☰
Explorar
Assinar em
Inscrever-se
Envio
×
Baixar
Sem categoria
eluzir pedrazzi chacon - Universidade Federal Fluminense
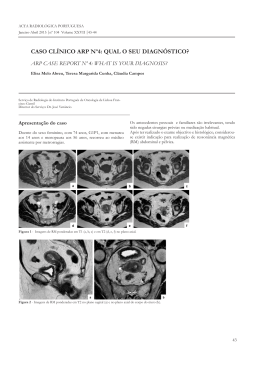
CASO CLÍNICO ARP Nº4: QUAL O SEU DIAGNÓSTICO?
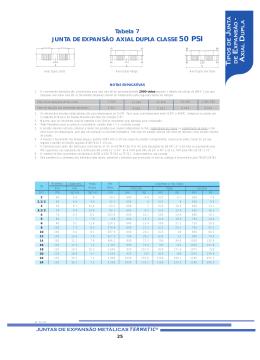
Junta de Expansão Axial Dupla classe 50 PSI
África – Quadro Natural
1. Qual a quantidade de calor necessária para que
Isometrias
xxsic4377 síntese de complexos de ouro(i)
A Circulação geral do ar
América Latina: quintal de atuação dos EUA
desenvolvimento de ligantes contendo braços piridínicos e