UNISALESIANO
Centro Universitário Católico Salesiano Auxilium
Bacharelado em Química
Ana Caroline Colombo Frastrone
Bianca Camila Pereira
Giovanna Amaral de Araújo
Karina Monteiro Soares
CONSTRUÇÃO DE UM FOTÔMETRO DE CHAMA PARA
FINS DIDÁTICOS
LINS – SP
2013
ANA CAROLINE COLOMBO FRASTRONE
BIANCA CAMILA PEREIRA
GIOVANNA AMARAL DE ARAÚJO
KARINA MONTEIRO SOARES
CONSTRUÇÃO DE UM FOTÔMETRO DE CHAMA PARA
FINS DIDÁTICOS
Trabalho de Conclusão de Curso
apresentado à Banca Examinadora
do Centro Universitário Católico
Salesiano
Auxilium,
curso
de
Bacharelado em Química, realizado
sob a orientação do Prof. Me. Olayr
Modesto Júnior.
LINS – SP
2013
Frastrone, Ana Caroline Colombo; Pereira, Bianca Camila; Araujo,
Giovanna Amaral; Soares, Karina Monteiro
Construção de um fotômetro de chama para fins didáticos / Ana
F921f
Caroline Frastrone; Bianca Camila Pereira; Giovanna Amaral Araujo;
Karina Monteiro Soares. -- --Lins, 2013.
65p. il. 31cm.
Monografia apresentada ao Centro Universitário Católico
Salesiano Auxilium – UNISALESIANO, Lins-SP, para graduação em
Bacharel Química, 2013.
Orientadores: Olayr Modesto Junior
1. Química. 2. Pesquisa e Desenvolvimento. 3. Fotômetro de
Chama. 4. Validação. I Título.
CDU 54
ANA CAROLINE COLOMBO FRASTRONE
BIANCA CAMILA PEREIRA
GIOVANNA AMARAL DE ARAÚJO
KARINA MONTEIRO SOARES
CONSTRUÇÃO DE UM FOTÔMETRO DE CHAMA PARA FINS DIDÁTICOS
Trabalho de Conclusão de Curso, apresentado ao Centro Universitário Católico
Salesiano Auxilium, como requisito obrigatório, para obtenção do título de
Bacharel em Química.
Aprovado em: _____/_____/________.
Banca Examinadora:
Professor Orientador: _____________________________________________.
Titulação: ______________________________________________________.
Assinatura: ______________________________.
1º Professor (a): _________________________________________________.
Titulação: ______________________________________________________.
Assinatura: ______________________________.
2º Professor (a): _________________________________________________.
Titulação: ______________________________________________________.
Assinatura: ______________________________.
DEDICATÓRIAS
Dedico esse trabalho com todo amor e carinho aos meus
pais, Sebastiana e Nelson, porque sem o apoio e o esforço deles
para que pudesse ter feito a faculdade, eu não teria conseguido
tornar meu sonho realidade... Dedico também a minha irmã, Erica,
por estar sempre do meu lado, incentivando a caminhar, e
também ao meu namorado.
Bianca
Dedico este trabalho realizado com tanto esforço, a minha
mãe Ivone Donizete Colombo Frastrone e ao meu irmão Diego
Colombo Frastrone, que estiveram ao meu lado me apoiando e
não medindo esforços para que eu conquistasse essa vitória. Em
especial, dedico a meu pai Nivaldo Frastrone que apesar de não
estar mais presente em minha vida foi minha estrutura, sem ele
eu não poderia ter chego aonde cheguei.
Ana Caroline
Dedico esse trabalho à Maria Ângela, minha mãe, a eterna
e incondicional incentivadora dos meus sonhos, a pessoa que
sempre está ao meu lado em todos os momentos. A meu pai,
João Augusto, por ter me proporcionado a oportunidade de um
futuro promissor, fazendo todos os esforços possíveis para dar
continuidade a essa jornada.
Giovanna
Dedico este trabalho, realizado com muito esforço e
perseverança, a mais bela razão de minha existência, minha mãe
Adeleusa, que sempre me apoiou e me incentivou. Às minhas
irmãs Iasmin e Emily e ao meu padrasto Célio.
Karina
AGRADECIMENTOS
A Deus pela oportunidade de viver cada dia com força e
saúde, por sua graça infinita, e seu amor incondicional, a todos
que contribuíram para a realização deste trabalho, assim como
nossas famílias pelo apoio, incentivo e principalmente paciência
nas horas mais difíceis durante a realização do mesmo. Em
especial agradecemos ao nosso orientador Professor Me. Olayr
Modesto Júnior que se mostrou muito atencioso proporcionando
toda a base necessária para a realização do mesmo, contribuindo
com suas experiências e conhecimentos, nos orientando com
toda paciência entendo nossas limitações, fazendo assim sua
concretização,
e
por
fim,
a
instituição
Unisalesiano
que
proporcionou o suporte e o aprendizado para finalizamos mais
essa etapa da vida, com suas instalações, materiais de pesquisas
e corpo discente.
EPÍGRAFE
“Que os vossos esforços desafiem as impossibilidades, lembrai-vos de que as
grandes coisas do homem foram conquistadas do que parecia impossível.”
Charles Chaplin
RESUMO
Este trabalho teve como objeto de estudo a construção e validação de
um fotômetro de chama com os conhecimentos adquiridos durante o curso de
Bacharelado em Química, numa ação interdisciplinar envolvendo Automação
Industrial, Estatística e Quimiometria, Física e Química Analítica. Cientes dos
altos custos dos equipamentos de laboratório químico e do fotômetro de chama
em particular, traçou-se como objetivo deste trabalho a obtenção de um
equipamento de baixo custo, porém, com qualidade suficiente para uso
didático. Para alcançar esse objetivo, estudou-se a luz e sua interação com a
matéria, bem como, cada parte que compõe esse equipamento e os
parâmetros necessários para a validação de seu uso. A escolha do fotômetro
para esta pesquisa se deu tanto pelo fato de seu custo elevado, quanto pela
sua versatilidade e potencial de utilização por diversos cursos da instituição.
Para dar um acabamento mais profissional ao equipamento, foi adquirida uma
carcaça de um fotômetro de chama sem condições de recuperação para
aproveitamento da caixa e nela adaptar os circuitos desenvolvidos. Os
principais componentes utilizados foram: um filtro de interferência específico
para os comprimentos de onda emitidos pelo sódio, um fotodiodo como sensor
para gerar o sinal analógico e o PIC 18F4520 como interpretador e conversor
do sinal analógico para digital. Após a construção, para a etapa de validação,
preparou-se cinco soluções-padrão de sódio nas concentrações entre 0,05 e
0,25 mol/L, as quais foram analisadas em replicatas para a avaliação dos
parâmetros de validação: precisão, exatidão, limite de detecção e
quantificação, robustez, linearidade e faixa linear, reprodutibilidade e
repetitividade, seletividade e especificidade. Os resultados obtidos mostraram
que o objetivo foi alcançado, o fotômetro embora tenha mostrado uma baixa
precisão, revelou-se ter exatidão e robustez satisfatórias.
Palavras chave: Química. Pesquisa e Desenvolvimento. Fotômetro de Chama.
Confiabilidade analítica. Validação.
ABSTRACT
This work had as object of study the construction and validation of a
flame photometer with the knowledge acquired during the course of Bachelor in
Chemistry, an interdisciplinary action involving Industrial Automation, Statistics
and Chemometrics, Physical and Analytical Chemistry. Aware of the high cost
of chemical laboratory equipments and of the flame photometer in particular,
traced as aim of this work to obtain low equipment cost, however, of sufficient
quality for didactic use. To obtain this objective, studied the light and its
interaction with matter, as well as, each part that makes up this equipment and
the parameters necessary for the validation of your use. The choice of the
photometer for this research, occurred both because of their high cost, as for its
versatility and potential for use by the various courses of the institution. To give
a more professional finish to the equipment, was acquired a carcass of a flame
photometer without recovery conditions for use of the box and on it adapt the
developed circuits. The main components used were: a filter of interference
specific for the wavelengths emitted by sodium, a photodiode as sensor to
generate the analog signal and the PIC 18F4520 as interpreter and analogue to
digital converter. After the construction, for the validation stage, prepared five
standard solutions of sodium at concentrations of between 0,05 and 0,25 mol/L,
which were analyzed in replicate for evaluating the validation parameters:
precision, accuracy, limit of detection and quantification, robustness, linearity
and linear range, reproducibility and repeatability, selectivity and specificity. The
results obtained showed that the goal was reached, the photometer although it
showed a low accuracy, have proved satisfactory robustness and accuracy.
Keywords: Chemistry. Research and Development, Flame Photometer,
Analytical Reliability, Validation.
