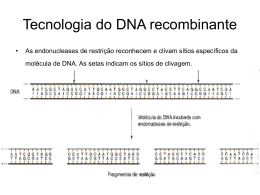
Mutações INSTABILIDADE DO GENOMA HUMANO REPARO DO DNA Genética Humana Profa. Dra. Ana Elizabete Silva SOBREVIVÊNCIA E REPRODUÇÃO ESTABILIDADE GENÉTICA REPLICAÇÃO PRECISA DO DNA REPARO DO DNA Falhas Aberrações cromossômicas: mudança no genoma Mutações no DNA: doenças genéticas (câncer) MUTAÇÃO: qualquer mudança na sequência de nucleotídeos ou arranjo do DNA Células germinativas: Mutação herdável Células somáticas: Mutação somática •Mutações genômicas: numéricas (aneuploidias) •Mutações cromossômicas: estruturais •Mutações gênicas: mutação de ponto MUTAÇÕES – LINHAGEM GERMINATIVA OVOCITOGÊNESE: ovócitos formados até 5o mês: número de divisões celulares do zigoto até oócito fertilizado constante: 24-31 divisões → erro de não-disjunção com aumento da idade (aneuploidias) ESPERMATOGÊNESE: 30-31 divisões celulares até puberdade + 5 div. p/espermatogênese (23 ciclos/ano): centenas de divisões celulares dependendo da idade: 30+5[23x(25-13)]= 310 divisões •divisões celulares contínuas → risco maior de erros de replicação do DNA → 1/10 espermatozóide → mutação deletéria nova Exemplos: Neurofibromatose Acondroplasia Hemofilia B (avô materno: fonte da mutação nova) BASE MOLECULAR DAS MUTAÇÕES GÊNICAS Mutações espontâneas naturais Mutações induzidas Agente mutagênico MUTAÇÃO ESPONTÂNEA ausência de um tratamento com mutágeno: fonte de variação genética Frequência baixa: uma célula em 105 a 108 Mecanismos: erros na replicação do DNA lesões espontâneas: depurinação, desaminação e danos oxidativos (radicais superóxido-O2; peróxido de hidrogênio- H2O2; radicais hidroxilaOH) elementos genéticos de transposição LESÕES ESPONTÂNEAS: ERROS DE REPLICAÇÃO DO DNA •PERDA DE BASES: Depurinação: 5.000 bases púricas (A e G)/dia/célula Desaminação: 100 bases C >U/dia/célula •REPLICAÇÃO: DNA pol: 1 base errada na cadeia filha/10 milhões pb •REVISÃO DE PROVA “PROOFREADING”: corrige 99,9% dos erros de replicação •TAXA DE MUTAÇÃO (erros de replicação): muito baixa 10-10 por pb/divisão •GENOMA DIPLÓIDE HUMANO: ~ 6x109 pb do DNA → menos 1 mutação de pb/divisão PREVENÇÃO DE ERROS Alguns sistemas enzimáticos neutralizam compostos potencialmente danosos, antes que reajam com o DNA Exemplos: desintoxicação de radicais superóxido produzidos durante os danos oxidativos a- Enzima superóxido dismutase catalisa a conversão dos radicais superóxido peróxido de hidrogênio b- Enzima catalase converte peróxido de hidrogênio em água MUTAÇÕES INDUZIDAS Agentes químicos: Agentes físicos: •Análogos de bases: 5-BrdU •Radiação UV •Alcilantes: EMS •Radiação ionizante: raios X, gama •Intercalantes: acridina orange Agentes biológicos: •Vírus mutação insercional RADIAÇÃO ULTRAVIOLETA Absorção da luz UV Estado excitado de uma base púrica ou pirimídica Ligação covalente entre duas pirimidinas adjacentes Anel ciclobutano: dímeros de timina (TT) mais estável dímeros menos estáveis: CT, CC, TU e UU TIPOS DE MUTAÇÕES: DOENÇAS GENÉTICAS HUMANAS 1. Substituição de Bases: Mutação de Ponto Mutação de sentido trocado (missense) Mutação sem sentido (nonsense) Mutação de processamento de RNA:destroem sítios consenso de corte; cap, poliadenilação, criam sítios crípticos: códons finalizadores e mudança de matriz de leitura (frameshift) Mutações reguladoras: afetam a ligação de fator de transcrição e controle transcricional TIPOS DE MUTAÇÕES: DOENÇAS GENÉTICAS HUMANAS 2. Deleções/Inserções: Adição/deleção: pouco no. bases Deleções maiores: Distrofina Inserção L1 ou Alu: hemofilia A (L1); neurofibromatose (Alu) Deleção/Duplicação (recombinação) Crossing-over desigual Expansão trinucleotídeos repetidos Repetição CAG: D. Huntington Repetição CGG: S. do X frágil POLIMORFISMO X MUTAÇÃO POLIMORFISMO: variações no DNA •ocorrência de dois ou mais alelos relativamente comuns para um único lócus. •Variação do DNA na qual