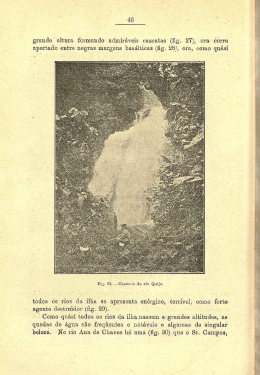
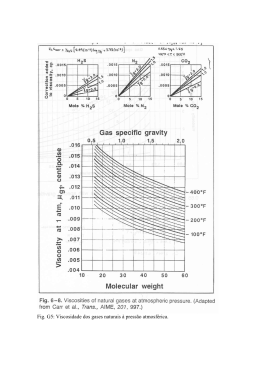
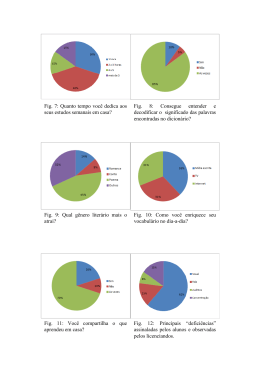
Desenvolvimento de uma metodologia de Bioinformática para a busca de riboswitches de tiamina pirofosfato no genoma humano NAN Jorge¹, CG Ferreira² , A Machado-Lima³ , F Passetti¹ ¹Laboratório de Bioinformática e Biologia Computacional, Coordenação de Pesquisa Clínica e Incorporação Tecnológica, Instituto Nacional de Câncer (INCA) ²Coordenação de Pesquisa Clínica e Incorporação Tecnológica, Instituto Nacional de Câncer (INCA) ³Escola de Artes, Ciência e Humanidades (EACH), Universidade de São Paulo (USP Leste) Apoio Financeiro: INCA/MS, CNPq-PIBIC, Swiss Bridge Foundation and Fundação do Câncer INTRODUÇÃO Riboswitches são estruturas encontradas nas regiões 5' e 3' UTR de alguns mRNAs de bactérias, fungos e plantas. Essas estruturas são formadas pelo aptâmero e plataforma de expressão, a ligação de um metabólito específico ao primeiro leva a mudanças estruturais que controlam a expressão do mRNA através do término prematuro da transcrição, inibição da tradução (Fig.1) ou alteração no splicing, sem participação protéica. Os genes regulados estão envolvidos na biossíntese ou transporte do metabólito alvo. O único riboswitch descrito em eucariotos liga-se a tiamina pirofosfato (TPP). A seqüência e estrutura secundária do aptâmero são bem conservadas em uma família de riboswitch¹, permitindo o uso de métodos de Bioinformática para sua identificação. Fig. 6. Porcentagem de seqüências classificadas de acordo com o e-value obtido com o Infernal nos conjuntos controles. 4. Genoma 4.1 HMMER 34 seqüências com e-value menor que 1x10-4 15 seqüências com score maior que zero 10 seqüências com genes anotados 08 seqüências em íntrons de regiões codificantes 01 em região de splicing alternativo 01 seqüência em UTR 01 seqüência em éxon 4.2 Infernal 05 seqüências com e-value menor que 1x10-4 Todos com score maior que 19,30 02 seqüências com genes anotados Todos em íntrons de regiões codificantes Fig. 1. Riboswitch hipotético: A)controle da transcrição. B) controle da tradução. (Figura adaptada de 2). OBJETIVO Avaliar a eficiência de softwares de cálculos probabilísticos a fim de desenvolver uma metodologia de bioinformática para a busca de riboswitches de TPP no genoma humano. METODOLOGIA Foram criados dois conjuntos controles: “positivo” e “negativo”. O primeiro com seqüências de aptâmeros obtidas no site Rfam³ (“Rfam”) e artigos publicados (“Conhecidos”). O segundo contém seqüências que não são riboswitches criadas através do embaralhamento trinta vezes de cada seqüência do conjunto “positivo” (“Embaralhado Rfam” e Embaralhado Conhecidos”) e outras famílias de RNA com estrutura secundária similar ao riboswitch de TPP (SL2, Glicina e Qrr). Foram avaliados três softwares: RNAshapes4, HMMER5 e Infernal6. Os softwares que apresentaram bons resultados com os conjuntos controles foram usados contra o UniGene7 (versão 207). As regiões candidatas obtidas foram comparadas com os contigs apresentados por Nakaya et al (2007). Fig.7. Regiões candidatas do genoma humano. A região do aptâmero é representada pelo retângulo rosa apontado pela seta de mesma cor. A) Região candidata do gene CHD8. B) Região candidata do gene HAPLN1. C) Região candidata do pseudogene TCRBV20S1A1N2. 