LISTA DE FIGURAS
Figura 1: Espectro eletromagnético .................................................................. 15
Figura 2: Espectro da luz solar que irradia a superfície do planeta .................. 17
Figura 3: Interações da luz com a matéria ........................................................ 18
Figura 4: Atenuação da luz incidente ................................................................ 20
Figura 5: Processo de excitação e emissão de luz ........................................... 20
Figura 6: Comparação dos espectros ............................................................... 22
Figura 7: Espectro de linha do sódio................................................................. 23
Figura 8: Experimento de Kirchoff e Bunsen .................................................... 24
Figura 9: Caminho da luz no monocromador. ................................................... 28
Figura 10: Fotodiodo típico ............................................................................... 29
Figura 11: Representação gráfica de precisão e exatidão ................................ 32
Figura 12: Curva analítica clássica ................................................................... 35
Figura 13: Fotômetro de chama, vista externa.................................................. 38
Figura 14: Compressor de um inalador ............................................................. 39
Figura 15: Câmara de nebulização e queimador .............................................. 40
Figura 16: Câmara de nebulização ................................................................... 41
Figura 17: Esquema do sistema nebulizador-queimador de um fotômetro de
chama .............................................................................................. 41
Figura 18: Fotômetro em funcionamento .......................................................... 42
Figura 19: Placa de circuito impresso, face dos componentes. ........................ 44
Figura 20: Fonte original do fotômetro de chama. ............................................ 46
Figura 21: Placa de circuito impresso (Modo Real World). ............................... 47
Figura 22: Placa de circuito integrado (Modo Normal). ..................................... 48
Figura 23: Esquema para simulação do circuito do fotômetro de chama no
Proteus............................................................................................. 51
Figura 24: Correlação entre concentração e emissão na análise de sódio ....... 57
Figura 25: Correlação entre concentração e emissão na análise de sódio
com método alterado. ...................................................................... 60
LISTA DE TABELAS
Tabela 1: Relação entre faixa espectral, comprimento de onda, frequência
e energia por fóton. ............................................................................ 16
Tabela 2: Preparo de soluções padrão de sódio............................................... 53
Tabela 3: Leituras das soluções-padrão ........................................................... 54
Tabela 4: Concentração de sódio nos padrões obtidos pela aplicação da
equação da reta. ................................................................................ 55
Tabela 5: Determinação da precisão do fotômetro de chama. ......................... 55
Tabela 6: Erro relativo nas análises das diferentes concentrações de sódio. ... 57
Tabela 7: Leituras do branco em 10 replicatas. ................................................ 49
Tabela 8: Dados referentes a análise de sódio pelo método 2.. ....................... 51
Tabela 9: Dados utilizados e valores obtidos para o teste t de Student. ........... 61
SUMÁRIO
CAPÍTULO I ..................................................................................................... 15
1 A LUZ NA QUÍMICA...................................................................................... 15
1.1
Propriedades da luz ...................................................................................... 15
1.2
Interação da luz com a matéria ................................................................... 17
1.3
Proporcionalidade entre concentração e absorção ou emissão de luz 19
1.4
Espectros de emissão e absorção .............................................................. 21
CAPÍTULO II .................................................................................................... 24
2 ESPECTROMETRIA DE EMISSÃO ATÔMICA ............................................ 24
2.1
Instrumentação para análise por emissão atômica de luz .................. 24
2.2
Sistema de nebulização................................................................................ 25
2.2.1
Pérola de vidro .............................................................................. 26
2.2.2
Mixing vanes ................................................................................. 26
2.2.3
Ultra-Som ...................................................................................... 26
2.3
Combustores .................................................................................................. 26
2.3.1
Combustor de queima total ........................................................... 27
2.3.2
Combustor de mistura prévia ........................................................ 27
2.4
Sistemas ópticos e monocromadores ........................................................ 28
2.5
Detectores ....................................................................................................... 28
2.5.1
Tubos fotomultiplicadores ............................................................. 29
2.5.2
Fotodiodos .................................................................................... 29
2.6
Dispositivos de Leitura .................................................................................. 30
CAPÍTULO III ................................................................................................... 31
3 VALIDAÇÃO DE EQUIPAMENTOS ANALÍTICOS ....................................... 31
3.1
Precisão e exatidão ....................................................................................... 31
3.2
Especificidade e seletividade ....................................................................... 33
3.3
Linearidade e faixa linear ............................................................................. 33
3.4
Limite de detecção e de quantificação ....................................................... 35
3.5
Robustez ......................................................................................................... 36
CAPÍTULO IV ................................................................................................... 38
4 CONSTRUÇÃO DO FOTOMÊTRO DE CHAMA ........................................... 38
4.1
Sistema de nebulização e queimador ........................................................ 39
4.2
Luz e sistema óptico...................................................................................... 42
4.3. Detecção e sistema eletrônico..............................................................34
4.4. Leitura, conversão e exibição ..............................................................38
CAPÍTULO V .................................................................................................... 53
5 VALIDAÇÃO DO FOTOMÊTRO DE CHAMA ............................................... 53
5.1
Preparação de padrões e metodologia de análise ................................... 53
5.2
Resultados analíticos obtidos com a leitura dos padrões ....................... 54
5.3
Validação do fotômetro de chama para análise de sódio ....................... 55
5.3.1
Precisão ........................................................................................ 55
5.3.2
Exatidão ........................................................................................ 56
5.3.3
Limite de Detecção e Quantificação .............................................. 58
5.3.4
Robustez ....................................................................................... 59
5.3.5
Seletividade e especificidade ........................................................ 61
CONCLUSÃO ................................................................................................... 62
REFERÊNCIAS ................................................................................................ 63
INTRODUÇÃO
Este trabalho teve como objeto de estudo a construção e validação de
um fotômetro de chama com os conhecimentos adquiridos durante o curso,
numa ação interdisciplinar envolvendo Automação Industrial, Estatística e
Quimiometria, Física e Química Analítica.
Devido ao alto custo de equipamentos laboratoriais, até mesmo os mais
simples, surgiu à idéia da construção de um equipamento alternativo, com um
custo bem reduzido, que proporcionasse análises confiáveis. Por esse motivo o
objetivo principal do trabalho foi o desenvolvimento deste equipamento e sua
validação para uso didático. É a validação que proporciona o conhecimento das
características analíticas do equipamento e por consequência a confiabilidade
na sua utilização. Escolheu-se o fotômetro de chama tanto pela facilidade na
sua montagem e aquisição de seus componentes, quanto a praticidade em
relação às análises, espaço e aperfeiçoamento.
Para a organização dos assuntos abordados elaborou-se cinco
capítulos. Nos três primeiros apresentou-se a fundamentação teórica
necessária para entender os princípios que regem o funcionamento do
equipamento, suas partes e funções, e por fim, os parâmetros necessários à
validação. Nos dois últimos abordou-se a parte prática da construção e
validação do equipamento. Nestes descreve-se detalhadamente a montagem
do equipamento incluindo o software escrito em linguagem C, responsável pelo
funcionamento do equipamento e tratamento do sinal gerado pelo sensor. Por
fim abordam-se cada um dos parâmetros de uma validação, com suas
equações e discussões dos resultados obtidos.
Dentre os vários autores utilizados neste trabalho se destacam Ribani
(2004), que apresenta informações necessárias para validação, Cienfuegos e
Vaitsman (2000), que abordam em sua obra vários equipamentos assim como
seus componentes e funções, e ainda Brown et al. (2005), que apesar de sua
obra tratar-se da Química Geral, ressalta significativamente as características
14
da luz, sua propagação e sua ação sobre os elétrons da camada de valência
dos átomos.
CAPÍTULO I
1 A LUZ NA QUÍMICA
1.1
Propriedades da luz
O espectro eletromagnético está dividido em várias faixas, sendo elas;
radiação gama, raios x, ultravioleta, visível, infravermelho, microondas e onda
de rádio, como mostra a Figura 1. Essas radiações se propagam no vácuo com
uma velocidade constante de aproximadamente 2,998 x 108 m/s (BROWN; et
al., 2005).
Essas radiações eletromagnéticas possuem comportamento ondulatório,
semelhante a uma onda do mar, com picos e depressões regulares. A distância
entre dois picos consecutivos se chama comprimento de onda. Quando um
determinado número de ondas passa por um ponto no tempo de um segundo
fala-se em frequência de onda (BROWN; et al., 2005).
Figura 1: Espectro eletromagnético
Fonte: Atkins; Jones, 2006, p. 114-115.
A relação entre características da radiação eletromagnética como
velocidade, comprimento de onda, frequência e energia, pode ser expressa
pelas equações (1) e (2) (BROWN; et al., 2005).
16
(1)
Onde:
= comprimento de onda,
= velocidade da luz,
= frequência
(2)
Onde:
-34
= energia do fóton,
= constante de Planck (6,626x10 J.s),
= frequência
Ao longo do tempo, cientistas como Bohr (1885 á 1962), perceberam
que a energia podia ser liberada ou absorvida por átomos em quantidades
discretas, Planck deu o nome de quantum a essa quantidade definida de
energia (BROWN; et al., 2005).
Em 1905 Einstein (1879 á 1955) usou a teoria de Planck para explicar o
efeito fotoelétrico. Ele observou que a luz ao incidir em uma superfície metálica
leva-a a emitir elétrons, sendo que essa energia quantizada fornecida pela luz
foi chamada de fóton (BROWN; et al., 2005).
Os fótons podem ser entendidos como pacotes de energia, estando
relacionado com a frequência da radiação, como mostra a equação (2)
(ATKINS; JONES, 2006).
A Tabela 1 mostra de forma resumida a relação entre faixa de radiação
eletromagnética, comprimentos de onda central, frequência e energia por fóton.
Tabela 1: Relação entre faixa espectral, comprimento de onda, frequência e
energia por fóton.
Tipo de radiação
raios x e raios γ
Frequência
14
(10 Hz)
3
Energia por fóton
-19
(10 J)
3
3
10
10
350
8,6
5,7
violeta
420
7,1
4,7
azul
470
6,4
4,2
verde
530
5,7
3,8
amarelo
580
5,2
3,4
laranja
620
4,8
3,2
vermelho
700
4,3
2,8
1.000
3,0
2,0
ultravioleta
luz
visível
Comprimento de onda
(nm)
infravermelho
Microondas
e
6
3x10
ondas de rádio
Fonte: Adaptado de Atkins; Jones, 2006, p. 114.
-3
10
-3
10
17
Os olhos humanos detectam apenas os comprimentos de onda entre
400 nm (violeta) e 800 nm (vermelha), é por esse motivo que o intervalo é
chamado de luz visível. A luz solar que chega à superfície do planeta é a
mistura de todos os comprimentos de onda da luz visível, mais uma pequena
fração da faixa do ultravioleta e do infravermelho, próximas à faixa visível,
como mostra a Figura 2 (ATKINS; JONES, 2006).
A radiação ultravioleta é a responsável por causar danos às células da
pele e por estimular a produção de melanina, provocando o bronzeamento. A
maior parte desse tipo de radiação, principalmente as de menor comprimento
de onda é bloqueada pela camada de ozônio, apenas a fração próxima à luz
visível consegue passar (ATKINS; JONES, 2006).
Já a radiação infravermelha, esta mais relacionada ao calor e provoca
queimaduras na pele humana, tem frequência menor do que luz vermelha,
portanto seu comprimento de onda é superior a 800 nm (ATKINS; JONES,
2006).
Figura 2: Espectro da luz solar que irradia a
superfície do planeta
Fonte: http://uvifusp.wordpress.com/o-que-e-a-radiacaoultra-violeta/
1.2
Interação da luz com a matéria
Avanços na eletrônica e na óptica têm melhorado a qualidade dos
equipamentos
denominados
espectrofotômetros,
melhorando
sua
18
sensibilidade, precisão, exatidão e produzindo menores limites de detecção e
quantificação (CIENFUEGOS; VAITSMAN, 2000).
A Figura 3 apresenta de forma resumida as diferentes interações da luz
com a matéria dependendo do comprimento de onda da radiação utilizada.
Figura 3: Interações da luz com a matéria
Fonte: LUZ; http://monikimica.files.wordpress.com/2011/09/fig3.jpg, 2013.
Considerando-se
as
faixas
ultravioleta
e
visível
do
espectro
eletromagnético de maior interesse neste trabalho, um feixe de radiação ao
passar por um meio pode ter sua intensidade atenuada, pois algumas de suas
frequências podem ser absorvidas por elementos ou compostos que o constitui,
os quais podem ser denominados analitos num processo de quantificação
química. Isto ocorre pelo que Skoog et al. (2007) denominam como transições
eletrônicas no nível de valência. Nesse processo o analito passa de seu estado
fundamental a um estado excitado, ou seja, elétrons de um nível de valência
com orbital 3s, por exemplo, são promovidos para orbitais 3p, ou mais
energéticos ainda, gerando uma situação instável, que tende ao relaxamento e
volta do elétron ao seu estado natural com a respectiva emissão de energia.
As outras formas de interação da luz com a matéria pode ser resumida
da seguinte forma:
A radiação de baixa energia, como as ondas de rádio são
utilizadas em ressonância nuclear magnética (RNM), e ressonância de spin
eletrônica (RSE), causam mudanças na orientação do spin (SKOOG; et al.,
2007).
A radiação na faixa de micro-ondas, segundo Cienfuegos e
Vaitsman (2000), interage com a matéria de três formas:
- Reflexão: quando os materiais refletem a radiação e não se aquecem;
19
- Transparência: quando o material é atingido por radiação e este é um
isolante e não se aquece;
- Absorção: quando o material é dielétrico ao receber radiação é
aquecido.