cada sequência possível está presente em pelo menos 1% da população. •Ocorre comumente e está associado a um fenótipo normal. Polimorfismo em sequência codificadora (5% DNA): variante protéica → fenótipos distintos Polimorfismos de proteínas: •Grupos sanguíneos ABO e RH •Sistema de alfa1-antitripsina (1-AT): doença pulmonar, vários alelos (frequência de 10-75%) •Proteínas de destoxificação de xenobióticos: CYP, GST, NAT •Proteínas de reparo do DNA: XPD, XRCC1, XRCC2, XRCC3 Polimorfismo em sequência não-codificadora (95% DNA): intergênica ou dentro de íntrons → sem consequência para o funcionamento do gene sem alterar proteínas ESTIMATIVA DE BASES POLIMÓRFICAS: 1:1000 pb TIPOS DE POLIMORFISMOS GENÉTICOS POLIMORFISMOS GENÉTICOS RFLP SNP SSLP Minissatélite Microssatélite VNTR STR •Utilização como marcador genético: análise de ligação •Diagnóstico pré-natal de doenças genéticas •Detecção de portador heterozigoto •Avaliação de pessoas com risco de predisposição a doenças complexas: câncer, doenças coronarianas e diabete •Teste de paternidade e aplicações forenses •Tipagem tissular para transplante de órgão •Alelos 1 e 2: diferentes polimorfismos alteram sítios de restrição Alelo 1: sítio R (cortado pela enzima restrição) Alelo 2: nucleotídeo X perda R (sem corte) •Amplificação (PCR): primers que flanqueiam o sítio de restrição Digestão do produto de PCR com enzima R •Eletroforese: gel de agarose RFLP: Polimorfismos de Comprimento de Fragmentos de Restrição POLIMORFISMO DE COMPRIMENTO DE SEQUÊNCIA SIMPLES (SSLP): - arranjos de sequências repetidas com variação no comprimento devido diferentes números de unidades repetidas (multi-alélicos). MINISSATÉLITES (VNTR): Número Variável de Repetições em Tandem -unidades repetidas de DNA com 10-100 pb (30pb), MICROSSATÉLITES (STR): Repetições em Tandem Curtas • unidades repetidas de 2-4 pb MINISSATÉLITES (VNTR): unidades repetidas de DNA com 10-100 pb (30pb), resultam de inserção em tandem. Encontrados preferencialmente nas regiões teloméricas (geralmente não transcritas) → sítio p/ recombinação homóloga. Utilizados como marcadores polimórficos: datiloscopia de DNA (DNA fingerprinting) http://www.fathom.com/course/21701758/session1.html Formação de unidades repetitivas http://www.usask.ca/biology/rank/316/genomics/genomics.htm Vítima de abuso sexual: suspeitos 1 e 2 comparados com DNA do esperma encontrado na vítima Família com filhos biológicos e adotivos: Teste de Paternidade Filha D2: o pai é de casamento anterior Filho S2: adotado http://www.scq.ubc.ca/?p=250 MICROSSATÉLITES (STR): •unidades repetidas de 2-4 pb: CACA...CA; CAACAA...CAA; AAATAAAT...AAAT. • Existem 6,5x105 microssatélites no genoma humano. •Lócus de microssatélite é multi-alélico: cada pessoa deve ser heterozigota em mais de 70% •Espalhados por todo o genoma Utilizados como marcadores polimórficos: datiloscopia de DNA http://www.usask.ca/biology/rank/316/genomics/genomics.htm Diagnóstico de Doença Genética Doença Ligada ao X (recombinação=0) POLIMORFISMO DE NUCLEOTÍDEO ÚNICO – SNP: •Mudanças de um único nucleotídeo distribuídas por todo o genoma. •Aproximadamente 1:1350 pb no genoma •Cerca de 1.42 milhões de SNPs no genoma •Polimorfismos com dois alelos •Aplicações: marcadores de mapeamento genético; antropologia molecular (evolução) e comparação entre diferentes populações (epidemiologia molecular) •Detecção: Chips de DNA (hibridização de nucleotídeos) REPARO DO DNA Reparar danos no DNA surgidos espontaneamente ou induzidos por mutágenos • Reparo de pareamento errôneo • Reparo direto • Reparo de excisão de bases •Reparo de excisão de nucleotídeos • Reparo de quebras de fita dupla no DNA LESÕES RETARDAM PROGRESSÃO DO CICLO CELULAR Reparo do DNA Bloqueio do Danos DNA ciclo celular (Checkpoint) Proteína ATM Apoptose Identifica lesão no DNA Sinal p/ p53 → Ativar Genes Reparo