95 s, ocasionando
batimentos cardíacos muito irregulares. De fato, esta irregularidade,
ilustrada pelo espaçamento muito variável dos batimentos cardíacos
no eletrocardiograma da Fig. 13.18, é um dos achados clínicos
utilizados para o diagnóstico
Fig. 13.18 Trajeto dos impulsos no flutter atrial e na fibrilação atrial.
127
Fig. 13.19 Fibrilação atrial ( derivação I)
Fig. 13.20 Fluter atrial – ritmos 2:1 e 3:1 ( derivação I)
da condição. Assim também, devido à rápida freqüência dos impulsos
fibrilatórios nos átrios, o ventrículo é em geral estimulado a manter
freqüência cardíaca rápida, geralmente entre 125 e 150 batimentos
por minuto.
Após parada cardíaca temporária, a ressuscitarão cardiopulmonar
geralmente é bastante eficaz no restabelecimento do ritmo cardíaco
normal. Em alguns pacientes, porém, uma miocardiopatia grave leva
a parada cardíaca permanente ou semipermanente, que pode,
obviamente, causar a morte imediata do indivíduo. Em muitos casos,
todavia, impulsos elétricos rítmicos de marcapasso cardíaco
eletrônico implantado têm sido eficazmente utilizados para manter
vivos os pacientes por muitos anos.
Tratamento da fibrilação atrial por eletro choque. Da mesma maneira
que a fibrilação ventricular pode ser reconvertida ao ritmo normal por
um choque elétrico, assim também a fibrilação atrial pode ser
convertida. O procedimento é basicamente o mesmo da conversão
ventricular, ou seja, a passagem de único e intenso choque elétrico
pelos átrios que faz todo o coração passar ao estado refratário por
alguns segundos; em geral vai sobrevir, então, ritmo normal, se o
coração for capaz disso.
FLUTTER ATRIAL
O flutter atrial é outra condição causada por movimentos circulares
nos átrios. Ele difere, porém, da fibrilação atrial porque o sinal
elétrico segue como uma só e grande onda, sempre na mesma direção
em torno da massa muscular atrial. Como é mostrado à esquerda na
Fig. 13.18, essa onda vai geralmente de cima para baixo em torno da
abertura das veias cava superior e inferior.
O flutter atrial causa contração muito rápida dos átrios, geralmente
entre 250 e 300 batimentos por minuto. Contudo, como um lado dos
átrios se contrai enquanto o outro se relaxa, a quantidade de sangue
bombeada pelos átrios é muito pequena. Além disso, os sinais
chegam ao nodo A-V de forma demasiado rápida para que todos
passem aos ventrículos, pois o período refratário do nodo A-V e do
feixe A-V é muito longo para deixar passar mais que uma fração dos
sinais atriais. Por esta razão, há em geral dois a três batimentos atriais
para cada batimento ventricular.
A Fig. 13.20 mostra o eletrocardiograma típico do flutter atrial. As
ondas P são fortes por causa da contração de massas musculares
semicoordenadas. Entretanto, veja no traçado que um complexo
QRS-T segue-se a uma onda P atrial uma vez a cada dois a três
batimentos atriais, dando ritmo 2:1 e 3:1.
PARADA CARDÍACA
A última anormalidade grave do sistema cardíaco de ritmicidade
condução é a parada cardíaca. Ela decorre da cessação de todos os
impulsos rítmicos para o coração. Ou seja, não resta absolutamente
nenhum ritmo espontâneo.
A parada cardíaca tende particularmente a ocorrer durante a anestesia
profunda, quando os pacientes apresentam com freqüência hipóxia
grave, devido à respiração inadequada. A hipóxia impede as fibras
musculares e as fibras condutoras de manter diferenciais normais de
concentração de eletrólitos através de sua membrana e sua
excitabilidade pode ser tão afetada que a ritmicidade automática
desaparece.