O
infravermelho
provoca
movimentos
moleculares
como
estiramento, contração, torção, e outros, dentro e fora do plano. Como esses
movimentos são característicos do grupo funcional e podem ser amplificados
por estímulo com frequência adequada, pode-se utilizar essas absorções para
identificar a presença ou ausência de um dado grupo funcional ou para
quantificação ou qualificação de um analito em uma amostra (CIENFUEGOS;
VAITSMAN, 2000);
Os raios x permitem identificar estruturas cristalinas e evidenciar a
presença de diversos compostos, e até determinar a concentração de átomos
específicos presentes em amostras de diferentes origens (CIENFUEGOS;
VAITSMAN, 2000);
Os raios y causam efeitos muito drásticos na matéria. São
capazes de provocar quebra de ligações químicas, alterando a estrutura das
moléculas sob seu efeito. Por isso não é utilizada em análises químicas
(CIENFUEGOS; VAITSMAN, 2000);
1.3
Proporcionalidade entre concentração e absorção ou emissão de luz
A lei de Lambert-Beer atesta que a atenuação da intensidade da luz que
atravessa uma amostra está relacionada com a concentração de absorventes e
com o percurso óptico que a luz faz ao atravessar a amostra (SKOOG; et al.,
2000).
Quanto mais concentrada estiver a solução ou mais longo for o caminho
por onde a luz passar mais centros absorventes terá, e portanto maior será a
atenuação da intensidade da luz transmitida (HARRIS, 2008).
A absorbância está relacionada à quantidade de analito presente na
solução com capacidade para absorver a luz incidente. Assim, como o
equipamento mede na realidade a luz que conseguiu atravessar a amostra e
chegou ao detector, quanto menor for à transmitância maior será a absorbância
(SKOOG; et al., 2007).
20
A equação (3) mostra como obter o valor da transmitância.
ou
(3)
Onde: T=transmitância; P0=Intensidade da luz incidente; P=Intensidade da luz transmitida.
A relação entre absorbância e transmitância pode ser observada na
equação (4) e, na Figura 4 mostra-se esquematicamente o fenômeno.
ou
(4)
Onde: A=absorbância; T=fração de luz transmitida; %T=percentual de luz transmitida.
Figura 4: Atenuação da luz incidente
Fonte: SKOOG; et al., 2007, p 678
De forma semelhante, pode-se avaliar a emissão de luz pelo analito
excitado, como mostra a Figura 5. Quanto maior a quantidade de espécies
excitadas mais intensa será a luz emitida pela amostra (SKOOG; et al., 2007).
Figura 5: Processo de excitação e emissão de luz
Fonte: CIENFUEGOS, 2000, p. 147
Cienfuegos e Vaitsman (2000) dizem que na medida em que aumenta o
21
número de átomos excitados, também aumenta a quantidade de átomos que
passam pelo processo inverso, ou seja, a relaxação com consequente emissão
de luz, mostrado na etapa 2 da Figura 5 (CIENFUEGOS; VAITSMAN, 2000).
Por meio de diversos processos, átomos, moléculas e íons, podem ser
excitados, promovendo elétrons para seus níveis de maior energia. A
durabilidade de uma espécie excitada é de (10-9 a 10-6 s), para o relaxamento,
os elétrons retornam aos níveis mais baixos, preenchendo as lacunas deixadas
com a excitação, para isso emitem a diferença de energia entre o nível que
estão e o nível que ocuparão (SKOOG; et al., 2007).
Para se calcular a população excitada em uma amostra pode-se aplicar
a equação (5) que representa a distribuição de Boltzmann.
⁄
Onde:
⁄
(5)
= razão de átomos excitados;
⁄
razão de degenerescência do estado excitado
em relação ao estado fundamental; ΔE = diferença de energia entre estado excitado e
-23
fundamental; K=constante de Boltzmann ( 1,381 x 10 J/K; T=temperatura em kelvin.
O estado excitado de mais baixa energia de um átomo de sódio segundo
Harris (2005), se localiza 3,371 x 10-19 J/átomo acima do estado fundamental.
Assim, como exemplo pode-se demonstrar que a 2.600 K menos de 0,02% dos
átomos estão no estado excitado:
[
1.4
⁄
]
Espectros de emissão e absorção
Quando determinados materiais são excitados por chama ou outra fonte
de estímulo, eles emitem luz de diferentes cores, e a luz emitida por eles é uma
característica específica de cada elemento, pois cada elemento possui um
espectro único que o distingue dos demais. Existem dois tipos de espectros; o
continuo e o de linhas (MENDHAN; et al., 2008).
O espectro continuo se baseia em um feixe de luz contendo todos os
22
comprimentos de onda. Não são todas as fontes que produzem um espectro
continuo (BROWN; et al., 2005).
A radiação de uma fonte caracteriza-se por meio de um espectro de
emissão, ou seja, um gráfico da intensidade da radiação, pelo comprimento de
onda ou frequência (SKOOG; et al., 2007). Existem três tipos de espectros:
contínuos, de linhas (emissão) e de bandas (absorção). Os contínuos são
emitidos por sólidos incandescentes, os de linhas por elementos isolados, e os
de bandas são emitidos por alguns tipos de moléculas. Estes são constituídos
por linhas que se aproximam, se unem e podem sobrepor-se parcialmente. Já
os espectros de absorção são formados por gráficos onde determinados
comprimentos de onda estão ausentes ou sua intensidade foi atenuada ao
atravessar um meio contendo o analito de interesse (MENDHAN; et al., 2008).
A Figura 6 mostra uma comparação entre os espectros existentes.
Figura 6: Comparação dos espectros
Fonte: http://portaldoprofessor.mec.gov.br/storage/discovirtual/galerias/im
agem/0000000677/0000006730.jpg
O espectro de emissão é formado pelo conjunto de comprimentos de
onda emitidos pelo conjunto de elétrons de um átomo, durante o processo de
relaxação, ou seja, de retorno dos elétrons aos orbitais de menor energia, esse
espectro também é chamado de espectro de linhas de emissão devido a
natureza dos comprimentos emitidos. A quantidade de linha no espectro de
emissão aumenta com o aumento da quantidade de elétrons no átomo
(CIENFUEGOS; VAITSMAN, 2000).
23
Foi o conhecimento de que as linhas do espectro de emissão são típicas
de cada elemento e que sua intensidade é proporcional à quantidade de
átomos excitados, que tornou possível a utilização dessas linhas espectrais
para a quantificação de um determinado elemento em uma matriz qualquer.
Na Figura 7 se pode observar que o espectro de linhas do sódio (Na) se
encontra na faixa visível do espectro com seu comprimento de onda de
aproximadamente de 590 nm (BROWN; et al., 2005).
Figura 7: Espectro de linha do sódio
Fonte: Brown; et al., 2005
CAPÍTULO II
2 ESPECTROMETRIA DE EMISSÃO ATÔMICA
2.1
Instrumentação para análise por emissão atômica de luz
O espectrômetro de emissão, estimulado por chama de gás natural-ar, é
utilizado para análises de metais alcalinos e alcalinos terrosos, devido à baixa
capacidade de excitação advinda da baixa temperatura da chama desta
combinação (1.200 a 1.500 ºC). Assim, normalmente, neste tipo de
equipamento, analisa-se sódio, potássio, lítio e cálcio, devido a fácil excitação
desses átomos. É um equipamento de custo moderado, de R$ 15.000,00
19.000,00 dependendo da marca, modelo e quantidade de íons analisados
(SKOOG; et al. 2007, 2013).
Figura 8: Experimento de Kirchoff e Bunsen
Fonte: CIENFUEGOS; VAITSMAN, 2000, p. 145.
Na Figura 8 pode-se observar o primeiro experimento feito por Kirchoff
Bunsen que deu origem ao fotômetro de chama. O processo se inicia com uma
fonte de estímulo, que poderá ser um arco elétrico ou de uma chama. A luz
emitida pelo elemento em análise passa por uma lente para focalização, por
25
um monocromador que separa a luz em seus diferentes comprimentos de onda
e por fim chega a uma tela, onde podem ser colocados detectores para medir a
intensidade de cada linha espectral. A intensidade dessa luz, comparada com
padrões, dará o teor do analito de interesse.
2.2
Sistema de nebulização
A primeira parte a ser considerada num equipamento para análise por
emissão atômica é o sistema de nebulização. Um sistema simples, porém, de
grande importância no processo de emissão atômica de luz, já que o analito de
interesse se encontra ou poderá ser transferido para uma matriz líquida. O
sistema de nebulização, também designado por Cienfuegos e Vaitsman (2000)
como sistema de nebulização-queima ou sistema de nebulização-combustor,
tem o papel de introduzir a amostra a ser analisada no equipamento, convertela em gotículas e selecionar as menores que formarão uma névoa chamada de
aerosol. É essa névoa que será passada pela chama para atomização e
excitação dos elétrons do analito de interesse (HARRIS, 2008).
Normalmente, através de gases oxidantes, a amostra a ser analisada é
arrastada para o nebulizador que é transformada em aerossol e ao passar pela
chama, essa nevoa é seca, ocorrendo o que se chama de dessolvatação,
seguida por uma atomização já que as partículas formadas se dispersarão em
átomos se a temperatura da chama for suficientemente alta (KRUG;
NÓBREGA; OLIVEIRA, 2004).
O nebulizador deve ser construído de material inerte, normalmente aço
inoxidável ou polímero, já que em alguns casos a matriz contém ácido nítrico
para favorecer a dissolução do analito, já o queimador, dependendo da
temperatura da chama deve ser em titânio (KRUG; NÓBREGA; OLIVEIRA,
2004).
Os
sistemas
de
nebulização
possuem
diferentes
câmaras
de
nebulização. Dentre elas as mais utilizadas são: pérola de vidro, mixing vanes,
e ultrassom (CIENFUEGOS; VAITSMAN, 2000).
26
2.2.1 Pérola de vidro
Nesse sistema de nebulização, a câmara contém uma esfera de vidro. O
spray gerado inicialmente pelo gás de arraste da amostra sofre impacto na
pérola de vidro, posicionada estrategicamente na frente do bico injetor
diminuindo ainda mais o tamanho das gotículas. As maiores são retidas,
encaminhadas para um dreno, e as menores conduzidas para o queimador.
Esse sistema possui grande facilidade de limpeza (CIENFUEGOS; VAITSMAN,
2000).
2.2.2 Mixing vanes
Nesse sistema de nebulização dentro da câmara existem duas
ventoinhas, que tem por finalidade criar barreiras à passagem das gotículas e
proporcionar uma separação das menores com mais eficiência que a câmara
com “pérola de vidro”. Neste as partículas pequenas passam pelos obstáculos
criados pelas ventoinhas e são carregadas para o queimador; as partículas
grandes são retidas, agregadas e levadas para o dreno. Apesar da maior
eficiência desse sistema, ele não possui a mesma facilidade da “pérola de
vidro” em relação a sua limpeza (CIENFUEGOS; VAITSMAN, 2000).
2.2.3 Ultra-Som
Diferente dos outros sistemas que utilizam gases a alta pressão para
nebulizar a amostra, o ultra-sônico utiliza uma célula piezoelétrica, em outras
palavras, uma lâmina vibracional. Esse sistema chega a ter até três vezes mais
eficiência para separar as gotículas menores das maiores, já que o fluxo de gás
pode ser extremamente baixo, já que será utilizado apenas para arrastar as
gotículas menores e não para gerá-las (CIENFUEGOS; VAITSMAN, 2000).
2.3
Combustores
Acoplado ao sistema de nebulização existem dois tipos de combustores
27
muito usados: combustor de queima total ou de escoamento turbulento e o
combustor de mistura prévia ou de escoamento laminar (JEFFERY, et al.,
1992).