4.3. Comparação com Nakaya et al (2007) Fig. 2. Porcentagem de seqüências classificadas de acordo com as probabilidades obtidas com RNAshapes nos conjuntos controles. Fig. 8. Região candidata em comum com Nakaya et al (2007). A região candidata a riboswitch é representada pelo retângulo rosa e o contig indicado por Nakaya et al (2007) é indicado pelo retângulo vermelho. DISCUSSÃO E CONCLUSÃO Fig. 3. Porcentagem de seqüências classificadas de acordo com os scores obtidos com o HMMER nos conjuntos controles Outros artigos relataram grande gasto computacional9 com o uso do RNAshapes, os resultados contraditórios nos conjuntos controles demonstraram que o software não é adequado para a busca no genoma humano. O HMMER apresentou resultados distintos para os conjuntos “negativo” e “positivo”, apesar de não considerar a estrutura secundária adotada pelo mRNA. Todas as seqüências do conjunto “positivo” avaliadas pelo Infernal apresentaram scores acima do valor de classificação do Rfam e nenhuma seqüência do conjunto “negativo” teve scores acima deste valor. Os softwares HMMER e Infernal provaram serem práticos e precisos para a busca de riboswitches durantes os testes realizados neste estudo, sendo assim adequados para a busca no genoma humano. A busca por riboswitch no genoma humano revelou candidatos com scores e e-values piores que os obtidos pelo conjunto controle “positivo” e diferentes dos valores obtidos com o conjunto controle “negativo”. A região candidata encontrada na região intronica transcrita (TIN) do genoma humano sugere um possível papel dos riboswitches no controle da expressão de ncRNAs. As regiões candidatas estão, atualmente, sendo validadas pelo Dr. Fabricio F. Costa no Children's Memorial Research Center em Chicago, USA. Fig. 4. Porcentagem de seqüências classificada de acordo com o e-value obtido com o HMMER nos conjuntos controles. BIBLIOGRAFIA Fig. 5. Porcentagem de seqüências classificadas de acordo com o score obtido com o Infernal nos conjuntos controles. 1.GARST, A. D. & BATEY, R. T., A switch in time: Detailing the life of a riboswitch. Biochim. Biophys. Acta, p1-8, 2009. 2.KIM, J.N. & BREAKER, R.R., Purine sensing by riboswitches. Biology of the cell, v.100, p.1-11, 2008. 3.GARDNER, P. P., et. al., Rfam: updates to the RNA families database., Nucleic Acids Res., 2009. 4.STEFFEN P. et al. RNAshapes: an integrated RNA analysis package based on abstract shapes. Bioinformatics, v.22, p.500-503, 2005. 5.EDDY, S. R., A probabilistic model of local sequence alignment that simplifies statistical significance estimation, PloS Comput Biol, 2008. 6.NAWROCKI, E. P., et. al., Infernal 1.0: inference of RNA alignments., Bioinformatics, 2009. 7. PONTIUS, J. U., WAGNER, L., SCHLUER, D. UniGene: A Unified view of the transcriptome. In: The NCBI Handbook. Bethesda (MD): National Center for Biotechnology Information, 2003. 8. NAKAYA, H. I., et al. Genome mapping and expression analyses of human intronic nonconding RNAs reveal tissue-specific patterns and enrichment in genes related to regulation of transcription. Genome Biol., v.8, n.3, p.R43.1-R43.25, 2007. 9.MACHADO LIMA, A., DEL PORTILLO, H. A., DURHAM, A. M. Computational methods in noncoding RNA search. J. Math. Biol., v.56, n.1-2, p.15-49, 2007. Projeto Gráfico: Serviço de Edição e Informação Técnico-Científica / CEDC / INCA RESULTADOS
Baixar