DNA REPARO DE PAREAMENTO ERRÔNEO MISMATCH REPAIR - MMR • reparo pós-replicação: bases incorporadas erroneamente no DNA durante a replicação (DNA pol: 1 base errada na cadeia filha/10 milhões pb) REVISÃO DE PROVA “PROOFREADING”: corrige 99,9% dos erros de replicação • eliminação de bases mal pareadas e alças de deleção/inserção • atua na cadeia recém-sintetizada •genes MSH, MLH e PMS Deslizamentos da DNA polimerase: regiões de microssatélite Câncer de cólon não-poliposo familial: instabilidade de microssatélites -70 a 85% de risco - mutações em MLH1 e MLH2 são mais comuns REPARO DE PAREAMENTO ERRÔNEO 5’ Base errada 3’ Cadeia filha G A Cadeia parental MSH3 MSH6 Reconhecimento do MSH2 MLH2 dano e corte na cadeia A PMS2 filha Reconhecimento do dano e corte na cadeia filha Excisão de fragmento G de 100 a 1000pb contendo a base A incorreta G DNA pol / A T A DNA ligase Síntese de DNA e ligação da fita reparada Fita reparada REPARO DIRETO • reversão ativa da lesão → sem retirar a base danificada • O6-metilguanina → transição G:C para A:T (produzida endogenamente ou mutagênicos químicos) •Enzima MGMT (metil guanina metiltransferase) → remove o grupo metil • alto custo energético → uma molécula da enzima é inativada para cada lesão corrigida REPARO DIRETO Agente alquilante O6-Metilguanina CH3 G G C C Reparo Direto Reconhecimento da base alterada e transferência do grupo metil para resíduo de cisteína da MGMT DNA restaurado e enzima inativa MGMT CH3 G C G C CH3 MGMT REPAROS DE EXCISÃO ETAPAS COMUNS etapa 1 = incisão (endonuclease) e excisão (exonuclease) etapa 2 = ressíntese do DNA (DNA polimerases β, δ, ε) etapa 3 = ligação das extremidades (DNA ligases) REPARO POR EXCISÃO DE BASES BER • reparo de danos causados por agentes endógenos (hidrólises, oxigênio reativo) que modificam a estrutura das bases • reparo de danos induzidos por radiação ionizante e agentes alcilantes • realizado pelas DNA glicosilases quebram ligações base-açúcar liberando as bases e gerando sítios apurínicos ou apirimidínicos (sítios AP) reparado por endonucleases específica •cada DNA glicosilase reconhece uma base alterada no DNA e catalisa sua remoção por hidrólise •Etapas: remoção da base danificada (DNA glicosilase) sítio AP incisão do sítio AP (endonuclease) excisão e remoção gerando lacuna síntese DNA (DNA polimerase) ligação da cadeia (DNA ligase) REPARO POR EXCISÃO DE BASES BER Quebra de cadeia simples Base danificada * DNA glicosilase APE1 DNA pol XRCC1 XRCC1 Reconhecimento e formação do sítio AP Reconhecimento e incisão 5’ do sítio AP Excisão da porção açúcar-P e síntese de DNA DNA Ligação da ligase III cadeia Short-patch 1 nucleotídeo XRCC1 PARP Reconhecimen to da quebra PNK DNA pol PCNA DNA ligase I Incisão 5’ FEN1 Síntese DNA e excisão Ligação da cadeia Long-patch 2-13 nucleotídeos REPARO POR EXCISÃO DE NUCLEOTÍDEOS NER • remoção de adutos no DNA distorção da dupla hélice • dímeros de pirimidina, HAP, cisplatina • fatores ambientais • principal mecanismo de reparo • deficiência: doenças genéticas •realizado pelo complexo multienzimático XPAXPG • remove 24-32 nucleotídeos Xeroderma Pigmentoso -Mutações em XPA-XPG - 1000 a 4000X o risco de câncer de pele exposição solar ou irradiação UV Reparo por excisão de nucleotídeos (NER) TFIIH: complexo da RNA pol II com atividade de helicase XPG: endonuclease que corta a fita em 3’ ERCC1-XPF: endonuclease que corta a fita em 5’ DNA pol, PCNA, RPA e RFC: síntese da fita nova Ligase REPARO DE QUEBRAS NO DNA FITA DUPLA • DSB (quebra fita dupla): lesão espontânea induzida por radicais de O2 livres, replicação do DNA e agentes genotóxicos como a radiação ionizante • causa de aberrações cromossômicas • Reparo homólogo (RH) pareamento com o cromossomo homólogo intacto • Reparo de ligação das extremidades nãohomologa (NHEJ) religação direta das extremidades quebradas