2.3.1 Combustor de queima total
Nesse combustor o gás combustível e o comburente são conduzidos á
chama por tubos capilares separados. Há um tubo central para a passagem da
amostra, e dois tubos capilares sobrepostos ao primeiro, onde um serve para a
passagem do comburente o outro para a passagem do combustível,
misturando-se no bico do combustor (JEFFERY, et al., 1992).
O processo de arraste dos gases nesse tipo de combustor é realizado
pelo efeito Venturi, que produz uma pressão no bico do capilar do queimador,
por onde a amostra passa, ao introduzir um gás no nebulizador. Suas
vantagens consistem no poder de queima total ou quase total da amostra, já
que a mesma chega ao combustor sem que seja fracionada pelo nebulizador;
não existe risco de explosão já que combustível, comburente e amostra são
mantidos separados até momento da queima. A desvantagem desse processo
é a produção de uma chama pequena, fria e mal formada pela grande
quantidade de amostra que chega até a mesma (CIENFUEGOS; VAITSMAN,
2000).
2.3.2 Combustor de mistura prévia
O combustor de mistura prévia homogeneíza a amostra com o os gases
de arraste, combustível e comburente, antes de chegar à chama. A amostra
nebulizada é arrastada pelos gases para um tubo que separara as gotículas
maiores das menores, as maiores são retidas e as menores são levadas á
chama (JEFFERY, et al., 1992).
Tem como vantagens, uma chama bem formada, mais quente e uma
atomização mais uniforme que a obtida com o combustor de queima total
(CIENFUEGOS; VAITSMAN, 2000). Sua maior desvantagem é o risco de
explosão pela mistura dos gases na câmara de nebulização, antes da chama
(JEFFERY, et al., 1992).
28
2.4
Sistemas ópticos e monocromadores
“Um espectroscópio é um instrumento óptico usado para a identificação
visual de linhas de emissão atômica.” (HOLLER; SKOOG; CROUCH, 2009. p.
217).
O sistema óptico tem como função captar, colimar, separar e direcionar
o feixe de luz isolado pelo monocromador, para ser quantificado. Antes do
monocromador é posicionada uma lente convergente que possui como função,
focalizar a luz em um ponto ou fenda, de forma que a fração da luz captada
pela lente sofra o menor percentual de perda possível até chegar ao detector
(YOUNG; FREEDMAN, 2004).
Figura 9: Caminho da luz no monocromador.
Fonte: HARRIS, 2005, p.458.
Como mostra a Figura 9, o monocromador possui uma fenda de entrada,
a luz captada incide inicialmente num espelho positivo côncavo, que a
direciona para um prisma ou rede de reflexão. Assim, ela é dispersa em linhas
de acordo com os comprimentos de onda que possuía. De forma semelhante
outro espelho côncavo redireciona a luz, agora monocromática, passa a fenda
de saída, posicionada em frente ao detector, para que ocorra sua leitura
(KRUG; NÓBREGA; OLIVEIRA, 2004).
2.5
Detectores
Existem vários tipos diferentes de detectores: tubos fotomultiplicadores,
29
fotodiodos e outros. Cienfuegos e Vaitsman (2000) argumentam que na
fotometria de chama o mais utilizado são os tubos fotomultiplicadores, devido
sua sensibilidade.
2.5.1 Tubos fotomultiplicadores
Os tubos fotomultiplicadores são tubos que contem em seu interior um
fotocatodo, diversos dinodos, geralmente nove, e um anodo. Um fóton da
radiação separada no monocromador ao atingir o fotocatodo provoca a
emissão de um elétron, que por sua vez colide com a superfície de um dinodo
produzindo a emissão de vários outros elétrons, que por sua vez esses elétrons
secundários (formados) também se colidem com outro dinodo dando origem á
mais elétrons, e assim por diante (HOLLER; SKOOG; CROUCH, 2009).
Por final, o fóton que entrou teve sua energia amplificada milhares de
vezes ao ser convertido em um fluxo de elétrons, esses elétrons terminam em
um anodo que terá como função converter todos esses elétrons em um sinal
analógico, expresso como diferença de potencial, cuja unidade é o Volt, para
posteriormente ser medida e analisada. (CIENFUEGOS; VAITSMAN, 2000).
2.5.2 Fotodiodos
Figura 10: Fotodiodo típico
Fonte:
http://teceletronica.up.com.br/painelgpa/uploads/
imagens/files/EngComputacao/Projetos%20Finais/2005/Manha/
2005_Espectrometro_Vanessa.pdf
30
Como pode ser visto na Figura 10, nesse dispositivo, os elétrons se
acumulam próximos a junção, que atua como um tipo de capacitor que
armazena os elétrons. Ao receber fótons de luz, os elétrons vão para as
regiões oposta, “descarregando o capacitor”, ou seja, gera uma corrente
elétrica que é proporcional à quantidade de fótons que atingem o fotodiodo e
pode ser medida, gerando um sinal analógico (HARRIS, 1999).
2.6
Dispositivos de Leitura
A consequência da passagem da luz pelos detectores é o aparecimento
uma tensão ou corrente elétrica proporcional à sua intensidade, que pode ser
medida, gerando um sinal analógico (CIENFUEGOS; VAITSMAN, 2000).
Como parte do dispositivo de leitura utiliza-se normalmente um
galvanômetro, dispositivo analógico usado para medir correntes e tensão
elétrica, o qual possui um imã permanente associado á uma bobina. A corrente
elétrica ao percorrer a bobina gera um campo magnético que se opõe ao do
imã movimentando um ponteiro a ele associado, num deslocamento de 90° á
120°. Este deslocamento é proporcional a intensidade da corrente que percorre
a bobina.
Para que a análise seja lida pelo operador num display ou
microcomputador, é necessário que o sinal analógico seja convertido para
digital. Para esta tarefa se utilizam microcontroladores, como o PIC 18F4520
da Microchip Technology Inc. que possui em sua estrutura interna um
conversor analógico-digital de 10 bits ou chips dedicados como o ICL7107 da
Maxim Integrated Products que proporciona uma leitura em 3 ½ dígitos
(LAMOGLIA, 2005).
CAPÍTULO III
3 VALIDAÇÃO DE EQUIPAMENTOS ANALÍTICOS
Um dos grandes intuitos da validação de um equipamento ou método
analítico é avaliar o grau de confiabilidade que se pode ter nos resultados com
ele obtido. Trata-se do anseio por resultados confiáveis e reprodutíveis, mesmo
com a existência de um percentual de erro inerente a todo processo de medida.
O aperfeiçoamento das técnicas, métodos e equipamentos leva a uma redução
do erro e aumenta a reprodutibilidade dos resultados (LEITE, 2002).
Para a validação de um equipamento ou método utilizado em análises
químicas deve-se utilizar apenas materiais de referência, cujo teor do analito de
interesse seja reconhecido como exato. Esses materiais de referência podem
ser adquiridos de empresas especializadas ou podem ser preparados pelo
interessado a partir de materiais de elevada pureza.
3.1
Precisão e exatidão
Normalmente, precisão e exatidão costumam ter seus significados
confundidos, porém, cada um fornece informações distintas que se
complementam. Juntas proporcionam uma avaliação concreta da qualidade do
equipamento ou método analítico em uso. Por isso, ambas devem ser
apresentadas
juntas,
entretanto
destacando-se
seus
significados
separadamente.
É considerado “exato” um método ou equipamento que apresente o valor
medido coerente com o valor real, e para ser considerado “preciso”, é
necessário que em repetidas análises da mesma amostra os resultados obtidos
sejam coerentes entre si. O ideal é que as análises feitas em replicata
proporcionem valores próximos uns dos outros e também o mais próximo
possível do valor verdadeiro. Resumidamente, “exatidão” representa o grau de
concordância de um valor medido em relação ao valor verdadeiro ou aceito
como referência e “precisão” representa o grau de concordância entre
32
diferentes medidas de uma mesma amostra (LEITE, 2002).
De acordo com a orientação DOQ-CGCRE-008 (INMETRO, 2011) a
repetitividade pode ser obtida através de diversas medições de uma mesma
amostra, realizadas com o menor intervalo de tempo possível. A precisão pode
ser verificada pela dispersão de resultados.
A Figura 11 traz uma representação gráfica mostrando a relação entre
precisão e exatidão.
Figura 11: Representação gráfica de precisão e exatidão
Fonte: AGROINFO, 2013.
A exatidão se relaciona com a reprodutibilidade dos resultados analíticos
e sua avaliação revela se há a presença de erros sistemáticos. Estes são
passíveis de eliminação com aquisição e instalação de equipamentos em
ambiente adequado, calibração dos mesmos e treinamento de pessoal. Já a
precisão se relaciona com a repetitividade dos resultados analíticos, e sua
avaliação revela se há a presença de erros aleatórios. Estes são passíveis
apenas de minimização, basicamente com as mesmas ações para a eliminação
dos erros sistemáticos (LEITE, 2002).
Os resultados obtidos com a avaliação da exatidão e da precisão
demonstram se um método ou equipamento é eficiente na repetitividade e
reprodutibilidade de medições analíticas, garantindo que estas sejam confiáveis
(OLIVEIRA, 2006).
De acordo com a ISO 5725-3 (1994), precisão intermediária pode ser
definida por resultados obtidos através de uma mesma amostra, porém sob
condições variadas, que podem resultar em diferentes analistas, equipamentos
33
ou tempo de ensaio. Este tipo de precisão é bastante utilizada em laboratórios,
para verificar a dispersão entre os analistas, equipamentos e condições
ambientais (AMSTALDEN, 2010).
A reprodutibilidade está ligada a análises interlaboratoriais sob os
mesmos aspectos da precisão intermediária, onde os resultados são expressos
através do desvio padrão amostral que determina se a diferença nos valores
obtidos na duplicata são significativos ou não (LEITE, 2002).
3.2
Especificidade e seletividade
De maneira geral, toda matriz pode ou não conter íons e compostos que
prejudicam a quantificação de um determinado analito nela presente, sendo por
isso, chamados interferentes (LEITE, 2002). A especificidade e a seletividade
são parâmetros muito importantes no processo de validação de um método ou
equipamento, eles permitem determinar a capacidade destes em selecionar
para quantificação apenas o analito de interesse, tornando os demais, íons e
compostos presentes na matriz, invisíveis durante a análise.
Especificidade pode ser definida como capacidade do método ou
equipamento fornecer um sistema exclusivo para identificar apenas o analito de
interesse na presença de interferentes; como exemplo pode-se citar os
métodos e equipamentos para espectrometrias de absorção atômica e de
massas. Já a seletividade pode ser definida como a capacidade do método em
reagir quimicamente apenas com o analito de interesse ou selecionar uma de
suas
propriedades
para
mensurá-lo.
Como
exemplo
pode-se
citar
espectrometrias UV-Vis e emissão atômica (LEITE, 2002).
De acordo com a orientação DOQ-CGCRE-008 (INMETRO, 2011), a
especificidade e a seletividade estão diretamente ligadas à linearidade,
exatidão e precisão, portanto se o método não for bem executado ou o
equipamento não estiver adequado os demais procedimentos serão ineficazes.
3.3
Linearidade e faixa linear
A linearidade faz referência à disposição de pontos de correlação entre o
34
teor de um analito da amostra e a leitura ou quantificação fornecida pelo
método ou equipamento (RIBANI; et al., 2004). De forma mais genérica podese dizer que a linearidade está diretamente ligada à curva de resposta que
pode ser elaborada a partir de um conjunto de padrões com diferentes teores
de analito e a resposta fornecida pelo método ou equipamento, conforme
descreve Leite (2002).