REFERENCIAS
Brown, H. F.: Electrophysiology of the sinoatrial node. Physiol Rev
6250 1982. Chung, E. K.: Electrocardiography: Self-Assessment.
East Norwalk, Conn. Appleton & Lange, 1988. Ohung, E. K.:
Principies of Cardiac Arrhythmias. 4th Ed. Baltimore Williams &
Wilkins, 1988. Chung, E. K.: Electrocardiography: Practical
Applications With Vectorial Principies. 2nd Ed. Hagerstown, Md.,
Harper & Row, 1980. Gilmour, R. F., Jr., and Zipes, D. P.: Slow
inward current and cardiac arrhyth mias. Am. J. Cardiol., 55:89B,
1985. Goldman, M. J.: Principies of Clinicai Electrocardiography.
12th Ed. East Norwatk, Conn., Appleton & Lange,
1986.Goldschlager, N., and Goldman, M. J.: Principies of Clinicai
Electrocardiography. 13th Ed. East Norwalk, Conn., Appleton &
Lange, 1986. Guyton, A. C, and Crowel), J. W.: A
stereovectorcardiograph J Lab Clin Med., 40:726, 1952. Hurst, J. W.,
et ai. (eds.): The Heart. 7th Ed. New York, McCraw-Hil] Book Co.,
1990. Johnson, R., and Swartz, M. H.: A Simpliried Approach to
Electrocardiography. Philadelphia, W. B. Saunders Co., 1986.
Josephson, M. E.: Clinicai Cardiac Electrophyaiology:
Techniquesand Interpretations. 2nd Ed. Philadelphia, Lea & Febiger,
1989. Kennedy, H. L.: Ambulatory Electrocardiography and Its
Technolngy. 2nd Ed. Philadelphia, Lea & Fehiger, 1989. Laiken,
N.,et ai,: Interpretation of Electrocardiograms: A Self-Instructional
Approach. 2nd Ed. New York, Raven Press, 1988. Lipman, B.:
Clinicai Electrocardiography. Chicago, Year Book Medicai
Publishers, 1984. Marriott, H. J. L., and Conover, M. B.: Advanced
Concepts in Arrhythmias. 2nd Ed. St. Louis, C. V. Mosby Co., 1989.
Marriott, H. J. L., and Wagner, G.: Practical Eleclrocardiography. Sth
Ed. Baltimore, Williams & Wilkins, 1988. McAnulty, J., and
Rahimtoola, S.: Prognosis in bundle branch block. Annu. Rev. Med,
32:499, 1981. Meijler, F. L.: At rio ventricular conduetion versus
heart size from mouse to whale. J. Am. Coll. Cardiol., 5:363, 1985.
Morgan roth.J.: Ambulatory Holter electrocardiography: Choiceof
technologies and clinicai uses. Ann. Intern. Med., 102:73, 1985. Pick,
A, and Langendorf, R.: Ini erpretation of Complex Arrhythmias.
Philadelphia, Lea & Febiger, 1980. Saksena, S., and Goldschlagfcr.
N. F. (ed.): Eíectrical Therapy for Cardiac Arrhythmias. Philadelphia,
W. B. Saunders Co., 1989. Scher, A. M., and Spach, M. S.: Cardiac
depolarization and repolarization and the electrocardiogram. In
Berne, R. M., et ai. (eds.): Handbook of Physiology. Sec. 2, Vol. I.
Baltimore, Williams & Wilkins, 1979, p. 357. Stein, E.: Interpretation
of Arrhythmias: A Self Study Program. Philadelphia, Lea & Febiger,
1988. Summerall, C. P.: Monitoring Heart Rhythm. New York, John
Wiley & Sons, 1982. Tamargo, J., et ai.: Modulated receptor
hypothesis: Selectivity and ínteractions of antiarrhythmir dmgs. News
Physiol. Sei., 4:88, 1989.
128
Baixar