Cromátides irmãs REPARO HOMÓLOGO Reparo DNA com fidelidade Radiação ionizante Quebra de cadeia dupla Rad50 MRE11 NBS1 DNA ligase DNA pol Síntese de DNA e ligação das cadeias Degradação 5’-3’ Rad52 BRCA2 Invasão da cadeia BRCA1 Rad54 Rad51 Uma das fitas vermelhas apresenta uma quebra DNA polimerase desloca a fita d. Reparo posterior à duplicação por recombinação semelhante à conversão gênica ligação dos fragmentos heteroduplex duplicação posterior do DNA resulta em 3 alelos e 1 alelo Resultado final: um alelo verde foi convertido em um vermelho LIGAÇÃO DAS EXTREMIDADES NÃO-HOMÓLOGAS Agente danificante DNA fita dupla Quebra de cadeia dupla DNA PKc1 KU80 KU70 KU80 KU70 DNA ligase IV XRCC4 Processamento das extremidades Ligação das extremidades DNA reparado com baixa fidelidade http://www.sbbq.org.br/revista/mtdidaticos/Env.pdf Xeroderma Pigmentoso Síndrome de Cockaine Tricotiodistrofia Anemia de Fanconi Ataxia telangiectasia SÍNDROMES DE INSTABILIDADE CROMOSSÔMICA Síndrome de Bloom defeitos nos mecanismos de reparo e replicação do DNA freqüência aberrações cromossômicas incidência câncer XERODERMA PIGMENTOSO (XP) AR Clínica manifestações cutâneas (1,5 anos) anomalias oculares (4 anos) anomalias neurológicas (6 meses) Xeroderma (pele seca) Incidência:1/250.000 (USA); 1/40.000 (Japão) Células XP: sensíveis a radiação UV indivíduos incapazes reparar dímeros TT: risco ↑ câncer de pele (2000x) XP: defeito no reparo de excisão de nucleotídeos (NER) grupos de complementação: XPA-XPG diferenças clínicas defeitos enzimáticos incapacidade de excisar danos induzidos pela luz UV e mutagênicos químicos diagnóstico exposição a luz solar tratamento proteção da pele da luz solar chapéus óculos que absorvem luz UV bloqueadores solares roupas protetoras acompanhamento periódico por dermatologista ANEMIA DE FANCONI (FA) AR Clínica anemia alterações da pigmentação da pele (64%) baixa estatura (62%) malformações do rádio (50%) anomalias oculares (41%), renais (34%), microcefalia (37%), deficiência mental (25%) 90% anemia aplástica incidência: 1/22.000 a 1/476.000 indivíduos 8 grupos de complementação: FANCA a FANCG (heterogeneidade genética) células FA: sensíveis à agentes químicos que causam ligações cruzadas entre as fitas de DNA: -mitomicina C (MMC), diepoxibutano (DEB), mostarda nitrogenada (NM), cisplatina , ... freqüência de neoplasias: leucemias e tumores hepáticos quebras cromossômicas espontâneas, figuras tri, quadri e multiradiais, figuras radiais endorreduplicação SÍNDROME DE BLOOM (SB) AR Clínica peso ao nascimento retardo de crescimento pré e pós-natal (145 cm H; 130 cm M) telangiectasias e fotossensibilidade (borboleta) cabeça alongada, microcefalia, inteligência normal imunodeficiência: infecções (respiratórias e gastrointestinais) Incidência: 1/58.000 judeus asquenazim figuras quadrirradiais mutações no gene BLM 15q26.1 DNA helicase ( RecQ) papel na replicação e reparo do DNA Risco maior de câncer: carcinomas, leucemias e linfomas ATAXIA TELANGIECTASIA (AT) SÍNDROME DE LOUIS-BAR AR Clínica -ataxia cerebelar (12-14 meses) disfunção neuromotora -telangiectasia olhos e pele (3 e 5 anos) retardo de crescimento (70%) incidência neoplasias (linfoma e leucemia linfóide) e imunodeficiências incidência: 1/40.000 risco câncer de mama heterozigotos AT células AT sensíveis a radiação ionizante e agentes radiomiméticos (N-acetoxi-N-2-acetil-2-aminofluoreno - 4-NQO) linfócitos AT rearranjos espontâneos dos cromossomos 7 e 14 4 grupos de complementação: A, C, D e E gene ATM (11q22-23) mutações gene ATM diversas vias controle transdução de sinal checkpoint ciclo celular resposta celular ao dano no DNA induzido por radiação gene ATM interage p53 na checagem G1-S e mutações no gene ATM abolem mecanismo de reparo pré-síntese de DNA defeito DNA mecanismo de reparo do DNA ou defeito na replicação
Download