A curva de resposta é a mais utilizada quando se fala em linearidade,
pois é através desta que se verifica a relação sinal/concentração. Nesta há dois
eixos, o x que representa a concentração do analito e o y a resposta obtida do
equipamento (LEITE, 2002).
Embora se possa obter realmente uma curva o que se busca é o
intervalo de concentração do analito no qual os pontos se alinhem, fornecendo
uma faixa cujo coeficiente de correlação (r²) entre os pontos seja maior que
0,90 (ou > 90%) (RIBANI; et al., 2004).
De acordo com Leite (2002) a partir dos dados analíticos pode-se obter a
equação da reta e o coeficiente de correlação empregando-se as equações (6),
(7), (8) e (9).
(6)
∑
̅
̅
∑
(7)
̅
(8)
Onde: a = coeficiente angular; b = coeficiente linear;
= valores discretos da concentração
e da leitura do equipamento, respectivamente; ̅ ̅ são as médias dos dados.
(
Onde:
∑
√∑
̅
̅
̅
̅
= coeficiente de correlação;
)
= valores discretos; ̅
(9)
̅ são as médias dos dados.
A equação (9) descreve um sistema linear simples, que na química só
será válida quando o valor de r² for maior que 0,90 (ou maior que 90%). A partir
deste intervalo define-se o limite inferior e superior de análise conhecida como
faixa linear (RIBANI; et al., 2004).
35
A acreditação DOQ-CGCRE-008 (INMETRO, 2011), diz que: “a faixa
linear sempre existirá independente do método quantitativo a ser utilizado. A
concentração da amostra deve situar-se sempre dentro da faixa linear”.
Figura 12: Curva analítica clássica
Fonte: Ribani, et al., 2004, p. 774
Skoog, et al (2007), diz que se algum dado se destacar da faixa linear,
conforme mostra a Figura 12 pode ser excluído, se a avaliação estatística de
sua condição confirmar ser um dado aberrante. Umas das causas para que
esta exclusão ocorra, é que este dado pode influenciar erroneamente os
valores da análise.
3.4
Limite de detecção e de quantificação
Limite de detecção é a menor massa de analito que o método tem a
capacidade de perceber sua presença em uma amostra, diferenciando seu
sinal do ruído por uma razão igual a 3. Porém, embora passível de detecção, a
quantificação do analito nessa concentração não é recomendada, tendo em
vista a presença de um percentual de erro muito alto (RIBANI; et al., 2004).
O limite de detecção pode ser realizado desde a análise por prova em
branco, quando a mesma expressar algum resultado, até através de curvas
analíticas, em que as amostras são expostas a fortificações, indicando assim a
partir de replicadas, a menor concentração detectável possível (RIBANI; et al.,
2004).
Já o limite de quantificação, expressa a menor concentração de analito
36
em uma amostra capaz de ser determinada com precisão considerável. Neste
caso a razão entre sinal e ruído situa-se entre 6 e 10, dependendo da
sensibilidade do método. Também obtido por prova em branco (RIBANI; et al.,
2004).
Em ambos os casos cálculos estatísticos são aplicados com intuito de
determinar esses limites (AMSTALDEN; 2010).
ou
ou
̅
̅
(10)
(11)
Onde: LD = limite de detecção; LQ = limite de quantificação; s= desvio padrão dos resíduos ou
dos brancos; S= sensibilidade do método, ou seja, o coeficiente angular; ̅ = média dos
brancos.
3.5
Robustez
De acordo com Amstalden (2010) é considerada robustez a baixa
sensibilidade que um método analítico apresenta mediante pequenas variações
ambientais e de uso.
A International Union of Pure and Applied Chemistry (IUPAC) utiliza a
palavra “ruggedness” para definir robustez. Já a USP faz uso do mesmo termo,
porém com o sentido voltado para a reprodutibilidade (RIBANI, et al., 2004).
A robustez de um método analítico é o nível de
reprodutibilidade dos resultados dos testes obtidos pelas
análises de algumas amostras sob uma variedade de
condições normais de teste, tais como diferentes laboratórios,
diferentes analistas, diferentes instrumentos, diferentes lotes de
reagentes, diferentes dias, etc.(RIBANI, et al., 2004 ).
Não faz muito tempo que a robustez foi incluída nas validações, um dos
motivos foi o fato de muitos equipamentos possuírem em suas estruturas
indicadores analógicos de leitura, popularmente conhecidos como ponteiros, e
tais indicadores sofriam oscilações em ocasiões inoportunas, alterando assim o
resultado das análises (LEITE; 2002).
Diz-se que uma metodologia possui robustez intrínseca, quando em
37
determinado ponto do processo há a troca de algum equipamento, alteração de
fornecedores ou afins, sem que haja grandes alterações nos resultados
analíticos (RIBANI, et al., 2004).
CAPÍTULO IV
4 CONSTRUÇÃO DO FOTOMÊTRO DE CHAMA
Nos primeiros capítulos foi abordada a parte teórica desse trabalho, ou
seja, foram descritas as informações básicas para o entendimento da estrutura,
do funcionamento e validação de um fotômetro de chama. Neste capítulo será
abordada
a
construção
do
equipamento,
descrevendo
as
partes
confeccionadas e as adaptações realizadas, e assim obter um equipamento
alternativo, porém, com características que o tornem didaticamente utilizável,
objetivo desse trabalho.
A partir da compreensão da estrutura de um equipamento de emissão
atômica, passa-se a abordar a construção dos dispositivos que compõem um
fotômetro de chama. A Figura 13 mostra uma vista externa de um fotômetro.
Figura 13: Fotômetro de chama, vista externa
Fonte: Autoras.
A proposta inicial desse trabalho era a confecção ou adaptação de todos
os dispositivos necessários à construção do equipamento, desde o queimador
até o seu sistema de leitura. No decorrer de sua construção optou-se por
39
adquirir uma carcaça original de um fotômetro de chama inutilizada como
mostrado na Figura 13, porém, algumas partes puderam ser reutilizadas,
facilitando e abreviando a construção do mesmo.
4.1
Sistema de nebulização e queimador
Na parte traseira do fotômetro de chama existe uma entrada para
conexão de um sistema de fornecimento de ar. Esse sistema provê o ar, que
será utilizado como comburente na queima do combustível e também é o
responsável pela nebulização da amostra e pelo arraste da mesma do
nebulizador até o combustor, ou seja, esse sistema deve fornecer ar
continuamente e com certa pressão para que não ocorra o apagamento da
chama ou perda de sua estabilidade, que é de suma importância para uma
queima completa durante as análises, e para que a amostra consiga chegar até
a chama para ser atomizada e excitada.
Figura 14: Compressor de um inalador
Fonte: Autoras
Para esta função foi adaptado um compressor de inalador. O
reservatório de soro fisiológico e a máscara foram descartados, aproveitandose apenas o compressor e a mangueira de ar, visto na Figura 14.
40
Adjacente à entrada de ar há uma para o combustível, nesse caso gás
liquefeito de petróleo (GLP). Ambas são conectadas ao nebulizador-queimador
por mangueiras de silicone, resistentes ao calor e a pressão.
Para a amostra ser nebulizada e levada ao queimador, na parte frontal
da câmara está acoplado um cateter de sucção da amostra. Esse cateter foi
confeccionado com a capa plástica utilizada para isolar fios de cobre. Uma das
extremidades é acoplada ao nebulizador e a outra fica livre para ser colocada
no tubo de ensaio contendo a amostra. Esse cateter tem a função de sugar a
amostra para dentro do nebulizador.
O conjunto câmara de nebulização-queimador feito de um metal duro e
inerte pode ser visto na Figura 15. O nebulizador vaporiza a amostra que se
encontra no estado líquido em um aerossol, que posteriormente sofrerá ainda a
seleção de gotículas, as menores serão arrastadas para o queimador para
serem atomizadas e excitadas pela chama, enquanto as gotículas maiores
serão descartadas pelo dreno da câmara de nebulização.
Figura 15: Câmara de nebulização e queimador
Fonte: Autoras
No capitulo II também foi descrito por Cienfuegos e Vaitsman (2000) os
tipos de câmara de nebulização existentes nas análises por emissão atômica,
que são elas: pérola de vidro, mixing vanes, e ultrassom. No fotômetro de
chama alternativo a câmara de nebulização, conectada ao queimador, conta
41
com um dispositivo mixing vanes, que são duas ventoinhas responsáveis pela
seleção das gotículas por tamanho (Figura 16).
Figura 16: Câmara de nebulização
Fonte: Autoras
Na Figura 17 têm-se uma visão geral do esquema do interior da câmara
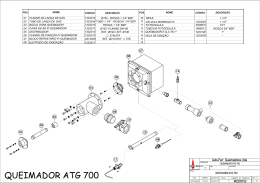
de nebulização de um fotômetro de chama.
Figura 17: Esquema do sistema nebulizador-queimador de um
fotômetro de chama
Fonte: http://www.digimed.ind.br/br/produtos/man/man_0000000066_br.pdf
42
Quando o equipamento é ligado, aciona-se o acendedor automático, que
possui um temporizador que mantêm o sinal do acendedor por alguns
segundos provocando um centelhamento entre um terminal de alta tensão
estrategicamente colocado e o bico do queimador até que o mesmo entre em
contado com a mistura gás-ar, produzindo assim uma chama que deverá ser
estabilizada, chama de coloração azul e branda, para que comece as análises.
O bico do queimador possui várias saídas para a chama como mostra a
Figura 15, proporcionando uma chama de maior área e auxiliando assim o
contato, chama/amostra aumentando a queima e a intensidade da luz emitida.
A carcaça do equipamento possui uma chaminé de dupla camada
(Figura 18) onde a chama produzida pelo queimador exala seus gases. A
externa de metal auxilia na dissipação do calor e possui um visor (orifício) para
auxiliar na regulagem da chama, e a interna um tubo removível de vidro borosilicato, protege o equipamento dos efeitos do calor da chama.
Figura 18: Fotômetro em funcionamento
Fonte: Autoras.
4.2
Luz e sistema óptico
Após a apresentação do funcionamento do nebulizador-queimador e de
como a luz é produzida, passa-se para a explicação de como essa luz,
43
específica de cada elemento analisado é medida e transformada em sinal
analógico.
Todo elemento possui um espectro diferente, ou seja, quando se excita
os elétrons da camada de valência, neste caso pela chama, os elétrons saltam
para níveis de energia mais externos e ao retornarem a seus níveis originais
emitem energia, e cada elemento emite essa energia com um comprimento de
onda diferente.
Em torno do queimador há um suporte metálico com orifícios (Figura 2)
que tem a finalidade de suportar sensores e filtros de interferência que
permitem exclusivamente a passagem da luz do elemento que se queira
analisar.
Os filtros são específicos para cada elemento, sendo eles de cores
diferentes, cada filtro possui a cor que é emitida pelo elemento em questão
quando o mesmo é queimado. Por exemplo, para validar a análise de sódio,
utilizou-se um filtro de cor amarelo/alaranjado, pois esta é a cor da luz emitida
pelo sódio ao ser queimado, as outras cores são refletidas pelo filtro para que
não interfiram na medida.
Diferente do capitulo II que o espectro de interesse deveria ser disperso
por um prisma ou rede de difração, quando a luz do elemento queimado
passasse por uma fenda na entrada do monocromador com o auxílio de uma
lente convergente. Os filtros substituem todo o sistema do monocromador.
Além de praticamente não ocupar espaço dentro do equipamento ao contrário
do monocromador, os filtros facilitam a separação do espectro de interesse
dispensando ajustes de posição.
4.3. Detecção e sistema eletrônico
O suporte já mencionado para os filtros, também terá a função de
acoplar um fotodiodo que será o responsável pela conversão de luz para sinal
analógico. O fotodiodo tem a capacidade de ao receber luz, converte-la em
corrente elétrica que será utilizada como sinal analógico da medida.
A luz transformada em sinal pelo fotodiodo possui uma tensão muito
pequena, o range total é da ordem de 0 á 400 milivolts (mV). Sem irradiação o
fotodiodo não produz diferença de tensão, ou seja, o sinal é de 0 mV. Na
44
intensidade máxima, ou seja, a plena luz o mesmo consegue gerar 400 mV.
Sendo assim, uma tensão muito pequena para ser lida diretamente pelo
microcontrolador, cujo range de leitura é de 0 á 5 V.
Além dessa pequena tensão teórica do fotodiodo, a luz ao ser filtrada
produz tensões ainda mais baixas, algo entre 50mV e 200mV. Isso por que,
mesmo no início das medidas, o fotodiodo não esta no escuro, 0V, pois a
chama já esta acesa e assim alguma luz também estará chagando ao mesmo,
e ao filtrarmos a luz da amostra reduz consequentemente a tensão máxima que
o fotodiodo consegue converter.
Por esse motivo foi colocado um amplificador operacional para aumentar
a tensão do fotodiodo de milivolts (mV) para volts (V). Esse amplificador foi
posicionado na placa onde se encontra o display de LCD, próximo ao
microcontrolador (Figura 19), já que o sinal amplificado pelo mesmo será
enviado ao micro. Na parte interna do equipamento foram montadas duas
placas, uma para a geração, estabilização e envio de tensões, e na outra foi
colocada o amplificador operacional, o micro controlador junto ao display de
LCD, e o temporizador, já mencionado anteriormente.
Figura 19: Placa de circuito impresso, face dos componentes.
Fonte: Autoras.
O amplificador operacional necessita para seu funcionamento de três
tipos de tensões diferentes, – 5 V (representada pelo fio de cor branco), + 5 V
45
(representada pelo fio de cor vermelho), e 0 V (representada pelo fio de cor
preta). Essas tensões são estabelecidas pelo padrão de operação do
amplificador, que exige uma alimentação simétrica.
Para que ocorra o funcionamento do equipamento este deve receber
energia suficientemente capaz de suprir todas as suas necessidades, que
deverá ser fornecida pela fonte de alimentação.
O temporizador é o responsável pelo tempo que o acendedor ficará
centelhando no queimador, e para que o mesmo funcione, carrega-se o
capacitor que descarrega via resistor. Se for desejável um tempo maior para o
acendimento do queimador basta aumentar o valor do capacitor, maior
capacitância maior o tempo de centelhamento. Outra forma de alterar o tempo
de centelhamento é alterar o valor do resistor de descarga, quanto maior a
resistência, maior o tempo para descarregar o capacitor, e assim maior será o
tempo do temporizador.
Quando o temporizador inicia seu trabalho mantêm um disparo de mais
ou menos 10 segundos, enviando para o flyback (transformador de alta tensão)
uma tensão de 12 V, ao passar esse tempo o temporizador é desligado
automaticamente, e só religado quando acionado o botão de disparo
novamente.
O flyback nada mais é que um transformador de alta tensão, ou seja, ele
recebe a tensão que chega do temporizador, 12 V, e converte a mesma para
cerca de 6.000V, isso é necessário pelo fato da tensão que chega do
temporizador ser insuficiente para vencer a resistência do ar (1000 V.mm-1) e
provocar o centelhamento entre o eletrodo que possui um envolto de cerâmica
isolante, que fica sobreposto ao bico do queimador e o próprio queimador.
Como explicado o flyback é um transformador de alta tensão, que aqui é
utilizado para gerar um centelhamento na grelha do queimador e provocar a
combustão dos gases. Na Figura 20 esse componente pode ser visto como
uma esfera negra ligada a um fio vermelho que vai da mesma até o eletrodo
sobre a grelha do queimador.
A fonte utilizada foi um dos componentes que pode ser restaurado e
reutilizado. A fonte do equipamento mostrada na Figura 20 é equivalente á uma
fonte de computador, com uma diferença, o sistema de alta tensão teria que
ser construído separadamente, já que este não existe na fonte de computador.
46
O fio azul escuro alimenta o sistema de alta tensão, é o retorno do
temporizador. O fio amarelo fornece +12 V, e alimenta o temporizador do
acendedor automático. Os fios pretos são “neutros” com 0 V. O fio de cor
vermelho fornece +5 V e o de cor branca – 5 V.
Figura 20: Fonte original do fotômetro de chama.
Fonte: Autoras
Nessa placa utilizada como fonte de energia, além do flyback que é um
transformador elevador de tensão, possui também um transformador para
reduzir a tensão da rede de alimentação com chave seletora para 110 ou 220 V
de acordo com a rede local. Esta fonte possui ainda fusível de segurança para
o caso de um curto ou sobrecarga.
A tensão rebaixada pelo transformador passa por diodos e capacitores
para ser retificada e filtrada, em seguida vai para os controladores de tensão,
circuitos integrados de controle de tensão, que nesse caso são os 7805 (+5V),
7905 (-5V) e o 7812 (+12V). Esses circuitos de controle de tensão simplificam a
confecção da fonte e evita a passagem de uma tensão maior em componentes
muito sensíveis, como o microcontrolador e o amplificador operacional, nestes
qualquer
oscilação
que
ocorra
na
alimentação
pode
prejudicar
seu
funcionamento até o ponto de sua perca, ou seja, os controladores mantêm a
47
tensão estabilizada e bem filtrada para todo o sistema elétrico. A corrente
utilizada e suficiente para alimentar todo o circuito elétrico é de apenas
1ampère.
4.5.
Leitura, conversão e exibição
Para que haja a leitura do sinal e sua conversão de sinal analógico para
digital, foi construída a placa mostrada na Figura 7, que ocupou o lugar da
placa original do equipamento, posicionando assim o display de LCD
(alfanumérico 16x2, configuração de 4 bits.) no mesmo lugar que o antigo, já
que a placa original não estava em condições de uso. O principal componente
dessa placa é o microcontrolador PIC 18F4520. Na mesma placa ainda podem
ser vistos o circuito para amplificação do sinal e o circuito do temporizador, este
embora não tenha conexão com o circuito principal foi colocado aí, tanto por
haver o espaço disponível quanto pela localização estratégica devido à
proximidade das conexões.
Figura 21: Placa de circuito impresso (Modo Real World).
Fonte: Autoras
A leitura e conversão do sinal analógico é realizado por um periférico do
48
próprio PIC, um conversor analógico-digital de 10 bits, integrado ao chip. Em
seguida o valor numérico é enviado para o display de LCD. As Figura 21 e 22
mostram o circuito da placa confeccionada no software PCB Wizard.
Figura 22: Placa de circuito integrado (Modo Normal).
Fonte: Autoras.
O sinal analógico que chega ao micro controlador (PIC 18F4520) pelo
amplificador operacional, produzido pelo fotodiodo, será enviado para um
pequeno micro controlador integrado ao display de LCD, que fará a conversão
do sinal analógico para o digital, fornecendo assim a possibilidade de leitura no
display. Para essa conversão o micro controlador (PIC 18F4520) que trabalha
de 0 a 5 volts deve passar para o micro do display que lê de 1 á 1023, ou seja,
as duas leituras devem ser equivalentes, onde o 0 é igual a 0 e 5 é igual a
1023.
Quando o sinal muda da leitura do micro controlador (PIC 18F4520), que
vai de 0 á 5 volts, para a leitura do display, que vai de 0 á 1023, essa mudança
transforma o sinal analógico para digital, enviando para a o display de LCD
para a leitura do operador.
Apesar da sua leitura teoria do micro controlador (PIC 18F4520) ser de 0
a 5 volts, na realidade do fotômetro alternativo existe uma perca de tensão,
49
tanto no começo quanto no final, isso pois, a tensão vinda do fotodiodo, como
mencionado anteriormente, não chega no seu potencial máximo que seria
400V, e nem o seu mínimo que é 0V.
Pensando nisso foram colocados referências (jumpers) para o micro
controlador
(PIC
18F4520),
onde
as
mesmas
não
influenciarão
no
funcionamento do equipamento, porém, no programa construído para rodar no
PIC, que é o programa responsável pelos comandos que o micro fará durante á
chegada no sinal analógico, as referências possuem os pinos 4 e 5, que
posteriormente poderão ser substituídos por potenciômetros, sem que precise
fazer qualquer alteração no programa que roda no PIC, para que os mesmos
limitem a faixa de tensão que chega no PIC reduzindo-a para que possamos ter
uma leitura mais precisa e sensível da amostra, já que existe uma perca, que
não se sabe qual, no começo do sistema.
O software que faz a leitura, a conversão do sinal analógico para digital
e envia o valor para o display de LCD foi escrito em linguagem C, utilizando-se
o compilador MikroC da empresa Mrikroelectronica, que disponibiliza uma
versão livre para aplicações didáticas, desde que o software gerado não
ultrapasse 2Kbytes. Após escrito o software foi compilado, obtendo-se o
arquivo em hexadecimal que foi gravado na memória do PIC 18F4520.
A seguir mostra-se o software em linguagem C delimitado por asteriscos.
Tudo que esta em verde devido à colocação de (//) ou por estarem entre (/* */)
o compilador entende ser comentários e não os inclui no arquivo hexadecimal.
/****************************FOTÔMETRO DE CHAMA*******************************
Objetivo: Captar o sinal analógico gerado por um fotodiodo e convertê-lo para
digital exibindo o resultado num display alfanumérico 16x2, numa configuração
de 4 bits.
Micro controlador: PIC 18F4520.
Freqüência: 4 MHz
******************************************************************************************/
//Definição dos pinos do PIC para conexão com o display de LCD:
Sbit LCD_RS at LATB4_bit;
Sbit LCD_EN at LATB5_bit;
Sbit LCD_D4 at LATB0_bit;
Sbit LCD_D5 at LATB1_bit;
Sbit LCD_D6 at LATB2_bit;
50
Sbit LCD_D7 at LATB3_bit;
Sbit LCD_RS_Direction at TRISB4_bit;
Sbit LCD_EN_Direction at TRISB5_bit;
Sbit LCD_D4_Direction at TRISB0_bit;
Sbit LCD_D5_Direction at TRISB1_bit;
Sbit LCD_D6_Direction at TRISB2_bit;
Sbit LCD_D7_Direction at TRISB3_bit;
/*****************************************************************************************/
// Criação de variáveis para armazenagem e manipulação de dados:
Int sensor;
//variável para armazenar a leitura do sensor
Int valor;
//variável para trabalhar o dado do sensor
Char texto [8];
/*variável tipo matriz para ser usada na conversão do
valor lido no sensor para texto. Os dados, para
serem enviados para o LCD precisam estar na forma
de texto (string)*/
/*****************************************************************************************/
//Criação do bloco principal do software
Void main() {
//função principal da linguagem C
ADCON1 = 0b00111010; /* Configuração do Registrador ADCON1 que
faz a conversão do sinal analógico para digital.
Desta forma aceita valores de tensão externa como
referência, e servirá para ajuste do 0 e 1000 na
aferição do equipamento. O Registrador ADCON0,
não precisa ser configurado devido ao uso da função
ADC_Read que lê o canal especificado e converte
para digital. Essa função configura automaticamente
esse registrador*/
TRISA.RA0 = 1;
TRISA.RA1 = 1;
TRISA.RA2 = 1;
TRISA.RA3 = 1;
TRISB = 0;
PORTB = 1;
/* Configura os pinos 2, 3, 4 e 5 (AN0, AN1, AN2 e
AN3) do PortA do PIC como entradas*/
//Configura os pinos do PortB usado no LCD como
saídas
//Configura os pinos do PortB como ativos
Lcd_Init();
Lcd_Cmd(_LCD_CLEAR);
Lcd_Cmd(_LCD_CURSOR_OFF);
Lcd_Out(1, 1, "**UNISALESIANO**");
Lcd_Out (2, 1, "Na = ");
// Initializa LCD
// Limpa o display
// Desativa o Cursor
/* Escreve no LCD o que esta
entre
aspas,
na
posição
indicada: linha 1, iniciando na
coluna 1*/
/* Escreve no LCD o que esta
entre aspas, na linha 2, coluna 1.
51
Como o display tem 16 colunas
em cada linha, o valor do sensor
será direcionado para iniciar na
coluna 6 */
/*****************************************************************************************/
/*O software deve estar sempre rodando, para isso deve ter um laço de
repetição, tudo que estiver dentro do laço {while} será repetido infinitamente ou
até que o software seja interrompido
While(1) {
// Criação do laço de repetição
sensor = ADC_Read (0);
// Lêr canal AN0 e atribuir a sensor
valor = sensor/10;
wordtostr (valor, texto);
// Converter sensor para texto
Lcd_Out (2, 8, texto);
// Escreve no LCD a variável texto
}
//Encerra a função while
}
//Encerra a função main
/*****************************************************************************************/
Figura 23: Esquema para simulação do circuito do fotômetro de chama no
Proteus.
Fonte: Autoras.
A Figura 23 exibe um circuito para simulação de operação do fotômetro
52
de chama no software Proteus, nele o fotodiodo foi substituído por um
potenciômetro linear, já que esta versão do software não dispõe desse
componente.
Este
software
foi
de
fundamental
importância
para
o
desenvolvimento do equipamento, pois através desse circuito pode-se avaliar
não só as conexões entre o PIC e seus periféricos, mas também, o software
escrito em linguagem C utilizando-se o software MikroC.
CAPÍTULO V
5 VALIDAÇÃO DO FOTOMÊTRO DE CHAMA
Como apresentado no capítulo 3 a validação de um método ou
equipamento se refere à avaliação de 7 parâmetros: precisão, exatidão,
linearidade ou faixa linear, especificidade ou seletividade; limite de detecção;
limite de quantificação e robustez.
5.1
Preparação de padrões e metodologia de análise
Para validação do fotômetro de chama com relação à análise de sódio,
foram preparados 5 soluções-padrão com concentrações distintas de cloreto de
sódio PA (para análise) .
A preparação dos padrões se iniciou com a preparação de uma solução
mãe ou solução estoque de cloreto de sódio na concentração de 0,5 mol/L.
A partir da solução mãe, por processo de diluição preparou-se o
conjunto de 5 soluções padrão nas seguintes concentrações: 0,05, 0,10, 0,15,
0,20 e 0,25 mol/L, como apresentado na Tabela 2.
Tabela 2: Preparo de soluções padrão de sódio
Soluções-padrão
mol/L
Volume de Solução Mãe
Água em qsp* 100 ml.
0,05
10 ml
0,10
20 ml
0,15
30 ml
0,20
40 ml
0,25
50 ml
Nota: *QSP – Quantidade suficiente para.
Fonte: Autoras.
Após o preparo das soluções padrão, foram realizadas as leituras das
mesmas seguindo-se o procedimento como descrito abaixo:
54
Separar e identificar 6 tubos de ensaio, cinco para as soluções
padrão e um para o branco, formado exclusivamente por água
demineralizada;
Ligar o equipamento, acender a chama, e aguardar alguns minutos
para estabilização do sistema.
Inicie as medidas lavando o sistema de nebulização com água
desmineralizada por 2 minutos;
Aferir o equipamento com a solução mais concentrada de modo que
o display mostre uma leitura de emissão em torno de 900 u.a.;
Lave novamente o sistema por 2 minutos e aguarde até obter uma
leitura estável. Considere o valor mostrado como sendo a leitura do
branco;
Leia a emissão dos padrões iniciando pelo de menor concentração.
Considere a emissão de maior intensidade, ou seja, a leitura mais
alta que o display mostrar;
Entre uma leitura e outra, passe água desmineralizada para eliminar
resíduos da leitura realizada e aguardar 2 minutos após a lavagem
para reestabilização do sistema.
5.2
Resultados analíticos obtidos com a leitura dos padrões
Tabela 3: Leituras das soluções-padrão
Intensidade de emissão do Na (u.a.)
Branco 0,05* 0,10* 0,15* 0,20* 0,25*
Replicata 1
144
629
691
845
917
Replicata 2
Replicata 3
144
144
361
311
716
389
897
494
980
805
Replicata 4
Replicata 5
144
144
500
464
669
560
775
756
864
867
R²
Equação da Reta
1000 0,9813
1000 0,8457
y = 1936X + 526
y = 3084X + 328,2
1000 0,9430
1000 0,9882
y = 3588X + 61,6
y = 2390X + 403,1
1000 0,9904
y = 2758X + 315,7
*Concentração de sódio (Na) em mol/L.
Fonte: Autoras.
As avaliações estatísticas exigem que análises químicas sejam feitas em
replicatas. Assim decidiu-se analisar cada amostra ou solução padrão 5 vezes.
Devido a fortes oscilações no sinal gerado os dados foram normalizados
para permitir comparação entre eles e a avaliação estatística desejada. A
55
tabela 3 apresenta os dados, já normalizados, das 5 replicatas.
As avaliações estatísticas que se seguiram foram realizadas com os
dados já normalizados.
Tabela 4: Concentração de sódio nos padrões obtidos pela aplicação da
equação da reta.
Concentração de sódio (mol/L)
Padrões
(mol/L) Repl. 1 Repl. 2 Repl. 3 Repl. 4 Repl. 5
0,05
0,053
0,011
0,070
0,041
0,054
0,10
0,085
0,126
0,091
0,111
0,089
0,15
0,165
0,184
0,121
0,156
0,160
0,20
0,202
0,211
0,207
0,193
0,200
0,25
0,245
0,218
0,262
0,250
0,248
Média
Desvio Padrão
0,046
0,100
0,157
0,203
0,244
0,022
0,017
0,023
0,007
0,016
Fonte: Autoras.
Embora haja grande oscilação entre os valores obtidos para a solução
de mais baixa concentração nas replicatas 2 e 3, estes resultados não puderam
ser descartados com a aplicação do teste Q para um nível de confiança de
95%.
5.3
Validação do fotômetro de chama para análise de sódio
5.3.1 Precisão
A precisão reflete a dispersão dos resultados de uma série de medidas
de uma mesma amostra ou solução padrão. Esta é obtida com a aplicação da
equação (12) que expressa o desvio padrão relativo.
(12)
̅
Onde: DPR = desvio padrão relativo das leituras; s = desvio padrão amostral; ̅ = média das
leituras
Tabela 5: Determinação da precisão do fotômetro de chama.
Padrões
(mol/L)
Média
mol/L
Desvio Padrão
DP
0,05
0,10
0,15
0,046
0,100
0,157
0,022
0,017
0,023
Desvio Padrão
Relativo
DPR (%)
48,4
17,3
14,8
(continua)
56
(conclusão)
Padrões
(mol/L)
Média
mol/L
Desvio Padrão
DP
0,20
0,25
0,203
0,244
0,007
0,016
Desvio Padrão
Relativo
DPR (%)
3,5
6,6
Fonte: Autoras.
Na Tabela 5, observam-se os valores para os desvios padrão relativos
para cada uma das cinco concentrações analisadas. Os resultados indicam que
o equipamento possui baixa precisão já que em quase todas as concentrações,
os valores obtidos possuem uma dispersão maior que 5% em relação à média.
Outro fato relevante é que a dispersão dos resultados aumenta com a
diminuição da concentração, com destaque para a concentração de 0,05 mol/L
com uma dispersão de 48,4%, esse fato se mostra coerente com os limites de
detecção e de quantificação calculados. A baixa precisão pode ser resultado da
falta de um sistema para controle de pressão para combustível e comburente.
5.3.2 Exatidão
Na figura 24, apresenta-se os dados relativos da análise de sódio nas
condições metodológicas 1.
A exatidão reflete a coerência das concentrações obtidas com as
concentrações reais ou admitidas como verdadeiras.
A exatidão é obtida com a aplicação da equação (13) que expressa o
erro relativo da medida.
(13)
Onde: %E = erro relativo;
= concentração calculada;
= concentração verdadeira.
A concentração calculada, necessária para o cálculo do erro relativo, é
obtida com a aplicação da equação da reta fornecida pelo gráfico mostrado na
Figura 24 que expressa à correlação entre as diferentes concentrações
analisadas e as médias das leituras de emissão do equipamento.
57
Figura 24: Correlação entre concentração e emissão na
análise de sódio
1200
y = 2751,2x + 326,92
R² = 0,9966
1000
800
600
400
200
0
0
0,05
0,1
0,15
0,2
0,25
0,3
Fonte: Autoras.
Na Figura 24 observa-se também que o coeficiente de correlação (R²),
nestas condições, mostra que há uma correlação maior que 99% entre
concentração das amostras e leituras de emissão atômica, tornando o método
aceitável.
As equações utilizadas para a obtenção das constantes da equação da
reta e para o cálculo do coeficiente de correlação são mostradas nas equações
de (6) a (9).
Tabela 6: Erro relativo nas análises das diferentes concentrações de sódio.
Padrões
(mol/L)
0,05
0,10
0,15
0,20
0,25
Média das
Concentração
Equação da Reta pela
leituras de
Calculada
média das leituras
emissão
(mol/L)
453
0,046
605
0,101
Y
=
2751,2X
+
326,92
753,4
0,155
886,6
0,203
1000
0,245
Resíduos
-0,0042
0,0011
0,0050
0,0034
-0,0054
Erro
Relativo
(%)
-8,35
1,08
3,34
1,72
-2,14
Fonte: Autoras
Os resultados apresentados na Tabela 6 indicam que embora o
equipamento esteja apresentando baixa precisão, a exatidão esta com um erro
aceitável se o padrão de menor concentração for desconsidera, já que o erro
nas demais concentrações foi de no máximo 3,34%, e de modo geral, admite-
58
se um erro de até 5%.
Segundo Gauss por mais que existam alguns outtliers dentro de uma
série de medidas estatísticas, a média tende sempre a estar próxima ao valor
verdadeiro (SARAIVA; 2013).
5.3.3 Limite de Detecção e Quantificação
Limite de detecção expressa a concentração mínima do analito presente
numa amostra que o equipamento é capaz de detectar, diferenciado seu sinal
do ruído natural por uma razão no mínimo igual a 3. Já o limite de quantificação
expressa a concentração mínima do analito que pode ser quantificada com
nível de confiabilidade aceitável. Para o limite de quantificação a razão
sinal/ruído se situa entre 6 e 10.
O ruído do equipamento é medido através da análise de replicatas do
branco. Assim, analisou-se 10 replicatas do branco para as determinações dos
limites de detecção e de quantificação.
Tabela 7: Leituras do branco em 10 replicatas.
Replicatas do branco
1
2
3
4
5
6
7
8
9
10
173
201
255
199
225
189
269
249
283
290
̅
237
41,6
Fonte: Autoras
Para a determinação dos limites de detecção e de quantificação, podese aplicar as equações como demonstrado abaixo:
̅
̅
=
59
Nota-se pelo cálculo que determina o limite para quantificação do analito
que a solução padrão de concentração mais baixa esta aquém desse limite,
portando deveria ser desconsiderada.
5.3.4 Robustez
A robustez de um método ou equipamento expressa a concordância
entre os resultados de análises feitas, com uma mesma amostra, sob
condições semelhantes. Pequenas variações nas condições ambientais ou de
execução do método não devem produzir resultados significativamente
diferentes.
A avaliação da robustez por ser feita aplicando-se o teste t de Student
para comparação de duas médias, mostrado na equação (14).
| ̅
̅ |
(14)
√
Onde:
= valor de t calculado para comparação com t crítico que é tabelado; ̅
̅ =
médias de cada conjunto de medidas;
= desvio padrão combinado;
= número de
replicatas em cada conjunto de medidas.
A equação do teste t exige entre os dados o desvio padrão combinado
dos dois conjuntos de dados. A equação (15) mostra como obter o desvio
padrão combinado, necessário para o teste t.
√
Onde:
medidas;
(15)
= desvio padrão combinado;
= desvios padrão de cada conjunto de
= número de replicatas em cada conjunto de medidas.
Para a aplicação do teste t, foi realizado um novo conjunto de medidas
para a comparação das análises em condições diferentes. Nestas as condições
ambientais e de condução do método foram alteradas.
No novo método não foi observado o tempo de eliminação de resíduos
da leitura anterior e de reestabilização do sistema. As leituras foram feitas de
forma contínua da menos concentrada para a mais concentrada aguardando-se
60
apenas o tempo de estabilização da leitura. Como foram feitas em dias
diferentes, considera-se que as condições ambientais também estavam
diferentes.
A Tabela 8 e a Figura 25 mostram os resultados obtidos com o novo
método.
Tabela 8: Dados referentes a análise de sódio pelo método 2.
Método
2
Média das Equação da Reta Concentração
Padrões
leituras de pela média das
Calculada
(mol/L)
emissão
leituras
(mol/L)
573
0,0406
0,05
711
0,1051
0,10
824
0,1573
0,15
932
0,2078
0,20
1000
0,2392
0,25
Erro
Relativo
-18,87
5,13
4,85
3,90
-4,31
Fonte: Autoras
Figura 25: Correlação entre concentração e emissão na análise de sódio com
método alterado.
1200
y = 2150,7x + 485,27
R² = 0,9866
1000
800
600
400
200
0
0
0,05
0,1
0,15
0,2
0,25
0,3
Fonte: Autoras.
Para comparação dos métodos, foi aplicado o teste t de Student. A partir
da tabela de distribuição estatística de Student obteve-se o tcrítico que é de 2,45
para um nível de confiança de 95%.
Abaixo segue um exemplo de aplicação do teste t.
61
| ̅
̅ |
|
|
√
√
Tabela 9: Dados utilizados e valores obtidos para o teste t de Student.
Método 1
̅
Método 2
Scomb
tcalc
̅
0,0455
0,0197
0,0406
0,0037
0,0066
1,0166
0,1004
0,0156
0,1051
0,0020
0,0052
1,2376
0,1570
0,0208
0,1573
0,0040
0,0070
0,2026
0,0063
0,2078
0,0106
0,0033
0,0587
2,1577
0,2444
0,0144
0,2392
0,0092
0,0053
1,3435
Fonte: Autoras.
Os resultados mostrados na Tabela 9 revelam que o equipamento se
mostrou robusto para as diferentes condições e métodos de análise, já que o
tcalculado foi menor que o tcrítico em todas as concentrações analisadas.
5.3.5 Seletividade e especificidade
Os parâmetros de seletividade e especificidade não foram avaliados,
pois o equipamento utiliza um filtro comercial de interferência, específico para
análise de sódio. Isto proporciona a leitura somente da linha espectral do sódio.
Outro fator que garante a especificidade da medida é a temperatura da chama;
mesmo que houvesse possíveis interferentes presentes, ou seja, outros
elementos que pudessem emitir luz de comprimento de onda muito próxima à
do sódio, a temperatura da chama produzida pela combinação gás natural-ar
não é suficiente para provocar excitação significativa desses elementos.
62
CONCLUSÃO
O intuito deste trabalho foi à construção de um fotômetro de chama de
baixo custo, porém capaz de realizar análises quantitativas com eficiência
satisfatória para uso didático. Assim, após a realização deste trabalho de
pesquisa e desenvolvimento pôde-se concluir que:
- Apesar da fotometria de chama ser um método analítico simples, é
bastante eficaz e confiável;
- O PIC 18F4520 se mostrou um microcontrolador extremamente versátil
e simplificou muito o trabalho de desenvolvimento do equipamento;
- A utilização do fotodiodo como sensor, numa configuração de célula
fotovoltaica, se mostrou satisfatório em termos de simplicidade e sensibilidade;
- A validação do equipamento para análise de sódio mostrou que o
equipamento apresenta-se com baixa precisão, entretanto, todos os demais
parâmetros avaliados na validação se mostraram satisfatórios, com destaque
para a exatidão de aproximadamente 97%.
- Como medidas para buscar a melhoria da precisão, sugere-se a
colocação de um sistema de controle de pressão para os gases, combustível e
comburente, bem como, a instalação de um segundo sensor para servir como
parâmetro de referência para reduzir as oscilações do sinal analítico.
Por fim conclui-se que o equipamento desenvolvido, com as sugestões
de melhorias implementadas, pode fornecer análises com resultados tão
satisfatórios quanto os equipamentos comerciais, porém, a confirmação desta
hipótese só virá com uma nova validação.
63
REFERÊNCIAS
AMSTALDEN, L. C. Validação e protocolos em análises químicas.
Rio Preto, 22 de mai. 2010. Disponível em: <http://www.crq4.org.br/sms/files/file
/validacao_protocolos_analises_quimicas_2010.pdf>. Acesso em: 10 jan. 2013.
ATKINS, P.; JONES, L. Princípios de Química: Questionando a Vida
Moderna e o Meio Ambiente. Tradução Ricardo Bicca de Alencastro. 3. ed.
São Paulo: Brookman, 2006. 968 p.
BROWN, T. L. et al. Química: a Ciência Central. 9. ed. São Paulo:
Pearson Prentice Hall, 2005. 972 p.
CERONI. Radiação ultravioleta, uma abordagem pela divulgação
científica. . [s.l.]. 27 de nov. 2009. Disponível em:
<http://uvifusp.wordpress.com/o-que-e-a-radiacao-ultra-violeta/> Acesso em 29:
nov. 2013.
CIENFUEGOS, F.; VAITSMAN, D. Análise Instrumental. 1. ed. Rio de
Janeiro: Interciência, 2000. 606 p.
DIGIMED. Manual de instruções: Fotômetro de chama. Disponível em:
<http://www.digimed.ind.br/br/produtos/man/man_0000000066
_br.pdf> Acesso em: 16 nov. 2013.
HARRIS, D. C.; Análise Química Quantitativa, 5 ed. Rio de Janeiro:
LTC- Livros Técnicos e Científicos , 1999. 886 p.
HARRIS; D C. Análise de Química Quantitativa. 7. ed. Rio de Janeiro:
LTC- Livros Técnicos e Científicos, 2008. 855 p.
HOLLER, F. J. ; SKOOG, D. A. ; CROUCH, S. R. Princípios de Análise
Instrumental. Tradução Célio Pasquini. 6. ed. Porto Alegre: Bookman, 2009,
1056p.
INMETRO. Orientação sobre validação de métodos de ensaios
químicos. DOQ-CGCRE-008 revisão 04, jul. 2011. Disponível em: <http://
www.inmetro.gov.br/sidoq/arquivos/Cgcre/DOQ/DOQ-Cgcre-8_04.pdf>. Acesso
em: 12 jan. 2013.
JEFFERY, G. H. et al. Vogel: Análise Química Quantitativa. Tradução
Horacio Macedo. 5. ed. Rio de Janeiro: LTC, 1992. 712 p.
64
KRUG, F. J.; NÓBREGA, J. A.; OLIVEIRA, P. V. Espectrometria de
Absorção Atômica: Parte1. Fundamentos e Atomização com chama. Artigo
Científico. jun. 2004. Disponível em: <http://apostilas.cena.usp.br/Krug/
AAS%20geral%20parte%201%20revisada. pdf >. Acesso em: 19 jan. 2013.
LAMOGLIA, V. Espectrômetro Acadêmico, Monografia. Curitiba. Nov.
2005. Disponível em:<http://teceletronica.up.com.br/painelgpa/uploads/imagens
/files/EngComputacao/Projetos%20Finais/2005/Manha/2005_Espectrometro_V
anessa.pdf > Acesso em: 21 mai. 2013.
LEITE, F. Validação em Análises Químicas. 4. ed. Campinas: Átomo,
2002. 360p.
LUZ, átomo e o tempo: o césio e o relógio atômico. Disponível em: <http
://monikimica.files.wordpress.com/2011/09/fig3.jpg>. Acesso em: 20 nov. 2013.
MENDHAN, J. et al. Vogel: Análise de Química Quantitativa. Tradução
Júlio C. A., Paula F. A., Ricardo B. A. 6. ed. Rio de Janeiro: LTC, 2008. 462 p.
OLIVEIRA, F. M. Controle de qualidade analítica. 2006. Disponível em:
<http://www.labwin.com.br/Artigos/ Curso_Cqa_Validacoes.pdf>. Acesso em:
10 jan. 2013.
PORTAL do professor. Disponivel em: <http://portaldoprofessor.
mec.gov.br/storage/discovirtual/galerias/imagem/0000000677/0000006730.jpg
>Acesso em 29 nov. 2013.
RIBANI, M. et al. Validação em Métodos Cromatográficos e
Eletroforéticos. 2004. Disponível em: <http://www.scielo.br/scielo.php?pid=S0
100-40422004000500017&script=sci_abstract&tlng=es>. Acesso em: 07 de jan.
de 2013.
SARAIVA, Celso P.; Expressão e Cálculo de Incerteza de Medição. São
Paulo: Remesp, 2013. 75 p.
SKOOG, D. A. et al. Fundamentos de Química Analítica. Tradução
Marco Grassi. 8. ed. São Paulo: Thomson Learning, 2007. 999 p.
YOUNG, H. D.; FREEDMAN, R. A. Física IV: Ótica e Física Moderna.
Tradução Adir Moysés Luiz. 10. ed. São Paulo: Pearson Addilson Wesley,
2004. 426 p.
Baixar